PART 7: Oncology and Hematology
SECTION 1 |
NEOPLASTIC DISORDERS |
99 |
Approach to the Patient with Cancer |
The application of current treatment techniques (surgery, radiation therapy, chemotherapy, and biologic therapy) results in the cure of nearly two of three patients diagnosed with cancer. Nevertheless, patients experience the diagnosis of cancer as one of the most traumatic and revolutionary events that has ever happened to them. Independent of prognosis, the diagnosis brings with it a change in a person’s self-image and in his or her role in the home and workplace. The prognosis of a person who has just been found to have pancreatic cancer is the same as the prognosis of the person with aortic stenosis who develops the first symptoms of congestive heart failure (median survival, ~8 months). However, the patient with heart disease may remain functional and maintain a self-image as a fully intact person with just a malfunctioning part, a diseased organ (“a bum ticker”). By contrast, the patient with pancreatic cancer has a completely altered self-image and is viewed differently by family and anyone who knows the diagnosis. He or she is being attacked and invaded by a disease that could be anywhere in the body. Every ache or pain takes on desperate significance. Cancer is an exception to the coordinated interaction among cells and organs. In general, the cells of a multicellular organism are programmed for collaboration. Many diseases occur because the specialized cells fail to perform their assigned task. Cancer takes this malfunction one step further. Not only is there a failure of the cancer cell to maintain its specialized function, but it also strikes out on its own; the cancer cell competes to survive using natural mutability and natural selection to seek advantage over normal cells in a recapitulation of evolution. One consequence of the traitorous behavior of cancer cells is that the patient feels betrayed by his or her body. The cancer patient feels that he or she, and not just a body part, is diseased.
THE MAGNITUDE OF THE PROBLEM
No nationwide cancer registry exists; therefore, the incidence of cancer is estimated on the basis of the National Cancer Institute’s Surveillance, Epidemiology, and End Results (SEER) database, which tabulates cancer incidence and death figures from 13 sites, accounting for about 10% of the U.S. population, and from population data from the U.S. Census Bureau. In 2014, 1.665 million new cases of invasive cancer (855,220 men, 810,320 women) were diagnosed, and 585,720 persons (310,010 men, 275,710 women) died from cancer. The percent distribution of new cancer cases and cancer deaths by site for men and women is shown in Table 99-1. Cancer incidence has been declining by about 2% each year since 1992. Cancer is the cause of one in four deaths in the United States.
|
DISTRIBUTION OF CANCER INCIDENCE AND DEATHS FOR 2014 |
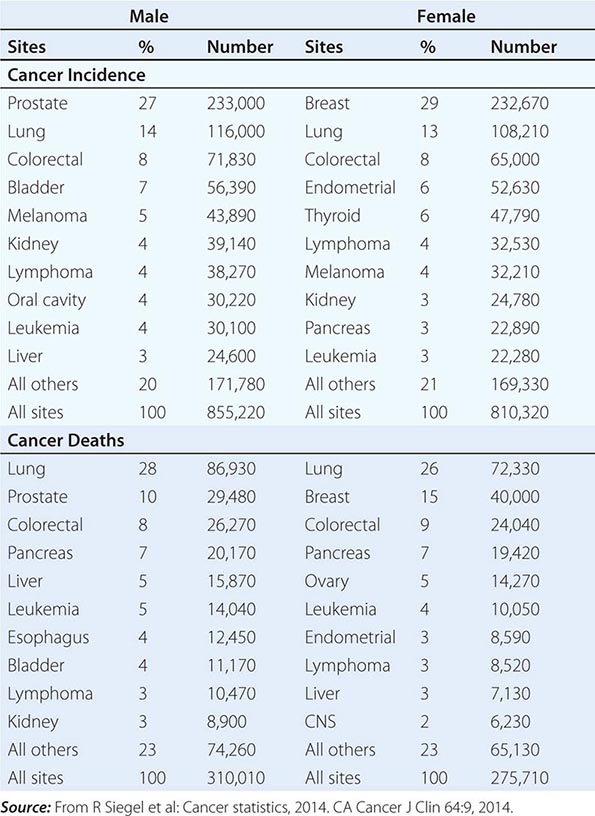
The most significant risk factor for cancer overall is age; two-thirds of all cases were in those older than age 65 years. Cancer incidence increases as the third, fourth, or fifth power of age in different sites. For the interval between birth and age 49 years, 1 in 29 men and 1 in 19 women will develop cancer; for the interval between ages 50 and 59 years, 1 in 15 men and 1 in 17 women will develop cancer; for the interval between ages 60 and 69 years, 1 in 6 men and 1 in 10 women will develop cancer; and for people age 70 and older, 1 in 3 men and 1 in 4 women will develop cancer. Overall, men have a 44% risk of developing cancer at some time during their lives; women have a 38% lifetime risk.
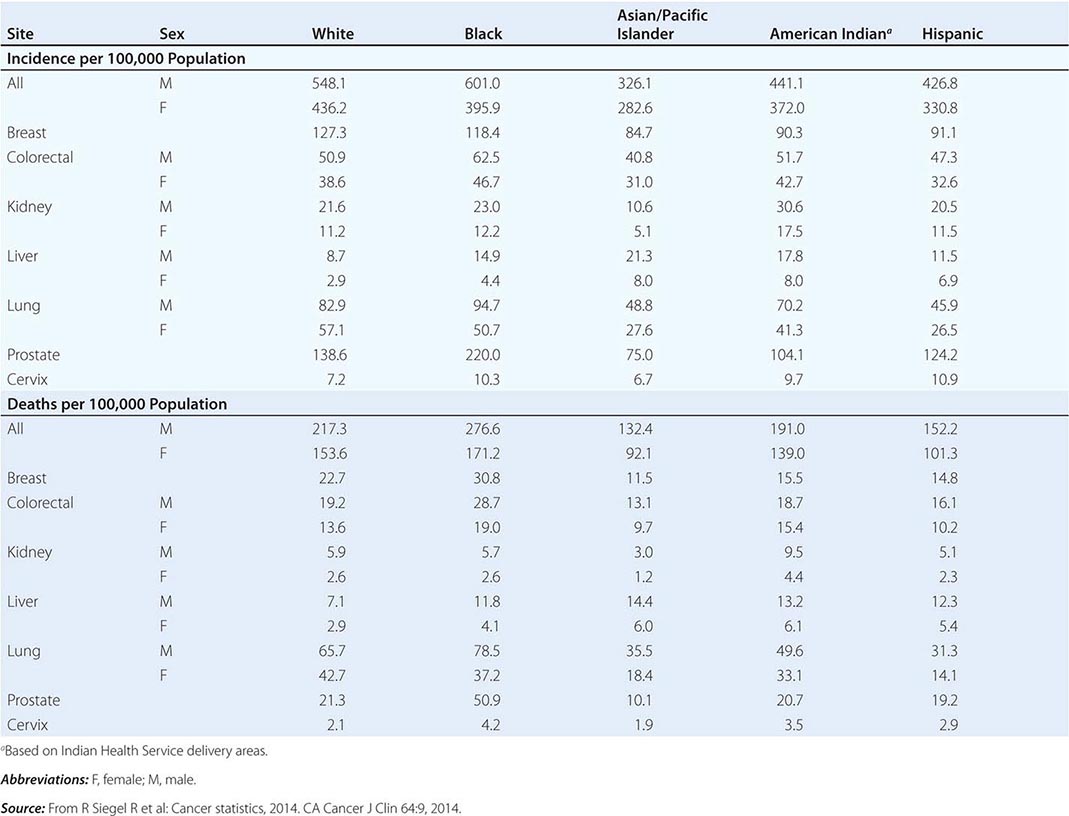
Cancer is the second leading cause of death behind heart disease. Deaths from heart disease have declined 45% in the United States since 1950 and continue to decline. Cancer has overtaken heart disease as the number one cause of death in persons younger than age 85 years. Incidence trends over time are shown in Fig. 99-1. After a 70-year period of increase, cancer deaths began to decline in 1990–1991 (Fig. 99-2). Between 1990 and 2010, cancer deaths decreased by 21% among men and 12.3% among women. The magnitude of the decline is illustrated in Fig. 99-3. The five leading causes of cancer deaths are shown for various populations in Table 99-2. The 5-year survival for white patients was 39% in 1960–1963 and 69% in 2003–2009. Cancers are more often deadly in blacks; the 5-year survival was 61% for the 2003–2009 interval; however, the racial differences are narrowing over time. Incidence and mortality vary among racial and ethnic groups (Table 99-3). The basis for these differences is unclear.
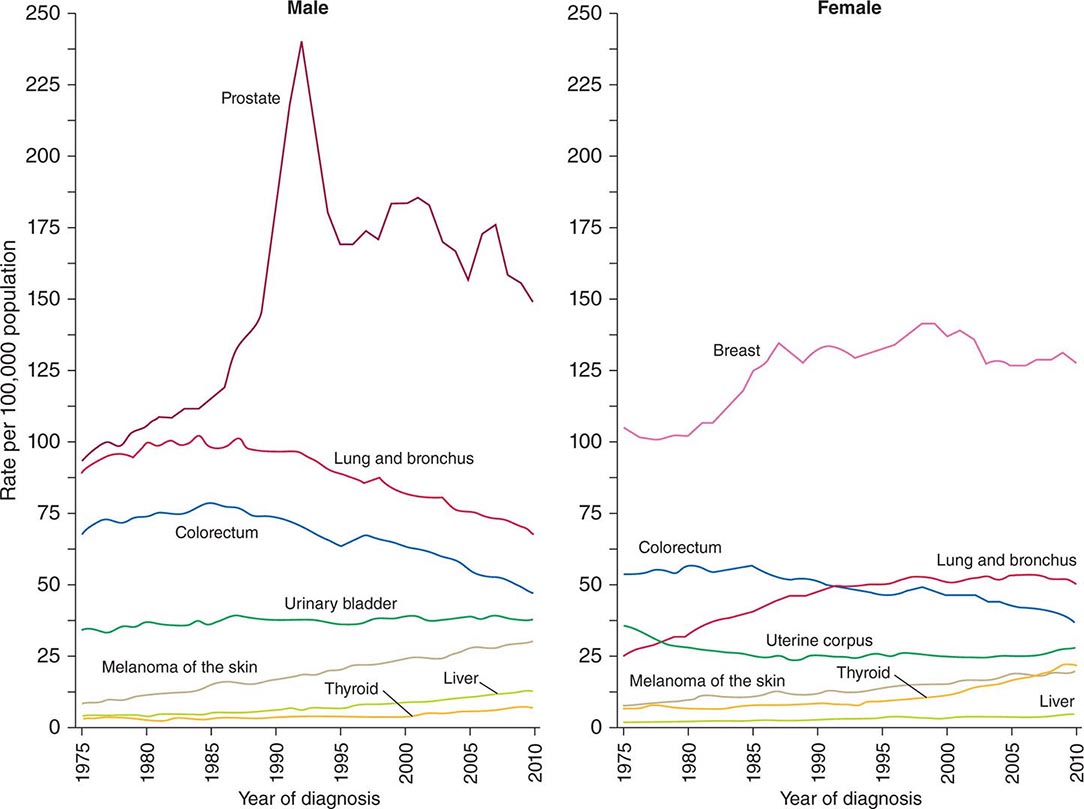
FIGURE 99-1 Incidence rates for particular types of cancer over the last 35 years in men (A) and women (B). (From R Siegel et al: CA Cancer J Clin 64:9, 2014.)
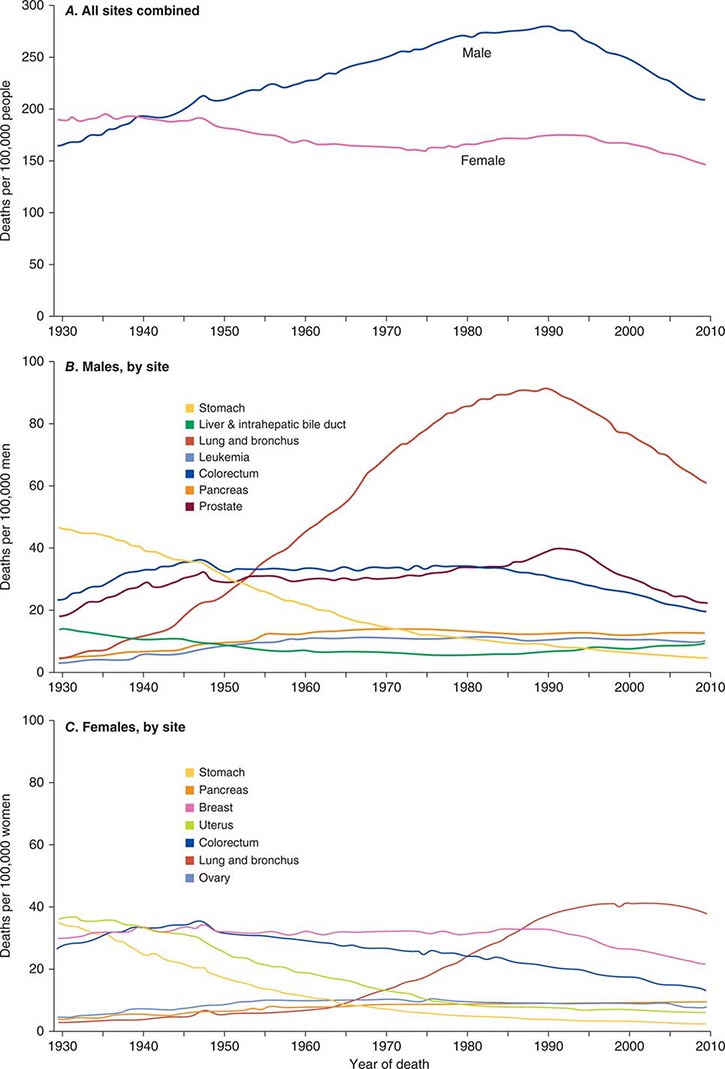
FIGURE 99-2 Eighty-year trend in cancer death rates for (A) women and (B) men by site in the United States, 1930–2010. Rates are per 100,000 age-adjusted to the 2000 U.S. standard population. All sites combined (A), individual sites in men (B) and individual sites in women (C) are shown. (From R Siegel et al: CA Cancer J Clin 64:9, 2014.)
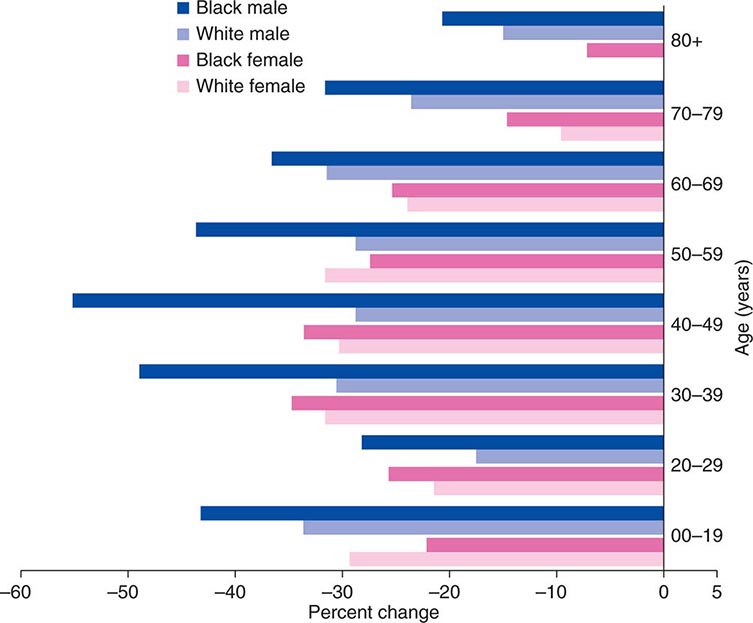
FIGURE 99-3 The decline in death rates from cancer is shown for different age ranges by sex and race for the 20-year period between 1991 and 2010 expressed as a percentage of the 1991 rate. (From R Siegel et al: CA Cancer J Clin 64:9, 2014.)
|
THE FIVE LEADING PRIMARY TUMOR SITES FOR PATIENTS DYING OF CANCER BASED ON AGE AND SEX IN 2010 |
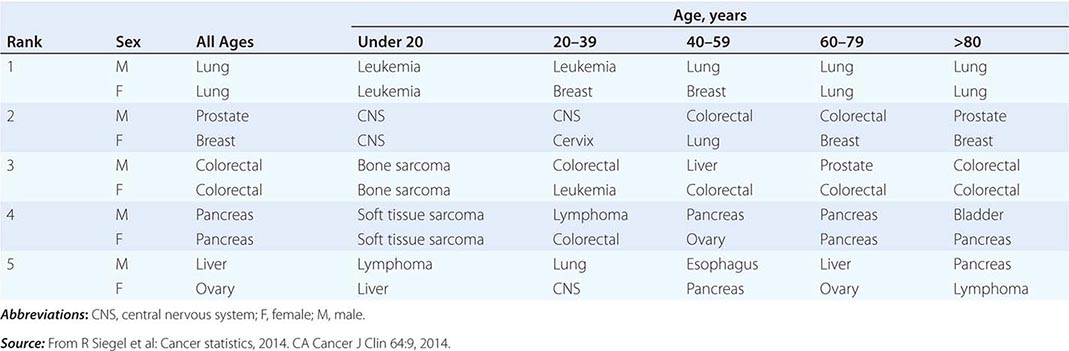
|
CANCER INCIDENCE AND MORTALITY IN RACIAL AND ETHNIC GROUPS, UNITED STATES, 2006–2010 |
CANCER AROUND THE WORLD
![]() In 2008, 12.7 million new cancer cases and 7.6 million cancer deaths were estimated worldwide, according to estimates of GLOBOCAN 2008, developed by the International Agency for Research on Cancer (IARC). When broken down by region of the world, ~45% of cases were in Asia, 26% in Europe, 14.5% in North America, 7.1% in Central/South America, 6% in Africa, and 1% in Australia/New Zealand (Fig. 99-4). Lung cancer is the most common cancer and the most common cause of cancer death in the world. Its incidence is highly variable, affecting only 2 per 100,000 African women but as many as 61 per 100,000 North American men. Breast cancer is the second most common cancer worldwide; however, it ranks fifth as a cause of death behind lung, stomach, liver, and colorectal cancer. Among the eight most common forms of cancer, lung (2-fold), breast (3-fold), prostate (2.5-fold), and colorectal (3-fold) cancers are more common in more developed countries than in less developed countries. By contrast, liver (2-fold), cervical (2-fold), and esophageal (2- to 3-fold) cancers are more common in less developed countries. Stomach cancer incidence is similar in more and less developed countries but is much more common in Asia than North America or Africa. The most common cancers in Africa are cervical, breast, and liver cancers. It has been estimated that nine modifiable risk factors are responsible for more than one-third of cancers worldwide. These include smoking, alcohol consumption, obesity, physical inactivity, low fruit and vegetable consumption, unsafe sex, air pollution, indoor smoke from household fuels, and contaminated injections.
In 2008, 12.7 million new cancer cases and 7.6 million cancer deaths were estimated worldwide, according to estimates of GLOBOCAN 2008, developed by the International Agency for Research on Cancer (IARC). When broken down by region of the world, ~45% of cases were in Asia, 26% in Europe, 14.5% in North America, 7.1% in Central/South America, 6% in Africa, and 1% in Australia/New Zealand (Fig. 99-4). Lung cancer is the most common cancer and the most common cause of cancer death in the world. Its incidence is highly variable, affecting only 2 per 100,000 African women but as many as 61 per 100,000 North American men. Breast cancer is the second most common cancer worldwide; however, it ranks fifth as a cause of death behind lung, stomach, liver, and colorectal cancer. Among the eight most common forms of cancer, lung (2-fold), breast (3-fold), prostate (2.5-fold), and colorectal (3-fold) cancers are more common in more developed countries than in less developed countries. By contrast, liver (2-fold), cervical (2-fold), and esophageal (2- to 3-fold) cancers are more common in less developed countries. Stomach cancer incidence is similar in more and less developed countries but is much more common in Asia than North America or Africa. The most common cancers in Africa are cervical, breast, and liver cancers. It has been estimated that nine modifiable risk factors are responsible for more than one-third of cancers worldwide. These include smoking, alcohol consumption, obesity, physical inactivity, low fruit and vegetable consumption, unsafe sex, air pollution, indoor smoke from household fuels, and contaminated injections.
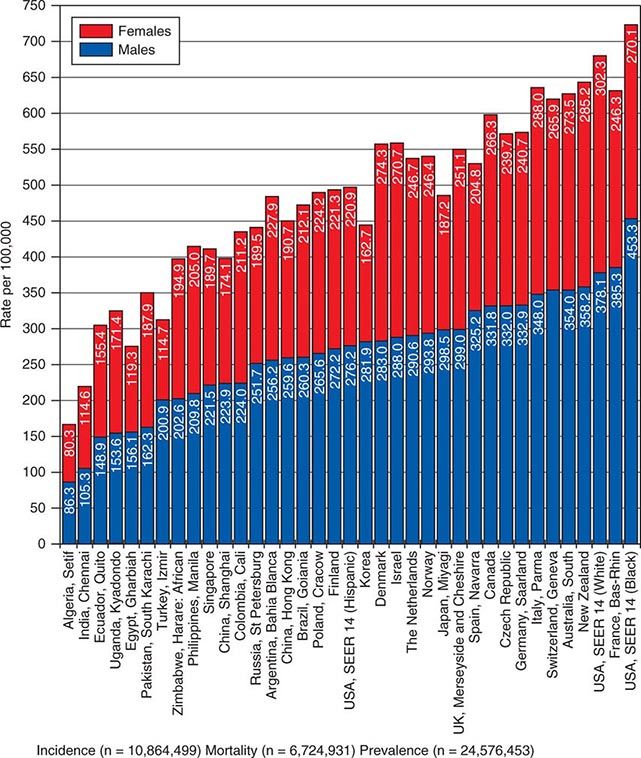
FIGURE 99-4 Worldwide overall annual cancer incidence, mortality, and 5-year prevalence for the period of 1993–2001. (Adapted from A Jemal et al: Cancer Epidemiol Biomarkers Prev 19:1893, 2010.)
PATIENT MANAGEMENT
Important information is obtained from every portion of the routine history and physical examination. The duration of symptoms may reveal the chronicity of disease. The past medical history may alert the physician to the presence of underlying diseases that may affect the choice of therapy or the side effects of treatment. The social history may reveal occupational exposure to carcinogens or habits, such as smoking or alcohol consumption, that may influence the course of disease and its treatment. The family history may suggest an underlying familial cancer predisposition and point out the need to begin surveillance or other preventive therapy for unaffected siblings of the patient. The review of systems may suggest early symptoms of metastatic disease or a paraneoplastic syndrome.
DIAGNOSIS
The diagnosis of cancer relies most heavily on invasive tissue biopsy and should never be made without obtaining tissue; no noninvasive diagnostic test is sufficient to define a disease process as cancer. Although in rare clinical settings (e.g., thyroid nodules), fine-needle aspiration is an acceptable diagnostic procedure, the diagnosis generally depends on obtaining adequate tissue to permit careful evaluation of the histology of the tumor, its grade, and its invasiveness and to yield further molecular diagnostic information, such as the expression of cell-surface markers or intracellular proteins that typify a particular cancer, or the presence of a molecular marker, such as the t(8;14) translocation of Burkitt’s lymphoma. Increasing evidence links the expression of certain genes with the prognosis and response to therapy (Chaps. 101e and 102e).
Occasionally a patient will present with a metastatic disease process that is defined as cancer on biopsy but has no apparent primary site of disease. Efforts should be made to define the primary site based on age, sex, sites of involvement, histology and tumor markers, and personal and family history. Particular attention should be focused on ruling out the most treatable causes (Chap. 120e).
Once the diagnosis of cancer is made, the management of the patient is best undertaken as a multidisciplinary collaboration among the primary care physician, medical oncologists, surgical oncologists, radiation oncologists, oncology nurse specialists, pharmacists, social workers, rehabilitation medicine specialists, and a number of other consulting professionals working closely with each other and with the patient and family.
DEFINING THE EXTENT OF DISEASE AND THE PROGNOSIS
The first priority in patient management after the diagnosis of cancer is established and shared with the patient is to determine the extent of disease. The curability of a tumor usually is inversely proportional to the tumor burden. Ideally, the tumor will be diagnosed before symptoms develop or as a consequence of screening efforts (Chap. 100). A very high proportion of such patients can be cured. However, most patients with cancer present with symptoms related to the cancer, caused either by mass effects of the tumor or by alterations associated with the production of cytokines or hormones by the tumor.
For most cancers, the extent of disease is evaluated by a variety of noninvasive and invasive diagnostic tests and procedures. This process is called staging. There are two types. Clinical staging is based on physical examination, radiographs, isotopic scans, computed tomography (CT) scans, and other imaging procedures; pathologic staging takes into account information obtained during a surgical procedure, which might include intraoperative palpation, resection of regional lymph nodes and/or tissue adjacent to the tumor, and inspection and biopsy of organs commonly involved in disease spread. Pathologic staging includes histologic examination of all tissues removed during the surgical procedure. Surgical procedures performed may include a simple lymph node biopsy or more extensive procedures such as thoracotomy, mediastinoscopy, or laparotomy. Surgical staging may occur in a separate procedure or may be done at the time of definitive surgical resection of the primary tumor.
Knowledge of the predilection of particular tumors for spreading to adjacent or distant organs helps direct the staging evaluation.
Information obtained from staging is used to define the extent of disease as localized, as exhibiting spread outside of the organ of origin to regional but not distant sites, or as metastatic to distant sites. The most widely used system of staging is the TNM (tumor, node, metastasis) system codified by the International Union Against Cancer and the American Joint Committee on Cancer. The TNM classification is an anatomically based system that categorizes the tumor on the basis of the size of the primary tumor lesion (T1–4, where a higher number indicates a tumor of larger size), the presence of nodal involvement (usually N0 and N1 for the absence and presence, respectively, of involved nodes, although some tumors have more elaborate systems of nodal grading), and the presence of metastatic disease (M0 and M1 for the absence and presence, respectively, of metastases). The various permutations of T, N, and M scores (sometimes including tumor histologic grade [G]) are then broken into stages, usually designated by the roman numerals I through IV. Tumor burden increases and curability decreases with increasing stage. Other anatomic staging systems are used for some tumors, e.g., the Dukes classification for colorectal cancers, the International Federation of Gynecologists and Obstetricians classification for gynecologic cancers, and the Ann Arbor classification for Hodgkin’s disease.
Certain tumors cannot be grouped on the basis of anatomic considerations. For example, hematopoietic tumors such as leukemia, myeloma, and lymphoma are often disseminated at presentation and do not spread like solid tumors. For these tumors, other prognostic factors have been identified (Chaps. 132–136).
In addition to tumor burden, a second major determinant of treatment outcome is the physiologic reserve of the patient. Patients who are bedridden before developing cancer are likely to fare worse, stage for stage, than fully active patients. Physiologic reserve is a determinant of how a patient is likely to cope with the physiologic stresses imposed by the cancer and its treatment. This factor is difficult to assess directly. Instead, surrogate markers for physiologic reserve are used, such as the patient’s age or Karnofsky performance status (Table 99-4) or Eastern Cooperative Oncology Group (ECOG) performance status (Table 99-5). Older patients and those with a Karnofsky performance status <70 or ECOG performance status ≥3 have a poor prognosis unless the poor performance is a reversible consequence of the tumor.
|
KARNOFSKY PERFORMANCE INDEX |
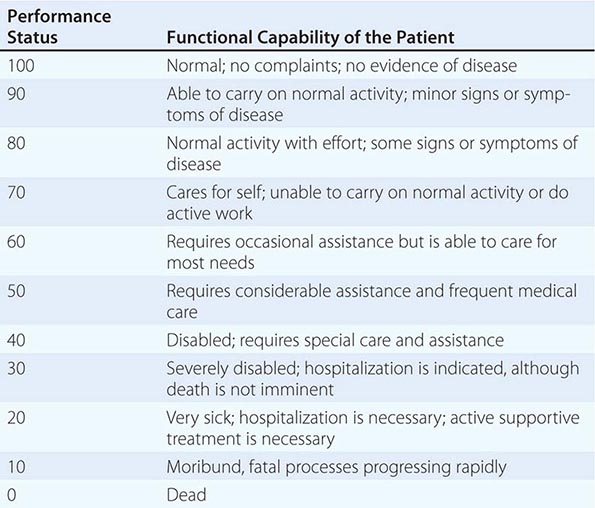
|
THE EASTERN COOPERATIVE ONCOLOGY GROUP (ECOG) PERFORMANCE SCALE |
Source: From MM Oken et al: Am J Clin Oncol 5:649, 1982.
Increasingly, biologic features of the tumor are being related to prognosis. The expression of particular oncogenes, drug-resistance genes, apoptosis-related genes, and genes involved in metastasis is being found to influence response to therapy and prognosis. The presence of selected cytogenetic abnormalities may influence survival. Tumors with higher growth fractions, as assessed by expression of proliferation-related markers such as proliferating cell nuclear antigen, behave more aggressively than tumors with lower growth fractions. Information obtained from studying the tumor itself will increasingly be used to influence treatment decisions. Host genes involved in drug metabolism can influence the safety and efficacy of particular treatments.
Enormous heterogeneity has been noted by studying tumors; we have learned that morphology is not capable of discerning certain distinct subsets of patients whose tumors have different sets of abnormalities. Tumors that look the same by light microscopy can be very different. Similarly, tumors that look quite different from one another histologically can share genetic lesions that predict responses to treatments. Furthermore, tumor cells vary enormously within a single patient even though the cells share a common origin.
MAKING A TREATMENT PLAN
From information on the extent of disease and the prognosis and in conjunction with the patient’s wishes, it is determined whether the treatment approach should be curative or palliative in intent. Cooperation among the various professionals involved in cancer treatment is of the utmost importance in treatment planning. For some cancers, chemotherapy or chemotherapy plus radiation therapy delivered before the use of definitive surgical treatment (so-called neoadjuvant therapy) may improve the outcome, as seems to be the case for locally advanced breast cancer and head and neck cancers. In certain settings in which combined-modality therapy is intended, coordination among the medical oncologist, radiation oncologist, and surgeon is crucial to achieving optimal results. Sometimes the chemotherapy and radiation therapy need to be delivered sequentially, and other times concurrently. Surgical procedures may precede or follow other treatment approaches. It is best for the treatment plan either to follow a standard protocol precisely or else to be part of an ongoing clinical research protocol evaluating new treatments. Ad hoc modifications of standard protocols are likely to compromise treatment results.
The choice of treatment approaches was formerly dominated by the local culture in both the university and the practice settings. However, it is now possible to gain access electronically to standard treatment protocols and to every approved clinical research study in North America through a personal computer interface with the Internet.1
The skilled physician also has much to offer the patient for whom curative therapy is no longer an option. Often a combination of guilt and frustration over the inability to cure the patient and the pressure of a busy schedule greatly limit the time a physician spends with a patient who is receiving only palliative care. Resist these forces. In addition to the medicines administered to alleviate symptoms (see below), it is important to remember the comfort that is provided by holding the patient’s hand, continuing regular examinations, and taking time to talk.
MANAGEMENT OF DISEASE AND TREATMENT COMPLICATIONS
Because cancer therapies are toxic (Chap. 103e), patient management involves addressing complications of both the disease and its treatment as well as the complex psychosocial problems associated with cancer. In the short term during a course of curative therapy, the patient’s functional status may decline. Treatment-induced toxicity is less acceptable if the goal of therapy is palliation. The most common side effects of treatment are nausea and vomiting (see below), febrile neutropenia (Chap. 104), and myelosuppression (Chap. 103e). Tools are now available to minimize the acute toxicity of cancer treatment.
New symptoms developing in the course of cancer treatment should always be assumed to be reversible until proven otherwise. The fatalistic attribution of anorexia, weight loss, and jaundice to recurrent or progressive tumor could result in a patient dying from a reversible intercurrent cholecystitis. Intestinal obstruction may be due to reversible adhesions rather than progressive tumor. Systemic infections, sometimes with unusual pathogens, may be a consequence of the immunosuppression associated with cancer therapy. Some drugs used to treat cancer or its complications (e.g., nausea) may produce central nervous system symptoms that look like metastatic disease or may mimic paraneoplastic syndromes such as the syndrome of inappropriate antidiuretic hormone. A definitive diagnosis should be pursued and may even require a repeat biopsy.
A critical component of cancer management is assessing the response to treatment. In addition to a careful physical examination in which all sites of disease are physically measured and recorded in a flow chart by date, response assessment usually requires periodic repeating of imaging tests that were abnormal at the time of staging. If imaging tests have become normal, repeat biopsy of previously involved tissue is performed to document complete response by pathologic criteria. Biopsies are not usually required if there is macroscopic residual disease. A complete response is defined as disappearance of all evidence of disease, and a partial response as >50% reduction in the sum of the products of the perpendicular diameters of all measurable lesions. The determination of partial response may also be based on a 30% decrease in the sums of the longest diameters of lesions (Response Evaluation Criteria in Solid Tumors [RECIST]). Progressive disease is defined as the appearance of any new lesion or an increase of >25% in the sum of the products of the perpendicular diameters of all measurable lesions (or an increase of 20% in the sums of the longest diameters by RECIST). Tumor shrinkage or growth that does not meet any of these criteria is considered stable disease. Some sites of involvement (e.g., bone) or patterns of involvement (e.g., lymphangitic lung or diffuse pulmonary infiltrates) are considered unmeasurable. No response is complete without biopsy documentation of their resolution, but partial responses may exclude their assessment unless clear objective progression has occurred.
