[level-membership-for-pulmolory-and-respiratory-category]
Chapter 57 Pulmonary Embolism
Epidemiology, Risk Factors, and Pathogenesis
Risk Factors
Inherited Thrombophilia
Inherited thrombophilias may result from qualitative or quantitative defects in coagulation factor inhibitors (antithrombin, protein C, protein S), increased levels or function of coagulation factors (activated protein C resistance, factor V Leiden mutation, prothrombin gene mutation, elevated factor VIII levels), hyperhomocysteinemia, defects in fibrolysis, or altered platelet function. Epidemiologic features of the common inherited thrombophilias are shown in Table 57-1.

Acquired Risk Factors
Acquired risk factors for VTE are far more prevalent than inherited thrombophilias. Box 57-1 lists common risk factors for acquired VTE.
Box 57-1
Acquired Risk Factors for Venous Thromboembolism
Clinical Features
The clinical consequences of PE range from incidental and clinically unimportant to circulatory collapse and sudden death. Equally challenging, the clinical signs and symptoms related to PE are diverse and nonspecific. Therefore, clinicians use a combination of history and examination findings in association with clinical prediction tools to determine appropriate diagnostic tests and the need for therapeutic interventions. Considerations in the differential diagnosis for acute PE are listed in Box 57-2.
Box 57-2
Differential Diagnosis for Acute Pulmonary Embolism
Medical History
Patients with PE often have one or more identifiable risk factors for the development of VTE at the time of clinical presentation (see earlier, under “Risk Factors”). Details should be sought regarding the patient’s personal and family history of prior VTE, coexisting medical conditions, functional status, travel history, and current medications. Major risk factors for VTE include surgery or trauma within the preceding 30 days, prolonged immobility, advanced age, malignancy, previous VTE, known thrombophilia, recent myocardial infarction or stroke (cerebrovascular accident), or indwelling venous catheter. Moderate risk factors include obesity, use of estrogen or hormone replacement therapy, and family history of VTE. Scoring systems such as the Wells score and the Geneva score have been devised to help assess the patient’s probability of being diagnosed with a PE (see under “Diagnosis” later in the chapter).
Symptoms and Signs
Acute PE embolism may manifest with a wide spectrum of signs and symptoms. The most common symptom in angiographically confirmed acute PE is dyspnea (Table 57-2). Less frequently, patients with acute PE present with hemoptysis, wheezing, or chest pain. Frequent findings on physical examination include tachypnea (respiratory rate greater than 20 breaths/minute), tachycardia (heart rate greater than 100 beats/minute), and crackles on lung auscultation (see Table 57-2). The presence of syncope, cyanosis, jugular venous distention, pulsatile liver, or parasternal heave or auscultation of an accentuated pulmonic component of the second heart sound, right-sided third heart sound, and/or an audible systolic murmur at the left sternal border may reflect significant right ventricular dysfunction.
Table 57-2 Frequency of Signs and Symptoms in Acute Pulmonary Embolism
| Manifestation | Frequency (%) |
|---|---|
| Symptoms | |
| Dyspnea | 73 |
| Pleuritic chest pain | 66 |
| Cough | 37 |
| Leg swelling | 33 |
| Hemoptysis | 13 |
| Wheezing | 9 |
| Chest pain | 4 |
| Signs | |
| Respiratory rate ≥20 breaths/min | 70 |
| Crackles | 51 |
| Heart rate ≥100 beats/min | 30 |
| Third or fourth heart sound | 26 |
| Loud pulmonary component of second heart sound | 23 |
| Temperature >38.5° C | 7 |
| Pleural rub | 3 |
Data from Stein PD, Terrin ML, Hales CA, et al: Clinical, laboratory, roentgenographic, and electrocardiographic findings in patients with acute pulmonary embolism and no pre-existing cardiac or pulmonary disease, Chest 100:598–603, 1991.
