[level-membership-for-internal-medicine-category]
Chapter 2 Principles of Myocardial Metabolism as They Relate to Imaging
INTRODUCTION
The metabolic cost on the heart of maintaining an adequate cardiac output is extremely high. Based on measurements of myocardial oxygen consumption1 and the degree of coupling between oxygen consumption and mitochondrial adenosine triphosphate (ATP) synthesis,2 the heart of a 70-kg individual would produce (and consume) 4.6 kg of ATP per day. As a result, the total content of ATP in the heart turns over approximately 8 times per minute to meet this high rate of flux. While the majority of this ATP expenditure is utilized for contractile activity, ATP is also required for maintaining cellular ionic homeostasis and supporting protein synthesis.
OVERVIEW OF METABOLIC REGULATION IN THE NORMAL HEART
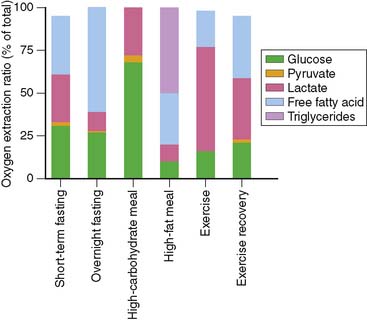
To understand the merits and drawbacks of various metabolic radiotracers, it is important to understand how cardiac metabolism is regulated. As noted above, the heart is an “omnivore,” synthesizing ATP through the metabolism of a variety of fuel substrates.3 Furthermore, the relative contribution of the different substrates varies, and the heart must adapt rapidly to changing sources of substrate. A good example of this plasticity of substrate selection by the heart is reflected in the changes that occur during different physiologic states based on nutritional status and degree of physical activity (Fig. 2-1). The relative contribution of a given substrate to myocardial ATP production is dependent on a variety of selection pressures, including the arterial concentration of the substrate, the availability of oxygen, hormonal stimulation, the workload imposed upon the heart, and the presence of pathologic conditions that affect myocardial utilization of substrates (e.g., coronary artery disease, heart failure, diabetes) through changes in the cardiac myocyte’s expression of regulatory proteins and enzymes.
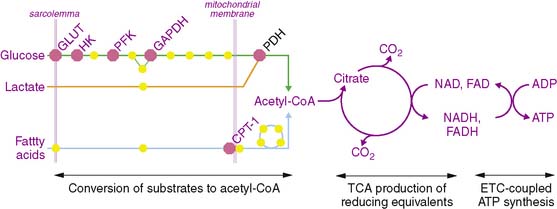
The metabolism of the primary fuels in the heart is graphically depicted in Fig. 2-2 and illustrates three major phases of metabolism. The first phase is involved in converting fatty acids, glucose, and lactate into a common substrate for entry into the tricarboxylic acid (TCA) cycle in the mitochondria. In the second phase, reducing equivalents in the form of reduced nicotinamide adenine dinucleotide (NADH2) and reduced flavin adenine dinucleotide (FADH2) are produced in the TCA cycle and provide electrons for the electron transport chain that ultimately are used to convert oxygen to water. In the third phase, a proton gradient across the inner mitochondrial membrane, which is generated by the proteins of the electron transport chain, drives ATP synthesis.
Fatty Acid Metabolism
Under normal conditions, fatty acids and triglycerides are the preferred substrate for the normal heart. Fatty acids are taken up by the cardiac myocyte through facilitative transport via fatty acid translocase (FAT/CD36).4 Once inside the myocyte, fatty acids are esterified to fatty acyl-CoA derivatives through a reaction mediated by fatty acyl-CoA synthetase, which utilizes the hydrolysis of ATP to adenosine monophosphate (AMP) to drive the reaction. This energy-requiring step may be of great importance in understanding the decrease in accumulation of the fatty acid analog, [123I]β-methyl-iodophenyl pentadecanoic acid (BMIPP), seen in ischemic myocardium. After this activation, fatty acyl-CoA may be transported into the mitochondria following transesterification with carnitine by carnitine palmitoyltransferase 1 (CPT-1). This step represents the rate-limiting step of fatty acid oxidation and is regulated by cytosolic concentrations of malonyl-CoA; this important regulatory step of fatty acid metabolism is discussed in detail later in the chapter. Once inside the mitochondria, CPT-2 converts the fatty acylcarnitine back into fatty acyl-CoA for entry into the β-oxidation pathway. Another metabolic fate of the cytosolic fatty acyl-CoA is incorporation into triglycerides. It is this fate that predominates for BMIPP because the β-methyl group inhibits it entry into β-oxidation in the mitochondria.5
Glucose Metabolism
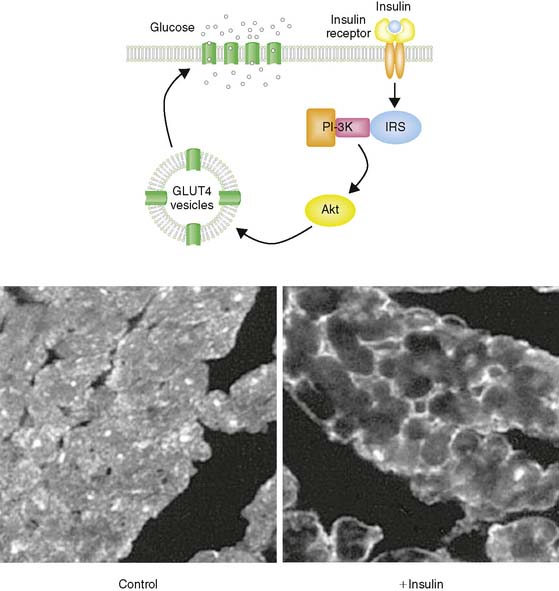
Glucose represents the other major fuel of the heart. The initial transport of glucose across the cell surface membrane represents the rate-limiting step of glucose metabolism and is mediated by facilitative glucose transporters (GLUTs).6 Of the 13 described GLUTs, only two, GLUT1 and GLUT4, are expressed to a significant degree in the heart. GLUT1 is present mostly on the cardiomyocyte cell surface and is responsible for basal glucose uptake. In contrast, GLUT4 exists both on the cell surface and in an intracellular pool of membrane vesicles that can translocate to the cell surface in response to insulin (Fig. 2-3). It is therefore GLUT4 translocation that is responsible for insulin-stimulated glucose uptake in the insulin-sensitive tissues of the heart, skeletal muscle, and adipose tissue. The translocation of GLUT4 is also responsible for the enhanced glycolysis observed during ischemia, although the mechanism of this translocation is independent of the insulin signaling pathway as described later in this chapter.7–9
PFK-1 is also inhibited by citrate, which increases when there is sufficient TCA cycle flux to meet the energetic needs of the cell. This inhibition of glycolysis at the level of PFK-1 by ATP and citrate is the basis of a critical aspect of myocardial metabolism that regulates substrate selection: the glucose/fatty acid, or Randle, cycle. The oxidation of fatty acids in the mitochondria results in an increase in both ATP and citrate, which inhibits PFK-1 and thereby reduces glucose uptake.10 The operation of the Randle cycle has important implications with respect to myocardial substrate utilization under physiologic conditions such as the transition from the postprandial state, in which insulin stimulation and abundant circulating glucose lead to increased reliance on glucose, to the fasting state, in which the greater concentration of free fatty acids increases fatty acid metabolism, and disease states such as diabetes, in which there is a persistent increase in the free fatty acid concentration.
Once glucose is metabolized to pyruvate, it is transported into the mitochondria, where it is converted to acetyl-CoA through the action of pyruvate dehydrogenase (PDH). PDH is a multienzyme complex that is regulated by the metabolic status of the cell. Specifically, PDH is inhibited by increased [NADH2]/[NAD] and [acetyl-CoA]/[CoASH] ratios, both of which occur when there is a relative overabundance of NADH2 and acetyl-CoA that outstrips the ability of the mitochondria to utilize these metabolites.11 This regulation of PDH activity is mediated by PDH kinase, which phosphorylates and thereby inactivates the PDH complex. In the setting of enhanced fatty acid oxidation, flux through PDH is inhibited by increased PDH kinase activity,12 providing another level of regulation of substrate selection in the heart. Conversely, PDH activity can be increased by dephosphorylation, which occurs in response to insulin stimulation.13 In addition, PDH can be activated by increases in workload through a calcium-dependent mechanism.
