CHAPTER 45 PROTHROMBOTIC STATES AND RELATED CONDITIONS
THE HEMOSTATIC SYSTEM
The human circulatory system does not act simply as an array of inert, lifeless pipes that convey blood between organs; it is an organ itself with functions beyond those of the various compounds and cells that pass through its conduits. One of these functions is the maintenance of its own structural integrity, essential for transmitting blood pressure, ensuring continuity of flow, and minimizing spread of infection. There has evolved a complex hemostatic system that enables on-line monitoring of a large area of endothelial space (approximately 600 m2, or the equivalent of three tennis courts), with rapid, local restoration of breaches as they occur. This system is composed of multifarious actions and interactions among solid-phase vessel wall, cellular platelets, and humoral coagulation factors; Figure 45-1 is a basic schema of the events that follow vessel injury. It is preferable to talk about this system as a whole rather than as a separate “coagulation system” because of the strong interdependence among these three components. An important attribute of hemostasis is the requirement of both “on knobs” and “off knobs,” delicately adjusted, to prevent a response to vessel wall injury from overshooting and resulting in a sealing of both the defect (which is desired) and the vessel lumen itself (which would cause local hypoperfusion). Endogenous anticoagulant systems are shown in Figure 45-2. As with most metabolic systems, the physiological state is optimized by both prohemostatic and antihemostatic reactions occurring simultaneously (i.e., a dynamic system); the net outcome depends on local vessel integrity.
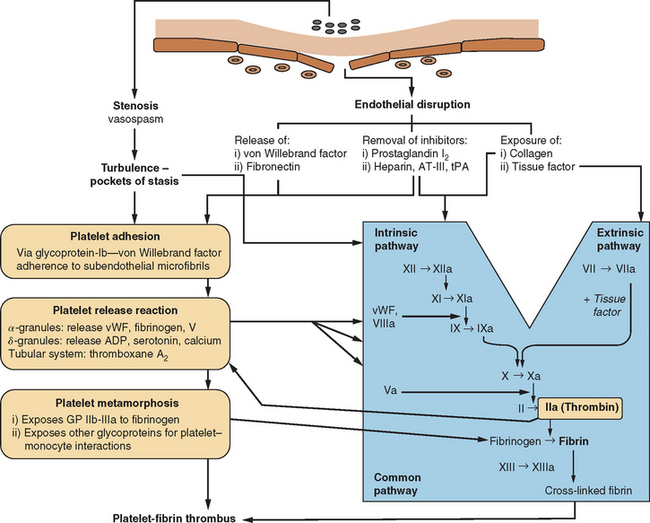
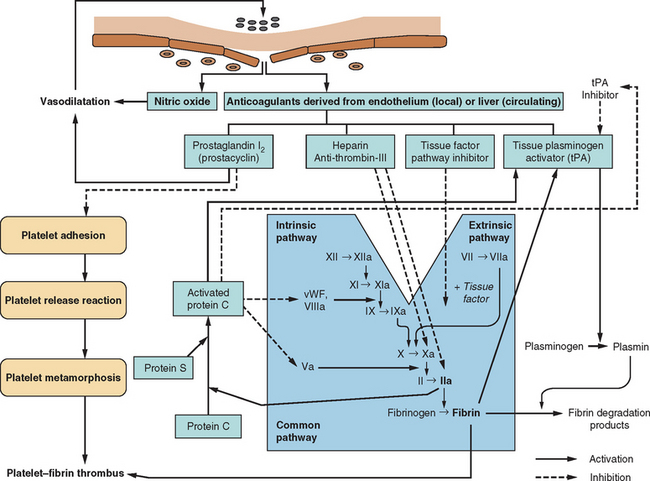
PATHOLOGY OF EXCESSIVE HEMOSTASIS
In the same way that the physiology of hemostasis is shared among the trio of vessel wall, platelets, and clotting factors, the pathophysiology of excessive hemostasis can be summarized by its own triad. This so-called Virchow’s triad represents three sets of factors, each of which may give rise to vessel closure, thrombosis being a final common pathway in most cases (Fig. 45-3).