Diagnosis
Clinical Assessment
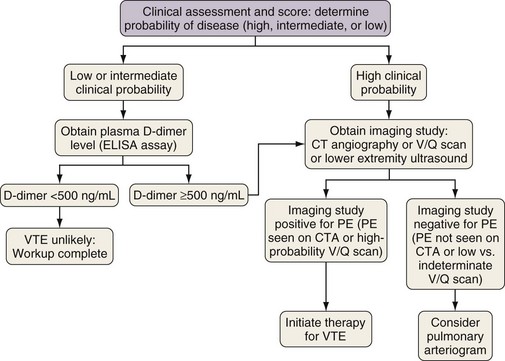
Typical clinical features of PE have already been described; here, it is important to stress that clinical judgment is an essential initial step in the evaluation of thromboembolic disease and figures prominently in diagnostic algorithms. The importance of the clinician’s assessment of the probability of PE initially was highlighted in the 1990 landmark Prospective Investigation of Pulmonary Embolism Diagnosis (PIOPED) study. Physicians in this study were asked to record their clinical impression (high, intermediate, or low probability) as to the likelihood of PE in patients they were treating before learning the results of the radiographic study (ventilation-perfusion scan or pulmonary arteriogram). The clinical impression was based on an agreed-on set of data but without standardized diagnostic algorithms. One very important finding of the PIOPED study was that diagnosis or exclusion of PE was possible only with clear and concordant clinical and radiographic findings. If the clinical impression did not match the findings on imaging (ventilation-perfusion scan in this study), pulmonary thromboembolic disease could not be confirmed or ruled out by that imaging study, and further investigation was necessary. Since the publication of PIOPED, numerous attempts have been made to standardize the definition of “clinical impression.” This has resulted in a variety of scoring systems, assigning points to historical, physical, and laboratory features of an individual patient. Patients receive scores on the basis of inherent risk factors and presenting signs that are then used to predict likelihood of disease. Currently, the two most commonly used scoring systems are the Wells criteria and the Geneva score (Table 57-3). These two scoring systems and subsequent modifications have been validated in a number of studies. By themselves, scoring systems lack adequate sensitivity or specificity to diagnose or exclude disease. Their true usefulness comes in conjunction with other laboratory or imaging studies, allowing the assessment of disease risk.
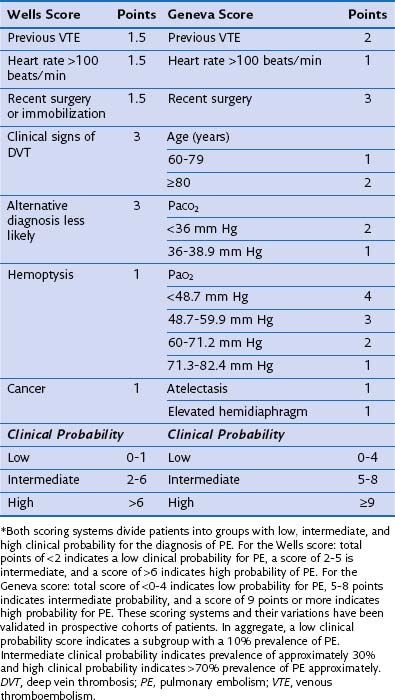
Table 57-3 Wells and Geneva Scoring Systems Used in Risk Assessment for the Diagnosis of Pulmonary Embolism (PE)*
Electrocardiograms and Chest Radiographs
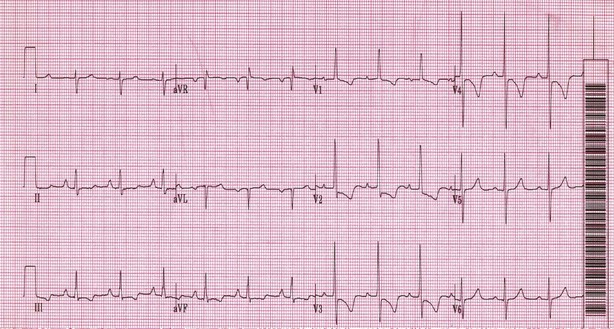
Electrocardiograms (ECG) and chest radiographs frequently are used in the evaluation of patients initially seen with dyspnea or chest pain. Although these studies are neither adequately sensitive nor specific to diagnose or exclude PE, they can suggest the diagnosis. ECG findings such as T wave inversions in the anterior leads, in particular V1 to V4, are typical of right ventricular strain and should raise suspicion for pulmonary thromboembolic disease (Figure 57-1). Other typical ECG changes include a deep S wave in lead I, a Q wave in lead III, and T wave inversions in lead III. Rhythm and conduction abnormalities such as new-onset atrial fibrillation or right bundle branch block occasionally are noted in association with acute PE.
