[level-membership-for-hematology-oncology-and-palliative-medicine-category]
4 Principles of systemic therapy
Introduction
This chapter gives an overview of the practical aspects of various systemic therapies (Box 4.1). A detailed pharmacological discussion is beyond the scope of this book but further reading is suggested for those interested.
Aim of systemic treatment
Before recommending or prescribing a systemic treatment the aim of the treatment has to be understood (Box 4.2). This, in addition to a knowledge of the specific disease and treatments that are effective for that tumour, will dictate the type of treatment offered and its likely intensity (Box 4.3). Treatment intensity should be greatest in those conditions where the intention is cure, and there is some evidence in certain tumours (e.g. bone tumours) that increased toxicity during chemotherapy is related to improved survival. When the treatment is not curative significant toxicity is unacceptable.
Box 4.2
Aim of systemic treatment
Curative
Chemotherapy is given as the definitive treatment for cure, e.g. acute leukaemia, choriocarcinoma.
In acute leukaemias the curative chemotherapy is given in various phases (Box 4.4).
Chemotherapy
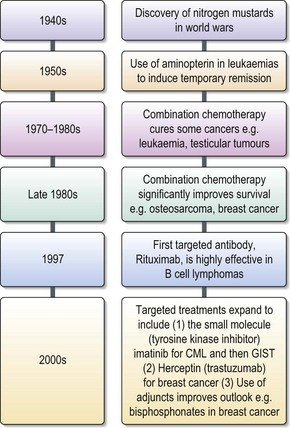
Chemotherapy involves the treatment with cytotoxic chemicals to kill cancer cells. Its benefit depends upon the high proliferation rate of cancer cells compared with the non cancer cells in the body. The discovery of nitrogen mustard and its effects on proliferating cells was discovered in both World Wars, but only put into practice to treat leukaemias and lymphomas in the 1940s (Figure 4.1). Later that decade aminopterin was shown to induce remissions in leukaemia, although the majority of patients relapsed and died. In the following 10 years different classes of chemotherapeutic agents were discovered. Most of these were directed against DNA synthesis or cell division. In the 1970s cytotoxic agents were seen to impact significantly on the cure of specific cancers such as testicular cancer and leukaemia (Box 4.5). This improvement in survival was due not only to the development of new drugs but also to the understanding of how to combine them.
Classes of chemotherapy drugs
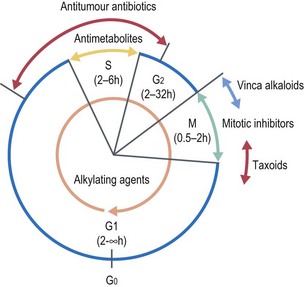
Chemotherapy agents are divided into different categories according to their mechanism of action. The rationale behind chemotherapy is to inhibit or kill rapidly dividing cancer cells. This may be due to the drug acting at a particular point in the cell cycle (cell cycle-specific) or is independent of the cell cycle (cell cycle-non specific) (Figure 4.2). Boxes 4.6 and 4.7 give examples of different classes of drugs in each of these categories with some specific examples.
Box 4.6
Cell cycle specific chemotherapy drugs
| Antimetabolites | S phase | e.g. methotraxate, capecitabine |
| Vinca alkaloids | M phase | e.g. vincristine, vinorelbine |
| Taxanes | M phase | e.g. paclitaxel, docetaxel |
| Epipodophyllotoxins | G2, S, premitotic, topo II | e.g. etoposide |
| Camptothecans | S phase, topo I | e.g. irinotecan, topotecan |
Box 4.7
Cell cycle non-specific chemotherapy drugs
| Antitumour antibiotics | e.g. doxorubicin, mitomycin-C |
| Alkylating agents | e.g. ifosfamide, chlorambucil |
| Nitrosoureas | e.g. lomustine, carmustine |
How does chemotherapy work?