Tricarboxylic Acid Cycle Metabolism and the Electron Transport Chain
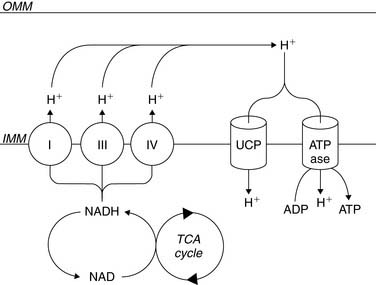
β-Oxidation of fatty acids, glycolysis of both exogenous glucose and endogenous glycogen, and uptake of exogenous lactate result in the conversion of these fuels to a common energetic currency, namely acetyl-CoA, which enters the TCA cycle by condensing with oxaloacetate to form citrate. The citrate that is formed undergoes subsequent oxidative and decarboxylating reactions in the TCA cycle, which results in the generation of five important compounds that not only help to drive mitochondrial ATP synthesis but are also important with respect to metabolic imaging. The first is the ultimate conversion of the 6-carbon citrate to the 4-carbon oxaloacetate, which is then available for another “turn” of the TCA. This is linked to the second important product, carbon dioxide (CO2), which is produced through two decarboxylation steps, one mediated by isocitrate dehydrogenase and the other mediated by α-ketoglutarate dehydrogenase. This CO2 is released from the cell and ultimately leaves the body through the lungs. The third is the production of NADH2 by isocitrate dehydrogenase, α-ketoglutarate dehydrogenase, and malate dehydrogenase. This NADH2 is used by the electron transport chain to generate the mitochondrial membrane potential required to power the mitochondrial F0F1-ATPase that converts ADP to ATP (Fig. 2-4). In addition, FADH2 is produced by succinate dehydrogenase and also contributes electrons to the electron transport chain for the conversion of oxygen to water, but because of the location of succinate dehydrogenase in the inner aspect of the inner mitochondrial membrane, it does not contribute to the mitochondrial membrane potential. Finally, the high-energy phosphate, guanosine triphosphate (GTP), is generated through substrate-level phosphorylation by succinyl-CoA synthetase, a reaction that becomes important to energy production during ischemia because, like glycolysis, it does not require oxygen to produce high-energy phosphates.14
Of great importance to the evaluation of myocardial mitochondrial function by nuclear methods is the fact that there is a direct relationship between the entry of acetyl-CoA into the tricarboxylic acid cycle and the conversion of oxygen to water through the electron transport chain. Because of this coupling of acetyl-CoA metabolism to oxygen consumption, it is possible to determine rates of myocardial oxygen consumption noninvasively using the positron emission tomography (PET) tracer [1-11C]-acetate.15 Furthermore, a coupling between energy demand and energy production translates to the coupling of TCA flux to ATP synthesis. Specifically, increases in workload result in an increase in cytosolic calcium; this increased cytosolic calcium concentration increases mitochondrial calcium content, which activates not only PDH but the calcium-dependent enzymes, isocitrate dehydrogenase and α-ketoglutarate dehydrogenase, that are part of the TCA cycle.
METABOLIC TRACERS
Radiotracers of metabolic pathways fall into two categories: those that are radioisotopes of the parent compound (e.g., [1-11C]glucose and [1-11C]palmitate) or those that are analogs of the parent compound (e.g., [2-18F]-2-fluoro-2-deoxyglucose [FDG] and BMIPP). The quantitative evaluation of metabolic pathways generally utilizes the former tracers because they follow the same metabolic fate of the parent compound, whereas the latter compounds are utilized for qualitative assessments of metabolism because they generally are retained by the tissue, making imaging easier. For example, because the PET tracer [1-11C]glucose is biochemically indistinguishable from glucose, it will follow the exact fate of glucose, including the eventual release from the cardiomyocyte as 11CO2. As a result, there is uptake, retention, and ultimately disappearance of radiotracer from the heart (Fig. 2-5). In contrast, FDG is taken up and phosphorylated by hexokinase, but it is not further metabolized in the cardiomyocyte because of the modification of the carbohydrate structure from glucose to deoxyglucose. As a result, FDG becomes trapped in the cell. Kinetic analysis of the time activity curves for FDG can be used to estimate the initial uptake and phosphorylation of glucose,16,17 but it offers no information about the oxidative fate of glucose.
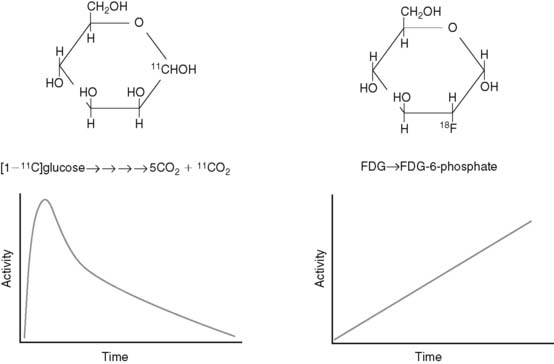
Although the kinetic analysis of a tracer such as FDG that demonstrates irreversible trapping would appear to be more straightforward than the analysis required for tracers that demonstrate accumulation and disappearance, there are two issues that must be kept in mind in translating information gained from irreversibly trapped radiotracers to conclusions about myocardial substrate utilization. First, as demonstrated earlier, these tracers only provide information about a portion of a given metabolic pathway. Second, differences in the structure of the parent compound and the radiotracer will alter the fidelity with which the tracer measures utilization of the parent compound, and this relationship between tracer and tracee can vary under different metabolic conditions.18
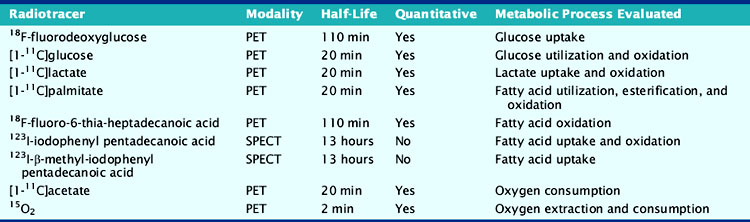
In addition to categorizing metabolic radiotracers based on their ability to trace metabolic pathways accurately and completely, they can also be grouped according to whether they are single photon–emitting or positron-emitting radiotracers (Table 2-1). Because of the coincidence detection of the two 511-keV photons produced by positron annihilation combined with the attenuation correction that is required for PET, kinetic analysis can be performed with the positron-emitting metabolic radiotracers, providing quantitative measurements of rates of substrate uptake and metabolism. In contrast, single photon–emitting metabolic radiotracers can only provide qualitative assessments of metabolic processes but have the advantage in that they do not require an on-site cyclotron, which is necessary for the production of short-lived C-11 and O-15 compounds. Because of this advantage, there is growing interest not only in the established I-123-labeled fatty acid analog, BMIPP, but newer Tc-99 m-labelled fatty acid analogs for metabolic imaging.19
METABOLIC RESPONSES TO DISEASE STATES
Myocardial Ischemia
With a decrease or cessation of blood flow, the cardiac myocyte rapidly switches from oxidative metabolism of fatty acids and glucose to a greater reliance on anaerobic glycolysis with the production of lactate (Fig. 2-6). The increase in glucose uptake is due in large part to the translocation of the facilitative glucose transporter, GLUT4, from intracellular storage vesicles to the surface of the cardiac myocyte.20 As discussed previously, GLUT4 translocates to the cell surface in response to stimulation of the insulin signaling pathway, which involves activation of key proteins that include phosphoinositidyl 3-kinase and Akt. However, the translocation of GLUT4 in response to myocardial ischemia is independent of the insulin signaling pathway.8,21
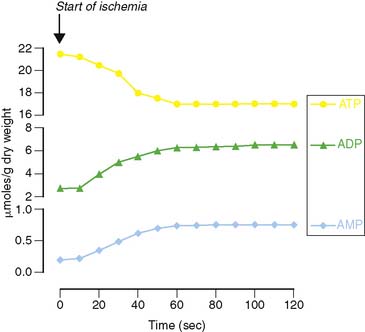
The molecular mechanisms responsible for the translocation of GLUT4 have recently been elucidated and start with the rapid hydrolysis of ATP to ADP and AMP that occurs with the onset of ischemia (Fig. 2-7). This change in the energy charge of the cell activates the metabolic stress protein, AMP-activated protein kinase (AMPK), both through phosphorylation of AMPK by upstream kinases and through noncovalent activation by binding of AMP to a regulatory subunit of AMPK.8,22
AMPK activation is a critical response to cellular metabolic stress, not only increasing GLUT4 translocation and thereby enhancing glucose uptake but also increasing the uptake and oxidation of fatty acids.23 The increase in fatty acid oxidation by AMPK is mediated indirectly through phosphorylation of acetyl-CoA carboxylase (ACC), which decreases the conversion of acetyl-CoA to malonyl-CoA. Malonyl-CoA is an inhibitor of CPT-1, the enzyme that regulates the entry of fatty acids into the mitochondria, thereby regulating fatty acid oxidation. Therefore, inactivation of ACC by AMPK relieves malonyl-CoA inhibition of CPT-1 to increase fatty acid oxidation. AMPK inhibition of ACC has no impact on fatty acid oxidation during ongoing ischemia, because the limitation of oxygen delivery to the myocyte inhibits β-oxidation of fatty acids and TCA cycle flux, AMPK remains activated during early reperfusion, which can result in enhanced postischemic fatty acid oxidation.23 This increase in fatty acid oxidation in the reperfused myocardium has been hypothesized to increase the production of lipid-derived free radicals and uncouple glucose uptake from glucose oxidation.
