[level-membership-for-cardiovascular-category]
Chapter 56 Pulmonary Arterial Hypertension
Definition and Classification of Pulmonary Arterial Hypertension
Pulmonary arterial hypertension (PAH) is formally defined as a mean pulmonary artery pressure (PAP) of 25 mmHg or greater at rest or 30 mmHg or greater during exercise. This is accompanied by a pulmonary vascular resistance (PVR) of greater than 3 Wood units (WU), with a normal pulmonary artery wedge pressure (< 15 mmHg) in the absence of any known cause of pulmonary hypertension (PH). Pulmonary arterial hypertension encompasses one of the five categories of PH, based on the most recent classification system.1 The pathophysiology and clinical characteristics of categories 2 through 5 of PH (i.e., PH associated with left-sided heart disease; lung disease/chronic hypoxia; thromboembolic PH; and other groups) are reviewed in Chapter 57. In this chapter, we will focus on category 1, PAH, which includes idiopathic and familial PAH as well as PAH associated with various other diseases (Box 56-1). Until improved therapeutic options became available during the past 2 decades, PAH was considered a rare but rapidly progressive and devastating condition, leading to death from right heart failure by a median of 2.8 years from the time of diagnosis.2 In recent years, an improved conceptual framework has been developed to better delineate the pathogenesis and clinical presentation of PAH, accompanied by improved directed therapeutic approaches. Nonetheless, PAH still carries a 15% annual mortality rate, and current therapeutics have been mostly effective at slowing illness progression rather than reversing or curing the disease. This chapter will highlight current molecular understanding of the complex disease, via the still incompletely characterized interplay of genetic and exogenous upstream stimuli with downstream vascular effectors. This framework will then be coupled to current understanding of the clinical presentation and progression of PAH and existing and evolving treatment modalities.
 Box 56-1 Clinical Classification of Pulmonary Arterial Hypertension
Box 56-1 Clinical Classification of Pulmonary Arterial Hypertension
Associated with significant venous or capillary involvement:
Adapted with permission from Simmoneau G, Galiè N, Rubin L, et al: Clinical classification of pulmonary hypertension. J Am Coll Cardiol 43:5 S–12 S, 2004.
Epidemiology
Early epidemiological data from the 1980s estimated the incidence of PAH at one to two cases per million people in the general U.S. population.2 More recent estimates in Europe3 place the incidence and prevalence of PAH, respectively, at 2.4 cases per million annually and 15 cases per million in France and 7.6 cases per million annually and 26 cases per million in Scotland. Globally, the prevalence may be much higher given that many risk factors for PAH, such as human immunodeficiency virus (HIV) and schistosomiasis, are also more prevalent in the developing world. An exact number is difficult to estimate on a global scale, given the difficulty of diagnosing PAH and overall limited access to health care worldwide.4 The disease affects women more frequently than men (1.7:1). This female predominance is exaggerated in the population of African descent (4.3:1), although the overall racial distribution of patients reflects that in the general population.
Primary PAH presents most commonly in the fourth decade of life; ages range from 1 to 81, with 9% of patients older than 60 years of age.5 Some sources cite a similar gender ratio among children diagnosed with the disease, whereas others note an equal distribution between male and female children.6 Primary PAH is typically difficult to diagnose, and the average time from onset of symptoms to diagnosis was 2 years in the National Institutes of Health (NIH) Registry.2 More recently, however, thanks to improved clinical awareness and more sophisticated invasive and noninvasive techniques for PAP measurements, time to diagnosis has decreased considerably, at least in the developed world.
Molecular Pathogenesis of Pulmonary Arterial Hypertension
In humans, the natural history of PAH lesions is unknown because patients usually present when the disease is advanced. The pathological appearance of severe PAH is similar regardless of cause and reflects the end stage of a common response to pulmonary vascular injury. Common histological features in nearly all types of PAH occur at the level of the small peripheral pulmonary arteries (100-1000 μm) (Fig. 56-1); these include intimal fibrosis, distal localization and proliferation of vascular smooth muscle, and pulmonary arterial occlusion.7 A hallmark of severe end-stage disease is the formation of a vessel “neointima,” characterized by increased deposition of extracellular matrix (ECM) and myofibroblasts. Plexiform lesions can predominate, characterized by over-proliferation of endothelial-like cells encroaching upon the vessel lumen.
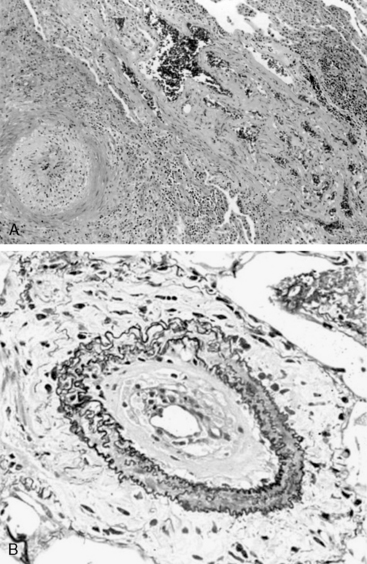
Figure 56-1 Histopathology of pulmonary arterial hypertension (PAH).
(Courtesy JL Faul, MD, Stanford University.)
Multiple cell types in the pulmonary arterial wall and pulmonary arterial circulation contribute to the specific response to injury and development of vessel remodeling8 (Fig. 56-2). The endothelium serves as a central sensor of injurious stimuli such as hypoxia, shear stress, inflammation, and toxins. Dysregulation of numerous downstream vascular effectors may be the result of initial endothelial cell (EC) injury or dysfunction. It has been hypothesized that endothelial apoptosis early in PAH initiates selection of apoptosis-resistant ECs that can further proliferate via monoclonal amplification in plexiform populations. This has led to speculation of a model of end-stage PAH similar to that of progression to cancer, with dysregulation of the cell cycle and apoptosis as predominant features. Similarly, dysregulated growth of pulmonary artery smooth muscle cells (SMCs) also plays a key role in PAH progression because apoptosis is suppressed while proliferation increases.
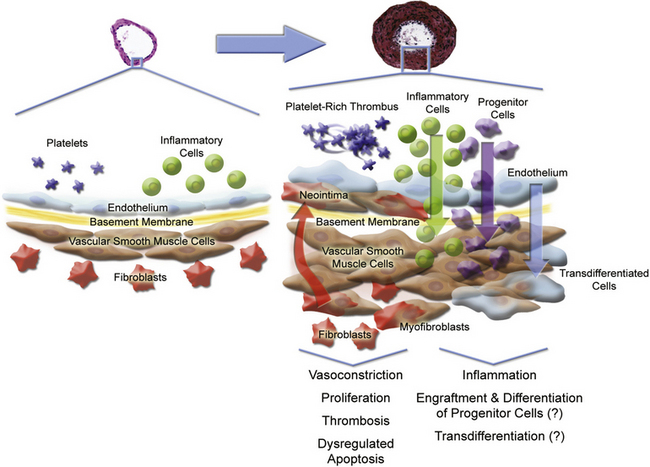
Figure 56-2 Pulmonary arterial pathobiology involves coordinate action of multiple cell types.
(Adapted with permission from Chan S, Loscalzo J: Pathogenic mechanisms of pulmonary arterial hypertension. J Mol Cell Cardiol 44:14–30, 2008. Micrographs of pulmonary arteries courtesy www.scleroderma.org and Humbert M, et al: Treatment of pulmonary arterial hypertension. N Engl J Med, 2004, 351:1425–1436, 2004. Copyright 2004, Massachusetts Medical Society. All rights reserved.)
Genetic Association
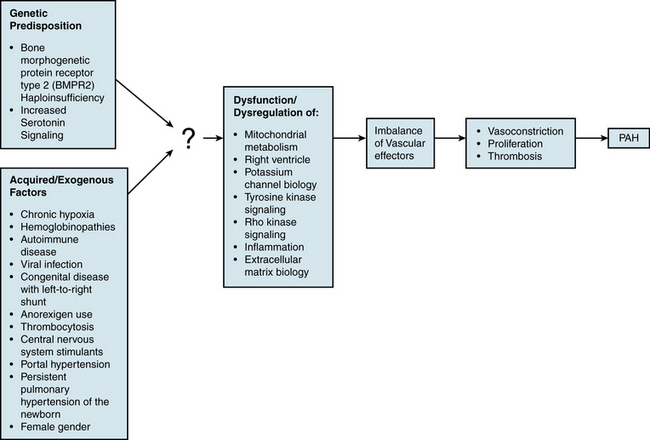
An understanding of the mechanism of genetic predisposition to PAH is of paramount importance for identifying the root pathogenesis (Fig. 56-3). The familial variety of idiopathic PAH accounts for at least 6% of all cases of PAH.7 Pedigree analysis has demonstrated an autosomal dominant inheritance but with variable penetrance; only 10% to 20% of putative genetic carriers develop clinical PAH. Genetic anticipation is present, since each successive generation of affected families is afflicted at a younger age and greater severity compared with the preceding generation.
Bone morphogenetic receptor-II
Mutations in the transforming growth factor (TGF)-β receptor superfamily have been genetically linked to PAH and likely play a causative role in disease development. A rare group of patients with hereditary hemorrhagic telangiectasia (HHT) and idiopathic PAH harbor specific mutations in ALK1 or endoglin, genes encoding for two such members of the TGF-β receptor superfamily.9,10 However, a more prevalent cohort of PAH patients carries mutations in the bone morphogenetic protein receptor type II (BMPR2 gene which encodes for BMPR2).11,12 Over 140 mutations in BMPR2 have been reported in patients with familial PAH,13 mainly located in the extracellular ligand-binding domain, cytoplasmic serine/threonine kinase domain, or long carboxyterminal domain. These account for 70% of all familial pedigrees of PAH and 10% to 30% of idiopathic PAH cases.8 BMPR2 loss-of-function mutations have only been found in the heterozygous state. The absence of BMPR2 mutations in some familial cohorts and in most sporadic cases indicates that additional unidentified genetic mutations can also predispose to development of PAH. Furthermore, the presence of incomplete penetrance (approximately 25% of carriers in affected families develop clinical PAH) and genetic anticipation suggests that BMPR2 mutations are necessary but insufficient alone to result in clinically significant disease.
The mechanism of action of BMPR2 is complex, and its role in PAH progression is still unclear (Fig. 56-4A). It functions as a receptor with serine/threonine kinase activity, and it activates a broad and complex range of intracellular signaling pathways (as reviewed in14). Upon binding one of many possible bone morphogenetic protein (BMP) ligands, BMPR2 forms a heterodimer with one of three type-I receptors. BMPR2 phosphorylates the bound type-I receptor, which in turn phosphorylates one of the Smad family of proteins to allow for nuclear translocation, binding to deoxyribonucleic acid (DNA), and regulation of gene transcription. Alternatively, BMPR2 activation can also lead to signaling via the LIM kinase pathway, p38/ mitogen-activated protein kinase/extracellular signal regulated kinase/c-jun NH 2-terminal kinase (MAPK/ERK/JNK) pathways, or c-Src pathway, independent of Smad activation.
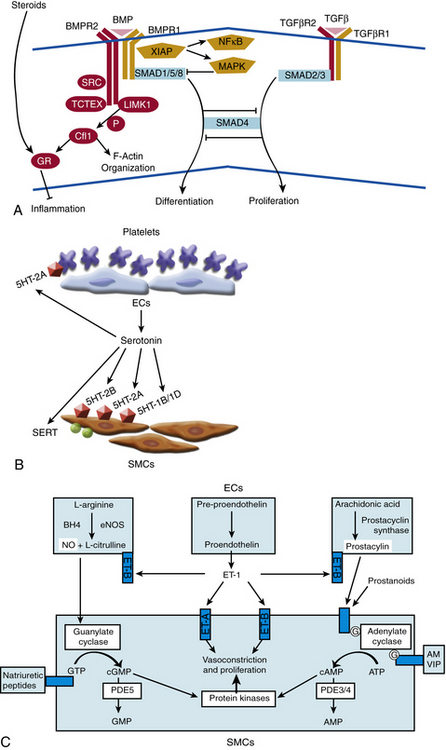
Figure 56-4 Molecular signaling mechanisms of pulmonary arterial hypertension (PAH).
(Adapted from Archer SL, Weir EK, Wilkins MR: Basic science of pulmonary arterial hypertension for clinicians: new concepts and experimental therapies. Circulation 121:2045–2066, 2010.)
Cellular effects of BMPR2 activation are multiform. In the adult, BMPR2 is expressed predominantly in pulmonary endothelium, medial SMCs, and macrophages.15 Under normal conditions, BMP ligands bind BMPR2 to suppress growth of VSMCs. In contrast, binding of BMP2 and BMP7 to BMPR2 in pulmonary endothelium protects against apoptosis. A widely held hypothesis contends that failure of the suppressive effects of BMP ligands on vascular smooth muscle and failure of the protective effects of BMP ligands on endothelium may trigger vascular proliferation and remodeling. Accordingly, in VSMCs derived from patients with familial PAH harboring BMPR2 mutations, exposure of BMP ligands does not suppress proliferation. Unlike the response in wild-type endothelium, exposure of ECs cultured from patients with idiopathic PAH to BMP2 does not protect against apoptosis.16 These dysfunctional signaling pathways have been corroborated in some rodent models of PAH. In correlation, pulmonary levels of BMPR2 are reduced both in familial cases of PAH without any BMPR2 mutation and in cases of secondary PAH.15 Thus, dysregulation of the BMP signaling pathway may be a common pathogenic finding in multiple types of PAH due to genetic or exogenous stimuli, but the definitive in vivo effects of these mutations have been difficult to decipher. Specifically, mouse models harboring specific BMPR2 heterozygous mutations have failed to exhibit robust PAH under static conditions,17,18 again suggesting that dysfunctional BMPR2 is likely insufficient alone to cause disease. As a result, a clear mechanistic explanation of the impact of BMPR2 mutations on pathogenesis remains elusive.
Additional genetic pathways
In addition to BMPR2 haploinsufficiency, alternative mechanisms involving complementary “modifier” genes may also contribute to a genetic predisposition to PAH. The most promising data identifying such modifier genes have analyzed the association of particular single nucleotide polymorphisms (SNPs) with the development of PAH. Thus far, SNP variants have suggested certain genes such as the serotonin transporter (SERT), Kv1.5, and the trp cation channel, subfamily C, member 6 (TRPC6).3 Such associations do not always suggest a causal relationship, so additional mechanistic data are necessary for proper interpretation.
In the case of SERT and serotonin signaling, supportive data are more prevalent. Serotonin is both a vasoconstrictor and mitogen that promotes smooth muscle hyperplasia and hypertrophy (see Fig. 56-4B). Primarily stored in platelet granules, secreted serotonin binds G protein–coupled serotonin receptors (GPCRs) present on pulmonary artery SMCs. Activation of these receptors leads to a decrease in adenylyl cyclase and cyclic adenosine monophosphate (cAMP), resulting in increased contraction. Furthermore, the cell-surface SERT allows for transport of extracellular serotonin into the cytoplasm of SMCs, thereby activating cellular proliferation directly through the action of serotonin or indirectly via potential pleiotropic mechanisms.
A number of observations in idiopathic and familial disease, congenital disease, and environmental exposure have implicated the proliferative effects of serotonin in PAH.19 In idiopathic PAH, pulmonary expression of serotonin receptors is increased, and plasma levels of serotonin are chronically elevated. A mouse model of hypoxic PH parallels these changes.20 A positive association has been noted among patients with congenital platelet defects in serotonin uptake (i.e., delta storage pool disease) and development of PAH. Chronic exposure to anorexigens, such dexfenfluramine (an inhibitor of serotonin reuptake and stimulator of serotonin secretion), leads to increased levels of circulating free serotonin. In mice, these changes are accompanied by increased 5HT receptor type 2B response and inhibition of SERT responses. In humans, these changes correlate with an increased risk for development of PAH. In addition, the L-allelic variant of SERT is associated with increased expression of the transporter and enhanced smooth muscle proliferation. In some human studies,21 this variant has been associated with an increased risk of PAH in the homozygous population.
Animal models of PH have also implicated the activated serotonin pathway in disease progression. Treatment with serotonin and chronic hypoxia in a rat model led to worsened hemodynamics and increased vessel remodeling.22 Exposure to increased serotonin led to worsened PH in a BMPR2+/− heterozygote murine model.23 Similarly, overexpression of SERT in mice resulted in spontaneous development of PAH in the absence of hypoxia and exaggeration of PH after a hypoxic stimulus.24 Conversely, vessel remodeling and hypoxic PH are reduced in a 5HT1B receptor–null mouse25 and are abrogated in a 5HT2B receptor–null mouse.20 As a result, serotonin signaling modulates pulmonary smooth muscle function in both normal and disease states and likely contributes to disease progression of PAH. However, attempts at using selective serotonin reuptake inhibitors (SSRIs) as a therapeutic approach have yielded mixed results3 to date. The exact contribution of this mechanism in PAH requires further clarification.
Acquired/Exogenous Factors
In addition to genetic predisposition, development of PAH likely depends on a variety of physiological, acquired, and/or exogenous stimuli. Some of these factors have been studied to a sufficient degree to hypothesize potential pathogenic mechanisms (see Fig. 56-3).
Chronic hypoxia
Pulmonary vascular response to hypoxia has been well studied in cell culture and animal models,26 but its impact on PAH is unclear. In general, pulmonary vascular responses in acute and chronic hypoxia likely allow for the propagation of PAH, and therefore may contribute to later stages of the disease. Acute hypoxia induces vasodilation in systemic vessels but induces vasoconstriction in pulmonary arteries. This acute and reversible effect is mediated in part by up-regulation of endothelin-1 (ET-1) and serotonin, and in part by hypoxia- and redox-sensitive potassium channel activity in pulmonary VSMCs. Coordinately, these events lead to membrane depolarization in SMCs, increase in cytosolic calcium, and vasoconstriction.27 In contrast, chronic hypoxia induces vascular remodeling and less reversible changes, including migration and proliferation of VSMCs and deposition of ECM. These cellular events in chronic hypoxia correlate with the remodeling events inend-stage PAH; however, because the histopathology of hypoxic PH does not recapitulate all aspects of PAH, some mechanistic differences in pathogenesis certainly exist but have not yet been fully identified. These are important considerations, especially in the context of interpreting studies of hypoxic PH and extrapolating those findings to the pathogenesis of PAH.
