73 Pulmonary Edema
 Definition
Definition Pulmonary Fluid Homeostasis
Pulmonary Fluid Homeostasis
Pulmonary fluid homeostasis is dependent upon the equilibrium between forces that drive fluid into the alveolar space and counterforces responsible for its clearance—primarily lymphatics. One of the main regulatory forces for this fluid balance is the microvascular pressure in the alveolar capillaries, as presented by modification of the Starling’s equation (Figure 73-1, A), which describes the balance between the hydrostatic pressure gradient and the oncotic pressure gradient. While the oncotic or osmolarity gradient is dependent mainly on protein concentrations, the pulmonary capillary hydrostatic pressure is dependent on pulmonary flow and resistance (see Figure 73-1, B). Pulmonary capillary pressure is regulated at the precapillary level by the arteriolar vasomotor tone, which determines the transmission of flow and pressures from the pulmonary artery to the capillary bed. By contrast, venous capillaries lack this protective mechanism, allowing unprotected transmission of elevated left ventricular pressure to the pulmonary capillary bed and excessive fluid accumulation.1

Protective mechanisms against fluid accumulation in the alveolar and interstitial space include both passive elements, such as the tight junctions between the alveolar epithelium, and active reabsorption of fluid from the airspace using Na+ and Cl− channels.2 The primary sites of sodium and chloride reabsorption are the epithelial ion channels located on the apical membrane of alveolar epithelial cells (both type I and II) and the distal airway epithelial cells. Water will follow the osmotic gradient created by the reabsorption of Na+ and Cl−, preventing edema formation. Pulmonary edema will occur when this delicate balance is overwhelmed by one of three pathologic processes: impaired clearance mechanisms, increased hydrostatic pressures resulting in excessive pressure gradients, or increased permeability of the capillary alveolar barrier. When the main cause is related to increased pulmonary venous pressure, pulmonary edema is said to be cardiogenic in origin. In contrast, when other factors such as increased permeability prevail, the term noncardiogenic pulmonary edema is used. The interstitial fluid content in each etiology is different, owing to the underlying pathophysiology. Increased pulmonary venous pressures causing cardiogenic pulmonary edema will yield fluid with low protein content. Increased permeability of microvascular epithelium in noncardiogenic pulmonary edema will result in fluid with relatively high protein content.
 Diagnosis and Assessment
Diagnosis and Assessment
History and Presenting Symptoms
The presenting signs and symptoms of pulmonary edema are dyspnea, tachypnea, and respiratory distress. Alveolar flooding can lead to cough and expectoration of frothy edema fluid. The history should focus on cardiogenic and noncardiogenic mechanisms contributing to pulmonary edema and elicit precipitating factors that might have led to edema formation. Common causes for cardiogenic pulmonary edema include ischemia, exacerbation of systolic or diastolic dysfunction (ischemia, infarct, or myopathic processes), severe valvular disease, or arrhythmias. A history of paroxysmal nocturnal dyspnea or progressive orthopnea usually indicates cardiogenic origin for pulmonary edema. However, silent ischemia may also present as pulmonary edema, with a paucity of clues provided by the history.2 Noncardiogenic pulmonary edema is usually preceded by specific predisposing clinical situations such as pneumonia, sepsis, multiple blood transfusions, or intravenous (IV) illicit drug usage.
Physical Examination
Physical findings on lung examination are quite similar for cardiogenic and noncardiogenic pulmonary edema. The patient is usually tachypneic, pale, and diaphoretic with wet inspiratory rales/crackles heard over both lung fields, and most notably the bases. Patients with cardiogenic causes may present with an S3 “gallop” on cardiac auscultation, indicating elevated left-ventricular diastolic pressures—a sign with high specificity (90%-97%) but low sensitivity (9%-51%).2 Stenotic or regurgitant valvular murmurs on auscultation may indicate a cardiac cause but are not always related to the primary cause of the edema. Peripheral edema, which may be a sign for coexisting right heart failure, is neither sensitive nor specific for a cardiogenic origin of pulmonary edema. Most patients with cardiogenic causes for pulmonary edema will have cold, clammy skin, but some patients with noncardiogenic causes will present with warm skin, indicating decreased peripheral resistance.
Auxiliary Tests
Plain chest radiography has been reported to be more sensitive than clinical examination3 for pulmonary edema, which makes it one of the cornerstones for this diagnosis. The first finding that indicates interstitial edema are “Kerley B” lines. These are 3- to 6-mm-long lines perpendicular to the pleural surface, usually at the bases (Figure 73-2). Another sign of interstitial edema is peribronchial cuffing resulting from edematous thickening of the bronchial wall. Redistribution of blood to the upper fields of the lungs results in upper-lobe blood vessel distension. When fluid eventually leaks to the alveoli, bilateral and diffuse opacities are seen, usually sparing the apices and extreme lung bases, causing a central “butterfly” distribution. As the process progresses, opacities may coalesce to produce a general “white-out” of the lungs.4 Chest radiographs may aid in distinguishing between cardiogenic and noncardiogenic etiologies for pulmonary edema. In one study, it was demonstrated that in 50% of patients with cardiogenic edema there was upper-lobe blood diversion, whereas in patients with increased permeability edema due to acute respiratory distress syndrome (ARDS), only 10% showed this inverted pattern. Normal or “balanced” patterns were more commonly seen in ARDS. A peripheral distribution of edema was absent in patients with cardiogenic edema but was the most common pattern seen in patients with ARDS5 (Figure 73-3). Unfortunately, about one out of five patients admitted for acute decompensated heart failure had no signs of congestion on chest radiograph6—a fact that emphasizes the importance of a holistic, integrative approach to the diagnosis of pulmonary edema.
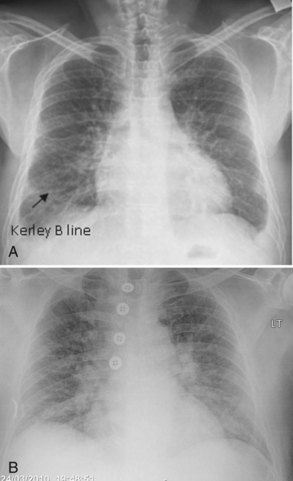
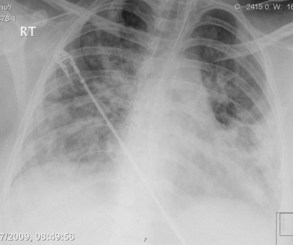
Figure 73-3 Chest x-ray images in ARDS. Diffuse bilateral opacities with involvement of peripheral lung fields.
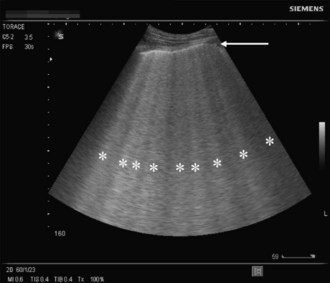
A novel approach utilizes ultrasound as a bedside tool to for the diagnosis of dyspnea and differentiation between pulmonary edema and other major dyspnea-causing diseases such as chronic obstructive pulmonary disease (COPD). Pulmonary edema induces abundant sonographic artifacts caused by interactions of water and air called B-lines or comet tails by some authors (Figure 73-4); these findings are usually not seen in other pulmonary diseases.7 Electrocardiograms are useful in diagnosing active myocardial ischemia or to provide other clues regarding organic cardiac disease leading the pulmonary congestion.
Biomarkers
In recent years, a variety of biomarkers have been used to enhance the diagnostic accuracy of cardiogenic pulmonary edema. Brain natriuretic peptide (BNP) and N-terminal proBNP (NT-proBNP) are both secreted from the ventricles and correlate with the left ventricular (LV) end-diastolic pressure; however, heart failure with preserved LV function usually results in much lower BNP levels than heart failure with impaired LV systolic function. These biomarkers can be used for several indications in the intensive care unit (ICU). Among others, it may aid in differentiating between cardiogenic pulmonary edema and acute lung injury (ALI), monitor volume load in septic patients, and differentiate between septic and cardiogenic shock. Several conflicting reports have addressed the use of BNP/NT-proBNP for the differentiation between ALI and cardiogenic pulmonary edema.8 Different cutoffs were used in the different trials, yielding various ranges of specificities and sensitivities for each diagnosis. It should be emphasized that these conflicting results arise in part from the fact that BNP increases with elevated right ventricular end-diastolic pressures and hypoxia, which are common properties of any severe lung disease. Based on currently available data, low levels of natriuretic peptide (BNP <100 pg/mL or NT-proBNP <250 pg/mL) may be used to exclude elevated cardiac filling pressures in patients presenting with respiratory failure with signs of pulmonary edema, whereas extremely elevated levels of these markers (BNP > 500 pg/mL and NT-proBNP > 1000 pg/mL) in the absence of signs and symptoms of septic shock will support a cardiogenic origin for pulmonary edema. The “gray zone” values between these extremes will necessitate further workup.8 Cardiac troponin (cTn) I or T measurement are highly sensitive for myocardial injury, which can aid in the diagnosis of cardiac origin of pulmonary edema, but in the setting of critical illness, various nonischemic conditions (sepsis, stroke, pulmonary embolism, acute renal failure, etc.) can also induce elevation of cTn and should be excluded before concluding that the cTn elevation is “ischemic.”
Echocardiography
Transthoracic echocardiography (TTE) is used to assist with the diagnosis of myocardial, valvular, and structural pathologies that contribute to pulmonary edema and thus should be performed in any patient presenting with pulmonary edema. While severe valvular stenosis or regurgitation is readily visible on echocardiographic exam, evaluation of cardiac function is more challenging. Decreased myocardial function in patients presenting with pulmonary edema can be due either to past myocardial ischemia/infarction or a current, ongoing ischemic event complicated by pulmonary edema. Furthermore, depressed myocardial function is often seen in other conditions associated with critical illness, such as sepsis. On the other hand, preserved systolic LV function cannot exclude a cardiac origin of pulmonary edema, since patients can present with heart failure and preserved LV systolic function (formerly diastolic dysfunction), thus necessitating further evaluation. Echo-Doppler can also provide semiquantitative measurements of ventricular filling pressures, cardiac output, stroke volume, and pulmonary artery pressures. TEE is used to enhance and refine evaluation of structural and valvular pathologies such as native or prosthetic valve dysfunction, cardiac origins of embolism, infective endocarditis, and congenital diseases.9 In one study performed in the ICU, TEE led to a significant change in management in 32% of cases,10 emphasizing its diagnostic value.