Another target of regulation by AMPK is PFK-1, one of the enzymes regulating glycolytic flux. Recent studies have demonstrated that PFK-1 activity is increased by fructose 2,6-bisphosphate, which is synthesized from fructose 6-phosphate by PFK-2. Furthermore, PFK-2 is activated through phosphorylation by AMPK.24 Therefore, the increase in glucose utilization in the ischemic heart is due to increased glucose uptake through GLUT4 translocation to the cell surface and increased glycolytic flux through activation of PFK-1, both of which are mediated by AMPK.
At the same time that AMPK activation increases the synthesis of ATP, it also decreases the consumption of ATP in energy-requiring cellular processes such as protein synthesis and cholesterol synthesis (Fig. 2-8). Because of these diverse actions, AMPK has been thought of as a metabolic fuel gauge for cells.25 Loss of AMPK function in transgenic mice has demonstrated the critical role of the protein in the heart’s response to ischemia, with loss of AMPK function resulting in a lack of an ischemia-mediated increase in glycolysis and increased myocyte damage and apoptosis resulting in decreased postischemic recovery of contractile function.9
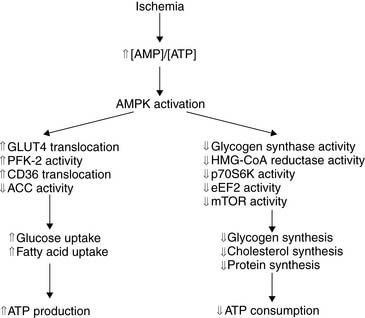
The duration of the acute translocation of GLUT4, and the concomitant increase in glucose uptake in response to myocardial ischemia, is variable and has been suggested to persist as much as 24 hours.26 As a result, there has been some interest in whether FDG could be used as a metabolic memory agent in patients presenting with a history of chest pain that has resolved. This concept is supported by the finding that increased FDG accumulation can be detected in patients with exercise-induced ischemia if they are injected with FDG either at the time of exercise or 1 hour after exercise.27,28 However, it is not known how long increased myocardial FDG uptake will persist after the resolution of ischemia. This is further complicated by the fact that GLUT4 translocation and increased glucose uptake can be mediated by insulin stimulation, making it difficult to identify increased FDG uptake in a patient who has either eaten or received insulin around the time of an ischemic episode. Given the fact that AMPK is not activated by insulin, this metabolic stress protein may be a better target for molecular imaging of a prior ischemic event, although the time course for the deactivation of AMPK requires further evaluation.
Another radiotracer that can be used to evaluate acute myocardial ischemia or a prior ischemic event is BMIPP. BMIPP has been in clinical use in Japan for more than a decade for the detection of myocardial ischemia. However, in contrast to FDG, which is a “hot-spot” imaging agent for myocardial ischemia, BMIPP is a “cold-spot” imaging agent. As a fatty acid analog, BMIPP is taken up and undergoes thioesterification with CoA, which requires a significant energy expenditure, with the hydrolysis of ATP to AMP. It has been demonstrated that the intracellular trapping of BMIPP is dependent on ATP content.29 Unlike dietary fatty acids that do not have a β-methyl substitution, BMIPP does not undergo significant mitochondrial β-oxidation. Rather, it is incorporated in the cytosolic triglyceride pool.30,31 However, the rate-determining enzyme of triglyceride synthesis, glycerol 3-phosphate acyltransferase, which esterifies fatty acyl-CoA to the glycerol backbone of triglycerides, is inhibited by ischemia.32 As a result of ATP depletion and decreased triglyceride synthesis in the setting of ischemia, free BMIPP can back-diffuse out of ischemic myocardium.33
Based on the decrease in BMIPP uptake in the ischemic myocardium, it has recently been shown that BMIPP defects may be seen from 4 to 30 hours after exercise-induced ischemia.34 Given this rather large window in which a prior ischemic insult may be detected, BMIPP may be superior to FDG for late imaging of ischemia. In addition, BMIPP does not require an on-site cyclotron for production, so its clinical use is more practical. Further studies defining the effects of patient characteristics such as diabetes and heart failure, duration of BMIPP defects, and prognostic impact of the results of BMIPP imaging of a previous ischemic insult remain to be defined.