Hemoglobin disorders
Pulmonary arterial hypertension is associated with hemoglobinopathies, especially thalassemias and sickle cell anemia.28 Hemolysis accompanying these disorders may lead to destruction of bioactive nitric oxide (NO) by free hemoglobin or reactive oxygen species (ROS). Furthermore, ROS may lead to increased levels of oxyhemoglobin, which further impairs delivery of NO to the vessel wall. As a result of the lack of available NO, an inflammatory and proliferative cascade may ensue, with culmination in PAH. Accordingly, decreased NO bioavailability with development of PH has been reported after hemolysis in a murine model of sickle cell disease (SCD),29 and acutely in a murine model of intravascular hemolysis.30 In vivo correlation to human disease is pending.
Portopulmonary hypertension
Portopulmonary hypertension is defined as PAH associated with portal hypertension (portal pressure >10 mmHg), with or without hepatic disease.31 Approximately 9% of patients with severe PAH are reported to have portal hypertension. Portopulmonary hypertension affects 4% to 6% of patients referred for liver transplantation. Liver transplantation perioperative mortality is significantly increased in patients with a mean PAP above 35 mmHg. Diagnosis of PH is usually made 4 to 7 years after the diagnosis of portal hypertension, but it has occasionally been reported to precede onset of portal hypertension. Risk of PH increases with duration of disease. The correlation between severity of portal hypertension and development of PH is debated. The female predominance of primary PAH is not seen in portopulmonary hypertension.
Collagen vascular disease
Pulmonary arteriopathy complicates autoimmune diseases, especially in the setting of the CREST (calcinosis, Raynaud phenomenon, esophageal dysfunction, sclerodactyly, telangiectasia) variant of limited systemic sclerosis and, to a lesser degree, in mixed connective tissue disease, systemic lupus erythematosus (SLE), and rheumatoid arthritis.32 Pulmonary arterial hypertension has been reported in approximately 10% to 30% of patients with mixed connective tissue disease, 5% to 10% of patients with SLE, and more rarely in the settings of rheumatoid arthritis, dermatomyositis, and polymyositis.33 Sjögren syndrome may rarely be complicated by rapidly progressive PAH. It is particularly important to distinguish between PAH and thromboembolic PH in patients with SLE and antiphospholipid syndrome. Occurrence of PAH in each disease has been associated with Raynaud phenomenon, suggesting at least some similarities in pathogenesis.34 Presence of interstitial lung disease and pulmonary fibrosis, seen at varying frequency in these autoimmune syndromes, may represent a common pathogenic factor in development of PAH. Accordingly, in the setting of pulmonary fibrosis and hypoxia, significant perivascular inflammation and deposition of ECM have been observed, which may increase vasoconstriction, proliferation, and vessel remodeling. Murine models of interstitial lung disease may prove important in further elucidating pathogenic mechanisms.
Human immunodeficiency virus
An association between HIV infection and PH has been noted in approximately 0.5% of all patients with HIV infection, a rate 6 to 12 times higher than the general population.35 Notably, HIV does not infect pulmonary arterial endothelium, but mechanisms of disease have been proposed that directly stem from effects of HIV infection.36 These include infection of SMCs with subsequent dysregulation of proliferation, imbalance of vascular mitogens in response to systemic HIV infection, and endothelial injury precipitated by HIV-infected T cells. The direct actions of HIV-encoded proteins may also factor into PAH development.8 The HIV gp120 protein may induce pulmonary endothelial dysfunction and apoptosis. In a macaque model of simian immunodeficiency virus infection, a pathogenic interaction of the viral Nef protein with the pulmonary vessel wall has been reported, leading to pulmonary arteriopathy. Cell culture studies have also demonstrated a role for the HIV Tat protein in repression of BMPR2 transcription, potentially provoking a proliferative response in the vessel wall.
It had been proposed that human herpesvirus 8 (HHV-8), the causative agent of Kaposi sarcoma and an opportunistic pathogen highly associated with HIV infection, may play a role in PAH development with progression to plexiform lesions. Although it was initially reported that HHV-8 infection is associated with idiopathic PAH,37 that link has not been consistently validated after study of additional populations. Nonetheless, PAH in the setting of HIV infection likely results from multifactorial effects, and the underlying pathogenesis may involve both direct results of viral infection and indirect consequences of associated pathogens.
Shunt physiology
Increased flow through the pulmonary circulation has long been associated with development of PAH. Certain types of congenital heart disease with functional systemic-to-pulmonary shunts, such as unrestricted ventricular septal defects (VSDs) and large patent ductus arteriosus (PDA), invariably lead to pulmonary vascular remodeling and the clinical syndrome of PAH during childhood (Eisenmenger’s syndrome).38 Approximately a third of patients with uncorrected VSDs and PDAs die from PAH. The timing of surgical repair greatly influences the outcome. If the shunt is repaired within the first 8 months of life, patients tend to have normal pulmonary pressures regardless of pathological findings; by contrast, patients operated on after age 2 tend to have persistent PAH. Importantly, when PVR equals or exceeds systemic vascular resistance, surgical correction of a shunt will increase the load on an already overburdened right ventricle (RV), worsen the patient’s clinical condition, and not reverse PAH.
The presence of atrial septal defects (ASD) with systemic-to-pulmonary shunts may also lead to PAH over time.39 Yet, in contrast to cases of unrestricted VSD and PDA, only 10% to 20% of all persons with ASDs progress to PAH. This observation may reflect differences in the response of the pulmonary vasculature to pressure overload (as seen in shunts with VSD and PDA) as compared to volume overload (as seen in shunts with ASD). Furthermore, patients with ASD may harbor a specific unidentified genetic predisposition to the development of PAH that may exacerbate the increased volume load to the pulmonary circulation.
At the molecular level, the physiological flow patterns of laminar shear stress, turbulent flow, and cyclic strain are all recognized by ECs, leading to transduction of intracellular signals and modulation of a wide variety of phenotypic changes.40 Significant prior work has focused mainly on the endothelium of the peripheral vasculature, suggesting that laminar flow induces a vasoprotective quiescent vascular state, whereas turbulent flow leads to a proinflammatory and thrombogenic state. It is unclear whether these flow-dependent phenotypes are recapitulated in the pulmonary vasculature. In part, this stems from the difficulty of directly studying directly the in vivo flow patterns at the anatomical level of the pulmonary arteriole. Ex vivo modeling of pulsatile flow with high levels of shear stress and chronic vascular endothelial growth factor (VEGF) inhibition has demonstrated apoptosis of pulmonary artery ECs, followed by outgrowth and selection for proliferating apoptosis-resistant cells.41 Therefore, chronically elevated flow may allow for selection of cells with dysregulated EC growth and resulting clonal or polyclonal expansion to plexiform lesions.
Persistent pulmonary hypertension of the newborn
Persistent pulmonary hypertension of the newborn (PPHN) is characterized by persistent elevation of PVR, right-to-left shunting, and severe hypoxemia. It can occur with pulmonary parenchymal diseases including sepsis, meconium aspiration, pneumonia, maladaptation of the pulmonary vascular bed, or without an apparent cause.42 Persistent PH in newborns may lead to death during the neonatal period, or it may be transient, leading to spontaneous and complete recovery. Inadequate production of NO may be an important contributor to persistent PH in infants, and inhaled NO is useful in treating this disorder.
Miscellaneous disorders
Pulmonary arterial hypertension is more rarely reported in patients suffering from other clinical syndromes such as pulmonary veno-occlusive disease, chronic myelodysplastic syndromes with thrombocytosis, and idiopathic thrombocythemia, as well as in persons exposed to stimulants of the central nervous system, such as methamphetamines and cocaine. Dysregulation of serotoninergic signaling may contribute8 but does not explain these associations entirely. Finally, idiopathic PAH demonstrates a gender predilection of unclear etiology, with a high predominance of affected females.43
Vascular Effectors
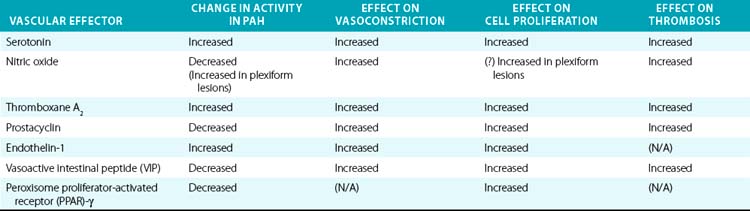
Downstream of the genetic and acquired triggers of PAH, the histopathological processes that predominate later stages of disease include vasoconstriction, cellular proliferation, and thrombosis. These processes are influenced by a complex and dysregulated balance of vascular effectors controlling vasodilation and vasoconstriction, growth suppressors and growth factors, and pro- vs. antithrombotic mediators. Most of these effectors have been described in previous comprehensive reviews and will be described here specifically in regard to their known roles in PAH pathogenesis (Table 56-1).
Nitric oxide
Gaseous vasoactive molecules regulate pulmonary vascular homeostasis, and alterations in their endogenous production have been linked to progression of PAH. Nitric oxide is the best described of these factors (see Fig. 56-4C).44 It is a potent pulmonary arterial vasodilator as well as a direct inhibitor of platelet activation and VSMC proliferation. Nitric oxide diffuses into recipient cells (e.g., vascular muscle), where it activates soluble guanylyl cyclase (sGC) to produce cyclic guanosine monophosphate (cGMP). In turn, cGMP interacts with at least three different groups of effectors: cGMP-dependent protein kinases, cGMP-regulated phosphodiesterase (PDE), and cyclic nucleotide-gated ion channels. Synthesis of NO is mediated by a family of NO synthase (NOS) enzymes. In the pulmonary vasculature, the endothelial (eNOS) isoform figures most prominently and is regulated by a multitude of vasoactive factors and physiological stimuli including hypoxia, inflammation, and oxidative stress. Reduced levels and reduced activity of eNOS, reduced NO bioavailability, dysregulated intracellular NO transport via caveolae, and increased degradation of cGMP all aggravate PAH progression in human studies and/or animal models.
Dysregulation of NO may also depend upon still incompletely characterized processes of NO transport in blood.45 Specific polymorphisms of NOS have also been associated with pulmonary hypertension. Together, these effects indicate a coordinated mechanism of dysregulated vasoconstriction. Correspondingly, murine models that genetically lack eNOS, GTP cyclohydrase-1 (the rate limiting enzyme for synthesis of an essential cofactor of NOS, tetrahydrobiopterin), or dimethylarginine dimethylaminohydrolase (DDAH, an enzyme important in degradation of NOS inhibitors) all display exaggerated susceptibility to developing PH in response to other endogenous stimuli.3 Finally, the impact of NO has been reflected in its therapeutic role in PAH, such as inhaled NO46 and the NO-dependent phosphodiesterase type-5 (PDE5) inhibitors (discussed later in detail).
Prostacyclin/thromboxanes
The arachidonic acid metabolites prostacyclin and thromboxane A2 (TxA2) also play crucial roles in vasoconstriction, thrombosis, and to a certain degree, vessel wall proliferation (see Fig. 56-4C). Prostacyclin (prostaglandin I2) activates cAMP-dependent pathways and serves as a vasodilator, antiproliferative agent for vascular smooth muscle, and inhibitor of platelet activation and aggregation. In contrast, TxA2 increases vasoconstriction and activates platelets.47 Protein levels of prostacyclin synthase are decreased in small and medium-sized pulmonary arteries in patients with PAH, particularly with the idiopathic form.48 Biochemical analysis of urine in patients with PAH has shown decreased levels of a breakdown product of prostacyclin (6-ketoprostacyclin F2α), accompanied by increased levels of a metabolite of TxA2 (thromboxane B2).49 Therefore, it appears that production of these effectors is coordinately regulated, with the imbalance toward TxA2 favored in the development of PAH. The mode of this regulation remains to be characterized. Nonetheless, recognition of this imbalance has led to the success of prostacyclin therapy and improvement of hemodynamics, clinical status, and survival in patients with severe PAH.50
Endothelin-1
Endothelin-1 is expressed by pulmonary ECs and has been identified as a significant vascular mediator in PAH (see Fig. 56-4C).51 It acts as both a potent pulmonary arterial vasoconstrictor and mitogen of pulmonary smooth muscle cells. The vasoconstrictor response relies upon binding to the endothelin receptor A (ET-A) on vascular smooth muscle cells. This leads to an increase in intracellular calcium, along with activation of protein kinase C (PKC), mitogen-activated protein kinase (MAPK), and the early growth response genes c-fos and c-jun.52 The mitogenic action of ET-1 on pulmonary VSMCs can occur through either the ET-A and/or the ET-B receptor subtype, depending on the anatomical location of cells. Endothelin receptor A predominantly mediates mitogenesis in the main pulmonary artery, whereas mitogenesis in resistance arteries relies upon contributions from both subtypes. The resulting vasoconstriction, mitogenesis, and vascular remodeling are thought to lead to significant hemodynamic changes in the pulmonary vasculature and to PAH. Plasma levels of ET-1 are increased in animal and human subjects suffering from PAH due to a variety of etiologies53 and correlate with disease severity.54 Again, improvement in hemodynamics, clinical status, and survival of PAH patients treated with chronic ET receptor antagonists highlights the significance of these effects.55
Vasoactive intestinal peptide
Down-regulation of vasoactive intestinal peptide (VIP) may also play a pathogenic role. Vasoactive intestinal peptide is a pulmonary vasodilator, an inhibitor of proliferation of VSMCs, an inhibitor of platelet aggregation, and free radical scavenger. Decreased concentrations of VIP and VIP receptors have been reported in serum and lung tissue of patients with PAH.56 Vasoactive intestinal peptide-null mice suffer from moderate pulmonary hypertension.57 Both pulmonary arterial pressure (PAP) and PVR in humans decrease after treatment with VIP.58,59 Key questions regarding the mode(s) of regulation of VIP expression and its putative causative role in PAH remain unanswered.
Peroxisome proliferator-activated receptor γ
Peroxisome proliferator-activated receptor gamma (PPAR-γ)and apolipoprotein E (apoE) may function as integral factors in PAH.60,61 PPARs are ligand-activated nuclear transcription factors that heterodimerize with the retinoid X receptor for subsequent binding to PPAR response elements in the promoters of target genes. Idiopathic PAH patients carry reduced pulmonary transcript levels of PPAR-γ and apoE. PPAR-γ in particular is a direct target of BMP2 in human PASMCs, leading to stimulation of apoE synthesis and downstream inhibition of vascular smooth muscle proliferation. PPAR-γ also regulates a host of protein kinases implicated in PASMC proliferation and migration. PPAR-γ agonists are antiinflammatory and induce pro-apoptotic phenotypes, both theoretically inhibitory to the pathogenesis of PAH. Mice deficient in smooth muscle–specific PPAR-γ are prone to PAH. Similarly, when fed a high-fat diet, male mice deficient in apoE develop PAH. This condition is reversed by rosiglitazone, a PPAR-γ agonist. Nonetheless, less robust results have been seen when using rosiglitazone hypoxic-PH rats, suggesting that these results offer a partial explanation to the complex molecular pathogenesis of BMPR2 signaling and PAH pathogenesis.
Natriuretic peptides
The atrial and brain natriuretic peptides (ANP and BNP) are produced by myocardium in response to stretch. They bind guanylyl cyclase receptors (NPR-A and NPR-B) that induce production of cGMP. Increased plasma concentrations of these peptides in PAH have been used as markers of the extent of RV dysfunction.62 Genetic inactivation of NPR-A in mice is associated with PH63; in contrast, administration of atrial natriuretic peptide (ANP) ameliorates PAH in rodent models.64 Additionally, inhibition of the metabolic breakdown of natriuretic peptides via neutral endopeptidase inhibitors has shown promise in animal studies of PAH. Human studies have yet to confirm efficacy.
Adrenomedullin
Adrenomedullin is an endogenous peptide that activates signaling pathways (i.e., cAMP, NO-cGMP, phosphatidylinositol-3-kinase/Akt) to induce vasodilation and inhibit proliferation. It decreases mean PAP and RV hypertrophy in hypoxic rats.65 A related peptide, adrenomedullin-2, binds the same cellular receptors as adrenomedullin, and it is elevated in some rodent forms of PAH.
Miscellaneous effectors
Other potential contributing factors include angiopoietin-1, the vasodilatory gases carbon monoxide and hydrogen sulfide, phosphodiesterase I, survivin, the calcium binding protein S100A4/Mts, the transient receptor potential channels, and Notch 3.66 These may represent important but as yet incompletely described pathogenic contributors.8 Other mitogenic and angiogenic growth factors, such as VEGF, platelet-derived growth factor (PDGF), basic fibroblast growth factor (bFGF)-2, insulin-like growth factor (IGF)-1, and epidermal growth factor (EGF) all may play downstream roles in later stages of PAH.
Pathogenic Pathways
Our understanding of specific actions of individual effectors has improved, but how they relate to upstream genetic or exogenous triggers of PAH remains unclear. In fact, none of these factors has yet been definitively linked to the root pathogenesis of disease. Insight into this topic is offered by the fact that known effectors are likely subject to upstream, overarching regulatory pathways that affect the action of multiple vasoactive molecules.67 Characterization of these regulatory mechanisms may eventually allow for identification of primary molecular triggers of disease and offer novel therapeutic targets for drug development (see Fig. 56-3).
Mitochondrial metabolism
Mitochondrial and metabolic dysfunction may be common in PAH. Endothelial and VSMCs from human PAH tissue display dysmorphic and hyperpolarized mitochondria.68 Metabolically, these cells preferentially exhibit a down-regulation of mitochondrial metabolism with an induction of glycolysis for energy production.69 Under hypoxic conditions, this so-called Pasteur effect is a normal adaptive event that improves cellular survival by optimizing adenosine triphosphate (ATP) production while reducing oxygen radicals generated from the mitochondrial electron transport chain. Under normoxic conditions, however, such a shift to glycolysis (the Warburg effect) is thought to confer inappropriate resistance to apoptosis and is prominently seen in various cancer lineages. Such metabolic changes in the mitochondria are dependent upon a master transcription factor of hypoxia, hypoxia inducible factor 1 alpha (HIF-1α), and its critical downstream targets such as pyruvate dehydrogenase kinase (PDK1), among others.70 In PAH, HIF-1α and PDK1 are dysregulated; conversely, treatment with dichloroacetate, a PDK inhibitor, ameliorates multiple forms of PAH in rodent models (chronic hypoxic PH, monocrotaline PAH, and fawn-hooded rat PAH).71 It is conceivable that therapies aimed at reversing this metabolic dysregulation may result in improvement and/or regression of the PAH phenotype.