Hemodynamic Assessment
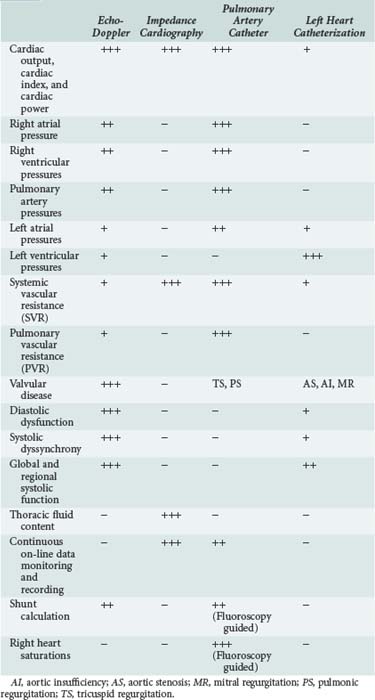
Pulmonary edema is a medical emergency and requires immediate medical therapy to alleviate symptoms. Dyspnea is the cardinal symptom of pulmonary edema and can be assessed subjectively (by analog scales of dyspnea severity) and objectively (by oxygen saturation, respiratory rate, alveolar-arterial difference, and acidemia). In most cases, the diagnosis, treatment, and monitoring of the patient with pulmonary edema is self-evident, but hemodynamic monitoring, either invasive or noninvasive, should be considered in selected patients. Since there are several techniques for hemodynamic assessment, the benefits and limitations of each technique should be considered prior to usage (Table 73-1).
Echo-Doppler
Pulmonary Artery Catheterization and Other Invasive Modalities
Insertion of a pulmonary artery catheter permits measurement of the pulmonary capillary wedge pressure (PCWP), a method first described in 1970 by Swan and Ganz12 and still considered to be the “gold standard” for diagnosis of pulmonary edema resulting from elevated LV diastolic filling pressures. Current monitoring systems that include cardiac output and systemic vascular resistance (SVR) calculators add further information and help distinguish cardiogenic pulmonary edema (high PCWP and high SVR) from noncardiac (low PCWP ± low/normal SVR). A wedge pressure of more than 18 mm Hg is indicative of elevated filling pressures of the left ventricle and usually indicates a cardiogenic origin of pulmonary edema. In addition to its utility in diagnosis, PCWP allows continuous monitoring of the LV filling pressure during treatment, facilitating the administration of appropriate therapy to alleviate pulmonary edema. It is recommended that pulmonary artery catheterization (PAC) be used in patients in whom a diagnostic dilemma exists, when echo-Doppler measurements are difficult to obtain, or in hemodynamically unstable patients not responding to conventional therapy.13
The clinical value and safety of PAC as a tool for hemodynamic assessment has been a subject of considerable debate. Gore14 and Connors15 demonstrated a neutral to negative effect of PAC on patient outcome. Meta-analyses assessing the effects of PAC on morbidity16 and mortality17 in clinical trials showed that mortality was unaffected, but morbidity was increased with the use of a PAC. There may be methodological issues in some of these studies; nevertheless, these publications resulted in a call for a moratorium18 on PAC. In 1997, a consensus conference19 attempted to reassess indications for PAC. Conditions that could be considered to benefit from PAC included myocardial infarction complicated by hypotension, shock, or mechanical complications, assessing and managing acute and chronic heart failure, and pulmonary hypertension. The ESCAPE study20 enrolled patients with established heart failure who did not require PAC for their diagnosis or management. The results of the study have demonstrated no benefit of right heart catheterization in the study’s primary endpoint (i.e., days alive out of hospital during the 6 months after randomization). Although the PAC is an invaluable tool for diagnostic, therapeutic, and prognostic assessment of PE, it should be used selectively by well-trained teams to address pertinent diagnostic and management issues.
Impedance Cardiography
Both thoracic impedance21 and total body impedance22,23 can accurately measure continuous CO and CI. ICG-derived COs appear to be less variable and more reproducible than CO measured by other techniques.24 Some bioimpedance systems, however, do not provide accurate CO values25 when compared to the gold standard of thermodilution.26 However, bioimpedance devices that can measure CO reliably can serve as tools for assessing pump performance by providing noninvasive measurements of CP and cardiac power index (CPI). None of these systems provide assessment of right side pressures or pulmonary vascular resistance (PVR).
Beat-to-Beat Pulse Contour
Semi-invasive techniques utilizing beat-to-beat pulse-contour analysis are becoming available for continuous cardiac output monitoring. At present, there are conflicting reports regarding the usefulness of these techniques compared to invasively measured PCWP in assessing the presence of pulmonary edema in different ICU populations,27–30 and no report has addressed the utility of pulse-contour analysis in differentiating between cardiac and noncardiac etiologies of pulmonary edema.
 Noncardiogenic Pulmonary Edema
Noncardiogenic Pulmonary Edema
A variety of etiologies may lead to noncardiogenic pulmonary edema (Table 73-2), with a final common pathway of fluid accumulation in the lung interstitium due to either increased permeability of capillaries or decreased fluid clearance mechanisms without evidence of elevation of LV end-diastolic pressure.
| Increased Capillary Permeability and Reduced Fluid Clearance |
Perioperative Pulmonary Edema
Perioperative pulmonary edema can result from a wide variety of etiologies including volume overload, negative pressure pulmonary edema (resulting from exaggerated negative intrathoracic pressure generated by an inspiratory effort against a closed glottis), and transfusions. In one large trial including 8159 patients undergoing major outpatient surgical procedures, an incidence of 7.6% of postoperative pulmonary edema was noted with approximately 12% mortality; of note, prior reports had reported lower rates of pulmonary edema and mortality.31,32 Excessive fluid administration during the postoperative period was associated with increased mortality, especially in patients without other comorbidities. Fluid overload during and after surgery can be attributed to exaggerated treatment for hypotension related to anesthesia, excessive blood loss, fluid shifts during surgery (“third spacing”), and postoperative fever. The relatively common incidence of pulmonary edema after surgery leads to a recommendation for close monitoring of fluid balance in the perioperative period, with special emphasis on monitoring patients at risk for developing pulmonary edema because of preexistent medical problems, including cardiac disease.
High-Altitude Pulmonary Edema
High-altitude pulmonary edema (HAPE) is the abnormal accumulation of edema involving the interstitial and alveolar spaces; it is due to a breakdown in the pulmonary blood-gas barrier. This is triggered by hypobaric hypoxia and rapid ascent to altitudes above 2500 m. Such hypoxia triggers a maladaptive mechanism including poor ventilatory response, increased sympathetic tone, exaggerated and uneven pulmonary vasoconstriction (pulmonary hypertension), and inadequate production of hormonal mediators (e.g., nitric oxide [NO]) that then lead to capillary leak and pulmonary edema.33 The risk for developing HAPE depends on individual susceptibility, altitude ascent rate, and time spent at the altitude. The incidence of HAPE increases at different heights, ranging from 0.2% to 6% at 4500 m to 2% to 15% at 5500 m.34 Clinical symptoms that precede presentation of pulmonary edema include shortness of breath, nonproductive cough, and difficulty in continuing to ascend to greater heights. such symptoms can easily be mistaken for exhaustion. The symptoms usually appear 2 to 4 days after arriving at a new altitude. It is unusual for HAPE to develop after more than 1 week at the same altitude. When symptoms progress, the patient becomes easily exhausted and may have productive pink sputum. In the later stages, the patient becomes severely hypoxemic, a situation that may be fatal without medical treatment. A favorable outcome depends on early recognition of the patient’s signs and symptoms, using supplementary oxygen, rapid descent to lower altitude, or the use of a hyperbaric chamber. No pharmacologic intervention beyond oxygen has been proven to be beneficial for HAPE, but several pharmacologic agents have been examined. Nifedipine (calcium channel blocker) may aid in both lowering the elevated pulmonary pressure and the systemic resistance. Tadalafil and sildenafil are phosphodiesterase-5 inhibitors acting on the pulmonary vasculature by increasing the amount of available nitric oxide. These agents have been shown to be beneficial for prophylactic treatment of HAPE but have not been examined in the treatment of this condition. Salmeterol, an inhaled β-agonist, has been proposed as a prophylactic drug for HAPE that may also be useful for treatment.
Pregnancy-Related Pulmonary Edema
Pregnancy causes significant hemodynamic changes in the cardiovascular system, including increase in plasma volume, cardiac output, heart rate, and capillary permeability, as well as decreased colloid osmotic pressure. In light of these and other factors, pulmonary edema may occur in pregnant women with preexisting cardiac conditions or abnormalities (cardiomyopathies and valvular disease) or with pregnancy-related abnormalities such as preeclampsia. The incidence of pulmonary edema ranges from 0.08% in normal pregnancies to 3.4% in preeclampsia and up to 5% in preterm labor.35 In a large survey including 62,917 women, the overall incidence of pulmonary edema was 0.08%. Among the pregnant women who developed pulmonary edema, the most common attributable causes or associated conditions were tocolytic use (13 patients [25.5%]), cardiac disease (13 patients [25.5%]), fluid overload (11 patients [21.5%]), and preeclampsia (9 patients [18%]).36 The diagnosis of pulmonary edema was made during the antepartum period in 24 patients (47%), the intrapartum period in 7 (14%), and the postpartum period in 20 (39%). The increased incidence of pulmonary edema in the intra- and postpartum period can be attributed to changes in the plasma colloid pressure. Plasma colloid pressure decreases from about 22 to 16 mm Hg at term after delivery in normal pregnancy, and from 18 to 14 mm Hg postpartum in preeclampsia complicated pregnancies. This reduction is attributed to blood loss and fluid shift due to increased vascular permeability, especially in pregnancies with preeclampsia, and leads to pulmonary edema occurring after delivery.37 Women with preeclampsia are at increased risk for the development of pulmonary edema due to underlying endothelial damage and decreased colloid osmotic pressure, which cause leakage into the pulmonary interstitium or alveolar space.