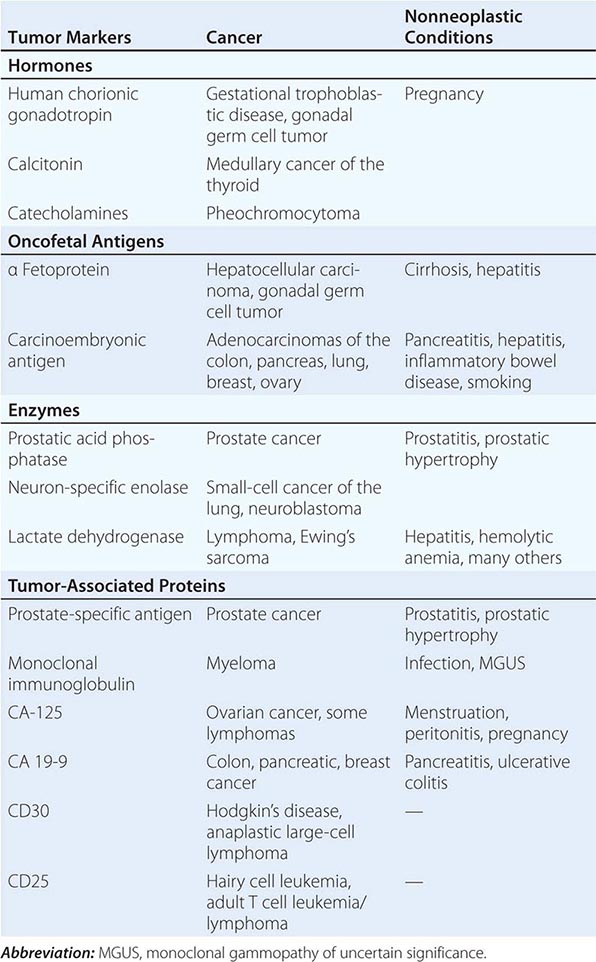
Tumor markers may be useful in patient management in certain tumors. Response to therapy may be difficult to gauge with certainty. However, some tumors produce or elicit the production of markers that can be measured in the serum or urine, and in a particular patient, rising and falling levels of the marker are usually associated with increasing or decreasing tumor burden, respectively. Some clinically useful tumor markers are shown in Table 99-6. Tumor markers are not in themselves specific enough to permit a diagnosis of malignancy to be made, but once a malignancy has been diagnosed and shown to be associated with elevated levels of a tumor marker, the marker can be used to assess response to treatment.
|
TUMOR MARKERS |
The recognition and treatment of depression are important components of management. The incidence of depression in cancer patients is ~25% overall and may be greater in patients with greater debility. This diagnosis is likely in a patient with a depressed mood (dysphoria) and/or a loss of interest in pleasure (anhedonia) for at least 2 weeks. In addition, three or more of the following symptoms are usually present: appetite change, sleep problems, psychomotor retardation or agitation, fatigue, feelings of guilt or worthlessness, inability to concentrate, and suicidal ideation. Patients with these symptoms should receive therapy. Medical therapy with a serotonin reuptake inhibitor such as fluoxetine (10–20 mg/d), sertraline (50–150 mg/d), or paroxetine (10–20 mg/d) or a tricyclic antidepressant such as amitriptyline (50–100 mg/d) or desipramine (75–150 mg/d) should be tried, allowing 4–6 weeks for response. Effective therapy should be continued at least 6 months after resolution of symptoms. If therapy is unsuccessful, other classes of antidepressants may be used. In addition to medication, psychosocial interventions such as support groups, psychotherapy, and guided imagery may be of benefit.
Many patients opt for unproven or unsound approaches to treatment when it appears that conventional medicine is unlikely to be curative. Those seeking such alternatives are often well educated and may be early in the course of their disease. Unsound approaches are usually hawked on the basis of unsubstantiated anecdotes and not only cannot help the patient but may be harmful. Physicians should strive to keep communications open and nonjudgmental, so that patients are more likely to discuss with the physician what they are actually doing. The appearance of unexpected toxicity may be an indication that a supplemental therapy is being taken.2
LONG-TERM FOLLOW-UP/LATE COMPLICATIONS
At the completion of treatment, sites originally involved with tumor are reassessed, usually by radiography or imaging techniques, and any persistent abnormality is biopsied. If disease persists, the multidisciplinary team discusses a new salvage treatment plan. If the patient has been rendered disease-free by the original treatment, the patient is followed regularly for disease recurrence. The optimal guidelines for follow-up care are not known. For many years, a routine practice has been to follow the patient monthly for 6–12 months, then every other month for a year, every 3 months for a year, every 4 months for a year, every 6 months for a year, and then annually. At each visit, a battery of laboratory and radiographic and imaging tests were obtained on the assumption that it is best to detect recurrent disease before it becomes symptomatic. However, where follow-up procedures have been examined, this assumption has been found to be untrue. Studies of breast cancer, melanoma, lung cancer, colon cancer, and lymphoma have all failed to support the notion that asymptomatic relapses are more readily cured by salvage therapy than symptomatic relapses. In view of the enormous cost of a full battery of diagnostic tests and their manifest lack of impact on survival, new guidelines are emerging for less frequent follow-up visits, during which the history and physical examination are the major investigations performed.
As time passes, the likelihood of recurrence of the primary cancer diminishes. For many types of cancer, survival for 5 years without recurrence is tantamount to cure. However, important medical problems can occur in patients treated for cancer and must be examined (Chap. 125). Some problems emerge as a consequence of the disease and some as a consequence of the treatment. An understanding of these disease- and treatment-related problems may help in their detection and management.
Despite these concerns, most patients who are cured of cancer return to normal lives.
SUPPORTIVE CARE
In many ways, the success of cancer therapy depends on the success of the supportive care. Failure to control the symptoms of cancer and its treatment may lead patients to abandon curative therapy. Of equal importance, supportive care is a major determinant of quality of life. Even when life cannot be prolonged, the physician must strive to preserve its quality. Quality-of-life measurements have become common endpoints of clinical research studies. Furthermore, palliative care has been shown to be cost-effective when approached in an organized fashion. A credo for oncology could be to cure sometimes, to extend life often, and to comfort always.
Pain Pain occurs with variable frequency in the cancer patient: 25–50% of patients present with pain at diagnosis, 33% have pain associated with treatment, and 75% have pain with progressive disease. The pain may have several causes. In ~70% of cases, pain is caused by the tumor itself—by invasion of bone, nerves, blood vessels, or mucous membranes or obstruction of a hollow viscus or duct. In ~20% of cases, pain is related to a surgical or invasive medical procedure, to radiation injury (mucositis, enteritis, or plexus or spinal cord injury), or to chemotherapy injury (mucositis, peripheral neuropathy, phlebitis, steroid-induced aseptic necrosis of the femoral head). In 10% of cases, pain is unrelated to cancer or its treatment.
Assessment of pain requires the methodical investigation of the history of the pain, its location, character, temporal features, provocative and palliative factors, and intensity (Chap. 18); a review of the oncologic history and past medical history as well as personal and social history; and a thorough physical examination. The patient should be given a 10-division visual analogue scale on which to indicate the severity of the pain. The clinical condition is often dynamic, making it necessary to reassess the patient frequently. Pain therapy should not be withheld while the cause of pain is being sought.
A variety of tools are available with which to address cancer pain. About 85% of patients will have pain relief from pharmacologic intervention. However, other modalities, including antitumor therapy (such as surgical relief of obstruction, radiation therapy, and strontium-89 or samarium-153 treatment for bone pain), neurostimulatory techniques, regional analgesia, or neuroablative procedures, are effective in an additional 12% or so. Thus, very few patients will have inadequate pain relief if appropriate measures are taken. A specific approach to pain relief is detailed in Chap. 10.
Nausea Emesis in the cancer patient is usually caused by chemotherapy (Chap. 103e). Its severity can be predicted from the drugs used to treat the cancer. Three forms of emesis are recognized on the basis of their timing with regard to the noxious insult. Acute emesis, the most common variety, occurs within 24 h of treatment. Delayed emesis occurs 1–7 days after treatment; it is rare, but, when present, usually follows cisplatin administration. Anticipatory emesis occurs before administration of chemotherapy and represents a conditioned response to visual and olfactory stimuli previously associated with chemotherapy delivery.
Acute emesis is the best understood form. Stimuli that activate signals in the chemoreceptor trigger zone in the medulla, the cerebral cortex, and peripherally in the intestinal tract lead to stimulation of the vomiting center in the medulla, the motor center responsible for coordinating the secretory and muscle contraction activity that leads to emesis. Diverse receptor types participate in the process, including dopamine, serotonin, histamine, opioid, and acetylcholine receptors. The serotonin receptor antagonists ondansetron and granisetron are the most effective drugs against highly emetogenic agents, but they are expensive.
As with the analgesia ladder, emesis therapy should be tailored to the situation. For mildly and moderately emetogenic agents, prochlorperazine, 5–10 mg PO or 25 mg PR, is effective. Its efficacy may be enhanced by administering the drug before the chemotherapy is delivered. Dexamethasone, 10–20 mg IV, is also effective and may enhance the efficacy of prochlorperazine. For highly emetogenic agents such as cisplatin, mechlorethamine, dacarbazine, and streptozocin, combinations of agents work best and administration should begin 6–24 h before treatment. Ondansetron, 8 mg PO every 6 h the day before therapy and IV on the day of therapy, plus dexamethasone, 20 mg IV before treatment, is an effective regimen. Addition of oral aprepitant (a substance P/neurokinin 1 receptor antagonist) to this regimen (125 mg on day 1, 80 mg on days 2 and 3) further decreases the risk of both acute and delayed vomiting. Like pain, emesis is easier to prevent than to alleviate.
Delayed emesis may be related to bowel inflammation from the therapy and can be controlled with oral dexamethasone and oral metoclopramide, a dopamine receptor antagonist that also blocks serotonin receptors at high dosages. The best strategy for preventing anticipatory emesis is to control emesis in the early cycles of therapy to prevent the conditioning from taking place. If this is unsuccessful, prophylactic antiemetics the day before treatment may help. Experimental studies are evaluating behavior modification.
Effusions Fluid may accumulate abnormally in the pleural cavity, pericardium, or peritoneum. Asymptomatic malignant effusions may not require treatment. Symptomatic effusions occurring in tumors responsive to systemic therapy usually do not require local treatment but respond to the treatment for the underlying tumor. Symptomatic effusions occurring in tumors unresponsive to systemic therapy may require local treatment in patients with a life expectancy of at least 6 months.
Pleural effusions due to tumors may or may not contain malignant cells. Lung cancer, breast cancer, and lymphomas account for ~75% of malignant pleural effusions. Their exudative nature is usually gauged by an effusion/serum protein ratio of ≥0.5 or an effusion/serum lactate dehydrogenase ratio of ≥0.6. When the condition is symptomatic, thoracentesis is usually performed first. In most cases, symptomatic improvement occurs for <1 month. Chest tube drainage is required if symptoms recur within 2 weeks. Fluid is aspirated until the flow rate is <100 mL in 24 h. Then either 60 units of bleomycin or 1 g of doxycycline is infused into the chest tube in 50 mL of 5% dextrose in water; the tube is clamped; the patient is rotated on four sides, spending 15 min in each position; and, after 1–2 h, the tube is again attached to suction for another 24 h. The tube is then disconnected from suction and allowed to drain by gravity. If <100 mL drains over the next 24 h, the chest tube is pulled, and a radiograph is taken 24 h later. If the chest tube continues to drain fluid at an unacceptably high rate, sclerosis can be repeated. Bleomycin may be somewhat more effective than doxycycline but is very expensive. Doxycycline is usually the drug of first choice. If neither doxycycline nor bleomycin is effective, talc can be used.
Symptomatic pericardial effusions are usually treated by creating a pericardial window or by stripping the pericardium. If the patient’s condition does not permit a surgical procedure, sclerosis can be attempted with doxycycline and/or bleomycin.
Malignant ascites is usually treated with repeated paracentesis of small volumes of fluid. If the underlying malignancy is unresponsive to systemic therapy, peritoneovenous shunts may be inserted. Despite the fear of disseminating tumor cells into the circulation, widespread metastases are an unusual complication. The major complications are occlusion, leakage, and fluid overload. Patients with severe liver disease may develop disseminated intravascular coagulation.
Nutrition Cancer and its treatment may lead to a decrease in nutrient intake of sufficient magnitude to cause weight loss and alteration of intermediary metabolism. The prevalence of this problem is difficult to estimate because of variations in the definition of cancer cachexia, but most patients with advanced cancer experience weight loss and decreased appetite. A variety of both tumor-derived factors (e.g., bombesin, adrenocorticotropic hormone) and host-derived factors (e.g., tumor necrosis factor, interleukins 1 and 6, growth hormone) contribute to the altered metabolism, and a vicious cycle is established in which protein catabolism, glucose intolerance, and lipolysis cannot be reversed by the provision of calories.
It remains controversial how to assess nutritional status and when and how to intervene. Efforts to make the assessment objective have included the use of a prognostic nutritional index based on albumin levels, triceps skinfold thickness, transferrin levels, and delayed-type hypersensitivity skin testing. However, a simpler approach has been to define the threshold for nutritional intervention as <10% unexplained body weight loss, serum transferrin level <1500 mg/L (150 mg/dL), and serum albumin <34 g/L (3.4 g/dL).
The decision is important, because it appears that cancer therapy is substantially more toxic and less effective in the face of malnutrition. Nevertheless, it remains unclear whether nutritional intervention can alter the natural history. Unless some pathology is affecting the absorptive function of the gastrointestinal tract, enteral nutrition provided orally or by tube feeding is preferred over parenteral supplementation. However, the risks associated with the tube may outweigh the benefits. Megestrol acetate, a progestational agent, has been advocated as a pharmacologic intervention to improve nutritional status. Research in this area may provide more tools in the future as cytokine-mediated mechanisms are further elucidated.
Psychosocial Support The psychosocial needs of patients vary with their situation. Patients undergoing treatment experience fear, anxiety, and depression. Self-image is often seriously compromised by deforming surgery and loss of hair. Women who receive cosmetic advice that enables them to look better also feel better. Loss of control over how one spends time can contribute to the sense of vulnerability. Juggling the demands of work and family with the demands of treatment may create enormous stresses. Sexual dysfunction is highly prevalent and needs to be discussed openly with the patient. An empathetic health care team is sensitive to the individual patient’s needs and permits negotiation where such flexibility will not adversely affect the course of treatment.
Cancer survivors have other sets of difficulties. Patients may have fears associated with the termination of a treatment they associate with their continued survival. Adjustments are required to physical losses and handicaps, real and perceived. Patients may be preoccupied with minor physical problems. They perceive a decline in their job mobility and view themselves as less desirable workers. They may be victims of job and/or insurance discrimination. Patients may experience difficulty reentering their normal past life. They may feel guilty for having survived and may carry a sense of vulnerability to colds and other illnesses. Perhaps the most pervasive and threatening concern is the ever-present fear of relapse (the Damocles syndrome).
Patients in whom therapy has been unsuccessful have other problems related to the end of life.
Death and Dying The most common causes of death in patients with cancer are infection (leading to circulatory failure), respiratory failure, hepatic failure, and renal failure. Intestinal blockage may lead to inanition and starvation. Central nervous system disease may lead to seizures, coma, and central hypoventilation. About 70% of patients develop dyspnea preterminally. However, many months usually pass between the diagnosis of cancer and the occurrence of these complications, and during this period, the patient is severely affected by the possibility of death. The path of unsuccessful cancer treatment usually occurs in three phases. First, there is optimism at the hope of cure; when the tumor recurs, there is the acknowledgment of an incurable disease, and the goal of palliative therapy is embraced in the hope of being able to live with disease; finally, at the disclosure of imminent death, another adjustment in outlook takes place. The patient imagines the worst in preparation for the end of life and may go through stages of adjustment to the diagnosis. These stages include denial, isolation, anger, bargaining, depression, acceptance, and hope. Of course, patients do not all progress through all the stages or proceed through them in the same order or at the same rate. Nevertheless, developing an understanding of how the patient has been affected by the diagnosis and is coping with it is an important goal of patient management.
It is best to speak frankly with the patient and the family regarding the likely course of disease. These discussions can be difficult for the physician as well as for the patient and family. The critical features of the interaction are to reassure the patient and family that everything that can be done to provide comfort will be done. They will not be abandoned. Many patients prefer to be cared for in their homes or in a hospice setting rather than a hospital. The American College of Physicians has published a book called Home Care Guide for Cancer: How to Care for Family and Friends at Home that teaches an approach to successful problem-solving in home care. With appropriate planning, it should be possible to provide the patient with the necessary medical care as well as the psychological and spiritual support that will prevent the isolation and depersonalization that can attend in-hospital death.
The care of dying patients may take a toll on the physician. A “burnout” syndrome has been described that is characterized by fatigue, disengagement from patients and colleagues, and a loss of self-fulfillment. Efforts at stress reduction, maintenance of a balanced life, and setting realistic goals may combat this disorder.
End-of-Life Decisions Unfortunately, a smooth transition in treatment goals from curative to palliative may not be possible in all cases because of the occurrence of serious treatment-related complications or rapid disease progression. Vigorous and invasive medical support for a reversible disease or treatment complication is assumed to be justified. However, if the reversibility of the condition is in doubt, the patient’s wishes determine the level of medical care. These wishes should be elicited before the terminal phase of illness and reviewed periodically. Information about advance directives can be obtained from the American Association of Retired Persons, 601 E Street, NW, Washington, DC 20049, 202-434-2277, or Choice in Dying, 250 West 57th Street, New York, NY 10107, 212-366-5540. Some states allow physicians to assist patients who choose to end their lives. This subject is challenging from an ethical and a medical point of view. Discussions of end-of-life decisions should be candid and involve clear informed consent, waiting periods, second opinions, and documentation. A full discussion of end-of-life management is in Chap. 10.
_____________________________
1The National Cancer Institute maintains a database called PDQ (Physician Data Query) that is accessible on the Internet under the name CancerNet at www.cancer.gov/cancertopics/pdq/cancerdatabase. Information can be obtained through a facsimile machine using CancerFax by dialing 301-402-5874. Patient information is also provided by the National Cancer Institute in at least three formats: on the Internet via CancerNet at www.cancer.gov, through the CancerFax number listed above, or by calling 1-800-4-CANCER. The quality control for the information provided through these services is rigorous.
2Information about unsound methods may be obtained from the National Council Against Health Fraud, Box 1276, Loma Linda, CA 92354, or from the Center for Medical Consumers and Health Care Information, 237 Thompson Street, New York, NY 10012.
100 |
Prevention and Early Detection of Cancer |
Improved understanding of carcinogenesis has allowed cancer prevention and early detection (also known as cancer control) to expand beyond the identification and avoidance of carcinogens. Specific interventions to prevent cancer in those at risk, and effective screening for early detection of cancer, are the goals.
Carcinogenesis is not an event but a process, a continuum of discrete tissue and cellular changes over time resulting in aberrant physiologic processes. Prevention concerns the identification and manipulation of the biologic, environmental, social, and genetic factors in the causal pathway of cancer.
EDUCATION AND HEALTHFUL HABITS
Public education on the avoidance of identified risk factors for cancer and encouraging healthy habits contributes to cancer prevention and control. The clinician is a powerful messenger in this process. The patient-provider encounter provides an opportunity to teach patients about the hazards of smoking, the features of a healthy lifestyle, use of proven cancer screening methods, and avoidance of excessive sun exposure.
SMOKING CESSATION
Tobacco smoking is a strong, modifiable risk factor for cardiovascular disease, pulmonary disease, and cancer. Smokers have an approximately 1 in 3 lifetime risk of dying prematurely from a tobacco-related cancer, cardiovascular, or pulmonary disease. Tobacco use causes more deaths from cardiovascular disease than from cancer. Lung cancer and cancers of the larynx, oropharynx, esophagus, kidney, bladder, pancreas, and stomach are all tobacco-related.
The number of cigarettes smoked per day and the level of inhalation of cigarette smoke are correlated with risk of lung cancer mortality. Light- and low-tar cigarettes are not safer, because smokers tend to inhale them more frequently and deeply.
Those who stop smoking have a 30–50% lower 10-year lung cancer mortality rate compared to those who continue smoking, despite the fact that some carcinogen-induced gene mutations persist for years after smoking cessation. Smoking cessation and avoidance would save more lives than any other public health activity.
The risk of tobacco smoke is not limited to the smoker. Environmental tobacco smoke, known as secondhand or passive smoke, causes lung cancer and other cardiopulmonary diseases in nonsmokers.
Tobacco use prevention is a pediatric issue. More than 80% of adult American smokers began smoking before the age of 18 years. Approximately 20% of Americans in grades 9 through 12 have smoked a cigarette in the past month. Counseling of adolescents and young adults is critical to prevent smoking. A clinician’s simple advice can be of benefit. Providers should query patients on tobacco use and offer smokers assistance in quitting.
Current approaches to smoking cessation recognize smoking as an addiction (Chap. 470). The smoker who is quitting goes through identifiable stages that include contemplation of quitting, an action phase in which the smoker quits, and a maintenance phase. Smokers who quit completely are more likely to be successful than those who gradually reduce the number of cigarettes smoked or change to lower-tar or lower-nicotine cigarettes. More than 90% of the Americans who have successfully quit smoking did so on their own, without participation in an organized cessation program, but cessation programs are helpful for some smokers. The Community Intervention Trial for Smoking Cessation (COMMIT) was a 4-year program showing that light smokers (<25 cigarettes per day) were more likely to benefit from simple cessation messages and cessation programs than those who did not receive an intervention. Quit rates were 30.6% in the intervention group and 27.5% in the control group. The COMMIT interventions were unsuccessful in heavy smokers (<25 cigarettes per day). Heavy smokers may need an intensive broad-based cessation program that includes counseling, behavioral strategies, and pharmacologic adjuncts, such as nicotine replacement (gum, patches, sprays, lozenges, and inhalers), bupropion, and/or varenicline.
The health risks of cigars are similar to those of cigarettes. Smoking one or two cigars daily doubles the risk for oral and esophageal cancers; smoking three or four cigars daily increases the risk of oral cancers more than eightfold and esophageal cancer fourfold. The risks of occasional use are unknown.
Smokeless tobacco also represents a substantial health risk. Chewing tobacco is a carcinogen linked to dental caries, gingivitis, oral leukoplakia, and oral cancer. The systemic effects of smokeless tobacco (including snuff) may increase risks for other cancers. Esophageal cancer is linked to carcinogens in tobacco dissolved in saliva and swallowed. The net effects of e-cigarettes on health are poorly studied. Whether they aid in smoking cessation or serve as a “gateway” for nonsmoking children to acquire a smoking habit is debated.
PHYSICAL ACTIVITY
Physical activity is associated with a decreased risk of colon and breast cancer. A variety of mechanisms have been proposed. However, such studies are prone to confounding factors such as recall bias, association of exercise with other health-related practices, and effects of preclinical cancers on exercise habits (reverse causality).
DIET MODIFICATION
International epidemiologic studies suggest that diets high in fat are associated with increased risk for cancers of the breast, colon, prostate, and endometrium. These cancers have their highest incidence and mortalities in Western cultures, where fat composes an average of one-third of the total calories consumed.
Despite correlations, dietary fat has not been proven to cause cancer. Case-control and cohort epidemiologic studies give conflicting results. In addition, diet is a highly complex exposure to many nutrients and chemicals. Low-fat diets are associated with many dietary changes beyond simple subtraction of fat. Other lifestyle changes are also associated with adherence to a low-fat diet.
In observational studies, dietary fiber is associated with a reduced risk of colonic polyps and invasive cancer of the colon. However, cancer-protective effects of increasing fiber and lowering dietary fat have not been proven in the context of a prospective clinical trial. The putative protective mechanisms are complex and speculative. Fiber binds oxidized bile acids and generates soluble fiber products, such as butyrate, that may have differentiating properties. Fiber does not increase bowel transit times. Two large prospective cohort studies of >100,000 health professionals showed no association between fruit and vegetable intake and risk of cancer.
The Polyp Prevention Trial randomly assigned 2000 elderly persons, who had polyps removed, to a low-fat, high-fiber diet versus routine diet for 4 years. No differences were noted in polyp formation.
The U.S. National Institutes of Health Women’s Health Initiative, launched in 1994, was a long-term clinical trial enrolling >100,000 women age 45–69 years. It placed women in 22 intervention groups. Participants received calcium/vitamin D supplementation; hormone replacement therapy; and counseling to increase exercise, eat a low-fat diet with increased consumption of fruits, vegetables, and fiber, and cease smoking. The study showed that although dietary fat intake was lower in the diet intervention group, invasive breast cancers were not reduced over an 8-year follow-up period compared to the control group. No reduction was seen in the incidence of colorectal cancer in the dietary intervention arm. The difference in dietary fat averaged ∼10% between the two groups. Evidence does not currently establish the anticarcinogenic value of vitamin, mineral, or nutritional supplements in amounts greater than those provided by a balanced diet.
ENERGY BALANCE
Risk of cancer appears to increase as body mass index increases beyond 25 kg/m2. Obesity is associated with increased risk for cancers of the colon, breast (female postmenopausal), endometrium, kidney (renal cell), and esophagus, although causality has not been established.
In observational studies, relative risks of colon cancer are increased in obesity by 1.5–2 for men and 1.2–1.5 for women. Obese postmenopausal women have a 30–50% increased relative risk of breast cancer. An unproven hypothesis for the association is that adipose tissue serves as a depot for aromatase that facilitates estrogen production.
SUN AVOIDANCE
Nonmelanoma skin cancers (basal cell and squamous cell) are induced by cumulative exposure to ultraviolet (UV) radiation. Intermittent acute sun exposure and sun damage have been linked to melanoma, but the evidence is inconsistent. Sunburns, especially in childhood and adolescence, may be associated with an increased risk of melanoma in adulthood. Reduction of sun exposure through use of protective clothing and changing patterns of outdoor activities can reduce skin cancer risk. Sunscreens decrease the risk of actinic keratoses, the precursor to squamous cell skin cancer, but melanoma risk may not be reduced. Sunscreens prevent burning, but they may encourage more prolonged exposure to the sun and may not filter out wavelengths of energy that cause melanoma.
Educational interventions to help individuals assess their risk of developing skin cancer have some impact. In particular, appearance-focused behavioral interventions in young women can decrease indoor tanning use and other UV exposures. Self-examination for skin pigment characteristics associated with skin cancer, such as freckling, may be useful in identifying people at high risk. Those who recognize themselves as being at risk tend to be more compliant with sun-avoidance recommendations. Risk factors for melanoma include a propensity to sunburn, a large number of benign melanocytic nevi, and atypical nevi.
CANCER CHEMOPREVENTION
Chemoprevention involves the use of specific natural or synthetic chemical agents to reverse, suppress, or prevent carcinogenesis before the development of invasive malignancy.
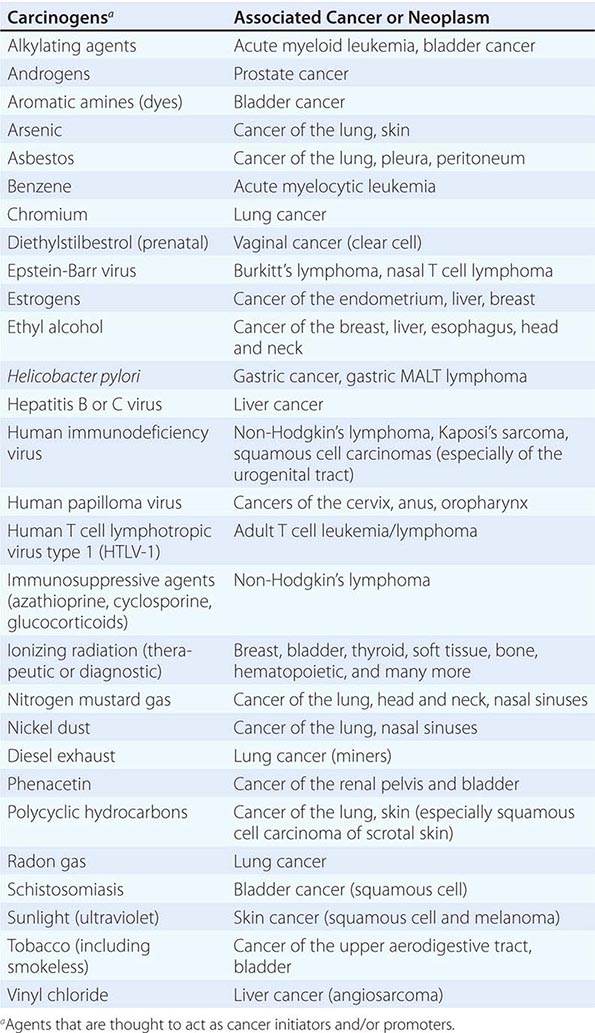
Cancer develops through an accumulation of tissue abnormalities associated with genetic and epigenetic changes, and growth regulatory pathways that are potential points of intervention to prevent cancer. The initial changes are termed initiation. The alteration can be inherited or acquired through the action of physical, infectious, or chemical carcinogens. Like most human diseases, cancer arises from an interaction between genetics and environmental exposures (Table 100-1). Influences that cause the initiated cell and its surrounding tissue microenvironment to progress through the carcinogenic process and change phenotypically are termed promoters. Promoters include hormones such as androgens, linked to prostate cancer, and estrogen, linked to breast and endometrial cancer. The distinction between an initiator and promoter is indistinct; some components of cigarette smoke are “complete carcinogens,” acting as both initiators and promoters. Cancer can be prevented or controlled through interference with the factors that cause cancer initiation, promotion, or progression. Compounds of interest in chemoprevention often have antimutagenic, hormone modulation, anti-inflammatory, antiproliferative, or proapoptotic activity (or a combination).
|
SUSPECTED CARCINOGENS |
CHEMOPREVENTION OF CANCERS OF THE UPPER AERODIGESTIVE TRACT
Smoking causes diffuse epithelial injury in the oral cavity, neck, esophagus, and lung. Patients cured of squamous cell cancers of the lung, esophagus, oral cavity, and neck are at risk (as high as 5% per year) of developing second cancers of the upper aerodigestive tract. Cessation of cigarette smoking does not markedly decrease the cured cancer patient’s risk of second malignancy, even though it does lower the cancer risk in those who have never developed a malignancy. Smoking cessation may halt the early stages of the carcinogenic process (such as metaplasia), but it may have no effect on late stages of carcinogenesis. This “field carcinogenesis” hypothesis for upper aerodigestive tract cancer has made “cured” patients an important population for chemoprevention of second malignancies.
Oral human papilloma virus (HPV) infection, particularly HPV-16, increases the risk for cancers of the oropharynx. This association exists even in the absence of other risk factors such as smoking or alcohol use (although the magnitude of increased risk appears greater than additive when HPV infection and smoking are both present). Oral HPV infection is believed to be largely sexually acquired. Although no direct evidence currently exists to confirm the hypothesis, the introduction of the HPV vaccine may eventually reduce oropharyngeal cancer rates.
Oral leukoplakia, a premalignant lesion commonly found in smokers, has been used as an intermediate marker of chemopreventive activity in smaller shorter-duration, randomized, placebo-controlled trials. Response was associated with upregulation of retinoic acid receptor-β (RAR-β). Therapy with high, relatively toxic doses of isotretinoin (13-cis-retinoic acid) causes regression of oral leukoplakia. However, the lesions recur when the therapy is withdrawn, suggesting the need for long-term administration. More tolerable doses of isotretinoin have not shown benefit in the prevention of head and neck cancer. Isotretinoin also failed to prevent second malignancies in patients cured of early-stage non-small cell lung cancer; mortality rates were actually increased in current smokers.
Several large-scale trials have assessed agents in the chemoprevention of lung cancer in patients at high risk. In the α-tocopherol/β-carotene (ATBC) Lung Cancer Prevention Trial, participants were male smokers, age 50–69 years at entry. Participants had smoked an average of one pack of cigarettes per day for 35.9 years. Participants received α-tocopherol, β-carotene, and/or placebo in a randomized, two-by-two factorial design. After median follow-up of 6.1 years, lung cancer incidence and mortality were statistically significantly increased in those receiving β-carotene. α-Tocopherol had no effect on lung cancer mortality, and no evidence suggested interaction between the two drugs. Patients receiving α-tocopherol had a higher incidence of hemorrhagic stroke.
The β-Carotene and Retinol Efficacy Trial (CARET) involved 17,000 American smokers and workers with asbestos exposure. Entrants were randomly assigned to one of four arms and received β-carotene, retinol, and/or placebo in a two-by-two factorial design. This trial also demonstrated harm from β-carotene: a lung cancer rate of 5 per 1000 subjects per year for those taking placebo and of 6 per 1000 subjects per year for those taking β-carotene.