Right ventricular dysfunction
After birth, PAPs typically decline, leading to involution of the RV to a thin-walled structure in the adult. During pathological conditions of increased PAPs in PAH, however, RV hypertrophy (RVH) and strain ensue, followed by RV failure if left untreated. Historically, the molecular pathways governing left ventricular (LV) failure, which have been better characterized, had been assumed to play primary roles in RV failure as well. Contemporary evidence suggests there may be distinct molecular and physiological differences between LV and RV failure that are just beginning to be explored.72 For instance, PDE5, which is expressed in the fetal RV, is selectively up-regulated in the hypertrophied RV. Interestingly, inhibition of PDE5 enhances RV contractility without affecting the LV, where PDE5 expression is absent.
Unique metabolic changes in the RV have also been reported. The normal RV can use either fatty acids or glucose as needed for energy production. In RVH, the RV nearly exclusively relies upon glycolysis. Adenosine monophosphate (AMP)-activated protein kinase is activated in RVH, which preserves ATP levels by increasing glucose transport and glycolysis while inhibiting the tricarboxylic acid (TCA) cycle via repression of acetyl-coenzyme A carboxylase.73 Furthermore, during RVH, PDK is activated, inducing a metabolic switch away from oxidative phosphorylation to glycolysis. As a result, the hypertrophied RV has been hypothesized to behave as “hibernating myocardium,” which may suggest that the RV can be targeted therapeutically in PAH.3
Potassium channel biology
Modulation of voltage-gated potassium channels (Kv) may also represent an overarching pathogenic mechanism of PAH.71 Kv channels are tetrameric membrane-spanning channels that selectively conduct potassium ions; in PASMCs, Kv channels aid in regulation of the resting membrane potential. In response to Kv inhibition or down-regulation, depolarization leads to the opening of voltage-gated calcium channels, increase in intracellular calcium, and initiation of a number of intracellular signaling cascades promoting vasoconstriction and proliferation and inhibiting apoptosis. Expression array analysis has demonstrated a depletion of Kv1.5 channels in pulmonary tissue derived from PAH patients.74 It is currently unknown whether these Kv channel abnormalities are congenital or acquired, but a number of polymorphisms in the Kv1.5 channel gene (KCNA5) have been described, which may suggest a genetic predisposition to channel depletion.75 Appetite suppressants (e.g., dexfenfluramine, aminorex) that are risk factors for development of PAH can also directly inhibit Kv1.5 and Kv2.1. A variety of transcription factors (HIF-1α, NFAT, and c-Jun) are increased in PAH, with resulting down-regulation of Kv1.5 expression in PASMCs. Inhibition of these factors consequently increases Kv1.5 expression, with resulting improvements in Kv current and in some cases, improved pulmonary arterial remodeling in hypoxic rodent models of PH. Inhibition of Kv currents in pulmonary SMCs may be regulated by serotonin, TxA2, and perhaps NO. Furthermore, BMP signaling can regulate Kv receptor expression. Taken together, the Kv pathway may represent a common point of regulation in pathogenesis. Accordingly, augmentation of Kv activation would be predicted to induce vasodilation and perhaps allow for regression of vessel remodeling. In vivo gene transfer of Kv channels in chronically hypoxic rats has led to improvement of PH and suggests its therapeutic potential.76
Tyrosine kinases
End stages of PAH are marked by exaggerated expression or activity of multiple growth factors, including PDGF, fibroblast growth factor 1 (FGF-1), FGF-2, EGF, and VEGF. All appear to contribute substantially to the overall obstructive arterial remodeling characteristic of PAH, via synergistic and combinatorial pathways. Some of the receptors for these growth factors are transmembrane receptor tyrosine kinases that activate a diverse and overlapping set of intracellular signaling pathways. Inhibition of EGF77 and PDGF78 receptors demonstrates improved hemodynamics and survival in PAH. Case reports exist of a beneficial effect of adding imatinib (a nonselective tyrosine kinase inhibitor)79 or sorafenib (a “multikinase inhibitor”)80 to baseline PAH therapy; however, further mechanistic clarity of the specific role of tyrosine kinases in the pathogenesis of PAH is needed.
Rho-kinase signaling
Multiple vascular cell types rely upon the rho kinase signaling pathway for homeostatic function and response to injury.81 Rho is a guanosine triphosphate (GTP) binding protein that activates its downstream target, rho-kinase, in response to activation of a variety of GPCRs (including those related to BMP/SMAD signaling and serotonin signaling). In vitro activation of these signaling cascades results in modulation of multiple cellular processes, including enhanced vasoconstriction, proliferation, impaired endothelial response to vasodilators, chronic pulmonary remodeling, and up-regulation of vasoactive cytokines via the nuclear factor kappa beta (NFκB) transcription pathway. Rho-kinase activity has also been linked specifically to a number of known effectors of PAH, including ET-1, serotonin, and eNOS, among others. Elevated rho-kinase activity has been demonstrated in animal models of PAH,82,83 and intravenous fasudil, a selective rho-kinase inhibitor, has induced pulmonary vasodilation and regression of PAH in various animal models (as reviewed in8), as well as in patients with severe PAH who were otherwise refractory to conventional therapies.84,85 Taken together, these data suggest that rho-kinase may control a master molecular “switch” in the pulmonary artery, initiating an activated state in disease from a quiescent state in health. As a result, rho-kinase represents an attractive and novel upstream therapeutic target for treatment of PAH.
Inflammation
As reflected by a strong association of PAH with various autoimmune and infectious states as well as a significant presence of T cells, B cells, and macrophages in plexiform lesions, a severe inflammatory state predominates end-stage PAH. Athymic nude rats, which lack functioning T cells, display greater sensitivity to PAH.86 T-cell dysregulation may figure prominently because regulatory T cells (Treg) are increased, whereas CD8+ cytotoxic cells are decreased, in idiopathic PAH.87 In addition, a number of soluble chemoattractants and proinflammatory cytokines from the pulmonary artery are up-regulated in human and animal models of severe PAH. These include interleukin (IL)-1β, TGF-β1, bradykinin, monocyte chemotactic protein-1, fractalkine, RANTES, and leukotrienes, among others. 5-Lipoxygenase (5-LO) regulates synthesis of leukotrienes, which in turn can promote cytokine release. 5-Lipoxygenase may represent a possible upstream factor involved in inciting this proinflammatory state, and elevated levels of 5-LO have been detected in macrophages and pulmonary endothelium derived from patients suffering from idiopathic PAH.88 In various murine models of PAH, overexpression of 5-LO has worsened PH and vascular remodeling,89 while 5-LO inhibitors have attenuated PH. It is thus tempting to speculate that 5-LO itself may possess an upstream regulatory role in PAH progression.
Extracellular matrix biology
Vascular-specific serine elastase activity has been implicated in PAH pathogenesis via regulation of the remodeling response in the extracellular matrix.90 In pulmonary arterioles, serine elastases are secreted into the extracellular space to activate matrix metalloproteinases (MMP) and inhibit tissue inhibitors of MMP (TIMP). Both MMP and elastases degrade most components of the ECM leading to an up-regulation of fibronectin (FN) and subsequent enhancement of cellular migration. Matrix degradation also increases integrin signaling, with resulting expression of the glycoprotein tenascin C. Tenascin C acts cooperatively with other factors to enhance smooth muscle proliferation. Increased degradation of elastin91 has been observed in pulmonary arteries from patients suffering from congenital heart disease and resultant PAH. In pulmonary tissue of PAH patients harboring BMPR2 mutations and rat models of PAH, increased production and activity of vascular elastases92 and tenascin C93 have been reported. This up-regulation of elastase function may be induced by a number of vascular effectors implicated in PAH, including NO, serotonin, and theoretically, the BMP pathway. Elastase inhibitors can induce apoptosis of SMCs in cell culture and can improve PAH in animal models.94,95 Thus, elastase function may also represent a novel therapeutic target.
Clinical Pathophysiology
The two most frequent causes of death in PAH are progressive RV failure and sudden death. Right ventricle failure may be exacerbated by pneumonia, and alveolar hypoxia can cause further vasoconstriction and greater impairment of CO. Sudden death may be caused by arrhythmias that arise in the setting of hypoxemia and acidosis, acute pulmonary emboli, massive pulmonary hemorrhage, and subendocardial RV ischemia.96
Diagnostic Evaluation
Initial Approach
There is no pathognomonic finding in PAH; thus the diagnosis is one of exclusion. A thorough evaluation must be performed to reveal potentially contributing factors, including causes of secondary forms of PH that require a different treatment approach. It is important to probe for a family history of PH, early unexplained deaths, congenital heart disease, and collagen vascular disease. A thorough history should also include all associated risk factors of PH to uncover a possible explanation for the onset of PAH and exclude secondary causes of PH. A functional assessment should be made on the basis of the New York Heart Association (NYHA) functional classification of heart failure adopted for PAH at the World Health Organization (WHO)-sponsored symposium.1 In addition to a comprehensive history, diagnostic evaluation should include physical examination, exercise testing (e.g., 6-minute walk), chest radiograph, electrocardiography, pulmonary function tests, arterial blood gas and other blood tests, noninvasive cardiac and pulmonary imaging, and cardiac catheterization with measurement of response to vasodilator administration (Fig. 56-5).
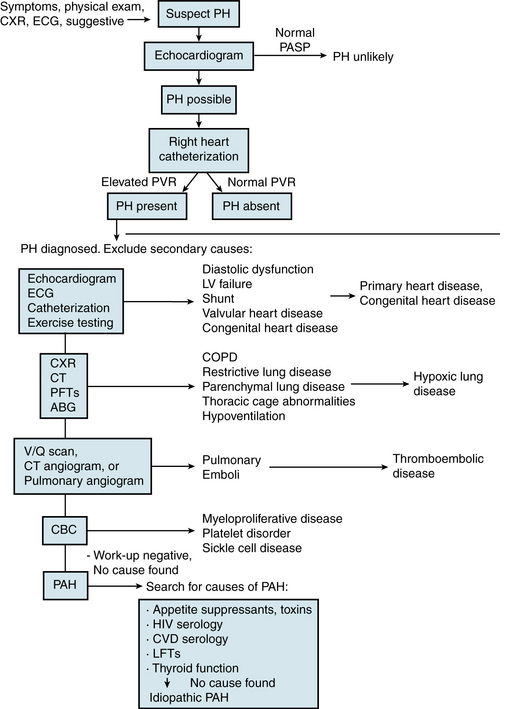
Figure 56-5 Diagnostic evaluation for suspected pulmonary arterial hypertension (PAH).
(See Chapter 58 for a discussion of secondary pulmonary hypertension.) ABG, arterial blood gas; CBC, complete blood cell count; COPD, chronic obstructive pulmonary disease; CPET, cardiopulmonary exercise test; CT, computed tomography scan; CVD, collagen vascular disease; CXR, chest x-ray; ECG, electrocardiogram; HIV, human immunodeficiency virus; LFTs, liver function tests; LV, left ventricle; PAH, pulmonary arterial hypertension; PASP, pulmonary arterial systolic pressure; PFTs, pulmonary function tests; PH, pulmonary hypertension; PVR, pulmonary vascular resistance; SCD, sickle cell disease; V/Q scan, ventilation/perfusion nucleotide scan.
Laboratory studies
Secondary causes of PAH should be sought with serology for HIV and collagen vascular diseases, liver function tests (LFTs), and toxic exposures. Thyroid function should be evaluated. Thrombocytopenia may be present in severe PAH and has multiple contributing causes, including platelet activation and aggregation, pulmonary vascular sequestration, hepatosplenomegaly with splenic sequestration, and an autoimmune-mediated syndrome similar to idiopathic thrombocytopenic purpura.97 Thrombocytopenia may accompany microangiopathic hemolysis when blood flows through fibrin deposits in plexiform lesions, with subsequent shearing of red blood cells and platelets. Prostacyclin may also induce thrombocytopenia. Thrombocytosis may be present in patients following splenectomy.
In patients with PH, high levels of ANP and BNP parallel decreased RV function. Levels of both peptides decrease with prostacyclin treatment and ensuing hemodynamic improvement. A subsequent increase in plasma BNP has been demonstrated to be an independent predictor of mortality.98
Radiographic studies
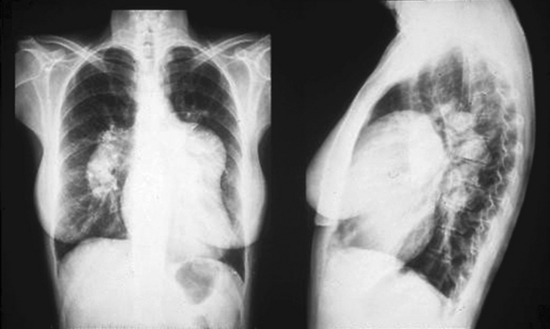
Chest radiographs in PH usually show an enlarged pulmonary trunk and hilar pulmonary arteries, pruning of peripheral vessels, and obliteration of the retrosternal clear space by the enlarged RV (Fig. 56-6). Occasionally the chest radiograph may appear normal.5 High-resolution computed tomography (CT) is used to evaluate lung parenchyma for interstitial lung disease. Helical CT is used to evaluate the central pulmonary arteries for presence of thrombi. Ventilation/perfusion scans are used to rule out chronic pulmonary thromboemboli. In patients with PAH, these scans are normal or show only patchy defects. If inconclusive, a pulmonary angiogram, which will show pruning of peripheral vessels in PAH, must be performed to definitively exclude thromboembolic disease. Use of pulmonary magnetic resonance angiography (MRA) has not been widely reported, but has recently been shown to identify patients with PAH with high sensitivity and negative predictive values.98
Electrocardiogram
The electrocardiogram (ECG) is not a sensitive or specific screening tool. In advanced disease, the ECG usually shows signs of right heart strain and enlargement, including right axis deviation and evidence of RV hypertrophy. Presence of a conduction abnormality is not typical of PAH. Electrocardiogram evidence of right heart strain has been associated with decreased survival.2
Pulmonary function tests
Pulmonary function tests (PFTs) are important in excluding secondary causes of PH, particularly chronic obstructive airways disease. Airway obstruction is not typical of PAH, although cases of bronchial obstruction due to enlarged pulmonary arteries have been reported. Pulmonary arterial hypertension patients typically demonstrate borderline restrictive physiology, a reduced diffusing capacity for carbon monoxide (DLCO), and hypoxemia with hypocapnea.5 Reduction in DLCO results from reduced blood volume in alveolar capillaries. In a study of pulmonary function in 79 patients presenting with PAH, significantly reduced DLCO (mean 68% of predicted values) was observed in 75% of patients, and mild restrictive physiology was seen in 50%.99
Echocardiography
Transthoracic echocardiography is a crucial diagnostic tool in evaluating patients for PH.96 It can determine the presence of left-sided heart disease, valvular disease, and intracardiac shunts, and it allows noninvasive measurement of PAP. A finding of elevated PAP must be further evaluated with pulmonary artery catheterization. Echocardiography of PAH patients frequently shows RV hypertrophy and dilation, right atrial enlargement, and decreased LV cavity size due to bowing of the interventricular septum in advanced disease. The inferior vena cava is typically distended and does not collapse during inspiration in advanced disease. Systolic PAP can be estimated using Doppler measurement of the tricuspid regurgitant flow velocity. The upper limit of normal systolic PAP is generally considered 40 to 50 mmHg at rest, corresponding to a tricuspid regurgitant velocity of 3 to 3.5 m/s (although this value varies with age, body mass index [BMI], and right atrial pressure).
Limitations to Doppler measurement of PAP do exist, however; false-negative studies are possible in patients with poor-quality views or moderate elevations in PAP. Doppler estimates of pressures are also operator dependent. Studies comparing Doppler-derived PAP values with pressures determined by catheterization yield varying results, with some reporting underestimation of systolic PAP by Doppler.98 Echocardiography is probably best employed for its negative predictive value, although a number of echocardiographic features of PAH suggest worsened prognosis.100 These mostly include echocardiographic markers of RV dysfunction (i.e., right atrial or ventricular size, septal shift toward the LV during diastole, tricuspid annular plane excursion to approximate RV ejection fraction, RV myocardial index). Notably, however, echocardiographic estimates of RV systolic pressure do not predict survival.101
Exercise echocardiography is a more sensitive test for the presence of early PAH, which is particularly valuable in pediatric cases where it may influence decisions regarding surgery. Exercise echocardiography has been studied as a screening tool to identify asymptomatic carriers of BMPR2 mutations in PAH families.102 A resting echocardiogram demonstrating normal PA pressure usually excludes a diagnosis of PH. When PAH is strongly suspected or a patient has unexplained dyspnea, however, exercise echocardiography performed during exercise may reveal exercise-induced PAH.
Exercise testing
Exercise testing is not required for a diagnosis of PH, but it may provide valuable information regarding prognosis. The most widely used and reproducible exercise test is the 6-minute walk test. This test is usually done after the diagnosis is confirmed by cardiac catheterization, and at regular intervals to monitor functional status. The distance walked in 6 minutes has been shown to decrease in proportion to the NYHA functional class and is a strong independent predictor of mortality. Patients with PAH who walk 300 or fewer meters and display decreased arterial oxygen saturation by 10% at maximal distance suffer from a significantly increased rate of mortality.98 Maximal exercise testing must be avoided because syncope and sudden death have been reported. Cardiopulmonary exercise testing with or without echocardiography has been increasingly used in research studies and can be helpful in distinguishing PAH from PH secondary to diastolic heart failure.