The development of pulmonary edema associated with pregnancy appears to be influenced by maternal age, parity, and preexisting essential hypertension. In a small study examining the role of echocardiography in the diagnosis of pulmonary edema in the setting of preeclampsia, 25% of the patients had decreased systolic function, and a significant number of the remaining patients had elevated diastolic pressures when compared to other pregnant hypertensive/normotensive women without preeclampsia, thereby indicating that elevated filling pressure may be a part of the pathologic process in preeclampasia.38 Tocolytics are also a major cause for pulmonary edema during pregnancy. Therapy using β-agonists can cause increased hydrostatic pressure and lead to pulmonary edema.39 Pulmonary edema has also been reported after usage of calcium channel blockers as tocolytics.40 As in most patients with pulmonary edema, the mainstay of treatment includes fluid restriction, diuretics, and cessation of tocolytics.
Postobstructive Pulmonary Edema
Postobstructive pulmonary edema (POPE) was first described in 1973 as sudden onset of pulmonary edema following relief of upper airway obstruction. The incidence may be up to 10% of cases after the relief of acute obstruction and up to 40% after relief of chronic obstruction.41 Two types are described: type I POPE follows a sudden, severe episode of upper airway obstruction such as postextubation laryngospasm, epiglottitis, or croup and is seen in strangulation and hanging; type II POPE develops after surgical relief of chronic upper airway obstruction.42 Type I POPE usually develops within 1 hour after the event, but it can be delayed up to 6 hours. In contrast, there is close proximity between the relief of the obstruction and the development of POPE in type II.
The etiology for type I POPE is multifactorial. Negative intrathoracic pressure is caused by inhaling against closed obstruction. This causes increased venous return, decreased cardiac output, and fluid transudation into the alveolar space.43 Risk factors for type I POPE are young age (owing to increased ability to generate increased negative pressure), direct suctioning of the endotracheal tube during thoracotomy, narcotics, short neck, oral or pharyngeal surgery or pathology, vocal cord paralysis, conditions leading to increased capillary-alveolar pressure gradients, endotracheal tube obstruction, and premature extubation. The etiology for type II POPE, which is less frequent than type I, is less clear. It is suggested that the obstructive lesion causes constant positive end-expiratory pressure (PEEP) with increased end-expiratory lung volume. Relief of the obstruction causes immediate reduction of the lung volume that is postulated to result in increased pulmonary permeability and transudation of fluid. The diagnosis usually is suggested by physical findings after surgery of tachypnea, tachycardia, agitation, and frothy pulmonary secretions. The diagnosis is confirmed by x-ray. Most patients will respond quickly to standard therapy with adjunct support of PEEP (5 mm H2O).
Reexpansion Pulmonary Edema
Reexpansion pulmonary edema (REPE) after spontaneous pneumothorax is a rare complication of tube thoracostomy, with reported mortality ranging from 0 to 20%.30,31,44,45 Most patients will present with symptoms as early as 1 hour after thoracostomy, but delayed presentation of up to 24 hours after thoracostomy has also been described. Tachypnea, tachycardia, and hypoxia are the main presenting signs and symptoms. The chest radiograph demonstrates unilateral pulmonary edema, although bilateral pulmonary edema has rarely been reported. In a recent study, many REPE cases were mild and asymptomatic and only diagnosed by computed tomography (CT) of the chest. Most cases will resolve within 24 to 72 hours.
The pathophysiology of REPE is unclear. The main hypothesis is that capillary leak is induced by a postexpansion inflammatory process. During reexpansion, mechanical injury to the alveolar-capillary membrane, together with reperfusion injury from the reinstitution of blood flow, initiates an acute inflammatory process. Predictive factors for REPE are age (20-39 years) and prolonged duration of pneumothorax prior to relief.46 It was also suggested that REPE may be related to the application of negative pressure to the chest tube. No human study has been performed prospectively to determine whether the incidence of REPE would be less if the chest tube is put to water seal only. Unfortunately, REPE can also occur in patients whose lungs are reexpanded without suction. REPE therefore appears to be related to three factors: longer duration of pneumothorax, greater size of the pneumothorax, and a rapid rate of expansion after tube thoracostomy. Controlling for one factor may not prevent the process if one or two of the other factors are present. In lieu of a randomized controlled trial, the American College of Chest Physicians (ACCP) recommends that in the presence of a spontaneous pneumothorax in clinically stable patients with a large (≥30% of the lung field) primary pneumothorax, either a small-bore (14F or smaller) catheter or 16 to 22F chest tube with the tube connected to Heimlich valve or a water-seal device be placed. However, if the lung fails to reexpand, application of negative pressure to the chest tube is deemed appropriate.47
Transfusion-Related Pulmonary Edema
Acute onset of dyspnea shortly after blood transfusion can be attributed to two main etiologies: transfusion-associated cardiac overload (TACO) and immune-mediated ALI resulting from transfusion of plasma-containing products (transfusion-related ALI, or TRALI).48 TRALI was defined by the National Heart, Lung, and Blood Institute Working Group as an ALI that develops within 6 hours after blood transfusion.49 TRALI is considered to be the leading cause for transfusion-related mortality. Virtually all blood products can lead to TRALI, but infusions of whole blood, platelets, packed red blood cells, and fresh frozen plasma are the most commonly identified precipitating causes. Owing to nonuniformity of definitions, the true incidence of TRALI is uncertain, but when uniform definitions are used, the incidence is reported to be 1 case for every 1000 to 2400 units transfused, with equal incidence between men and women and wide age variability.50 Risk factors for TRALI are prolonged storage of blood products, fresh frozen plasma infusion, and underlying conditions such as recent surgery, thrombocytopenia, and massive transfusions. The pathogenesis of TACO is similar to other causes of acute congestive heart failure: volume overload leading to increased central and pulmonary pressures resulting in increased hydrostatic pressure and extravasation of fluid into the alveolar space. The pathogenesis of TRALI is less obvious. Three hypotheses are proposed: (1) antigranulocyte antibodies in the donor’s plasma (or less commonly, in the recipient’s plasma) react with antigens on the recipient’s (or less commonly, donor’s) granulocytes to initiate an inflammatory response within the pulmonary microvasculature; (2) biologically active substances such as lipids and cytokines contained within the transfusions prime granulocytes in the pulmonary vasculature, contributing to increased vascular permeability; or (3) a “two-hit” hypothesis wherein the primary stimulus causes granulocyte sequestration in the pulmonary capillaries, and a secondary stimulus causes the granulocytes to “activate,” damaging the endothelial layer such that fluid and protein leak into the alveolar space. Surgery, infections, and other situations can serve as the initial primer for this process.51
The mainstay of TACO treatment is discontinuation of blood-product transfusion, respiratory support as needed, and diuretics. It has been suggested that subsequent blood products should be infused at a slower rate after the appearance of TACO, but no solid evidence supports this suggestion. TRALI treatment is mainly supportive: mechanical positive-pressure invasive ventilation and high concentrations of oxygen and PEEP. Although some authors have advocated the use of steroids for TRALI, this approach is still considered anecdotal.52 The mortality rate for TRALI varies between 5% and 8%, but rates of up to 47% in critically ill patients have also been reported.48 Most survivors recover completely with appropriate treatment. It is recommended that patients who recover from TRALI should not receive any other blood products from the same donor, but it seems they are not at increased risk for TRALI when receiving blood products from other donors.
Drug Toxicities
Development of pulmonary edema has been linked to a number of drugs and substances.
Opiates
Opiate overdose can induce pulmonary edema due to increased capillary permeability. Interstitial protein content in this setting is similar to the plasma protein content. The pulmonary capillary wedge pressure is generally within the normal range. Direct toxic and hypoxic etiologies have also been suggested as contributing mechanisms in this clinical setting. The incidence of pulmonary edema in patients with heroin overdose is 0.8% to 2.4%,53 with most of the symptoms developing over the first 2 hours of admission. The entire opiate family shares the ability to induce pulmonary edema, and even overdose of codeine has been linked to this condition.54 Most patients require mechanical ventilation to correct severe hypoxia and respond within 24 to 36 hours to supportive care.
Salicylates
Salicylate intoxication can induce pulmonary edema. The mainstay of treatment for aspirin intoxication is volume resuscitation and bicarbonate, which can lead to volume overload and pulmonary edema that cannot be easily differentiated from ALI. Development of pulmonary edema in the setting of aspirin intoxication is an indication for immediate hemodialysis.55
Neurogenic Pulmonary Edema
Acute central nervous system (CNS) injury may lead to a clinical presentation similar to ARDS.56 Symptoms develop within minutes to several hours after the offending injury. Classic signs and symptoms of pulmonary edema include tachycardia, tachypnea, basilar rales on auscultation, and bilateral infiltrates on chest radiograph. Both cardiac output and pulmonary capillary wedge pressure are normal in this situation. These signs and symptoms together with evidence of acute CNS injury establish the diagnosis. The most common causes for neurogenic pulmonary edema (NPE) are epileptic seizures, head injury, and cerebral hemorrhage, but any intracranial or spinal injury can be associated with this condition.56
There are several theories describing the precipitating factors leading to NPE. Excessive stimulation of the autonomic nervous system can result in pulmonary venous vasoconstriction, causing elevations in hydrostatic pressure and extravasation of fluid into the pulmonary interstitium. This mechanism is supported by data showing the ability of α-adrenergic agonists to alleviate pulmonary edema caused by cerebral stimulation in rats.57 Furthermore, rapid elevation of pulmonary venous pressure may cause microvascular injury and excessive capillary permeability, leading to ALI. Two etiologies must be differentiated from NPE in the setting of an intubated head injured patient: aspiration pneumonia and ventilator-associated pneumonia (VAP). The treatment of NPE must first focus on treatment of the offending head/spinal injury. It is essential that hematomas are evacuated, intracranial pressure (ICP) decreased, and convulsions controlled. Other supportive therapies include ventilation that meets the oxygenation needs of the patient, with permissive hypercapnia allowed only in patients with ICP monitoring, and avoidance of high PEEP that may reduce cerebral perfusion. Hemodynamic support should aim to maintain low cardiac filling pressures without compromising cerebral perfusion. Invasive hemodynamic monitoring may be required. The exact place of α- and β-adrenergic agents in the therapy of NPE is not established. Most NPE episodes will resolve within 48 to 72 hours.
