[level-membership-for-cardiovascular-category]
Chapter 8 Pulmonary circulation
The pulmonary circulation is contained wholly within the thorax. It is, therefore, much shorter than most regional beds of the systemic circulation. As well, all segments of the pulmonary vessels are slightly larger in radius than the corresponding segments of the systemic vasculature. The net result of these two factors is that the pulmonary circulation exerts a resistance to flow only around 15% of that in the systemic circuit. Hence it requires a correspondingly far lower pressure gradient in order to move the same cardiac output. At rest, pulmonary arterial pressure is typically 25/8 mmHg, giving a mean arterial pressure of the order of 14 mmHg, and mean pulmonary capillary pressure is 7–8 mmHg rather than the 25–30 mmHg seen in systemic vascular beds.
FUNCTIONAL CONSEQUENCES OF LOW PULMONARY BLOOD PRESSURE
Gas diffusion
In systemic capillaries the balance between oncotic and hydrostatic pressures mean that small increases in hydrostatic pressure caused, for example, by reduced arteriolar resistance will result in significant movement of plasma water into the interstitium. When a large tissue mass, such as skeletal muscle, is involved, this movement will cause a substantial reduction of plasma volume (see Chapter 9, p. 112), but the increased interstitial volume does not prejudice diffusion of solutes between cells and bloodstream because the intercellular connective tissue minimizes tissue expansion. In the lung, by contrast, there is little supporting tissue. If water moves into the interstitial space separating the pulmonary capillaries from the alveoli, it pushes these two structures further apart and increases the distance over which gases must diffuse between air and plasma.
The practising exercise physiologist is most likely to encounter pulmonary oedema as a consequence of high altitude, since it occurs in many healthy individuals who ascend to heights greater than 3000 m, well below the altitude of many ski resorts and permanent settlements. We will return to examine the reasons for this response to altitude in Chapter 12 (p. 151).
Right ventricular function
Left ventricular perfusion occurs only during diastole because during systole the left coronary vessels are compressed by the surrounding cardiac muscle (see Chapter 2, p. 6). By contrast, since right ventricular pressure does not normally rise above 25 mmHg, the coronary vasculature supplying the right ventricle is not compressed at any stage of the cardiac cycle and coronary flow is continuous. The increased efficiency of coronary oxygen delivery is reflected in a lower density of coronary blood vessels in the right heart. However, this design feature has potentially disadvantageous results if left atrial pressure becomes chronically elevated and right ventricular pressure rises in response to this higher afterload.
Under these circumstances, right coronary perfusion during systole becomes progressively reduced and the metabolic demands of the right-side cardiac muscle cells are less efficiently serviced. Over time, the right ventricle will respond to the high afterload by muscle hypertrophy. This will exaggerate the coronary insufficiency, both by providing greater metabolic demand for oxygen delivery and by producing more coronary compression. As a result, chronic hypoxic damage to the muscle of the right heart, with an inability to increase contraction appropriately in response to increased filling (right cardiac failure) is a common long-term effect of elevated pulmonary blood pressure (see Chapter 12, p. 148).
REGIONAL MATCHING OF VENTILATION AND PERFUSION
Vertical variations
Although both ventilation and perfusion rise towards the base of the lungs, the gravitational effect on regional blood flow is rather more powerful than the vertical variation in ventilation, with the result that the ratio of ventilation  to perfusion
to perfusion  is around 3 at the apices and around 0.5 at the bases. Exact matching of the two is achieved only over a relatively narrow region of lung corresponding to about the mid-sternal level (Fig. 8.1).
is around 3 at the apices and around 0.5 at the bases. Exact matching of the two is achieved only over a relatively narrow region of lung corresponding to about the mid-sternal level (Fig. 8.1).
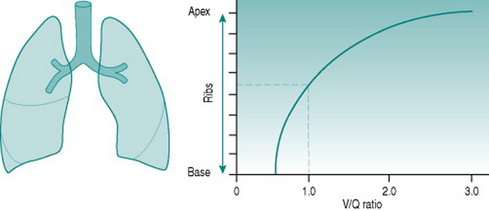
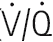 ) ratio variation in the vertical lung. At rest, optimal matching (
) ratio variation in the vertical lung. At rest, optimal matching (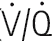 = 1.0) occurs only over a relatively narrow region around the mid-sternal area.