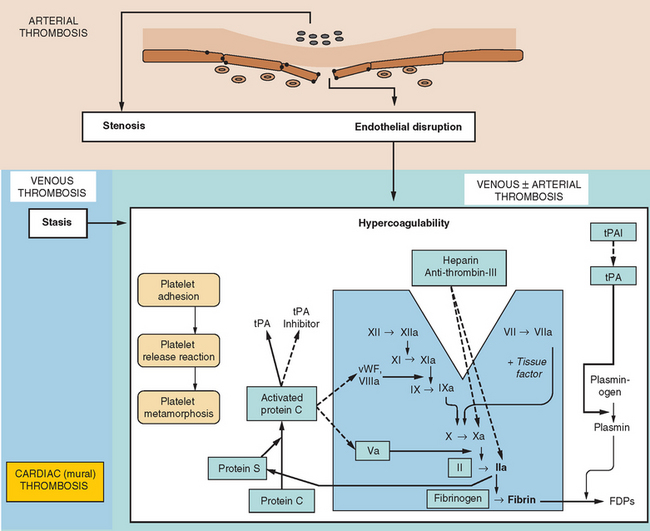
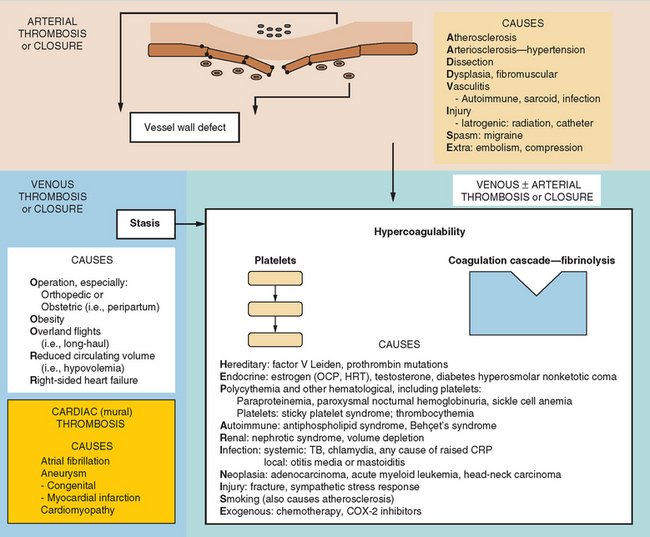
A modern-day listing of Virchow’s triad has changed little since its original formulation in 1860:
Figure 45-4 lists the various causes within each of these categories, including states predisposing to thrombosis or vessel closure by other means (e.g., vasospasm). These causes can be remembered by the mnemonic “ADVISE OR HEPARINISE.”
A thrombosis consists of a solid aggregation of platelets and erythrocytes within a fibrin mesh adherent to the vessel wall. Microscopic observation reveals that thrombi within arteries have a higher platelet composition (in relation to fibrin) than do those in veins. Because fibrin tends to adhere to red blood cells, arterial thrombi appear relatively white, whereas venous thromboses, with a higher fibrin content, appear red.
NEUROLOGICAL CONSEQUENCES OF EXCESSIVE HEMOSTASIS
Thromboembolism represents the commonest cause of ischemic stroke; other pathological processes may also result in vessel closure to produce regional ischemia within the nervous system. Examples of such pathological processes are vasospasm (that in the brain results in migrainous auras), arteriosclerotic lipohyalinosis (commonly caused by hypertension and resulting in isolated “lacunar” strokes or, when diffuse, a subcortical dementia), and vasculitis. Figure 45-5 depicts the varied neurological effects of ischemia secondary to thromboembolism or other processes resulting in vessel closure.