Other Noncardiogenic Etiologies of Acute Pulmonary Edema
Massive pulmonary embolism (PE) can cause pulmonary edema due to elevated hydrostatic pressure and injury to adjacent pleural and pulmonary vasculature. Viral infections can also induce pulmonary edema, as demonstrated in severe cases of Hanta virus infection.58 Other viruses, including enteroviruses and coronavirus, can also lead to pulmonary edema.
There are reports of pulmonary edema in trained athletes after strenuous exercise such as marathon running or swimming. The theories that explain this phenomenon point to preexisting ventilation/perfusion mismatch in combination with increased cardiac output, causing elevated hydrostatic pressure that leads to pulmonary edema.59,60 Cold water immersion can induce pulmonary edema by both increasing cardiac output and elevating pulmonary vascular resistance and pressures.
 Cardiogenic Pulmonary Edema
Cardiogenic Pulmonary Edema
Definition and Pathophysiology
Pulmonary edema is a life-threatening presentation of acute heart failure (AHF). AHF is defined as rapid onset or change in the signs or symptoms of heart failure, resulting in the need for urgent therapy. It may be new or worsening of a preexisting condition.13 During 2006 there were over 1 million admissions in the United States alone with AHF as the primary diagnosis and more than 3 million admissions with heart failure as a secondary diagnosis, with a direct and indirect cost of 25 million and 37.2 million U.S. dollars, respectively.
AHF is predominantly a disease of the elderly. The primary cardiac pathologies that predispose the patient to develop AHF can be related to ischemic, myocardial, valvular, pericardial, or rhythm disorders. Noncardiac factors may also contribute to the development of AHF by increasing pressure (hypertension) and volume overload (Table 73-3). The precipitating insult leading to the appearance of signs and symptoms of AHF are diverse and include (among others) active ischemia, increased afterload (hypertensive emergencies), increased preload (volume overload), circulatory failure due to high output state (sepsis, thyrotoxicosis, anemia), and drugs (NSAIDs or discontinuation of prescribed drugs).
TABLE 73-3 Common Precipitating Factors for Acute Heart Failure
| Noncompliance with medical regimen, sodium and/or fluid restriction | Atrial fibrillation and other arrhythmias |
| Acute myocardial ischemia or ischemia | Recent addition of negative inotropic drugs (e.g., verapamil, nifedipine, diltiazem, beta-blockers) |
| Uncorrected high blood pressure | Pulmonary embolism |
| Nonsteroidal antiinflammatory drugs | Excessive alcohol or illicit drug use |
| Stress related cardiomyopathy | Concurrent infections (pneumonia, viral illnesses) |
| Cardiac toxicity: chemotherapy | Worsening lung disease (respiratory insufficiency or failure) |
| Anemia | Acute renal failure |
Adapted from Hunt et al. 2009 focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2009;119:e391-479.
Cardiogenic pulmonary edema results from transudation of protein-poor fluid from the alveolar interstitium into the alveolar space as a result of rapid increase in pulmonary capillary pressure overwhelming alveolar fluid reabsorption mechanisms. Mild elevations in LV and left atrial pressures (18-25 mm Hg) cause edema in the perimicrovascular and peribronchovascular interstitial spaces. As left atrial pressure rises further (>25 mm Hg), edema fluid floods the alveoli with protein-poor fluid, leading to the full-blown presentation of pulmonary edema.2 Cardiovascular failure leading to pulmonary edema may be the result of reduced LV contractility and increased systemic vascular resistance, or from impaired LV filling due to abnormal relaxation or excessive stiffness.
Traditionally it is thought that volume overload, nonadherence to medical therapy, ischemia, and arrhythmia can all induce decrease in cardiac contractility and progressive volume overload. It seems that all these factors may serve as triggers for cardiogenic pulmonary edema, but other factors are also crucial for the initiation of an acute episode of decompensated heart failure.61 Vascular resistance and afterload mismatch are probably the predominant mechanisms in a substantial proportion of these events. Invasive monitoring of patients in AHF episodes often reveals decreased cardiac contractility compared to baseline and increased systemic vascular resistance (SVR)—a mismatch between rapidly increasing afterload (or SVR) and impaired systolic performance resulting in an acute elevation of LV end-diastolic pressures and decrease in cardiac output.
Cardiogenic pulmonary edema due to AHF can be considered a two-step process: induction and amplification (Figure 73-5). The initiation phase, or “cardiac pathway,” is caused by low cardiac reserve in the cardiac pathway due to factors such as prior myocardial infarction or nonadherence to medications; such factors can be amplified by an acute decrease in contractility. This decrease is then exacerbated by an acute decrease in contractility due to arrhythmia, ischemia, or inflammatory activation. In contrast, the “vascular pathway” is activated in individuals with mild to moderate impairment in contractile reserve, but it rarely leads to AHF by itself. Here, a variety of offenders (neurohormonal activation, inflammation, aging processes) will abruptly lead to afterload mismatch, resulting in forward heart failure without a significant change in the LV systolic function (as assessed by LVEF). In most patients, both pathways coexist, and their combination may lead to the combination of excessive pulmonary venous pressure and pulmonary edema, along with reduced cardiac output, resulting in reduction of perfusion of vital organs. AHF can then be further exacerbated through additional mechanisms, including:
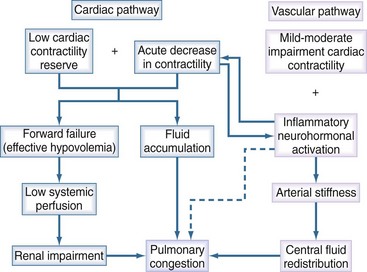
Classification
One of the most clinically applicable classifications of AHF is the modified Forrester classification62 (Table 73-4). This classification utilizes a 4-square table to define the clinical status of the patient and establish treatment strategy. Most patients will present in category B (warm and wet) and will respond favorably to medical therapy (composed predominantly of loop diuretics and vasodilators). Patients in category C (cold and wet) will require inotropic agents and vasodilators to improve tissue perfusion and promote diuresis. Category A (warm and dry) is found in heart failure patients who present with dyspnea or edema that appears to be unrelated to the heart failure. In this setting, other causes such as respiratory disease or sepsis should be sought. Category L stands for “light,” representing either a rare situation of overdiuresis of category-B patients or patients who are free of symptoms at rest but develop symptoms with exercise.

Diagnosis
The etiology of cardiogenic pulmonary edema presenting as acute decompensated heart failure includes decreased contractility, increased systemic vascular resistance, or a combination of the two. Along with providing the immediate treatment needed for stabilization, initial assessment should focus on volume status, adequacy of vital organ perfusion, delineation of the cardiac pathology, and determination of the role of precipitating factors (see Table 73-3).
Heart Failure with Preserved Ejection Fraction
Close to 50% of patients admitted with AHF have relatively preserved LV systolic ejection fraction (LVEF > 45%). Increasing age, female gender, hypertension, small size heart on chest radiograph, and an ischemia- or infarction-free ECG may suggest the diagnosis of heart failure with preserved ejection fraction (HFPEF; formally known as diastolic dysfunction). Pulmonary edema in this setting is related to complex pathophysiologic processes that are only partially elucidated. Stressors lead to increased venous vasoconstriction, which in turn increases the blood flow to the right ventricle, lung, and eventually the left ventricle. Owing to limitations in LV compliance, this excessive flow can not be accommodated by the left ventricle without considerable rise in left ventricular, left atrial, and pulmonary venous pressures. The elevated pulmonary venous and arterial pressures lead to neurohormonal activation that increases the systemic vascular resistance, which further increases venous return and systemic blood pressure and amplifies the development of pulmonary edema.63
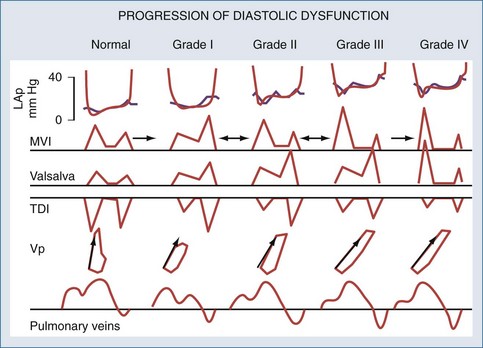
Various echo-Doppler indices are used for assessing the severity of diastolic function (Figure 73-6). Doppler measurements made in diastole across the mitral valve are useful in characterizing and quantifying diastolic dysfunction. However, these measurements may be affected by heart rate, afterload, and preload. E wave represents the early filling and the active relaxation of the LV, after which comes a plateau with absence of flow. The second wave, called the A wave, represents flow produced by atrial contraction. Measurements of isovolumic relaxation time (IVRT), E-wave deceleration time, the E wave, and the A wave peak velocity and ratio, as well as the pulmonary venous flow patterns, allow the clinician to define the nature and severity of “diastolic dysfunction.” Tissue Doppler (TD) measures tissue velocity relative to the transducer, with high spatial (1 mm) and temporal resolution (>100 s-1). The most frequently used modality of TD is measurement of LV basal (“annular”), longitudinal myocardial shortening. The early diastolic (E′) lengthening velocities are considered sensitive for diastolic dysfunction and E/E′ ratios correlate closely with LV filling pressures.64
Exclusion of Active Myocardial Etiologies as Causative or Aggravating Factors for AHF
Valvular and structural heart disease must be excluded as causative factors for pulmonary edema, using echocardiography. Echo-Doppler provides definitive diagnosis of abnormal flow velocities and pressure gradients over stenotic lesions as well as accurate assessment of LV function, the presence and degree of hypertrophy or ventricular dilatation, hypertrophic obstructive cardiomyopathy, sub-/supravalvular LV outflow obstruction, and prosthetic valves (Table 73-5). Some of these pathologies will necessitate prompt surgical intervention.