= 1.0) occurs only over a relatively narrow region around the mid-sternal area.Effects of intrapulmonary gas tensions
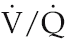 matching, it is clearly more efficient if the pulmonary blood flow bypasses these alveoli and is directed entirely to regions in which the concentration gradients for gas exchange are optimal.
matching, it is clearly more efficient if the pulmonary blood flow bypasses these alveoli and is directed entirely to regions in which the concentration gradients for gas exchange are optimal.To fulfil this purpose, hypoxia sensors exist within the pulmonary arteriolar walls that monitor the oxygen concentration in adjacent alveoli (the PAO2). We have seen previously that this receptor type is found in systemic arterioles, where it mediates a hyperpolarizing vasodilator response to hypoxia (Chapter 6, p. 68). In pulmonary arterioles, by contrast, hypoxia receptor activation causes muscle cell depolarization and vasoconstriction. The subcellular mechanisms that transduce this process are still the subject of debate; oxygen-sensitive potassium channels appear to be involved, but evidence exists also for release of some endothelium-derived vasoactive factor (Moudgil et al 2005). The terminal airways also possess receptors that detect locally inefficient ventilation – mainly by recognizing carbon dioxide build-up rather than oxygen depletion – and this dilates the airways and so helps re-oxygenate the under-ventilated alveoli. The interaction of arteriolar receptors for hypoxia and airway receptors for hypercapnia means that the regions of poor perfusion do not remain constant but oscillate in response to the oscillations in regional ventilation.
When hypoxic vasoconstriction affects small regions of the lung during normal air breathing, it helps optimize the efficiency of gas exchange. If, however, one were to breathe air that contained less oxygen than usual, then there would be widespread vasoconstriction and an increase in total pulmonary vascular resistance that would increase right heart workload. We shall look at this situation further when we examine the circulatory effects of high altitude in Chapter 12 (p. 143).
CHANGES IN PULMONARY EFFICIENCY WITH EXERCISE
In addition, the increased depth of breathing results in greater intrathoracic negative pressure during inspiration, which increases right atrial filling, right stroke volume and pulmonary arterial pressure. This produces more efficient perfusion of lung areas that are gravitationally above the heart. Collectively, these factors transform the variable regional 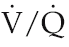 relationship to one that is virtually independent of vertical position (Fig. 8.2). In addition, distribution of blood flow over a greater number of pulmonary capillaries reduces the velocity of flow through each of these and so increases the time for oxygen loading.
relationship to one that is virtually independent of vertical position (Fig. 8.2). In addition, distribution of blood flow over a greater number of pulmonary capillaries reduces the velocity of flow through each of these and so increases the time for oxygen loading.
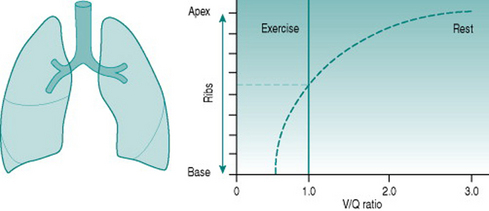
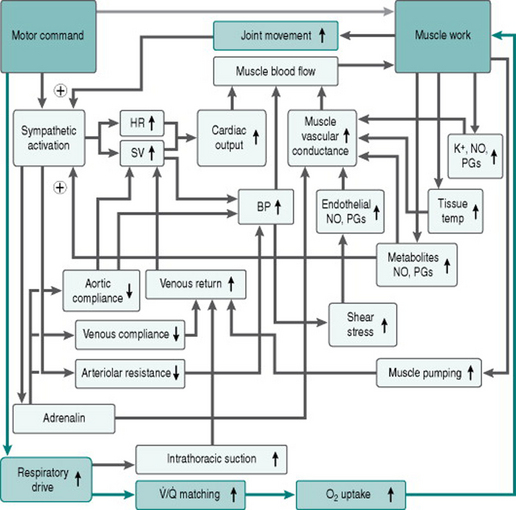
Figure 8.3 adds the contributions of pulmonary circulation and lungs to our flow chart of the exercise response.
Although the processes outlined above are able in most individuals to preserve optimal uptake of oxygen, even at the highest work intensities, there is one population in whom pulmonary gas exchange does limit work capacity under normoxic conditions. In a proportion of elite athletes with work capacities of the order of 70 ml O2/min/kg, arterial oxygen saturation falls from the normal of 96–97% to as low as around 90% at peak exercise (Stewart & Pickering 2006). It appears that these individuals generate cardiac outputs during exercise which are so high that, even with optimal 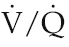 matching, there is not enough time for equilibration of oxygen between alveolar air and bloodstream. In addition, they can be predicted to be relatively more susceptible to any circumstances that limit inspired oxygen levels, such as those associated with ascent to high altitude (see Chapter 12, p. 144). Some sports scientists have made the case that these athletes should be classified as having an abnormal arterial oxygenation response to exercise. A more realistic interpretation would be that they simply have an extremely efficient cardiovascular response.
matching, there is not enough time for equilibration of oxygen between alveolar air and bloodstream. In addition, they can be predicted to be relatively more susceptible to any circumstances that limit inspired oxygen levels, such as those associated with ascent to high altitude (see Chapter 12, p. 144). Some sports scientists have made the case that these athletes should be classified as having an abnormal arterial oxygenation response to exercise. A more realistic interpretation would be that they simply have an extremely efficient cardiovascular response.
