Chapter 4 Pulmonary Circulation
Circulatory Structure
Pulmonary Circulation
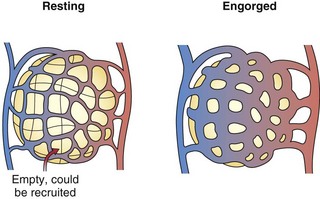
The pulmonary arteries lie near and branch in unison with the airways in the bronchovascular bundle. They are much thinner than systemic arteries and have proportionately more elastic tissue in their walls. The walls of the arterioles, with a diameter less than 100 µm, are so thin relative to those of their systemic counterparts that fluid and gas can move across them. Within the gas-exchanging zone, the arterioles give rise to a network of pulmonary capillaries in the alveolar walls that is continuous throughout the lungs. They are so numerous that, when distended, blood flows almost as an unbroken sheet between the air spaces (Figure 4-1). “Sheet flow” reduces vascular resistance and optimizes gas exchange by creating a very large surface area, estimated at over 100 m2. When the transmural pressure difference between the inside and outside of the vessels is low, many of the capillary segments are closed, but flow switches among segments frequently as some open and others close. Nonflowing segments are rapidly recruited into the pulmonary vascular bed as needed to accommodate increased flow and may be further distended by an increase in transmural pressure. Both recruitment and distention of the pulmonary capillary bed reduce resistance to blood flow and help to maintain a low pressure in the face of increased blood flow. This low pressure allows the capillary-alveolar membrane to be very thin (approximately 1 µm), facilitating diffusion of respiratory gases between blood and alveoli. A red cell that follows a capillary path from the pulmonary artery to a vein may cross several alveoli, with the average transit time through the vessels engaged in gas exchange calculated to be approximately 0.75 second. The capillaries unite to form larger alveolar microvessels, which become venules and then veins that run between the lobules toward the hila, where upper and lower pulmonary veins from each lung empty into the left atrium.
Circulatory Physiology
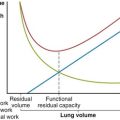
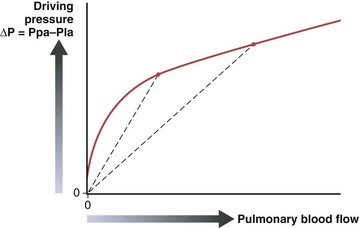
The pulmonary vascular resistance, PVR, is calculated as transvascular driving pressure, ΔP (mean upstream Ppa minus mean downstream Pla), divided by the flow: PVR = ΔP/Q. The calculated resistance must be interpreted in the context of flow, because the relationship of driving pressure to flow usually is not linear and its plotted curve does not pass through zero. As shown in Figure 4-2, pulmonary vascular resistance decreases as flow and pressure increase with the attendant recruitment and distention of vessels.
The resistance to flow through a vessel increases with its length, with the viscosity of the fluid, and, most important, with the inverse of the radius to the fourth power. In addition to muscle activity in the wall, the caliber of a distensible vessel depends passively on the transmural pressure difference between intravascular and extravascular pressures. This mechanism is particularly important in the lungs, where the vessels are embedded in expandable parenchyma. It is convenient to consider separately the effect of lung expansion on the extraalveolar arterial and venous vessels, which differs from the effect on the microvessels of the alveolar zone. With lung volume increase, extraalveolar vessels are distended as the pressure is lowered in the expanding perivascular space around them (Figure 4-3), and they are elongated as the lung expands.
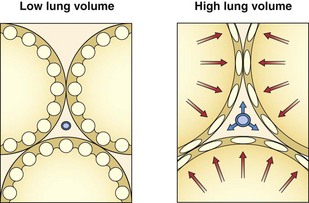
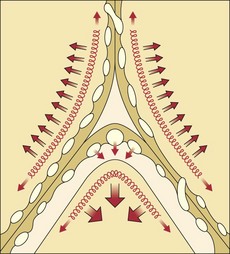
By contrast, the alveolar microvessels in the alveolar walls are elongated but partially collapsed by lung inflation, because the alveolar pressure that surrounds them tends to increase relative to the intravascular pressure. This effect is easy to recognize with positive-pressure ventilation, but it also occurs with spontaneous inspiration, because intravascular pressures fall relative to atmospheric and alveolar pressure. The sheets of capillaries in the alveolar walls are protected from the full compressive force of the alveolar pressure by the surface tension of the fluid that lines curved portions of the alveolar surface. Microvessels in the “corners” where alveolar walls meet are more fully protected from compression by the sharper curvature of the surface film and perhaps by local distending forces, analogous to the situation with extraalveolar vessels (Figure 4-4). The pulmonary vascular resistance is the sum of that through alveolar and extraalveolar vessels and thus has a complex relationship with lung volume. It is lowest at approximately the normal resting lung volume (functional residual capacity) but increases at higher and lower volumes.