TABLE 73-5 Common Mechanical/Valvular Abnormalities Causing Heart Failure Decompensation
Determining Prognostic Factors and Assessing Severity
CCU, cardiac care unit; ED, emergency department; ICU, intensive care unit.
* Adapted from Dickstein K, Cohen-Solal A, Filippatos G et al. ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2008: the Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2008 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association of the ESC (HFA) and endorsed by the European Society of Intensive Care Medicine (ESICM. European heart journal. Eur Heart J 2008;29(19):2388-442.
Initial Stabilization
Stabilization measures include establishment/maintenance of the airway, oxygenation, and ventilation. Vital signs should be continuously monitored, with emphasis on oxygen saturation and blood pressure while following heart rate and watching for arrhythmias. When arrhythmias or conduction abnormalities are diagnosed, they should be treated promptly, especially atrial fibrillation and other hemodynamically significant arrhythmias. Ischemia and major severe valvular diseases should be sought and treated. Fluid-balance monitoring is best achieved by daily weight and closely following input and output. Hypoxemic patients should be treated with supplemental oxygen therapy to achieve the goal of oxygen saturation above 95% (>90% in COPD patients). Patients with respiratory distress, respiratory acidosis, or persisting hypoxemia should receive assisted ventilation using noninvasive positive-pressure ventilation (NIPPV). NIPPV should be considered as early as possible, since it improves LV function by reducing afterload (by decreasing systolic wall stress) and preload (by decreasing venous return). NIPPV should not be used in patients with cardiogenic shock or right ventricular involvement. Three meta-analyses reported short-term mortality benefit and decrease in need for intubation in patients who were treated early with NIPPV, but the benefit on mortality was equivocal.13 Patients who fail NIPPV or do not tolerate it should undergo endotracheal intubation and conventional mechanical ventilation using PEEP.
Loop Diuretics
Loop diuretics have been the mainstay of AHF therapy for more than 200 years despite lack of adequate knowledge regarding their efficacy, safety, and dosing. Loop diuretics initially produce a rapid fall in both left and right heart pressures via venodilatation, resulting in improved cardiac function and symptom relief. However, diuretics activate the renin-angiotensin-aldosterone system. In later stages, by promoting fluid removal, loop diuretics serve as the mainstay of treatment in patients with volume overload. In most patients presenting with volume overload, diuretic therapy should be initiated in the emergency department (ED) without delay.65 These agents should not be used in hypotensive patients and should be used cautiously in patients with hyponatremia and aortic stenosis. Diuretic dosing should be sufficient to cause a rate of diuresis that will cause relief of volume overload and signs of congestion without inducing complications. An initial dose of 20-40 mg of IV furosemide should be given in the ED, and further treatment should be guided according to renal function and prior usage of oral diuretics. The total furosemide dose should be less than 100 mg in the first 6 hours and 240 mg during the first 24 hours.13 Further treatment should include multiple doses or continuous infusion of loop diuretics, with the goal of relieving signs of congestion. A debate exists regarding the best approach for diuresis: continuous versus boluses. A recent trial showed no superiority for continuous diuresis.66 Response to diuretic treatment may be optimized by a strict limitation of sodium intake. Urinary output, body weight, volume status, and laboratory indices should be monitored continuously both for signs and symptoms of resolution of heart failure and for complications of treatment such as deterioration of renal function and electrolyte imbalance. If a patient’s status remains unchanged with this strategy, a second type of diuretic should be added, usually a thiazide (oral metolazone/IV chlorothiazide) or spironolactone. When these fail and the patient is still symptomatic, ultrafiltration should be considered.
Morphine
Morphine reduces patient anxiety and decreases the work of breathing, causing decreased sympathetic tone and leading to both arterial and venous dilatation and reduced filling pressures. Although this drug is used frequently in patients with pulmonary edema, its long-term benefits are controversial, and some authors have reported high rates of adverse effects, including the necessity of mechanical ventilation and increased mortality, highlighting the need to use this drug cautiously.67,68
Vasopressin Antagonists
Vasopressin (antidiuretic hormone [ADH]) is a peptide hormone secreted from the posterior pituitary gland that promotes vasoconstriction through interaction with V1 receptors and water retention through V2 receptors. Blockage of these receptors has the potential for augmenting the effect of diuretics through the increase in free water clearance. Tolvaptan is the most studied vasopressin antagonist. It is an oral V2-selective receptor antagonist. In the EVEREST trial, tolvaptan promoted weight loss and relieved symptoms of pulmonary edema, but without decrease in morbidity and mortality after 1 year.69 Use of this and other vasopressin antagonists is still under investigation.
Ultrafiltration
Ultrafiltration is a mode of continuous renal replacement therapy that prompts fluid loss with minimal solute loss. Since the benefits of this method over diuretic therapy are not well established, it should be considered for patients resistant to medical therapy and those with severe renal insufficiency.13,65
Vasodilators
Nitrates
Inotropic Agents
Assist Devices
Intraaortic Balloon Pump
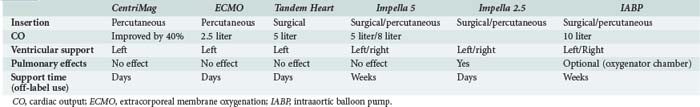
The intraaortic balloon pump (IABP) is one of the most commonly used mechanical assistance devices. Between 1996 and 2001, more than 22,000 IABPs were used in 250 centers worldwide for various indications.76 Apart from cardiogenic shock, the use of this device is supported by evidence in postinfarct angina, refractory ventricular arrhythmia, ventricular septal rupture, acute mitral insufficiency, and post acute myocardial infarction. Use of IABP in the setting of acute myocardial infarction complicated by hypotension unresponsive to other interventions is listed as a class I indication in both the American Heart Association (AHA)77 and the European Society of Cardiology (ESC) guidelines. The IABP is a polyethylene balloon mounted on a catheter, which is inserted into the aorta through the femoral artery. The pump is available in a wide range of sizes (2.5 cc to 50 cc) that will fit patients of any age and size. The balloon is guided into the descending aorta and positioned approximately 2 cm from the left subclavian artery. Inflation of the IABP occurs at the beginning of diastole, on the dicrotic notch on the arterial waveform, causing augmentation of blood perfusion to the coronary arteries. Deflation of the balloon should occur at the beginning of systole, immediately prior to the arterial upstroke, augmenting coronary perfusion. As the balloon deflates, blood is ejected from the left ventricle against a decreased afterload, causing an increase of cardiac output by as much as 40% and decrease in the LV stroke work and myocardial oxygen requirements.
Despite the guideline recommendations, the efficacy of routine IABP use adjunctive to primary percutaneous coronary intervention in cardiogenic shock was questioned in a meta-analysis.78 The principal findings of the meta-analysis of randomized clinical trials of IABP therapy in myocardial infarction with ST-T wave abnormalities showed no efficacy benefit of adjunctive IABP therapy, including lack of 30-day survival benefit or improved LVEF. Instead, IABP therapy was associated with a significant increase in the rates of stroke and bleeding. These clinically relevant higher complication rates are not outweighed by any clinical benefit. Currently, only one prospective randomized study has been performed,79 but it was underpowered to demonstrate any benefit of adding IABP to optimal medical therapy in reducing short-term morbidity in acute myocardial infarction patients with cardiogenic shock.
Ventricular Assist Devices
Impella
The Impella LP 2.5 (Abiomed Europe GmbH, Aachen, Germany) is a catheter-based, axial-flow pump with a maximal flow of 2.5 L/min. The pump is inserted via a 13F sheath in the femoral artery and placed in retrograde fashion through the aortic valve. The microaxial pump continuously aspirates blood from the left ventricle and expels it to the ascending aorta, with a maximal flow of 2.5 L/min (Figure 73-7). The ISAR-SHOCK study prospectively followed 26 patients with cardiogenic shock treated with either IABP or Impella 2.5. Though cardiac index significantly increased in patients with the Impella LP2.5 as compared with patients with IABP, mortality at 30 days was similar.80 Impella 5, which can generate flows of up to 5 L/min is also available. The device is implanted via a cutdown (femoral or subclavian) and is used for the same indications as the Impella 2.5. Contraindications to use of the Impella devices include mechanical aortic valve, aortic valve stenosis/calcification, moderate to severe aortic insufficiency, and severe peripheral arterial obstructive disease. Complications of Impella device use include aortic valve injury, arrhythmia, bleeding, hemolysis, thrombocytopenia, infection, limb ischemia, and vascular injury.
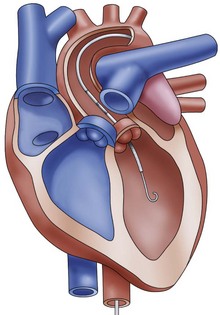
Tandem Heart
The Tandem Heart system (Cardiac Assist Technologies Inc., Pittsburgh, Pennsylvania) is a percutaneous ventricular assist device (pVAD) indicated for the hemodynamic stabilization of patients with cardiogenic shock. The Tandem Heart largely serves to unload the left ventricle by providing a bypass circuit drawing blood from the left atrium and then perfusing the withdrawn blood into the descending aorta (Figure 73-8). A transseptally introduced left atrial cannula with multiple side holes withdraws blood to a centrifugal pump placed outside the patient’s body. Using adjustable rotation, it then injects blood through an arterial cannula to the iliac artery or descending aorta. The size of the left atrial cannula is 21F, whereas the size of the arterial cannula ranges from 15- to 17F, capable of delivering up to 5 L/min of blood flow. As with the Impella device, the hemodynamic and metabolic parameters in cardiogenic shock can be reversed more effectively by Tandem Heart support as compared to standard IABP treatment.81,82 However, there were more complications encountered by the Tandem system. Complications of the Tandem Heart support include puncture of the aortic root, coronary sinus, or posterior free wall of the right atrium, and thromboembolism, systemic hypothermia, canula dislodgment, bleeding, and infection.