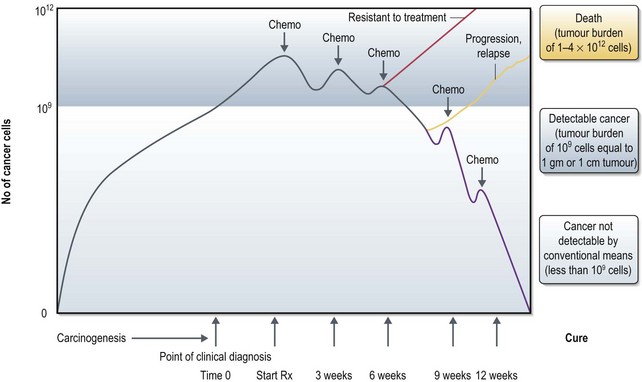
Tumour cells are detectable by conventional means at 109 cells (equivalent to 1 g tumour or 1 cm tumour) and continue to grow without treatment. Patients usually die if they remain untreated or after unsuccessful treatment when the tumour load reaches 1012 cells. When a chemotherapy regime is given to a sensitive tumour it causes cell death in a proportion of the cancer cells (log kill). In a chemo-sensitive tumour, each course of chemotherapy (see Box 4.9) results in a proportional cell kill and with a few courses of chemotherapy, tumour may not be detectable by conventional means (called complete response to treatment). At this point there is a possibility of <109 cells remaining and stopping treatment at this point may lead to an early progression/relapse of disease. Hence patients with chemo-sensitive tumours receive additional courses of chemotherapy to bring down the number of tumour cells to an absolute minimum. However this does not necessarily result in complete removal of tumour cells at the end of chemotherapy and it is believed that the normal body immunosurveillance will help to achieve a cure in some instances. In some patients, cancer can grow back at any point of time (relapse) during the conventionally undetectable phase (see Box 4.8). In some other patients, the tumours do not respond to chemotherapy (resistance to treatment) which requires change of treatment (Figure 4.3).
Box 4.9
Cycles of chemotherapy
How many cycles of chemotherapy?
For curative treatment, further cycles of chemotherapy are needed after a conventional clinical response, to tackle the clinically undetectable tumour burden (Figure 4.3). Hence the amount of chemotherapy may vary from a few cycles to months to years depending on the specific cancer (e.g. acute lymphatic leukaemia needs more than a year of chemotherapy, whereas in choriocarcinoma two more courses of chemotherapy is given after a complete tumour marker response).
Box 4.8
Why not give treatment more often to prevent tumour regrowth?
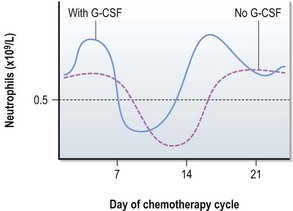
Figure 4.4 shows response of the bone marrow to chemotherapy after chemotherapy given on day 1. Neutrophils fall to their minimum between days 10 and 14 in a standard 21-day cycle. If G-CSF is given the neutrophils may not drop to such a low number.
Rationale for combining chemotherapy agents
In clinical practice cancer cells tend to develop resistance to a single drug by further gene mutations, or development of cellular pumps which reduce the dose of drug received by the tumour cells. Consequently the tumour will have a period of sensitivity followed by a rebound tumour regrowth. This may be partly due to the fact that not all tumour cells are passing through the same point of the cell cycle at the same time. Combining drugs allows the oncologist to direct agents with different activities or against different parts of the cell cycle simultaneously. The idea is that this increases cell kill at the time of each treatment but also reduces the development of drug resistance.
Planning a combination chemotherapy regimen requires some knowledge of the mechanisms of action of the individual drugs, their dosing for particular cancers and their toxicities (see Box 4.10).
Box 4.10
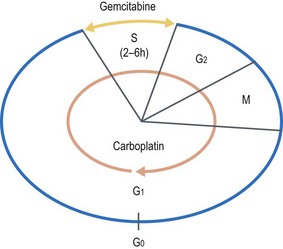
How do you decide which drugs to combine for a given cancer?
e.g. Combination of gemcitabine and carboplatin in lung cancer:
Actions of specific drug classes (Figure 4.6)
Cell cycle phase specific drugs act on cells within a particular phase of the cell cycle.