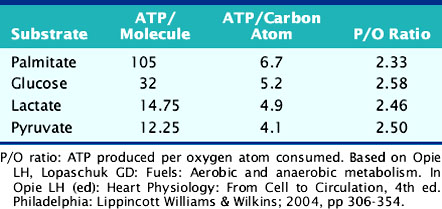
Chronic myocardial ischemia results in metabolic remodeling of the heart to increase glucose utilization and decrease fatty acid utilization.35–38 This alteration in substrate utilization is due chiefly to the greater reliance on anaerobic glycolysis in the setting of reduced oxygen delivery. However, there is also a theoretical advantage to the oxidation of glucose over fatty acid oxidation, owing to the fact that a greater amount of ATP is produced per mole of oxygen consumed for glucose oxidation compared to fatty acid oxidation (Table 2-2). These changes in myocardial metabolism are due in part to increased expression of key proteins involved in glucose metabolism, including GLUT1, hexokinase, pyruvate kinase, lactate dehydrogenase, and pyruvate dehydrogenase.39–42 The increase in expression of the GLUT1 and lactate dehydrogenase genes may be due to chronic AMPK activation43 and is mediated by the transcription factor, hypoxia inducible factor (HIF)-1α.44
Congestive Heart Failure
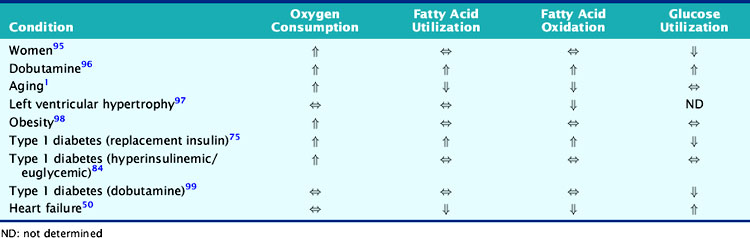
The development of heart failure is associated with a variety of alterations in the expression of proteins involved in contraction, metabolism, ionic homeostasis, and transcriptional regulation.45,46 In general, these alterations result in a reversion to a more fetal pattern of gene expression.47–49 The metabolic implications of these changes in gene expression in the setting of heart failure include a greater reliance on glucose utilization and a decrease in fatty acid oxidation, which has been demonstrated in both animal models of heart failure and patients with nonischemic cardiomyopathy (Table 2-3).50–52
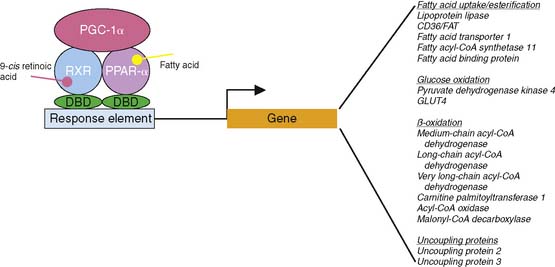
These changes are due in part to alterations in the expression of the key transcriptional regulators, peroxisome proliferator activated receptor (PPAR)-α and peroxisome proliferator activated receptor-γ coactivator (PGC)-1α. PPAR-α and PGC-1α interact in the nucleus to increase the transcription of a variety of genes that are primarily involved in mitochondrial biogenesis and fatty acid metabolism (Fig. 2-9).53,54 In the setting of heart failure, the expression and activity of these regulators of metabolic gene transcription are decreased in association with decreased expression of key proteins involved in fatty acid metabolism.55,56 In transgenic mice lacking PPAR-α, stimulating glucose utilization improves left ventricular function.57 Interestingly, overexpression of PPAR-α has been demonstrated to cause a cardiomyopathy, likely related to excessive utilization of fatty acids.58
Metabolic imaging in patients with heart failure is already a standard of care for the identification of viable myocardium based on the uptake of FDG in hypoperfused but viable myocardium. As discussed previously, FDG is transported into intact cells and phosphorylated by hexokinase in a manner similar to glucose. The phosphorylation of FDG to FDG-6-phosphate by hexokinase requires ATP, which is only found in viable cells. In the setting of persistent low-flow ischemia under resting conditions, perfusion defects may appear fixed, suggesting the presence of scar tissue. However, viable cardiac myocytes in the region utilize anaerobic glycolysis to generate ATP to maintain survival and will therefore take up FDG, producing the classic mismatch pattern between blood flow, determined by PET blood flow tracers such as N-13 ammonia or Rb-82, and metabolism. Numerous studies have demonstrated that regions of the heart with flow/metabolism mismatch will benefit from revascularization, with improvement in left ventricular function.35,59–68
In addition to the identification of viable myocardium in patients with presumed ischemic myocardium using FDG, metabolic imaging may provide insights into the metabolic efficiency of the failing myocardium.69 Using [1-11C]acetate to evaluate myocardial oxygen consumption, abnormalities in mitochondrial function can be evaluated70 and may be used to evaluate the response to therapies such as cardiac resynchronization therapy.71
Because congestive heart failure is an increasingly common entity, understanding the basic pathophysiology, including the metabolic (mal)adaptations of the failing heart, becomes increasingly important. Specifically, is the switch to a greater reliance on glucose a protective response in the failing heart? It has been demonstrated that β-blocker treatment of heart failure patients, a therapy that has been demonstrated to reduce mortality, is associated with increased myocardial glucose utilization and decreased fatty acid oxidation.72 Furthermore, treatment with the fatty acid oxidation inhibitor, ranolazine, has been shown to improve left ventricular performance.73 In contrast, increased activation of PPAR-α with fenofibrate in a canine model of heart failure increases fatty acid metabolism but does not result in a significant improvement in left ventricular function.74 Based on the metabolic changes that occur in the failing heart, combined with potential metabolically based therapies, there may be a greater role for metabolic imaging for the assessment of patients with congestive heart failure and evaluation of the response to therapeutic interventions.
Diabetes
The growing worldwide problems of obesity, insulin resistance, and diabetes have helped to draw greater attention to the cardiac manifestations of these metabolic disorders and the alterations in myocardial energetics. Diabetes, especially type 2 diabetes, represents not only an abnormality of glucose homeostasis but also an abnormality of fatty acid metabolism. The diabetic heart is characterized by increased fatty acid utilization and decreased glucose utilization (see Table 2-3).75–77 This increased reliance on fatty acids is due to a variety of factors. First, increases in circulating concentrations of free fatty acids and triglycerides in diabetic individuals result in greater uptake and utilization of fatty acids in the cardiac myocyte. While circulating glucose concentrations are also increased in the setting of diabetes, there is down-regulation of GLUT1 and GLUT4 protein expression so that basal glucose uptake is decreased.78 Furthermore, because of the defects in the insulin signaling cascade that are responsible for insulin resistance, the translocation of GLUT4 to the cardiomyocyte surface is blunted, resulting in decreased insulin-stimulated glucose uptake.79,80 In addition to the decreased uptake of glucose in the diabetic heart, the conversion of glucose to pyruvate through the glycolytic pathway and the oxidation of pyruvate are inhibited by the enhanced fatty acid oxidation through the effects of the Randle cycle discussed earlier.
Just as the decreases in glucose utilization are determined in part by changes in protein expression, the enhanced fatty acid metabolism is due to altered expression of genes involved in fatty acid metabolism. Fatty acids regulate the expression of key enzymes involved in fatty acid transport and β-oxidation through PPAR-α-mediated changes in gene expression.58,81 Therefore, in the diabetic patient, the elevated plasma concentrations of free fatty acids increase the expression of PPAR-α-regulated genes.
These alterations in the utilization of fatty acids have been hypothesized to cause lipotoxicity in the diabetic heart and may in part be responsible for the entity of diabetic cardiomyopathy.82 In fact, triglyceride accumulation in the myocardium precedes the development of left ventricular systolic dysfunction in patients with diabetes,83 although it remains to be established if it is triglyceride or another fatty acid metabolite that is responsible for alterations in contractile function. However, measures that normalize myocardial utilization of substrates may be beneficial in preventing changes in cardiac function in patients with diabetes, and studies using PET assessment of glucose utilization have been able to demonstrate improvements in myocardial glucose utilization in diabetic patients in response to insulin, sulfonylureas, and thiazolidinediones.84–87 As with future therapies for heart failure, metabolic imaging may play an important role in identifying diabetic patients at risk of developing cardiomyopathy and may also play a role in guiding therapy and determining the response to metabolic interventions.
Renal Disease
Patients with renal disease are at increased risk for left ventricular dysfunction and cardiac death.88 This is certainly attributable in large part to the development of atherosclerosis due to factors such as the cause of renal impairment (e.g., diabetes) and the development of hypertension in the setting of renal disease. However, renal failure and uremia have also been shown to alter cardiac metabolism in both in vivo and in vitro animal studies,89–92 which may contribute to the cardiac manifestations of renal disease. Recent studies have demonstrated that BMIPP imaging can identify hemodialysis patients who are at high risk for cardiac death.93 Interestingly, this finding extends to hemodialysis patients who have undergone coronary revascularization.94 It was hypothesized that such patients most likely have extensive microvascular disease and repetitive ischemia, but metabolic derangements may also contribute to the abnormalities in fatty acid imaging using BMIPP and to cardiovascular morbidity and mortality.
1. Kates A.M., Herrero P., Dence C., et al. Impact of aging on substrate metabolism by the human heart. J Am Coll Cardiol. 2003;41(2):293-299.
2. Kingsley-Hickman P.B., Sako E.Y., Ugurbil K., et al. 31P NMR measurement of mitochondrial uncoupling in isolated rat hearts. J Biol Chem. 1990;265(3):1545-1550.
3. Taegtmeyer H. Energy metabolism of the heart: From basic concepts to clinical applications. Curr Prob Cardiol. 1994;19:57-116.
4. Schaffer J.E. Fatty acid transport: the roads taken. Am J Physiol. 2002;282(2):E239-E246.
5. Morishita S., Kusuoka H., Yamamichi Y., et al. Kinetics of radioiodinated species in subcellular fractions from rat hearts following administration of iodine-123-labelled 15-(p-iodophenyl)-3-(R,S)-methylpentadecanoic acid (123I-BMIPP). Eur J Nucl Med. 1996;23(4):383-389.