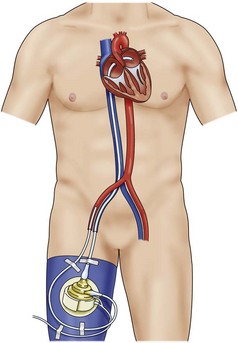
Venous-Arterial Extracorporeal Membrane Oxygenation
The extracorporeal membrane oxygenation (ECMO) device is an easily applicable and widely accepted option for temporary mechanical circulatory support, allowing cardiac and pulmonary recovery or bridging until further therapeutic alternatives can be considered. There are two cannulation types: VA cannulation (femoral artery/axillary artery to femoral vein), which is used in patients who require cardiac support in addition to respiratory support (Figure 73-9). In patients with pure respiratory failure, VV cannulation (usually via the femoral vein and internal jugular vein) is preferred.
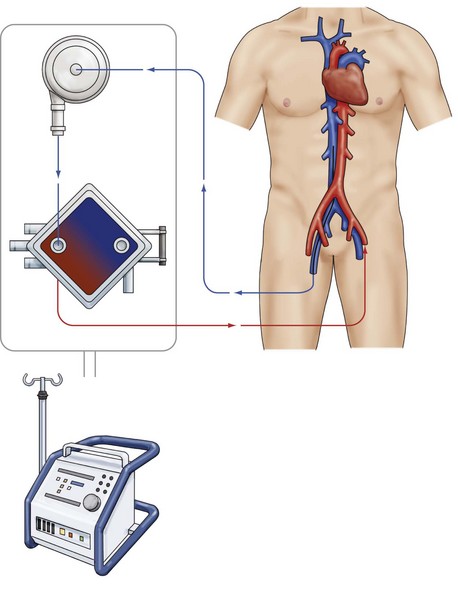
Figure 73-9 VA ECMO.
Venous-arterial extracorporeal membrane oxygenation device. Femoral vein to femoral artery.
Historically, ECMO has been used most frequently for support of respiratory failure,83,84 but recently the use of ECMO has been evaluated in other patient populations. In a series of 517 patients with refractory postcardiotomy shock treated with ECMO, the overall hospital survival was 24.8%.85 Given the poor prognosis of patients who have undergone ECMO for the treatment of postcardiotomy shock, ECMO may at best only function as salvage therapy in this setting. In pediatric patients undergoing CPR, ECMO has recently been demonstrated to be associated with survival rates to hospital discharge of 34% to 38%.86
CentriMag
The CentriMag Blood Pumping System (Levitronix LLC, Waltham, Massachusetts) is one of a new generation of magnetically levitated centrifugal pumps that produce unidirectional flow. The device is unique in that the absence of rotating seals or bearings allows for minimal friction and shear stress, resulting in lower levels of complement activation. This device also has the potential to produce higher flows (up to 10 L/min) at lower rotations per minute (rpm). The CentriMag system can be inserted in the operating room87 or via a percutaneous approach.88 Beside cardiocirculatory support of up to 10 L/min, the device provides the possibility to function as an ECMO. CentriMag has been used for postcardiotomy shock with encouraging results, achieving a survival rate of around 50%.89 Complications with this system include vascular injury as well as peripheral embolization and infection.
In summary, although percutaneous VAD provides superior hemodynamic support in patients with cardiogenic shock compared with IABP, there is no evidence of improved early survival with the use of these more powerful devices (Table 73-7). The results of studies therefore do not yet support percutaneous VAD as a first-choice approach in the mechanical management of cardiogenic shock.
Treatment of Heart Failure with Normal Systolic Function
Outcome
Pulmonary edema is a severe presentation of AHF, with short-term mortality reported between 12% and 45%.91 As stated earlier, several prognostic factors can be identified at presentation, such as advanced age, altered renal function, and diminished level of oxygenation. Reported rates of short-term mortality in patients with cardiogenic pulmonary edema and myocardial infarction ranged between 46% and 80%, while patients without infarction had a significantly lower rate of short-term mortality. Only a few studies in specific populations have addressed the long-term prognosis of patients treated for pulmonary edema.92,93 These trials documented mortality rates as high as 40% at 1 year.
Noveanu M, Mebazaa A, Mueller C. Cardiovascular biomarkers in the ICU. Curr Opin Crit Care. 2009;15:377-383. Available at: http://www.ncbi.nlm.nih.gov/pubmed/19606027
A most important manuscript explaining in detail the value of biomarkers in the ICU.
Swan HJ, Ganz W, Forrester J, et al. Catheterization of the heart in man with use of a flow-directed balloon-tipped catheter. N Engl J Med. 1970;283:447-451. Available at: http://www.ncbi.nlm.nih.gov/pubmed/5434111
Dickstein K, Cohen-Solal A, Filippatos G, et al. ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2008: the Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2008 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association of the ESC (HFA) and endorsed by the European Society of Intensive Care Medicine (ESICM. Eur Heart J. 2008;29:2388-2442.
Stream JO, Grissom CK. Update on high-altitude pulmonary edema: pathogenesis, prevention, and treatment. Wilderness Environ Med. 2008;19:293-303. Available at: http://www.ncbi.nlm.nih.gov/pubmed/19099331
The most up-to-date and complete manuscript describing high-altitude pulmonary edema.
Sciscione AC, Ivester T, Largoza M, et al. Acute pulmonary edema in pregnancy. Obstet Gynecol. 2003;101:511-515. Available at: http://www.ncbi.nlm.nih.gov/pubmed/12636955
Paulus WJ, Tschöpe C, Sanderson JE, et al. How to diagnose diastolic heart failure: a consensus statement on the diagnosis of heart failure with normal left ventricular ejection fraction by the Heart Failure and Echocardiography Associations of the European Society of Cardiology. Eur Heart J. 2007;28:2539-2550. Available at: http://www.ncbi.nlm.nih.gov/pubmed/17428822
Hunt SA, Abraham WT, Chin MH, et al. 2009 focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation. 2009;119:e391-e479. Available at: http://www.ncbi.nlm.nih.gov/pubmed/19324966
1 Rimoldi SF, Yuzefpolskaya M, Allemann Y, Messerli F. Flash pulmonary edema. Prog Cardiovasc Dis. 2009;52(3):249-259.
2 Ware LB, Matthay MA. Clinical practice. Acute pulmonary edema. N Engl J Med. 2005;353(26):2788-2796.
3 Harrison MO, Conte PJ, Heitzman ER. Radiological detection of clinically occult cardiac failure following myocardial infarctionl. Br J Radiol. 1971;44(520):265-272. Available at: http://www.ncbi.nlm.nih.gov/pubmed/5552145
4 Praveen Peddu SR. Airspace Diseases. In: Adam: Grainger & Allison’s Diagnostic Radiology 5th ed.
5 Milne EN, Pistolesi M, Miniati M, Giuntini C. The radiologic distinction of cardiogenic and noncardiogenic edema. AJR. Am J Roentgenol. 1985;144(5):879-894. Available at: http://www.ncbi.nlm.nih.gov/pubmed/3872571
6 Collins SP, Lindsell CJ, Storrow AB, Abraham WT. Prevalence of negative chest radiography results in the emergency department patient with decompensated heart failure. Ann Emerg Med. 2006;47(1):13-18.
7 Volpicelli G, Cardinale L, Garofalo G, Veltri A. Usefulness of lung ultrasound in the bedside distinction between pulmonary edema and exacerbation of COPD. Emerg Radiol. 2008;15(3):145-151. Available at: http://www.ncbi.nlm.nih.gov/pubmed/18236088
8 Noveanu M, Mebazaa A, Mueller C. Cardiovascular biomarkers in the ICU. Curr Opin Crit Care. 2009;15(5):377-383. Available at: http://www.ncbi.nlm.nih.gov/pubmed/19606027
9 Glassberg H, Kirkpatrick J, Ferrari VA. Imaging studies in patients with heart failure: current and evolving technologies. Crit Care Med. 2008;36(Suppl. 1):S28-S39. Available at: http://www.ncbi.nlm.nih.gov/pubmed/18158474
10 Chenzbraun A, Pinto FJ, Schnittger I. Transesophageal echocardiography in the intensive care unit: impact on diagnosis and decision-making. Clin Cardiol. 1994;17(8):438-444.
11 Kim W, Poulsen J, Terp K, Staalsen N. A new doppler method for quantification of volumetric flow: In vivo validation using color doppler. J Am Coll Cardiol. 1996;27(1):182-192. Available at: http://www.ncbi.nlm.nih.gov/pubmed/8522693
12 Swan HJ, Ganz W, Forrester J, et al. Catheterization of the heart in man with use of a flow-directed balloon-tipped catheter. N Engl J Med. 1970;283(9):447-451. Available at: http://www.ncbi.nlm.nih.gov/pubmed/5434111
13 Dickstein K, Cohen-Solal A, Filippatos G, et al. ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2008: the Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2008 of the European Society of Cardiology. Developed in collaboration with the Heart. Eur Heart J. 2008;29(19):2388-2442.