The ATBC and CARET results demonstrate the importance of testing chemoprevention hypotheses thoroughly before their widespread implementation because the results contradict a number of observational studies. The Physicians’ Health Trial showed no change in the risk of lung cancer for those taking β-carotene; however, fewer of its participants were smokers than those in the ATBC and CARET studies.
CHEMOPREVENTION OF COLON CANCER
Many colon cancer prevention trials are based on the premise that most colorectal cancers develop from adenomatous polyps. These trials use adenoma recurrence or disappearance as a surrogate endpoint (not yet validated) for colon cancer prevention. Early clinical trial results suggest that nonsteroidal anti-inflammatory drugs (NSAIDs), such as piroxicam, sulindac, and aspirin, may prevent adenoma formation or cause regression of adenomatous polyps. The mechanism of action of NSAIDs is unknown, but they are presumed to work through the cyclooxygenase pathway. Although two randomized controlled trials (the Physicians’ Health Study and the Women’s Health Study) did not show an effect of aspirin on colon cancer or adenoma incidence in persons with no previous history of colonic lesions after 10 years of therapy, these trials did show an approximately 18% relative risk reduction for colonic adenoma incidence in persons with a previous history of adenomas after 1 year. Pooled findings from observational cohort studies do demonstrate a 22% and 28% relative reduction in colorectal cancer and adenoma incidence, respectively, with regular aspirin use, and a well-conducted meta-analysis of four randomized controlled trials (albeit primarily designed to examine aspirin’s effects on cardiovascular events) found that aspirin at doses of at least 75 mg resulted in a 24% relative reduction in colorectal cancer incidence after 20 years, with no clear increase in efficacy at higher doses. Cyclooxygenase-2 (COX-2) inhibitors have also been considered for colorectal cancer and polyp prevention. Trials with COX-2 inhibitors were initiated, but an increased risk of cardiovascular events in those taking the COX-2 inhibitors was noted, suggesting that these agents are not suitable for chemoprevention in the general population.
Epidemiologic studies suggest that diets high in calcium lower colon cancer risk. Calcium binds bile and fatty acids, which cause proliferation of colonic epithelium. It is hypothesized that calcium reduces intraluminal exposure to these compounds. The randomized controlled Calcium Polyp Prevention Study found that calcium supplementation decreased the absolute risk of adenomatous polyp recurrence by 7% at 4 years; extended observational follow-up demonstrated a 12% absolute risk reduction 5 years after cessation of treatment. However, in the Women’s Health Initiative, combined use of calcium carbonate and vitamin D twice daily did not reduce the incidence of invasive colorectal cancer compared with placebo after 7 years.
The Women’s Health Initiative demonstrated that postmenopausal women taking estrogen plus progestin have a 44% lower relative risk of colorectal cancer compared to women taking placebo. Of >16,600 women randomized and followed for a median of 5.6 years, 43 invasive colorectal cancers occurred in the hormone group and 72 in the placebo group. The positive effect on colon cancer is mitigated by the modest increase in cardiovascular and breast cancer risks associated with combined estrogen plus progestin therapy.
A case-control study suggested that statins decrease the incidence of colorectal cancer; however, several subsequent case-control and cohort studies have not demonstrated an association between regular statin use and a reduced risk of colorectal cancer. No randomized controlled trials have addressed this hypothesis. A meta-analysis of statin use showed no protective effect of statins on overall cancer incidence or death.
CHEMOPREVENTION OF BREAST CANCER
Tamoxifen is an antiestrogen with partial estrogen agonistic activity in some tissues, such as endometrium and bone. One of its actions is to upregulate transforming growth factor β, which decreases breast cell proliferation. In randomized placebo-controlled trials to assess tamoxifen as adjuvant therapy for breast cancer, tamoxifen reduced the number of new breast cancers in the opposite breast by more than a third. In a randomized placebo-controlled prevention trial involving >13,000 pre- and postmenopausal women at high risk, tamoxifen decreased the risk of developing breast cancer by 49% (from 43.4 to 22 per 1000 women) after a median follow-up of nearly 6 years. Tamoxifen also reduced bone fractures; a small increase in risk of endometrial cancer, stroke, pulmonary emboli, and deep vein thrombosis was noted. The International Breast Cancer Intervention Study (IBIS-I) and the Italian Randomized Tamoxifen Prevention Trial also demonstrated a reduction in breast cancer incidence with tamoxifen use. A trial comparing tamoxifen with another selective estrogen receptor modulator, raloxifene, in postmenopausal women showed that raloxifene is comparable to tamoxifen in cancer prevention. This trial only included postmenopausal women. Raloxifene was associated with more invasive breast cancers and a trend toward more noninvasive breast cancers, but fewer thromboembolic events than tamoxifen; the drugs are similar in risks of other cancers, fractures, ischemic heart disease, and stroke. Both tamoxifen and raloxifene (the latter for postmenopausal women only) have been approved by the U.S. Food and Drug Administration (FDA) for reduction of breast cancer in women at high risk for the disease (1.66% risk at 5 years based on the Gail risk model: http://www.cancer.gov/bcrisktool/).
Because the aromatase inhibitors are even more effective than tamoxifen in adjuvant breast cancer therapy, it has been hypothesized that they would be more effective in breast cancer prevention. A randomized, placebo-controlled trial of exemestane reported a 65% relative reduction (from 5.5 to 1.9 per 1000 women) in the incidence of invasive breast cancer in women at elevated risk after a median follow-up of about 3 years. Common adverse effects included arthralgias, hot flashes, fatigue, and insomnia. No trial has directly compared aromatase inhibitors with selective estrogen receptor modulators for breast cancer chemoprevention.
CHEMOPREVENTION OF PROSTATE CANCER
Finasteride and dutasteride are 5-α-reductase inhibitors. They inhibit conversion of testosterone to dihydrotestosterone (DHT), a potent stimulator of prostate cell proliferation. The Prostate Cancer Prevention Trial (PCPT) randomly assigned men age 55 years or older at average risk of prostate cancer to finasteride or placebo. All men in the trial were being regularly screened with prostate-specific antigen (PSA) levels and digital rectal examination. After 7 years of therapy, the incidence of prostate cancer was 18.4% in the finasteride arm, compared with 24.4% in the placebo arm, a statistically significant difference. However, the finasteride group had more patients with tumors of Gleason score 7 and higher compared with the placebo arm (6.4 vs 5.1%). Reassuringly, long-term (10–15 years) follow-up did not reveal any statistically significant differences in overall mortality between all men in the finasteride and placebo arms or in men diagnosed with prostate cancer; differences in prostate cancer in favor of finasteride persisted.
Dutasteride has also been evaluated as a preventive agent for prostate cancer. The Reduction by Dutasteride of Prostate Cancer Events (REDUCE) trial was a randomized double-blind trial in which approximately 8200 men with an elevated PSA (2.5–10 ng/mL for men age 50–60 years and 3–10 ng/mL for men age 60 years or older) and negative prostate biopsy on enrollment received daily 0.5 mg of dutasteride or placebo. The trial found a statistically significant 23% relative risk reduction in the incidence of biopsy-detected prostate cancer in the dutasteride arm at 4 years of treatment (659 cases vs 858 cases, respectively). Overall, across years 1 through 4, there was no difference between the arms in the number of tumors with a Gleason score of 7 to 10; however, during years 3 and 4, there was a statistically significant difference in tumors with Gleason score of 8 to 10 in the dutasteride arm (12 tumors vs 1 tumor, respectively).
The clinical importance of the apparent increased incidence of higher-grade tumors in the 5-α-reductase inhibitor arms of these trials is controversial. It may likely represent an increased sensitivity of PSA and digital rectal exam for high-grade tumors in men receiving these agents. The FDA has analyzed both trials, and it determined that the use of a 5-α-reductase inhibitor for prostate cancer chemoprevention would result in one additional high-grade (Gleason score 8 to 10) prostate cancer for every three to four lower-grade (Gleason score <6) tumors averted. Although it acknowledged that detection bias may have accounted for the finding, it stated that it could not conclusively dismiss a causative role for 5-α-reductase inhibitors. These agents are therefore not FDA-approved for prostate cancer prevention.
Because all men in both the PCPT and REDUCE trials were being screened and because screening approximately doubles the rate of prostate cancer, it is not known if finasteride or dutasteride decreases the risk of prostate cancer in men who are not being screened.
Several favorable laboratory and observational studies led to the formal evaluation of selenium and α-tocopherol (vitamin E) as potential prostate cancer preventives. The Selenium and Vitamin E Cancer Prevention Trial (SELECT) assigned 35,533 men to receive 200 μg/d selenium, 400 IU/d α-tocopherol, selenium plus vitamin E, or placebo. After a median follow-up of 7 years, a trend toward an increased risk of developing prostate cancer was observed for those men taking vitamin E alone as compared to the placebo arm (hazard ratio 1.17; 95% confidence interval, 1.004–1.36).
VACCINES AND CANCER PREVENTION
A number of infectious agents cause cancer. Hepatitis B and C are linked to liver cancer; some HPV strains are linked to cervical, anal, and head and neck cancer; and Helicobacter pylori is associated with gastric adenocarcinoma and gastric lymphoma. Vaccines to protect against these agents may reduce the risk of their associated cancers.
The hepatitis B vaccine is effective in preventing hepatitis and hepatomas due to chronic hepatitis B infection.
A quadrivalent HPV vaccine (covering HPV strains 6, 11, 16, and 18) and a bivalent vaccine (covering HPV strains 16 and 18) are available for use in the United States. HPV types 16 and 18 cause cervical and anal cancer; reduction in these HPV types could prevent >70% of cervical cancers worldwide. HPV types 6 and 11 cause genital papillomas. For individuals not previously infected with these HPV strains, the vaccines demonstrate high efficacy in preventing persistent strain-specific HPV infections; however, the trials and substudies that evaluated the vaccines’ ability to prevent cervical and anal cancer relied on surrogate outcome measures (cervical or anal intraepithelial neoplasia [CIN/AIN] I, II, and III), and the degree of durability of the immune response beyond 5 years is not currently known. The vaccines do not appear to impact preexisting infections and the efficacy appears to be markedly lower for populations that had previously been exposed to vaccine-specific HPV strains. The vaccine is recommended in the United States for females and males age 9–26 years.
SURGICAL PREVENTION OF CANCER
Some organs in some individuals are at such high risk of developing cancer that surgical removal of the organ at risk may be considered. Women with severe cervical dysplasia are treated with laser or loop electrosurgical excision or conization and occasionally even hysterectomy. Colectomy is used to prevent colon cancer in patients with familial polyposis or ulcerative colitis.
Prophylactic bilateral mastectomy may be chosen for breast cancer prevention among women with genetic predisposition to breast cancer. In a prospective series of 139 women with BRCA1 and BRCA2 mutations, 76 chose to undergo prophylactic mastectomy and 63 chose close surveillance. At 3 years, no cases of breast cancer had been diagnosed in those opting for surgery, but eight patients in the surveillance group had developed breast cancer. A larger (n = 639) retrospective cohort study reported that three patients developed breast cancer after prophylactic mastectomy compared with an expected incidence of 30–53 cases: a 90–94% reduction in breast cancer risk. Postmastectomy breast cancer–related deaths were reduced by 81–94% for high-risk women compared with sister controls and by 100% for moderate-risk women when compared with expected rates.
Prophylactic oophorectomy may also be employed for the prevention of ovarian and breast cancers among high-risk women. A prospective cohort study evaluating the outcomes of BRCA mutation carriers demonstrated a statistically significant association between prophylactic oophorectomy and a reduced incidence of ovarian or primary peritoneal cancer (36% relative risk reduction, or a 4.5% absolute difference). Studies of prophylactic oophorectomy for prevention of breast cancer in women with genetic mutations have shown relative risk reductions of approximately 50%; the risk reduction may be greatest for women having the procedure at younger (i.e., <50 years) ages.
All of the evidence concerning the use of prophylactic mastectomy and oophorectomy for prevention of breast and ovarian cancer in high-risk women has been observational in nature; such studies are prone to a variety of biases, including case selection bias, family relationships between patients and controls, and inadequate information about hormone use. Thus, they may give an overestimate of the magnitude of benefit.
CANCER SCREENING
Screening is a means of detecting disease early in asymptomatic individuals, with the goal of decreasing morbidity and mortality. While screening can potentially reduce disease-specific deaths and has been shown to do so in cervical, colon, lung, and breast cancer, it is also subject to a number of biases that can suggest a benefit when actually there is none. Biases can even mask net harm. Early detection does not in itself confer benefit. Cause-specific mortality, rather than survival after diagnosis, is the preferred endpoint (see below).
Because screening is done on asymptomatic, healthy persons, it should offer substantial likelihood of benefit that outweighs harm. Screening tests and their appropriate use should be carefully evaluated before their use is widely encouraged in screening programs, as a matter of public policy.
A large and increasing number of genetic mutations and nucleotide polymorphisms have been associated with an increased risk of cancer. Testing for these genetic mutations could in theory define a high-risk population. However, most of the identified mutations have very low penetrance and individually provide minimal predictive accuracy. The ability to predict the development of a particular cancer may some day present therapeutic options as well as ethical dilemmas. It may eventually allow for early intervention to prevent a cancer or limit its severity. People at high risk may be ideal candidates for chemoprevention and screening; however, efficacy of these interventions in the high-risk population should be investigated. Currently, persons at high risk for a particular cancer can engage in intensive screening. While this course is clinically reasonable, it is not known if it reduces mortality in these populations.
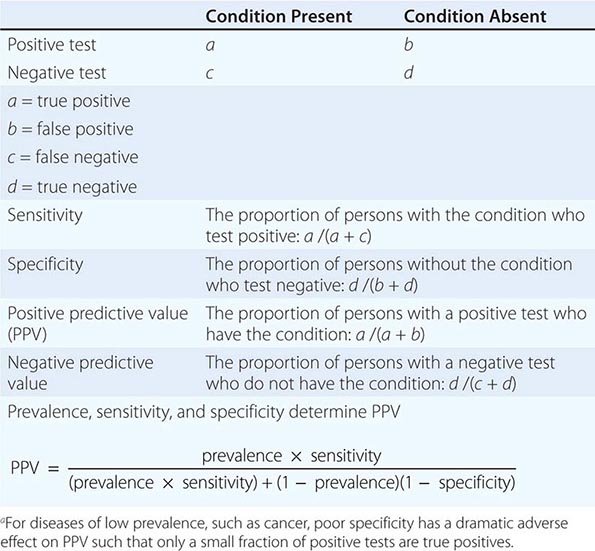
The Accuracy of Screening A screening test’s accuracy or ability to discriminate disease is described by four indices: sensitivity, specificity, positive predictive value, and negative predictive value (Table 100-2). Sensitivity, also called the true-positive rate, is the proportion of persons with the disease who test positive in the screen (i.e., the ability of the test to detect disease when it is present). Specificity, or 1 minus the false-positive rate, is the proportion of persons who do not have the disease that test negative in the screening test (i.e., the ability of a test to correctly identify that the disease is not present). The positive predictive value is the proportion of persons who test positive that actually have the disease. Similarly, negative predictive value is the proportion testing negative that do not have the disease. The sensitivity and specificity of a test are independent of the underlying prevalence (or risk) of the disease in the population screened, but the predictive values depend strongly on the prevalence of the disease.
|
ASSESSMENT OF THE VALUE OF A DIAGNOSTIC TESTa |
Screening is most beneficial, efficient, and economical when the target disease is common in the population being screened. Specificity is at least as important to the ultimate feasibility and success of a screening test as sensitivity.
Potential Biases of Screening Tests Common biases of screening are lead time, length-biased sampling, and selection. These biases can make a screening test seem beneficial when actually it is not (or even causes net harm). Whether beneficial or not, screening can create the false impression of an epidemic by increasing the number of cancers diagnosed. It can also produce a shift in the proportion of patients diagnosed at an early stage and inflate survival statistics without reducing mortality (i.e., the number of deaths from a given cancer relative to the number of those at risk for the cancer). In such a case, the apparent duration of survival (measured from date of diagnosis) increases without lives being saved or life expectancy changed.
Lead-time bias occurs whether or not a test influences the natural history of the disease; the patient is merely diagnosed at an earlier date. Survival appears increased even if life is not really prolonged. The screening test only prolongs the time the subject is aware of the disease and spends as a patient.
Length-biased sampling occurs because screening tests generally can more easily detect slow-growing, less aggressive cancers than fast-growing cancers. Cancers diagnosed due to the onset of symptoms between scheduled screenings are on average more aggressive, and treatment outcomes are not as favorable. An extreme form of length bias sampling is termed overdiagnosis, the detection of “pseudo disease.” The reservoir of some undetected slow-growing tumors is large. Many of these tumors fulfill the histologic criteria of cancer but will never become clinically significant or cause death. This problem is compounded by the fact that the most common cancers appear most frequently at ages when competing causes of death are more frequent.
Selection bias must be considered in assessing the results of any screening effort. The population most likely to seek screening may differ from the general population to which the screening test might be applied. In general, volunteers for studies are more health conscious and likely to have a better prognosis or lower mortality rate, irrespective of the screening result. This is termed the healthy volunteer effect.
Potential Drawbacks of Screening Risks associated with screening include harm caused by the screening intervention itself, harm due to the further investigation of persons with positive tests (both true and false positives), and harm from the treatment of persons with a true-positive result, whether or not life is extended by treatment (e.g., even if a screening test reduces relative cause-specific mortality by 20–30%, 70–80% of those diagnosed still go on to die of the target cancer). The diagnosis and treatment of cancers that would never have caused medical problems can lead to the harm of unnecessary treatment and give patients the anxiety of a cancer diagnosis. The psychosocial impact of cancer screening can also be substantial when applied to the entire population.
Assessment of Screening Tests Good clinical trial design can offset some biases of screening and demonstrate the relative risks and benefits of a screening test. A randomized controlled screening trial with cause-specific mortality as the endpoint provides the strongest support for a screening intervention. Overall mortality should also be reported to detect an adverse effect of screening and treatment on other disease outcomes (e.g., cardiovascular disease). In a randomized trial, two like populations are randomly established. One is given the usual standard of care (which may be no screening at all) and the other receives the screening intervention being assessed. The two populations are compared over time. Efficacy for the population studied is established when the group receiving the screening test has a better cause-specific mortality rate than the control group. Studies showing a reduction in the incidence of advanced-stage disease, improved survival, or a stage shift are weaker (and possibly misleading) evidence of benefit. These latter criteria are early indicators but not sufficient to establish the value of a screening test.
Although a randomized, controlled screening trial provides the strongest evidence to support a screening test, it is not perfect. Unless the trial is population-based, it does not remove the question of generalizability to the target population. Screening trials generally involve thousands of persons and last for years. Less definitive study designs are therefore often used to estimate the effectiveness of screening practices. However, every nonrandomized study design is subject to strong confounders. In descending order of strength, evidence may also be derived from the findings of internally controlled trials using intervention allocation methods other than randomization (e.g., allocation by birth date, date of clinic visit); the findings of analytic observational studies; or the results of multiple time series studies with or without the intervention.
Screening for Specific Cancers Screening for cervical, colon, and breast cancer is beneficial for certain age groups. Depending on age and smoking history, lung cancer screening can also be beneficial in specific settings. Special surveillance of those at high risk for a specific cancer because of a family history or a genetic risk factor may be prudent, but few studies have assessed the influence on mortality. A number of organizations have considered whether or not to endorse routine use of certain screening tests. Because these groups have not used the same criteria to judge whether a screening test should be endorsed, they have arrived at different recommendations. The American Cancer Society (ACS) and the U.S. Preventive Services Task Force (USPSTF) publish screening guidelines (Table 100-3); the American Academy of Family Practitioners (AAFP) generally follow/endorse the USPSTF recommendations; and the American College of Physicians (ACP) develops recommendations based on structured reviews of other organizations’ guidelines.
|
SCREENING RECOMMENDATIONS FOR ASYMPTOMATIC SUBJECTS NOT KNOWN TO BE AT INCREASED RISK FOR THE TARGET CONDITIONa |
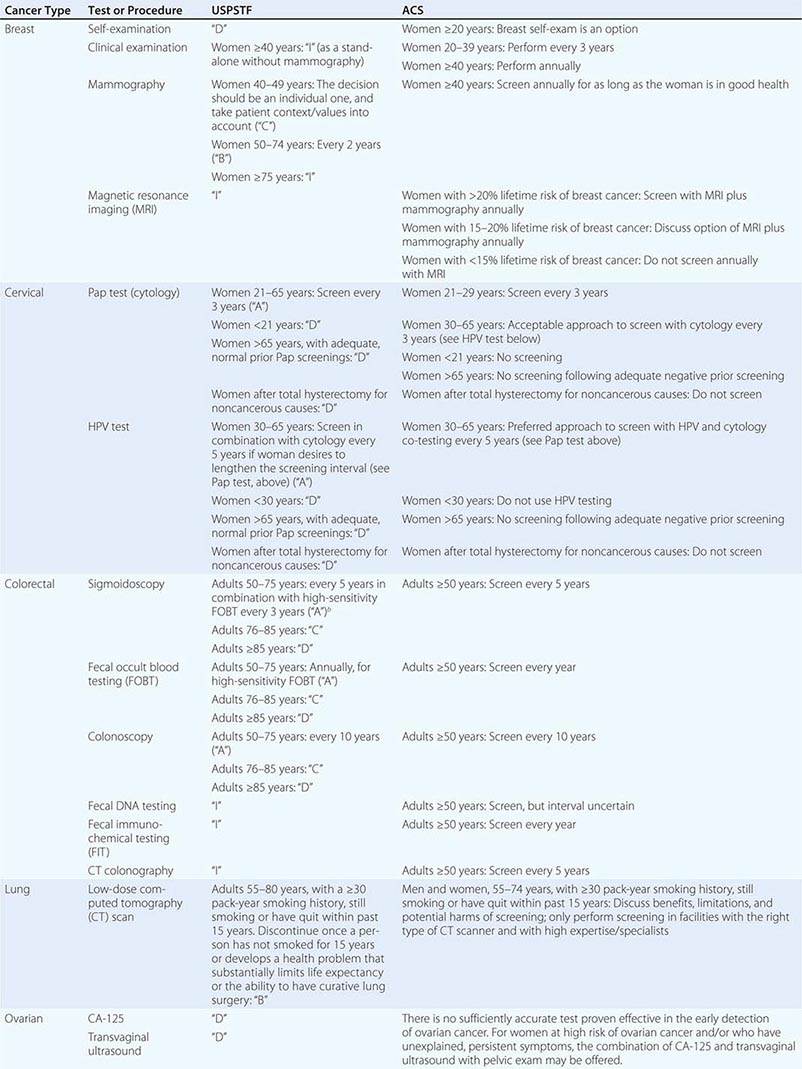

BREAST CANCER Breast self-examination, clinical breast examination by a caregiver, mammography, and magnetic resonance imaging (MRI) have all been variably advocated as useful screening tools.
A number of trials have suggested that annual or biennial screening with mammography or mammography plus clinical breast examination in normal-risk women older than age 50 years decreases breast cancer mortality. Each trial has been criticized for design flaws. In most trials, breast cancer mortality rate is decreased by 15–30%. Experts disagree on whether average-risk women age 40–49 years should receive regular screening (Table 100-3). The U.K. Age Trial, the only randomized trial of breast cancer screening to specifically evaluate the impact of mammography in women age 40–49 years, found no statistically significant difference in breast cancer mortality for screened women versus controls after about 11 years of follow-up (relative risk 0.83; 95% confidence interval 0.66–1.04); however, <70% of women received screening in the intervention arm, potentially diluting the observed effect. A meta-analysis of eight large randomized trials showed a 15% relative reduction in mortality (relative risk 0.85; 95% confidence interval 0.75–0.96) from mammography screening for women age 39–49 years after 11–20 years of follow-up. This is equivalent to a number needed to invite to screening of 1904 over 10 years to prevent one breast cancer death. At the same time, nearly half of women age 40–49 years screened annually will have false-positive mammograms necessitating further evaluation, often including biopsy. Estimates of overdiagnosis range from 10 to 40% of diagnosed invasive cancers. In the United States, widespread screening over the last several decades has not been accompanied by a reduction in incidence of metastatic breast cancer despite a large increase in early-stage disease, suggesting a substantial amount of overdiagnosis at the population level.
No study of breast self-examination has shown it to decrease mortality. A randomized controlled trial of approximately 266,000 women in China demonstrated no difference in breast cancer mortality between a group that received intensive breast self-exam instruction and reinforcement/reminders and controls at 10 years of follow-up. However, more benign breast lesions were discovered and more breast biopsies were performed in the self-examination arm.
Genetic screening for BRCA1 and BRCA2 mutations and other markers of breast cancer risk has identified a group of women at high risk for breast cancer. Unfortunately, when to begin and the optimal frequency of screening have not been defined. Mammography is less sensitive at detecting breast cancers in women carrying BRCA1 and BRCA2 mutations, possibly because such cancers occur in younger women, in whom mammography is known to be less sensitive. MRI screening may be more sensitive than mammography in women at high risk due to genetic predisposition or in women with very dense breast tissue, but specificity may be lower. An increase in overdiagnosis may accompany the higher sensitivity. The impact of MRI on breast cancer mortality with or without concomitant use of mammography has not been evaluated in a randomized controlled trial.
CERVICAL CANCER Screening with Papanicolaou (Pap) smears decreases cervical cancer mortality. The cervical cancer mortality rate has fallen substantially since the widespread use of the Pap smear. With the onset of sexual activity comes the risk of sexual transmission of HPV, the fundamental etiologic factor for cervical cancer. Screening guidelines recommend regular Pap testing for all women who have reached the age of 21 (prior to this age, even in individuals that have begun sexual activity, screening may cause more harm than benefit). The recommended interval for Pap screening is 3 years. Screening more frequently adds little benefit but leads to important harms, including unnecessary procedures and overtreatment of transient lesions. Beginning at age 30, guidelines also offer the alternative of combined Pap smear and HPV testing for women. The screening interval for women who test normal using this approach may be lengthened to 5 years.
An upper age limit at which screening ceases to be effective is not known, but women age 65 years with no abnormal results in the previous 10 years may choose to stop screening. Screening should be discontinued in women who have undergone a hysterectomy for noncancerous reasons.
Although the efficacy of the Pap smear in reducing cervical cancer mortality has never been directly confirmed in a randomized, controlled setting, a clustered randomized trial in India evaluated the impact of one-time cervical visual inspection and immediate colposcopy, biopsy, and/or cryotherapy (where indicated) versus counseling on cervical cancer deaths in women age 30–59 years. After 7 years of follow-up, the age-standardized rate of death due to cervical cancer was 39.6 per 100,000 person-years in the intervention group versus 56.7 per 100,000 person-years in controls.
COLORECTAL CANCER Fecal occult blood testing (FOBT), digital rectal examination (DRE), rigid and flexible sigmoidoscopy, colonoscopy, and computed tomography (CT) colonography have been considered for colorectal cancer screening. A meta-analysis of four randomized controlled trials demonstrated a 15% relative reduction in colorectal cancer mortality with FOBT. The sensitivity for FOBT is increased if specimens are rehydrated before testing, but at the cost of lower specificity. The false-positive rate for rehydrated FOBT is high; 1–5% of persons tested have a positive test. Only 2–10% of those with occult blood in the stool have cancer. The high false-positive rate of FOBT dramatically increases the number of colonoscopies performed.
Fecal immunochemical tests appear to have higher sensitivity for colorectal cancer than nonrehydrated FOBT tests. Fecal DNA testing is an emerging testing modality; it may have increased sensitivity and comparable specificity to FOBT and could potentially reduce harms associated with follow-up of false-positive tests. The body of evidence on the operating characteristics and effectiveness of fecal DNA tests in reducing colorectal cancer mortality is limited.
Two meta-analyses of five randomized controlled trials of sigmoidoscopy (i.e., the NORCCAP, SCORE, PLCO, Telemark, and U.K. trials) found an 18% relative reduction in colorectal cancer incidence and a 28% relative reduction in colorectal cancer mortality. Participant ages ranged from 50 to 74 years, with follow-up ranging from 6 to 13 years. Diagnosis of adenomatous polyps by sigmoidoscopy should lead to evaluation of the entire colon with colonoscopy. The most efficient interval for screening sigmoidoscopy is unknown, but an interval of 5 years is often recommended. Case-control studies suggest that intervals of up to 15 years may confer benefit; the U.K. trial demonstrated benefit with one-time screening.
One-time colonoscopy detects ∼25% more advanced lesions (polyps >10 mm, villous adenomas, adenomatous polyps with high-grade dysplasia, invasive cancer) than one-time FOBT with sigmoidoscopy; comparative programmatic performance of the two modalities over time is not known. Perforation rates are about 3/1000 for colonoscopy and 1/1000 for sigmoidoscopy. Debate continues on whether colonoscopy is too expensive and invasive and whether sufficient provider capacity exists to be recommended as the preferred screening tool in standard-risk populations. Some observational studies suggest that efficacy of colonoscopy to decrease colorectal cancer mortality is primarily limited to the left side of the colon.
CT colonography, if done at expert centers, appears to have a sensitivity for polyps ≥6 mm comparable to colonoscopy. However, the rate of extracolonic findings of abnormalities of uncertain significance that must nevertheless be worked up is high (∼15–30%); the long-term cumulative radiation risk of repeated colonography screenings is also a concern.
LUNG CANCER Chest x-ray and sputum cytology have been evaluated in several randomized lung cancer screening trials. The most recent and largest (n = 154,901) of these, one substudy of the Prostate, Lung, Colorectal, and Ovarian (PLCO) cancer screening trial, found that, compared with usual care, annual chest x-ray did not reduce the risk of dying from lung cancer (relative risk 0.99; 95% confidence interval 0.87–1.22) after 13 years. Low-dose CT has also been evaluated in several randomized trials. The largest and longest of these, the National Lung Screening Trial (NLST), was a randomized controlled trial of screening for lung cancer in approximately 53,000 persons age 55–74 years with a 30+ pack-year smoking history. It demonstrated a statistically significant relative reduction of about 15–20% in lung cancer mortality in the CT arm compared to the chest x-ray arm (or about 3 fewer deaths per 1000 people screened with CT). However, the harms include the potential radiation risks associated with multiple scans, the discovery of incidental findings of unclear significance, and a high rate of false-positive test results. Both incidental findings and false-positive tests can lead to invasive diagnostic procedures associated with anxiety, expense, and complications (e.g., pneumo- or hemothorax after lung biopsy). The NLST was performed at experienced screening centers, and the balance of benefits and harms may differ in the community setting at less experienced centers.
OVARIAN CANCER Adnexal palpation, transvaginal ultrasound (TVUS), and serum CA-125 assay have been considered for ovarian cancer screening. A large randomized controlled trial has shown that an annual screening program of TVUS and CA-125 in average-risk women does not reduce deaths from ovarian cancer (relative risk 1.21; 95% confidence interval 0.99–1.48). Adnexal palpation was dropped early in the study because it did not detect any ovarian cancers that were not detected by either TVUS or CA-125. The risks and costs associated with the high number of false-positive results are impediments to routine use of these modalities for screening. In the PLCO trial, 10% of participants had a false-positive result from TVUS or CA-125, and one-third of these women underwent a major surgical procedure; the ratio of surgeries to screen-detected ovarian cancer was approximately 20:1.
PROSTATE CANCER The most common prostate cancer screening modalities are DRE and serum PSA assay. An emphasis on PSA screening has caused prostate cancer to become the most common nonskin cancer diagnosed in American males. This disease is prone to lead-time bias, length bias, and overdiagnosis, and substantial debate continues among experts as to whether screening should be offered unless the patient specifically asks to be screened. Virtually all organizations stress the importance of informing men about the uncertainty regarding screening efficacy and the harms associated with screening. Prostate cancer screening clearly detects many asymptomatic cancers, but the ability to distinguish tumors that are lethal but still curable from those that pose little or no threat to health is limited, and randomized trials indicate that the effect of PSA screening on prostate cancer mortality across a population is, at best, small. Men older than age 50 years have a high prevalence of indolent, clinically insignificant prostate cancers (about 30–50% of men, increasing further as men age).
Two major randomized controlled trials of the impact of PSA screening on prostate cancer mortality have been published. The PLCO Cancer Screening Trial was a multicenter U.S. trial that randomized almost 77,000 men age 55–74 years to receive either annual PSA testing for 6 years or usual care. At 13 years of follow-up, no statistically significant difference in the number of prostate cancer deaths were noted between the arms (rate ratio 1.09; 95% confidence interval 0.87–1.36). Approximately 50% of men in the control arm received at least one PSA test during the trial, which may have potentially diluted a small effect.
The European Randomized Study of Screening for Prostate Cancer (ERSPC) was a multinational study that randomized approximately 182,000 men between age 50 and 74 years (with a predefined “core” screening group of men age 55–69 years) to receive PSA testing or no screening. Recruitment and randomization procedures, as well as actual frequency of PSA testing, varied by country. After a median follow-up of 11 years, a 20% relative reduction in the risk of prostate cancer death in the screened arm was noted in the “core” screening group. The trial found that 1055 men would need to be invited to screening, and 37 cases of prostate cancer detected, to avert 1 death from prostate cancer. Of the seven countries included in the mortality analysis, two demonstrated statistically significant reductions in prostate cancer deaths, whereas five did not. There was also an imbalance in treatment between the two study arms, with a higher proportion of men with clinically localized cancer receiving radical prostatectomy in the screening arm and receiving it at experienced referral centers.