Blood Flow Distribution
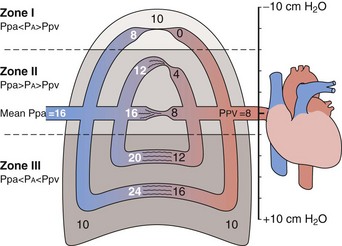
The gravitational effect has been conceptualized by dividing the lung into four zones, one above another, on the basis of the relationship of vascular and alveolar pressures (Figure 4-5). Intravascular pressures are higher at the bottom of the lung than at the top by an amount equal to a vertical hydrostatic column as high as the lung. Near the lung apex, zone I, the pressure in the alveoli (PA) exceeds that in both the pulmonary arteries (Ppa) and pulmonary vein (Ppv) and collapses the alveolar vessels, except those in the alveolar corners, which remain patent and allow some flow to continue. Below this, in zone II, Ppa exceeds PA, but PA is greater than Ppv, so flow depends on the pressure difference between Ppa and PA. The vessels remain open but are critically narrowed at the downstream end, where venous pressure is lower than alveolar pressure. This condition creates independence of flow from the downstream venous pressure, analogous to a waterfall in which a stream that flows over a precipice is unaffected by a rising level in the pool below until it rises above the level of the lip. In the middle to lower portion of the lung, zone III, both Ppa and Ppv exceed PA, the vessels are distended, and blood flow is the highest. Zone IV is restricted to a small area in the most dependent region, where flow diminishes. It has been postulated that this reduction is the result of increased vascular resistance secondary to low lung volume or perivascular edema in this area.
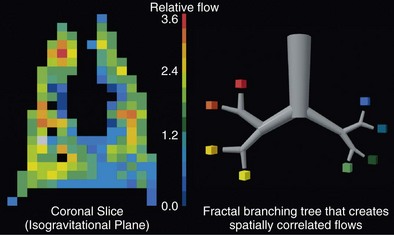
Although the gravitational effect expressed in the vertical zone concept contributes to the average increase in flow down the lung, it does not explain the observed large variability in flow within an isogravitational slice, which implies that other anatomic or vasoregulatory factors are important at this level. More recent studies have determined that the heterogeneous distribution of blood flow within horizontal (isogravitational) planes is due to asymmetric branching geometries (and hence resistances) of the vascular tree. Because the vascular tree is largely a dichotomous branching structure, differences in resistances between daughter branches cause flow to be distributed unevenly between the branches. With differences in resistances occurring at every bifurcation in the vascular tree, blood flow becomes progressively more heterogeneous, resulting in a broad distribution of flows at the terminal branches. Owing to the shared heritage up the vascular tree, neighboring lung regions have similar magnitudes of flow, with high-flow regions near other high-flow regions and low-flow regions neighboring other low-flow regions. Hence, the spatial distribution of pulmonary blood flow is not random but rather exhibits a clear pattern of high and low flows (Figure 4-6). Studies have demonstrated that the pattern of perfusion distribution is very stable over time and with growth, and that the pattern is genetically determined. These insights provide a new perspective on blood flow distribution in the lung. The traditional model of vertically stacked zones needs to be replaced by one in which the multiple zones can exist within horizontal planes. In addition, the large degree of heterogeneity within isogravitational planes suggests that mechanisms other than gravity must be responsible for the tight matching between regional ventilation and blood flow.
Regulation of Pulmonary Blood Flow
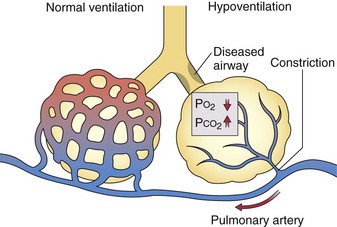
Although the role of nasal nitric oxide in ventilation-perfusion matching is still speculative, the role of alveolar hypoxia in vasoregulation has been recognized for more than 50 years, but the mechanisms involved are still uncertain. Pulmonary arterioles constrict when the PO2 in the alveoli they serve falls, and additional vasoconstriction results if alveolar PCO2 rises (Figure 4-7). Thus, when ventilation is decreased by an obstructed airway or other injury, local hypoxic pulmonary vasoregulation decreases blood flow to the affected region, which tends to restore the local ventilation-perfusion ratio (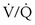 ) toward normal and thereby improve the PO2 of the blood flowing through that area. The diverted blood flow can be directed to better-ventilated regions, which further contributes to an improvement in overall matching. This hypoxic vasoconstriction seems to be a response to a low PO2 in the air spaces, rather than in the intraluminal blood, which normally is desaturated in these prealveolar vessels. The effector cell is thought to be pulmonary artery smooth muscle located at the entrance to the acinus, and the sensor may be the oxygen-consuming mitochondria within these cells. Several candidate signaling pathways to generate the increase in intracellular calcium necessary for muscle contraction are under investigation.