14 Gore JM, Goldberg RJ, Spodick DH, Alpert JS, Dalen JE. A community-wide assessment of the use of pulmonary artery catheters in patients with acute myocardial infarction. Chest.. 1987;92(4):721-727. Available at: http://www.ncbi.nlm.nih.gov/pubmed/3652758
15 Connors AF, Speroff T, Dawson NV, et al. The effectiveness of right heart catheterization in the initial care of critically ill patients. SUPPORT Investigators. JAMA. 1996;276(11):889-897. Available at: http://www.ncbi.nlm.nih.gov/pubmed/8782638
16 Ivanov R, Allen J, Calvin JE. The incidence of major morbidity in critically ill patients managed with pulmonary artery catheters: a meta-analysis. Crit Care Med. 2000;28(3):615-619. Available at: http://www.ncbi.nlm.nih.gov/pubmed/10752803
17 Ivanov RI, Allen J, Sandham JD, Calvin JE. Pulmonary artery catheterization: a narrative and systematic critique of randomized controlled trials and recommendations for the future. New Horizons. 1997;5(3):268-276. Available at: http://www.ncbi.nlm.nih.gov/pubmed/9259342
18 Dalen JE, Bone RC. Is it time to pull the pulmonary artery catheter? JAMA. 1996;276(11):916-918. Available at: http://www.ncbi.nlm.nih.gov/pubmed/8782643
19 Pulmonary Artery Catheter Consensus Conference. Consensus statement. Crit Care Med. 1997;25:910-925.
20 Shah MR, Hasselblad V, Stevenson LW, et al. Impact of the pulmonary artery catheter in critically ill patients: meta-analysis of randomized clinical trials. JAMA. 2005;294(13):1664-1670. Available at: http://www.ncbi.nlm.nih.gov/pubmed/16204666
21 Sageman WS, Riffenburgh RH, Spiess BD. Equivalence of bioimpedance and thermodilution in measuring cardiac index after cardiac surgery. J Cardiothorac Vasc Anesth. 2002;16(1):8-14. Available at: http://www.ncbi.nlm.nih.gov/pubmed/11854871
22 Cotter G, Moshkovitz Y, Kaluski E, et al. Accurate, noninvasive continuous monitoring of cardiac output by whole-body electrical bioimpedance. Chest. 2004;125(4):1431-1440. Available at: http://www.ncbi.nlm.nih.gov/pubmed/15078756
23 Wong KL, Hou PC. The accuracy of bioimpedance cardiography in the measurement of cardiac output in comparison with thermodilution method. Acta Anaesthesiol Sin. 1996;34(2):55-59. Available at: http://www.ncbi.nlm.nih.gov/pubmed/9084523
24 Van De Water JM, Miller TW, Vogel RL, Mount BE, Dalton ML. Impedance cardiography: the next vital sign technology? Chest. 2003;123(6):2028-2033. Available at: http://www.ncbi.nlm.nih.gov/pubmed/12796185
25 Atallah MM, Demain AD. Cardiac output measurement: lack of agreement between thermodilution and thoracic electric bioimpedance in two clinical settings. J Clin Anesth. 1995;7(3):182-185. Available at: http://www.ncbi.nlm.nih.gov/pubmed/7669305
26 Barry B, Mallick A, Bodenham A, Vucevic M. Lack of agreement between bioimpedance and continuous thermodilution measurement of cardiac output in intensive care unit patients. Crit Care. 1997;1(2):71-74. Available at: http://www.ncbi.nlm.nih.gov/pubmed/11056698
27 Gödje O, Höke K, Goetz AE, et al. Reliability of a new algorithm for continuous cardiac output determination by pulse-contour analysis during hemodynamic instability. Crit Care Med. 2002;30(1):52-58. Available at: http://www.ncbi.nlm.nih.gov/pubmed/11902287
28 Mayer J, Boldt J, Schöllhorn T, et al. Semi-invasive monitoring of cardiac output by a new device using arterial pressure waveform analysis: a comparison with intermittent pulmonary artery thermodilution in patients undergoing cardiac surgery. Br J Anaesth. 2007;98(2):176-182. Available at: http://www.ncbi.nlm.nih.gov/pubmed/17218375
29 Mayer J, Boldt J, Wolf MW, Lang J, Suttner S. Cardiac output derived from arterial pressure waveform analysis in patients undergoing cardiac surgery: validity of a second generation device. Anesth Analg. 2008;106(3):867-872. table of contents. Available at: http://www.ncbi.nlm.nih.gov/pubmed/18292432
30 Pittman J, Bar-Yosef S, SumPing J, Sherwood M, Mark J. Continuous cardiac output monitoring with pulse contour analysis: a comparison with lithium indicator dilution cardiac output measurement. Crit Care Med. 2005;33(9):2015-2021. Available at: http://www.ncbi.nlm.nih.gov/pubmed/16148474
31 Baggish MS, Sze EH. Endometrial ablation: a series of 568 patients treated over an 11-year period. Am J Obstet Gynecol. 1996;174(3):908-913.
32 Khuri SF, Daley J, Henderson W, et al. The National Veterans Administration Surgical Risk Study: risk adjustment for the comparative assessment of the quality of surgical care. J Am Coll Surg. 1995;180(5):519-531. Available at: http://www.ncbi.nlm.nih.gov/pubmed/7749526
33 Stream JO, Grissom CK. Update on high-altitude pulmonary edema: pathogenesis, prevention, and treatment. Wilderness Environ Med. 2008;19(4):293-303. Available at: http://www.ncbi.nlm.nih.gov/pubmed/19099331
34 Hackett PH, Rennie D, Levine HD. The incidence, importance, and prophylaxis of acute mountain sickness. Lancet.. 1976;2(7996):1149-1155. Available at: http://www.ncbi.nlm.nih.gov/pubmed/62991
35 Poggi SH, Barr S, Cannum R, et al. Risk factors for pulmonary edema in triplet pregnancies. J Perinatol. 2003;23(6):462-465. Available at: http://www.ncbi.nlm.nih.gov/pubmed/13679932
36 Sciscione AC, Ivester T, Largoza M, et al. Acute pulmonary edema in pregnancy. Obstet Gynecol. 2003;101(3):511-515. Available at: http://www.ncbi.nlm.nih.gov/pubmed/12636955
37 Bauer ST, Cleary KL. Cardiopulmonary complications of pre-eclampsia. Semin Perinatol. 2009;33(3):158-165.
38 Desai DK, Moodley J, Naidoo DP, Bhorat I. Cardiac abnormalities in pulmonary oedema associated with hypertensive crises in pregnancy. Br J Obstet Gynaecol. 1996;103(6):523-528.
39 Pisani RJ, Rosenow EC. Pulmonary edema associated with tocolytic therapy. Ann Intern Med. 1989;110(9):714-718. Available at: http://www.ncbi.nlm.nih.gov/pubmed/2648928
40 Vaast P, Dubreucq-Fossaert S, Houfflin-Debarge V, et al. Acute pulmonary oedema during nicardipine therapy for premature labour; Report of five cases. Eur J Obstet, Gynecol Reprod Biol. 2004;113(1):98-99. Available at: http://www.ncbi.nlm.nih.gov/pubmed/15036720
41 Galvis AG. Pulmonary edema complicating relief of upper airway obstruction. Am J Emerg Med. 1987;5(4):294-297. Available at: http://www.ncbi.nlm.nih.gov/pubmed/3593494
42 Van Kooy MA, Gargiulo RF. Postobstructive pulmonary edema. Am Fam Physician. 2000;62(2):401-404. Available at: http://www.ncbi.nlm.nih.gov/pubmed/10929702
43 Fremont RD, Kallet RH, Matthay MA, Ware LB. Postobstructive pulmonary edema: a case for hydrostatic mechanisms. Chest.. 2007;131(6):1742-1746.
44 Sherman SC. Reexpansion pulmonary edema: a case report and review of the current literature. J Emerg Med. 2003;24(1):23-27. Available at: http://www.ncbi.nlm.nih.gov/pubmed/12554036
45 Kim YK, Kim H, Lee CC, et al. New classification and clinical characteristics of reexpansion pulmonary edema after treatment of spontaneous pneumothorax. Am J Emerg Med. 2009;27(8):961-967. Available at: http://www.ncbi.nlm.nih.gov/pubmed/19857415
46 Matsuura Y, Nomimura T, Murakami H, et al. Clinical analysis of reexpansion pulmonary edema. Chest.. 1991;100(6):1562-1566. Available at: http://www.ncbi.nlm.nih.gov/pubmed/1959396
47 Baumann MH, Strange C, Heffner JE, et al. Management of spontaneous pneumothorax: an American College of Chest Physicians Delphi consensus statement. Chest.. 2001;119(2):590-602.
48 Rana R, Fernández-Pérez ER, Khan SA, et al. Transfusion-related acute lung injury and pulmonary edema in critically ill patients: a retrospective study. Transfusion.. 2006;46(9):1478-1483. Available at: http://www.ncbi.nlm.nih.gov/pubmed/16965572
49 Toy P, Popovsky MA, Abraham E, et al. Transfusion-related acute lung injury: definition and review. Crit Care Med. 2005;33(4):721-726. Available at: http://www.ncbi.nlm.nih.gov/pubmed/15818095
50 Finlay HE, Cassorla L, Feiner J, Toy P. Designing and testing a computer-based screening system for transfusion-related acute lung injury. Am J Clin Pathol. 2005;124(4):601-609.
51 Skeate RC, Eastlund T. Distinguishing between transfusion related acute lung injury and transfusion associated circulatory overload. Curr Opin Hematol. 2007;14(6):682-687. Available at: http://www.ncbi.nlm.nih.gov/pubmed/17898575
52 Reissman P, Manny N, Shapira SC, Shapira Y, Cotev S. Transfusion-related adult respiratory distress syndrome. Israel J Med Sci. 1993;29(5):303-307. Available at: http://www.ncbi.nlm.nih.gov/pubmed/8314693
53 Sporer KA. Acute heroin overdose. Ann Intern Med. 1999;130(7):584-590. Available at: http://www.ncbi.nlm.nih.gov/pubmed/10189329
54 Sklar J, Timms RM. Codeine-induced pulmonary edema. Chest.. 1977;72(2):230-231. Available at: http://www.ncbi.nlm.nih.gov/pubmed/884987
55 O’Malley GF. Emergency department management of the salicylate-poisoned patient. Emerg Med Clin North Am. 2007;25(2):333-346. abstract viii. Available at: http://www.ncbi.nlm.nih.gov/pubmed/17482023
56 Baumann A, Audibert G, McDonnell J, Mertes PM. Neurogenic pulmonary edema. Acta Anaesthesiol Scand. 2007;51(4):447-455.
57 Schraufnagel DE, Thakkar MB. Pulmonary venous sphincter constriction is attenuated by alpha-adrenergic antagonism. Am REv Respir Dis. 1993;148(2):477-482. Available at: http://www.ncbi.nlm.nih.gov/pubmed/8393639
58 Peters CJ, Khan AS. Hantavirus pulmonary syndrome: the new American hemorrhagic fever. Clin Infect Dis. 2002;34(9):1224-1231. Available at: http://www.ncbi.nlm.nih.gov/pubmed/11941549
59 Hopkins SR, Sheel AW, McKenzie DC. Point: Counterpoint “Pulmonary edema does/does not occur in human athletes performing heavy sea-level exercise.”. J Appl Physiol. 2010 Jan 7. [Epub ahead of print]. Available at: http://www.ncbi.nlm.nih.gov/pubmed/20056844
60 Lund KL, Mahon RT, Tanen DA, Bakhda S. Swimming-induced pulmonary edema. Ann Emerg Med. 2003;41(2):251-256. Available at: http://www.ncbi.nlm.nih.gov/pubmed/12548277
61 Cotter G, Felker GM, Adams KF, Milo-Cotter O, O’Connor CM. The pathophysiology of acute heart failure–is it all about fluid accumulation? Am Heart J. 2008;155(1):9-18.