Treatments for low-stage prostate cancer, such as surgery and radiation therapy, can cause significant morbidity, including impotence and urinary incontinence. In a trial conducted in the United States after the initiation of widespread PSA testing, random assignment to radical prostatectomy compared with “watchful waiting” did not result in a statistically significant decrease in prostate cancer deaths (absolute risk reduction 2.7%; 95% confidence interval–1.3 to 6.2%).
SKIN CANCER Visual examination of all skin surfaces by the patient or by a health care provider is used in screening for basal and squamous cell cancers and melanoma. No prospective randomized study has been performed to look for a mortality decrease. Unfortunately, screening is associated with a substantial rate of overdiagnosis.
101e |
Cancer Genetics |
CANCER IS A GENETIC DISEASE
Cancer arises through a series of somatic alterations in DNA that result in unrestrained cellular proliferation. Most of these alterations involve actual sequence changes in DNA (i.e., mutations). They may originate as a consequence of random replication errors, exposure to carcinogens (e.g., radiation), or faulty DNA repair processes. While most cancers arise sporadically, familial clustering of cancers occurs in certain families that carry a germline mutation in a cancer gene.
HISTORICAL PERSPECTIVE
The idea that cancer progression is driven by sequential somatic mutations in specific genes has only gained general acceptance in the past 25 years. Before the advent of the microscope, cancer was believed to be composed of aggregates of mucus or other noncellular matter. By the middle of the nineteenth century, it became clear that tumors were masses of cells and that these cells arose from the normal cells of the tissue from which the cancer originated. However, the molecular basis for the uncontrolled proliferation of cancer cells was to remain a mystery for another century. During that time, a number of theories for the origin of cancer were postulated. The great biochemist Otto Warburg proposed the combustion theory of cancer, which stipulated that cancer was due to abnormal oxygen metabolism. In addition, some believed that all cancers were caused by viruses, and that cancer was in fact a contagious disease.
In the end, observations of cancer occurring in chimney sweeps, studies of x-rays, and the overwhelming data demonstrating cigarette smoke as a causative agent in lung cancer, together with Ames’s work on chemical mutagenesis, provided convincing evidence that cancer originated through changes in DNA. Although the viral theory of cancer did not prove to be generally accurate (with the exception of human papillomaviruses, which can cause cervical and other cancers in human), the study of retroviruses led to the discovery of the first human oncogenes in the late 1970s. Soon after, the study of families with genetic predisposition to cancer was instrumental in the discovery of tumor-suppressor genes. The field that studies the type of mutations, as well as the consequence of these mutations in tumor cells, is now known as cancer genetics.
THE CLONAL ORIGIN AND MULTISTEP NATURE OF CANCER
Nearly all cancers originate from a single cell; this clonal origin is a critical discriminating feature between neoplasia and hyperplasia. Multiple cumulative mutational events are invariably required for the progression of a tumor from normal to fully malignant phenotype. The process can be seen as Darwinian microevolution in which, at each successive step, the mutated cells gain a growth advantage resulting in an increased representation relative to their neighbors (Fig. 101e-1). Based on observations of cancer frequency increases during aging, as well as molecular genetics work, it is believed that 5 to 10 accumulated mutations are necessary for a cell to progress from the normal to the fully malignant phenotype.
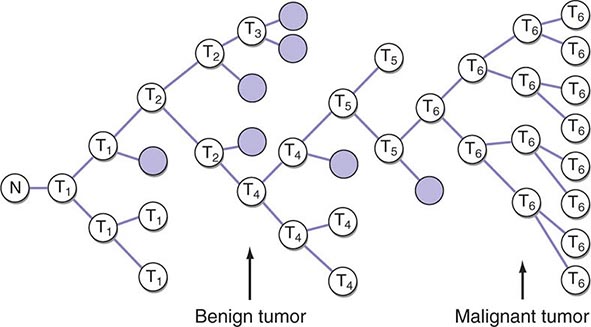
FIGURE 101e-1 Multistep clonal development of malignancy. In this diagram a series of five cumulative mutations (T1, T2, T4, T5, T6), each with a modest growth advantage acting alone, eventually results in a malignant tumor. Note that not all such alterations result in progression; for example, the T3 clone is a dead end. The actual number of cumulative mutations necessary to transform from the normal to the malignant state is unknown in most tumors. (After P Nowell: Science 194:23, 1976, with permission.)
We are beginning to understand the precise nature of the genetic alterations responsible for some malignancies and to get a sense of the order in which they occur. The best-studied example is colon cancer, in which analyses of DNA from tissues extending from normal colon epithelium through adenoma to carcinoma have identified some of the genes mutated in the process (Fig. 101e-2). Other malignancies are believed to progress in a similar stepwise fashion, although the order and identity of genes affected may be different.
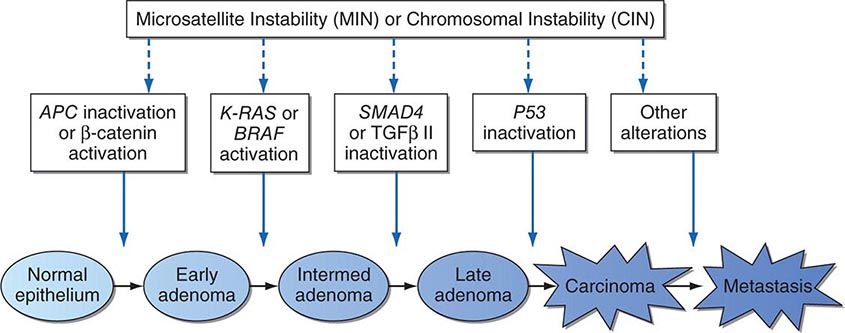
FIGURE 101e-2 Progressive somatic mutational steps in the development of colon carcinoma. The accumulation of alterations in a number of different genes results in the progression from normal epithelium through adenoma to full-blown carcinoma. Genetic instability (microsatellite or chromosomal) accelerates the progression by increasing the likelihood of mutation at each step. Patients with familial polyposis are already one step into this pathway, because they inherit a germline alteration of the APC gene. TGF, transforming growth factor.
TWO TYPES OF CANCER GENES: ONCOGENES AND TUMOR-SUPPRESSOR GENES
There are two major types of cancer genes. The first type comprises genes that positively influence tumor formation and are known as oncogenes. The second type of cancer genes negatively impact tumor growth and have been named tumor-suppressor genes. Both oncogenes and tumor-suppressor genes exert their effects on tumor growth through their ability to control cell division (cell birth) or cell death (apoptosis), although the mechanisms can be extremely complex. While tightly regulated in normal cells, oncogenes acquire mutations in cancer cells, and the mutations typically relieve this control and lead to increased activity of the gene products. This mutational event typically occurs in a single allele of the oncogene and acts in a dominant fashion. In contrast, the normal function of tumor-suppressor genes is usually to restrain cell growth, and this function is lost in cancer. Because of the diploid nature of mammalian cells, both alleles must be inactivated for a cell to completely lose the function of a tumor-suppressor gene, leading to a recessive mechanism at the cellular level. From these ideas and studies on the inherited form of retinoblastoma, Knudson and others formulated the two-hit hypothesis, which in its modern version states that both copies of a tumor-suppressor gene must be inactivated in cancer.
There is a subset of tumor-suppressor genes, the caretaker genes, that do not affect cell growth directly, but rather control the ability of the cell to maintain the integrity of its genome. Cells with a deficiency in these genes have an increased rate of mutations throughout their genomes, including in oncogenes and tumor-suppressor genes. This “mutator” phenotype was first hypothesized by Loeb to explain how the multiple mutational events required for tumorigenesis can occur in the lifetime of an individual. A mutator phenotype has now been observed in some forms of cancer, such as those associated with deficiencies in DNA mismatch repair. The great majority of cancers, however, do not harbor repair deficiencies, and their rate of mutation is similar to that observed in normal cells. Many of these cancers, however, appear to harbor a different kind of genetic instability, affecting the loss or gains of whole chromosomes or large parts thereof (as explained in more detail below).
ONCOGENES IN HUMAN CANCER
Work by Peyton Rous in the early 1900s revealed that a chicken sarcoma could be transmitted from animal to animal in cell-free extracts, suggesting that cancer could be induced by an agent acting positively to promote tumor formation. The agent responsible for the transmission of the cancer was a retrovirus (Rous sarcoma virus, RSV) and the oncogene responsible was identified 75 years later as v-src. Other oncogenes were also discovered through their presence in the genomes of retroviruses that are capable of causing cancers in chickens, mice, and rats. The cellular homologues of these viral genes are called protooncogenes and are often targets of mutation or aberrant regulation in human cancer. Whereas many oncogenes were discovered because of their presence in retroviruses, other oncogenes, particularly those involved in translocations characteristic of particular leukemias and lymphomas, were isolated through genomic approaches. Investigators cloned the sequences surrounding the chromosomal translocations observed cytogenetically and then deduced the nature of the genes that were the targets of these translocations (see below). Some of these were oncogenes known from retroviruses (like ABL, involved in chronic myeloid leukemia [CML]), whereas others were new (like BCL2, involved in B cell lymphoma). In the normal cellular environment, protooncogenes have crucial roles in cell proliferation and differentiation. Table 101e-1 is a partial list of oncogenes known to be involved in human cancer.
|
COMMON ONCOGENES ALTERED IN HUMAN CANCERS |
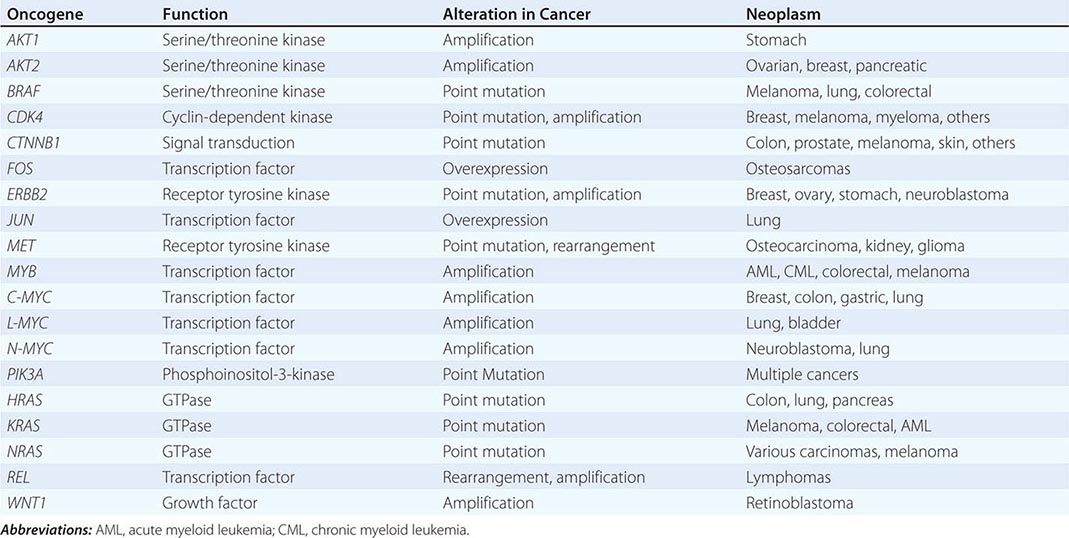
The normal growth and differentiation of cells is controlled by growth factors that bind to receptors on the surface of the cell. The signals generated by the membrane receptors are transmitted inside the cells through signaling cascades involving kinases, G proteins, and other regulatory proteins. Ultimately, these signals affect the activity of transcription factors in the nucleus, which regulate the expression of genes crucial in cell proliferation, cell differentiation, and cell death. Oncogene products have been found to function at critical steps in these pathways (Chap. 102e), and inappropriate activation of these pathways can lead to tumorigenesis.
MECHANISMS OF ONCOGENE ACTIVATION
POINT MUTATION
Point mutation is a common mechanism of oncogene activation. For example, mutations in one of the RAS genes (HRAS, KRAS, or NRAS) are present in up to 85% of pancreatic cancers and 45% of colon cancers but are less common in other cancer types, although they can occur at significant frequencies in leukemia, lung, and thyroid cancers. Remarkably—and in contrast to the diversity of mutations found in tumor-suppressor genes (see below)—most of the activated RAS genes contain point mutations in codons 12, 13, or 61 (these mutations reduce RAS GTPase activity, leading to constitutive activation of the mutant RAS protein). The restricted pattern of mutations observed in oncogenes compared to that of tumor-suppressor genes reflects the fact that gain-of-function mutations are less likely to occur than mutations that simply lead to loss of activity. Indeed, inactivation of a gene can in theory be accomplished through the introduction of a stop codon anywhere in the coding sequence, whereas activations require precise substitutions at residues that can somehow lead to an increase in the activity of the encoded protein. Importantly, the specificity of oncogene mutations provides diagnostic opportunities, as tests that identify mutations at defined positions are easier to design than tests aimed at detecting random changes in a gene.
DNA AMPLIFICATION
The second mechanism for activation of oncogenes is DNA sequence amplification, leading to overexpression of the gene product. This increase in DNA copy number may cause cytologically recognizable chromosome alterations referred to as homogeneous staining regions (HSRs) if integrated within chromosomes, or double minutes (dmins) if extrachromosomal. The recognition of DNA amplification is accomplished through various cytogenetic techniques such as comparative genomic hybridization (CGH) or fluorescence in situ hybridization (FISH), which allow the visualization of chromosomal aberrations using fluorescent dyes. In addition, noncytogenetic, microarray-based approaches are now available for identifying changes in copy number at high resolution. Newer short-tag–based sequencing approaches have been used to evaluate amplifications. When paired with next-generation sequencing, this approach offers the highest degree of resolution and quantification available. With both microarray and sequencing technologies, the entire genome can be surveyed for gains and losses of DNA sequences, thus pinpointing chromosomal regions likely to contain genes important in the development or progression of cancer.
Numerous genes have been reported to be amplified in cancer. Several of these genes, including NMYC and LMYC, were identified through their presence within the amplified DNA sequences of a tumor and had homology to known oncogenes. Because the region amplified often includes hundreds of thousands of base pairs, multiple oncogenes may be amplified in a single amplicon in some cancers (particularly in sarcomas). Indeed, MDM2, GLI, CDK4, and SAS at chromosomal location 12q13-15 have been shown to be simultaneously amplified in several types of sarcomas and other tumors. Amplification of a cellular gene is often a predictor of poor prognosis; for example, ERBB2/HER2 and NMYC are often amplified in aggressive breast cancers and neuroblastoma, respectively.
CHROMOSOMAL REARRANGEMENT
Chromosomal alterations provide important clues to the genetic changes in cancer. The chromosomal alterations in human solid tumors such as carcinomas are heterogeneous and complex and occur as a result of the frequent chromosomal instability (CIN) observed in these tumors (see below). In contrast, the chromosome alterations in myeloid and lymphoid tumors are often simple translocations, i.e., reciprocal transfers of chromosome arms from one chromosome to another. Consequently, many detailed and informative chromosome analyses have been performed on hematopoietic cancers. The breakpoints of recurring chromosome abnormalities usually occur at the site of cellular oncogenes. Table 101e-2 lists representative examples of recurring chromosome alterations in malignancy and the associated gene(s) rearranged or deregulated by the chromosomal rearrangement. Translocations are particularly common in lymphoid tumors, probably because these cell types have the capability to rearrange their DNA to generate antigen receptors. Indeed, antigen receptor genes are commonly involved in the translocations, implying that an imperfect regulation of receptor gene rearrangement may be involved in the pathogenesis. An interesting example is Burkitt’s lymphoma, a B cell tumor characterized by a reciprocal translocation between chromosomes 8 and 14. Molecular analysis of Burkitt’s lymphomas demonstrated that the breakpoints occurred within or near the MYC locus on chromosome 8 and within the immunoglobulin heavy chain locus on chromosome 14, resulting in the transcriptional activation of MYC. Enhancer activation by translocation, although not universal, appears to play an important role in malignant progression. In addition to transcription factors and signal transduction molecules, translocation may result in the overexpression of cell cycle regulatory proteins or proteins such as cyclins and of proteins that regulate cell death.
|
REPRESENTATIVE ONCOGENES AT CHROMOSOMAL TRANSLOCATIONS |
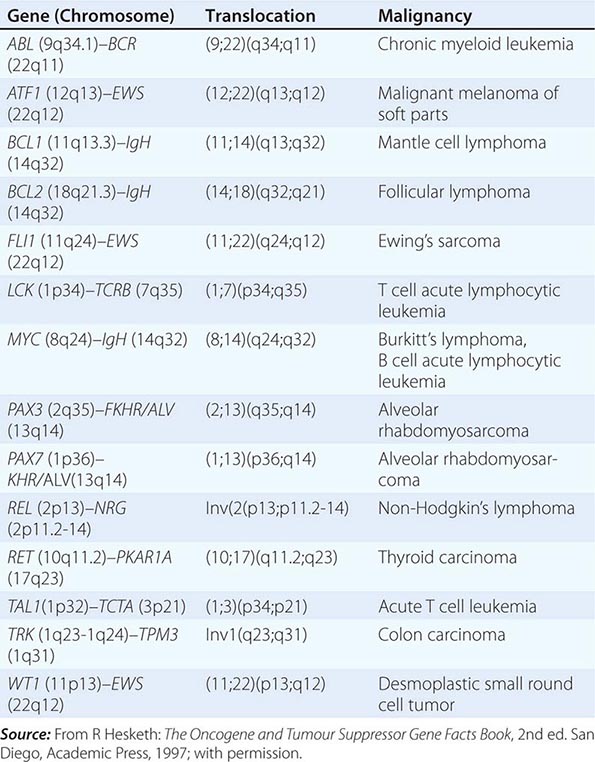
The first reproducible chromosome abnormality detected in human malignancy was the Philadelphia chromosome detected in CML. This cytogenetic abnormality is generated by reciprocal translocation involving the ABL oncogene on chromosome 9, encoding a tyrosine kinase, being placed in proximity to the BCR (breakpoint cluster region) gene on chromosome 22. Figure 101e-3 illustrates the generation of the translocation and its protein product. The consequence of expression of the BCR–ABL gene product is the activation of signal transduction pathways leading to cell growth independent of normal external signals. Imatinib (marketed as Gleevec), a drug that specifically blocks the activity of Abl tyrosine kinase, has shown remarkable efficacy with little toxicity in patients with CML. It is hoped that knowledge of genetic alterations in other cancers will likewise lead to mechanism-based design and development of a new generation of chemotherapeutic agents.
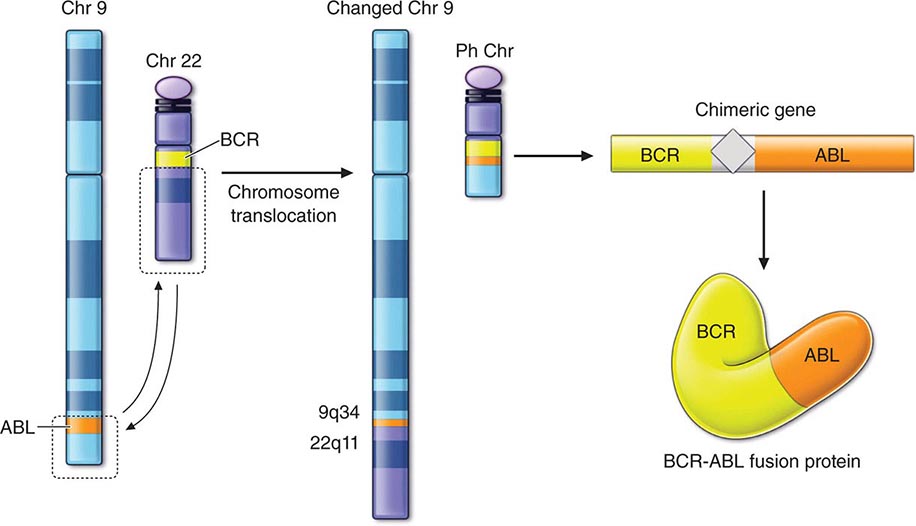
FIGURE 101e-3 Specific translocation seen in chronic myeloid leukemia (CML). The Philadelphia chromosome (Ph) is derived from a reciprocal translocation between chromosomes 9 and 22 with the breakpoint joining the sequences of the ABL oncogene with the BCR gene. The fusion of these DNA sequences allows the generation of an entirely novel fusion protein with modified function.
CHROMOSOMAL INSTABILITY IN SOLID TUMORS
Solid tumors are generally highly aneuploid, containing an abnormal number of chromosomes; these chromosomes also exhibit structural alterations such as translocations, deletions, and amplifications. These abnormalities are collectively referred to as chromosomal instability (CIN). Normal cells possess several cell cycle checkpoints, essentially quality-control requirements that have to be met before subsequent events are allowed to take place. The mitotic checkpoint, which ensures proper chromosome attachment to the mitotic spindle before allowing the sister chromatids to separate, is altered in certain cancers. The molecular basis of CIN remains unclear, although a number of mitotic checkpoint genes are found mutated or abnormally expressed in various tumors. The exact effects of these changes on the mitotic checkpoint are unknown, and both weakening and overactivation of the checkpoint have been proposed. The identification of the cause of CIN in tumors will likely be a formidable task, considering that several hundred genes are thought to control the mitotic checkpoint and other cellular processes ensuring proper chromosome segregation. Regardless of the mechanisms underlying CIN, the measurement of the number of chromosomal alterations present in tumors is now possible with both cytogenetic and molecular techniques, and several studies have shown that this information can be useful for prognostic purposes. In addition, because the mitotic checkpoint is essential for cellular viability, it may become a target for novel therapeutic approaches.
TUMOR-SUPPRESSOR GENE INACTIVATION IN CANCER
The first indication of the existence of tumor-suppressor genes came from experiments showing that fusion of mouse cancer cells with normal mouse fibroblasts led to a nonmalignant phenotype in the fused cells. The normal role of tumor-suppressor genes is to restrain cell growth, and the function of these genes is inactivated in cancer. The two major types of somatic lesions observed in tumor-suppressor genes during tumor development are point mutations and large deletions. Point mutations in the coding region of tumor-suppressor genes will frequently lead to truncated protein products or otherwise nonfunctional proteins. Similarly, deletions lead to the loss of a functional product and sometimes encompass the entire gene or even the entire chromosome arm, leading to loss of heterozygosity (LOH) in the tumor DNA compared to the corresponding normal tissue DNA (Fig. 101e-4). LOH in tumor DNA is considered a hallmark for the presence of a tumor-suppressor gene at a particular chromosomal location, and LOH studies have been useful in the positional cloning of many tumor-suppressor genes.
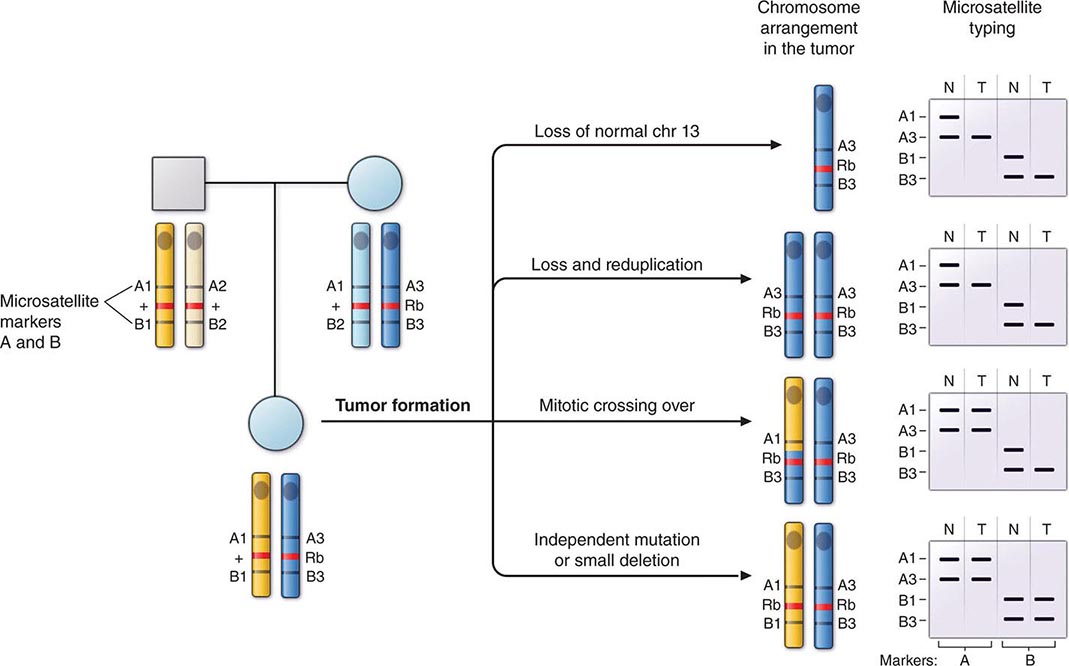
FIGURE 101e-4 Diagram of possible mechanisms for tumor formation in an individual with hereditary (familial) retinoblastoma. On the left is shown the pedigree of an affected individual who has inherited the abnormal (Rb) allele from her affected mother. The normal allele is shown as a (+). The four chromosomes of her two parents are drawn to indicate their origin. Flanking the retinoblastoma locus are microsatellite markers (A and B) also analyzed in this family. Markers A3 and B3 are on the chromosome carrying the retinoblastoma disease gene. Tumor formation results when the normal allele, which this patient inherited from her father, is inactivated. On the right are shown four possible ways in which this could occur. In each case, the resulting chromosome 13 arrangement is shown, as well as the results of PCR typing using the microsatellite markers comparing normal tissue (N) with tumor tissue (T). Note that in the first three situations, the normal allele (B1) has been lost in the tumor tissue, which is referred to as loss of heterozygosity (LOH) at this locus.
Gene silencing, an epigenetic change that leads to the loss of gene expression and occurs in conjunction with hypermethylation of the promoter and histone deacetylation, is another mechanism of tumor-suppressor gene inactivation. (An epigenetic modification refers to a change in the genome, heritable by cell progeny, that does not involve a change in the DNA sequence. The inactivation of the second × chromosome in female cells is an example of an epigenetic silencing that prevents gene expression from the inactivated chromosome.) During embryologic development, regions of chromosomes from one parent are silenced and gene expression proceeds from the chromosome of the other parent. For most genes, expression occurs from both alleles or randomly from one allele or the other. The preferential expression of a particular gene exclusively from the allele contributed by one parent is called parental imprinting and is thought to be regulated by covalent modifications of chromatin protein and DNA (often methylation) of the silenced allele.
The role of epigenetic control mechanisms in the development of human cancer is unclear. However, a general decrease in the level of DNA methylation has been noted as a common change in cancer. In addition, numerous genes, including some tumor-suppressor genes, appear to become hypermethylated and silenced during tumorigenesis. VHL and p16INK4 are well-studied examples of such tumor-suppressor genes. Overall, epigenetic mechanisms may be responsible for reprogramming the expression of a large number of genes in cancer and, together with the mutation of specific genes, are likely to be crucial in the development of human malignancies. The use of drugs that can reverse epigenetic changes in cancer cells may represent a novel therapeutic option in certain cancers or premalignant conditions. For example, demethylating agents (azacitidine or decitabine) are now approved by the U.S. Food and Drug Administration (FDA) for the treatment of patients with high-risk myelodysplastic syndrome (MDS).
FAMILIAL CANCER SYNDROMES
A small fraction of cancers occur in patients with a genetic predisposition. In these families, the affected individuals have a predisposing loss-of-function mutation in one allele of a tumor-suppressor gene. The tumors in these patients show a loss of the remaining normal allele as a result of somatic events (point mutations or deletions), in agreement with the two-hit hypothesis (Fig. 101e-4). Thus, most cells of an individual with an inherited loss-of-function mutation in a tumor-suppressor gene are functionally normal, and only the rare cells that develop a mutation in the remaining normal allele will exhibit uncontrolled regulation.
Roughly 100 syndromes of familial cancer have been reported, although many are rare. The majority are inherited as autosomal dominant traits, although some of those associated with DNA repair abnormalities (xeroderma pigmentosum, Fanconi’s anemia, ataxia telangiectasia) are autosomal recessive. Table 101e-3 shows a number of cancer predisposition syndromes and the responsible genes. The current paradigm states that the genes mutated in familial syndromes can also be targets for somatic mutations in sporadic (noninherited) tumors. The study of cancer syndromes has thus provided invaluable insights into the mechanisms of progression for many tumor types. This section examines the case of inherited colon cancer in detail, but similar lessons can be applied to many of the cancer syndromes listed in Table 101e-3. In particular, the study of inherited colon cancer will clearly illustrate the difference between two types of tumor-suppressor genes: the gatekeepers, which directly regulate the growth of tumors, and the caretakers, which, when mutated, lead to genetic instability and therefore act indirectly on tumor growth.
|
CANCER PREDISPOSITION SYNDROMES AND ASSOCIATED GENES |
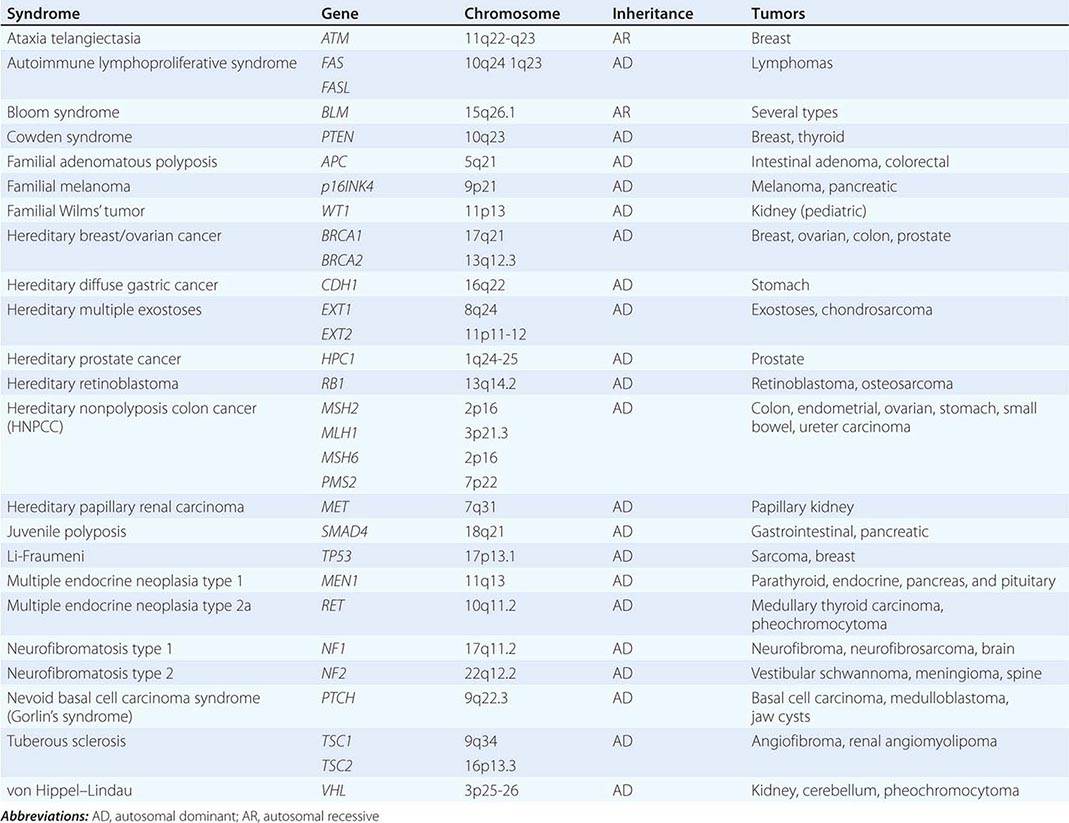
Familial adenomatous polyposis (FAP) is a dominantly inherited colon cancer syndrome due to germline mutations in the adenomatous polyposis coli (APC) tumor-suppressor gene on chromosome 5. Patients with this syndrome develop hundreds to thousands of adenomas in the colon. Each of these adenomas has lost the normal remaining allele of APC but has not yet accumulated the required additional mutations to generate fully malignant cells (Fig. 101e-2). The loss of the second functional APC allele in tumors from FAP families often occurs through loss of heterozygosity. However, out of these thousands of benign adenomas, several will invariably acquire further abnormalities and a subset will even develop into fully malignant cancers. APC is thus considered to be a gatekeeper for colon tumorigenesis: in the absence of mutation of this gatekeeper (or a gene acting within the same pathway), a colorectal tumor simply cannot form. Figure 101e-5 shows germline and somatic mutations found in the APC gene. The function of the APC protein is still not completely understood, but it likely provides differentiation and apoptotic cues to colonic cells as they migrate up the crypts. Defects in this process may lead to abnormal accumulation of cells that should normally undergo apoptosis.