6. Mueckler M. Facilitative glucose transporters. Eur J Biochem. 1994;219(3):713-725.
7. Sun D., Nguyen N., DeGrado T., et al. Ischemia induces translocation of the insulin-responsive glucose transporter GLUT4 to the plasma membrane of cardiac myocytes. Circulation. 1994;89:793-798.
8. Russell R.R., Bergeron R., Shulman G.I., Young L.H. Translocation of myocardial GLUT4 and increased glucose uptake through activation of AMP-activated protein kinase by AICAR. Am J Physiol. 1999;277:H643-H649.
9. Russell R.R.III, Li J., Coven D.L., et al. AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J Clin Invest. 2004;114(4):495-503.
10. Newsholme E., Randle P., Manchester K. Inhibition of the phosphofructokinase reaction in perfused rat heart by respiration of ketone bodies, fatty acids and pyruvate. Nature. 1962;193:270-271.
11. Olson M., Dennis S., DeBuysere M., Padma A. The regulation of pyruvate dehydrogenase in the isolated perfused rat heart. J Biol Chem. 1978;253:7369-7375.
12. Holness M.J., Smith N.D., Bulmer K., et al. Evaluation of the role of peroxisome-proliferator-activated receptor alpha in the regulation of cardiac pyruvate dehydrogenase kinase 4 protein expression in response to starvation, high-fat feeding and hyperthyroidism. Biochem J. 2002;364(Pt 3):687-694.
13. Sugden M.C., Holness M.J. Therapeutic potential of the mammalian pyruvate dehydrogenase kinases in the prevention of hyperglycaemia. Curr Drug Targets Immune Endocr Metabol Disord. 2002;2(2):151-165.
14. Taegtmeyer H., Russell R. Glutamate metabolism in rabbit heart: Augmentation by ischemia and inhibition with acetoacetate. J Appl Cardiol. 1987;2:231-249.
15. Dence C.S., Herrero P., Schwarz S.W., et al. Imaging myocardium enzymatic pathways with carbon-11 radiotracers. Methods Enzymol. 2004;385:286-315.
16. Reivich M., Kuhl D., Wolf A., et al. Measurement of local cerebral glucose metabolism in man with [18-F]-2-fluoro-2-deoxy-D- glucose. Acta Neurol Scand. 1977;56(Suppl 64):190-191.
17. Patlak C., Blasberg R., Fenstermacher J. Graphical evaluation of blood-to-brain transfer constants from multiple-time uptake data. J Cereb Blood Flow Metab. 1983;3:1-7.
18. Russell R., Mrus J., Mommessin J., Taegtmeyer H. Compartmentation of hexokinase in rat heart. A critical factor for tracer kinetic analysis of myocardial glucose metabolism. J Clin Invest. 1992;90:1972-1977.
19. Heintz A.C., Jung C.M., Stehr S.N., et al. Myocardial uptake and biodistribution of newly designed technetium-labelled fatty acid analogues. Nucl Med Commun. 2007;28(8):637-645.
20. Young L.H., Renfu Y., Russell R., et al. Low flow ischemia leads to translocation of canine heart GLUT-4 and GLUT-1 glucose transporters to the sarcolemma in vivo. Circulation. 1997;95:415-422.
21. Egert S., Nguyen N., Brosius F.III, Schwaiger M. Effects of wortmannin on insulin- and ischemia-induced stimulation of GLUT4 translocation and FDG uptake in perfused rat hearts. Cardiovasc Res. 1997;35(2):283-293.
22. Baron S.J., Li J., Russell R.R.III, et al. Dual mechanisms regulating AMPK kinase action in the ischemic heart. Circ Res. 2005;96(3):337-345.
23. Kudo N., Barr A.J., Barr R.L., et al. High rates of fatty acid oxidation during reperfusion of ischemic hearts are associated with a decrease in malonyl-CoA levels due to an increase in 5 ′-AMP-activated protein kinase inhibition of acetyl-CoA carboxylase. J Biol Chem. 1995;270(29):17513-17520.
24. Marsin A.S., Bertrand L., Rider M.H., et al. Phosphorylation and activation of heart PFK-2 by AMPK has a role in the stimulation of glycolysis during ischaemia. Curr Biol. 2000;10(20):1247-1255.
25. Hardie D., Carling D. The AMP-activated protein kinase: Fuel gauge of the mammalian cell? Eur J Biochem. 1997;246:259-273.
26. McNulty P.H., Jagasia D., Cline G.W., et al. Persistent changes in myocardial glucose metabolism in vivo during reperfusion of a limited-duration coronary occlusion. Circulation. 2000;101(8):917-922.
27. Abbott B.G., Liu Y.H., Arrighi J.A. [18F]Fluorodeoxyglucose as a memory marker of transient myocardial ischaemia. Nucl Med Commun. 2007;28(2):89-94.
28. He Z.X., Shi R.F., Wu Y.J., et al. Direct imaging of exercise-induced myocardial ischemia with fluorine-18-labeled deoxyglucose and Tc-99 m-sestamibi in coronary artery disease. Circulation. 2003;108(10):1208-1213.
29. Fujibayashi Y., Yonekura Y., Takemura Y., et al. Myocardial accumulation of iodinated beta-methyl-branched fatty acid analogue, iodine-125–15-(p-iodophenyl)-3-(R,S)methylpentadecanoic acid (BMIPP), in relation to ATP concentration. J Nucl Med. 1990;31(11):1818-1822.
30. Yamamichi Y., Kusuoka H., Morishita K., et al. Metabolism of iodine-123-BMIPP in perfused rat hearts. J Nucl Med. 1995;36(6):1043-1050.
31. Nohara R., Hosokawa R., Hirai T., et al. Basic kinetics of 15-(p-iodophenyl)-3-R,S-methylpentadecanoic acid (BMIPP) in canine myocardium. Int J Card Imaging. 1999;15:11-20.
32. Heathers G.P., Brunt R.V. The effect of coronary artery occlusion and reperfusion on the activities of triglyceride lipase and glycerol 3-phosphate acyl transferase in the isolated perfused rat heart. J Mol Cell Cardiol. 1985;17(9):907-916.
33. Hosokawa R., Nohara R., Fujibayashi Y., et al. Myocardial kinetics of iodine-123-BMIPP in canine myocardium after regional ischemia and reperfusion: implications for clinical SPECT. J Nucl Med. 1997;38:1857-1863.
34. Dilsizian V., Bateman T.M., Bergmann S.R., et al. Metabolic imaging with β-methyl-p-[123I]-iodophenyl-pentadecanoic acid identifies ischemic memory after demand ischemia. Circulation. 2005;112(14):2169-2174.
35. Gerber B.L., Vanoverschelde J.L., Bol A., et al. Myocardial blood flow, glucose uptake, and recruitment of inotropic reserve in chronic left ventricular ischemic dysfunction. Implications for the pathophysiology of chronic myocardial hibernation. Circulation. 1996;94(4):651-659.
36. McFalls E.O., Baldwin D., Palmer B., et al. Regional glucose uptake within hypoperfused swine myocardium as measured by positron emission tomography. Am J Physiol. 1997;272(1 Pt 2):H343-349.
37. Kim S.J., Peppas A., Hong S.K., et al. Persistent stunning induces myocardial hibernation and protection: flow/function and metabolic mechanisms. Circ Res. 2003;92(11):1233-1239.
38. Liedtke A., DeMaison L., Eggelston A., et al. Changes in substrate metabolism and effects of excess fatty acids in reperfused myocardium. Circ Res. 1988;62:535-542.
39. Brosius F.III, Liu Y., Nguyen N., et al. Persistent myocardial ischemia increases GLUT1 glucose transporter expression in both ischemic and non-ischemic heart regions. J Mol Cell Cardiol. 1997;29(6):1675-1685.
40. Hammond G.L., Nadal-Ginard B., Talner N.S., Markert C.L. Myocardial LDH isozyme distribution in the ischemic and hypoxic heart. Circulation. 1976;53(4):637-643.