62 Stevenson LW. Tailored therapy to hemodynamic goals for advanced heart failure. Eur Heart J. 1999;1(3):251-257. Available at: http://www.ncbi.nlm.nih.gov/pubmed/10935671
63 Kumar R, Gandhi SK, Little WC. Acute heart failure with preserved systolic function. Crit Care Med. 2008;36(Suppl. 1):S52-S56. Available at: http://www.ncbi.nlm.nih.gov/pubmed/18158478
64 Paulus WJ, Tschöpe C, Sanderson JE, et al. How to diagnose diastolic heart failure: a consensus statement on the diagnosis of heart failure with normal left ventricular ejection fraction by the Heart Failure and Echocardiography Associations of the European Society of Cardiology. Eur Heart J. 2007;28(20):2539-2550. Available at: http://www.ncbi.nlm.nih.gov/pubmed/17428822
65 Hunt SA, Abraham WT, Chin MH, et al. 2009 focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation.. 2009;119(14):e391-e479. Available at: http://www.ncbi.nlm.nih.gov/pubmed/19324966
66 Diuretic dosing trial to be published. Available at. http://www.cardiosource.com/clinicaltrials/trial.asp?trialID=1924.
67 Sosnowski MA. Review article: lack of effect of opiates in the treatment of acute cardiogenic pulmonary oedema. Emerg Med Australas. 2008;20(5):384-390. Available at: http://www.ncbi.nlm.nih.gov/pubmed/18973635
68 Peacock WF, Hollander JE, Diercks DB, et al. Morphine and outcomes in acute decompensated heart failure: an ADHERE analysis. Emerg Med J. 2008;25(4):205-209. Available at: http://www.ncbi.nlm.nih.gov/pubmed/18356349
69 Konstam MA, Gheorghiade M, Burnett JC, et al. Effects of oral tolvaptan in patients hospitalized for worsening heart failure: the EVEREST Outcome Trial. JAMA. 2007;297(12):1319-1331. Available at: http://www.ncbi.nlm.nih.gov/pubmed/17384437
70 Sackner-Bernstein JD, Skopicki HA, Aaronson KD. Risk of worsening renal function with nesiritide in patients with acutely decompensated heart failure. Circulation.. 2005;111(12):1487-1491. Available at: http://www.ncbi.nlm.nih.gov/pubmed/15781736
71 Sackner-Bernstein JD, Kowalski M, Fox M, Aaronson K. Short-term risk of death after treatment with nesiritide for decompensated heart failure: a pooled analysis of randomized controlled trials. JAMA. 2005;293(15):1900-1905. Available at: http://www.ncbi.nlm.nih.gov/pubmed/15840865
72 Schulz R, Rose J, Martin C, Brodde OE, Heusch G. Development of short-term myocardial hibernation. Its limitation by the severity of ischemia and inotropic stimulation. Circulation.. 1993;88(2):684-695. Available at: http://www.ncbi.nlm.nih.gov/pubmed/8393390
73 Oliva F, Latini R, Politi A, et al. Intermittent 6-month low-dose dobutamine infusion in severe heart failure: DICE multicenter trial. Am Heart J. 1999;138(2 Pt 1):247-253. Available at: http://www.ncbi.nlm.nih.gov/pubmed/10426835
74 Thackray S, Easthaugh J, Freemantle N, Cleland JG. The effectiveness and relative effectiveness of intravenous inotropic drugs acting through the adrenergic pathway in patients with heart failure-a meta-regression analysis. Eur J Heart Fail. 2002;4(4):515-529. Available at: http://www.ncbi.nlm.nih.gov/pubmed/12167393
75 Mebazaa A, Nieminen MS, Packer M, et al. Levosimendan vs dobutamine for patients with acute decompensated heart failure: the SURVIVE Randomized Trial. JAMA. 2007;297(17):1883-1891. Available at: http://www.ncbi.nlm.nih.gov/pubmed/17473298
76 Stone GW, Ohman EM, Miller MF, et al. Contemporary utilization and outcomes of intra-aortic balloon counterpulsation in acute myocardial infarction: the benchmark registry. J Am Coll Cardiol. 2003;41(11):1940-1945. Available at: http://www.ncbi.nlm.nih.gov/pubmed/12798561
77 Antman EM, Anbe DT, Armstrong PW, et al. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction–executive summary. A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to revise the 1999. J Am Coll Cardiol. 2004;44(3):671-719. Available at: http://www.ncbi.nlm.nih.gov/pubmed/15358045
78 Sjauw KD, Engström AE, Vis MM, et al. A systematic review and meta-analysis of intra-aortic balloon pump therapy in ST-elevation myocardial infarction: should we change the guidelines? Eur Heart J. 2009;30(4):459-468. Available at: http://www.ncbi.nlm.nih.gov/pubmed/19168529
79 Prondzinsky R, Lemm H, Swyter M, et al. Intra-aortic balloon counterpulsation in patients with acute myocardial infarction complicated by cardiogenic shock: the prospective, randomized IABP SHOCK Trial for attenuation of multiorgan dysfunction syndrome. Crit Care Med. 2010;38(1):152-160. Available at: http://www.ncbi.nlm.nih.gov/pubmed/19770739
80 Seyfarth M, Sibbing D, Bauer I, et al. A randomized clinical trial to evaluate the safety and efficacy of a percutaneous left ventricular assist device versus intra-aortic balloon pumping for treatment of cardiogenic shock caused by myocardial infarction. J Am Coll Cardiol. 2008;52(19):1584-1588. Available at: http://www.ncbi.nlm.nih.gov/pubmed/19007597
81 Thiele H, Sick P, Boudriot E, et al. Randomized comparison of intra-aortic balloon support with a percutaneous left ventricular assist device in patients with revascularized acute myocardial infarction complicated by cardiogenic shock. Eur Heart J. 2005;26(13):1276-1283. Available at: http://www.ncbi.nlm.nih.gov/pubmed/15734771
82 Burkhoff D, Cohen H, Brunckhorst C, O’Neill WW. A randomized multicenter clinical study to evaluate the safety and efficacy of the TandemHeart percutaneous ventricular assist device versus conventional therapy with intraaortic balloon pumping for treatment of cardiogenic shock. Am Heart J. 2006;152(3):469.e1-469.e8. Available at: http://www.ncbi.nlm.nih.gov/pubmed/16923414
83 Hill JD, O’Brien TG, Murray JJ, et al. Prolonged extracorporeal oxygenation for acute post-traumatic respiratory failure (shock-lung syndrome). Use of the Bramson membrane lung. N Engl J Med. 1972;286(12):629-634. Available at: http://www.ncbi.nlm.nih.gov/pubmed/5060491
84 Schuerer DJ, Kolovos NS, Boyd KV, Coopersmith CM. Extracorporeal membrane oxygenation: current clinical practice, coding, and reimbursement. Chest. 2008;134(1):179-184. Available at: http://www.ncbi.nlm.nih.gov/pubmed/18628221
85 Rastan AJ, Dege A, Mohr M, et al. Early and late outcomes of 517 consecutive adult patients treated with extracorporeal membrane oxygenation for refractory postcardiotomy cardiogenic shock. J Thorac Cardiovasc Surg. 2010;139(2):302-311. 311.e1. Available at: http://www.ncbi.nlm.nih.gov/pubmed/20106393
86 Thiagarajan RR, Laussen PC, Rycus PT, Bartlett RH, Bratton SL. Extracorporeal membrane oxygenation to aid cardiopulmonary resuscitation in infants and children. Circulation. 2007;116(15):1693-1700. Available at: http://www.ncbi.nlm.nih.gov/pubmed/17893278
87 Gregoric ID, Cohn WE, Akay MH, et al. CentriMag left ventricular assist system: cannulation through a right minithoracotomy. Tex Heart Inst J. 2008;35(2):184-185. Available at: http://www.ncbi.nlm.nih.gov/pubmed/18612493
88 Aziz TA, Singh G, Popjes E, et al. Initial experience with CentriMag extracorporal membrane oxygenation for support of critically ill patients with refractory cardiogenic shock. J Heart Lung Transplant. 2010;29(1):66-71. Available at: http://www.ncbi.nlm.nih.gov/pubmed/19837609
89 De Robertis F, Birks EJ, Rogers P, et al. Clinical performance with the Levitronix Centrimag short-term ventricular assist device. J Heart Lung Transplant. 2006;25(2):181-186. Available at: http://www.ncbi.nlm.nih.gov/pubmed/16446218
90 Fonarow GC, Abraham WT, Albert NM, et al. Influence of beta-blocker continuation or withdrawal on outcomes in patients hospitalized with heart failure: findings from the OPTIMIZE-HF program. J Am Coll Cardiol. 2008;52(3):190-199.
91 Fiutowski M, Waszyrowski T, Krzemińska-Pakula M, Kasprzak JD. Pulmonary edema prognostic score predicts in-hospital mortality risk in patients with acute cardiogenic pulmonary edema. Heart Lung. 2008;37(1):46-53.
92 Goldberger JJ, Peled HB, Stroh JA, Cohen MN, Frishman WH. Prognostic factors in acute pulmonary edema. Arch Intern Med. 1986;146(3):489-493. Available at: http://www.ncbi.nlm.nih.gov/pubmed/3954520
93 Plotnick GD, Kelemen MH, Garrett RB, Randall W, Fisher ML. Acute cardiogenic pulmonary edema in the elderly: factors predicting in-hospital and one-year mortality. South Med J. 1982;75(5):565-569. Available at: http://www.ncbi.nlm.nih.gov/pubmed/7079813