Screening
Screening asymptomatic patients at high risk for PAH is recommended.103 The exact population recommended for screening is controversial, since the prevalence of disease is low even in categories of patients at increased risk. Patient groups who likely benefit from screening include those with already known genetic mutations predisposing to PAH, first-degree relatives in a familial PAH cluster, patients with scleroderma, portal hypertension prior to liver transplantation, and patients with congenital systemic-to-pulmonary shunts.104 Some patients should be evaluated for PAH only if they present with symptoms suggestive of the disease, including those with collagen vascular disease other than scleroderma, HIV, intravenous drug users, patients exposed to appetite-suppressant drugs, and patients with portal hypertension who are not being considered for transplantation. Screening asymptomatic or minimally symptomatic patients should begin with a thorough history and physical examination to elicit symptoms or signs consistent with PH, followed by diagnostic testing if inconclusive. Transthoracic echocardiogram is the best noninvasive test used for screening patients.
Genetic testing
Genetic testing of asymptomatic individuals is not currently advocated as an effective method for diagnosis, considering the low penetrance of disease manifestation even if a predisposing mutation is present. The risk for disease in first-degree relatives of a patient with PAH is relatively low, which has led to uncertainty in genetic screening of their family members. Advocates suggest that screening asymptomatic patients may enhance knowledge of the prevalence of familial PAH and shed light on whether early treatment influences disease pathogenesis. Screening also allows at-risk individuals to be aware of known risks that theoretically may augment penetrance of the disease.105 However, genetic testing results can be confusing; even if family members have inherited a predisposing mutation, about an 80% chance exists that no discernible disease phenotype will manifest. Notably, such circumstances may result in detrimental psychological, employment, and insurance effects and, if pursued, must be supported by appropriate genetic counseling.
Treatment
There is no known cure for PAH, but current therapeutic options have dramatically improved survival and quality of life for PAH patients. Recently the American Heart Association (AHA) and American College of Cardiology, in collaboration with the American College of Chest Physicians, American Thoracic Society, and Pulmonary Hypertension Association, released consensus recommendations for treatment of PAH103 (Fig. 56-7).
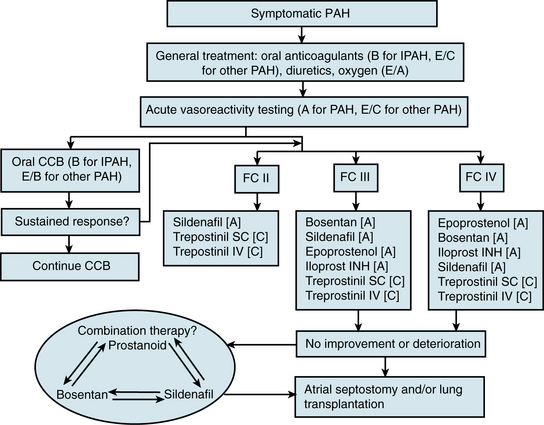
Figure 56-7 Algorithm for treatment of pulmonary arterial hypertension (PAH), adapted from 2007 AHA/ACC/ACCP Guidelines.
(Adapted from Badesch DB, Abman SH, Simonneau G, et al: Medical therapy for pulmonary arterial hypertension: updated ACCP evidence-based clinical practice guidelines. Chest 131:1917–1928, 2007.)
Anticoagulation
Anticoagulation has been incorporated into PAH treatment on the basis of presence of thrombosis in small PAs, risk of compounding PAH with a thromboembolic component, and increased risk of deep venous thrombosis (DVT) in the setting of a low CO. No controlled trial of anticoagulation in PAH currently exists, but anticoagulation is based on improved survival of patients who received warfarin in historical smaller studies.106 The usual recommended International Normalized Ratio (INR) is 2 to 2.5.107 Although prostacyclin inhibits platelet aggregation, additional anticoagulation is typically used in the absence of a contraindication. Since patients with congenital systemic-to-pulmonary shunts are at greater risk for hemoptysis, however, some practitioners do not recommend anticoagulation in those cases.108
Pulmonary Vascular–Specific Therapies
Initial screening for vasodilator reserve
Acute vasoreactivity testing is generally used as an initial screen to assess for vasodilator reserve and potential response to vasodilator therapy.109 A significant response to vasodilator testing is generally accepted as a decrease in PVR by at least 25%, with a decrease in mean PAP to less than 40 mmHg. Other parameters include a greater than 20% decrease in systolic PAP, a greater than 10 mmHg drop in mean PAP, or an increase in cardiac index by 30% or more. The most widely used drugs for acute vasoreactivity testing include inhaled NO and intravenous prostacyclin (epoprostenol), adenosine, and iloprost, a prostacyclin analog with less effect on the systemic vasculature. Testing more than one drug offers no advantage. Vasodilator challenge must be performed with care because drug-induced systemic hypotension (e.g., with prostacyclin) may reduce RV coronary blood flow and cause RV ischemia.
Most studies on PAH report the proportion of responders to vasodilators as between 12% and 25%.96 There is no particular clinical or disease characteristic that reliably predicts vasodilator response. Lack of response to acute vasodilators predicts response to oral vasodilator therapy (i.e., nonresponders to inhaled NO do not respond to oral calcium channel blockers). Response to acute vasodilators, however, does not predict a response to prostacyclin. A trial of long-term calcium channel blocker therapy is usually recommended in PAH patients who respond to vasodilator challenge with a decrease in PVR of 50 or more.
Calcium channel blockers
Calcium channel blockers are the oral drugs of choice for treating patients who have a significant response to acute vasodilators.109 These agents have been shown to improve hemodynamics, RV function, and survival in the subpopulation of PAH patients responding to acute vasoreactivity testing.110 Empirical therapy without vasodilator testing is not recommended because of the high rate of treatment failure. Both nifedipine and diltiazem are effective in appropriate patients, and the choice between these two drugs is guided by resting heart rate. Doses of these drugs required to lower PVR are much higher than those used for other indications, which makes systemic side effects a significant problem, particularly hypotension and lower-extremity edema. Verapamil is not used because of its greater negative inotropic effects. Acute administration of amlodipine causes pulmonary vasodilation, but its long-term efficacy has not been studied.
Endothelin-1 receptor antagonists
Presently, three endothelin receptor antagonists are approved for oral use in PAH: two in the United States (bosentan [Tracleer] and ambrisentan [Letairis]) and one in Europe and Canada (sitaxsentan [Thelin]).111 Bosentan is a nonspecific antagonist recognizing both endothelin receptor subtypes A (ET-A) and B (ET-B), and ambrisentan and sitaxsentan are specific for ET-A. It is thought that specific inhibition of ET-A may provide more benefit by decreasing the vasoconstrictor effects of ET-A while allowing the vasodilator and ET-1 clearance functions of ET-B receptors. All three agents have been found to improve exercise capacity and hemodynamics in 12- to 16-week clinical trials.112–114 Bosentan has also improved survival in open-label studies and comparison with historical control data.96 Long-term survival data for the selective endothelin inhibitors appear favorable as well, with some preliminary data suggesting that time to clinical worsening favors sitaxsentan over bosentan. No clear survival data are yet available from properly designed clinical trials. Because of its efficacy and ease of use, bosentan is currently recommended for stable functional class III PAH patients as first- or second-line agents.
Phosphodiesterase inhibitors
Originally marketed for erectile dysfunction (ED), oral PDE5 inhibitors such as sildenafil (Revatio) selectively inhibit the cGMP-specific enzyme PDE5, enabling endogenous NO to exert a more sustained effect.115 This PDE is highly abundant in pulmonary vasculature, making such medications relatively selective pulmonary vasodilators. A more recently approved PDE5 inhibitor for use in PAH includes tadalafil. In patients with lung fibrosis and PAH, acute administration of sildenafil surpasses the vasodilating effect of NO. The effect is similar to intravenous epoprostenol, except sildenafil is more selective for better ventilated areas of the lung, resulting in improved gas exchange. Clinical efficacy has been reported with use of sildenafil in patients with primary PAH and PAH associated with congenital shunts, SLE, and HIV. Benefits with treatment include improved symptoms and hemodynamics, and increased 6-minute walk distance. In a multicenter double-blinded, randomized, placebo-controlled study of sildenafil treatment for PAH, significant improvements in exercise ability and quality of life have been observed; in the open-label follow-up study, 95% survival was measured at 1 year.116
Prostacyclin and prostacyclin analogs
Prostacyclin is an endogenous prostaglandin (prostaglandin I2) that causes vasodilation and inhibits platelet aggregation. It also has antiproliferative and weak fibrinolytic activities. Intravenous prostacyclin (epoprostenol [Flolan]) was first used to treat PAH in the early 1980s. Since then, multiple trials of epoprostenol have demonstrated improved survival, exercise capacity, and hemodynamics in patients with PAH compared with conventional treatment.117 Epoprostenol was approved for use in PAH in 1995; it is now considered the treatment of choice in patients with functional class IV PAH and is an alternative to lung transplantation.107 A chronic, perhaps remodeling, effect of epoprostenol has additionally been suggested by several findings. Patients who have no acute response may demonstrate a delayed hemodynamic improvement. Hemodynamic improvement increases in patients who show an initial response, and RV dysfunction improves after long-term therapy.
Major limitations to use of epoprostenol include the need for permanent central venous access, with the associated small risk of catheter-related infection or air embolism, the capacity to handle the catheter and pump, and the high cost of the drug. Complexities of epoprostenol administration have led investigators to search for alternative agents.118
The prostacyclin analogs treprostinil (Remodulin), iloprost (Ventavis), and beraprost have been tested in 12-week placebo-controlled trials.117 All these agents have demonstrated significant improvements in mean exercise capacity. Treprostinil (Remodulin, UT-15) has a longer half-life (≈︀ 4 hours) than epoprostenol and can be delivered intravenously or subcutaneously with a pump system similar to that used with subcutaneous insulin. Treprostinil does not require reconstitution or cold temperature. Pain at the site of subcutaneous infusion is a frequent problem that requires cessation of the drug in 8% to 12% of patients. Iloprost is another chemically stable analog that can be given intravenously and by inhaled routes. Iloprost can be used in a nebulized form that must be inhaled 6 to 9 times daily for a continuous effect. Beraprost is an oral analog taken four times daily. Treprostinil and iloprost are approved for PAH therapy—treprostinil for functional class II-IV and iloprost for functional class II-IV.
Treatment of Pulmonary Arterial Hypertension Associated with Specific Diseases
Current treatment guidelines do not distinguish between PAH subgroups in initial therapy recommendations, but most of the cited data supporting vasodilator therapies have originated from patient populations suffering from idiopathic PAH. Additionally, vasodilator therapy in PAH patients with collagen vascular diseases has been substantially studied with good evidence for benefit.119 As in idiopathic PAH, baseline hemodynamic data do not predict a response to epoprostenol in PAH associated with scleroderma. The acute vasodilator response is present in an even smaller proportion of patients with collagen vascular diseases than with idiopathic PAH. Accordingly, calcium channel blockers are not beneficial in this group of patients.120 Nonetheless, epoprostenol can be effective in improving symptoms, hemodynamics, and survival. Skin lesions in patients with scleroderma may also improve substantially with this treatment.121 Case reports of successful epoprostenol treatment of PAH in patients with SLE also exist, but treatment in this group has been complicated by severe thrombocytopenia. Patient outcomes with CREST/scleroderma have also been studied, with improved symptoms and hemodynamics after use of oral endothelin receptor antagonists.
Efficacy of such treatment in other PAH subgroups has been studied, but in smaller numbers of patients. Epoprostenol has been shown to improve functional capacity, oxygen saturation, and hemodynamics in a group of adults with Eisenmenger’s syndrome who were suffering from NYHA class III and IV heart failure.122 Small numbers of adults with repaired or unrepaired congenital heart disease have also been included in clinical trials of endothelin receptor blockers and PDE5 inhibitors; improvement in hemodynamics, exercise capacity, and symptoms have been demonstrated.123–125 Epoprostenol has also been used successfully in patients with PAH secondary to HIV infection. In patients suffering from portopulmonary hypertension, such treatment has facilitated successful liver transplantation.96 There are additional reports of improvement in PAH secondary to portopulmonary hypertension with the use of β-blockers and nitrates, which may also decrease the incidence of variceal bleeding. Yet, caveats to PAH treatment exist for this special patient population. Anticoagulation in these patients is controversial because of the risk of hemorrhage. Furthermore, the liver toxicity associated with bosentan and sitaxsentan make these unacceptable choices for patients with hepatic disease.
Disease Course, Prognosis, and Monitoring
Prior to the introduction of vasodilator therapies, median survival among patients diagnosed with PAH in the United States between 1981 and 1985 was 2.8 years, with single-year survival rates of 68% at 1 year, 48% at 3 years, and 34% at 5 years.2 Given such a bleak prognosis, most patients were referred for lung transplantation at the time of diagnosis. With the advent of effective medical therapies to ameliorate disease, survival and quality of life have improved substantially. Currently, at the time of diagnosis, patients begin medical therapy with serial reassessments. Five-year survival among epoprostenol-treated patients is now 47% to 55%, with better than 70% 5-year survival among those improving to functional class I or II.126,127
After starting medical therapy, only a small subset of patients benefit from immediate hemodynamic improvement.96 Most, however, display a slow improvement in hemodynamics over several months, as evidenced by increases in 6-minute walk distance and reduced symptoms. These improvements typically peak around 12 to 16 weeks of treatment and can be maintained for years, although significant variability and deterioration is not unexpected.
Frequent evaluations at 3-month intervals, with detailed evaluation of PAP and RV function annually, are necessary to identify patients failing therapy and consider lung transplantation. Patients who develop signs or symptoms of right heart failure have much lower survival rates than those with only elevations of PAP. Evidence of poor prognosis after treatment include elevated right atrial pressure, low cardiac index, low mixed venous oxygen saturation, continued functional class III/IV symptoms, poor exercise capacity, pericardial effusion, or elevated B-type natriuretic peptide levels.100,126,128 In early NIH registry data, lower DLCO and presence of Raynaud phenomenon also portended a worse outcome.2 Increased PAP itself may also predict worse outcomes when RV function is still relatively normal. Finally, failure to demonstrate improvements after treatment in symptoms, hemodynamics, and exercise capacity correlate with worse outcomes. In these cases, consideration for combination therapy, atrial septostomy, or lung transplantation should be pursued.
Management of Refractory Pulmonary Arterial Hypertension
Combination Therapy
Combination therapy using at least two separate classes of PAH medications has been advocated, based on the potential for additive or synergistic effects. Improvement in hemodynamics, exercise capacity, and symptoms over 12- to 16-week periods have been reported in nonresponders who failed standard drug regimens.129 Typically, improvement in 6-minute walk distance is modest (20-26 meters) compared to initial response to monotherapy. A single long-term but uncontrolled trial studying protocol-driven combination therapy for patients failing to achieve exercise test goals while on monotherapy reported good survival rates (93%, 83%, and 80% at 1, 2, and 3 years) that are better than historical controls on monotherapy.130 More rigorous and better controlled long-term studies comparing rates of survival and morbidity indices are ongoing. Currently, combination therapy can be considered for patients with signs of right heart failure, 6-minute walk distance less than 380 meters, and persistent functional class III-IV symptoms despite monotherapy for more than 6 months.
Atrial Septostomy
The rationale for surgical creation of an interatrial orifice to improve survival is based on the observation that survival in PAH depends critically upon RV function, and relieving stress on the RV theoretically improves PAH in general.131 For example, PAH patients with a patent foramen ovale or Eisenmenger’s syndrome tend to carry better cardiac function and survive longer compared with patients without intracardiac defects. A review of 64 cases who underwent atrial septostomy prior to contemporary treatment regimens reported improved clinical status in 47 of 50 patients. Median survival of the 54 patients who survived the procedure was 19.5 months (range, 2-96 months), which represents an improvement compared to treatments available before epoprostenol. However, because of the improved efficacy of currently available vasodilators, atrial septostomy is only considered as a palliative procedure for patients with severe PAH and clinical deterioration despite maximal medical therapy, or as a bridge to transplantation. It should only be attempted in centers with experience in the procedure.
Lung Transplantation
Before the introduction of epoprostenol, transplantation was the treatment of choice for severe PAH. It is still the last option in treating severe PAH and the only available cure.132 Patients with persistent functional class III or IV symptoms, right atrial pressure above 15 mmHg, or cardiac index below 2 L/min/m2 should be considered for lung transplantation. Hemodynamic responses to a 3-month epoprostenol infusion may also aid in identifying a subset of PAH patients who might benefit from being considered earlier for lung transplantation. Poor survival has been noted in patients with NYHA functional classes III and IV who remained in that class or failed to achieve a 30% decrease in PVR after 3 months on continuous IV epoprostenol.
Survival in the early postoperative period is lower for PAH patients than other patients undergoing lung transplantation. However, long-term outcome in patients with PAH is similar to general transplantation patients.133 Survival rates at 1, 3, and 5 years after transplantation have been reported as 65%, 55%, and 44%, respectively. Pulmonary arterial hypertension has not been reported to recur after transplantation, although a careful examination of those with genetic predisposition has not been performed.
Experimental Therapies
In addition to the more established therapies for PAH, additional intriguing yet still developing therapeutic approaches are under investigation (Table 56-2). Unlike current medical therapies that at best ameliorate disease, there is optimism that regression of pulmonary vascular remodeling and cure for PAH is possible with these evolving treatment paradigms.