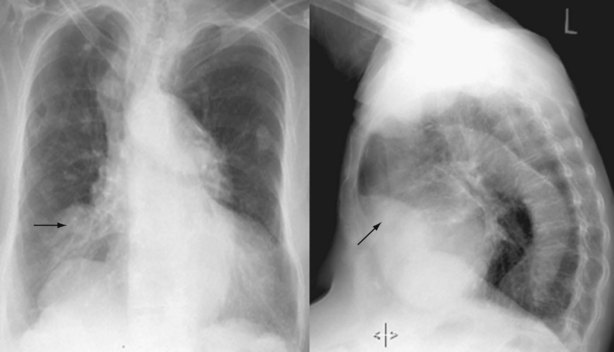
In the evaluation for PE, chest radiographs predominantly serve to exclude other potential explanations for the patient’s symptoms (e.g., a lobar infiltrate consistent with pneumonia). Occasionally, the chest radiograph will demonstrate changes suggestive of PE, such as focal oligemia (Westermark sign), a peripheral wedge-shaped density that indicates infarct (Hampton hump) (Figure 57-2), or an enlarged right descending pulmonary artery (Palla sign).
Ventilation-Perfusion Lung Scan
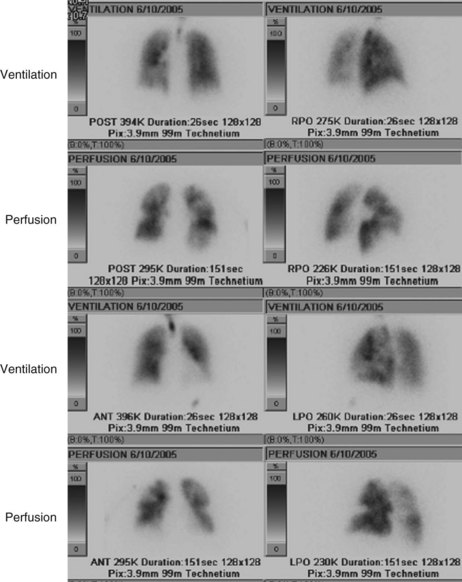
For many years, the ventilation-perfusion lung scan was considered the imaging study of choice to evaluate for PE (Figure 57-3). Recently, computed tomography angiography (CTA) has replaced the ventilation-perfusion lung scan as the predominant diagnostic test. However, the ventilation-perfusion scan maintains an important place in the evaluation of patients for thromboembolic disease who have contraindications to CTA such as renal insufficiency or contrast allergy. As mentioned previously, the PIOPED study correlated the clinical probability of a PE (high, intermediate, or low probability as assessed by history and clinical findings) with the interpretation of the ventilation-perfusion scan (high, intermediate, or low probability or normal perfusion). With concordance of the clinical assessment and the interpretation of the V/Q scan in the high or low probability range, PE can be diagnosed or excluded with reasonable certainty. When the clinical assessment and the interpretation of the ventilation-perfusion scan are discordant (i.e., high clinical probability but low-probability ventilation-perfusion scan, or vice versa), the possibility of PE cannot be adequately assessed, and other studies are required. A normal-appearing ventilation-perfusion scan (with a normal perfusion component) essentially excludes the diagnosis of PE.
Echocardiogram
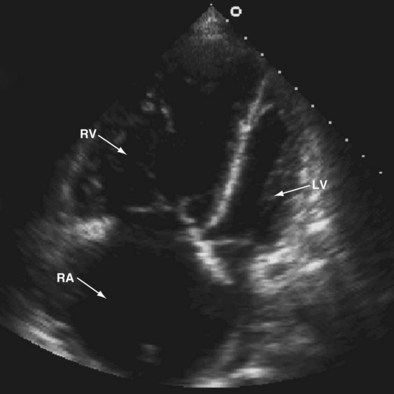
Transthoracic and transesophageal echocardiography have limited use in the diagnosis of PE. The sensitivity and specificity of these tests are inadequate for diagnosis, because the offending emboli are rarely proximal enough to be visualized. Echocardiography can assist in acute care management decisions for those patients too unstable to be moved from a critical care setting for more definitive imaging studies. Although it is rare to visualize a thrombus within the pulmonary arteries by echocardiogram, changes in right ventricular size and function and increases in tricuspid regurgitation imply acute right heart strain. In the appropriate clinical scenario, these changes in the right ventricle can suggest the diagnosis of acute PE (Figure 57-4).