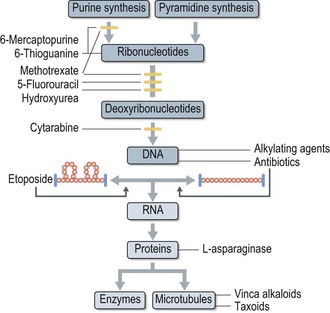
Antimetabolites
These drugs are developed to mimic naturally produced metabolites, purines, pyrimidines or folates essential to the synthesis of nucleic acids and DNA (S phase) preventing the correct incorporation of purines or pyrimidines in the DNA structure. They therefore prevent DNA replication and lead to cell death. They are most effective against tumours with a high growth fraction and similarly have predominant toxicities on normal tissues with high cell turn over (e.g. mucosal epithelium and gastrointestinal tract). Examples are methotrexate, cytosine arabinoside, capecitabine, gemcitabine and 5-fluorouracil (5-FU).
Nitrosoureas
Nitrosoureas are lipid soluble alkylating agents which also destroy DNA preventing any further DNA synthesis. Due to their lipid solubility they can cross the blood–brain barrier into the CNS (see Box 4.11) and are used in several CNS protocols. Examples are lomustine and carmustine.
Delivery of chemotherapy
Consent
Prior to the prescribing and administration of chemotherapy, a definite histological diagnosis (exceptions include germ cell tumours and choriocarcinoma where elevated tumour markers in the appropriate clinical setting is sufficient) and informed consent is necessary. Informed consent is obtained after detailed discussion of benefits and side effects of the treatment (Box 4.12).
Prescribing chemotherapy
Prescribing and reviewing chemotherapy charts is an important role of an oncologist. Many hospitals now have charts which have preprinted chemotherapeutic and supportive drugs on the charts (Figure 4.7) which require only a dose calculation, patient details and test results to be added. Others have electronic systems with that information included. Some of these charts are very simple with all supportive drugs included. In some hospitals however the entire chart has to be written by hand and safety checks are especially important in that case. Before the chart is written, given that chemotherapy is toxic, a number of calculations and checks have to be made. These are summarized in Box 4.13.
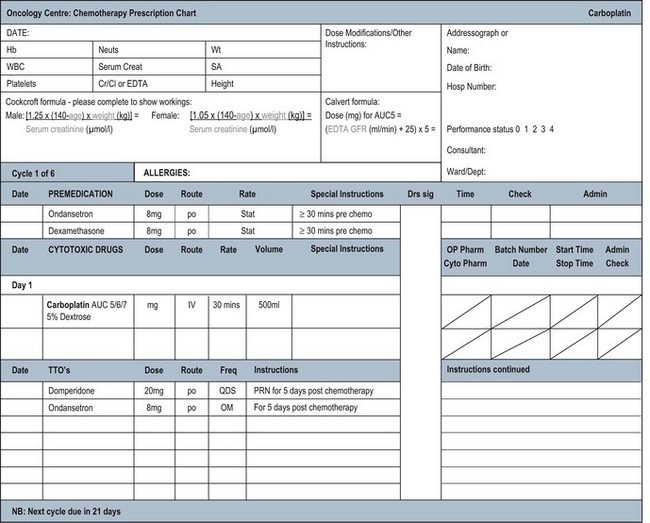
Box 4.13
Pre-prescribing chemotherapy checks
Some drugs are not metabolized in the same way. Carboplatin is the classical example of this type of drug. During the original phase I studies the dose limiting toxicity of carboplatin was thrombocytopenia. However, unlike other drugs, there appeared to be no relationship between the dose administered according to mg/m2 and the degree of thrombocytopenia. It was known that carboplatin is excreted almost entirely by the kidney and Calvert and colleagues then realized that the drug’s toxicity was related to the glomerular filtration rate (GFR). This gave rise to the Calvert formula which is used to prescribe carboplatin (Box 4.14). It requires knowledge of the GFR, ideally calculated by nuclear medicine scan.
Box 4.14
Prescribing carboplatin: AUC and the Calvert formula
The required dose is calculated by the following formula:
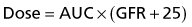
where AUC is the area under the curve. It refers to increasing dosages which have an increasing effect until the effect plateaus and any increase in dose results only in increased toxicity. In general AUC values are in the range 5–7 in adults but up to 9 in children.