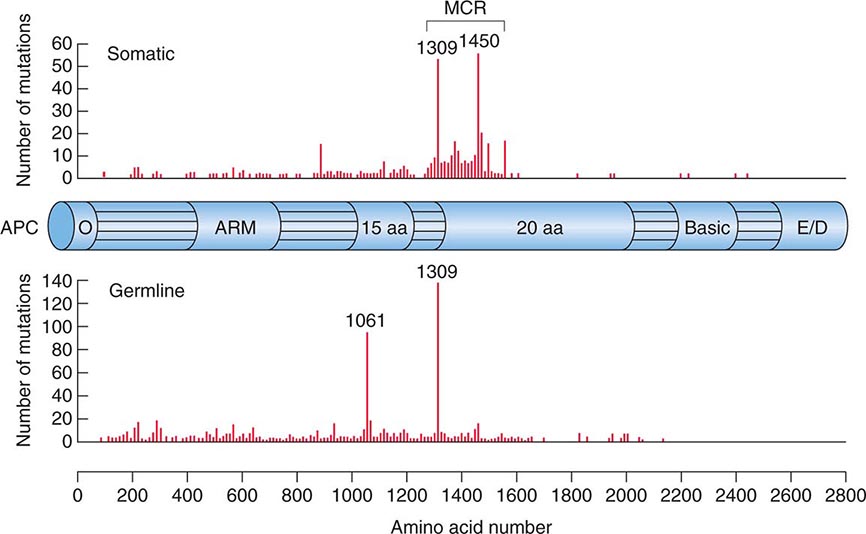
FIGURE 101e-5 Germline and somatic mutations in the tumor-suppressor gene APC. APC encodes a 2843-amino-acid protein with six major domains: an oligomerization region (O), armadillo repeats (ARM), 15-amino-acid repeats (15 aa), 20-amino-acid repeats (20 aa), a basic region, and a domain involved in binding EB1 and the Drosophila discs large homologue (E/D). Shown are the positions within the APC gene of a total of 650 somatic and 826 germline mutations (from the APC database at http://www.umd.be/APC). The vast majority of these mutations result in the truncation of the APC protein. Germline mutations are found to be relatively evenly distributed up to codon 1600 except for two mutation hotspots at amino acids 1061 and 1309, which together account for one-third of the mutations found in familial adenomatous polyposis (FAP) families. Somatic APC mutations in colon tumors cluster in an area of the gene known as the mutation cluster region (MCR). The location of the MCR suggests that the 20-amino-acid domain plays a crucial role in tumor suppression.
In contrast to patients with FAP, patients with hereditary nonpolyposis colon cancer (HNPCC, or Lynch’s syndrome) do not develop multiple polyposis, but instead develop only one or a small number of adenomas that rapidly progress to cancer. Most HNPCC cases are due to mutations in one of four DNA mismatch repair genes (Table 101e-3), which are components of a repair system that is normally responsible for correcting errors in freshly replicated DNA. Germline mutations in MSH2 and MLH1 account for more than 90% of HNPCC cases, whereas mutations in MSH6 and PMS2 are much less frequent. When a somatic mutation inactivates the remaining wild-type allele of a mismatch repair gene, the cell develops a hypermutable phenotype characterized by profound genomic instability, especially for the short repeated sequences called microsatellites. This microsatellite instability (MSI) favors the development of cancer by increasing the rate of mutations in many genes, including oncogenes and tumor-suppressor genes (Fig. 101e-2). These genes can thus be considered caretakers. Interestingly, CIN can also be found in colon cancer, but MSI and CIN appear to be mutually exclusive, suggesting that they represent alternative mechanisms for the generation of a mutator phenotype in this cancer (Fig. 101e-2). Other cancer types rarely exhibit MSI, but most exhibit CIN.
Although most autosomal dominant inherited cancer syndromes are due to mutations in tumor-suppressor genes (Table 101e-3), there are a few interesting exceptions. Multiple endocrine neoplasia type 2, a dominant disorder characterized by pituitary adenomas, medullary carcinoma of the thyroid, and (in some pedigrees) pheochromocytoma, is due to gain-of-function mutations in the protooncogene RET on chromosome 10. Similarly, gain-of-function mutations in the tyrosine kinase domain of the MET oncogene lead to hereditary papillary renal carcinoma. Interestingly, loss-of-function mutations in the RET gene cause a completely different disease, Hirschsprung’s disease (aganglionic megacolon [Chaps. 353 and 408]).
Although the Mendelian forms of cancer have taught us much about the mechanisms of growth control, most forms of cancer do not follow simple patterns of inheritance. In many instances (e.g., lung cancer), a strong environmental contribution is at work. Even in such circumstances, however, some individuals may be more genetically susceptible to developing cancer, given the appropriate exposure, due to the presence of modifier alleles.
GENETIC TESTING FOR FAMILIAL CANCER
The discovery of cancer susceptibility genes raises the possibility of DNA testing to predict the risk of cancer in individuals of affected families. An algorithm for cancer risk assessment and decision making in high-risk families using genetic testing is shown in Fig. 101e-6. Once a mutation is discovered in a family, subsequent testing of asymptomatic family members can be crucial in patient management. A negative gene test in these individuals can prevent years of anxiety in the knowledge that their cancer risk is no higher than that of the general population. On the other hand, a positive test may lead to alteration of clinical management, such as increased frequency of cancer screening and, when feasible and appropriate, prophylactic surgery. Potential negative consequences of a positive test result include psychological distress (anxiety, depression) and discrimination, although the Genetic Information Nondiscrimination Act (GINA) makes it illegal for predictive genetic information to be used to discriminate in health insurance or employment. Testing should therefore not be conducted without counseling before and after disclosure of the test result. In addition, the decision to test should depend on whether effective interventions exist for the particular type of cancer to be tested. Despite these caveats, genetic cancer testing for some cancer syndromes already appears to have greater benefits than risks. Companies offer genetic testing for many of the cancer syndromes listed in Table 83-3, including FAP (APC gene), hereditary breast and ovarian cancer syndrome (BRCA1 and BRCA2 genes), Lynch’s syndrome (mismatch repair genes), Li-Fraumeni syndrome (TP53 gene), Cowden syndrome (PTEN gene), hereditary retinoblastoma (RB1 gene), and others.
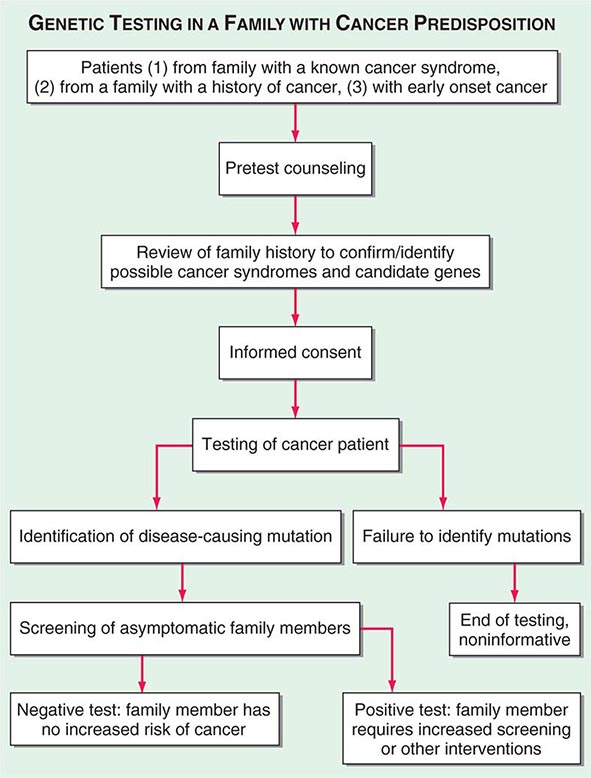
FIGURE 101e-6 Algorithm for genetic testing in a family with cancer predisposition. The key step is the identification of a mutation in a cancer patient, which allows testing of asymptomatic family members. Asymptomatic family members who test positive may require increased screening or surgery, whereas others are at no greater risk for cancer than the general population.
Because of the inherent problems of genetic testing such as cost, specificity, and sensitivity, it is not yet appropriate to offer these tests to the general population. However, testing may be appropriate in some subpopulations with a known increased risk, even without a defined family history. For example, two mutations in the breast cancer susceptibility gene BRCA1, 185delAG and 5382insC, exhibit a sufficiently high frequency in the Ashkenazi Jewish population that genetic testing of an individual of this ethnic group may be warranted.
As noted above, it is important that genetic test results be communicated to families by trained genetic counselors, especially for high-risk high-penetrance conditions such as the hereditary breast and ovarian cancer syndrome (BRCA1/BRCA2). To ensure that the families clearly understand its advantages and disadvantages and the impact it may have on disease management and psyche, genetic testing should never be done before counseling. Significant expertise is needed to communicate the results of genetic testing to families. For example, one common mistake is to misinterpret the result of negative genetic tests. For many cancer predisposition genes, the sensitivity of genetic testing is less than 70% (i.e., of 100 kindreds tested, disease-causing mutations can be identified in 70 at most). Therefore, such testing should in general begin with an affected member of the kindred (the youngest family member still alive who has had the cancer of interest). If a mutation is not identified in this individual, then the test should be reported as noninformative (Fig. 101e-6) rather than negative (because it is possible that, for technical reasons, the mutation in this individual is not detectable by standard genetic assays). On the other hand, if a mutation can be identified in this individual, then testing of other family members can be performed, and the sensitivity of such subsequent tests will be 100% (because the mutation in the family is in this case known to be detectable by the method used).
MICRORNAs AND CANCER
MicroRNAs (miRNAs) are small noncoding RNAs 20–22 nucleotides in length that are involved in posttranscriptional gene regulation. Studies in chronic lymphocytic leukemia first suggested a link between miRNAs and cancer when miR-15 and miR-16 were found to be deleted or downregulated in the vast majority of tumors. Various miRNAs have since been found abnormally expressed in several human malignancies. Aberrant expression of miRNAs in cancer has been attributed to several mechanisms, such as chromosomal rearrangements, genomic copy number change, epigenetic modifications, defects in miRNA biogenesis pathway, and regulation by transcriptional factors. Somatic mutations of miRNAs have been identified in many cancers, but the exact functional consequences of these changes on cancer development remain to be determined. The SomaMir database (http://compbio.uthsc.edu/SomamiR) catalogs somatic and germline miRNA mutations that have been identified in cancer.
Functionally, miRNAs have been suggested to contribute to tumorigenesis through their ability to regulate oncogenic signaling pathways. For example, miR-15 and miR-16 have been shown to target the BCL2 oncogene, leading to its downregulation in leukemic cells and apoptosis. As another example of miRNAs’ involvement in oncogenic pathways, the p53 tumor suppressor can transcriptionally induce miR-34 following genotoxic stress, and this induction is important in mediating p53 function. The expression of miRNAs is extremely specific, and there is evidence that miRNA expression patterns may be useful in distinguishing lineage and differentiation state, as well as cancer diagnosis and outcome prediction.
VIRUSES IN HUMAN CANCER
Certain human malignancies are associated with viruses. Examples include Burkitt’s lymphoma (Epstein-Barr virus; Chap. 218), hepatocellular carcinoma (hepatitis viruses), cervical cancer (human papillomavirus [HPV]; Chap. 222), and T cell leukemia (retroviruses; Chap. 225e). The mechanisms of action of these viruses are varied but always involve activation of growth-promoting pathways or inhibition of tumor-suppressor products in the infected cells. For example, HPV proteins E6 and E7 bind and inactivate cellular tumor suppressors p53 and pRB, respectively. There are several HPV types, and some of these types have been associated with the development of several malignancies, including cervical, vulvar, vaginal, penile, anal, and oropharyngeal cancer. Viruses are not sufficient for cancer development, but constitute one alteration in the multistep process of cancer progression.
GENE EXPRESSION IN CANCER
The tumorigenesis process, driven by alterations in tumor suppressors, oncogenes, and epigenetic regulation, is accompanied by changes in gene expression. The advent of powerful techniques for high-throughput gene expression profiling, based on sequencing or microarrays, has allowed the comprehensive study of gene expression in neoplastic cells. It is indeed possible to identify the expression levels of thousands of genes expressed in normal and cancer tissues. Figure 101e-7 shows a typical microarray experiment examining gene expression in cancer. This global knowledge of gene expression allows the identification of differentially expressed genes and, in principle, the understanding of the complex molecular circuitry regulating normal and neoplastic behaviors. Such studies have led to molecular profiling of tumors, which has suggested general methods for distinguishing tumors of various biologic behaviors (molecular classification), elucidating pathways relevant to the development of tumors, and identifying molecular targets for the detection and therapy of cancer. The first practical applications of this technology have suggested that global gene expression profiling can provide prognostic information not evident from other clinical or laboratory tests. The Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) is a searchable online repository for expression profiling data.
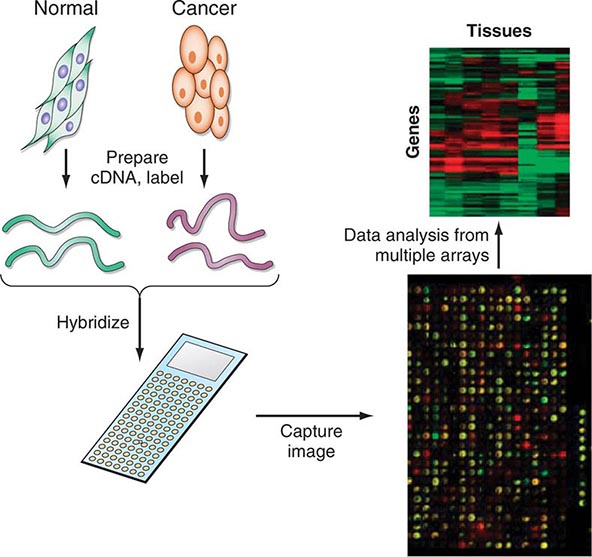
FIGURE 101e-7 A microarray experiment. RNA is prepared from cells, reverse transcribed to cDNA, and labeled with fluorescent dyes (typically green for normal cells and red for cancer cells). The fluorescent probes are mixed and hybridized to a cDNA array. Each spot on the array is an oligonucleotide (or cDNA fragment) that represents a different gene. The image is then captured with a fluorescence camera; red spots indicate higher expression in tumor cells compared with reference, while green spots represent the lower expression in tumor cells. Yellow signals indicate equal expression levels in normal and tumor specimens. After clustering analysis of multiple arrays, the results are typically represented graphically using a visualization software, which shows, for each sample, a color-coded representation of gene expression for every gene on the array.
GENOMEWIDE MUTATIONAL PROFILING IN CANCER
With the completion of the Human Genome Project and advances in sequencing technologies, systematic mutational analysis of the cancer genome has become possible. In fact, whole genome sequencing of cancer cells is now possible, and this technology has the potential to revolutionize our approach to cancer prevention, diagnosis, and treatment. The International Cancer Genome Consortium (http://icgc.org/) was developed by leading cancer agencies worldwide, genome and cancer scientists, and statisticians with the goal to launch and coordinate cancer genomics research projects worldwide and to disseminate the data. Hundreds of cancer genomes from at least 25 cancer types have been sequenced through various collaborative efforts. In addition, exome sequencing (sequencing all the coding regions of the genome) has also been performed on a large number of tumors. These sequencing data have been used to elucidate the mutational profile of cancer, including the identification of driver mutations that are functionally involved in tumor development. There are generally 40 to 100 genetic alterations that affect protein sequence in a typical cancer, although statistical analyses suggest that only 8–15 are functionally involved in tumorigenesis. The picture that emerges from these studies is that most genes found mutated in tumors are actually mutated at relatively low frequencies (<5%), whereas a small number of genes (such as p53, KRAS) are mutated in a large proportion of tumors (Fig. 101e-8). In the past, the focus of research has been on the frequently mutated genes, but it appears that the large number of genes that are infrequently mutated in cancer are major contributors to the cancer phenotype. Understanding the signaling pathways altered by mutations in these genes, as well as the functional relevance of these different mutations, represents the next challenge in the field. Moreover, a detailed knowledge of the genes altered in a particular tumor may allow for a new era of personalized treatment in cancer medicine (see below). A major effort in the United States, The Cancer Genome Atlas (http://cancergenome.nih.gov) is a coordinated effort from the National Cancer Institute and the National Human Genome Research Institute to systematically characterize the entire spectrum of genomic changes involved in human cancers. Similarly, COSMIC (Catalogue of Somatic Mutations in Cancer) is an initiative from the Welcome Trust Sanger Institute to store and display somatic mutation information and related details regarding human cancers (http://cancer.sanger.ac.uk/).
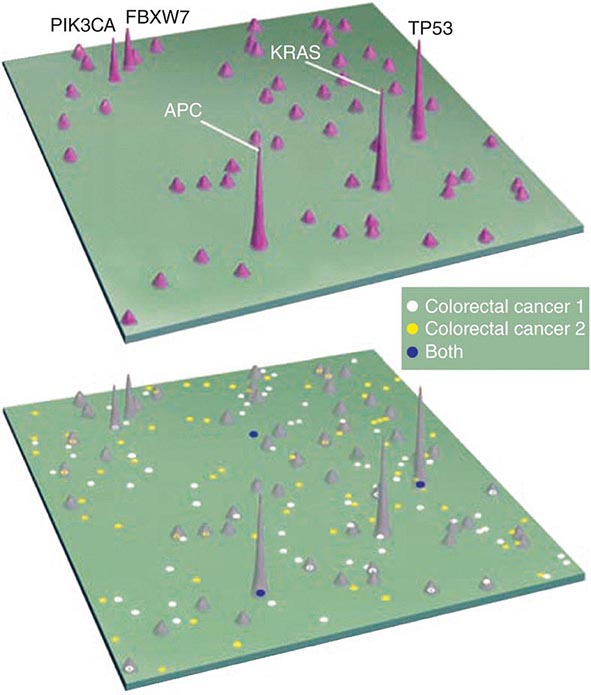
FIGURE 101e-8 A two-dimensional maps of genes mutated in colorectal cancer. The two-dimensional landscape represents the positions of the RefSeq genes along the chromosomes and the height of the peaks represents the mutation frequency. On the top map, the taller peaks represent the genes that are commonly mutated in colon cancer, while the large number of smaller hills indicates the genes that are mutated at lower frequency. On the lower map, the mutations of two individual tumors are indicated. Note that there is little overlap between the mutated genes of the two colorectal tumors shown. These differences may represent the basis for the heterogeneity in terms of behavior and responsiveness to therapy observed in human cancer. (From LD Wood et al: Science 318:1108, 2007, with permission.)
PERSONALIZED CANCER TREATMENT BASED ON MOLECULAR PROFILES: PRECISION THERAPY
Gene expression profiling and genomewide sequencing approaches have allowed for an unprecedented understanding of cancer at the molecular level. It has been suggested that individualized knowledge of pathways or genes deregulated in a given tumor (personalized genomics) may provide a guide for therapeutic options on the tumor, thus leading to personalized therapy (also called precision medicine). Because tumor behavior is highly heterogeneous, even within a tumor type, personalized information-based medicine will likely supplement or perhaps one day supplant the current histology-based therapy, especially in the case of tumors resistant to conventional therapeutic approaches. Molecular nosology has revealed similarities in tumors of diverse histotype. The success of this approach will be dependent on the identification of sufficient actionable changes (mutations or pathways that can be targeted with a specific drug). Examples of currently actionable changes include mutations in BRAF (targeted by the drug vemurafenib) and RET (targeted by sunitinib and sorafenib), and ALK rearrangements (targeted by crizotinib). Interestingly, studies have reported that 20% of triple-negative breast cancers and 60% of lung cancers have potentially actionable genetic changes. Gene expression also offers the potential to predict drug sensitivities as well as provide prognostic information. Commercial diagnostic tests, such as Mammaprint and Oncotype DX for breast cancer, are available to help the patients and their physicians make treatment decisions. Personalized medicine is an exciting new avenue for cancer treatment based on matching the unique features of a tumor to an effective therapy, and this concept is in the process of changing our approach to cancer therapy in fundamental ways. On a cautionary note, gene expression can vary enormously within a single person’s cancer and at different anatomic sites in the patient. We have not yet determined whether such clonal variation within an individual tumor will interfere with the goal tailoring therapy to a particular patient’s tumor.
THE FUTURE
A revolution in cancer genetics has occurred in the past 25 years. Identification of cancer genes has led to a deep understanding of the tumorigenesis process and has had important repercussions on all fields of cancer biology. In particular, the advancement of powerful techniques for genomewide expression profiling and mutation analyses has provided a detailed picture of the molecular defects present in individual tumors. Individualized treatment based on the specific genetic alterations within a given tumor has already become possible. Although these advances have not yet translated into overall changes in cancer prevention, prognosis, or treatment, it is expected that breakthroughs in these areas will continue to emerge and be applicable to an ever-increasing number of cancers.
102e |
Cancer Cell Biology |
Cancers are characterized by unregulated cell division, avoidance of cell death, tissue invasion, and the ability to metastasize. A neoplasm is benign when it grows in an unregulated fashion without tissue invasion. The presence of unregulated growth and tissue invasion is characteristic of malignant neoplasms. Cancers are named based on their origin: those derived from epithelial tissue are called carcinomas, those derived from mesenchymal tissues are sarcomas, and those derived from hematopoietic tissue are leukemias, lymphomas, and plasma cell dyscrasias (including multiple myeloma).
Cancers nearly always arise as a consequence of genetic alterations, the vast majority of which begin in a single cell and therefore are monoclonal in origin. However, because a wide variety of genetic and epigenetic changes can occur in different cells within malignant tumors over time, most cancers are characterized by marked heterogeneity in the populations of cells. This heterogeneity significantly complicates the treatment of most cancers because it is likely that there are subsets of cells that will be resistant to therapy and will therefore survive and proliferate even if the majority of cells are killed.
A few cancers appear to, at least initially, be primarily driven by an alteration in a dominant gene that produces uncontrolled cell proliferation. Examples include chronic myeloid leukemia (abl), about half of melanomas (braf), Burkitt’s lymphoma (c-myc), and subsets of lung adenocarcinomas (egfr, alk, ros1, and ret). The genes that can promote cell growth when altered are often called oncogenes. They were first identified as critical elements of viruses that cause animal tumors; it was subsequently found that the viral genes had normal counterparts with important functions in the cell and had been captured and mutated by viruses as they passed from host to host.
However, the vast majority of human cancers are characterized by a multiple-step process involving many genetic abnormalities, each of which contributes to the loss of control of cell proliferation and differentiation and the acquisition of capabilities, such as tissue invasion, the ability to metastasize, and angiogenesis. These properties are not found in the normal adult cell from which the tumor is derived. Indeed, normal cells have a large number of safeguards against uncontrolled proliferation and invasion. Many cancers go through recognizable steps of progressively more abnormal phenotypes: hyperplasia, to adenoma, to dysplasia, to carcinoma in situ, to invasive cancer with the ability to metastasize (Table 102e-1). For most cancers, these changes occur over a prolonged period of time, usually many years.
|
PHENOTYPIC CHARACTERISTICS OF MALIGNANT CELLS |
Abbreviations: FGF, fibroblast growth factor; IL, interleukin; MHC, major histocompatibility complex; VEGF, vascular endothelial growth factor.
In most organs, only primitive undifferentiated cells are capable of proliferating and the cells lose the capacity to proliferate as they differentiate and acquire functional capability. The expansion of the primitive cells is linked to some functional need in the host through receptors that receive signals from the local environment or through hormonal and other influences delivered by the vascular supply. In the absence of such signals, the cells are at rest. The signals that keep the primitive cells at rest remain incompletely understood. These signals must be environmental, based on the observations that a regenerating liver stops growing when it has replaced the portion that has been surgically removed after partial hepatectomy and regenerating bone marrow stops growing when the peripheral blood counts return to normal. Cancer cells clearly have lost responsiveness to such controls and do not recognize when they have overgrown the niche normally occupied by the organ from which they are derived. A better understanding of the mechanisms of growth regulation is evolving.
CELL CYCLE CHECKPOINTS
Normal cells have a number of control mechanisms that are targeted by specific genetic alterations in cancer. Critical proteins in these control processes that are frequently mutated or otherwise inactivated in cancers are called tumor-suppressor genes. Examples include p53 and Rb (discussed below). The progression of a cell through the cell division cycle is regulated at a number of checkpoints by a wide array of genes. In the first phase, G1, preparations are made to replicate the genetic material. The cell stops before entering the DNA synthesis phase, or S phase, to take inventory. Are we ready to replicate our DNA? Is the DNA repair machinery in place to fix any mutations that are detected? Are the DNA replicating enzymes available? Is there an adequate supply of nucleotides? Is there sufficient energy? The main brake on the process is the retinoblastoma protein, Rb. When the cell determines that it is prepared to move ahead, sequential activation of cyclin-dependent kinases (CDKs) results in the inactivation of the brake, Rb, by phosphorylation. Phosphorylated Rb releases the S phase–regulating transcription factor, E2F/DP1, and genes required for S phase progression are expressed. If the cell determines that it is unready to move ahead with DNA replication, a number of inhibitors are capable of blocking the action of the CDKs, including p21Cip2/Waf1, p16Ink4a, and p27Kip1. Nearly every cancer has one or more genetic lesions in the G1 checkpoint that permits progression to S phase.
At the end of S phase, when the cell has exactly duplicated its DNA content, a second inventory is taken at the S checkpoint. Have all of the chromosomes been fully duplicated? Were any segments of DNA copied more than once? Do we have the right number of chromosomes and the right amount of DNA? If so, the cell proceeds to G2, in which the cell prepares for division by synthesizing mitotic spindle and other proteins needed to produce two daughter cells. When DNA damage is detected, the p53 pathway is normally activated. Called the guardian of the genome, p53 is a transcription factor that is normally present in the cell in very low levels. Its level is generally regulated through its rapid turnover. Normally, p53 is bound to mdm2, a ubiquitin ligase, that both inhibits p53 transcriptional activation and also targets p53 for degradation in the proteasome. When damage is sensed, the ATM (ataxia-telangiectasia mutated) pathway is activated; ATM phosphorylates mdm2, which no longer binds to p53, and p53 then stops cell cycle progression, directs the synthesis of repair enzymes, or if the damage is too great, initiates apoptosis of the cell to prevent the propagation of a damaged cell (Fig. 102e-1).
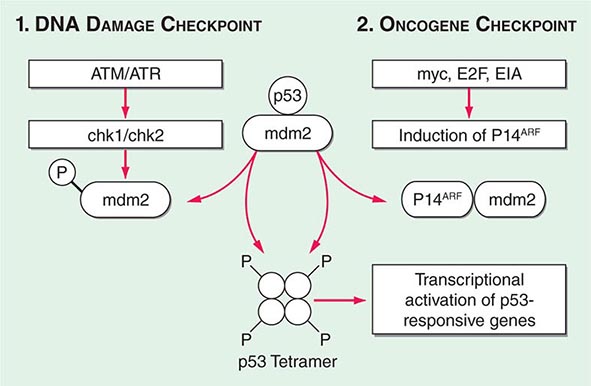
FIGURE 102e-1 Induction of p53 by the DNA damage and oncogene checkpoints. In response to noxious stimuli, p53 and mdm2 are phosphorylated by the ataxia-telangiectasia mutated (ATM) and related (ATR) serine/threonine kinases, as well as the immediate downstream checkpoint kinases, Chk1 and Chk2. This causes dissociation of p53 from mdm2, leading to increased p53 protein levels and transcription of genes leading to cell cycle arrest (p21Cip1/Waf1) or apoptosis (e.g., the proapoptotic Bcl-2 family members Noxa and Puma). Inducers of p53 include hypoxemia, DNA damage (caused by ultraviolet radiation, gamma irradiation, or chemotherapy), ribonucleotide depletion, and telomere shortening. A second mechanism of p53 induction is activated by oncogenes such as Myc, which promote aberrant G1/S transition. This pathway is regulated by a second product of the Ink4a locus, p14ARF (p19 in mice), which is encoded by an alternative reading frame of the same stretch of DNA that codes for p16Ink4a. Levels of ARF are upregulated by Myc and E2F, and ARF binds to mdm2 and rescues p53 from its inhibitory effect. This oncogene checkpoint leads to the death or senescence (an irreversible arrest in G1 of the cell cycle) of renegade cells that attempt to enter S phase without appropriate physiologic signals. Senescent cells have been identified in patients whose premalignant lesions harbor activated oncogenes, for instance, dysplastic nevi that encode an activated form of BRAF (see below), demonstrating that induction of senescence is a protective mechanism that operates in humans to prevent the outgrowth of neoplastic cells.
A second method of activating p53 involves the induction of p14ARF by hyperproliferative signals from oncogenes. p14ARF competes with p53 for binding to mdm2, allowing p53 to escape the effects of mdm2 and accumulate in the cell. Then p53 stops cell cycle progression by activating CDK inhibitors such as p21 and/or initiating the apoptosis pathway. Not surprisingly given its critical role in controlling cell cycle progression, mutations in the gene for p53 on chromosome 17p are found in more than 50% of human cancers. Most commonly these mutations are acquired in the malignant tissue in one allele and the second allele is deleted, leaving the cell unprotected from DNA-damaging agents or oncogenes. Some environmental exposures produce signature mutations in p53; for example, aflatoxin exposure leads to mutation of arginine to serine at codon 249 and leads to hepatocellular carcinoma. In rare instances, p53 mutations are in the germline (Li-Fraumeni syndrome) and produce a familial cancer syndrome. The absence of p53 leads to chromosome instability and the accumulation of DNA damage including the acquisition of properties that give the abnormal cell a proliferative and survival advantage. Like Rb dysfunction, most cancers have mutations that disable the p53 pathway. Indeed, the importance of p53 and Rb in the development of cancer is underscored by the neoplastic transformation mechanism of human papillomavirus. This virus has two main oncogenes, E6 and E7. E6 acts to increase the rapid turnover of p53, and E7 acts to inhibit Rb function; inhibition of these two targets is required for transformation of epithelial cells.
Another cell cycle checkpoint exists when the cell is undergoing division, the spindle checkpoint. The details of this checkpoint are still being discovered; however, it appears that if the spindle apparatus does not properly align the chromosomes for division, if the chromosome number is abnormal (i.e., greater or less than 4n), or if the centromeres are not properly paired with their duplicated partners, then the cell initiates a cell death pathway to prevent the production of aneuploid progeny (having an altered number of chromosomes). Abnormalities in the spindle checkpoint facilitate the development of aneuploidy. In some tumors, aneuploidy is a predominant genetic feature. In others, a defect in the cells’ ability to repair errors in the DNA due to mutations in genes coding for the proteins critical for mismatched DNA repair is the primary genetic lesion. This is usually detected by finding alterations in repeat sequences of DNA (called microsatellites) or microsatellite instability in malignant cells. In general, tumors either have defects in chromosome number or microsatellite instability, but not both. Defects that lead to cancer include abnormal cell cycle checkpoints, inadequate DNA repair, and failure to preserve genome integrity.
Efforts are under way to therapeutically restore the defects in cell cycle regulation that characterize cancer, although this remains a challenging problem because it is much more difficult to restore normal biologic function than to inhibit abnormal function of proteins driving cell proliferation, such as oncogenes.
CANCER AS AN ORGAN THAT IGNORES ITS NICHE
The fundamental cellular defects that create a malignant neoplasm act at the cellular level. However, that is not the entire story. Cancers behave as organs that have lost their specialized function and stopped responding to signals that normally limit their growth. Human cancers usually become clinically detectable when a primary mass is at least 1 cm in diameter—such a mass consists of about 109 cells. More commonly, patients present with tumors that are 1010 cells or greater. A lethal tumor burden is about 1012 to 1013 cells. If all tumor cells were dividing at the time of diagnosis, patients would reach a lethal tumor burden in a very short time. However, human tumors grow by Gompertzian kinetics—this means that not every daughter cell produced by a cell division is itself capable of dividing. The growth fraction of a tumor declines exponentially with time. The growth fraction of the first malignant cell is 100%, and by the time a patient presents for medical care, the growth fraction is 2–3% or less. This fraction is similar to the growth fraction of normal bone marrow and normal intestinal epithelium, the most highly proliferative normal tissues in the human body, a fact that may explain the dose-limiting toxicities of agents that target dividing cells.
The implication of these data is that the tumor is slowing its own growth over time. How does it do this? The tumor cells have multiple genetic lesions that tend to promote proliferation, yet by the time the tumor is clinically detectable, its capacity for proliferation has declined. We need to better understand how a tumor slows its own growth. A number of factors are known to contribute to the failure of tumor cells to proliferate in vivo. Some cells are hypoxemic and have inadequate supply of nutrients and energy. Some have sustained too much genetic damage to complete the cell cycle but have lost the capacity to undergo apoptosis and therefore survive but do not proliferate. However, an important subset is not actively dividing but retains the capacity to divide and can start dividing again under certain conditions such as when the tumor mass is reduced by treatments. Just as the bone marrow increases its rate of proliferation in response to bone marrow–damaging agents, the tumor also seems to sense when tumor cell numbers have been reduced and can respond by increasing growth rate. However, the critical difference is that the marrow stops growing when it has reached its production goals, whereas tumors do not.
Additional tumor cell vulnerabilities are likely to be detected when we learn more about how normal cells respond to “stop” signals from their environment and why and how tumor cells fail to heed such signals.
IS IN VITRO SENESCENCE RELEVANT TO CARCINOGENESIS?