Computed Tomography Angiography
CT pulmonary angiography (i.e., CTA) has become a favored study for the evaluation of PE over the past decade (Figure 57-5, B). CTA provides a number of potential advantages over other imaging modalities in the diagnosis of PE, including (1) direct visualization of the embolus, (2) the ability to assess for other potential causes for the patient’s complaints such as pneumonia, and (3) imaging algorithms that scan through the pelvis and lower extremities, as well as the chest, allowing simultaneous evaluation for PE and for DVT. The ability to evaluate for other thoracic disease is of no small consequence, because up to two thirds of patients initially suspected to have PE eventually receive another diagnosis for their symptoms. Many of these subsequently diagnosed disorders (i.e., pneumonia, thoracic aorta dissection, pneumothorax) are associated with lung changes that can be visualized on CT scan. The interobserver agreement for CT is better than that for ventilation-perfusion scan. The initial hardware used for assessment of PE was single-detector scanners that provided high specificity for the diagnosis of PE (greater than 90%), but their sensitivity was unacceptable (approximately 72%) for the exclusion of this potentially life-threatening diagnosis. Multidetector (40-, 64-, and 96-slice) scanners in current use, however, have significantly improved the sensitivity and specificity of CTA for the diagnosis of PE. The very high spatial resolution of these studies allows rapid evaluation of pulmonary vessels down to the sixth-order branches during a single breath hold, with consequent increased detection rate for segmental and subsegmental PEs.
PIOPED II also attempted to associate clinical probability with imaging studies to assess positive and negative predictive values. The results were reminiscent of the original PIOPED study in that the positive predictive value of the CT studies was 96% with a concordantly high or low clinical probability, 92% with an intermediate clinical probability, and nondiagnostic with a discordant clinical probability. As discussed earlier, however, a number of large outcome studies demonstrated the efficacy of CTA in the evaluation of PE and the safety of withholding anticoagulation in patients with negative CTA findings. CTA now plays the predominant role in diagnostic algorithms for the evaluation of PE (Figure 57-6).
Pulmonary Arteriography
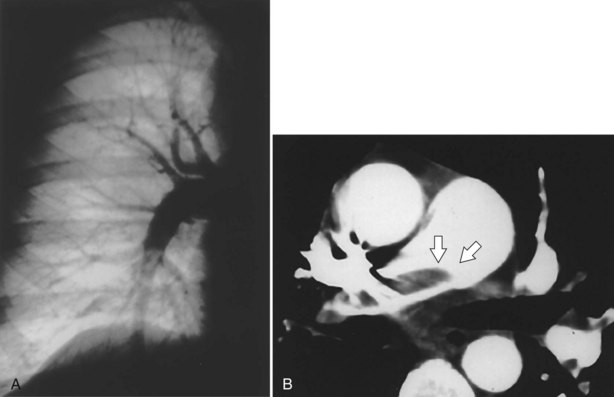
Pulmonary angiography or digital subtraction angiography (DSA) previously had been the “gold standard” modality for the diagnosis of PE (see Figure 57-5, A). Because of its invasive nature, it also is associated with the most inherent risk. Arrhythmias, hypotension, bleeding, and nephrotoxicity from contrast dye are potential complications. The mortality rate associated with pulmonary angiography has been estimated at 0.5%, with major nonfatal adverse events occurring with a frequency of 1%. Angiography also is more expensive than the noninvasive means of evaluating for PE and not always immediately available. As other imaging modalities, such as CTA, have gained popularity in the assessment of PE, angiography has become less used, so less experience with its application has been accrued. Approximately 1% of patients with a normal-appearing pulmonary angiogram will be diagnosed with a VTE at 6 months, implying that the result on angiography was falsely negative. Although large, segmental embolic events are readily appreciated, interobserver variability can be significant in evaluating smaller subsegmental emboli. The main role for DSA at present is for the evaluation of patients with chronic thromboembolic disease being considered for pulmonary endarterectomy (PEA) to assess surgical resectability.