41. Liedtke A., Lynch M. Alteration of gene expression for glycolytic enzymes in aerobic and ischemic myocardium. Am J Physiol. 1999;277:H1435-H1440.
42. Feldhaus L.M., Liedtke A.J. mRNA expression of glycolytic enzymes and glucose transporter proteins in ischemic myocardium with and without reperfusion. J Mol Cell Cardiol. 1998;30(11):2475-2485.
43. Abbud W., Habinowski S., Zhang J.Z., et al. Stimulation of AMP-activated protein kinase (AMPK) is associated with enhancement of Glut1-mediated glucose transport. Arch Biochem Biophys. 2000;380(2):347-352.
44. Chen C., Pore N., Behrooz A., et al. Regulation of glut1 mRNA by hypoxia-inducible factor-1. Interaction between H-ras and hypoxia. J Biol Chem. 2001;276(12):9519-9525.
45. Hwang J.J., Allen P.D., Tseng G.C., et al. Microarray gene expression profiles in dilated and hypertrophic cardiomyopathic end-stage heart failure. Physiol Genomics. 2002;10(1):31-44.
46. Chen Y., Park S., Li Y., et al. Alterations of gene expression in failing myocardium following left ventricular assist device support. Physiol Genomics. 2003;14(3):251-260.
47. Depré C., Shipley G.L., Chen W., et al. Unloaded heart in vivo replicates fetal gene expression of cardiac hypertrophy. Nat Med. 1998;4(11):1269-1275.
48. Razeghi P., Young M.E., Alcorn J.L., et al. Metabolic gene expression in fetal and failing human heart. Circulation. 2001;104(24):2923-2931.
49. Sack M.N., Rader T.A., Park S., et al. Fatty acid oxidation enzyme gene expression is downregulated in the failing heart. Circulation. 1996;94(11):2837-2842.
50. Davila-Roman V.G., Vedala G., Herrero P., et al. Altered myocardial fatty acid and glucose metabolism in idiopathic dilated cardiomyopathy. J Am Coll Cardiol. 2002;40(2):271-277.
51. Osorio J.C., Stanley W.C., Linke A., et al. Impaired myocardial fatty acid oxidation and reduced protein expression of retinoid X receptor-alpha in pacing-induced heart failure. Circulation. 2002;106(5):606-612.
52. Stanley W.C., Recchia F.A., Lopaschuk G.D. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev. 2005;85(3):1093-1129.
53. Huss J.M., Kelly D.P. Nuclear receptor signaling and cardiac energetics. Circ Res. 2004;95(6):568-578.
54. Lehman J.J., Barger P.M., Kovacs A., et al. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J Clin Invest. 2000;106(7):847-856.
55. Barger P.M., Brandt J.M., Leone T.C., et al. Deactivation of peroxisome proliferator-activated receptor-alpha during cardiac hypertrophic growth. J Clin Invest. 2000;105(12):1723-1730.
56. Garnier A., Fortin D., Delomenie C., et al. Depressed mitochondrial transcription factors and oxidative capacity in rat failing cardiac and skeletal muscles. J Physiol. 2003;551(Pt 2):491-501.
57. Luptak I., Balschi J.A., Xing Y., et al. Decreased contractile and metabolic reserve in peroxisome proliferator-activated receptor-{alpha}-null hearts can be rescued by increasing glucose transport and utilization. Circulation. 2005;112(15):2339-2346.
58. Finck B.N., Han X., Courtois M., et al. A critical role for PPARα-mediated lipotoxicity in the pathogenesis of diabetic cardiomyopathy: Modulation by dietary fat content. Proc Natl Acad Sci U S A. 2003;100(3):1226-1231.
59. Tillisch J., Brunken R., Marshall R., et al. Reversibility of cardiac wall-motion abnormalities predicted by positron tomography. N Engl J Med. 1986;314(14):884-888.
60. Marwick T.H., MacIntyre W.J., Lafont A., et al. Metabolic responses of hibernating and infarcted myocardium to revascularization. A follow-up study of regional perfusion, function, and metabolism. Circulation. 1992;85(4):1347-1353.
61. Tamaki N., Yonekura Y., Yamashita K., et al. Positron emission tomography using fluorine-18 deoxyglucose in evaluation of coronary artery bypass grafting. Am J Cardiol. 1989;64(14):860-865.
62. Gropler R.J., Geltman E.M., Sampathkumaran K., et al. Comparison of carbon-11-acetate with fluorine-18-fluorodeoxyglucose for delineating viable myocardium by positron emission tomography. J Am Coll Cardiol. 1993;22(6):1587-1597.
63. Maes A.F., Borgers M., Flameng W., et al. Assessment of myocardial viability in chronic coronary artery disease using technetium-99m sestamibi SPECT. Correlation with histologic and positron emission tomographic studies and functional follow-up. J Am Coll Cardiol. 1997;29(1):62-68.
64. Tamaki N., Kawamoto M., Tadamura E., et al. Prediction of reversible ischemia after revascularization. Perfusion and metabolic studies with positron emission tomography. Circulation. 1995;91(6):1697-1705.
65. Knuuti M.J., Saraste M., Nuutila P., et al. Myocardial viability: fluorine-18-deoxyglucose positron emission tomography in prediction of wall motion recovery after revascularization. Am Heart J. 1994;127(4 Pt 1):785-796.
66. Baer F.M., Voth E., Deutsch H.J., et al. Predictive value of low dose dobutamine transesophageal echocardiography and fluorine-18 fluorodeoxyglucose positron emission tomography for recovery of regional left ventricular function after successful revascularization. J Am Coll Cardiol. 1996;28(1):60-69.
67. Lucignani G., Paolini G., Landoni C., et al. Presurgical identification of hibernating myocardium by combined use of technetium-99m hexakis 2-methoxyisobutylisonitrile single photon emission tomography and fluorine-18 fluoro-2-deoxy-D-glucose positron emission tomography in patients with coronary artery disease. Eur J Nucl Med. 1992;19(10):874-881.
68. Carrel T., Jenni R., Haubold-Reuter S., et al. Improvement of severely reduced left ventricular function after surgical revascularization in patients with preoperative myocardial infarction. Eur J Cardiothorac Surg. 1992;6(9):479-484.
69. Bengel F.M., Permanetter B., Ungerer M., et al. Non-invasive estimation of myocardial efficiency using positron emission tomography and carbon-11 acetate: comparison between the normal and failing human heart. Eur J Nucl Med. 2000;27(3):319-326.
70. Kronenberg M.W., Cohen G.I., Leonen M.F., et al. Myocardial oxidative metabolic supply-demand relationships in patients with nonischemic dilated cardiomyopathy. J Nucl Cardiol. 2006;13(4):544-553.
71. Lindner O., Sorensen J., Vogt J., et al. Cardiac efficiency and oxygen consumption measured with 11C-acetate PET after long-term cardiac resynchronization therapy. J Nucl Med. 2006;47(3):378-383.
72. Stanley W.C., Chandler M.P. Energy metabolism in the normal and failing heart: potential for therapeutic interventions. Heart Fail Rev. 2002;7(2):115-130.
73. Chandler M.P., Stanley W.C., Morita H., et al. Short-term treatment with ranolazine improves mechanical efficiency in dogs with chronic heart failure. Circ Res. 2002;91(4):278-280.
74. Labinskyy V., Bellomo M., Chandler M.P., et al. Chronic activation of peroxisome proliferator-activated receptor-alpha with fenofibrate prevents alterations in cardiac metabolic phenotype without changing the onset of decompensation in pacing-induced heart failure. J Pharmacol Exp Ther. 2007;321(1):165-171.
75. Herrero P., Peterson L.R., McGill J.B., et al. Increased myocardial fatty acid metabolism in patients with type 1 diabetes mellitus. J Am Coll Cardiol. 2006;47(3):598-604.
76. Chatham J.C., Forder J.R. Relationship between cardiac function and substrate oxidation in hearts of diabetic rats. Am J Physiol. 1997;273(1 Pt 2):H52-58.