Table 56-2 Established and Experimental Therapies in Pulmonary Arterial Hypertension
| Target | Goal | Drug |
|---|---|---|
| L-type Ca2 + channels | Decrease SMC Ca2 + | L-type Ca2 + channel blockers |
| Coagulation cascade | Decrease thrombosis | Warfarin |
| Prostacyclin receptors | Increase cAMP | Epoprostenol |
| Endothelin receptors A and B | Inhibit constriction and proliferation | Bosentan |
| Endothelin receptor A | Inhibit constriction and proliferation | Sitaxsentan |
| PDE5 inhibitors | Increase cGMP | Sildenafil |
| Guanylate cyclase activators | Increase cGMP | iNO |
| Novel Targets | ||
| Rho kinase | Decrease Ca2 + sensitivity in SMCs | Rho kinase inhibitors: fasudil |
| Rho A prenylation | Decrease Ca2 + sensitivity in SMCs | Statins |
| Serine elastases | Decrease MMP activation | Elastase inhibitors |
| Kinase-associated receptors | Inhibit PDGF or EGF activity | Tyrosine kinase inhibitors |
| PDF kinase | Normalize mitochondrial function | Dichloroacetate |
| NFAT | Decrease antiapoptotic bcl-2; slow proliferation | Cyclosporine A |
| Immune system | Immunosuppression | Mycophenolate mofetil |
| Survivin | Inhibits antiapoptotic effect of survivin | Transfection of dominant negative |
| Guanylate cyclase | Increase cGMP | Direct sGC activators (e.g., Riociguat [BAY 63-2521]- DHEA) |
| COX | Inhibit TxA2 | Aspirin |
| Ornithine decarboxylase | Inhibit polyamine synthesis | α-Difluoromethylornithine |
| CDKI γ27 | Inhibit SMC proliferation | Heparin |
| PPAR-γ | Increase PPAR-γ activity | Rosiglitazone |
| Angiopoietin and TIE2 | Inhibit SMC proliferation | Adenoviral gene transfection |
| Serotonin transporter | Inhibit SMC proliferation | SSRI |
| VPAC 1 and 2 receptors | Inhibit SMC proliferation | VIP, inhaled |
| Adrenomedullin receptors | Inhibit SMC proliferation, vasodilation | Adrenomedullin |
| BMPR2 | Enhance BMPR2 signaling | Adenoviral transfection and/or enhancing receptor trafficking to membrane |
| eNOS | Increase cGMP and NO signaling | eNOS-transfected EPCs |
| eNOS | Increase cGMP and NO signaling | VEGF-transfected fibroblasts |
Alternative nitric oxide–based therapy
In addition to PDE5 inhibitors, a number of therapeutic approaches designed to improve NO production, release, and activity in the pulmonary vasculature are under investigation. Inhaled NO gas has been successfully used to decrease PAP and improve oxygenation and hemodynamics in diverse forms of PAH. However, long-term NO therapy is cumbersome, necessitates continuous inhalation, and must be monitored carefully to avoid toxicity with nitrogen oxides and methemoglobin. Alternative strategies to deliver NO include the use of NONOates, compounds that release predictable levels of NO when exposed to physiological pH. Such compounds have been used by daily nebulization in rodent PAH models, with reduction of PAP without systemic hypotension.134
Another avenue of increasing NO levels in PAH relies upon enhancing NOS activity. Tetrahydrobiopterin (BH4) is an important NOS cofactor that enzymatically links oxygenation of L-arginine to create NO and L-citrulline. BH4 levels may be decreased in PAH patients, so administration of BH4 in its synthetic form (sapropterin) may hold promise in PAH to increase NOS activity and NO levels in the pulmonary vasculature.3
Finally, NO-independent sGC activators have been developed for PAH patients.44 Both heme-dependent (BAY 41-2272) and heme-independent (BAY 58-2667 and HMR-1766) sGC activators exist.135 BAY 41-2272 stimulates sGC directly and sensitizes sGC to NO, resulting in synergism. It improves pulmonary hemodynamics in models of PH. NO-independent sGC activators appear to provide an additive rather than synergistic effect when combined with NO donors. Both BAY 41-2272 and BAY 58-2667 ameliorate PAH in rodent models, yet this benefit is partially dependent on endogenous NOS activity. BAY 63-2521 is an oral agent, has a favorable safety profile, and is currently under phase III clinical study in PAH. Interestingly, dehydroepiandrosterone (DHEA), an endogenous steroid, also increases sGC expression, activates vasodilatory calcium-sensitive potassium channels (BKCa) in vascular smooth muscle, and inhibits hypoxic PH in rats.136 More data are necessary before testing in human trials.
Serotonin and serotonin transporter inhibitors
Terguride is a potent antagonist of 5-hydroxytryptamine receptors and is currently in a phase II study for PAH patients.3 It has already received orphan drug status in Europe. PRX-08066 is a selective 5-hydroxytryptamine 2B receptor antagonist that inhibits hypoxia-mediated increases in PAP in humans and is also under phase II trial study. Given the importance of SERT in the biology of serotonin signaling, synergistic inhibition of both SERT and serotonin receptors may represent a potent future strategy for therapeutic benefit.
BMPR2-targeted therapies
Recent advances linking BMPR2 haploinsufficiency to PAH suggests that enhancing BMPR2 expression or activity may benefit or reverse PAH progression. Gene replacement therapy has been proposed but has yet to show success in animal models. It has also been found that some mutant forms of BMPR2 in PAH result in impaired trafficking to the cell surface rather than decreased expression.137 In that case, medications that improve protein trafficking (e.g., sodium 4-phenybutyrate as used in cystic fibrosis) may be useful; however, it remains uncertain how much mutant BMPR2 must reach the cell membrane to result in a relevant effect in vivo. Finally, dysfunction of BMPR2 can lead to increased signaling via other TGF receptor superfamily members such as the TGF-β/activin receptor-like kinase 5. Notably, SB525334, an inhibitor of TGF-β/activin receptor-like kinase 5, reverses PAH and RVH in a rodent model and may have therapeutic benefit.138
Tyrosine kinase inhibitors
Initial experience with the tyrosine kinase inhibitor imatinib and the “multikinase inhibitor” sorafenib in PAH patients have been encouraging but results are still preliminary. In humans, there are case reports of beneficial effects in adding imatinib to conventional therapy.79 Sorafenib reverses PAH and RV remodeling in monocrotaline-treated rats.80 Phase I clinical trials studying both medications have been performed recently.
Rho kinase inhibitors
Rho kinase inhibitors (i.e., Y-27632, fasudil) have been developed for study in the PAH population.139 These medications reduce PAP in multiple rodent models of PAH. In humans, fasudil induces modest but immediate reductions in PVR. However, a common adverse side effect includes systemic vasodilation and hypotension. Alternative delivery options such as inhaled or nebulized formulations may allow circumvention of this side effect.
Interestingly, one of the pleiotropic effects of HMG CoA reductase inhibitors (statins) includes inhibition of rho kinase activity. In some rodent studies, simvastatin reduces PAH and demonstrates a significant improvement in survival140,141; however, these beneficial effects have not been replicated in all rodent models.142 Nonetheless, several statin trails in human PAH are underway.
Miscellaneous pathways
Therapeutic targets in several additional pathogenic pathways have been studied to varying degrees. Inhaled adrenomedullin modestly reduces PVR and increases peak oxygen consumption during exercise in humans with PAH.143 Nebulized VIP improves pulmonary hemodynamics in PAH to a larger degree, and if continued for 3 months, reduces PVR and improves 6-minute walk distance56; however, VIP is susceptible to rapid degradation. A sustained-release liposomal VIP preparation is currently under development. Heparins and inhibitors of angiopoietin 1, STAT3, polyamines, survivin, and the cell cycle have also been proposed for therapeutic benefit, but with varying degrees of success.3
Endothelial progenitor cells
Progenitor cell infusion represents a potentially powerful, but currently nebulous, therapeutic possibility. Specifically, endothelial progenitor cells (EPCs) arise from mesodermal stem cells or hemangioblasts in the bone marrow. From there, they are released and circulate in blood until they engraft into sites of ischemia or injury, leading to angiogenesis and revascularization. Circulating EPCs (defined by CD34+/KDR+ and CD34+/CD133+/KDR+ cells) are lower in patients with Eisenmenger’s syndrome.144 Some reports have indicated reduced levels of EPCs in idiopathic PAH patients.145 Furthermore, the functions of EPCs in cell culture (i.e., colony forming activity, adherence, migration, apoptotic potential) differ from those isolated from PAH patients as compared with normal controls.144–146 Administration of EPCs in rats has resulted in improvement in pulmonary hemodynamics, vascular remodeling, and survival in models of PAH.147 Additionally, two small studies in which adults148 and children149 with idiopathic PAH were given a single infusion of autologous mononuclear cells provides support for the therapeutic potential of such therapy. A therapeutic trial has begun to assess the safety of administering such cells in patients with PAH.
Conclusions
Pulmonary arterial hypertension is a clinical syndrome of vascular disease with a stereotyped pattern of histopathology and is related to a variety of secondary disease states. It has become increasingly clear that development of PAH entails a complex multifactorial pathophysiology. Although genetic mutations, exogenous exposures, and acquired disease states can predispose to PAH, no one factor identified thus far is sufficient alone to fully drive the pathogenic process. Similar to carcinogenesis, in which a susceptible person with a specific genetic mutation requires additional injuries to manifest disease, a “multiple-hit” hypothesis has emerged to explain progression to clinical PAH (Fig. 56-8). More delineated mechanisms of disease have focused on the end-stage condition and the effects of the imbalance of multiple vascular effectors. Some unifying mechanisms of disease have become more apparent, linking regulation of these effectors into a more cohesive model. As a result, effective therapeutic approaches have been developed to ameliorate disease and improve survival.
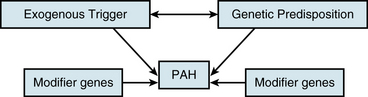
Figure 56-8 Paradigm for the “multiple-hit” hypothesis promoting pulmonary arterial hypertension (PAH).
Additionally, a challenge of research in PAH is lack of a small animal model that can be followed and genetically manipulated from initiation through progression to severe disease. There has been some success in physiological study of PAH in larger mammals, but identification of contributors in the molecular process of pulmonary remodeling has been difficult, owing to incomplete genetic data and tools specific for these models.150,151 Conversely, although genetic and molecular studies in the mouse are more tractable, induction of significant PAH in murine models has been challenging. Nonetheless, recent advances have hinted at the possibility of more tractable animal models of PAH, which would be useful for combining genetic study and pathophysiological exposures.
1. Simonneau G., Robbins IM., Beghetti M., et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2009;54(1 Suppl):S43–S54.
2. D’Alonzo GE., Barst RJ., Ayres SM., et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann Intern Med. 1991;115(5):343–349.
3. Archer SL., Weir EK., Wilkins MR. Basic science of pulmonary arterial hypertension for clinicians: new concepts and experimental therapies. Circulation. 2010;121(18):2045–2066.
4. Butrous G., Ghofrani HA., Grimminger F. Pulmonary vascular disease in the developing world. Circulation. 2008;118(17):1758–1766.
5. Rich S., Dantzker DR., Ayres SM., et al. Primary pulmonary hypertension. A national prospective study. Ann Intern Med. 1987;107(2):216–223.
6. Widlitz A., Barst RJ. Pulmonary arterial hypertension in children. Eur Respir J. 2003;21(1):155–176.
7. Farber H., Loscalzo J. Pulmonary arterial hypertension. N Engl J Med. 2004;351(16):1655–1665.
8. Chan SY., Loscalzo J. Pathogenic mechanisms of pulmonary arterial hypertension. J Mol Cell Cardiol. 2008;44(1):14–30.
9. Trembath R., Thomson J., Machado R., et al. Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N Engl J Med. 2001;345(5):325–334.
10. Chaouat A., Coulet F., Favre C., et al. Endoglin germline mutation in a patient with hereditary haemorrhagic telangiectasia and dexfenfluramine associated pulmonary arterial hypertension. Thorax. 2004;59(5):446–448.
11. Lane K., Machado R., Pauciulo M., et al. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. The International PPH Consortium. Nat Genet. 2000;26(1):81–84.
12. Newman J., Wheeler L., Lane K., et al. Mutation in the gene for bone morphogenetic protein receptor II as a cause of primary pulmonary hypertension in a large kindred. N Engl J Med. 2001;345(5):319–324. [Erratum, N Engl J Med 2345:1506, 2001, 2346:1258, 2002]
13. Aldred M, Machado R, James V, et al: Characterization of the BMPR2 5′-untranslated region and a novel mutation in pulmonary hypertension, Am J Respir Crit Care Med Epublication.
14. Shi Y., Massagueé J. Mechanisms of TGF-beta signaling from cell membrane to nucleus. Cell. 2003;113(6):685–700.
15. Atkinson C., Stewart S., Upton P., et al. Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation. 2002;105(14):1672–1678.
16. Teichert-Kuliszewska K., Kutryk M., Kuliszewski M., et al. Bone morphogenetic protein receptor-2 signaling promotes pulmonary arterial endothelial cell survival: implications for loss-of-function mutations in the pathogenesis of pulmonary hypertension. Circ Res. 2006;98(2):209–217.
17. Beppu H., Ichinose F., Kawai N., et al. BMPR-II heterozygous mice have mild pulmonary hypertension and an impaired pulmonary vascular remodeling response to prolonged hypoxia. Am J Physiol Lung Cell Mol Physiol. 2004;287(6):L1241–L1247.
18. West J., Fagan K., Steudel W., et al. Pulmonary hypertension in transgenic mice expressing a dominant-negative BMPRII gene in smooth muscle. Circ Res. 2004;94(8):1109–1114.
19. Morrell NW., Adnot S., Archer SL., et al. Cellular and molecular basis of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54(1 Suppl):S20–S31.
20. Launay J., Herveé P., Peoc’h KT., et al. Function of the serotonin 5-hydroxytryptamine 2B receptor in pulmonary hypertension. Nat Med. 2002;8(10):1129–1135.
21. Eddahibi S., Chaouat A., Morrell N., et al. Polymorphism of the serotonin transporter gene and pulmonary hypertension in chronic obstructive pulmonary disease. Circulation. 2003;108(15):1839–1844.
22. Eddahibi S., Raffestin B., Launay J., et al. Effect of dexfenfluramine treatment in rats exposed to acute and chronic hypoxia. Am J Respir Crit Care Med. 1998;157(4 Pt 1):1111–1119.
23. Long L., MacLean M., Jeffery T., et al. Serotonin increases susceptibility to pulmonary hypertension in BMPR2-deficient mice. Circ Res. 2006;98(6):818–827.
24. MacLean M., Deuchar G., Hicks M., et al. Overexpression of the 5-hydroxytryptamine transporter gene: effect on pulmonary hemodynamics and hypoxia-induced pulmonary hypertension. Circulation. 2004;109(17):2150–2155.
25. Keegan A., Morecroft I., Smillie D., et al. Contribution of the 5-HT(1B) receptor to hypoxia-induced pulmonary hypertension: converging evidence using 5-HT(1B)-receptor knockout mice and the 5-HT(1B/1D)-receptor antagonist GR127935. Circ Res. 2001;89(12):1231–1239.
26. Moudgil R., Michelakis E., Archer S. Hypoxic pulmonary vasoconstriction. J Appl Physiol. 2005;98(1):390–403.
27. Sweeney M., Yuan J. Hypoxic pulmonary vasoconstriction: role of voltage-gated potassium channels. Respir Res. 2000;1(1):40–48.
28. Vij R, Machado RF: Pulmonary complications of hemoglobinopathies, Chest 138(4):973–983.
29. Hsu L., Champion H., Campbell-Lee S., et al. Hemolysis in sickle cell mice causes pulmonary hypertension due to global impairment in nitric oxide bioavailability. Blood. 2007;109(7):3088–3098.
30. Hu W, Jin R, Zhang J, et al: The critical roles of platelet activation and reduced NO bioavailability in fatal pulmonary arterial hypertension in a murine hemolysis model, Blood 116(9):1613–1622.
31. Stauber RE, Olschewski H: Portopulmonary hypertension: short review, Eur J Gastroenterol Hepatol 22(4):385–390.
32. Le Pavec J, Humbert M, Mouthon L, et al: Systemic sclerosis-associated pulmonary arterial hypertension, Am J Respir Crit Care Med 181(12):1285–1293.
33. Hassoun PM. Pulmonary arterial hypertension complicating connective tissue diseases. Semin Respir Crit Care Med. 2009;30(4):429–439.
34. Peacock A. Primary pulmonary hypertension. Thorax. 1999;54(12):1107–1118. [Thorax 1155(1103):1254, 2000]
35. Degano B., Sitbon O., Simonneau G. Pulmonary arterial hypertension and HIV infection. Semin Respir Crit Care Med. 2009;30(4):440–447.
36. Galiè N., Manes A., Uguccioni L., et al. Primary pulmonary hypertension: insights into pathogenesis from epidemiology. Chest. 1998;114(3 Suppl):184S–194S.
37. Cool C., Rai P., Yeager M., et al. Expression of human herpesvirus 8 in primary pulmonary hypertension. N Engl J Med. 2003;349(12):1113–1122.
38. Beghetti M., Tissot C. Pulmonary arterial hypertension in congenital heart diseases. Semin Respir Crit Care Med. 2009;30(4):421–428.
39. Webb G., Gatzoulis M. Atrial septal defects in the adult: recent progress and overview. Circulation. 2006;114(15):1645–1653.
40. Topper J., Gimbrone MJ. Blood flow and vascular gene expression: fluid shear stress as a modulator of endothelial phenotype. Mol Med Today. 1999;5(1):40–46.
41. Sakao S., Taraseviciene-Stewart L., Lee J., et al. Initial apoptosis is followed by increased proliferation of apoptosis-resistant endothelial cells. FASEB J. 2005;19(9):1178–1180.
42. Cool CD., Deutsch G. Pulmonary arterial hypertension from a pediatric perspective. Pediatr Dev Pathol. 2008;11(3):169–177.
43. Robles A., Shure D. Gender issues in pulmonary vascular disease. Clin Chest Med. 2004;25(2):373–377.
44. Coggins MP., Bloch KD. Nitric oxide in the pulmonary vasculature. Arterioscler Thromb Vasc Biol. 2007;27(9):1877–1885.
45. Gladwin M., Schechter A. NO contest: nitrite versus S-nitroso-hemoglobin. Circ Res. 2004;94(7):851–855.
46. Griffiths M., Evans T. Inhaled nitric oxide therapy in adults. N Engl J Med. 2005;353(25):2683–2695.
47. Gerber J., Voelkel N., Nies A., et al. Moderation of hypoxic vasoconstriction by infused arachidonic acid: role of PGI2. J Appl Physiol Respir Env Exercise Physiol. 1980;49:107–112.
48. Tuder R., Cool C., Geraci M., et al. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am J Respir Crit Care Med. 1999;159(6):1925–1932.
49. Christman B., McPherson C., Newman J., et al. An imbalance between the excretion of thromboxane and prostacyclin metabolites in pulmonary hypertension. N Engl J Med. 1992;327(2):70–75.