Treatment
Treatment of Acute Pulmonary Embolism
Prophylaxis Against Recurrent Venous Thromboembolism (Secondary Prevention)
Clear evidence has shown that “early” discontinuation of anticoagulation after an acute VTE results in a substantially increased risk for symptomatic extension of the thrombus, embolization, or recurrence of clot. The difficulty, however, is defining early. Most studies examining optimal duration of anticoagulation have found that the longer a person receives anticoagulation after a DVT or PE, the less likely they are to have a repeat VTE. Furthermore, when anticoagulation is discontinued, the risk of VTE increases substantially and is significantly above that in persons without a history of VTE. This elevation in risk is reflected in the clinical scoring systems (Wells score, Geneva score) discussed earlier. Of note, however, chronic anticoagulation presents its own inherent risks, cost, and requirements for lifestyle modification. The challenge then becomes balancing the inherent risk of anticoagulation with the individual patient’s risk of recurrent disease. The ACCP has published consensus statement recommendations regarding the duration of chronic anticoagulation to prevent recurrent VTE by considering patient risk factors and presentation (Table 57-4). These recommendations by necessity are directed at broad categories of patients. When applying these standards, therefore, clinicians must consider the individual patient’s risk of adverse outcomes with anticoagulation. As discussed earlier, however, the recent availability of the oral Xa inhibitors may change the risk-benefit ratio, resulting in longer periods of anticoagulation after VTE. The EINSTEIN investigators demonstrated that continuation of the oral Xa inhibitor rivaroxaban for an additional 6 to 12 months after completion of 6 to 12 months of anticoagulation resulted in significantly fewer episodes of recurrent VTE (1.3% versus 7.1%). Only a slight increase in nonfatal major bleeding (0.7%) was observed in the treatment cohort compared with the placebo cohort (0%).
Table 57-4 Recommendations for Duration of Anticoagulation in Patients Diagnosed With Venous Thromboembolism (VTE)
| Indication for Anticoagulation | Duration of Therapy |
|---|---|
| First VTE with reversible or transient risk factor | Minimum of 3 months |
| First episode of idiopathic VTE | Minimum of 6-12 months; consider use for indefinite period |
| VTE associated with malignancy | LMWH for the first 3-6 months; then indefinitely or until the malignancy resolves |
| First episode of VTE associated with hypercoagulable state | 12 months; suggest indefinitely |
| Two or more documented episodes of VTE | Indefinite |
LMWH, low-molecular-weight heparin.
Treatment of Chronic Thromboembolic Disease
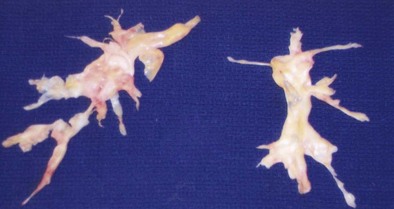
The treatment for CTEPH differs significantly from that for acute PE. Although these patients require anticoagulation to prevent further embolic events, the endothelialized clot is not accessible to these medications. Therapy, therefore, revolves around either removing the thrombus surgically or treating the elevation in pulmonary artery pressures medically. Surgical resection, termed pulmonary endarterectomy (PEA), is the treatment of choice in eligible patients (Figure 57-7). It is performed by dissecting away the endothelialized thrombus through careful separation of the thrombus from the pulmonary artery wall. This procedure is associated with significant operative and postoperative risk (as reflected by reported 5% to 10% mortality rates) and should be performed only in experienced centers. When PEA is successful, outcomes include significant improvement in pulmonary artery pressure, right-sided heart function, cardiac output, and functional class. A substantial number of patients (10% to 50%) with CTEPH referred for PEA, however, are deemed to be ineligible because of inaccessible distal thrombus or other serious comorbid conditions. Furthermore, persistent PH after successful PEA is frequent, with substantial small-vessel occlusion or arteriopathy. For these reasons, medical therapy for CTEPH has been applied. These treatments include nonspecific therapies such as administration of diuretics to improve fluid status, long-term oxygen therapy for hypoxemia, and digoxin to improve right ventricular contractility. More recently, however, novel therapies more specific for the treatment of PAH and approved for the treatment of idiopathic pulmonary artery hypertension (IPAH) have garnered attention as potentially useful in the medical management of CTEPH. Such therapies include use of the prostacyclin analogues (epoprostenol, treprostinil, and iloprost), endothelin receptor antagonists (bosentan), and the phosphodiesterase-5 (PDE-5) inhibitors (sildenafil). Evidence for the success of these medications, however, is limited to case series, retrospective studies, and prospective cohort studies. The only randomized controlled clinical trial to date that has included patients with CTEPH, as well as patients with other causes of PAH, is the Aerosolized Iloprost Randomized (AIR) study. Iloprost is an inhaled prostacyclin analogue approved for the treatment of PAH. This study did not demonstrate significant beneficial effects of inhaled iloprost in the CTEPH population, however.