Routes of chemotherapy administration
Systemic treatments can be given by a number of routes. Chemotherapy is usually given intravenously (IV) but some drugs can be given orally and some as both IV and orally. For some intravenous drugs there are several ways to administer it. For example, 5-fluorouracil (5-FU) can be given as a daily bolus for 5 days every 3 weeks (Mayo regimen) or as a 48 hour continuous infusion every 2 weeks (modified de Gramont) (p. 165). This drug needs prolonged or repeated exposure as it acts in S phase but these two methods produce different toxicity profiles with the Mayo regimen producing more significant stomatitis and myelosuppression whereas modified de Gramont produces significant palmo-planter erythema. Although many clinicians believe the two methods to be equally effective, randomized trials have not been performed for all cancer types so that some oncologists will only prescribe the method used in the original trials.
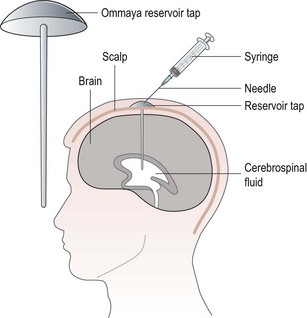
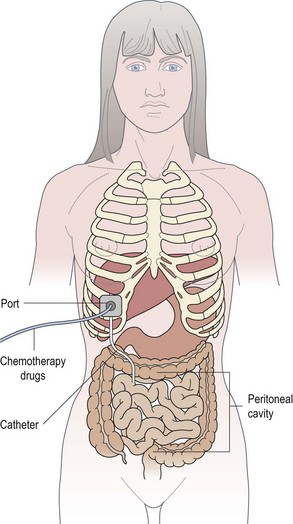
Some drugs can also given either intrathecally (Box 4.15 and Figure 4.8) or via the intraperitoneal route (Box 4.16 and Figure 4.9).
Toxicity recording
In order to minimize side effects and to evaluate how changes in treatment have affected these side effects or the patient’s symptoms due to the cancer, accurate recordings of drug toxicity (Box 4.17) must be made. Ideally this should be recorded on a flow chart so that progress can be monitored. It also allows the clinician to record dates of future scans which can be reviewed prior to further treatment. Toxicity should be scored according to an internationally accepted system such as the National Cancer Institute Common Toxicity Criteria and Adverse Events version 3 (NCI CTCAE v3) which can be found at: http://www.fda.gov/cder/cancer/toxicityframe.htm.
Box 4.17
Chemotherapy toxicities
Acute toxicities are those which occur within a treatment cycle in contrast to late effects which can occur many years later and are covered in (p. 58). Each drug will have its own specific acute toxicities but the following are common to many classes of drugs:
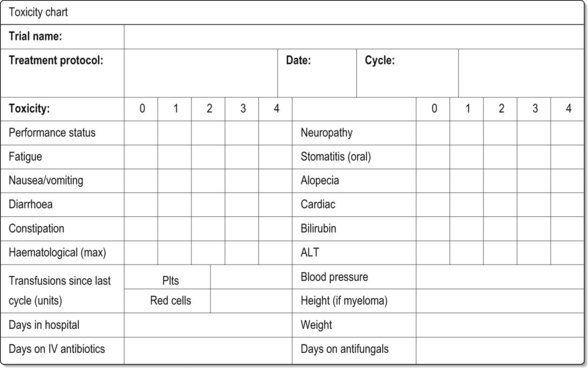
When consenting patients, do not forget the late effects (p. 58) as these can be significant. Figure 4.10 gives an example of a toxicity chart.
Dose adjustment
Although supportive drugs to minimize side effects are usually given prophylactically significant toxicity can occur. Initially the aim would be to change or add to the supportive drugs to reduce toxicity, e.g. different or additional anti-emetics for nausea control, or use of G-CSF to prevent neutropenic sepsis. If the toxicity has been significant, such that it has not resolved with adjustment of supportive drugs, a dose reduction should be made. For example recurrent neutropaenic sepsis which occurs even after prophylactic G-CSF requires dose reduction. For clinical trials there will also be specific instances when this should occur and the protocol must be followed in this case. In general, a reduction of 20–25% is made but this depends on the drugs being given, whether all or only one of them is likely to be the cause. It also depends on the intended outcome of the treatment and the likely effect that dose reduction will have on the outcome. For example it is known that the survival benefit from adjuvant chemotherapy can be reduced by a reduction in dose intensity. However if the patient has suffered a life-threatening infection associated with the treatment, the risk of death as a complication of continuing the same dose of treatment may be greater than that of recurrence of the cancer.