When normal cells are placed in culture in vitro, most are not capable of sustained growth. Fibroblasts are an exception to this rule. When they are cultured, fibroblasts may divide 30–50 times and then they undergo what has been termed a “crisis” during which the majority of cells stop dividing (usually due to an increase in p21 expression, a CDK inhibitor), many die, and a small fraction emerge that have acquired genetic changes that permit their uncontrolled growth. The cessation of growth of normal cells in culture has been termed “senescence,” and whether this phenomenon is relevant to any physiologic event in vivo is debated.
Among the cellular changes during in vitro propagation is telomere shortening. DNA polymerase is unable to replicate the tips of chromosomes, resulting in the loss of DNA at the specialized ends of chromosomes (called telomeres) with each replication cycle. At birth, human telomeres are 15- to 20-kb pairs long and are composed of tandem repeats of a six-nucleotide sequence (TTAGGG) that associates with specialized telomere-binding proteins to form a T-loop structure that protects the ends of chromosomes from being mistakenly recognized as damaged. The loss of telomeric repeats with each cell division cycle causes gradual telomere shortening, leading to growth arrest (called senescence) when one or more critically short telomeres trigger a p53-regulated DNA-damage checkpoint response. Cells can bypass this growth arrest if pRb and p53 are nonfunctional, but cell death usually ensues when the unprotected ends of chromosomes lead to chromosome fusions or other catastrophic DNA rearrangements. The ability to bypass telomere-based growth limitations is thought to be a critical step in the evolution of most malignancies. This occurs by the reactivation of telomerase expression in cancer cells. Telomerase is an enzyme that adds TTAGGG repeats onto the 3′ ends of chromosomes. It contains a catalytic subunit with reverse transcriptase activity (hTERT) and an RNA component that provides the template for telomere extension. Most normal somatic cells do not express sufficient telomerase to prevent telomere attrition with each cell division. Exceptions include stem cells (such as those found in hematopoietic tissues, gut and skin epithelium, and germ cells) that require extensive cell division to maintain tissue homeostasis. More than 90% of human cancers express high levels of telomerase that prevent telomere shortening to critical levels and allow indefinite cell proliferation. In vitro experiments indicate that inhibition of telomerase activity leads to tumor cell apoptosis. Major efforts are under way to develop methods to inhibit telomerase activity in cancer cells. For example, the protein component of telomerase (hTERT) may act as one of the most widely expressed tumor-associated antigens and be targeted by vaccine approaches.
Although most of the functions of telomerase relate to cell division, it also has several other effects including interfering with the differentiated functions of at least certain stem cells, although the impact on differentiated function of normal non-stem cells is less clear. Nevertheless, a major growth industry in medical research has been discovering an association between short telomeres and human diseases ranging from diabetes and coronary artery disease to Alzheimer’s disease. The picture is further complicated by the fact that rare genetic defects in the telomerase enzyme seem to cause pulmonary fibrosis, aplastic anemia, or dyskeratosis congenita (characterized by abnormalities in skin, nails, and oral mucosa with increased risk for certain malignancies) but not defects in nutrient absorption in the gut, a site that might be presumed to be highly sensitive to defective cell proliferation. Much remains to be learned about how telomere shortening and telomere maintenance are related to human illness in general and cancer in particular.
SIGNAL TRANSDUCTION PATHWAYS IN CANCER CELLS
Signals that affect cell behavior come from adjacent cells, the stroma in which the cells are located, hormonal signals that originate remotely, and from the cells themselves (autocrine signaling). These signals generally exert their influence on the receiving cell through activation of signal transduction pathways that have as their end result the induction of activated transcription factors that mediate a change in cell behavior or function or the acquisition of effector machinery to accomplish a new task. Although signal transduction pathways can lead to a wide variety of outcomes, many such pathways rely on cascades of signals that sequentially activate different proteins or glycoproteins and lipids or glycolipids, and the activation steps often involve the addition or removal of one or more phosphate groups on a downstream target. Other chemical changes can result from signal transduction pathways, but phosphorylation and dephosphorylation play a major role. The proteins that add phosphate groups to proteins are called kinases. There are two major distinct classes of kinases; one class acts on tyrosine residues, and the other acts on serine/threonine residues. The tyrosine kinases often play critical roles in signal transduction pathways; they may be receptor tyrosine kinases, or they may be linked to other cell-surface receptors through associated docking proteins (Fig. 102e-2).
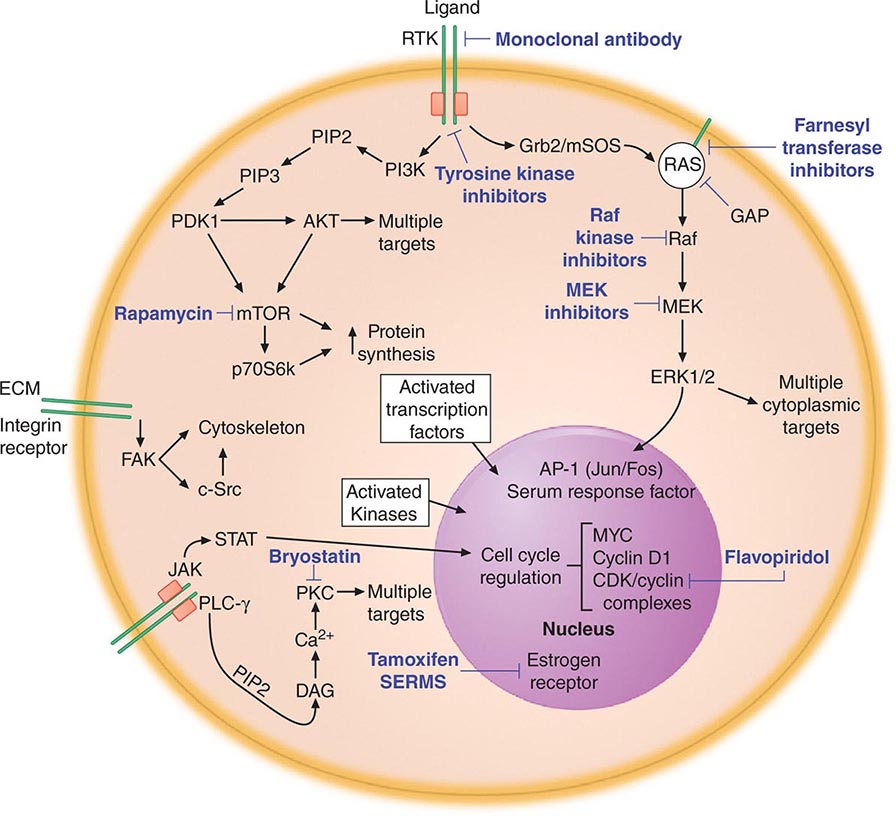
FIGURE 102e-2 Therapeutic targeting of signal transduction pathways in cancer cells. Three major signal transduction pathways are activated by receptor tyrosine kinases (RTK). 1. The protooncogene Ras is activated by the Grb2/mSOS guanine nucleotide exchange factor, which induces an association with Raf and activation of downstream kinases (MEK and ERK1/2). 2. Activated PI3K phosphorylates the membrane lipid PIP2 to generate PIP3, which acts as a membrane-docking site for a number of cellular proteins including the serine/threonine kinases PDK1 and Akt. PDK1 has numerous cellular targets, including Akt and mTOR. Akt phosphorylates target proteins that promote resistance to apoptosis and enhance cell cycle progression, whereas mTOR and its target p70S6K upregulate protein synthesis to potentiate cell growth. 3. Activation of PLC-γ leads to the formation of diacylglycerol (DAG) and increased intracellular calcium, with activation of multiple isoforms of PKC and other enzymes regulated by the calcium/calmodulin system. Other important signaling pathways involve non-RTKs that are activated by cytokine or integrin receptors. Janus kinases (JAK) phosphorylate STAT (signal transducer and activator of transcription) transcription factors, which translocate to the nucleus and activate target genes. Integrin receptors mediate cellular interactions with the extracellular matrix (ECM), inducing activation of FAK (focal adhesion kinase) and c-Src, which activate multiple downstream pathways, including modulation of the cell cytoskeleton. Many activated kinases and transcription factors migrate into the nucleus, where they regulate gene transcription, thus completing the path from extracellular signals, such as growth factors, to a change in cell phenotype, such as induction of differentiation or cell proliferation. The nuclear targets of these processes include transcription factors (e.g., Myc, AP-1, and serum response factor) and the cell cycle machinery (CDKs and cyclins). Inhibitors of many of these pathways have been developed for the treatment of human cancers. Examples of inhibitors that are currently being evaluated in clinical trials are shown in purple type.
Normally, tyrosine kinase activity is short-lived and reversed by protein tyrosine phosphatases (PTPs). However, in many human cancers, tyrosine kinases or components of their downstream pathways are activated by mutation, gene amplification, or chromosomal translocations. Because these pathways regulate proliferation, survival, migration, and angiogenesis, they have been identified as important targets for cancer therapeutics.
Inhibition of kinase activity is effective in the treatment of a number of neoplasms. Lung cancers with mutations in the epidermal growth factor receptor are highly responsive to erlotinib and gefitinib (Table 102e-2). Lung cancers with activation of anaplastic lymphoma kinase (ALK) or ROS1 by translocations respond to crizotinib, an ALK and ROS1 inhibitor. A BRAF inhibitor is highly effective in melanomas and thyroid cancers in which BRAF is mutated. Targeting a protein (MEK) downstream of BRAF also has activity against BRAF mutant melanomas. Janus kinase inhibitors are active in myeloproliferative syndromes in which JAK2 activation is a pathogenetic event. Imatinib (which targets a number of tyrosine kinases) is an effective agent in tumors that have translocations of the c-Abl and BCR gene (such as chronic myeloid leukemia), mutant c-Kit (gastrointestinal stromal cell tumors), or mutant platelet-derived growth factor receptor (PDGFR; chronic myelomonocytic leukemia); second-generation inhibitors of BCR-Abl, dasatinib, and nilotinib are even more effective. The third-generation agent bosutinib has activity in some patients who have progressed on other inhibitors, whereas the third-generation agent ponatinib has activity against the T315I mutation, which is resistant to the other agents. Sorafenib and sunitinib, agents that inhibit a large number of kinases, have shown antitumor activity in a number of malignancies, including renal cell cancer (RCC) (both), hepatocellular carcinoma (sorafenib), thyroid cancer (sorafenib), gastrointestinal stromal tumor (GIST) (sunitinib), and pancreatic neuroendocrine tumors (sunitinib). Inhibitors of the mammalian target of rapamycin (mTOR) are active in RCC, pancreatic neuroendocrine tumors, and breast cancer. The list of active agents and treatment indications is growing rapidly. These new agents have ushered in a new era of personalized therapy. It is becoming more routine for resected tumors to be assessed for specific molecular changes that predict response and to have clinical decision-making guided by those results.
|
SOME FDA-APPROVED MOLECULARLY TARGETED AGENTS FOR THE TREATMENT OF CANCER |
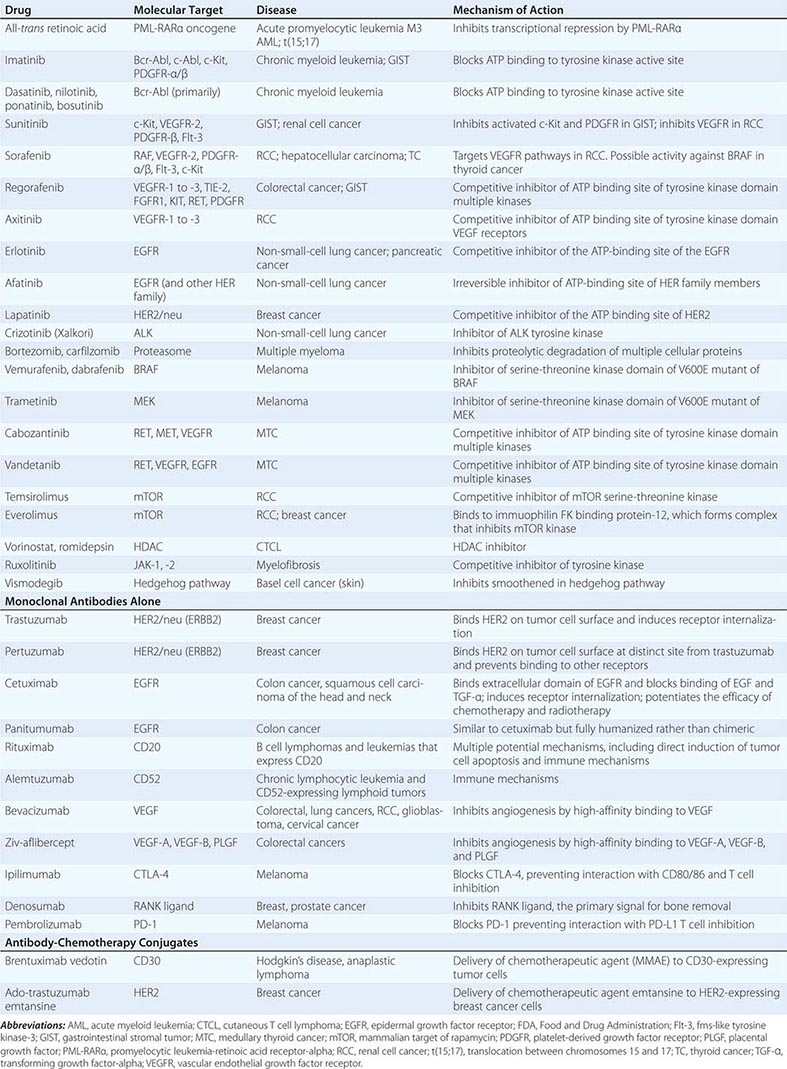
However, none of these therapies has yet been curative by themselves for any malignancy, although prolonged periods of disease control lasting many years frequently occur in chronic myeloid leukemia. The reasons for the failure to cure are not completely defined, although resistance to the treatment ultimately develops in most patients. In some tumors, resistance to kinase inhibitors is related to an acquired mutation in the target kinase that inhibits drug binding. Many of these kinase inhibitors act as competitive inhibitors of the ATP-binding pocket. ATP is the phosphate donor in these phosphorylation reactions. Mutation in the BCR-ABL kinase in the ATP-binding pocket (such as the threonine to isoleucine change at codon 315 [T315I]) can prevent imatinib binding. Other resistance mechanisms include altering other signal transduction pathways to bypass the inhibited pathway. As resistance mechanisms become better defined, rational strategies to overcome resistance will emerge. In addition, many kinase inhibitors are less specific for an oncogenic target than was hoped, and toxicities related to off-target inhibition of kinases limit the use of the agent at a dose that would optimally inhibit the cancer-relevant kinase.
Targeted agents can also be used to deliver highly toxic compounds. An important component of the technology for developing effective conjugates is the design of the linker between the two, which needs to be stable. Currently approved antibody drug conjugates include brentuximab vedotin, which links the microtubule toxin monomethyl auristatin E (MMAE) to an antibody targeting the cell surface antigen CD30, which is expressed on a number of malignant cells but especially in Hodgkin’s disease and anaplastic lymphoma. The linker in this case is cleavable, which allows diffusion of the drug out of the cell after delivery. The second approved conjugate is ado-trastuzumab emtansine, which links the microtubule formation inhibitor mertansine and the monoclonal antibody trastuzumab targeted against human epidermal growth factor receptor 2 (HER2) on breast cancer cells. In this case, the linker is noncleavable, thus trapping the chemotherapeutic agent within the cells. There are theoretical pluses and minuses to having either cleavable or noncleavable linkers, and it is likely that both will be used in future developments of antibody-drug conjugates.
Another strategy to enhance the antitumor effects of targeted agents is to use them in rational combinations with each other and in empiric combinations with chemotherapy agents that kill cells in ways distinct from targeted agents. Combinations of trastuzumab (a monoclonal antibody that targets the HER2 receptor [member of the epidermal growth factor receptor (EGFR) family]) with chemotherapy have significant activity against breast and stomach cancers that have high levels of expression of the HER2 protein. The activity of trastuzumab and chemotherapy can be enhanced further by combinations with another targeted monoclonal antibody (pertuzumab), which prevents dimerization of the HER2 receptor with other HER family members including HER3.
Although targeted therapies have not yet resulted in cures when used alone, their use in the adjuvant setting and when combined with other effective treatments has substantially increased the fraction of patients cured. For example, the addition of rituximab, an anti-CD20 antibody, to combination chemotherapy in patients with diffuse large B cell lymphoma improves cure rates by 15–20%. The addition of trastuzumab, antibody to HER2, to combination chemotherapy in the adjuvant treatment of HER2-positive breast cancer reduces relapse rates by 50%.
A major effort is under way to develop targeted therapies for mutations in the ras family of genes, which are the most common mutations in oncogenes in cancers (especially kras) but have proved to be very difficult targets for a number of reasons related to how RAS proteins are activated and inactivated. Targeted therapies against proteins downstream of RAS (including mitogen-activated protein [MAP] kinase and ERK) are currently being studied, both individually and in combination. A large number of inhibitors of phospholipid signaling pathways such as the phosphatidylinositol-3-kinase (PI3K) and phospholipase C-gamma pathways, which are involved in a large number of cellular processes that are important in cancer development and progression, are being evaluated. The targeting of a variety of other pathways that are activated in malignant cells, such as the MET pathway, hedgehog pathway, and various angiogenesis pathways, is also being explored.
One of the strategies for new drug development is to take advantage of so-called oncogene addiction. This situation (Fig. 102e-3) is created when a tumor cell develops an activating mutation in an oncogene that becomes a dominant pathway for survival and growth with reduced contributions from other pathways, even when there may be abnormalities in those pathways. This dependency on a single pathway creates a cell that is vulnerable to inhibitors of that oncogene pathway. For example, cells harboring mutations in BRAF are very sensitive to MEK inhibitors that inhibit downstream signaling in the BRAF pathway.
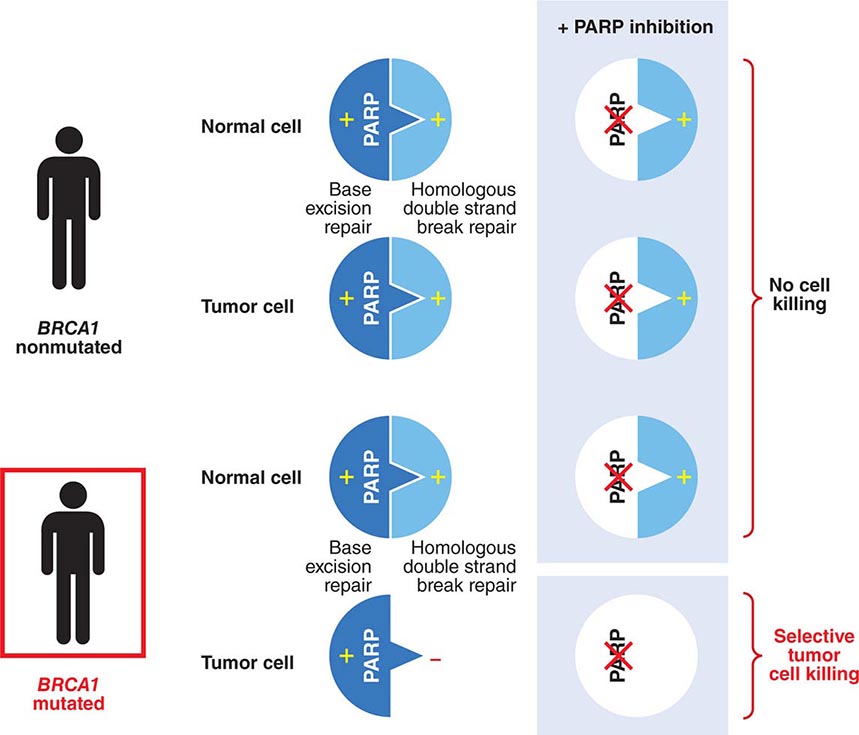
FIGURE 102e-3 Synthetic lethality. Genes are said to have a synthetic lethal relationship when mutation of either gene alone is tolerated by the cell but mutation of both genes leads to lethality, as originally noted by Bridges and later named by Dobzhansky. Thus, mutant gene a and gene b have a synthetic lethal relationship, implying that the loss of one gene makes the cell dependent on the function of the other gene. In cancer cells, loss of function of a DNA repair gene like BRCA1, which repairs double-strand breaks, makes the cell dependent on base excision repair mediated in part by PARP. If the PARP gene product is inhibited, the cell attempts to repair the break using the error-prone nonhomologous end-joining method, which results in tumor cell death. High-throughput screens can now be performed using isogenic cell line pairs in which one cell line has a defined defect in a DNA repair pathway. Compounds can be identified that selectively kill the mutant cell line; targets of these compounds have a synthetic lethal relationship to the repair pathway and are potentially important targets for future therapeutics.
Targeting proteins critical for transcription of proteins vital for malignant cell survival or proliferation provides another potential target for treating cancers. The transcription factor nuclear factor-κB (NF-κB) is a heterodimer composed of p65 and p50 subunits that associate with an inhibitor, IκB, in the cell cytoplasm. In response to growth factor or cytokine signaling, a multi-subunit kinase called IKK (IκB kinase) phosphorylates IκB and directs its degradation by the ubiquitin/proteasome system. NF-κB, free of its inhibitor, translocates to the nucleus and activates target genes, many of which promote the survival of tumor cells. Novel drugs called proteasome inhibitors block the proteolysis of IκB, thereby preventing NF-κB activation. For unexplained reasons, this is selectively toxic to tumor cells. The antitumor effects of proteasome inhibitors are more complicated and involve the inhibition of the degradation of multiple cellular proteins. Proteasome inhibitors (e.g., bortezomib [Velcade]) have activity in patients with multiple myeloma, including partial and complete remissions. Inhibitors of IKK are also in development, with the hope of more selectively blocking the degradation of IκB, thus “locking” NF-κB in an inhibitory complex and rendering the cancer cell more susceptible to apoptosis-inducing agents. Many other transcription factors are activated by phosphorylation, which can be prevented by tyrosine kinase inhibitors or serine/threonine kinase inhibitors, a number of which are currently in clinical trials.
Estrogen receptors (ERs) and androgen receptors (ARs), members of the steroid hormone family of nuclear receptors, are targets of inhibition by drugs used to treat breast and prostate cancers, respectively. Tamoxifen, a partial agonist and antagonist of ER function, can mediate tumor regression in metastatic breast cancer and can prevent disease recurrence in the adjuvant setting. Tamoxifen binds to the ER and modulates its transcriptional activity, inhibiting activity in the breast but promoting activity in bone and uterine epithelium. Selective ER modulators (SERMs) have been developed with the hope of a more beneficial modulation of ER activity, i.e., antiestrogenic activity in the breast, uterus, and ovary, but estrogenic for bone, brain, and cardiovascular tissues. Aromatase inhibitors, which block the conversion of androgens to estrogens in breast and subcutaneous fat tissues, have demonstrated improved clinical efficacy compared with tamoxifen and are often used as first-line therapy in patients with ER-positive disease. A number of approaches have been developed for blocking androgen stimulation of prostate cancer, including decreasing production (e.g., orchiectomy, luteinizing hormone–releasing hormone agonists or antagonists, estrogens, ketoconazole, and inhibitors of enzymes such as CYP17 involved in androgen production) and AR blockers (Chap. 108).
ONCOGENE ADDICTION AND SYNTHETIC LETHALITY
The concepts of oncogene addiction and synthetic lethality have spurred new drug development targeting oncogene- and tumor-suppressor pathways. As discussed earlier in this chapter and outlined in Fig. 102e-3, cancer cells can become dependent on signaling pathways containing activated oncogenes; this can effect proliferation (i.e., mutated Kras, Braf, overexpressed Myc, or activated tyrosine kinases), DNA repair (loss of BRCA1 or BRCA2 gene function), survival (overexpression of Bcl-2 or NF-κB), cell metabolism (as occurs when mutant Kras enhances glucose uptake and aerobic glycolysis), and perhaps angiogenesis (production of VEGF in response to HIF-2α in RCC). In such cases, targeted inhibition of the pathway can lead to specific killing of the cancer cells. However, targeting defects in tumor-suppressor genes has been much more difficult, both because the target of mutation is often deleted and because it is much more difficult to restore normal function than to inhibit abnormal function of a protein. Synthetic lethality occurs when loss of function in either of two genes alone has limited effects on cell survival but loss of function in both genes leads to cell death. Identifying genes that have a synthetic lethal relationship to tumor-suppressor pathways that have been mutated in tumor cells may allow targeting of proteins required uniquely by those cells (Fig. 102e-3). Several examples of this have been identified. For instance, cells with mutations in the BRCA1 or BRCA2 tumor-suppressor genes (e.g., a subset of breast and ovarian cancers) are unable to repair DNA damage by homologous recombination. PARP are a family of proteins important for single-strand break (SSB) DNA repair. PARP inhibition results in selective killing of cancer cells with BRCA1 or BRCA2 loss. Preliminary trials have suggested some effectiveness of PARP inhibition, especially in combination with chemotherapy; clinical trials are ongoing. The concept of synthetic lethality provides a framework for genetic screens to identify other synthetic lethal combinations involving known tumor-suppressor genes and development of novel therapeutic agents to target dependent pathways.
EPIGENETIC INFLUENCES ON CANCER GENE TRANSCRIPTION
Chromatin structure regulates the hierarchical order of sequential gene transcription that governs differentiation and tissue homeostasis. Disruption of chromatin remodeling (the process of modifying chromatin structure to control exposure of specific genes to transcriptional proteins, thereby controlling the expression of those genes) leads to aberrant gene expression and can induce proliferation of undifferentiated cells. Epigenetics is defined as changes that alter the pattern of gene expression that persist across at least one cell division but are not caused by changes in the DNA code. Epigenetic changes include alterations of chromatin structure mediated by methylation of cytosine residues in CpG dinucleotides, modification of histones by acetylation or methylation, or changes in higher-order chromosome structure (Fig. 102e-4). The transcriptional regulatory regions of active genes often contain a high frequency of CpG dinucleotides (referred to as CpG islands), which are normally unmethylated. Expression of these genes is controlled by transient association with repressor or activator proteins that regulate transcriptional activation. However, hypermethylation of promoter regions is a common mechanism by which tumor-suppressor loci are epigenetically silenced in cancer cells. Thus one allele may be inactivated by mutation or deletion (as occurs in loss of heterozygosity), while expression of the other allele is epigenetically silenced, usually by methylation.
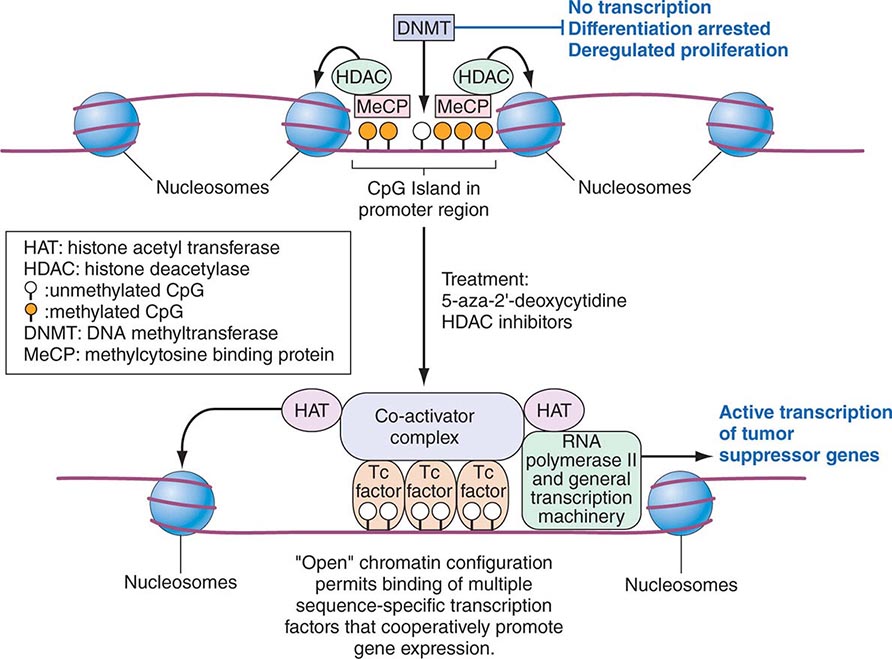
FIGURE 102e-4 Epigenetic regulation of gene expression in cancer cells. Tumor-suppressor genes are often epigenetically silenced in cancer cells. In the upper portion, a CpG island within the promoter and enhancer regions of the gene has been methylated, resulting in the recruitment of methyl-cytosine binding proteins (MeCP) and complexes with histone deacetylase (HDAC) activity. Chromatin is in a condensed, nonpermissive conformation that inhibits transcription. Clinical trials are under way using the combination of demethylating agents such as 5-aza-2′-deoxycytidine plus HDAC inhibitors, which together confer an open, permissive chromatin structure (lower portion). Transcription factors bind to specific DNA sequences in promoter regions and, through protein-protein interactions, recruit coactivator complexes containing histone acetyl transferase (HAT) activity. This enhances transcription initiation by RNA polymerase II and associated general transcription factors. The expression of the tumor-suppressor gene commences, with phenotypic changes that may include growth arrest, differentiation, or apoptosis.
Acetylation of the amino terminus of the core histones H3 and H4 induces an open chromatin conformation that promotes transcription initiation. Histone acetylases are components of coactivator complexes recruited to promoter/enhancer regions by sequence-specific transcription factors during the activation of genes (Fig. 102e-4). Histone deacetylases (HDACs; at least 17 are encoded in the human genome) are recruited to genes by transcriptional repressors and prevent the initiation of gene transcription. Methylated cytosine residues in promoter regions become associated with methyl cytosine–binding proteins that recruit protein complexes with HDAC activity. The balance between permissive and inhibitory chromatin structure is therefore largely determined by the activity of transcription factors in modulating the “histone code” and the methylation status of the genetic regulatory elements of genes.
The pattern of gene transcription is aberrant in all human cancers, and in many cases, epigenetic events are responsible. Unlike genetic events that alter DNA primary structure (e.g., deletions), epigenetic changes are potentially reversible and appear amenable to therapeutic intervention. In certain human cancers, including pancreatic cancer and multiple myeloma, the p16Ink4a promoter is inactivated by methylation, thus permitting the unchecked activity of CDK4/cyclin D and rendering pRb nonfunctional. In sporadic forms of renal, breast, and colon cancer, the von Hippel–Lindau (VHL), breast cancer 1 (BRCA1), and serine/threonine kinase 11 (STK11) genes, respectively, are epigenetically silenced. Other targeted genes include the p15Ink4b CDK inhibitor, glutathione-S-transferase (which detoxifies reactive oxygen species), and the E-cadherin molecule (important for junction formation between epithelial cells). Epigenetic silencing can occur in premalignant lesions and can affect genes involved in DNA repair, thus predisposing to further genetic damage. Examples include MLH1 (mut L homologue) in hereditary nonpolyposis colon cancer (HNPCC, also called Lynch’s syndrome), which is critical for repair of mismatched bases that occur during DNA synthesis, and O6-methylguanine-DNA methyltransferase, which removes alkylated guanine adducts from DNA and is often silenced in colon, lung, and lymphoid tumors.
Human leukemias often have chromosomal translocations that code for novel fusion proteins with enzymatic activities that alter chromatin structure. The promyelocytic leukemia–retinoic acid receptor (PML-RAR) fusion protein, generated by the t(15;17) observed in most cases of acute promyelocytic leukemia (APL), binds to promoters containing retinoic acid response elements and recruits HDAC to these promoters, effectively inhibiting gene expression. This arrests differentiation at the promyelocyte stage and promotes tumor cell proliferation and survival. Treatment with pharmacologic doses of all-trans retinoic acid (ATRA), the ligand for RARα, results in the release of HDAC activity and the recruitment of coactivators, which overcome the differentiation block. This induced differentiation of APL cells has improved treatment of these patients but also has led to a novel treatment toxicity when newly differentiated tumor cells infiltrate the lungs. However, ATRA represents a treatment paradigm for the reversal of epigenetic changes in cancer. For other leukemia-associated fusion proteins, such as acute myeloid leukemia (AML)-eight-twenty-one (ETO) and the MLL fusion proteins seen in AML and acute lymphocytic leukemia, no ligand is known. Therefore, efforts are ongoing to determine the structural basis for interactions between translocation fusion proteins and chromatin-remodeling proteins and to use this information to rationally design small molecules that will disrupt specific protein-protein associations, although this has proven to be technically difficult. Drugs that block the enzymatic activity of HDAC are being tested. HDAC inhibitors have demonstrated antitumor activity in clinical studies against cutaneous T cell lymphoma (e.g., vorinostat) and some solid tumors. HDAC inhibitors may target cancer cells via a number of mechanisms, including upregulation of death receptors (DR4/5, FAS, and their ligands) and p21Cip1/Waf1, as well as inhibition of cell cycle checkpoints.
Efforts are also under way to reverse the hypermethylation of CpG islands that characterizes many malignancies. Drugs that induce DNA demethylation, such as 5-aza-2′-deoxycytidine, can lead to reexpression of silenced genes in cancer cells with restoration of function, and 5-aza-2′-deoxycytidine is approved for use in myelodysplastic syndrome (MDS). However, 5-aza-2′-deoxycytidine has limited aqueous solubility and is myelosuppressive. Other inhibitors of DNA methyltransferases are in development. In ongoing clinical trials, inhibitors of DNA methylation are being combined with HDAC inhibitors. The hope is that by reversing coexisting epigenetic changes, the deregulated patterns of gene transcription in cancer cells will be at least partially reversed.