Prevention of Pulmonary Embolism
Inferior Vena Cava Filters
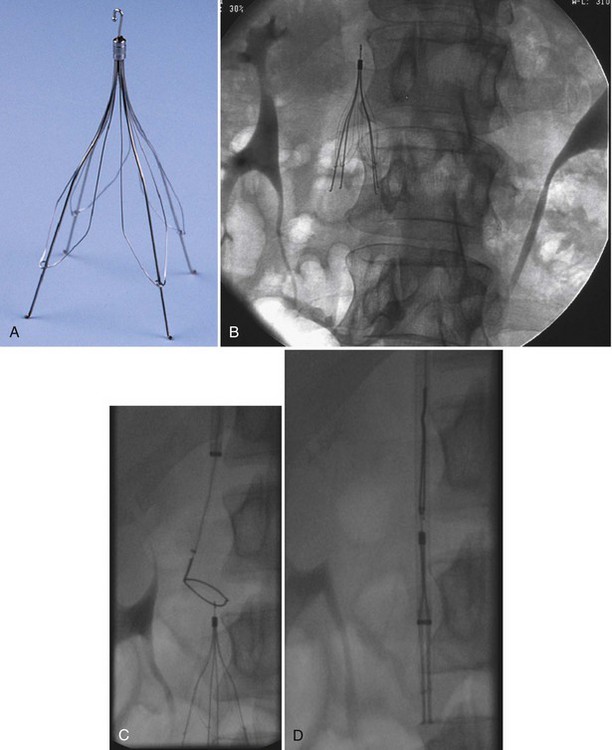
No randomized trials have been conducted to examine the incidence of PE in patients who received an IVC filter but did not receive anticoagulation. Retrievable filters are a potential option in patients who have only a transiently increased risk for VTE (Figure 57-8). The filters should be removed before endothelization of the struts occurs, which usually is within 7 to 21 days of placement. An increasing number of case reports and case series, however, have demonstrated the ability to remove retrievable filters months after their placement. The risk of complications rises with delayed removal. Randomized controlled trials demonstrating the efficacy of retrievable filters in terms of outcomes have yet to be performed.
Nonthrombotic Pulmonary Emboli
Although most pulmonary emboli arise from DVT, other clinically significant forms of emboli may have an impact on the lung vasculature, as summarized in Box 57-3.
Controversies and Pitfalls
Bauer KA. The thrombophilias: well-defined risk factors with uncertain therapeutic implications. Ann Intern Med. 2001;135:367–373.
Buller HR, Agnelli G, Hull RD, et al. Antithrombotic therapy for venous thromboembolic disease: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest. 2004;126:401–428.
Cohn D, Vansenne F, de Borgie C, Middeldorp S. Thrombophilia testing for prevention of recurrent venous thromboembolism. Cochrane Database Syst Rev. 1, 2009. CD007069
Decousus H, Leizorovicz A, Parent F, et al. A clinical trial of vena caval filters in the prevention of pulmonary embolism in patients with proximal deep-vein thrombosis. Prevention du Risque d’Embolie Pulmonaire par Interruption Cave Study Group. N Engl J Med. 1998;338:409–415.