) toward normal and thereby improve the PO2 of the blood flowing through that area. The diverted blood flow can be directed to better-ventilated regions, which further contributes to an improvement in overall matching. This hypoxic vasoconstriction seems to be a response to a low PO2 in the air spaces, rather than in the intraluminal blood, which normally is desaturated in these prealveolar vessels. The effector cell is thought to be pulmonary artery smooth muscle located at the entrance to the acinus, and the sensor may be the oxygen-consuming mitochondria within these cells. Several candidate signaling pathways to generate the increase in intracellular calcium necessary for muscle contraction are under investigation.
Nonrespiratory Functions of the Pulmonary Circulation
Fluid Exchange in the Pulmonary Circulation
The fluid flux across the pulmonary vascular endothelium is influenced by the same pressure relationship as in the systemic capillaries, summarized in the modified Starling equation presented in Table 4-1. The hydrostatic pressure in the pulmonary microvessels (Pmv) exceeds the interstitial hydrostatic pressure outside the microvessels, i.e., the perimicrovascular pressure (Ppmv). This effect favors filtration. The interstitial tissue fluid protein osmotic pressure is approximately two thirds that in the vessel; thus, the net osmotic force is absorptive and inward. The components of this equation make it convenient to categorize abnormal fluid flux into the lung into two broad types: hydrostatic edema, when the primary abnormality is an increase in Pmv minus Ppmv, and permeability edema, when endothelial injury increases fluid conductivity across the membrane (incorporated into the permeability factor) and decreases the osmotic reflection coefficient and osmotic gradient. The terms cardiogenic and noncardiogenic also are commonly used for these two mechanisms of edema formation.
| F = Kf [(Pmv − Ppmv) − σ(pv − pt)] | |
| Symbol | Description |
| F | Net fluid flux out of vessels |
| Kf | Permeability factor (constant) |
| σ | Reflection coefficient for oncotic agents |
| Pmv | Pressure in microvessels |
| Ppmv | Perimicrovascular pressure |
| pv | Osmotic pressure in vessels |
| pt | Osmotic pressure of tissues |
Surface tension in the fluid film that lines the alveoli opposes alveolar pressure and tends to lower the interstitial pressure around pulmonary microvessels, particularly in corner areas (see Figure 4-4). An increase in surface tension may contribute to edema when surfactant is lost in an injured lung. Interstitial pressure around the extraalveolar vessels is close to intrathoracic (pleural) pressure and falls as the lungs are distended, which favors relatively more leakage from them at high rather than low lung volumes (see Figure 4-3).
Alveolar Edema
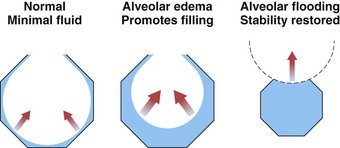
The epithelial cells that line the air spaces have tight junctions along their apical surface, so this membrane normally is even less permeable than the endothelial membrane, protecting alveolar spaces as interstitial edema increases. After total lung water has increased by approximately 50%, the edema fluid appears in the alveoli. A structural failure, at the epithelial cell junctions or elsewhere, is suspected, because there is no protein gradient between interstitial and alveolar edema fluid. Fluid initially is seen only in the corners of the alveoli, where surface tension causes the pressure below the curved fluid film to be lowest. As more fluid accumulates, the alveoli rapidly become completely filled, again because of surface tension effects. As alveoli fill, the radius of the curvature of the meniscus of the fluid becomes shorter, and the effect of surface tension becomes greater (Laplace’s law), which pulls fluid in more strongly (Figure 4-8). Thus, the sequence of edema development progresses from the perimicrovascular interstitium to peribronchovascular “sump” to patchy alveolar flooding.