Epigenetic gene regulation can also occur via microRNAs or long non-coding RNAs (lncRNAs). MicroRNAs are short (average 22 nucleotides in length) RNA molecules that silence gene expression after transcription by binding and inhibiting the translation or promoting the degradation of mRNA transcripts. It is estimated that more than 1000 microRNAs are encoded in the human genome. Each tissue has a distinctive repertoire of microRNA expression, and this pattern is altered in specific ways in cancers. However, specific correlations between microRNA expression and tumor biology and clinical behavior are just now emerging. Therapies targeting microRNAs are not currently at hand but represent a novel area of treatment development. lncRNAs are longer than 200 nucleotides and compose the largest group of noncoding RNAs. Some of them have been shown to play important roles in gene regulation. The potential for altering these RNAs for therapeutic benefit is an area of active investigation, although much more needs to be learned before this will be feasible.
APOPTOSIS AND OTHER MECHANISMS OF CELL DEATH
Tissue homeostasis requires a balance between the death of aged, terminally differentiated cells or severely damaged cells and their renewal by proliferation of committed progenitors. Genetic damage to growth-regulating genes of stem cells could lead to catastrophic results for the host as a whole. Thus, genetic events causing activation of oncogenes or loss of tumor suppressors, which would be predicted to lead to unregulated cell proliferation unless corrected, usually activate signal transduction pathways that block aberrant cell proliferation. These pathways can lead to a form of programmed cell death (apoptosis) or irreversible growth arrest (senescence). Much as a panoply of intra- and extracellular signals impinge upon the core cell cycle machinery to regulate cell division, so too are these signals transmitted to a core enzymatic machinery that regulates cell death and survival.
Apoptosis is induced by two main pathways (Fig. 102e-5). The extrinsic pathway of apoptosis is activated by cross-linking members of the tumor necrosis factor (TNF) receptor superfamily, such as CD95 (Fas) and death receptors DR4 and DR5, by their ligands, Fas ligand or TRAIL (TNF-related apoptosis-inducing ligand), respectively. This induces the association of FADD (Fas-associated death domain) and procaspase-8 to death domain motifs of the receptors. Caspase-8 is activated and then cleaves and activates effector caspases-3 and -7, which then target cellular constituents (including caspase-activated DNAse, cytoskeletal proteins, and a number of regulatory proteins), inducing the morphologic appearance characteristic of apoptosis, which pathologists term “karyorrhexis.” The intrinsic pathway of apoptosis is initiated by the release of cytochrome c and SMAC (second mitochondrial activator of caspases) from the mitochondrial intermembrane space in response to a variety of noxious stimuli, including DNA damage, loss of adherence to the extracellular matrix (ECM), oncogene-induced proliferation, and growth factor deprivation. Upon release into the cytoplasm, cytochrome c associates with dATP, procaspase-9, and the adaptor protein APAF-1, leading to the sequential activation of caspase-9 and effector caspases. SMAC binds to and blocks the function of inhibitor of apoptosis proteins (IAP), negative regulators of caspase activation.
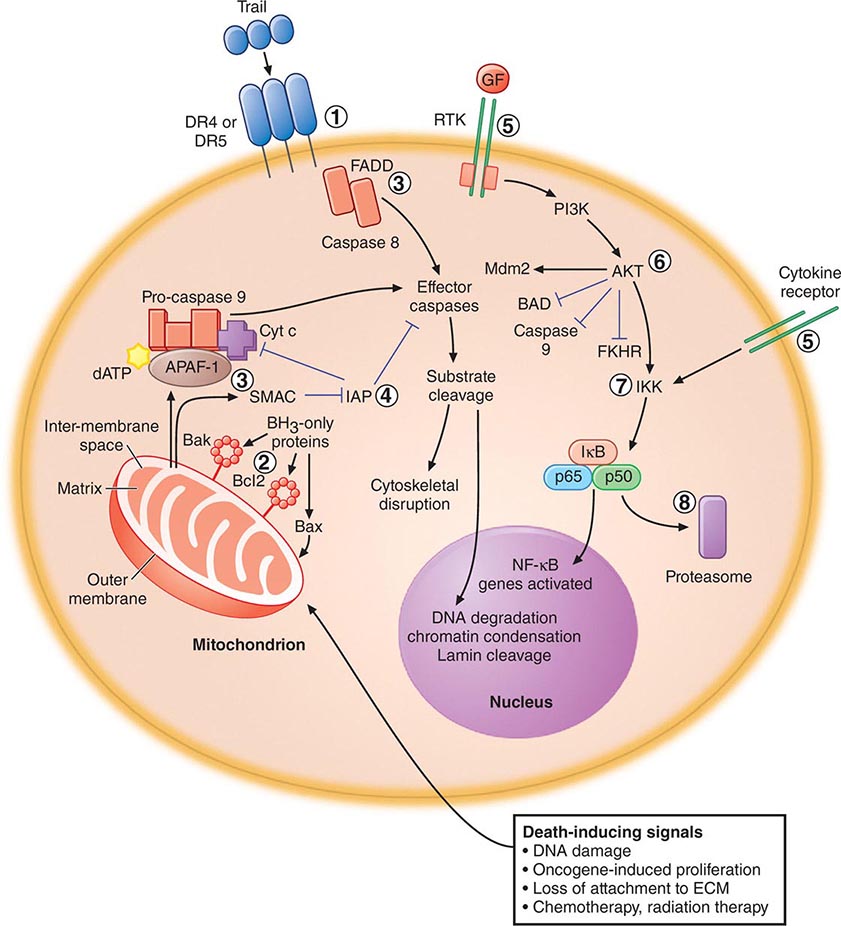
FIGURE 102e-5 Therapeutic strategies to overcome aberrant survival pathways in cancer cells. 1. The extrinsic pathway of apoptosis can be selectively induced in cancer cells by TRAIL (the ligand for death receptors 4 and 5) or by agonistic monoclonal antibodies. 2. Inhibition of antiapoptotic Bcl-2 family members with antisense oligonucleotides or inhibitors of the BH3-binding pocket will promote formation of Bak- or Bax-induced pores in the mitochondrial outer membrane. 3. Epigenetic silencing of APAF-1, caspase-8, and other proteins can be overcome using demethylating agents and inhibitors of histone deacetylases. 4. Inhibitor of apoptosis proteins (IAP) blocks activation of caspases; small-molecule inhibitors of IAP function (mimicking SMAC action) should lower the threshold for apoptosis. 5. Signal transduction pathways originating with activation of receptor tyrosine kinase receptors (RTKs) or cytokine receptors promote survival of cancer cells by a number of mechanisms. Inhibiting receptor function with monoclonal antibodies, such as trastuzumab or cetuximab, or inhibiting kinase activity with small-molecule inhibitors can block the pathway. 6. The Akt kinase phosphorylates many regulators of apoptosis to promote cell survival; inhibitors of Akt may render tumor cells more sensitive to apoptosis-inducing signals; however, the possibility of toxicity to normal cells may limit the therapeutic value of these agents. 7 and 8. Activation of the transcription factor NF-κB (composed of p65 and p50 subunits) occurs when its inhibitor, IκB, is phosphorylated by IκB kinase (IKK), with subsequent degradation of IκB by the proteasome. Inhibition of IKK activity should selectively block the activation of NF-κB target genes, many of which promote cell survival. Inhibitors of proteasome function are Food and Drug Administration approved and may work in part by preventing destruction of IκB, thus blocking NF-κB nuclear localization. NF-κB is unlikely to be the only target for proteasome inhibitors.
The release of apoptosis-inducing proteins from the mitochondria is regulated by pro- and antiapoptotic members of the Bcl-2 family. Antiapoptotic members (e.g., Bcl-2, Bcl-XL, and Mcl-1) associate with the mitochondrial outer membrane via their carboxyl termini, exposing to the cytoplasm a hydrophobic binding pocket composed of Bcl-2 homology (BH) domains 1, 2, and 3 that is crucial for their activity. Perturbations of normal physiologic processes in specific cellular compartments lead to the activation of BH3-only proapoptotic family members (such as Bad, Bim, Bid, Puma, Noxa, and others) that can alter the conformation of the outer-membrane proteins Bax and Bak, which then oligomerize to form pores in the mitochondrial outer membrane resulting in cytochrome c release. If proteins composed only of BH3 domains are sequestered by Bcl-2, Bcl-XL, or Mcl-1, pores do not form and apoptosis-inducing proteins are not released from the mitochondria. The ratio of levels of antiapoptotic Bcl-2 family members and the levels of proapoptotic BH3-only proteins at the mitochondrial membrane determines the activation state of the intrinsic pathway. The mitochondrion must therefore be recognized not only as an organelle with vital roles in intermediary metabolism and oxidative phosphorylation but also as a central regulatory structure of the apoptotic process.
The evolution of tumor cells to a more malignant phenotype requires the acquisition of genetic changes that subvert apoptosis pathways and promote cancer cell survival and resistance to anticancer therapies. However, cancer cells may be more vulnerable than normal cells to therapeutic interventions that target the apoptosis pathways that cancer cells depend on. For instance, overexpression of Bcl-2 as a result of the t(14;18) translocation contributes to follicular lymphoma. Upregulation of Bcl-2 expression is also observed in prostate, breast, and lung cancers and melanoma. Targeting of antiapoptotic Bcl-2 family members has been accomplished by the identification of several low-molecular-weight compounds that bind to the hydrophobic pockets of either Bcl-2 or Bcl-XL and block their ability to associate with death-inducing BH3-only proteins. These compounds inhibit the antiapoptotic activities of Bcl-2 and Bcl-XL at nanomolar concentrations in the laboratory and are entering clinical trials.
Preclinical studies targeting death receptors DR4 and DR5 have demonstrated that recombinant, soluble, human TRAIL or humanized monoclonal antibodies with agonist activity against DR4 or DR5 can induce apoptosis of tumor cells while sparing normal cells. The mechanisms for this selectivity may include expression of decoy receptors or elevated levels of intracellular inhibitors (such as FLIP, which competes with caspase-8 for FADD) by normal cells but not tumor cells. Synergy has been shown between TRAIL-induced apoptosis and chemotherapeutic agents. For instance, some colon cancers encode mutated Bax protein as a result of mismatch repair (MMR) defects and are resistant to TRAIL. However, upregulation of Bak by chemotherapy restores the ability of TRAIL to activate the mitochondrial pathway of apoptosis. However, clinical studies have not yet shown significant activity of approaches targeting the TRAIL pathway.
Many of the signal transduction pathways perturbed in cancer promote tumor cell survival (Fig. 102e-5). These include activation of the PI3K/Akt pathway, increased levels of the NF-κB transcription factor, and epigenetic silencing of genes such as APAF-1 and caspase-8. Each of these pathways is a target for therapeutic agents that, in addition to affecting cancer cell proliferation or gene expression, may render cancer cells more susceptible to apoptosis, thus promoting synergy when combined with other chemotherapeutic agents.
Some tumor cells resist drug-induced apoptosis by expression of one or more members of the ABC family of ATP-dependent efflux pumps that mediate the multidrug-resistance (MDR) phenotype. The prototype, P-glycoprotein (PGP), spans the plasma membrane 12 times and has two ATP-binding sites. Hydrophobic drugs (e.g., anthracyclines and vinca alkaloids) are recognized by PGP as they enter the cell and are pumped out. Numerous clinical studies have failed to demonstrate that drug resistance can be overcome using inhibitors of PGP. However, ABC transporters have different substrate specificities, and inhibition of a single family member may not be sufficient to overcome the MDR phenotype. Efforts to reverse PGP-mediated drug resistance continue.
Cells, including cancer cells, can also undergo other mechanisms of cell death including autophagy (degradation of proteins and organelles by lysosomal proteases) and necrosis (digestion of cellular components and rupturing of the cell membrane). Necrosis usually occurs in response to external forces resulting in release of cellular components, which leads to inflammation and damage to surrounding tissues. Although necrosis was thought to be unprogrammed, evidence now suggests that at least some aspects may be programmed. The exact role of necrosis in cancer cell death in various settings is still being determined. In addition to its role in cell death, autophagy can serve as a homeostatic mechanism to promote survival for the cell by recycling cellular components to provide necessary energy. The mechanisms that control the balance between enhancing survival versus leading to cell death are still not fully understood. Autophagy appears to play conflicting roles in the development and survival of cancer. Early in the carcinogenic process, it can act as a tumor suppressor by preventing the cell from accumulating abnormal proteins and organelles. However, in established tumors, it may serve as a mechanism of survival for cancer cells when they are stressed by damage such as from chemotherapy. Inhibition of this process can enhance the sensitivity of cancer cells to chemotherapy. Better understanding of the factors that control the survival-promoting versus death-inducing aspects of autophagy is required in order to know how to best manipulate it for therapeutic benefit.
METASTASIS
The metastatic process accounts for the vast majority of deaths from solid tumors, and therefore, an understanding of this process is critical. The biology of metastasis is complex and requires multiple steps. The three major features of tissue invasion are cell adhesion to the basement membrane, local proteolysis of the membrane, and movement of the cell through the rent in the membrane and the ECM. Cells that lose contact with the ECM normally undergo programmed cell death (anoikis), and this process has to be suppressed in cells that metastasize. Another process important for metastasizing epithelial cancer cells is epithelial-mesenchymal transition (EMT). This is a process by which cells lose their epithelial properties and gain mesenchymal properties. This normally occurs during the developmental process in embryos, allowing cells to migrate to their appropriate destinations in the embryo. It also occurs in wound healing, tissue regeneration, and fibrotic reactions, but in all of these processes, cells stop proliferating when the process is complete. Malignant cells that metastasize undergo EMT as an important step in that process but retain the capacity for unregulated proliferation. Malignant cells that gain access to the circulation must then repeat those steps at a remote site, find a hospitable niche in a foreign tissue, avoid detection by host defenses, and induce the growth of new blood vessels. The rate-limiting step for metastasis is the ability for tumor cells to survive and expand in the novel microenvironment of the metastatic site, and multiple host-tumor interactions determine the ultimate outcome (Fig. 102e-6). Few drugs have been developed to attempt to directly target the process of metastasis, in part because the specifics of the critical steps in the process that would be potentially good targets for drugs are still being identified. However, a number of potential targets are known. HER2 can enhance the metastatic potential of breast cancer cells, and as discussed above, the monoclonal antibody trastuzumab, which targets HER2, improves survival in the adjuvant setting for HER2-positive breast cancer patients. Other potential targets that increase metastatic potential of cells in preclinical studies include HIF-1 and -2, transcription factors induced by hypoxia within tumors; growth factors (e.g., cMET and VEGFR); oncogenes (e.g., SRC); adhesion molecules (e.g., focal adhesion kinase [FAK]); ECM proteins (e.g., matrix metalloproteinases-1 and -2); and inflammatory molecules (e.g., COX-2).
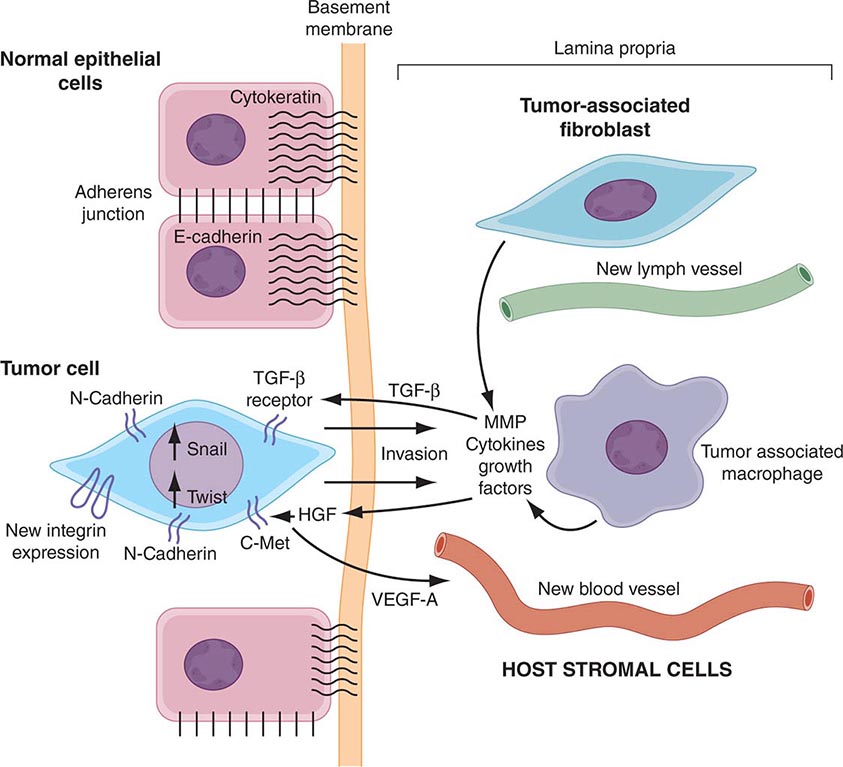
FIGURE 102e-6 Oncogene signaling pathways are activated during tumor progression and promote metastatic potential. This figure shows a cancer cell that has undergone epithelial to mesenchymal transition (EMT) under the influence of several environmental signals. Critical components include activated transforming growth factor β (TGF-β) and the hepatocyte growth factor (HGF)/c-Met pathways, as well as changes in the expression of adhesion molecules that mediate cell-cell and cell–extracellular matrix interactions. Important changes in gene expression are mediated by the Snail and Twist family of transcriptional repressors (whose expression is induced by the oncogenic pathways), leading to reduced expression of E-cadherin, a key component of adherens junctions between epithelial cells. This, in conjunction with upregulation of N-cadherin, a change in the pattern of expression of integrins (which mediate cell–extracellular matrix associations that are important for cell motility), and a switch in intermediate filament expression from cytokeratin to vimentin, results in the phenotypic change from adherent highly organized epithelial cells to motile and invasive cells with a fibroblast or mesenchymal morphology. EMT is thought to be an important step leading to metastasis in some human cancers. Host stromal cells, including tumor-associated fibroblasts and macrophages, play an important role in modulating tumor cell behavior through secretion of growth factors and proangiogenic cytokines, and matrix metalloproteinases that degrade the basement membrane. VEGF-A, -C, and -D are produced by tumor cells and stromal cells in response to hypoxemia or oncogenic signals and induce production of new blood vessels and lymphatic channels through which tumor cells metastasize to lymph nodes or tissues.
The metastatic phenotype is likely restricted to a small fraction of tumor cells (Fig. 102e-6). A number of genetic and epigenetic changes are required for tumor cells to be able to metastasize, including activation of metastasis-promoting genes and inhibition of genes that suppress the metastatic ability. Cells with metastatic capability frequently express chemokine receptors that are likely important in the metastatic process. A number of candidate metastasis-suppressor genes have been identified, including genes coding for proteins that enhance apoptosis, suppress cell division, are involved in the interactions of cells with each other or the ECM, or suppress cell migration. The loss of function of these genes enhances metastasis. Gene expression profiling is being used to study the metastatic process and other properties of tumor cells that may predict susceptibilities.
An example of the ability of malignant cells to survive and grow in a novel microenvironment is bone metastasis. Bone metastases are extremely painful, cause fractures of weight-bearing bones, can lead to hypercalcemia, and are a major cause of morbidity for cancer patients. Osteoclasts and their monocyte-derived precursors express the surface receptor RANK (receptor activator of NF-κB), which is required for terminal differentiation and activation of osteoclasts. Osteoblasts and other stromal cells express RANK ligand (RANKL), as both a membrane-bound and soluble cytokine. Osteoprotegerin (OPG), a soluble receptor for RANKL produced by stromal cells, acts as a decoy receptor to inhibit RANK activation. The relative balance of RANKL and OPG determines the activation state of RANK on osteoclasts. Many tumors increase osteoclast activity by secretion of substances such as parathyroid hormone (PTH), PTH-related peptide, interleukin (IL)-1, or Mip1 that perturb the homeostatic balance of bone remodeling by increasing RANK signaling. One example is multiple myeloma, where tumor cell–stromal cell interactions activate osteoclasts and inhibit osteoblasts, leading to the development of multiple lytic bone lesions. Inhibition of RANKL by an antibody (denosumab) can prevent further bone destruction. Bisphosphonates are also effective inhibitors of osteoclast function that are used in the treatment of cancer patients with bone metastases.
CANCER STEM CELLS
Only a small proportion of the cells within a tumor are capable of initiating colonies in vitro or forming tumors at high efficiency when injected into immunocompromised NOD/SCID mice. Acute and chronic myeloid leukemias (AML and CML) have a small population of cells (<1%) that have properties of stem cells, such as unlimited self-renewal and the capacity to cause leukemia when serially transplanted in mice. These cells have an undifferentiated phenotype (Thy1–CD34+CD38– and do not express other differentiation markers) and resemble normal stem cells in many ways, but are no longer under homeostatic control (Fig. 102e-7). Solid tumors may also contain a population of stem cells. Cancer stem cells, like their normal counterparts, have unlimited proliferative capacity and paradoxically traverse the cell cycle at a very slow rate; cancer growth occurs largely due to expansion of the stem cell pool, the unregulated proliferation of an amplifying population, and failure of apoptosis pathways (Fig. 102e-7). Slow cell cycle progression and high levels of expression of antiapoptotic Bcl-2 family members and drug efflux pumps of the MDR family render cancer stem cells less vulnerable to cancer chemotherapy or radiation therapy. Implicit in the cancer stem cell hypothesis is the idea that failure to cure most human cancers is due to the fact that current therapeutic agents do not kill the stem cells. If cancer stem cells can be identified and isolated, then aberrant signaling pathways that distinguish these cells from normal tissue stem cells can be identified and targeted. Evidence that cells with stem cell properties can arise from other epithelial cells within the cancer by processes such as epithelial mesenchymal transition also implies that it is essential to treat all of the cancer cells, and not just those with current stem cell-like properties, in order to eliminate the self-renewing cancer cell population. The exact nature of cancer stem cells remains an area of investigation. One of the unanswered questions is the exact origin of cancer stem cells for the different cancers.
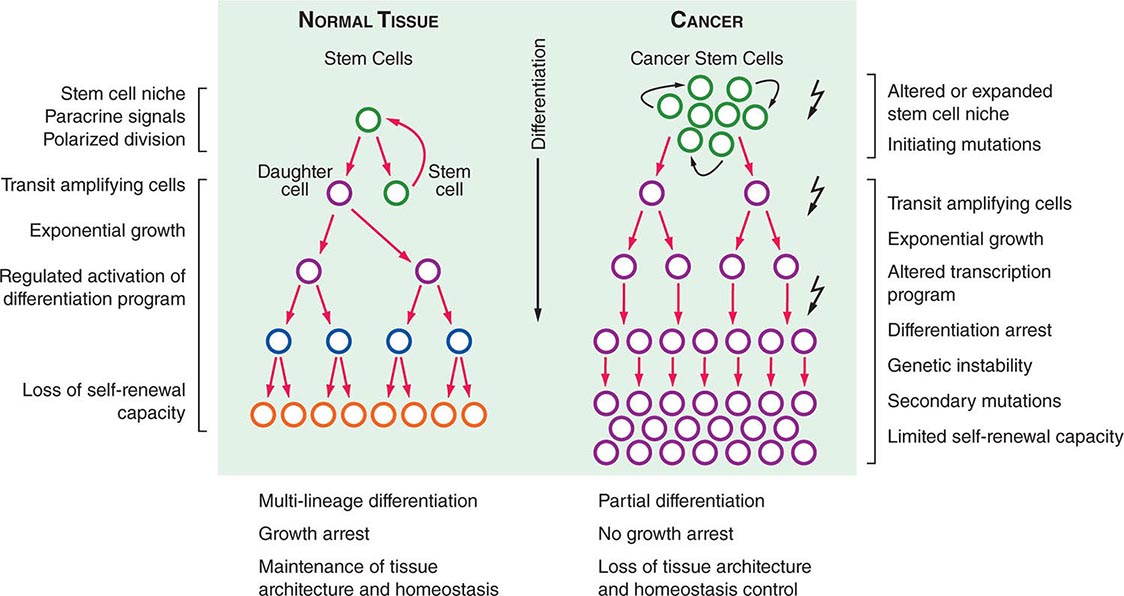
FIGURE 102e-7 Cancer stem cells play a critical role in the initiation, progression, and resistance to therapy of malignant neoplasms. In normal tissues (left), homeostasis is maintained by asymmetric division of stem cells, leading to one progeny cell that will differentiate and one cell that will maintain the stem cell pool. This occurs within highly specific niches unique to each tissue, such as in close apposition to osteoblasts in bone marrow, or at the base of crypts in the colon. Here, paracrine signals from stromal cells, such as sonic hedgehog or Notch ligands, as well as upregulation of β-catenin and telomerase, help to maintain stem cell features of unlimited self-renewal while preventing differentiation or cell death. This occurs in part through upregulation of the transcriptional repressor Bmi-1 and inhibition of the p16Ink4a/Arf and p53 pathways. Daughter cells leave the stem cells niche and enter a proliferative phase (referred to as transit-amplifying) for a specified number of cell divisions, during which time a developmental program is activated, eventually giving rise to fully differentiated cells that have lost proliferative potential. Cell renewal equals cell death, and homeostasis is maintained. In this hierarchical system, only stem cells are long-lived. The hypothesis is that cancers harbor stem cells that make up a small fraction (i.e., 0.001–1%) of all cancer cells. These cells share several features with normal stem cells, including an undifferentiated phenotype, unlimited self-renewal potential, and a capacity for some degree of differentiation; however, due to initiating mutations (mutations are indicated by lightning bolts), they are no longer regulated by environmental cues. The cancer stem cell pool is expanded, and rapidly proliferating progeny, through additional mutations, may attain stem cell properties, although most of this population is thought to have a limited proliferative capacity. Differentiation programs are dysfunctional due to reprogramming of the pattern of gene transcription by oncogenic signaling pathways. Within the cancer transit-amplifying population, genomic instability generates aneuploidy and clonal heterogeneity as cells attain a fully malignant phenotype with metastatic potential. The cancer stem cell hypothesis has led to the idea that current cancer therapies may be effective at killing the bulk of tumor cells but do not kill tumor stem cells, leading to a regrowth of tumors that is manifested as tumor recurrence or disease progression. Research is in progress to identify unique molecular features of cancer stem cells that can lead to their direct targeting by novel therapeutic agents.
PLASTICITY AND RESISTANCE
Cancer cells, and especially stem cells, have the capacity for significant plasticity, allowing them to alter multiple aspects of cell biology in response to external factors (e.g., chemotherapy, inflammation, immune response). Thus, a major problem in cancer therapy is that malignancies have a wide spectrum of mechanisms for both initial and adaptive resistance to treatments. These include inhibiting drug delivery to the cancer cells, blocking drug uptake and retention, increasing drug metabolism, altering levels of target proteins, acquiring mutations in target proteins, modifying metabolism and cell signaling pathways, using alternate signaling pathways, adjusting the cell replication process including mechanisms by which the cell deals with DNA damage, inhibiting apoptosis, and evading the immune system. Thus, most metastatic cancers (except those curable with chemotherapy such as germ cell tumors) eventually become resistant to the therapy being used. Overcoming resistance is a major area of research.
CANCER METABOLISM
One of the distinguishing characteristics of cancer cells is that they have altered metabolism as compared with normal cells in supporting survival and their high rates of proliferation. These cells must focus a significant fraction of their energy resources on synthesis of proteins and other molecules while still maintaining sufficient ATP production to survive and grow. Although normal proliferating cells also have similar needs, there are differences in how cancer cells metabolize glucose and a number of other compounds, including glutamine, as compared to normal cells. Many cancer cells use aerobic glycolysis (the Warburg effect) (Fig. 102e-8) to metabolize glucose, leading to increased lactic acid production, whereas normal cells use oxidative phosphorylation in mitochondria under aerobic conditions, a much more efficient process. One consequence is increased glucose uptake by cancer cells, a fact used in fluorodeoxyglucose (FDG) positron emission tomography (PET) scanning to detect tumors. A number of proteins in cancer cells, including CMYC, HIF1, RAS, p53, pRB, and AKT, are all involved in modulating glycolytic processes and controlling the Warburg effect. Although these pathways remain difficult to target therapeutically, both the PI3 kinase pathway with signaling through mTOR and the AMP-activated kinase (AMPK) pathway, which inhibits mTOR complex 1 (mTORC1; a protein complex that includes mTOR), are important in controlling the glycolytic process and thus provide potential targets for inhibiting this process. The inefficient utilization of glucose also leads to a need for alternative metabolic pathways for other compounds as well, one of which is glutamine. Similar to glucose, this provides both a source for structural molecules as well as energy production. Glutamine is also inefficiently used by cancer cells.
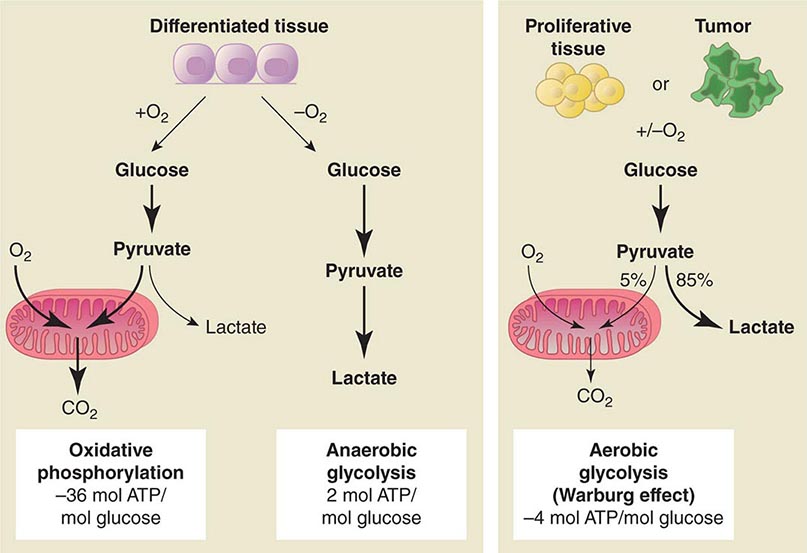
FIGURE 102e-8 Warburg effect versus oxidative phosphorylation. In most normal tissues, the vast majority of cells are differentiated and dedicated to a particular function within the organ in which they reside. The metabolic needs are mainly for energy and not for building blocks for new cells. In these tissues, ATP is generated by oxidative phosphorylation that efficiently generates about 36 molecules of ATP for each molecule of glucose metabolized. By contrast, proliferative tumor tissues, especially in the setting of hypoxia, a typical condition within tumors, use aerobic glycolysis to generate energy for cell survival and generation of building blocks for new cells.
Mutations in genes involved in the metastatic process occur in a number of cancers. Among the most frequently found to date are mutations in isocitrate dehydrogenases 1 and 2 (IDH1 and IDH2). These have been most commonly seen in gliomas, AML, and intrahepatic cholangiocarcinomas. These mutations lead to the production of an oncometabolite (2-hydroxyglutarate [2HG]) instead of the normal product α-ketoglutarate. Although the exact mechanisms of oncogenesis by 2HG are still being elucidated, α-ketoglutarate is a key cofactor for a number of dioxygenases involved in controlling DNA methylation. 2HG can act as a competitive inhibitor for α-ketoglutarate, leading to alterations in methylation status (primarily hypermethylation) of genes (epigenetic changes) that can have profound effects on a number of cellular processes including differentiation. Inhibitors of mutant IDH1 and IDH2 are being developed.
Much needs to be learned about the specific differences in metabolism between cancer cells and normal cells; however, modulators of metabolism are being tested clinically. The first of these is the antidiabetic agent metformin, both alone and in combination with chemotherapeutic agents. Metformin inhibits gluconeogenesis and may have direct effects on tumor cells by activating the 5′-adenosine monophosphate-activated kinase (AMPK), a serine/threonine protein kinase that is downstream of the LKB1 tumor suppressor, and thus inhibiting mTORC1. This leads to decreased protein synthesis and proliferation. A second approach being tested involves dichloracetate (DCA), an inhibitor of pyruvate dehydrogenase kinase (PDK). PDK inhibits pyruvate dehydrogenase in cancer cells, leading to a switch from mitochondrial oxidative phosphorylation of glucose to cytoplasmic glycolysis (the Warburg effect). By blocking PDK, DCA inhibits glycolysis. Additional approaches targeting tumor metabolism will likely emerge.
TUMOR MICROENVIRONMENT, ANGIOGENESIS, AND IMMUNE EVASION
Tumors consist not only of malignant cells but also of a complex microenvironment including many other types of cells (e.g., inflammatory cells), ECM, secreted factors (e.g., growth factors), reactive oxygen and nitrogen species, mechanical factors, blood vessels, and lymphatics. This microenvironment is not static but rather is dynamic and continually evolving. Both the complexity and dynamic nature of the microenvironment enhance the difficulty of treating tumors. There are also a number of mechanisms by which the microenvironment can contribute to resistance to anticancer therapies.