EINSTEIN InvestigatorsBauersachs R, Berkowitz SD, et al. Oral rivaroxaban for symptomatic venous thromboembolism. N Engl J Med. 2010;363:2499–2510.
Geerts WH, Pineo GF, Heit JA, et al. Prevention of venous thromboembolism: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest. 2004;126:338S–400S.
Goldhaber SZ. Pulmonary embolism. Lancet. 2004;363:1295–1305.
Goldhaber SZ. Thrombolysis in pulmonary embolism: a debatable indication. Thromb Haemost. 2001;86:444–451.
Jerjes-Sanchez C, Ramirez-Rivera A, de Lourdes GM, et al. Streptokinase and heparin versus heparin alone in massive pulmonary embolism: a randomized controlled trial. J Thromb Thrombolysis. 1995;2:227–229.
Konstantinides S, Geibel A, Heusel G, et al. Heparin plus alteplase compared with heparin alone in patients with submassive pulmonary embolism. N Engl J Med. 2002;347:1143–1150.
Park B, Messina L, Dargon P, et al. Recent trends in clinical outcomes and resource utilization for pulmonary embolism in the United States: findings from the nationwide inpatient sample. Chest. 2009;136:983–990.
Pengo V, Lensing AW, Prins MH, et al. Incidence of chronic thromboembolic pulmonary hypertension after pulmonary embolism. N Engl J Med. 2004;350:2257–2264.
PREPIC Study Group. Eight year follow-up of patients with permanent vena cava filters in the prevention of pulmonary embolism: the PREPIC (Prevention du Risque d’Embolie Pulmonaire par Interruption Cave) randomized study. Circulation. 2005;112:416–422.
Stein PD, Chenevert TL, Fowler SE, et al. Gadolinium-enhanced magnetic resonance angiography for pulmonary embolism: a multicenter prospective study (PIOPED III). Ann Intern Med. 2010;152:434–443.
Stein PD, Woodard PK, Weg JG, et al. Diagnostic pathways in acute pulmonary embolism: recommendations of the PIOPED II investigators. Am J Med. 2006;119:1048–1055.
1990 Value of the ventilation/perfusion scan in acute pulmonary embolism. Results of the prospective investigation of pulmonary embolism diagnosis (PIOPED). The PIOPED Investigators. JAMA. 1990;263:2753–2759.
Van Belle A, Buller HR, Huisman MV, et al. Effectiveness of managing suspected pulmonary embolism by use of an algorithm combining clinical probability, D-dimer testing, and computed tomography. JAMA. 2006;295:172–179.
[/level-membership-for-pulmolory-and-respiratory-category][not-level-membership-for-pulmolory-and-respiratory-category]
Chapter 57 Pulmonary Embolism
Epidemiology, Risk Factors, and Pathogenesis
Risk Factors
Inherited Thrombophilia
Inherited thrombophilias may result from qualitative or quantitative defects in coagulation factor inhibitors (antithrombin, protein C, protein S), increased levels or function of coagulation factors (activated protein C resistance, factor V Leiden mutation, prothrombin gene mutation, elevated factor VIII levels), hyperhomocysteinemia, defects in fibrolysis, or altered platelet function. Epidemiologic features of the common inherited thrombophilias are shown in Table 57-1.
Acquired Risk Factors
Acquired risk factors for VTE are far more prevalent than inherited thrombophilias. Box 57-1 lists common risk factors for acquired VTE.
Box 57-1
Acquired Risk Factors for Venous Thromboembolism
Clinical Features
The clinical consequences of PE range from incidental and clinically unimportant to circulatory collapse and sudden death. Equally challenging, the clinical signs and symptoms related to PE are diverse and nonspecific. Therefore, clinicians use a combination of history and examination findings in association with clinical prediction tools to determine appropriate diagnostic tests and the need for therapeutic interventions. Considerations in the differential diagnosis for acute PE are listed in Box 57-2.
Box 57-2
Differential Diagnosis for Acute Pulmonary Embolism
Asthma or exacerbation of chronic obstructive lung disease
Pericarditis/cardiac tamponade
Pulmonary edema/congestive heart failure
[/not-level-membership-for-pulmolory-and-respiratory-category]