Respiratory-Circulatory Interactions
Positive-Pressure Ventilation
When patients are mechanically ventilated with positive inspiratory pressure, the same mechanisms seen in spontaneous breathing are involved, but the pressure effects shift phase in the tidal cycle. For example, the pressure outside the left ventricle rises during inspiration, so the same contraction yields a higher blood pressure early in the inspiratory phase. This increase may be augmented by blood pushed out of the capillaries by the positive alveolar pressure (see Figure 4-3). During late inspiration or early expiration, the blood pressure decreases as the effect of an inspiratory decrease in venous return to the right heart reaches the left side. If the expiratory phase is long enough, the blood pressure will begin to rise, reflecting enhanced venous return to the right heart earlier in expiration.
When an increase in end-expiratory lung volume is recruited by PEEP, the chest wall also must be passively expanded, and its pressure-volume relationship (see Chapter 3, Figure 3-7) would predict at least a modest increase in pleural pressure. As shown by direct measurement with suitable flat devices, however, when the lungs are distended with PEEP, the pressure in the cardiac fossa may rise more than that measured by an esophageal balloon, and the pressure in the pericardium may be still higher. Bedside measurements of a decreased cardiac output accompanied by a higher pulmonary arterial occlusion pressure may suggest a decrease in cardiac function or contractility, but when accurate measurements of juxtacardiac pressure or left ventricular end-diastolic volume are made, the ventricle is seen to be operating at a lower preload on the same function curve. The same phenomenon may be seen when patients with severe airflow obstruction develop dynamic hyperinflation with an associated increase in cardiac fossa pressure.
Butler J, ed. The bronchial circulation, vol 57 of Lenfant C, editor: Lung biology in health and disease. New York: Marcel Dekker, 1992.
Culver BH, Butler J. Mechanical influences on the pulmonary circulation. Annu Rev Physiol. 1980;42:187–198.
Paredi P, Barnes PJ. The airway vasculature: recent advances and clinical implications. Thorax. 2009;64:444–450.
Weibel ER. What makes a good lung? The morphometric basis of lung function. Swiss Med Wkly. 2009;139:375–386.
Blood Flow Regulation and Distribution
Baumgartner WAJr, Jaryszak EM, Peterson AJ, et al. Heterogeneous capillary recruitment among adjoining alveoli. J Appl Physiol. 2003;95:469–476.
Glenny RW. Determinants of regional ventilation and blood flow in the lung. Intensive Care Med. 2009;35:1833–1842.
Glenny RW, Bernard S, Robertson HT, Hlastala MP. Gravity is an important but secondary determinant of regional pulmonary blood flow in upright primates. J Appl Physiol. 1999;86:623–632.
Glenny RW, Robertson HT. Regional differences in the lung: a changing perspective on blood flow distribution. In: Hlastala MP, Robertson HT. Complexity in structure and function of the lung, vol 121 of Lenfant C, editor: Lung biology in health and disease. New York: Marcel Dekker; 1998:461–481.
Prisk GK, Yamada K, Henderson AC, et al. Pulmonary perfusion in the prone and supine postures in the normal human lung. J Appl Physiol. 2007;103:883–894.
Sommer N, Dietrich A, Schermuly RT, et al. Regulation of hypoxic pulmonary vasoconstriction: basic mechanisms. Eur Respir J. 2008;32:1639–1651.
Bhattacharya J. Physiological basis of pulmonary edema. In: Matthay M, Ingbar D. Pulmonary edema, vol 116 of Lenfant C, editor: Lung biology in health and disease. New York: Marcel Dekker, 1998.
Effros RM, Parker JC. Pulmonary vascular heterogeneity and the Starling hypothesis. Microvasc Res. 2009;78:71–77.
Matthay M, Folkesson HG, Clerici C. Lung epithelial fluid transport and the resolution of pulmonary edema. Physiol Rev. 2002;82:569–600.
Ware LB, Matthay MA. Clinical practice. Acute pulmonary edema. N Engl J Med. 2005;353:2788–2796.
Bartsch P, Mairbaurl H, Maggiorini M, Swenson E. Physiological aspects of high-altitude pulmonary edema. J Appl Physiol. 2005;98:1101–1110.
Scherrer U, Rexhaj E, Jayet PY, et al. New insights in the pathogenesis of high-altitude pulmonary edema. Prog Cardiovasc Dis. 2010;52:485–492.
Respiratory-Circulatory Interactions
Feihl F, Broccard AF. Interactions between respiration and systemic hemodynamics. Part 1: basic concepts. Intensive Care Med. 2009;35:45–54.
Marini JJ, Culver BH, Butler J. Mechanical effects of lung distension with positive pressure on cardiac function. Am Rev Respir Dis. 1981;124:382–386.
Tyberg JV, Grant DA, Kingma I, et al. Effects of positive intrathoracic pressure on pulmonary and systemic hemodynamics. Respir Physiol. 2000;119:163–171.