One of the critical elements of tumor cell proliferation is delivery of oxygen, nutrients, and circulating factors important for growth and survival. The diffusion limit for oxygen in tissues is ~100–200 μm, and thus, a critical aspect in the growth of tumors is the development of new blood vessels, or angiogenesis. The growth of primary and metastatic tumors to larger than a few millimeters requires the recruitment of blood vessels and vascular endothelial cells to support their metabolic requirements. Thus, a critical element in growth of primary tumors and formation of metastatic sites is the angiogenic switch: the ability of the tumor to promote the formation of new capillaries from preexisting host vessels. The angiogenic switch is a phase in tumor development when the dynamic balance of pro- and antiangiogenic factors is tipped in favor of vessel formation by the effects of the tumor on its immediate environment. Stimuli for tumor angiogenesis include hypoxemia, inflammation, and genetic lesions in oncogenes or tumor suppressors that alter tumor cell gene expression. Angiogenesis consists of several steps, including the stimulation of endothelial cells (ECs) by growth factors, degradation of the ECM by proteases, proliferation and migration of ECs into the tumor, and the eventual formation of new capillary tubes.
Tumor blood vessels are not normal; they have chaotic architecture and blood flow. Due to an imbalance of angiogenic regulators such as VEGF and angiopoietins (see below), tumor vessels are tortuous and dilated with an uneven diameter, excessive branching, and shunting. Tumor blood flow is variable, with areas of hypoxemia and acidosis leading to the selection of variants that are resistant to hypoxemia-induced apoptosis (often due to the loss of p53 expression). Tumor vessel walls have numerous openings, widened interendothelial junctions, and discontinuous or absent basement membrane; this contributes to the high vascular permeability of these vessels and, together with lack of functional intratumoral lymphatics, causes increased interstitial pressure within the tumor (which also interferes with the delivery of therapeutics to the tumor; Figs. 102e-9, 102e-10, and 102e-11). Tumor blood vessels lack perivascular cells such as pericytes and smooth-muscle cells that normally regulate flow in response to tissue metabolic needs.
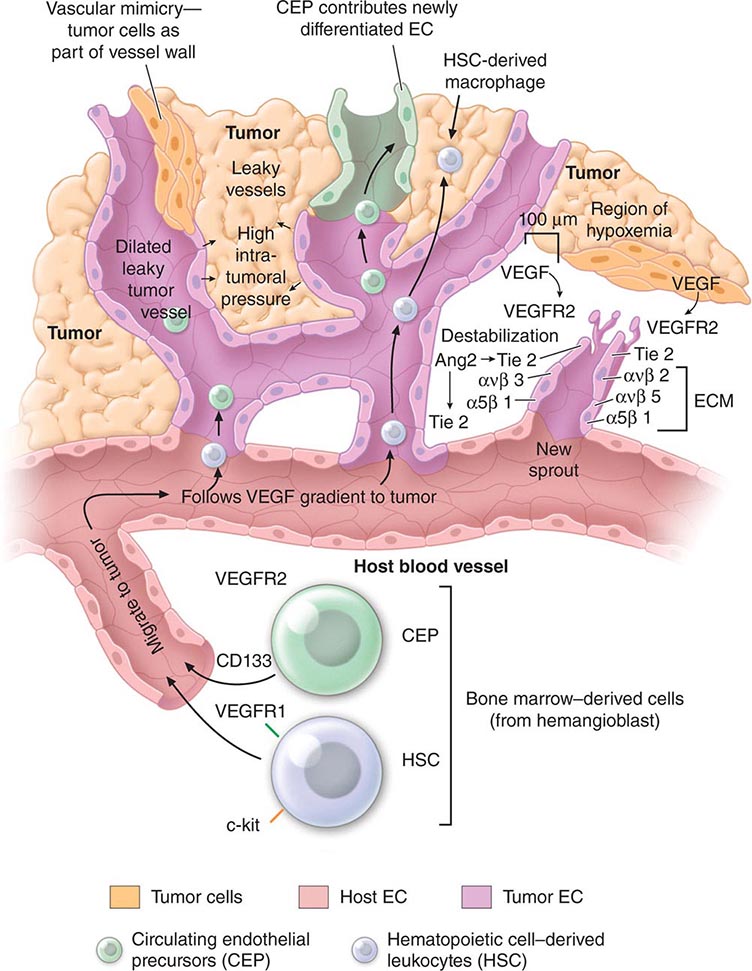
FIGURE 102e-9 Tumor angiogenesis is a complex process involving many different cell types that must proliferate, migrate, invade, and differentiate in response to signals from the tumor microenvironment. Endothelial cells (ECs) sprout from host vessels in response to VEGF, bFGF, Ang2, and other proangiogenic stimuli. Sprouting is stimulated by VEGF/VEGFR2, Ang2/Tie2, and integrin/extracellular matrix (ECM) interactions. Bone marrow–derived circulating endothelial precursors (CEPs) migrate to the tumor in response to VEGF and differentiate into ECs, while hematopoietic stem cells differentiate into leukocytes, including tumor-associated macrophages that secrete angiogenic growth factors and produce matrix metalloproteinases (MMPs) that remodel the ECM and release bound growth factors. Tumor cells themselves may directly form parts of vascular channels within tumors. The pattern of vessel formation is haphazard: vessels are tortuous, dilated, and leaky and branch in random ways. This leads to uneven blood flow within the tumor, with areas of acidosis and hypoxemia (which stimulate release of angiogenic factors) and high intratumoral pressures that inhibit delivery of therapeutic agents.
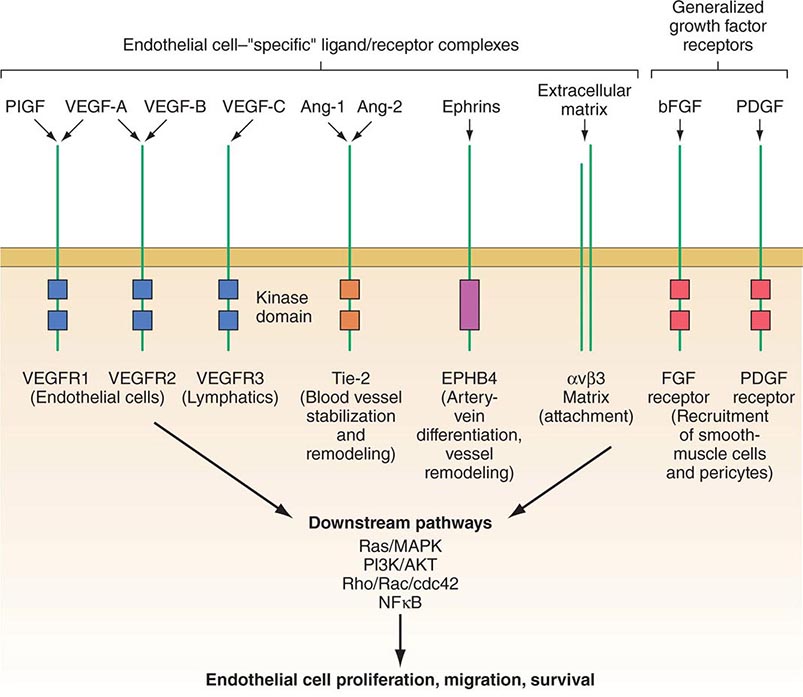
FIGURE 102e-10 Critical molecular determinants of endothelial cell biology. Angiogenic endothelium expresses a number of receptors not found on resting endothelium. These include receptor tyrosine kinases (RTKs) and integrins that bind to the extracellular matrix and mediate endothelial cell (EC) adhesion, migration, and invasion. ECs also express RTK (i.e., the FGF and PDGF receptors) that are found on many other cell types. Critical functions mediated by activated RTK include proliferation, migration, and enhanced survival of endothelial cells, as well as regulation of the recruitment of perivascular cells and bloodborne circulating endothelial precursors and hematopoietic stem cells to the tumor. Intracellular signaling via EC-specific RTK uses molecular pathways that may be targets for future antiangiogenic therapies.
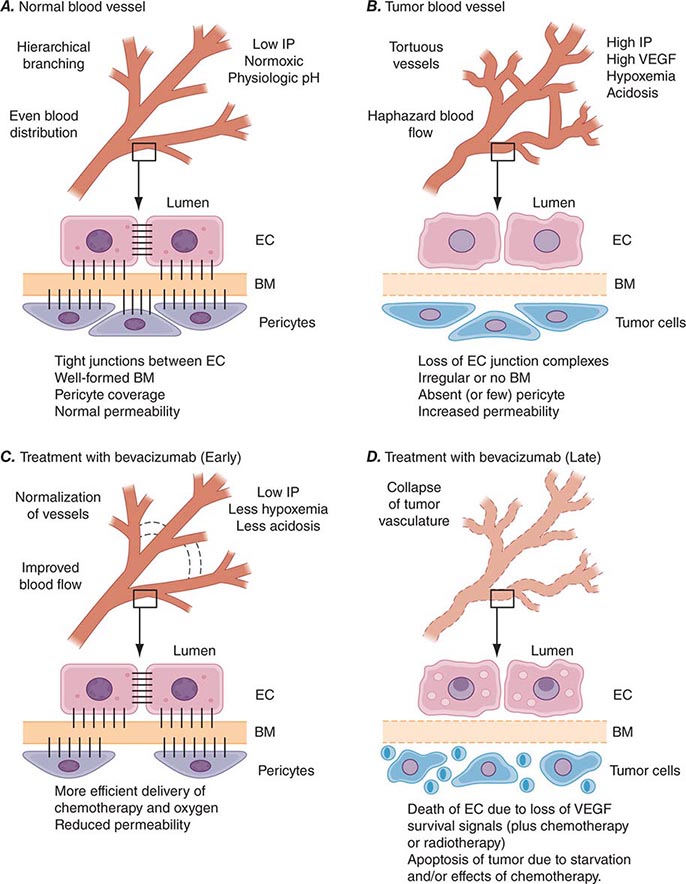
FIGURE 102e-11 Normalization of tumor blood vessels due to inhibition of VEGF signaling. A. Blood vessels in normal tissues exhibit a regular hierarchical branching pattern that delivers blood to tissues in a spatially and temporally efficient manner to meet the metabolic needs of the tissue (top). At the microscopic level, tight junctions are maintained between endothelial cells (ECs), which are adherent to a thick and evenly distributed basement membrane (BM). Pericytes form a surrounding layer that provides trophic signals to the EC and helps maintain proper vessel tone. Vascular permeability is regulated, interstitial fluid pressure is low, and oxygen tension and pH are physiologic. B. Tumors have abnormal vessels with tortuous branching and dilated, irregular interconnecting branches, causing uneven blood flow with areas of hypoxemia and acidosis. This harsh environment selects genetic events that result in resistant tumor variants, such as the loss of p53. High levels of VEGF (secreted by tumor cells) disrupt gap junction communication, tight junctions, and adherens junctions between EC via src-mediated phosphorylation of proteins such as connexin 43, zonula occludens-1, VE-cadherin, and α/β-catenins. Tumor vessels have thin, irregular BM, and pericytes are sparse or absent. Together, these molecular abnormalities result in a vasculature that is permeable to serum macromolecules, leading to high tumor interstitial pressure, which can prevent the delivery of drugs to the tumor cells. This is made worse by the binding and activation of platelets at sites of exposed BM, with release of stored VEGF and microvessel clot formation, creating more abnormal blood flow and regions of hypoxemia. C. In experimental systems, treatment with bevacizumab or blocking antibodies to VEGFR2 leads to changes in the tumor vasculature that has been termed vessel normalization. During the first week of treatment, abnormal vessels are eliminated or pruned (dotted lines), leaving a more normal branching pattern. ECs partially regain features such as cell-cell junctions, adherence to a more normal BM, and pericyte coverage. These changes lead to a decrease in vascular permeability, reduced interstitial pressure, and a transient increase in blood flow within the tumor. Note that in murine models, this normalization period lasts only for ~5–6 days. D. After continued anti-VEGF/VEGFR therapy (which is often combined with chemo- or radiotherapy), ECs die, leading to tumor cell death (either due to direct effects of the chemotherapy or lack of blood flow).
Unlike normal blood vessels, the vascular lining of tumor vessels is not a homogeneous layer of ECs but often consists of a mosaic of ECs and tumor cells with upregulated genes seen in ECs and vessel formation that can occur in hypoxic conditions because of their plasticity; the concept of cancer cell–derived vascular channels, which may be lined by ECM secreted by the tumor cells, is referred to as vascular mimicry. During tumor angiogenesis, ECs are highly proliferative and express a number of plasma membrane proteins that are characteristic of activated endothelium, including growth factor receptors and adhesion molecules such as integrins.
MECHANISMS OF TUMOR VESSEL FORMATION
Tumors use a number of mechanisms to promote vascularization, subverting normal angiogenic processes for this purpose (Fig. 102e-9). Primary or metastatic tumor cells sometimes arise in proximity to host blood vessels and grow around these vessels, parasitizing nutrients by co-opting the local blood supply. However, most tumor blood vessels arise by the process of sprouting, in which tumors secrete trophic angiogenic molecules, the most potent being vascular endothelial growth factors (VEGF), that induce the proliferation and migration of host ECs into the tumor. Sprouting in normal and pathogenic angiogenesis is regulated by three families of transmembrane receptor tyrosine kinases (RTKs) expressed on ECs and their ligands (VEGFs, angiopoietins, ephrins; Fig. 102e-10), which are produced by tumor cells, inflammatory cells, or stromal cells in the tumor microenvironment.
When tumor cells arise in or metastasize to an avascular area, they grow to a size limited by hypoxemia and nutrient deprivation. Hypoxemia, a key regulator of tumor angiogenesis, causes the transcriptional induction of the gene encoding VEGF. VEGF and its receptors are required for embryonic vasculogenesis (development of new blood vessels when none preexist) and normal (wound healing, corpus luteum formation) and pathologic angiogenesis (tumor angiogenesis, inflammatory conditions such as rheumatoid arthritis). VEGF-A is a heparin-binding glycoprotein with at least four isoforms (splice variants) that regulates blood vessel formation by binding to the RTKs VEGFR1 and VEGFR2, which are expressed on all ECs in addition to a subset of hematopoietic cells (Fig. 102e-9). VEGFR2 regulates EC proliferation, migration, and survival, whereas VEGFR1 may act as an antagonist of R2 in ECs but is probably also important for angioblast differentiation during embryogenesis. Tumor vessels may be more dependent on VEGFR signaling for growth and survival than normal ECs. Although VEGF signaling is a critical initiator of angiogenesis, this is a complex process regulated by additional signaling pathways (Fig. 102e-10). The angiopoietin, Ang1, produced by stromal cells, binds to the EC RTK Tie2 and promotes the interaction of ECs with the ECM and perivascular cells, such as pericytes and smooth-muscle cells, to form tight, nonleaky vessels. Platelet-derived growth factor (PDGF) and basic fibroblast growth factor (bFGF) help to recruit these perivascular cells. Ang1 is required for maintaining the quiescence and stability of mature blood vessels and prevents the vascular permeability normally induced by VEGF and inflammatory cytokines.
For tumor cell–derived VEGF to initiate sprouting from host vessels, the stability conferred by the Ang1/Tie2 pathway must be perturbed; this occurs by the secretion of Ang2 by ECs that are undergoing active remodeling. Ang2 binds to Tie2 and is a competitive inhibitor of Ang1 action: under the influence of Ang2, preexisting blood vessels become more responsive to remodeling signals, with less adherence of ECs to stroma and associated perivascular cells and more responsiveness to VEGF. Therefore, Ang2 is required at early stages of tumor angiogenesis for destabilizing the vasculature by making host ECs more sensitive to angiogenic signals. Because tumor ECs are blocked by Ang2, there is no stabilization by the Ang1/Tie2 interaction, and tumor blood vessels are leaky, hemorrhagic, and have poor association of ECs with underlying stroma. Sprouting tumor ECs express high levels of the transmembrane protein ephrin-B2 and its receptor, the RTK EPH, whose signaling appears to work with the angiopoietins during vessel remodeling. During embryogenesis, EPH receptors are expressed on the endothelium of primordial venous vessels while the transmembrane ligand ephrin-B2 is expressed by cells of primordial arteries; the reciprocal expression may regulate differentiation and patterning of the vasculature.
A number of ubiquitously expressed host molecules play critical roles in normal and pathologic angiogenesis. Proangiogenic cytokines, chemokines, and growth factors secreted by stromal cells or inflammatory cells make important contributions to neovascularization, including bFGF, transforming growth factor α (TGF-α), TNF-α, and IL-8. In contrast to normal endothelium, angiogenic endothelium overexpresses specific members of the integrin family of ECM-binding proteins that mediate EC adhesion, migration, and survival. Specifically, expression of integrins αvβ3, αvβ5, and α5β1 mediates spreading and migration of ECs and is required for angiogenesis induced by VEGF and bFGF, which in turn can upregulate EC integrin expression. The αvβ3 integrin physically associates with VEGFR2 in the plasma membrane and promotes signal transduction from each receptor to promote EC proliferation (via focal adhesion kinase, src, PI3K, and other pathways) and survival (by inhibition of p53 and increasing the Bcl-2/Bax expression ratio). In addition, αvβ3 forms cell-surface complexes with matrix metalloproteinases (MMPs), zinc-requiring proteases that cleave ECM proteins, leading to enhanced EC migration and the release of heparin-binding growth factors, including VEGF and bFGF. EC adhesion molecules can be upregulated (i.e., by VEGF, TNF-α) or downregulated (by TGF-β); this, together with chaotic blood flow, explains poor leukocyte-endothelial interactions in tumor blood vessels and may help tumor cells avoid immune surveillance.
Lymphatic vessels also exist within tumors. Development of tumor lymphatics is associated with expression of VEGFR3 and its ligands VEGF-C and VEGF-D. The role of these vessels in tumor cell metastasis to regional lymph nodes remains to be determined. However, VEGF-C levels correlate significantly with metastasis to regional lymph nodes in lung, prostate, and colorectal cancers.
ANTIANGIOGENIC THERAPY
Angiogenesis inhibitors function by targeting the critical molecular pathways involved in EC proliferation, migration, and/or survival, many of which are unique to the activated endothelium in tumors. Inhibition of growth factor and adhesion-dependent signaling pathways can induce EC apoptosis with concomitant inhibition of tumor growth. Different types of tumors can use distinct combinations of molecular mechanisms to activate the angiogenic switch. Therefore, it is doubtful that a single antiangiogenic strategy will suffice for all human cancers; rather, a number of agents or combinations of agents will be needed, depending on distinct programs of angiogenesis used by different human cancers. Despite this, experimental data indicate that for some tumor types, blockade of a single growth factor (e.g., VEGF) may inhibit tumor-induced vascular growth.
Bevacizumab, an antibody that binds VEGF, appears to potentiate the effects of a number of different types of active chemotherapeutic regimens used to treat a variety of different tumor types including colon cancer, lung cancer, cervical cancer, and RCC.
Bevacizumab is administered IV every 2–3 weeks (its half-life is nearly 20 days) and is generally well tolerated. Hypertension is the most common side effect of inhibitors of VEGF (or its receptors), but can be treated with antihypertensive agents and rarely requires discontinuation of therapy. Rare but serious potential risks include arterial thromboembolic events, including stroke and myocardial infarction, and hemorrhage. Another serious complication is bowel perforation, which has been observed in 1–3% of patients (mainly those with colon and ovarian cancers). Inhibition of wound healing is also seen.
Several small-molecule inhibitors (SMIs) that target VEGFR tyrosine kinase activity but are also inhibitory to other kinases have also been approved to treat certain cancers. Sunitinib (see above and Table 102e-2) has activity directed against mutant c-Kit receptors (approved for GIST), but also targets VEGFR and PDGFR, and has shown significant antitumor activity against metastatic RCC, presumably on the basis of its antiangiogenic activity. Similarly, sorafenib, originally developed as a Raf kinase inhibitor but with potent activity against VEGFR and PDGFR, has activity against RCC, thyroid cancer, and hepatocellular cancer. Other inhibitors of VEGFR approved for the treatment of RCC include axitinib and pazopanib.
The success in targeting tumor angiogenesis has led to enhanced enthusiasm for the development of drugs that target other aspects of the angiogenic process; some of these therapeutic approaches are outlined in Fig. 102e-12.
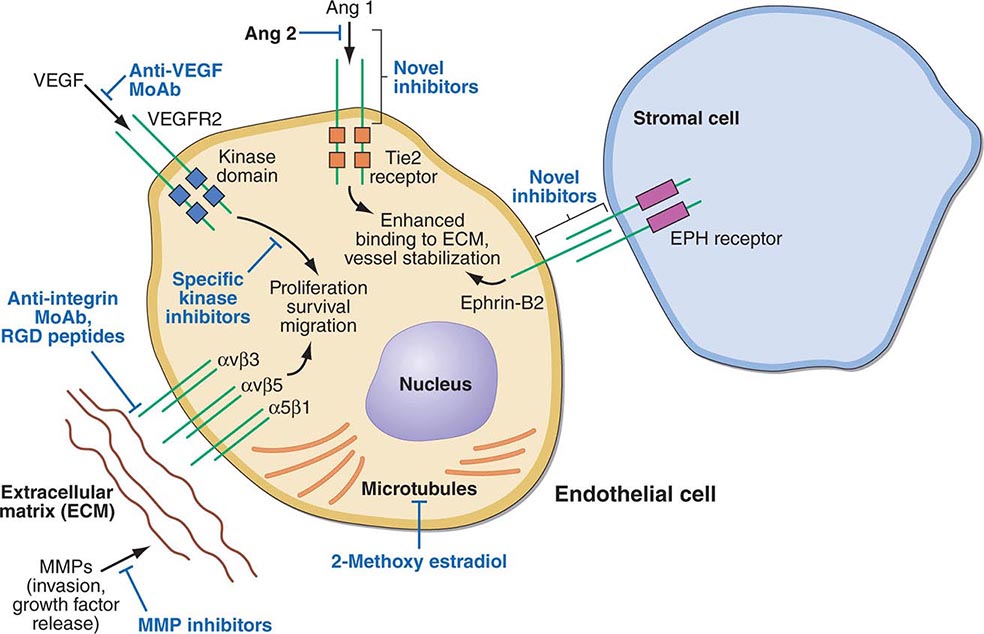
FIGURE 102e-12 Knowledge of the molecular events governing tumor angiogenesis has led to a number of therapeutic strategies to block tumor blood vessel formation. The successful therapeutic targeting of VEGF is described in the text. Other endothelial cell–specific receptor tyrosine kinase pathways (e.g., angiopoietin/Tie2 and ephrin/EPH) are likely targets for the future. Ligation of the αvβ3 integrin is required for endothelial cell (EC) survival. Integrins are also required for EC migration and are important regulators of matrix metalloproteinase (MMP) activity, which modulates EC movement through the extracellular matrix (ECM) as well as release of bound growth factors. Targeting of integrins includes development of blocking antibodies, small peptide inhibitors of integrin signaling, and arg-gly-asp–containing peptides that prevent integrin:ECM binding. Peptides derived from normal proteins by proteolytic cleavage, including endostatin and tumstatin, inhibit angiogenesis by mechanisms that include interfering with integrin function. Signal transduction pathways that are dysregulated in tumor cells indirectly regulate EC function. Inhibition of EGF-family receptors, whose signaling activity is upregulated in a number of human cancers (e.g., breast, colon, and lung cancers), results in downregulation of VEGF and IL-8, while increasing expression of the antiangiogenic protein thrombospondin-1. The Ras/MAPK, PI3K/Akt, and Src kinase pathways constitute important antitumor targets that also regulate the proliferation and survival of tumor-derived EC. The discovery that ECs from normal tissues express tissue-specific “vascular addressins” on their cell surface suggests that targeting specific EC subsets may be possible.
EVASION OF THE IMMUNE SYSTEM BY CANCERS
Cancers have a number of mechanisms that allow them to evade detection and elimination by the immune system. These include downregulation of cell surface proteins involved in immune recognition (including MHC proteins and tumor-specific antigens), expression of other cell surface proteins that inhibit immune function (including members of the B7 family of proteins such as PD-L1), secretion of proteins and other molecules that are immunosuppressive, recruitment and expansion of immunosuppressive cells such as regulatory T cells, and induction of T cell tolerance. In addition, the inflammatory effects of some of the immune mediator cells in the tumor microenvironment (especially tissue-associated macrophages and myeloid-derived suppressor cells) can suppress T cell responses to the tumor as well as stimulate inflammation that can enhance tumor growth.
Immunotherapy approaches to treat cancer aimed at activating the immune response against tumors using immunostimulatory molecules such as interferons, IL-2, and monoclonal antibodies have had some successes. Another approach that has shown particular clinical promise is the targeting of proteins or cells (such as regulatory T cells) involved in normal homeostatic control to prevent autoimmune damage to the host but that malignant cells and their stroma can also use to inhibit the immune response directed against them. The approach that is furthest along clinically has involved targeting CTLA-4, PD-1, and PDL-1, co-inhibitory molecules that are expressed on the surface of cancer cells, cells of the immune system, and/or stromal cells and are involved in inhibiting the immune response against cancer (Fig. 102e-13). Monoclonal antibodies directed against CTLA-4 and PD-1 are approved for the treatment of melanoma, and additional antibodies targeting PD-1 or PDL-1 have shown activity against melanoma, RCC, and lung cancer and continue to be evaluated against other malignancies as well. Combination approaches targeting more than one protein or involving other anticancer approaches (targeted agents, chemotherapy, radiation therapy) are also being explored and have shown promise in early studies. An important aspect of these approaches is balancing sufficient release of the negative control of the immune response to allow immune-mediated attack on the tumors while not allowing too much release and inducing severe autoimmune effects (such as against skin, thyroid, pituitary gland, or the gastrointestinal tract).
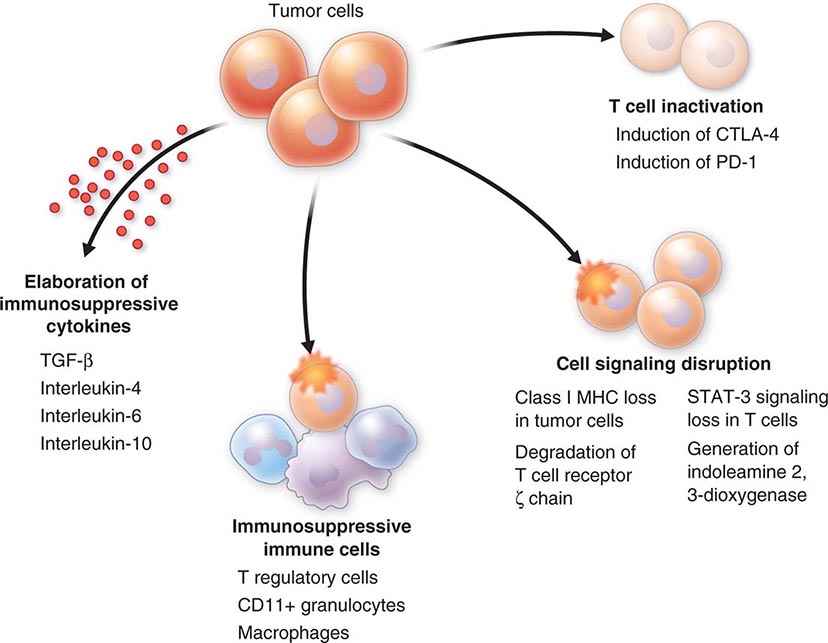
FIGURE 102e-13 Tumor-host interactions that suppress the immune response to the tumor.
SUMMARY
The explosion of information on tumor cell biology, metastasis, and tumor-host interactions (including angiogenesis and immune evasion by tumors) has ushered in a new era of rational targeted therapy for cancer. Furthermore, it has become clear that specific molecular factors detected in individual tumors (specific gene mutations, gene-expression profiles, microRNA expression, overexpression of specific proteins) can be used to tailor therapy and maximize antitumor effects.
ACKNOWLEDGMENT
Robert G. Fenton contributed to this chapter in prior editions, and important material from those prior chapters has been included here.
103e |
Principles of Cancer Treatment |
CANCER PRESENTATION
Cancer in a localized or systemic state is a frequent item in the differential diagnosis of a variety of common complaints. Although not all forms of cancer are curable at diagnosis, affording patients the greatest opportunity for cure or meaningful prolongation of life is greatly aided by diagnosing cancer at the earliest point possible in its natural history and defining treatments that prevent or retard its systemic spread. Indeed, certain forms of cancer, notably breast, colon, and possibly lung cancers in certain patients, can be prevented by screening appropriately selected asymptomatic patients; screening is arguably the earliest point in the spectrum of possible cancer-related interventions where cure is possible (Table 103e-1).
|
SPECTRUM OF CANCER-RELATED INTERVENTIONS |
DETECTION OF A CANCER
The term cancer, as used here, is synonymous with the term tumor, whose original derivation from Latin simply meant “swelling,” not otherwise specified. We now understand that the swelling that is a common physical manifestation of a tumor derives from increased interstitial fluid pressure and increased cellular and stromal mass per volume, compared to normal tissue. Tumors historically were referred to as carcinomas, or “crab-like” infiltrating tumors, or sarcomas, or “fleshy tumors,” derived from the Greek terms for “crab” and “flesh,” respectively. Leukemias are a special case of a cancer of the blood-forming tissues presenting in a disseminated form frequently without definable tumor masses. In addition to localized swelling, tumors present by altered function of the organ they afflict, such as dyspnea on exertion from the anemia caused by leukemia replacing normal hematopoietic cells, cough from lung cancers, jaundice from tumors disrupting the hepatobiliary tree, or seizures and neurologic signs from brain tumors. Hemorrhage is also a frequent presenting sign of tumors involving hollow viscera, as are decreases in the number of platelets and inappropriate inhibition of blood coagulation. Thus, although statistically the fraction of patients with cancer underlying a particular presenting sign or symptom may be low, the implications for a patient with cancer of missing an early-stage tumor call for vigilance; therefore, persistent signs or symptoms should be evaluated as possibly coming from an early-stage tumor.
Evidence of a tumor’s existence can objectively be established by careful physical examination, such as enlarged lymph nodes in lymphomas or a palpable mass in a breast or soft tissue site. A mass may also be detected or confirmed by an imaging modality, such as plain x-ray, computed tomography (CT) scan, ultrasound, positron emission tomography (PET) imaging, or nuclear magnetic resonance approaches. Sensitivity of these technologies varies considerably, and the index of suspicion for a tumor should match the technology chosen. For example, low-dose helical CT scans are superior to plain chest radiographs in detecting lung cancers. Another way of initially establishing the existence of a possible tumor is through direct visualization of an afflicted organ by endoscopy.
ESTABLISHING A CANCER DIAGNOSIS
Once the existence of a likely tumor is defined, unequivocally establishing the diagnosis is the next step in the spectrum of correctly addressing a patient’s needs. This is usually accomplished by a biopsy procedure and the emergence after pathologic examination of an unequivocal statement that cancer is present. The underlying principle in cancer diagnosis is to obtain as much tissue as safely as possible. Due to tumor heterogeneity, pathologists are better able to make the diagnosis when they have more tissue to examine. In addition to light microscopic inspection of a tumor for pattern of growth, degree of cellular atypia, invasiveness, and morphologic features that aid in the differential diagnosis, sufficient tissue is of value in searching for genetic abnormalities and protein expression patterns, such as hormone receptor expression in breast cancers, that may aid in the differential diagnosis or provide information about prognosis or likely response to treatment. Efforts to define “personalized” information from the biology of each patient’s tumor and pertinent to each patient’s treatment plan are becoming increasingly important in selecting treatment options. The general internist should make sure that a patient’s cancer biopsy is appropriately referred from the surgical suite for important molecular studies that can advise the best treatment (Table 103e-2).
|
DIAGNOSTIC BIOPSY: STANDARD OF CARE MOLECULAR AND SPECIAL STUDIES |
Similar-appearing tumors by microscopic morphology may have very different gene expression patterns when assessed by such techniques as microarray analysis for gene expression patterns using gene chips, with important differences in biology and response to treatment. Such testing requires that the tissue be handled properly (e.g., immunologic detection of proteins is more effective in fresh-frozen tissue rather than in formalin-fixed tissue). Coordination among the surgeon, pathologist, and primary care physician is essential to ensure that the amount of information learned from the biopsy material is maximized. These goals are best met by an excisional biopsy in which the entire tumor mass is removed with a small margin of normal tissue surrounding it. If an excisional biopsy cannot be performed, incisional biopsy is the procedure of second choice. A wedge of tissue is removed, and an effort is made to include the majority of the cross-sectional diameter of the tumor in the biopsy to minimize sampling error. Biopsy techniques that involve cutting into tumor carry with them a risk of facilitating the spread of the tumor, and consideration of whether the biopsy might be the prelude to a curative surgery if certain diagnoses are established should inform the actual approach taken. Core-needle biopsy usually obtains considerably less tissue, but this procedure often provides enough information to plan a definitive surgical procedure. Fine-needle aspiration generally obtains only a suspension of cells from within a mass. This procedure is minimally invasive, and if positive for cancer, it may allow inception of systemic treatment when metastatic disease is evident, or it can provide a basis for planning a more meticulous and extensive surgical procedure. However, a negative fine-needle aspiration for a neoplastic diagnosis cannot be taken as definitive evidence that a tumor is absent or make a definitive diagnosis in someone not known to have a cancer.