77. Belke D.D., Larsen T.S., Gibbs E.M., Severson D.L. Altered metabolism causes cardiac dysfunction in perfused hearts from diabetic (db/db) mice. Am J Physiol. 2000;279(5):E1104-E1113.
78. Depre C., Young M.E., Ying J., et al. Streptozotocin-induced changes in cardiac gene expression in the absence of severe contractile dysfunction. J Mol Cell Cardiol. 2000;32(6):985-996.
79. Fischer Y., Thomas J., Rosen P., Kammermeier H. Action of metformin on glucose transport and glucose transporter GLUT1 and GLUT4 in heart muscle cells from healthy and diabetic rats. Endocrinology. 1995;136(2):412-420.
80. Voipio-Pulkki L.M., Nuutila P., Knuuti M.J., et al. Heart and skeletal muscle glucose disposal in type 2 diabetic patients as determined by positron emission tomography. J Nucl Med. 1993;34(12):2064-2067.
81. Brandt J.M., Djouadi F., Kelly D.P. Fatty acids activate transcription of the muscle carnitine palmitoyltransferase I gene in cardiac myocytes via the peroxisome proliferator-activated receptor alpha. J Biol Chem. 1998;273(37):23786-23792.
82. Young M.E., McNulty P., Taegtmeyer H. Adaptation and maladaptation of the heart in diabetes: Part II: potential mechanisms. Circulation. 2002;105(15):1861-1870.
83. McGavock J.M., Lingvay I., Zib I., et al. Cardiac steatosis in diabetes mellitus: a 1H-magnetic resonance spectroscopy study. Circulation. 2007;116(10):1170-1175.
84. Peterson L.R., Herrero P., McGill J., et al. Fatty acids and insulin modulate myocardial substrate metabolism in humans with type 1 diabetes. Diabetes. 2008;57(1):32-40.
85. Yokoyama I., Inoue Y., Moritan T., et al. Myocardial glucose utilisation in type II diabetes mellitus patients treated with sulphonylurea drugs. Eur J Nucl Med Mol Imaging. 2006;33(6):703-708.
86. Lautamaki R., Airaksinen K.E., Seppanen M., et al. Rosiglitazone improves myocardial glucose uptake in patients with type 2 diabetes and coronary artery disease: a 16-week randomized, double-blind, placebo-controlled study. Diabetes. 2005;54(9):2787-2794.
87. Hallsten K., Virtanen K.A., Lonnqvist F., et al. Enhancement of insulin-stimulated myocardial glucose uptake in patients with Type 2 diabetes treated with rosiglitazone. Diabet Med. 2004;21(12):1280-1287.
88. Parfrey P.S., Foley R.N., Harnett J.D., et al. Outcome and risk factors for left ventricular disorders in chronic uraemia. Nephrol Dial Transplant. 1996;11(7):1277-1285.
89. Scheuer J., Stezoski W. The effects of uremic compounds on cardiac function and metabolism. J Mol Cell Cardiol. 1973;5(3):287-300.
90. Penpargkul S., Scheuer J. Regulation of glycogen metabolism in acute uremic hearts. Metabolism. 1974;23(7):631-644.
91. Williams E.S., Luft F.C. The effect of chronic uremia on fatty acid metabolism in the heart. J Lab Clin Med. 1978;92(4):548-555.
92. Smogorzewski M., Perna A.F., Borum P.R., Massry S.G. Fatty acid oxidation in the myocardium: effects of parathyroid hormone and CRF. Kidney Int. 1988;34(6):797-803.
93. Nishimura M., Tsukamoto K., Hasebe N., et al. Prediction of cardiac death in hemodialysis patients by myocardial fatty acid imaging. J Am Coll Cardiol. 2008;51(2):139-145.
94. Nishimura M., Tokoro T., Nishida M., et al. Myocardial fatty acid imaging identifies a group of hemodialysis patients at high risk for cardiac death after coronary revascularization. Kidney Int. 2008;74(4):513-520.
95. Peterson L.R., Soto P.F., Herrero P., et al. Sex differences in myocardial oxygen and glucose metabolism. J Nucl Cardiol. 2007;14(4):573-581.
96. Soto P.F., Herrero P., Kates A.M., et al. Impact of aging on myocardial metabolic response to dobutamine. Am J Physiol. 2003;285(5):H2158-H2164.
97. de las Fuentes L., Soto P.F., Cupps B.P., et al. Hypertensive left ventricular hypertrophy is associated with abnormal myocardial fatty acid metabolism and myocardial efficiency. J Nucl Cardiol. 2006;13(3):369-377.
98. Peterson L.R., Herrero P., Schechtman K.B., et al. Effect of obesity and insulin resistance on myocardial substrate metabolism and efficiency in young women. Circulation. 2004;109(18):2191-2196.
99. Herrero P., McGill J., Lesniak D.S., et al. PET detection of the impact of dobutamine on myocardial glucose metabolism in women with type 1 diabetes mellitus. J Nucl Cardiol. 2008;15(6):791-799.
[/level-membership-for-internal-medicine-category][not-level-membership-for-internal-medicine-category]
Chapter 2 Principles of Myocardial Metabolism as They Relate to Imaging
INTRODUCTION
The metabolic cost on the heart of maintaining an adequate cardiac output is extremely high. Based on measurements of myocardial oxygen consumption1 and the degree of coupling between oxygen consumption and mitochondrial adenosine triphosphate (ATP) synthesis,2 the heart of a 70-kg individual would produce (and consume) 4.6 kg of ATP per day. As a result, the total content of ATP in the heart turns over approximately 8 times per minute to meet this high rate of flux. While the majority of this ATP expenditure is utilized for contractile activity, ATP is also required for maintaining cellular ionic homeostasis and supporting protein synthesis.
OVERVIEW OF METABOLIC REGULATION IN THE NORMAL HEART
To understand the merits and drawbacks of various metabolic radiotracers, it is important to understand how cardiac metabolism is regulated. As noted above, the heart is an “omnivore,” synthesizing ATP through the metabolism of a variety of fuel substrates.3 Furthermore, the relative contribution of the different substrates varies, and the heart must adapt rapidly to changing sources of substrate. A good example of this plasticity of substrate selection by the heart is reflected in the changes that occur during different physiologic states based on nutritional status and degree of physical activity (Fig. 2-1). The relative contribution of a given substrate to myocardial ATP production is dependent on a variety of selection pressures, including the arterial concentration of the substrate, the availability of oxygen, hormonal stimulation, the workload imposed upon the heart, and the presence of pathologic conditions that affect myocardial utilization of substrates (e.g., coronary artery disease, heart failure, diabetes) through changes in the cardiac myocyte’s expression of regulatory proteins and enzymes.
The metabolism of the primary fuels in the heart is graphically depicted in Fig. 2-2 and illustrates three major phases of metabolism. The first phase is involved in converting fatty acids, glucose, and lactate into a common substrate for entry into the tricarboxylic acid (TCA) cycle in the mitochondria. In the second phase, reducing equivalents in the form of reduced nicotinamide adenine dinucleotide (NADH2) and reduced flavin adenine dinucleotide (FADH2) are produced in the TCA cycle and provide electrons for the electron transport chain that ultimately are used to convert oxygen to water. In the third phase, a proton gradient across the inner mitochondrial membrane, which is generated by the proteins of the electron transport chain, drives ATP synthesis.