Other classes of systemic therapy
Hormone treatments (Table 4.1)
The observation that breast cancer can regress after oophorectomy and that prostate cancer can be controlled by orchidectomy suggested that some cancers may be hormone sensitive. Since the observation that surgical measures to reduce hormone levels were effective, drugs were developed which would achieve a similar effect. In the 1990s dramatic improvements in breast cancer survival were seen with the introduction of the anti-oestrogen, tamoxifen, to women with oestrogen receptor positive breast cancer. This led to the developments of other drugs which target the production of oestrogen (such as the aromatase inhibitors) as well as examining the role of hormone treatments in other hormone sensitive cancers such as prostate cancer. These are discussed in more detail in the breast and prostate chapters (Chapters 10 and 12). In general these treatments are taken for many years to prevent recurrence or control the disease and can be effective without chemotherapy. In fact there is evidence to suggest that they should not be given alongside chemotherapy, particularly for anti-oestrogen therapies in breast cancer as it is thought that these cytostatic agents (e.g. tamoxifen) causes cell cycle arrest preventing the action of chemotherapy which is dependent on rapidly dividing cells for its action.
| Example | Mechanism of action | |
|---|---|---|
| Breast cancer | ||
Immune therapy
In addition to antibodies, which may be considered an immune treatment, there are also a group of drugs which act by modifying the immune response of the host (patient) in general, rather than targeting a specific molecule or receptor. Examples in this group include interferons and interleukins. High dose interferon has been used in melanoma (p. 235) and interleukin-2 has some efficacy in renal cell carcinoma (p. 176). Interferon and interleukin both produce systemic side effects of an immune response which resemble a viral infection, e.g. fever, fatigue, myalgia and headache.
Small molecules
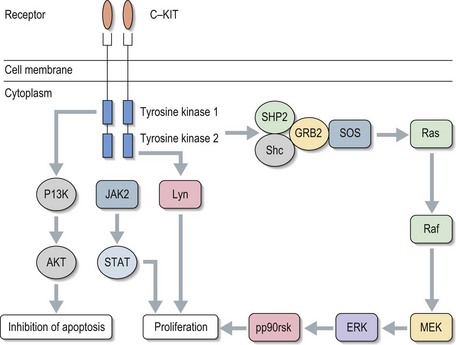
Small molecules are drugs developed with a specific target, usually to a cellular pathway or molecule within that pathway. Examples include tyrosine kinase inhibitors (TKIs) and mTOR inhibitors. TKIs can be specific to a particular tyrosine kinase, e.g. imatinib, or may have activity across a number of kinases, e.g. sunitinib. Figure 4.11 shows an example of a tyrosine kinase signalling pathway using c-KIT. Once the receptor is bound it sets off a cascade leading to further cell proliferation. Some of the cascade steps are common to pathways involved in many cancers such that effective inhibitors to one protein could be useful drugs in a range of cancers.
Systemic radiotherapy
In general radiotherapy is directed against a specific site in the body. Generally a radioisotope scan is done to assess the uptake by the specific tumour prior to giving definitive radioablative dose (for example, see p. 282). Doses can be repeated several times but there is a risk of late myelodysplastic syndromes.
Assessment of response to treatment
Systemic treatments are often given to treat metastatic disease or neoadjuvantly prior to surgery. In these cases an assessment needs to be made to ensure that the treatment is effective (Figure 4.12) so that an alternative treatment may be considered and to avoid inappropriate toxicity. Most often this assessment is made radiologically (e.g. by CT or MRI) by Response Evaluation Criteria in Solid Tumours (RECIST) criteria (Boxes 4.18 and 4.19, Table 4.2). An understanding of these terms is necessary in the management of all cancers, even if alternative means of assessment may also be used (e.g. see section on gastrointestinal stromal tumours, p. 261). However for some tumours or in the adjuvant setting RECIST terms may not adequately describe response. In adjuvant treatment where the aim is to cure, the overall survival and sometimes the progression free survival are the key indicators of success. For clinical trials this may take many years to assess so sometimes other indicators are used. These include pathological response for tumours such as bone tumours or breast cancer in whom the patients have been given neoadjuvant treatment. The degree of cell necrosis or pathological response reflects prognosis in these groups. For gastrointestinal stromal tumours which have been treated with a targeted treatment, imatinib, the response assessed by RECIST criteria may not reflect the true response. For these tumours, and other sarcomas there may not be an obvious reduction in the size of the tumour, and yet the patient feels better. Radiologically this is usually accompanied by a change in the density of the tumour becoming more cystic and less dense. For gastrointestinal stromal tumours new criteria (Choi criteria) have been applied to these tumours to take these changes into account (p. 263).