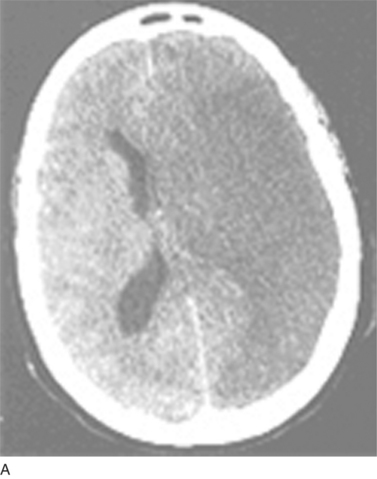




(B, From http://www.aic.cuhk.edu.hk/web8/0294_Cerebral_venous_haemorrhagic_infarction.jpg. Copyright © Chanles Gomersall. C, From http://www.kup.at/kup/images/thumbs/336.jpg. E, From The Robert Bendheim Digital Atlas of Ophthalmology, The New York Eye and Ear Infirmary, http://www.nyee.edu/page_deliv.html?page_no=50. F, From http://medicine.ucsd.edu/clinicalmed/eyes-cn6-palsy3.jpg.)
VESSEL WALL DEFECTS
Atherosclerosis
Postmortem appearances of arteries from people who died from nonischemic causes have shown that atherosclerotic lesions are virtually universal after middle age, especially in residents of industrialized nations. In fact, the strong geographical dependence of atherosclerosis risk corresponds to recognized environmental factors that may initiate atherogenesis (Fig. 45-6). The most important predisposing state is a high circulating triglyceride (lipid) load, especially during the postprandial period—which for many residents of the industrialized world represents most of the waking day. Because triglyceride-rich particles (very-low-density lipoprotein) continuously exchange their triglycerides with cholesterol found within high-density lipoprotein particles, the level of high-density lipoprotein-cholesterol is inversely correlated with, and an accurate predictor of, atherosclerosis risk. The level of low-density lipoprotein (LDL)-cholesterol is less predictive of atherosclerosis, although a subset of LDL-cholesterol particles formed under high triglyceride conditions, called small, dense LDL-cholesterol, are highly atherogenic (after oxidation or glycation).
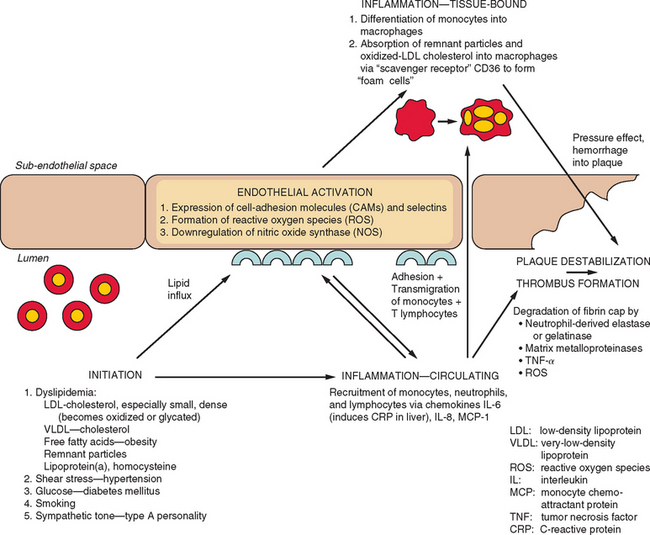
Figure 45-6 Pathogenesis of atherosclerosis and subsequent plaque destabilization with thrombus formation.
Although diet is the leading global factor in accounting for the distribution of atherosclerosis-related diseases, there are multiple other associations. Some of these factors act through secondary effects on lipid metabolism. Diabetes mellitus results in both a high circulating triglyceride level and glycation of small, dense LDL-cholesterol, both predisposing to premature atherosclerosis. Of interest, most genetic types of hypertriacylglyceridemia are not associated with atherosclerosis, whereas familial hypercholesterolemia (usually caused by a LDL-cholesterol receptor mutation) is strongly associated with premature atherosclerosis; homozygous patients suffer ischemic heart disease or strokes in their 20s. There are other genetic factors for atherosclerosis that may or may not result from interactions with lipid metabolism (Table 45-1). Both lipoprotein(a) and homocysteine levels are risk factors for atherosclerosis that also have strong genetic influences; these factors also have prothrombotic effects as a result of interactions with the coagulation cascade (see later discussion). Certain factors, such as hypertension, smoking, and epinephrine (present with, e.g., “easily stressed” personalities) act as synergistic factors with lipid metabolism for atherogenesis. Exercise and fish oils rich in omega-3 fatty acids (e.g., eicosapentaenoic acid) engender a more favorable lipid profile and correspondingly reduce atherosclerosis.