Fatty Acid Metabolism
Under normal conditions, fatty acids and triglycerides are the preferred substrate for the normal heart. Fatty acids are taken up by the cardiac myocyte through facilitative transport via fatty acid translocase (FAT/CD36).4 Once inside the myocyte, fatty acids are esterified to fatty acyl-CoA derivatives through a reaction mediated by fatty acyl-CoA synthetase, which utilizes the hydrolysis of ATP to adenosine monophosphate (AMP) to drive the reaction. This energy-requiring step may be of great importance in understanding the decrease in accumulation of the fatty acid analog, [123I]β-methyl-iodophenyl pentadecanoic acid (BMIPP), seen in ischemic myocardium. After this activation, fatty acyl-CoA may be transported into the mitochondria following transesterification with carnitine by carnitine palmitoyltransferase 1 (CPT-1). This step represents the rate-limiting step of fatty acid oxidation and is regulated by cytosolic concentrations of malonyl-CoA; this important regulatory step of fatty acid metabolism is discussed in detail later in the chapter. Once inside the mitochondria, CPT-2 converts the fatty acylcarnitine back into fatty acyl-CoA for entry into the β-oxidation pathway. Another metabolic fate of the cytosolic fatty acyl-CoA is incorporation into triglycerides. It is this fate that predominates for BMIPP because the β-methyl group inhibits it entry into β-oxidation in the mitochondria.5
Glucose Metabolism
Glucose represents the other major fuel of the heart. The initial transport of glucose across the cell surface membrane represents the rate-limiting step of glucose metabolism and is mediated by facilitative glucose transporters (GLUTs).6 Of the 13 described GLUTs, only two, GLUT1 and GLUT4, are expressed to a significant degree in the heart. GLUT1 is present mostly on the cardiomyocyte cell surface and is responsible for basal glucose uptake. In contrast, GLUT4 exists both on the cell surface and in an intracellular pool of membrane vesicles that can translocate to the cell surface in response to insulin (Fig. 2-3). It is therefore GLUT4 translocation that is responsible for insulin-stimulated glucose uptake in the insulin-sensitive tissues of the heart, skeletal muscle, and adipose tissue. The translocation of GLUT4 is also responsible for the enhanced glycolysis observed during ischemia, although the mechanism of this translocation is independent of the insulin signaling pathway as described later in this chapter.7–9
PFK-1 is also inhibited by citrate, which increases when there is sufficient TCA cycle flux to meet the energetic needs of the cell. This inhibition of glycolysis at the level of PFK-1 by ATP and citrate is the basis of a critical aspect of myocardial metabolism that regulates substrate selection: the glucose/fatty acid, or Randle, cycle. The oxidation of fatty acids in the mitochondria results in an increase in both ATP and citrate, which inhibits PFK-1 and thereby reduces glucose uptake.10 The operation of the Randle cycle has important implications with respect to myocardial substrate utilization under physiologic conditions such as the transition from the postprandial state, in which insulin stimulation and abundant circulating glucose lead to increased reliance on glucose, to the fasting state, in which the greater concentration of free fatty acids increases fatty acid metabolism, and disease states such as diabetes, in which there is a persistent increase in the free fatty acid concentration.
Once glucose is metabolized to pyruvate, it is transported into the mitochondria, where it is converted to acetyl-CoA through the action of pyruvate dehydrogenase (PDH). PDH is a multienzyme complex that is regulated by the metabolic status of the cell. Specifically, PDH is inhibited by increased [NADH2]/[NAD] and [acetyl-CoA]/[CoASH] ratios, both of which occur when there is a relative overabundance of NADH2 and acetyl-CoA that outstrips the ability of the mitochondria to utilize these metabolites.11 This regulation of PDH activity is mediated by PDH kinase, which phosphorylates and thereby inactivates the PDH complex. In the setting of enhanced fatty acid oxidation, flux through PDH is inhibited by increased PDH kinase activity,12 providing another level of regulation of substrate selection in the heart. Conversely, PDH activity can be increased by dephosphorylation, which occurs in response to insulin stimulation.13 In addition, PDH can be activated by increases in workload through a calcium-dependent mechanism.
Tricarboxylic Acid Cycle Metabolism and the Electron Transport Chain
β-Oxidation of fatty acids, glycolysis of both exogenous glucose and endogenous glycogen, and uptake of exogenous lactate result in the conversion of these fuels to a common energetic currency, namely acetyl-CoA, which enters the TCA cycle by condensing with oxaloacetate to form citrate. The citrate that is formed undergoes subsequent oxidative and decarboxylating reactions in the TCA cycle, which results in the generation of five important compounds that not only help to drive mitochondrial ATP synthesis but are also important with respect to metabolic imaging. The first is the ultimate conversion of the 6-carbon citrate to the 4-carbon oxaloacetate, which is then available for another “turn” of the TCA. This is linked to the second important product, carbon dioxide (CO2), which is produced through two decarboxylation steps, one mediated by isocitrate dehydrogenase and the other mediated by α-ketoglutarate dehydrogenase. This CO2 is released from the cell and ultimately leaves the body through the lungs. The third is the production of NADH2 by isocitrate dehydrogenase, α-ketoglutarate dehydrogenase, and malate dehydrogenase. This NADH2 is used by the electron transport chain to generate the mitochondrial membrane potential required to power the mitochondrial F0F1-ATPase that converts ADP to ATP (Fig. 2-4). In addition, FADH2 is produced by succinate dehydrogenase and also contributes electrons to the electron transport chain for the conversion of oxygen to water, but because of the location of succinate dehydrogenase in the inner aspect of the inner mitochondrial membrane, it does not contribute to the mitochondrial membrane potential. Finally, the high-energy phosphate, guanosine triphosphate (GTP), is generated through substrate-level phosphorylation by succinyl-CoA synthetase, a reaction that becomes important to energy production during ischemia because, like glycolysis, it does not require oxygen to produce high-energy phosphates.14
Of great importance to the evaluation of myocardial mitochondrial function by nuclear methods is the fact that there is a direct relationship between the entry of acetyl-CoA into the tricarboxylic acid cycle and the conversion of oxygen to water through the electron transport chain. Because of this coupling of acetyl-CoA metabolism to oxygen consumption, it is possible to determine rates of myocardial oxygen consumption noninvasively using the positron emission tomography (PET) tracer [1-11C]-acetate.15 Furthermore, a coupling between energy demand and energy production translates to the coupling of TCA flux to ATP synthesis. Specifically, increases in workload result in an increase in cytosolic calcium; this increased cytosolic calcium concentration increases mitochondrial calcium content, which activates not only PDH but the calcium-dependent enzymes, isocitrate dehydrogenase and α-ketoglutarate dehydrogenase, that are part of the TCA cycle.
METABOLIC TRACERS
Radiotracers of metabolic pathways fall into two categories: those that are radioisotopes of the parent compound (e.g., [1-11C]glucose and [1-11C]palmitate) or those that are analogs of the parent compound (e.g., [2-18F]-2-fluoro-2-deoxyglucose [FDG] and BMIPP). The quantitative evaluation of metabolic pathways generally utilizes the former tracers because they follow the same metabolic fate of the parent compound, whereas the latter compounds are utilized for qualitative assessments of metabolism because they generally are retained by the tissue, making imaging easier. For example, because the PET tracer [1-11C]glucose is biochemically indistinguishable from glucose, it will follow the exact fate of glucose, including the eventual release from the cardiomyocyte as 11CO2. As a result, there is uptake, retention, and ultimately disappearance of radiotracer from the heart (Fig. 2-5). In contrast, FDG is taken up and phosphorylated by hexokinase, but it is not further metabolized in the cardiomyocyte because of the modification of the carbohydrate structure from glucose to deoxyglucose. As a result, FDG becomes trapped in the cell. Kinetic analysis of the time activity curves for FDG can be used to estimate the initial uptake and phosphorylation of glucose,16,17 but it offers no information about the oxidative fate of glucose.
Although the kinetic analysis of a tracer such as FDG that demonstrates irreversible trapping would appear to be more straightforward than the analysis required for tracers that demonstrate accumulation and disappearance, there are two issues that must be kept in mind in translating information gained from irreversibly trapped radiotracers to conclusions about myocardial substrate utilization. First, as demonstrated earlier, these tracers only provide information about a portion of a given metabolic pathway. Second, differences in the structure of the parent compound and the radiotracer will alter the fidelity with which the tracer measures utilization of the parent compound, and this relationship between tracer and tracee can vary under different metabolic conditions.18
In addition to categorizing metabolic radiotracers based on their ability to trace metabolic pathways accurately and completely, they can also be grouped according to whether they are single photon–emitting or positron-emitting radiotracers (Table 2-1
[/not-level-membership-for-internal-medicine-category]