50. Strauss W., Edelman J. Prostanoid therapy for pulmonary arterial hypertension. Clin Chest Med. 2007;28(1):127–142.
51. Davie NJ., Schermuly RT., Weissmann N., et al. The science of endothelin-1 and endothelin receptor antagonists in the management of pulmonary arterial hypertension: current understanding and future studies. Eur J Clin Invest. 2009;39(Suppl 2):38–49.
52. Jeffery T., Morrell N. Molecular and cellular basis of pulmonary vascular remodeling in pulmonary hypertension. Prog Cardiovasc Dis. 2002;45(3):173–202.
53. Allen S., Chatfield B., Koppenhafer S., et al. Circulating immunoreactive endothelin-1 in children with pulmonary hypertension. Association with acute hypoxic pulmonary vasoreactivity. Am Rev Respir Dis. 1993;148(2):519–522.
54. Giaid A., Yanagisawa M., Langleben D., et al. Expression of endothelin-1 in the lungs of patients with pulmonary hypertension. N Engl J Med. 1993;328(24):1732–1739.
55. Langleben D. Endothelin receptor antagonists in the treatment of pulmonary arterial hypertension. Clin Chest Med. 2007;28(1):117–125.
56. Petkov V., Mosgoeller W., Ziesche R., et al. Vasoactive intestinal peptide as a new drug for treatment of primary pulmonary hypertension. J Clin Invest. 2003;111(9):1339–1346.
57. Said S., Hamidi S., Dickman K., et al. Moderate pulmonary arterial hypertension in male mice lacking the vasoactive intestinal peptide gene. Circulation. 2007;115(10):1260–1268.
58. Söderman C., Eriksson L., Juhlin-Dannfelt A., et al. Effect of vasoactive intestinal polypeptide (VIP) on pulmonary ventilation-perfusion relationships and central haemodynamics in healthy subjects. Clin Physiol. 1993;13(6):677–685.
59. Haydar S., Sarti J., Grisoni E. Intravenous vasoactive intestinal polypeptide lowers pulmonary-to-systemic vascular resistance ratio in a neonatal piglet model of pulmonary arterial hypertension. J Pediatr Surg. 2007;42(5):758–764.
60. Hansmann G., Wagner R., Schellong S., et al. Pulmonary arterial hypertension is linked to insulin resistance and reversed by peroxisome proliferator-activated receptor-gamma activation. Circulation. 2007;115(10):1275–1284.
61. Hansmann G., de Jesus Perez VA., Alastalo TP., et al. An antiproliferative BMP-2/PPARgamma/apoE axis in human and murine SMCs and its role in pulmonary hypertension. J Clin Invest. 2008;118(5):1846–1857.
62. Nagaya N., Nishikimi T., Okano Y., et al. Plasma brain natriuretic peptide levels increase in proportion to the extent of right ventricular dysfunction in pulmonary hypertension. J Am Coll Cardiol. 1998;31(1):202–208.
63. Zhao L., Winter RJ., Krausz T., et al. Effects of continuous infusion of atrial natriuretic peptide on the pulmonary hypertension induced by chronic hypoxia in rats. Clin Sci (Lond). 1991;81(3):379–385.
64. Baliga RS., Zhao L., Madhani M., et al. Synergy between natriuretic peptides and phosphodiesterase 5 inhibitors ameliorates pulmonary arterial hypertension. Am J Respir Crit Care Med. 2008;178(8):861–869.
65. Qi JG., Ding YG., Tang CS., et al. Chronic administration of adrenomedullin attenuates hypoxic pulmonary vascular structural remodeling and inhibits proadrenomedullin N-terminal 20-peptide production in rats. Peptides. 2007;28(4):910–919.
66. Li X., Zhang X., Leathers R., et al. Notch3 signaling promotes the development of pulmonary arterial hypertension. Nat Med. 2009;15(11):1289–1297.
67. Loscalzo J., Kohane I., Barabasi A. Human disease classification in the postgenomic era: complex systems approach to human pathobiology. Mol Syst Biol. 2007;3:124.
68. Bonnet S., Michelakis ED., Porter CJ., et al. An abnormal mitochondrial-hypoxia inducible factor-1alpha-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: similarities to human pulmonary arterial hypertension. Circulation. 2006;113(22):2630–2641.
69. Xu W., Koeck T., Lara AR., et al. Alterations of cellular bioenergetics in pulmonary artery endothelial cells. Proc Natl Acad Sci U S A. 2007;104(4):1342–1347.
70. Semenza GL. Hypoxia-inducible factor 1: regulator of mitochondrial metabolism and mediator of ischemic preconditioning. Biochim Biophys Acta. 2010. Aug 21 [Epub ahead of print]
71. Archer SL., Gomberg-Maitland M., Maitland ML., et al. Mitochondrial metabolism, redox signaling, and fusion: a mitochondria-ROS-HIF-1alpha-Kv1.5 O2-sensing pathway at the intersection of pulmonary hypertension and cancer. Am J Physiol Heart Circ Physiol. 2008;294(2):H570–H578.
72. Sharma S., Taegtmeyer H., Adrogue J., et al. Dynamic changes of gene expression in hypoxia-induced right ventricular hypertrophy. Am J Physiol Heart Circ Physiol. 2004;286(3):H1185–H1192.
73. Young LH., Li J., Baron SJ., et al. AMP-activated protein kinase: a key stress signaling pathway in the heart. Trends Cardiovasc Med. 2005;15(3):110–118.
74. Yuan X., Wang J., Juhaszova M., et al. Attenuated K+ channel gene transcription in primary pulmonary hypertension. Lancet. 1998;351(9104):726–727.
75. Remillard C., Tigno D., Platoshyn O., et al. Function of Kv1.5 channels and genetic variations of KCNA5 in patients with idiopathic pulmonary arterial hypertension. Am J Physiol Cell Physiol. 2007;292(5):C1837–C1853.
76. Pozeg Z., Michelakis E., McMurtry M., et al. In vivo gene transfer of the O2-sensitive potassium channel Kv1.5 reduces pulmonary hypertension and restores hypoxic pulmonary vasoconstriction in chronically hypoxic rats. Circulation. 2003;107(15):2037–2044.
77. Merklinger SL., Jones PL., Martinez EC., et al. Epidermal growth factor receptor blockade mediates smooth muscle cell apoptosis and improves survival in rats with pulmonary hypertension. Circulation. 2005;112(3):423–431.
78. Schermuly RT., Dony E., Ghofrani HA., et al. Reversal of experimental pulmonary hypertension by PDGF inhibition. J Clin Invest. 2005;115(10):2811–2821.
79. Ghofrani HA., Seeger W., Grimminger F. Imatinib for the treatment of pulmonary arterial hypertension. N Engl J Med. 2005;353(13):1412–1413.
80. Moreno-Vinasco L., Gomberg-Maitland M., Maitland ML., et al. Genomic assessment of a multikinase inhibitor, sorafenib, in a rodent model of pulmonary hypertension. Physiol Genomics. 2008;33(2):278–291.
81. Shimokawa H., Takeshita A. Rho-kinase is an important therapeutic target in cardiovascular medicine. Arterioscler Thromb Vasc Biol. 2005;25(9):1767–1775.
82. Nagaoka T., Morio Y., Casanova N., et al. Rho/Rho kinase signaling mediates increased basal pulmonary vascular tone in chronically hypoxic rats. Am J Physiol Lung Cell Mol Physiol. 2004;287(4):L665–L672.
83. Oka M., Homma N., Taraseviciene-Stewart L., et al. Rho kinase-mediated vasoconstriction is important in severe occlusive pulmonary arterial hypertension in rats. Circ Res. 2007;100(6):923–929.
84. Fukumoto Y., Matoba T., Ito A., et al. Acute vasodilator effects of a Rho-kinase inhibitor, fasudil, in patients with severe pulmonary hypertension. Heart. 2005;91(3):391–392.
85. Ishikura K., Yamada N., Ito M., et al. Beneficial acute effects of rho-kinase inhibitor in patients with pulmonary arterial hypertension. Circulation J. 2006;70(2):174–178.
86. Taraseviciene-Stewart L., Nicolls MR., Kraskauskas D., et al. Absence of T cells confers increased pulmonary arterial hypertension and vascular remodeling. Am J Respir Crit Care Med. 2007;175(12):1280–1289.
87. Ulrich S., Nicolls MR., Taraseviciene L., et al. Increased regulatory and decreased CD8 + cytotoxic T cells in the blood of patients with idiopathic pulmonary arterial hypertension. Respiration. 2008;75(3):272–280.
88. Wright L., Tuder R., Wang J., et al. 5-Lipoxygenase and 5-lipoxygenase activating protein (FLAP) immunoreactivity in lungs from patients with primary pulmonary hypertension. Am J Respir Crit Care Med. 1998;157(1):219–229.
89. Song Y., Coleman L., Shi J., et al. Inflammation, endothelial injury, and persistent pulmonary hypertension in heterozygous BMPR2-mutant mice. Am J Physiol Heart Circ Physiol. 2008;295(2):H677–H690.
90. Rabinovitch M. Pathobiology of pulmonary hypertension. Extracellular matrix. Clin Chest Med. 2001;22(3):433–449.
91. Rabinovitch M. Elastase and the pathobiology of unexplained pulmonary hypertension. Chest. 1998;114(3 Suppl):213S–224S.
92. Zhu L., Wigle D., Hinek A., et al. The endogenous vascular elastase that governs development and progression of monocrotaline-induced pulmonary hypertension in rats is a novel enzyme related to the serine proteinase adipsin. J Clin Invest. 1994;94(3):1163–1171.
93. Jones P., Rabinovitch M. Tenascin-C is induced with progressive pulmonary vascular disease in rats and is functionally related to increased smooth muscle cell proliferation. Circ Res. 1996;79(6):1131–1142.
94. Cowan K., Jones P., Rabinovitch M. Elastase and matrix metalloproteinase inhibitors induce regression, and tenascin-C antisense prevents progression, of vascular disease. J Clin Invest. 2000;105(1):21–34.
95. Zaidi S., You X., Ciura S., et al. Overexpression of the serine elastase inhibitor elafin protects transgenic mice from hypoxic pulmonary hypertension. Circulation. 2002;105(4):516–521.
96. Chin KM., Rubin LJ. Pulmonary arterial hypertension. J Am Coll Cardiol. 2008;51(16):1527–1538.
97. Nef HM., Mollmann H., Hamm C., et al. Pulmonary hypertension: updated classification and management of pulmonary hypertension. Heart. 2010;96(7):552–559.
98. Chemla D., Castelain V., Herve P., et al. Haemodynamic evaluation of pulmonary hypertension. Eur Respir J. 2002;20(5):1314–1331.
99. Sun XG., Hansen JE., Oudiz RJ., et al. Pulmonary function in primary pulmonary hypertension. J Am Coll Cardiol. 2003;41(6):1028–1035.
100. Raymond RJ., Hinderliter AL., Willis PW., et al. Echocardiographic predictors of adverse outcomes in primary pulmonary hypertension. J Am Coll Cardiol. 2002;39(7):1214–1219.
101. McLaughlin VV., Presberg KW., Doyle RL., et al. Prognosis of pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines. Chest. 2004;126(1 Suppl):78S–92S.
102. Grunig E., Janssen B., Mereles D., et al. Abnormal pulmonary artery pressure response in asymptomatic carriers of primary pulmonary hypertension gene. Circulation. 2000;102(10):1145–1150.
103. McLaughlin VV., Archer SL., Badesch DB., et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: developed in collaboration with the American College of Chest Physicians, American Thoracic Society, Inc., and the Pulmonary Hypertension Association. Circulation. 2009;119(16):2250–2294.
104. McGoon M., Gutterman D., Steen V., et al. Screening, early detection, and diagnosis of pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines. Chest. 2004;126(1 Suppl):14S–34S.
105. Humbert M., Trembath RC. Genetics of pulmonary hypertension: from bench to bedside. Eur Respir J. 2002;20(3):741–749.
106. Johnson SR., Mehta S., Granton JT. Anticoagulation in pulmonary arterial hypertension: a qualitative systematic review. Eur Respir J. 2006;28(5):999–1004.
107. Badesch DB., Abman SH., Simonneau G., et al. Medical therapy for pulmonary arterial hypertension: updated ACCP evidence-based clinical practice guidelines. Chest. 2007;131(6):1917–1928.
108. Herveé P., Humbert M., Sitbon O., et al. Pathobiology of pulmonary hypertension: the role of platelets and thrombosis. Clin Chest Med. 2001;22(3):451–458.
109. Tonelli AR, Alnuaimat H, Mubarak K: Pulmonary vasodilator testing and use of calcium channel blockers in pulmonary arterial hypertension, Respir Med 104(4):481–496
110. Sitbon O., Humbert M., Jais X., et al. Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation. 2005;111(23):3105–3111.
111. Price LC., Howard LS. Endothelin receptor antagonists for pulmonary arterial hypertension: rationale and place in therapy. Am J Cardiovasc Drugs. 2008;8(3):171–185.
112. Channick R., Badesch DB., Tapson VF., et al. Effects of the dual endothelin receptor antagonist bosentan in patients with pulmonary hypertension: a placebo-controlled study. J Heart Lung Transplant. 2001;20(2):262–263.
113. Rubin LJ., Badesch DB., Barst RJ., et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med. 2002;346(12):896–903.
114. Barst RJ., Langleben D., Badesch D., et al. Treatment of pulmonary arterial hypertension with the selective endothelin-A receptor antagonist sitaxsentan. J Am Coll Cardiol. 2006;47(10):2049–2056.
115. Archer SL., Michelakis ED. Phosphodiesterase type 5 inhibitors for pulmonary arterial hypertension. N Engl J Med. 2009;361(19):1864–1871.
116. Galiè N., Ghofrani H., Torbicki A., et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med. 2005;353(20):2148–2157.
117. Mubarak KK. A review of prostaglandin analogs in the management of patients with pulmonary arterial hypertension. Respir Med. 2010;104(1):9–21.
118. Boutet K., Montani D., Jais X., et al. Therapeutic advances in pulmonary arterial hypertension. Ther Adv Respir Dis. 2008;2(4):249–265.
119. Badesch DB., Tapson VF., McGoon MD., et al. Continuous intravenous epoprostenol for pulmonary hypertension due to the scleroderma spectrum of disease. A randomized, controlled trial. Ann Intern Med. 2000;132(6):425–434.
120. Galie N., Manes A., Farahani KV., et al. Pulmonary arterial hypertension associated to connective tissue diseases. Lupus. 2005;14(9):713–717.
121. Mathai SC., Hassoun PM. Therapy for pulmonary arterial hypertension associated with systemic sclerosis. Curr Opin Rheumatol. 2009;21(6):642–648.
122. Rame JE. Pulmonary hypertension complicating congenital heart disease. Curr Cardiol Rep. 2009;11(4):314–320.
123. Galie N., Beghetti M., Gatzoulis MA., et al. Bosentan therapy in patients with Eisenmenger syndrome: a multicenter, double-blind, randomized, placebo-controlled study. Circulation. 2006;114(1):48–54.
124. Singh TP., Rohit M., Grover A., et al. A randomized, placebo-controlled, double-blind, crossover study to evaluate the efficacy of oral sildenafil therapy in severe pulmonary artery hypertension. Am Heart J. 2006;151(4):851. e851–e855
125. Apostolopoulou SC., Manginas A., Cokkinos DV., et al. Long-term oral bosentan treatment in patients with pulmonary arterial hypertension related to congenital heart disease: a 2-year study. Heart. 2007;93(3):350–354.
126. Sitbon O., Humbert M., Nunes H., et al. Long-term intravenous epoprostenol infusion in primary pulmonary hypertension: prognostic factors and survival. J Am Coll Cardiol. 2002;40(4):780–788.
127. McLaughlin VV., Shillington A., Rich S. Survival in primary pulmonary hypertension: the impact of epoprostenol therapy. Circulation. 2002;106(12):1477–1482.
128. McLaughlin VV., Sitbon O., Badesch DB., et al. Survival with first-line bosentan in patients with primary pulmonary hypertension. Eur Respir J. 2005;25(2):244–249.
129. Abraham T., Wu G., Vastey F., et al. Role of combination therapy in the treatment of pulmonary arterial hypertension. Pharmacotherapy. 2010;30(4):390–404.
130. Hoeper MM., Markevych I., Spiekerkoetter E., et al. Goal-oriented treatment and combination therapy for pulmonary arterial hypertension. Eur Respir J. 2005;26(5):858–863.
131. Corris PA. Atrial septostomy and transplantation for patients with pulmonary arterial hypertension. Semin Respir Crit Care Med. 2009;30(4):493–501.
132. Keogh AM., Mayer E., Benza RL., et al. Interventional and surgical modalities of treatment in pulmonary hypertension. J Am Coll Cardiol. 2009;54(1 Suppl):S67–S77.
133. Trulock EP., Christie JD., Edwards LB., et al. Registry of the International Society for Heart and Lung Transplantation: twenty-fourth official adult lung and heart-lung transplantation report-2007. J Heart Lung Transplant. 2007;26(8):782–795.
134. Hampl V., Tristani-Firouzi M., Hutsell TC., et al. Nebulized nitric oxide/nucleophile adduct reduces chronic pulmonary hypertension. Cardiovasc Res. 1996;31(1):55–62.
135. Evgenov OV., Pacher P., Schmidt PM., et al. NO-independent stimulators and activators of soluble guanylate cyclase: discovery and therapeutic potential. Nat Rev Drug Discov. 2006;5(9):755–768.
136. Oka M., Karoor V., Homma N., et al. Dehydroepiandrosterone upregulates soluble guanylate cyclase and inhibits hypoxic pulmonary hypertension. Cardiovasc Res. 2007;74(3):377–387.
137. Sobolewski A., Rudarakanchana N., Upton PD., et al. Failure of bone morphogenetic protein receptor trafficking in pulmonary arterial hypertension: potential for rescue. Hum Mol Genet. 2008;17(20):3180–3190.
138. Thomas M., Docx C., Holmes AM., et al. Activin-like kinase 5 (ALK5) mediates abnormal proliferation of vascular smooth muscle cells from patients with familial pulmonary arterial hypertension and is involved in the progression of experimental pulmonary arterial hypertension induced by monocrotaline. Am J Pathol. 2009;174(2):380–389.