Discussion
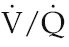 ratio of 1.0, minute ventilation would be expected to be similar in magnitude. In fact, however, minute ventilation was (0.75.14) or 10.5 l/min.
ratio of 1.0, minute ventilation would be expected to be similar in magnitude. In fact, however, minute ventilation was (0.75.14) or 10.5 l/min.This degree of hyperventilation might be expected as a result of peripheral chemoreceptor activation, since Robert’s arterial oxygen saturation of 87% equates to a PaO2 of around 60 mmHg – but what was the cause of the poor oxygenation? It could be because a proportion of the pulmonary vasculature was not perfused and the entire right cardiac output supplied a reduced volume of lung. This would increase the velocity of capillary blood flow and so reduce the time available for oxygen uptake. Alternatively, a proportion of the alveoli might not be ventilated, so that blood passing through some pulmonary capillaries remained unoxygenated. These possibilities can be distinguished by breathing pure oxygen. If the limitation is time for oxygen uptake, then this would be overcome by increasing the diffusional concentration gradient. On the other hand, dilution of normal arterial blood by deoxygenated blood returning from unventilated lung would persist regardless of efficiency of oxygen uptake in the remaining areas. In Robert’s case, the data clearly indicated presence of an unperfused area of lung.
PRACTICAL CONSEQUENCES OF LOW PULMONARY ARTERIAL PRESSURE
Kinetic pressure
In Chapter 3 (p. 28), we saw that intravascular pressures measured in the moving bloodstream can have a component that is due to the kinetic energy of fluid movement, depending on the orientation of the catheter tip. The value of this kinetic pressure is around 5 mmHg at rest, which is only a minor contribution to the total pressure in the systemic circulation. It is, on the other hand, a substantial proportion of pulmonary arterial pressure and very different values for pulmonary blood pressure would be recorded if a catheter had an end-opening or a side-opening tip. Remember also that the magnitude of the kinetic pressure component rises dramatically as cardiac output rises. If one is interpreting data on pulmonary pressures, especially during exercise, it is, therefore, important to know which catheter type was used, and it is equally important for the catheter type to be standardized between experiments where one wishes to compare pulmonary pressures.
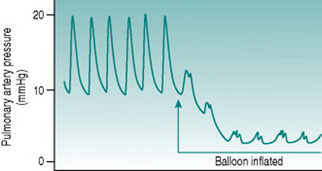
Pulmonary wedge pressure
Recording of this so-called wedge pressure is the only practicable way in which mechanical function in the left heart can be measured, because it is not technically possible to pass a catheter retrogradely up the aorta and back into the heart. In practice, the catheter used for recording wedge pressure (called a Swann-Ganz catheter) has a small balloon just behind the tip that can be inflated so as to guarantee that the wedged tip is isolated from the pressure upstream (Fig. 8.4).
IMPLICATIONS OF DEVELOPMENTAL CHANGES IN PULMONARY PERFUSION
Before birth, all foetal gas exchange takes place in the placenta where foetal venous and maternal arterial bloodstreams are separated only by thin membranes. As well, the foetal lungs are filled with relatively hypoxic amniotic fluid, so that the pulmonary microcirculation is compressed externally and is also constricted by activation of airways hypoxia receptors. At this time, therefore, the pulmonary circulation not only has no functional role, but also is a site of much higher vascular resistance than is the situation after birth. This creates higher systolic pressures in the fetal right atrium and ventricle, and pulmonary arteries than those in the corresponding parts of the left circulation. To prevent massive overwork of the right-side myocardium, most of the right cardiac output has to be short-circuited around the pulmonary circuit.