Although the influences of lipid and glucose metabolism on atheroma formation are well established, the contributions played by the immune system on both atherogenesis and plaque destabilization are becoming increasingly appreciated. Inflammation is triggered at the first sign of vessel wall injury and results in upregulation of cell-adhesion molecules, release of chemotaxins and reactive oxygen species, and facilitation of lipid influx and storage. Eventually, the fibrinous cap and endothelial lining of an atherosclerotic plaque becomes degraded by proteolytic enzymes derived from the inflammatory infiltrate, and at this point, rapid thrombus development is triggered. The effects of local inflammation within the vessel wall can often be observed through levels of circulating inflammatory markers. Hence, levels of C-reactive protein, an acute-phase reactant, and of CD3 provide indexes of the risk of ischemic heart disease or stroke. In addition to mirroring the inflammatory turnover within atherosclerotic lesions, these proteins act together to accelerate atheroma formation. Of interest, statins have been found to have anti-inflammatory effects, including reduction of C-reactive protein levels, and these effects have been associated with reduction of stroke or myocardial infarction risk, independent of the effect that these drugs have through lowering cholesterol.
Arteriosclerosis
Arteriosclerosis is the progressive replacement of smooth muscle cells with proteinaceous material (predominantly collagen) within the media of small arteries and arterioles (40 to 400 μm in diameter). The microscopic appearance is of concentric layers of a hyaline substance in the same location where smooth muscle normally occurs. The most strongly associated risk factors are hypertension and diabetes. As the process continues, the lumen constricts, which results in progressive tissue hypoperfusion. The latter is exacerbated during periods of hypotension, because the loss of smooth muscle prevents vasodilation. In certain cases, especially with accelerated hypertension, the same vessels develop a more disorganized process, which results in patchy fibrinoid accumulation, necrosis, and foam cell formation. When this accumulation is rapid, the end result may be acute small-vessel or lacunar infarction. From a therapeutic point of view, this knowledge is important, because it explains why anticoagulation and thrombolysis have no effect on stroke from this cause.
STASIS
Wherever the circulation becomes sluggish, activated clotting factors may accumulate to reach a concentration that surpasses a critical threshold, potentiating formation of an expanding fibrin core and, ultimately, thrombus. The most common predisposing circumstance is prolonged immobility, when thrombosis tends to form in the deep leg veins and usually when an additional hypercoagulability risk factor, such as infection or genetic status, prevails. The usual clinical consequences of this are local pressure effects in the leg and, more devastatingly, pulmonary embolism. However, in patients with a right-to-left cardiopulmonary shunt, a dislodged venous thrombus may course into the arterial circulation to cause, among other outcomes, cerebral embolism. This “paradoxical embolism” occurs most commonly in patients with either congenital cyanotic heart disease or a pulmonary arteriovenous malformation (e.g., as part of hereditary hemorrhagic telangiectasia), but in rare cases, it occurs in patients with a patent foramen ovale (present in up to 25% of the population) during, for example, inadvertent performance of the Valsalva maneuver.