139. McMurtry IF., Abe K., Ota H., et al. Rho kinase-mediated vasoconstriction in pulmonary hypertension. Adv Exp Med Biol. 2010;661:299–308.
140. Nishimura T., Faul J., Berry G., et al. Simvastatin attenuates smooth muscle neointimal proliferation and pulmonary hypertension in rats. Am J Respir Crit Care Med. 2002;166(10):1403–1408.
141. Hu H., Sung A., Zhao G., et al. Simvastatin enhances bone morphogenetic protein receptor type II expression. Biochem Biophys Res Commun. 2006;339(1):59–64.
142. McMurtry MS., Bonnet S., Michelakis ED., et al. Statin therapy, alone or with rapamycin, does not reverse monocrotaline pulmonary arterial hypertension: the rapamycin-atorvastatin-simvastatin study. Am J Physiol Lung Cell Mol Physiol. 2007;293(4):L933–L940.
143. Nagaya N., Kyotani S., Uematsu M., et al. Effects of adrenomedullin inhalation on hemodynamics and exercise capacity in patients with idiopathic pulmonary arterial hypertension. Circulation. 2004;109(3):351–356.
144. Diller GP., van Eijl S., Okonko DO., et al. Circulating endothelial progenitor cells in patients with Eisenmenger syndrome and idiopathic pulmonary arterial hypertension. Circulation. 2008;117(23):3020–3030.
145. Junhui Z., Xingxiang W., Guosheng F., et al. Reduced number and activity of circulating endothelial progenitor cells in patients with idiopathic pulmonary arterial hypertension. Respir Med. 2008;102(7):1073–1079.
146. Asosingh K., Aldred MA., Vasanji A., et al. Circulating angiogenic precursors in idiopathic pulmonary arterial hypertension. Am J Pathol. 2008;172(3):615–627.
147. Zhao YD., Courtman DW., Ng DS., et al. Microvascular regeneration in established pulmonary hypertension by angiogenic gene transfer. Am J Respir Cell Mol Biol. 2006;35(2):182–189.
148. Wang XX., Zhang FR., Shang YP., et al. Transplantation of autologous endothelial progenitor cells may be beneficial in patients with idiopathic pulmonary arterial hypertension: a pilot randomized controlled trial. J Am Coll Cardiol. 2007;49(14):1566–1571.
149. Zhu JH., Wang XX., Zhang FR., et al. Safety and efficacy of autologous endothelial progenitor cells transplantation in children with idiopathic pulmonary arterial hypertension: open-label pilot study. Pediatr Transplant. 2008;12(6):650–655.
150. Medhora M., Bousamra MN., Zhu D., et al. Upregulation of collagens detected by gene array in a model of flow-induced pulmonary vascular remodeling. Am J Physiol Heart Circ Physiol. 2002;282(2):H414–H422.
151. Botney M. Role of hemodynamics in pulmonary vascular remodeling: implications for primary pulmonary hypertension. Am J Respir Crit Care Med. 1999;159(2):361–364.
[/level-membership-for-cardiovascular-category][not-level-membership-for-cardiovascular-category]
Chapter 56 Pulmonary Arterial Hypertension
Definition and Classification of Pulmonary Arterial Hypertension
Pulmonary arterial hypertension (PAH) is formally defined as a mean pulmonary artery pressure (PAP) of 25 mmHg or greater at rest or 30 mmHg or greater during exercise. This is accompanied by a pulmonary vascular resistance (PVR) of greater than 3 Wood units (WU), with a normal pulmonary artery wedge pressure (< 15 mmHg) in the absence of any known cause of pulmonary hypertension (PH). Pulmonary arterial hypertension encompasses one of the five categories of PH, based on the most recent classification system.1 The pathophysiology and clinical characteristics of categories 2 through 5 of PH (i.e., PH associated with left-sided heart disease; lung disease/chronic hypoxia; thromboembolic PH; and other groups) are reviewed in Chapter 57. In this chapter, we will focus on category 1, PAH, which includes idiopathic and familial PAH as well as PAH associated with various other diseases (Box 56-1). Until improved therapeutic options became available during the past 2 decades, PAH was considered a rare but rapidly progressive and devastating condition, leading to death from right heart failure by a median of 2.8 years from the time of diagnosis.2 In recent years, an improved conceptual framework has been developed to better delineate the pathogenesis and clinical presentation of PAH, accompanied by improved directed therapeutic approaches. Nonetheless, PAH still carries a 15% annual mortality rate, and current therapeutics have been mostly effective at slowing illness progression rather than reversing or curing the disease. This chapter will highlight current molecular understanding of the complex disease, via the still incompletely characterized interplay of genetic and exogenous upstream stimuli with downstream vascular effectors. This framework will then be coupled to current understanding of the clinical presentation and progression of PAH and existing and evolving treatment modalities.
Box 56-1 Clinical Classification of Pulmonary Arterial Hypertension
Associated with significant venous or capillary involvement:
Adapted with permission from Simmoneau G, Galiè N, Rubin L, et al: Clinical classification of pulmonary hypertension. J Am Coll Cardiol 43:5 S–12 S, 2004.
Epidemiology
Early epidemiological data from the 1980s estimated the incidence of PAH at one to two cases per million people in the general U.S. population.2 More recent estimates in Europe3 place the incidence and prevalence of PAH, respectively, at 2.4 cases per million annually and 15 cases per million in France and 7.6 cases per million annually and 26 cases per million in Scotland. Globally, the prevalence may be much higher given that many risk factors for PAH, such as human immunodeficiency virus (HIV) and schistosomiasis, are also more prevalent in the developing world. An exact number is difficult to estimate on a global scale, given the difficulty of diagnosing PAH and overall limited access to health care worldwide.4 The disease affects women more frequently than men (1.7:1). This female predominance is exaggerated in the population of African descent (4.3:1), although the overall racial distribution of patients reflects that in the general population.
Primary PAH presents most commonly in the fourth decade of life; ages range from 1 to 81, with 9% of patients older than 60 years of age.5 Some sources cite a similar gender ratio among children diagnosed with the disease, whereas others note an equal distribution between male and female children.6 Primary PAH is typically difficult to diagnose, and the average time from onset of symptoms to diagnosis was 2 years in the National Institutes of Health (NIH) Registry.2 More recently, however, thanks to improved clinical awareness and more sophisticated invasive and noninvasive techniques for PAP measurements, time to diagnosis has decreased considerably, at least in the developed world.
Molecular Pathogenesis of Pulmonary Arterial Hypertension
In humans, the natural history of PAH lesions is unknown because patients usually present when the disease is advanced. The pathological appearance of severe PAH is similar regardless of cause and reflects the end stage of a common response to pulmonary vascular injury. Common histological features in nearly all types of PAH occur at the level of the small peripheral pulmonary arteries (100-1000 μm) (Fig. 56-1); these include intimal fibrosis, distal localization and proliferation of vascular smooth muscle, and pulmonary arterial occlusion.7 A hallmark of severe end-stage disease is the formation of a vessel “neointima,” characterized by increased deposition of extracellular matrix (ECM) and myofibroblasts. Plexiform lesions can predominate, characterized by over-proliferation of endothelial-like cells encroaching upon the vessel lumen.
Figure 56-1 Histopathology of pulmonary arterial hypertension (PAH).
(Courtesy JL Faul, MD, Stanford University.)
Multiple cell types in the pulmonary arterial wall and pulmonary arterial circulation contribute to the specific response to injury and development of vessel remodeling8 (Fig. 56-2). The endothelium serves as a central sensor of injurious stimuli such as hypoxia, shear stress, inflammation, and toxins. Dysregulation of numerous downstream vascular effectors may be the result of initial endothelial cell (EC) injury or dysfunction. It has been hypothesized that endothelial apoptosis early in PAH initiates selection of apoptosis-resistant ECs that can further proliferate via monoclonal amplification in plexiform populations. This has led to speculation of a model of end-stage PAH similar to that of progression to cancer, with dysregulation of the cell cycle and apoptosis as predominant features. Similarly, dysregulated growth of pulmonary artery smooth muscle cells (SMCs) also plays a key role in PAH progression because apoptosis is suppressed while proliferation increases.
Figure 56-2 Pulmonary arterial pathobiology involves coordinate action of multiple cell types.
(Adapted with permission from Chan S, Loscalzo J: Pathogenic mechanisms of pulmonary arterial hypertension. J Mol Cell Cardiol 44:14–30, 2008. Micrographs of pulmonary arteries courtesy www.scleroderma.org and Humbert M, et al: Treatment of pulmonary arterial hypertension. N Engl J Med, 2004, 351:1425–1436, 2004. Copyright 2004, Massachusetts Medical Society. All rights reserved.)
Genetic Association
An understanding of the mechanism of genetic predisposition to PAH is of paramount importance for identifying the root pathogenesis (Fig. 56-3). The familial variety of idiopathic PAH accounts for at least 6% of all cases of PAH.7 Pedigree analysis has demonstrated an autosomal dominant inheritance but with variable penetrance; only 10% to 20% of putative genetic carriers develop clinical PAH. Genetic anticipation is present, since each successive generation of affected families is afflicted at a younger age and greater severity compared with the preceding generation.
Bone morphogenetic receptor-II
Mutations in the transforming growth factor (TGF)-β receptor superfamily have been genetically linked to PAH and likely play a causative role in disease development. A rare group of patients with hereditary hemorrhagic telangiectasia (HHT) and idiopathic PAH harbor specific mutations in ALK1 or endoglin, genes encoding for two such members of the TGF-β receptor superfamily.9,10 However, a more prevalent cohort of PAH patients carries mutations in the bone morphogenetic protein receptor type II (BMPR2 gene which encodes for BMPR2).11,12 Over 140 mutations in BMPR2 have been reported in patients with familial PAH,13 mainly located in the extracellular ligand-binding domain, cytoplasmic serine/threonine kinase domain, or long carboxyterminal domain. These account for 70% of all familial pedigrees of PAH and 10% to 30% of idiopathic PAH cases.8 BMPR2 loss-of-function mutations have only been found in the heterozygous state. The absence of BMPR2 mutations in some familial cohorts and in most sporadic cases indicates that additional unidentified genetic mutations can also predispose to development of PAH. Furthermore, the presence of incomplete penetrance (approximately 25% of carriers in affected families develop clinical PAH) and genetic anticipation suggests that BMPR2 mutations are necessary but insufficient alone to result in clinically significant disease.
The mechanism of action of BMPR2 is complex, and its role in PAH progression is still unclear (Fig. 56-4A). It functions as a receptor with serine/threonine kinase activity, and it activates a broad and complex range of intracellular signaling pathways (as reviewed in14). Upon binding one of many possible bone morphogenetic protein (BMP) ligands, BMPR2 forms a heterodimer with one of three type-I receptors. BMPR2 phosphorylates the bound type-I receptor, which in turn phosphorylates one of the Smad family of proteins to allow for nuclear translocation, binding to deoxyribonucleic acid (DNA), and regulation of gene transcription. Alternatively, BMPR2 activation can also lead to signaling via the LIM kinase pathway, p38/ mitogen-activated protein kinase/extracellular signal regulated kinase/c-jun NH 2-terminal kinase (MAPK/ERK/JNK) pathways, or c-Src pathway, independent of Smad activation.
Figure 56-4 Molecular signaling mechanisms of pulmonary arterial hypertension (PAH).
(Adapted from Archer SL, Weir EK, Wilkins MR: Basic science of pulmonary arterial hypertension for clinicians: new concepts and experimental therapies. Circulation 121:2045–2066, 2010.)
Cellular effects of BMPR2 activation are multiform. In the adult, BMPR2 is expressed predominantly in pulmonary endothelium, medial SMCs, and macrophages.15 Under normal conditions, BMP ligands bind BMPR2 to suppress growth of VSMCs. In contrast, binding of BMP2 and BMP7 to BMPR2 in pulmonary endothelium protects against apoptosis. A widely held hypothesis contends that failure of the suppressive effects of BMP ligands on vascular smooth muscle and failure of the protective effects of BMP ligands on endothelium may trigger vascular proliferation and remodeling. Accordingly, in VSMCs derived from patients with familial PAH harboring BMPR2 mutations, exposure of BMP ligands does not suppress proliferation. Unlike the response in wild-type endothelium, exposure of ECs cultured from patients with idiopathic PAH to BMP2 does not protect against apoptosis.16 These dysfunctional signaling pathways have been corroborated in some rodent models of PAH. In correlation, pulmonary levels of BMPR2 are reduced both in familial cases of PAH without any BMPR2 mutation and in cases of secondary PAH.15 Thus, dysregulation of the BMP signaling pathway may be a common pathogenic finding in multiple types of PAH due to genetic or exogenous stimuli, but the definitive in vivo effects of these mutations have been difficult to decipher. Specifically, mouse models harboring specific BMPR2 heterozygous mutations have failed to exhibit robust PAH under static conditions,17,18 again suggesting that dysfunctional BMPR2 is likely insufficient alone to cause disease. As a result, a clear mechanistic explanation of the impact of BMPR2 mutations on pathogenesis remains elusive.
Additional genetic pathways
In addition to BMPR2 haploinsufficiency, alternative mechanisms involving complementary “modifier” genes may also contribute to a genetic predisposition to PAH. The most promising data identifying such modifier genes have analyzed the association of particular single nucleotide polymorphisms (SNPs) with the development of PAH. Thus far, SNP variants have suggested certain genes such as the serotonin transporter (SERT), Kv1.5, and the trp cation channel, subfamily C, member 6 (TRPC6).3 Such associations do not always suggest a causal relationship, so additional mechanistic data are necessary for proper interpretation.
In the case of SERT and serotonin signaling, supportive data are more prevalent. Serotonin is both a vasoconstrictor and mitogen that promotes smooth muscle hyperplasia and hypertrophy (see Fig. 56-4B). Primarily stored in platelet granules, secreted serotonin binds G protein–coupled serotonin receptors (GPCRs) present on pulmonary artery SMCs. Activation of these receptors leads to a decrease in adenylyl cyclase and cyclic adenosine monophosphate (cAMP), resulting in increased contraction. Furthermore, the cell-surface SERT allows for transport of extracellular serotonin into the cytoplasm of SMCs, thereby activating cellular proliferation directly through the action of serotonin or indirectly via potential pleiotropic mechanisms.
A number of observations in idiopathic and familial disease, congenital disease, and environmental exposure have implicated the proliferative effects of serotonin in PAH.19 In idiopathic PAH, pulmonary expression of serotonin receptors is increased, and plasma levels of serotonin are chronically elevated. A mouse model of hypoxic PH parallels these changes.20 A positive association has been noted among patients with congenital platelet defects in serotonin uptake (i.e., delta storage pool disease) and development of PAH. Chronic exposure to anorexigens, such dexfenfluramine (an inhibitor of serotonin reuptake and stimulator of serotonin secretion), leads to increased levels of circulating free serotonin. In mice, these changes are accompanied by increased 5HT receptor type 2B response and inhibition of SERT responses. In humans, these changes correlate with an increased risk for development of PAH. In addition, the L-allelic variant of SERT is associated with increased expression of the transporter and enhanced smooth muscle proliferation. In some human studies,21 this variant has been associated with an increased risk of PAH in the homozygous population.
Animal models of PH have also implicated the activated serotonin pathway in disease progression. Treatment with serotonin and chronic hypoxia in a rat model led to worsened hemodynamics and increased vessel remodeling.22 Exposure to increased serotonin led to worsened PH in a BMPR2+/− heterozygote murine model.23 Similarly, overexpression of SERT in mice resulted in spontaneous development of PAH in the absence of hypoxia and exaggeration of PH after a hypoxic stimulus.24 Conversely, vessel remodeling and hypoxic PH are reduced in a 5HT1B receptor–null mouse25 and are abrogated in a 5HT2B receptor–null mouse.20 As a result, serotonin signaling modulates pulmonary smooth muscle function in both normal and disease states and likely contributes to disease progression of PAH. However, attempts at using selective serotonin reuptake inhibitors (SSRIs) as a therapeutic approach have yielded mixed results3 to date. The exact contribution of this mechanism in PAH requires further clarification.
Acquired/Exogenous Factors
In addition to genetic predisposition, development of PAH likely depends on a variety of physiological, acquired, and/or exogenous stimuli. Some of these factors have been studied to a sufficient degree to hypothesize potential pathogenic mechanisms (see Fig. 56-3).
Chronic hypoxia
Pulmonary vascular response to hypoxia has been well studied in cell culture and animal models,26 but its impact on PAH is unclear. In general, pulmonary vascular responses in acute and chronic hypoxia likely allow for the propagation of PAH, and therefore may contribute to later stages of the disease. Acute hypoxia induces vasodilation in systemic vessels but induces vasoconstriction in pulmonary arteries. This acute and reversible effect is mediated in part by up-regulation of endothelin-1 (ET-1) and serotonin, and in part by hypoxia- and redox-sensitive potassium channel activity in pulmonary VSMCs. Coordinately, these events lead to membrane depolarization in SMCs, increase in cytosolic calcium, and vasoconstriction.27 In contrast, chronic hypoxia induces vascular remodeling and less reversible changes, including migration and proliferation of VSMCs and deposition of ECM. These cellular events in chronic hypoxia correlate with the remodeling events inend-stage PAH; however, because the histopathology of hypoxic PH does not recapitulate all aspects of PAH, some mechanistic differences in pathogenesis certainly exist but have not yet been fully identified. These are important considerations, especially in the context of interpreting studies of hypoxic PH and extrapolating those findings to the pathogenesis of PAH.
Hemoglobin disorders
Pulmonary arterial hypertension is associated with hemoglobinopathies, especially thalassemias and sickle cell anemia.28 Hemolysis accompanying these disorders may lead to destruction of bioactive nitric oxide (NO) by free hemoglobin or reactive oxygen species (ROS). Furthermore, ROS may lead to increased levels of oxyhemoglobin, which further impairs delivery of NO to the vessel wall. As a result of the lack of available NO, an inflammatory and proliferative cascade may ensue, with culmination in PAH. Accordingly, decreased NO bioavailability with development of PH has been reported after hemolysis in a murine model of sickle cell disease (SCD),29 and acutely in a murine model of intravascular hemolysis.30 In vivo correlation to human disease is pending.