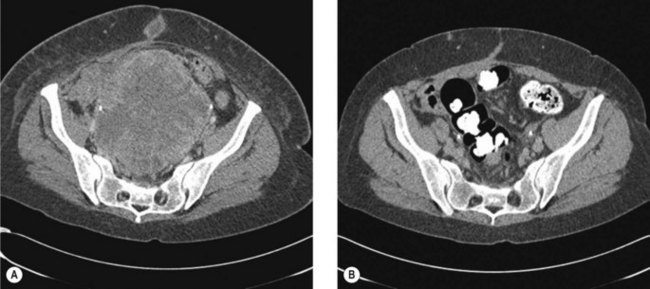
Box 4.18
Definition of lesions for RECIST evaluation
Box 4.19
Confirmation of response by RECIST criteria
| Evaluation of target lesions | |
|---|---|
| Complete response (CR): | Disappearance of all target lesions |
| Partial response (PR): | At least a 30% decrease in the sum of the longest diameter (LD) of target lesions, taking as reference the baseline sum LD |
| Progressive disease (PD): | At least a 20% increase in the sum of the LD of target lesions, taking as reference the smallest sum LD recorded since the treatment started or the appearance of one or more new lesions |
| Stable disease (SD): | Neither sufficient shrinkage to qualify for PR nor sufficient increase to qualify for PD, taking as reference the smallest sum LD since the treatment started |
| Evaluation of non-target lesions | |
|---|---|
| Complete response (CR): | Disappearance of all non-target lesions and normalization of tumour marker level |
| Incomplete response/stable disease (SD): | Persistence of one or more non-target lesion(s) or/and maintenance of tumour marker level above the normal limits |
| Progressive disease (PD): | Appearance of one or more new lesions and/or unequivocal progression of existing non-target lesions |
In addition, particularly in the context of clinical trials the term Clinical Benefit is used. This takes into account not only the obvious CR and PR RECIST responses but also those tumours which may be stable by RECIST, but treatment has also been associated with an improvement in symptoms. For palliative treatments this is clearly important.
Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45:228-247.
Brighton D, Wood M. The Royal Marsden Hospital Book of Cancer Chemotherapy. Edinburgh: Elsevier; 2005.
Arkenau H, Carden C, De Bono S. Targeted agents in cancer therapy. Medicine. 2007;36(1):33-37.
[/level-membership-for-hematology-oncology-and-palliative-medicine-category][not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
4 Principles of systemic therapy
Introduction
This chapter gives an overview of the practical aspects of various systemic therapies (Box 4.1). A detailed pharmacological discussion is beyond the scope of this book but further reading is suggested for those interested.
Aim of systemic treatment
Before recommending or prescribing a systemic treatment the aim of the treatment has to be understood (Box 4.2). This, in addition to a knowledge of the specific disease and treatments that are effective for that tumour, will dictate the type of treatment offered and its likely intensity (Box 4.3). Treatment intensity should be greatest in those conditions where the intention is cure, and there is some evidence in certain tumours (e.g. bone tumours) that increased toxicity during chemotherapy is related to improved survival. When the treatment is not curative significant toxicity is unacceptable.
Box 4.2
Aim of systemic treatment
Curative
Chemotherapy is given as the definitive treatment for cure, e.g. acute leukaemia, choriocarcinoma.
In acute leukaemias the curative chemotherapy is given in various phases (Box 4.4).
Chemotherapy
Chemotherapy involves the treatment with cytotoxic chemicals to kill cancer cells. Its benefit depends upon the high proliferation rate of cancer cells compared with the non cancer cells in the body. The discovery of nitrogen mustard and its effects on proliferating cells was discovered in both World Wars, but only put into practice to treat leukaemias and lymphomas in the 1940s (Figure 4.1). Later that decade aminopterin was shown to induce remissions in leukaemia, although the majority of patients relapsed and died. In the following 10 years different classes of chemotherapeutic agents were discovered. Most of these were directed against DNA synthesis or cell division. In the 1970s cytotoxic agents were seen to impact significantly on the cure of specific cancers such as testicular cancer and leukaemia (Box 4.5). This improvement in survival was due not only to the development of new drugs but also to the understanding of how to combine them.