The shunting takes place in two ways (Moore 2003). The embryonic heart has only one atrium and one ventricle, which by around 8 weeks’ gestation has been partitioned into left and right sides by formation of intervening walls. The inter-atrial wall grows from both upper and lower extremities of the common atrium, with the two leaves overlapping each other but not fusing. This forms a valve (called the foramen ovale) by which a proportion of the venous blood returning to the right atrium can flow directly into the left atrium. The second shunting pathway is a short, muscular blood vessel that runs between the aortic arch and the pulmonary artery and is known as the ductus arteriosus. This vessel is maintained in a dilated state during fetal life by cells in its wall synthesizing large amounts of relaxant prostaglandins. In consequence, all of the relatively small right stroke volume of blood that was not shunted through the foramen ovale flows from pulmonary artery to aorta, bypassing the pulmonary microcirculation.
With any persistent ductus arteriosus and with some cases of persistent foramen ovale, there is chronic left–right shunting resulting in right heart overload. Intense exercise may not be advisable in such individuals and will certainly not be associated with entirely normal cardiorespiratory responses to exercise. The presence of these abnormalities is very likely to cause systolic murmurs and so, even though they are rare, checking for murmurs is a sensible safeguard in any naïve subject being admitted to an exercise study. The Case History in Chapter 12 (p. 151) returns to this theme.
Questions for revision
[/level-membership-for-cardiovascular-category][not-level-membership-for-cardiovascular-category]
Chapter 8 Pulmonary circulation
The pulmonary circulation is contained wholly within the thorax. It is, therefore, much shorter than most regional beds of the systemic circulation. As well, all segments of the pulmonary vessels are slightly larger in radius than the corresponding segments of the systemic vasculature. The net result of these two factors is that the pulmonary circulation exerts a resistance to flow only around 15% of that in the systemic circuit. Hence it requires a correspondingly far lower pressure gradient in order to move the same cardiac output. At rest, pulmonary arterial pressure is typically 25/8 mmHg, giving a mean arterial pressure of the order of 14 mmHg, and mean pulmonary capillary pressure is 7–8 mmHg rather than the 25–30 mmHg seen in systemic vascular beds.
FUNCTIONAL CONSEQUENCES OF LOW PULMONARY BLOOD PRESSURE
Gas diffusion
In systemic capillaries the balance between oncotic and hydrostatic pressures mean that small increases in hydrostatic pressure caused, for example, by reduced arteriolar resistance will result in significant movement of plasma water into the interstitium. When a large tissue mass, such as skeletal muscle, is involved, this movement will cause a substantial reduction of plasma volume (see Chapter 9, p. 112), but the increased interstitial volume does not prejudice diffusion of solutes between cells and bloodstream because the intercellular connective tissue minimizes tissue expansion. In the lung, by contrast, there is little supporting tissue. If water moves into the interstitial space separating the pulmonary capillaries from the alveoli, it pushes these two structures further apart and increases the distance over which gases must diffuse between air and plasma.
The practising exercise physiologist is most likely to encounter pulmonary oedema as a consequence of high altitude, since it occurs in many healthy individuals who ascend to heights greater than 3000 m, well below the altitude of many ski resorts and permanent settlements. We will return to examine the reasons for this response to altitude in Chapter 12 (p. 151).
Right ventricular function
Left ventricular perfusion occurs only during diastole because during systole the left coronary vessels are compressed by the surrounding cardiac muscle (see Chapter 2, p. 6). By contrast, since right ventricular pressure does not normally rise above 25 mmHg, the coronary vasculature supplying the right ventricle is not compressed at any stage of the cardiac cycle and coronary flow is continuous. The increased efficiency of coronary oxygen delivery is reflected in a lower density of coronary blood vessels in the right heart. However, this design feature has potentially disadvantageous results if left atrial pressure becomes chronically elevated and right ventricular pressure rises in response to this higher afterload.
Under these circumstances, right coronary perfusion during systole becomes progressively reduced and the metabolic demands of the right-side cardiac muscle cells are less efficiently serviced. Over time, the right ventricle will respond to the high afterload by muscle hypertrophy. This will exaggerate the coronary insufficiency, both by providing greater metabolic demand for oxygen delivery and by producing more coronary compression. As a result, chronic hypoxic damage to the muscle of the right heart, with an inability to increase contraction appropriately in response to increased filling (right cardiac failure) is a common long-term effect of elevated pulmonary blood pressure (see Chapter 12, p. 148).
REGIONAL MATCHING OF VENTILATION AND PERFUSION
Vertical variations
Although both ventilation and perfusion rise towards the base of the lungs, the gravitational effect on regional blood flow is rather more powerful than the vertical variation in ventilation, with the result that the ratio of ventilation to perfusion is around 3 at the apices and around 0.5 at the bases. Exact matching of the two is achieved only over a relatively narrow region of lung corresponding to about the mid-sternal level (Fig. 8.1).