Portopulmonary hypertension
Portopulmonary hypertension is defined as PAH associated with portal hypertension (portal pressure >10 mmHg), with or without hepatic disease.31 Approximately 9% of patients with severe PAH are reported to have portal hypertension. Portopulmonary hypertension affects 4% to 6% of patients referred for liver transplantation. Liver transplantation perioperative mortality is significantly increased in patients with a mean PAP above 35 mmHg. Diagnosis of PH is usually made 4 to 7 years after the diagnosis of portal hypertension, but it has occasionally been reported to precede onset of portal hypertension. Risk of PH increases with duration of disease. The correlation between severity of portal hypertension and development of PH is debated. The female predominance of primary PAH is not seen in portopulmonary hypertension.
Collagen vascular disease
Pulmonary arteriopathy complicates autoimmune diseases, especially in the setting of the CREST (calcinosis, Raynaud phenomenon, esophageal dysfunction, sclerodactyly, telangiectasia) variant of limited systemic sclerosis and, to a lesser degree, in mixed connective tissue disease, systemic lupus erythematosus (SLE), and rheumatoid arthritis.32 Pulmonary arterial hypertension has been reported in approximately 10% to 30% of patients with mixed connective tissue disease, 5% to 10% of patients with SLE, and more rarely in the settings of rheumatoid arthritis, dermatomyositis, and polymyositis.33 Sjögren syndrome may rarely be complicated by rapidly progressive PAH. It is particularly important to distinguish between PAH and thromboembolic PH in patients with SLE and antiphospholipid syndrome. Occurrence of PAH in each disease has been associated with Raynaud phenomenon, suggesting at least some similarities in pathogenesis.34 Presence of interstitial lung disease and pulmonary fibrosis, seen at varying frequency in these autoimmune syndromes, may represent a common pathogenic factor in development of PAH. Accordingly, in the setting of pulmonary fibrosis and hypoxia, significant perivascular inflammation and deposition of ECM have been observed, which may increase vasoconstriction, proliferation, and vessel remodeling. Murine models of interstitial lung disease may prove important in further elucidating pathogenic mechanisms.
Human immunodeficiency virus
An association between HIV infection and PH has been noted in approximately 0.5% of all patients with HIV infection, a rate 6 to 12 times higher than the general population.35 Notably, HIV does not infect pulmonary arterial endothelium, but mechanisms of disease have been proposed that directly stem from effects of HIV infection.36 These include infection of SMCs with subsequent dysregulation of proliferation, imbalance of vascular mitogens in response to systemic HIV infection, and endothelial injury precipitated by HIV-infected T cells. The direct actions of HIV-encoded proteins may also factor into PAH development.8 The HIV gp120 protein may induce pulmonary endothelial dysfunction and apoptosis. In a macaque model of simian immunodeficiency virus infection, a pathogenic interaction of the viral Nef protein with the pulmonary vessel wall has been reported, leading to pulmonary arteriopathy. Cell culture studies have also demonstrated a role for the HIV Tat protein in repression of BMPR2 transcription, potentially provoking a proliferative response in the vessel wall.
It had been proposed that human herpesvirus 8 (HHV-8), the causative agent of Kaposi sarcoma and an opportunistic pathogen highly associated with HIV infection, may play a role in PAH development with progression to plexiform lesions. Although it was initially reported that HHV-8 infection is associated with idiopathic PAH,37 that link has not been consistently validated after study of additional populations. Nonetheless, PAH in the setting of HIV infection likely results from multifactorial effects, and the underlying pathogenesis may involve both direct results of viral infection and indirect consequences of associated pathogens.
Shunt physiology
Increased flow through the pulmonary circulation has long been associated with development of PAH. Certain types of congenital heart disease with functional systemic-to-pulmonary shunts, such as unrestricted ventricular septal defects (VSDs) and large patent ductus arteriosus (PDA), invariably lead to pulmonary vascular remodeling and the clinical syndrome of PAH during childhood (Eisenmenger’s syndrome).38 Approximately a third of patients with uncorrected VSDs and PDAs die from PAH. The timing of surgical repair greatly influences the outcome. If the shunt is repaired within the first 8 months of life, patients tend to have normal pulmonary pressures regardless of pathological findings; by contrast, patients operated on after age 2 tend to have persistent PAH. Importantly, when PVR equals or exceeds systemic vascular resistance, surgical correction of a shunt will increase the load on an already overburdened right ventricle (RV), worsen the patient’s clinical condition, and not reverse PAH.
The presence of atrial septal defects (ASD) with systemic-to-pulmonary shunts may also lead to PAH over time.39 Yet, in contrast to cases of unrestricted VSD and PDA, only 10% to 20% of all persons with ASDs progress to PAH. This observation may reflect differences in the response of the pulmonary vasculature to pressure overload (as seen in shunts with VSD and PDA) as compared to volume overload (as seen in shunts with ASD). Furthermore, patients with ASD may harbor a specific unidentified genetic predisposition to the development of PAH that may exacerbate the increased volume load to the pulmonary circulation.
At the molecular level, the physiological flow patterns of laminar shear stress, turbulent flow, and cyclic strain are all recognized by ECs, leading to transduction of intracellular signals and modulation of a wide variety of phenotypic changes.40 Significant prior work has focused mainly on the endothelium of the peripheral vasculature, suggesting that laminar flow induces a vasoprotective quiescent vascular state, whereas turbulent flow leads to a proinflammatory and thrombogenic state. It is unclear whether these flow-dependent phenotypes are recapitulated in the pulmonary vasculature. In part, this stems from the difficulty of directly studying directly the in vivo flow patterns at the anatomical level of the pulmonary arteriole. Ex vivo modeling of pulsatile flow with high levels of shear stress and chronic vascular endothelial growth factor (VEGF) inhibition has demonstrated apoptosis of pulmonary artery ECs, followed by outgrowth and selection for proliferating apoptosis-resistant cells.41 Therefore, chronically elevated flow may allow for selection of cells with dysregulated EC growth and resulting clonal or polyclonal expansion to plexiform lesions.
Persistent pulmonary hypertension of the newborn
Persistent pulmonary hypertension of the newborn (PPHN) is characterized by persistent elevation of PVR, right-to-left shunting, and severe hypoxemia. It can occur with pulmonary parenchymal diseases including sepsis, meconium aspiration, pneumonia, maladaptation of the pulmonary vascular bed, or without an apparent cause.42 Persistent PH in newborns may lead to death during the neonatal period, or it may be transient, leading to spontaneous and complete recovery. Inadequate production of NO may be an important contributor to persistent PH in infants, and inhaled NO is useful in treating this disorder.
Miscellaneous disorders
Pulmonary arterial hypertension is more rarely reported in patients suffering from other clinical syndromes such as pulmonary veno-occlusive disease, chronic myelodysplastic syndromes with thrombocytosis, and idiopathic thrombocythemia, as well as in persons exposed to stimulants of the central nervous system, such as methamphetamines and cocaine. Dysregulation of serotoninergic signaling may contribute8 but does not explain these associations entirely. Finally, idiopathic PAH demonstrates a gender predilection of unclear etiology, with a high predominance of affected females.43
Vascular Effectors
Downstream of the genetic and acquired triggers of PAH, the histopathological processes that predominate later stages of disease include vasoconstriction, cellular proliferation, and thrombosis. These processes are influenced by a complex and dysregulated balance of vascular effectors controlling vasodilation and vasoconstriction, growth suppressors and growth factors, and pro- vs. antithrombotic mediators. Most of these effectors have been described in previous comprehensive reviews and will be described here specifically in regard to their known roles in PAH pathogenesis (Table 56-1).
Nitric oxide
Gaseous vasoactive molecules regulate pulmonary vascular homeostasis, and alterations in their endogenous production have been linked to progression of PAH. Nitric oxide is the best described of these factors (see Fig. 56-4C).44 It is a potent pulmonary arterial vasodilator as well as a direct inhibitor of platelet activation and VSMC proliferation. Nitric oxide diffuses into recipient cells (e.g., vascular muscle), where it activates soluble guanylyl cyclase (sGC) to produce cyclic guanosine monophosphate (cGMP). In turn, cGMP interacts with at least three different groups of effectors: cGMP-dependent protein kinases, cGMP-regulated phosphodiesterase (PDE), and cyclic nucleotide-gated ion channels. Synthesis of NO is mediated by a family of NO synthase (NOS) enzymes. In the pulmonary vasculature, the endothelial (eNOS) isoform figures most prominently and is regulated by a multitude of vasoactive factors and physiological stimuli including hypoxia, inflammation, and oxidative stress. Reduced levels and reduced activity of eNOS, reduced NO bioavailability, dysregulated intracellular NO transport via caveolae, and increased degradation of cGMP all aggravate PAH progression in human studies and/or animal models.
Dysregulation of NO may also depend upon still incompletely characterized processes of NO transport in blood.45 Specific polymorphisms of NOS have also been associated with pulmonary hypertension. Together, these effects indicate a coordinated mechanism of dysregulated vasoconstriction. Correspondingly, murine models that genetically lack eNOS, GTP cyclohydrase-1 (the rate limiting enzyme for synthesis of an essential cofactor of NOS, tetrahydrobiopterin), or dimethylarginine dimethylaminohydrolase (DDAH, an enzyme important in degradation of NOS inhibitors) all display exaggerated susceptibility to developing PH in response to other endogenous stimuli.3 Finally, the impact of NO has been reflected in its therapeutic role in PAH, such as inhaled NO46 and the NO-dependent phosphodiesterase type-5 (PDE5) inhibitors (discussed later in detail).
Prostacyclin/thromboxanes
The arachidonic acid metabolites prostacyclin and thromboxane A2 (TxA2) also play crucial roles in vasoconstriction, thrombosis, and to a certain degree, vessel wall proliferation (see Fig. 56-4C). Prostacyclin (prostaglandin I2) activates cAMP-dependent pathways and serves as a vasodilator, antiproliferative agent for vascular smooth muscle, and inhibitor of platelet activation and aggregation. In contrast, TxA2 increases vasoconstriction and activates platelets.47 Protein levels of prostacyclin synthase are decreased in small and medium-sized pulmonary arteries in patients with PAH, particularly with the idiopathic form.48 Biochemical analysis of urine in patients with PAH has shown decreased levels of a breakdown product of prostacyclin (6-ketoprostacyclin F2α), accompanied by increased levels of a metabolite of TxA2 (thromboxane B2).49 Therefore, it appears that production of these effectors is coordinately regulated, with the imbalance toward TxA2 favored in the development of PAH. The mode of this regulation remains to be characterized. Nonetheless, recognition of this imbalance has led to the success of prostacyclin therapy and improvement of hemodynamics, clinical status, and survival in patients with severe PAH.50
Endothelin-1
Endothelin-1 is expressed by pulmonary ECs and has been identified as a significant vascular mediator in PAH (see Fig. 56-4C).51 It acts as both a potent pulmonary arterial vasoconstrictor and mitogen of pulmonary smooth muscle cells. The vasoconstrictor response relies upon binding to the endothelin receptor A (ET-A) on vascular smooth muscle cells. This leads to an increase in intracellular calcium, along with activation of protein kinase C (PKC), mitogen-activated protein kinase (MAPK), and the early growth response genes c-fos and c-jun.52 The mitogenic action of ET-1 on pulmonary VSMCs can occur through either the ET-A and/or the ET-B receptor subtype, depending on the anatomical location of cells. Endothelin receptor A predominantly mediates mitogenesis in the main pulmonary artery, whereas mitogenesis in resistance arteries relies upon contributions from both subtypes. The resulting vasoconstriction, mitogenesis, and vascular remodeling are thought to lead to significant hemodynamic changes in the pulmonary vasculature and to PAH. Plasma levels of ET-1 are increased in animal and human subjects suffering from PAH due to a variety of etiologies53 and correlate with disease severity.54 Again, improvement in hemodynamics, clinical status, and survival of PAH patients treated with chronic ET receptor antagonists highlights the significance of these effects.55
Vasoactive intestinal peptide
Down-regulation of vasoactive intestinal peptide (VIP) may also play a pathogenic role. Vasoactive intestinal peptide is a pulmonary vasodilator, an inhibitor of proliferation of VSMCs, an inhibitor of platelet aggregation, and free radical scavenger. Decreased concentrations of VIP and VIP receptors have been reported in serum and lung tissue of patients with PAH.56 Vasoactive intestinal peptide-null mice suffer from moderate pulmonary hypertension.57 Both pulmonary arterial pressure (PAP) and PVR in humans decrease after treatment with VIP.58,59 Key questions regarding the mode(s) of regulation of VIP expression and its putative causative role in PAH remain unanswered.
Peroxisome proliferator-activated receptor γ
Peroxisome proliferator-activated receptor gamma (PPAR-γ)and apolipoprotein E (apoE) may function as integral factors in PAH.60,61 PPARs are ligand-activated nuclear transcription factors that heterodimerize with the retinoid X receptor for subsequent binding to PPAR response elements in the promoters of target genes. Idiopathic PAH patients carry reduced pulmonary transcript levels of PPAR-γ and apoE. PPAR-γ in particular is a direct target of BMP2 in human PASMCs, leading to stimulation of apoE synthesis and downstream inhibition of vascular smooth muscle proliferation. PPAR-γ also regulates a host of protein kinases implicated in PASMC proliferation and migration. PPAR-γ agonists are antiinflammatory and induce pro-apoptotic phenotypes, both theoretically inhibitory to the pathogenesis of PAH. Mice deficient in smooth muscle–specific PPAR-γ are prone to PAH. Similarly, when fed a high-fat diet, male mice deficient in apoE develop PAH. This condition is reversed by rosiglitazone, a PPAR-γ agonist. Nonetheless, less robust results have been seen when using rosiglitazone hypoxic-PH rats, suggesting that these results offer a partial explanation to the complex molecular pathogenesis of BMPR2 signaling and PAH pathogenesis.
Natriuretic peptides
The atrial and brain natriuretic peptides (ANP and BNP) are produced by myocardium in response to stretch. They bind guanylyl cyclase receptors (NPR-A and NPR-B) that induce production of cGMP. Increased plasma concentrations of these peptides in PAH have been used as markers of the extent of RV dysfunction.62 Genetic inactivation of NPR-A in mice is associated with PH63; in contrast, administration of atrial natriuretic peptide (ANP) ameliorates PAH in rodent models.64 Additionally, inhibition of the metabolic breakdown of natriuretic peptides via neutral endopeptidase inhibitors has shown promise in animal studies of PAH. Human studies have yet to confirm efficacy.
Adrenomedullin
Adrenomedullin is an endogenous peptide that activates signaling pathways (i.e., cAMP, NO-cGMP, phosphatidylinositol-3-kinase/Akt) to induce vasodilation and inhibit proliferation. It decreases mean PAP and RV hypertrophy in hypoxic rats.65 A related peptide, adrenomedullin-2, binds the same cellular receptors as adrenomedullin, and it is elevated in some rodent forms of PAH.
Miscellaneous effectors
Other potential contributing factors include angiopoietin-1, the vasodilatory gases carbon monoxide and hydrogen sulfide, phosphodiesterase I, survivin, the calcium binding protein S100A4/Mts, the transient receptor potential channels, and Notch 3.66 These may represent important but as yet incompletely described pathogenic contributors.8 Other mitogenic and angiogenic growth factors, such as VEGF, platelet-derived growth factor (PDGF), basic fibroblast growth factor (bFGF)-2, insulin-like growth factor (IGF)-1, and epidermal growth factor (EGF) all may play downstream roles in later stages of PAH.
Pathogenic Pathways
Our understanding of specific actions of individual effectors has improved, but how they relate to upstream genetic or exogenous triggers of PAH remains unclear. In fact, none of these factors has yet been definitively linked to the root pathogenesis of disease. Insight into this topic is offered by the fact that known effectors are likely subject to upstream, overarching regulatory pathways that affect the action of multiple vasoactive molecules.67 Characterization of these regulatory mechanisms may eventually allow for identification of primary molecular triggers of disease and offer novel therapeutic targets for drug development (see Fig. 56-3).
Mitochondrial metabolism
Mitochondrial and metabolic dysfunction may be common in PAH. Endothelial and VSMCs from human PAH tissue display dysmorphic and hyperpolarized mitochondria.68 Metabolically, these cells preferentially exhibit a down-regulation of mitochondrial metabolism with an induction of glycolysis for energy production.69 Under hypoxic conditions, this so-called Pasteur effect is a normal adaptive event that improves cellular survival by optimizing adenosine triphosphate (ATP) production while reducing oxygen radicals generated from the mitochondrial electron transport chain. Under normoxic conditions, however, such a shift to glycolysis (the Warburg effect) is thought to confer inappropriate resistance to apoptosis and is prominently seen in various cancer lineages. Such metabolic changes in the mitochondria are dependent upon a master transcription factor of hypoxia, hypoxia inducible factor 1 alpha (HIF-1α), and its critical downstream targets such as pyruvate dehydrogenase kinase (PDK1), among others.70 In PAH, HIF-1α and PDK1 are dysregulated; conversely, treatment with dichloroacetate, a PDK inhibitor, ameliorates multiple forms of PAH in rodent models (chronic hypoxic PH, monocrotaline PAH, and fawn-hooded rat PAH).71 It is conceivable that therapies aimed at reversing this metabolic dysregulation may result in improvement and/or regression of the PAH phenotype.
Right ventricular dysfunction
After birth, PAPs typically decline, leading to involution of the RV to a thin-walled structure in the adult. During pathological conditions of increased PAPs in PAH, however, RV hypertrophy (RVH) and strain ensue, followed by RV failure if left untreated. Historically, the molecular pathways governing left ventricular (LV) failure, which have been better characterized, had been assumed to play primary roles in RV failure as well. Contemporary evidence suggests there may be distinct molecular and physiological differences between LV and RV failure that are just beginning to be explored.72 For instance, PDE5, which is expressed in the fetal RV, is selectively up-regulated in the hypertrophied RV. Interestingly, inhibition of PDE5 enhances RV contractility without affecting the LV, where PDE5 expression is absent.
[/not-level-membership-for-cardiovascular-category]