HYPERCOAGULABILITY
Hereditary
Many of the recognized genetic causes of hypercoagulability (and ischemic stroke) can be appreciated by reference to the endogenous coagulation and anticoagulation pathways (see Fig. 45-3 and Table 45-1). Hence, one of the most common abnormalities, factor V Leiden mutation, can be explained as an insensitivity of factor V to deactivation by protein C, which leads to deregulation of the production of thrombin. Mutations of endogenous anticoagulant/coagulant proteins are associated most strongly with venous thrombosis, although certain mutations (mainly factor V Leiden and prothrombin mutations and protein S deficiency) are also associated with arterial thrombosis, including stroke. The odds ratio associated with these conditions is approximately 10 for development of venous thrombosis and less than 3 for development of an arterial thrombosis. Whether a particular mutation results in an ischemic event depends on whether other prothrombotic or vascular risk factors coexist. Factor V Leiden and prothrombin mutations occur in approximately 10% and 5% of the population, respectively. Most affected people are heterozygous for the mutations although the relative risk for thrombosis is much greater in people with homozygous mutations. Deficiencies of protein C, protein S, and antithrombin III occur in fewer than 1% of the population for each. Most of these mutations are autosomal dominant, except for plasminogen deficiency, which is autosomal recessive.
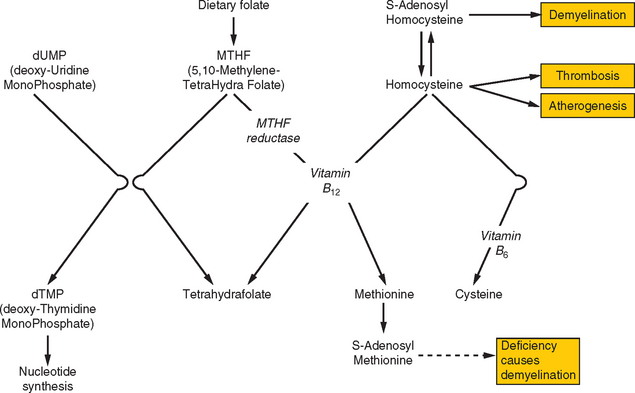
Both hyperhomocysteinemia and elevated lipoprotein(a) levels are strong risk factors for ischemic stroke in either arterial or venous circulations, which indicates that these conditions incur risks of both atherosclerosis and hypercoagulability. The main determinant of the population’s distribution of homocysteine levels lies with the methylene-tetrahydrofolate reductase (MTHFR) genotype: high levels are predicted by the TT genotype and low levels by the CC wild-type (abbreviations refer to a single nucleic acid polymorphism within the gene). A high homocysteine level may be exacerbated by deficiencies of folate or vitamin B12, both of which are required for the metabolism of homocysteine (Fig. 45-7). Homocysteine interacts with all three components of the hemostatic system: increasing platelet adhesiveness, activating coagulation factors, and resulting in endothelial disruption; it is also involved in the conversion of LDL-cholesterol into proatherogenic forms. Hyperhomocysteinemia may be treated with supplemental folic acid and vitamins B12 and B6, although there is insufficient evidence at present to support this form of treatment routinely for prophylaxis against arterial events. Lipoprotein(a) is a cholesterol-rich lipoprotein that also has close homology with plasminogen, the latter fact helping explain its influence on coagulation.
Renal Disorders
Nephrotic syndrome predisposes to venous thrombosis because of preferential renal loss of anticoagulants, such as antithrombin III, and by encouraging hypovolemia. In the long term, it is associated with hypercholesterolemia and accelerated atherosclerosis as a result of filtering of apolipoproteins.
Exogenous
Bogousslavsky J, Caplan LR. Uncommon Causes of Stroke. Cambridge, UK: Cambridge University Press, 2001.
International Congress on Thrombosis. 18th Congress, Ljubljana, June 2004: Reports [special issue]. Pathophysiol Haemost Thromb. 2003/2004;33:233-506.
Kullo IJ, Ballantyne CM. Conditional risk factors for atherosclerosis. Mayo Clin Proc. 2005;80:219-230.
Meschia JF, Brott TG, Brown RDJr. Genetics of cerebrovascular disorders. Mayo Clin Proc. 2005;80:122-132.
Stam J. Thrombosis of the cerebral veins and sinuses. N Engl J Med. 2005;352:1791-1798.






{kind=link}
{kind=link}
{kind=link}