Classes of chemotherapy drugs
Chemotherapy agents are divided into different categories according to their mechanism of action. The rationale behind chemotherapy is to inhibit or kill rapidly dividing cancer cells. This may be due to the drug acting at a particular point in the cell cycle (cell cycle-specific) or is independent of the cell cycle (cell cycle-non specific) (Figure 4.2). Boxes 4.6 and 4.7 give examples of different classes of drugs in each of these categories with some specific examples.
Box 4.6
Cell cycle specific chemotherapy drugs
| Antimetabolites | S phase | e.g. methotraxate, capecitabine |
| Vinca alkaloids | M phase | e.g. vincristine, vinorelbine |
| Taxanes | M phase | e.g. paclitaxel, docetaxel |
| Epipodophyllotoxins | G2, S, premitotic, topo II | e.g. etoposide |
| Camptothecans | S phase, topo I | e.g. irinotecan, topotecan |
Box 4.7
Cell cycle non-specific chemotherapy drugs
| Antitumour antibiotics | e.g. doxorubicin, mitomycin-C |
| Alkylating agents | e.g. ifosfamide, chlorambucil |
| Nitrosoureas | e.g. lomustine, carmustine |
How does chemotherapy work?
Tumour cells are detectable by conventional means at 109 cells (equivalent to 1 g tumour or 1 cm tumour) and continue to grow without treatment. Patients usually die if they remain untreated or after unsuccessful treatment when the tumour load reaches 1012 cells. When a chemotherapy regime is given to a sensitive tumour it causes cell death in a proportion of the cancer cells (log kill). In a chemo-sensitive tumour, each course of chemotherapy (see Box 4.9) results in a proportional cell kill and with a few courses of chemotherapy, tumour may not be detectable by conventional means (called complete response to treatment). At this point there is a possibility of <109 cells remaining and stopping treatment at this point may lead to an early progression/relapse of disease. Hence patients with chemo-sensitive tumours receive additional courses of chemotherapy to bring down the number of tumour cells to an absolute minimum. However this does not necessarily result in complete removal of tumour cells at the end of chemotherapy and it is believed that the normal body immunosurveillance will help to achieve a cure in some instances. In some patients, cancer can grow back at any point of time (relapse) during the conventionally undetectable phase (see Box 4.8). In some other patients, the tumours do not respond to chemotherapy (resistance to treatment) which requires change of treatment (Figure 4.3).
Box 4.9
Cycles of chemotherapy
How many cycles of chemotherapy?
For curative treatment, further cycles of chemotherapy are needed after a conventional clinical response, to tackle the clinically undetectable tumour burden (Figure 4.3). Hence the amount of chemotherapy may vary from a few cycles to months to years depending on the specific cancer (e.g. acute lymphatic leukaemia needs more than a year of chemotherapy, whereas in choriocarcinoma two more courses of chemotherapy is given after a complete tumour marker response).
Box 4.8
Why not give treatment more often to prevent tumour regrowth?
Figure 4.4 shows response of the bone marrow to chemotherapy after chemotherapy given on day 1. Neutrophils fall to their minimum between days 10 and 14 in a standard 21-day cycle. If G-CSF is given the neutrophils may not drop to such a low number.
Rationale for combining chemotherapy agents
In clinical practice cancer cells tend to develop resistance to a single drug by further gene mutations, or development of cellular pumps which reduce the dose of drug received by the tumour cells. Consequently the tumour will have a period of sensitivity followed by a rebound tumour regrowth. This may be partly due to the fact that not all tumour cells are passing through the same point of the cell cycle at the same time. Combining drugs allows the oncologist to direct agents with different activities or against different parts of the cell cycle simultaneously. The idea is that this increases cell kill at the time of each treatment but also reduces the development of drug resistance.
Planning a combination chemotherapy regimen requires some knowledge of the mechanisms of action of the individual drugs, their dosing for particular cancers and their toxicities (see Box 4.10).
Box 4.10
How do you decide which drugs to combine for a given cancer?
e.g. Combination of gemcitabine and carboplatin in lung cancer:
