[level-membership-for-pediatrics-category]Chapter 64
Principles of Adjuvant Therapy in Childhood Cancer
Historical Overview
Early strides in improving oncologic outcomes through the use of single chemotherapeutic agents were first reported by Farber in 19481 and Li in 1956.2 As additional chemotherapeutic agents were developed, they were combined in multidrug regimens that demonstrated both significantly improved response rates and response duration.3,4 By the late 1970s, multimodal therapy was shown to improve cure rates in children with Wilms tumor5 and was being adopted for the treatment of rhabdomyosarcoma, Ewing sarcoma, lymphoma, and other solid tumors.6 The close collaboration of multidisciplinary cooperative groups and the development of improved supportive care measures added impetus to progress. During the 1990s, dose-intensive chemotherapy programs were shown to be successful in improving outcome for patients with Burkitt lymphoma, neuroblastoma, and other advanced-stage solid tumors.7–9 In addition, improvements in outcome were achieved either by alternating effective groups of chemotherapeutic agents to overcome or prevent resistance,10 or by administering agents by continuous infusion rather than bolus.11
By 2001, noncytotoxic biologic therapies (e.g., signal transduction inhibitors, various tissue growth factor receptor inhibitors, antiangiogenesis agents, tumor-targeted antibodies, and adoptive immunotherapy techniques) were developed to target specific biologic pathways.12 In addition, improvements in radiation therapy have led to the development of intraoperative radiation therapy and radiosurgery techniques. Collectively, these advancements have resulted in the continually increasing survival of children with solid tumors and a profound improvement in their quality of life.
Incidence and Survival Rates
Although childhood cancers account for only 2% of all reported cancer cases in the general population, they account for 10% of all deaths in children.13 On average, 1 to 2 of every 10,000 children in the USA develop cancer each year.14 The distribution of the types of cancer in childhood is different than in adults. Whereas most cancers in adults have an epithelial cell origin, <10% of childhood cancers fall into this category.
The incidence of specific cancers varies by age, gender, and race. Overall, however, it rose from 11.5 cases per 100,000 children in 1975 to 14.8 per 100,000 in 2004.14 The peak incidence (>200 cases per million) is in children younger than 2 years of age. The incidence then decreases to a low of 82.5 cases per million by age 9 years, at which point it begins to rise again through adolescence. In children younger than age 2, central nervous system malignancies, neuroblastoma, acute myeloid leukemia (AML), Wilms tumor, and retinoblastoma account for the majority of diagnoses. In children 2 to 4 years of age, acute lymphoblastic leukemia (ALL) is the most common childhood cancer. After age 9 years, the incidence of Hodgkin disease, osteosarcoma, and Ewing sarcoma begins to increase sharply.13
Over the past several decades, the mortality from childhood cancers has declined dramatically (40%),15 while the 5-year survival rate has risen from 58% in 1975–1977 to close to 80% in 1996–2003.14
Tumor Biology
Understanding normal cell growth and regulation is a prerequisite to understanding both the genetic basis for the development of childhood cancer and the mechanisms of action of chemotherapeutic agents designed to kill rapidly proliferating cancer cells (Fig. 64-1). Normal cell growth occurs by the regulated progression of the cell through the cell cycle of DNA replication and mitosis. This cycle is separated by two intervening growth phases, referred to as G1 and G2. Cells can temporarily leave the cell cycle and enter a resting state referred to as G0. They are programmed to proceed through the cell cycle by a series of external and internal stimuli. Binding of proteins (growth factors) to cell surface receptors stimulates a cascade of cytoplasmic signaling proteins (membrane kinases and signal transducers) that carries the stimuli to the nucleus. Other proteins (transcription factors) then bind to the DNA, resulting in the expression of growth-regulating genes. When functioning normally, these genes promote or prevent cell division, direct the cell to differentiate, or initiate apoptosis, the process of programmed cell death.
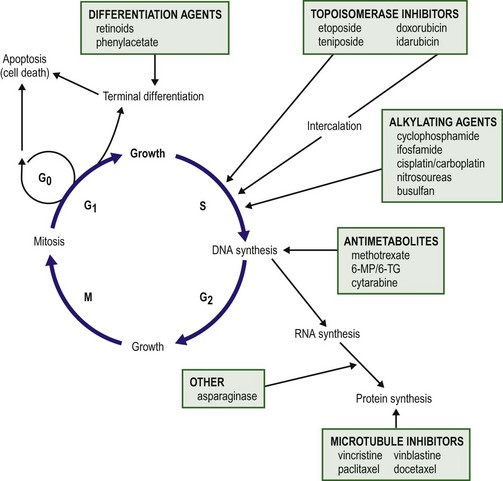
FIGURE 64-1 The cell cycle. Normal cell growth proceeds through DNA replication (S) and mitosis (M), separated by two growth phases (G1 and G2). Cells leave the cell cycle to enter a resting phase (G0) to differentiate or to die. Chemotherapy agents act at specific sites along the cell cycle, as indicated. (Adapted from Balis FRM, Holcenberg JS, Poplack DG. General principles of chemotherapy. In Pizzo P, Poplack D, editors. Principles and Practice of Pediatric Oncology. 3rd ed. Philadelphia: Lippincott-Raven; 1997. p. 219.)
Cancer Cytogenetics
The presence of consistent cytogenetic abnormalities associated with a specific childhood leukemia or solid tumor is useful in both cancer diagnosis and prognosis. Specific cytogenetic aberrations have been identified in rhabdomyosarcoma, Ewing sarcoma, synovial sarcoma, germ cell tumors, medulloblastoma, neuroblastoma, retinoblastoma, and Wilms tumor.16 Chromosomal aberrations can also be helpful in predicting prognosis. For example, the finding of a chromosome 1q deletion, the presence of double-minute chromatin bodies, or the presence of homogeneous staining regions in neuroblastoma confer a poor prognosis.17
The treatment of Wilms tumor based on risk stratification determined by multiple tumor characteristics that impact prognosis (including loss of heterozygosity at 1p and 16q) is a topic of ongoing study. In the future, specific tumors may be identified by a specific ‘fingerprint’ determined by microarray analysis that can simultaneously analyze expression of thousands of genes on a single chip.18 The ability to tailor therapy to individual patients based on the genetic characteristics of their particular tumor is quickly becoming a reality.
Clincial Trials
Combination chemotherapy remains the mainstay of treatment. The likelihood of cure is maximized when all available active agents are administered simultaneously after local control measures have been undertaken and the tumor burden is as low as possible.19 This approach has led to increased survival rates in children with neuroblastoma, Ewing sarcoma, anaplastic Wilms tumor, and osteosarcoma.
Adjuvant and Neoadjuvant Chemotherapy
The use of adjuvant chemotherapy is supported by the finding that less than 20% of sarcoma and lymphoma patients with initially nonmetastatic solid tumors can be cured by operative or radiation therapy alone or combined.20 Tumor recurrence is generally at a distant site, lending support to the hypothesis that micrometastatic disease exists at the time of presentation for most patients with clinically nonmetastatic disease. As many as 40% of patients with Wilms tumor can be cured with resection or radiation therapy alone. However, survival increases to 90% with the addition of combination chemotherapy.21
As the goal of adjuvant chemotherapy is to prevent the growth of metastatic disease, it is vital that chemotherapy begins as soon as possible after local control measures are employed. For this reason, most current chemotherapy protocols for childhood solid tumors advise that chemotherapy is initiated within 2 weeks of operation.22 In children with Wilms tumor, rapid assessment of tumor biology and risk stratification is important for determining the appropriate chemotherapy regimen.
Neoadjuvant chemotherapy has become standard in the treatment of Ewing sarcoma and osteosarcoma, and has the theoretical advantage of minimizing resistance to chemotherapy.10,23,24 Delayed surgical intervention may allow a more complete or less morbid resection, as well as histologic assessment of tumor responsiveness to the chemotherapy agents. Neoadjuvant chemotherapy is beneficial only in tumors for which a known highly effective combination chemotherapy program limits the risk of tumor progression at the primary site. For example, diagnostic biopsy followed by neoadjuvant chemotherapy and delayed resection of the primary tumor for complex neuroblastoma reduces the operative complication rate without compromising survival.25
Dose Intensity and Duration of Chemotherapy
Advances in supportive care have enabled increased dose intensity of active chemotherapy agents in pediatric clinical trials. These advances decrease or minimize the toxic effects of higher-dose chemotherapy on normal tissues. The use of cytokines (granulocyte colony-stimulating factor [G-CSF] and interleukin-11 [IL-11]) to speed recovery of white blood cells and platelets,26,27 and the use of cardioprotectant agents to allow use of a higher cumulative dose of doxorubicin have helped in the development of new dose-intensive therapy for solid tumors.28 Similarly, progress in bone marrow and stem cell transplantation have allowed dose intensities to be pushed to the upper limits.29–32
The positive impact of increasing the dose intensity on improving response rate and survival has been demonstrated for Burkitt lymphoma, osteosarcoma, Ewing sarcoma, testicular cancer, and advanced ovarian cancer.7–9,33–36 The duration of chemotherapy programs for most pediatric solid tumors has been 1 year. However, as the dose intensity of chemotherapy programs has increased, the duration of therapy has concomitantly decreased. This downward trend is likely to continue.
Chemotherapeutic Agents
Chemotherapy drugs are divided into classes by their mechanism of action. These classes include alkylating agents (cisplatin and its analogs), antimetabolites, topoisomerase inhibitors, antimicrotubule agents, differentiation agents, miscellaneous nonclassified agents, and biologic agents. Understanding the mechanism of action for each of these agents helps in establishing combination therapies with synergistic antitumor effects. The most common agents from each class, their mechanism of action, common side effects, and tumors in which they are active are listed in Table 64-1.
TABLE 64-1
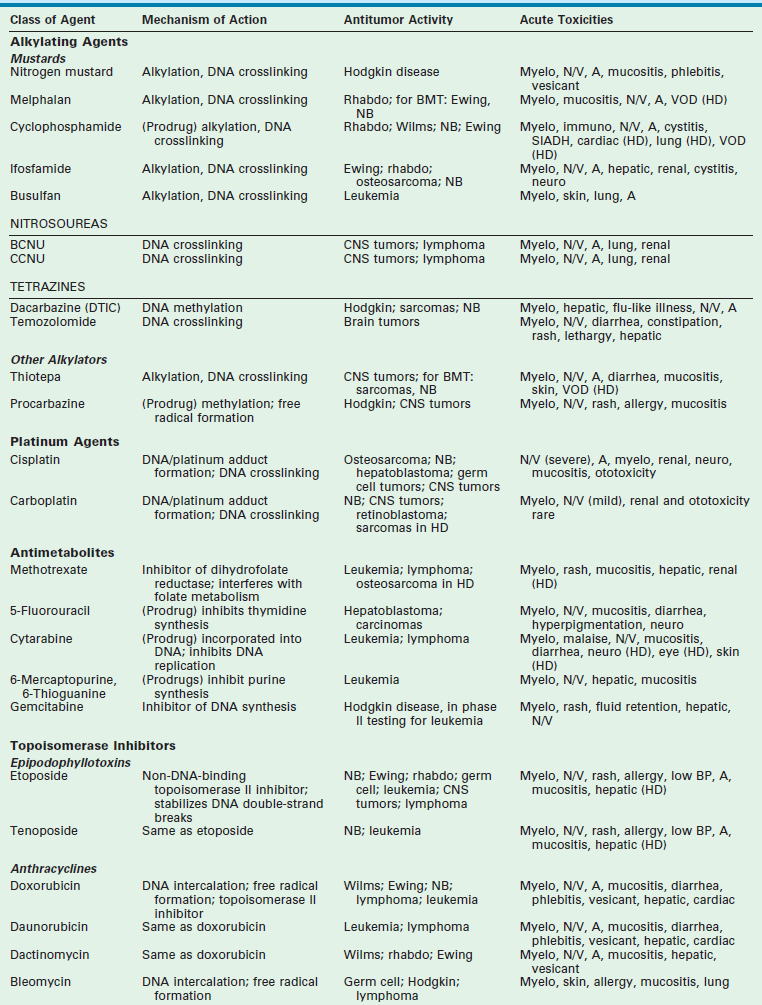
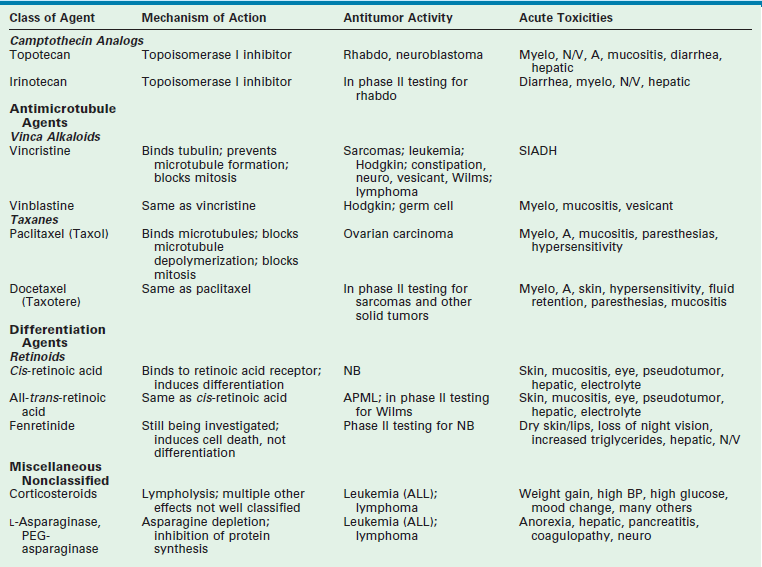
Data from Balis FM, Holcenberg JS, Poplack DG. General principles of chemotherapy. In: Pizzo P, Poplack D, editors. Principles and Practice of Pediatric Oncology. 3rd ed. Philadelphia: Lippincott-Raven; 1997. p. 215–72; Ratain M, Teicher B, O’Dwyer P, et al. Pharmacology of cancer chemotherapy. In: DeVita V, Hellman S, Rosenberg S, editors. Cancer: Principles and Practice of Oncology. Philadelphia: Lippincott-Raven; 1997. p. 375–85; Dorr R, Von Hoff D. Drug monographs. In: Dorr R, Von Hoff D, editors. Cancer Chemotherapy Handbook. Norwalk, CT: Appleton & Lange; 1994. p. 129.
Acute Chemotherapy Toxicity and Supportive Care
Myelosuppression is an expected side effect of almost all treatment protocols for solid tumors. Transfusions of packed cells and platelets are frequently needed. Of greatest concern is the risk of severe life-threatening bacterial or fungal infections that occur during episodes of neutropenia. In dose-intensive regimens, more than 75% of chemotherapy courses result in hospitalization for fever, with the incidence of bacteremia ranging from 10– 20% per course.37
Success in improving treatment outcomes is partially attributed to advances in supportive care. The routine use of hematopoietic growth factors, specifically G-CSF, results in more rapid granulocyte recovery and shorter hospitalizations for fever and neutropenia.26 In addition, IL-11 enhances platelet recovery, decreases the depth of the platelet nadir, and decreases platelet transfusion requirements.27,38,39 It is well tolerated and is beneficial in combination regimens that induce severe thrombocytopenia.40,41
The gastrointestinal tract is injured by cytarabine, anthracyclines, and high-dose methotrexate; however, the folate derivative Leucovorin can be given to rescue normal mucosal and bone marrow cells from the effects of the latter. No rescue is known for the mucositis and diarrhea which occur with other agents. In addition to enhancing platelet production, IL-11 may help speed recovery from gastrointestinal injury after chemotherapy.42
Renal toxicity can occur from the use of cisplatin, ifosfamide, and high-dose methotrexate. Cisplatin causes renal tubular damage, leading to elevation of levels of blood urea nitrogen and creatinine; this effect is generally reversible. Both ifosfamide and cisplatin cause renal electrolyte wasting in which hypokalemia, hypocalcemia, hypophosphatemia, and hypomagnesemia can occur. Renal injury from these agents can be improved by hyperhydration and forced diuresis. Mesna can prevent hemorrhagic cystitis resulting from cyclophosphamide and ifosfamide by binding to the bladder-toxic acrolein metabolites.40
Additionally, a recent study demonstrates that amifostine administered prior to and during cisplatin infusion significantly reduces the risk of severe ototoxicity in children with average risk medulloblastoma who are undergoing treatment with dose-intense chemotherapy.43
Targeting Biologic Pathways
Signal Transduction Inhibitors
Cellular signaling is a basic biologic function of all normal cells. Signaling can be extracellular (e.g., growth factor receptor tyrosine kinase) or through multiple intracellular effector and survival pathways (e.g., RAS, RAF, TP53, BCL-2). Cancer cells differ from normal cells as they are associated with chromosomal mutations. These mutations result in cancer cells being dependent on several hyperactive signaling pathways. The rationale behind signal transduction therapy is that blocking the hyperactive pathways induces cancer cell apoptosis. By contrast, normal cells have redundant signaling pathways that protect them against cell death. Multiple new signal transduction inhibitors are in development.44,45
Biologic Response Modifiers
T-lymphocytes directly interact with specific cell surface antigens on a target cell, causing cell lysis through cytotoxic granule release or programmed cell death. These cytotoxic lymphocytes are involved with the killing of tumor cells. To initiate this response, antigens must be presented to the T cell by antigen-presenting cells (APCs) that express the antigens bound to major histocompatibility complex proteins in the presence of stimulatory cytokines. Cytokines or interleukins (e.g., IL-2, interferon-α, tumor necrosis factor) are proteins produced by helper T-lymphocytes and monocytes that help recruit other effector cells, including APCs, as well as regulate antibody production. The effector cells of the immune system (e.g., granulocytes, monocytes, macrophages, eosinophils, dendritic cells) can become tumor selective when activated by a specific antibody, a process called antibody-dependent cell-mediated cytotoxicity.
Adoptive Immunotherapy
Adoptive immunotherapy involves the use of tumor vaccines made from autologous or allogeneic tumor-associated antigens. Specific purified tumor antigens can be made more immunogenic by attachment to carrier proteins (adjuvants). Clinical trials using this type of tumor vaccine have been performed primarily in melanoma patients.46 Other forms of immunotherapy involve the use of cytokine infusions such as interferon-α, IL-2, and tumor necrosis factor to stimulate immune reaction against tumor cells.47
Tumor-Targeted Antibody Therapy
Chimeric human/mouse antibodies have been produced that decrease the risk of human/anti-mouse antibody generation.48 Recombinant chimeric antibodies are produced by linking the constant region of human antibodies to the variable combining region of a mouse mAb. These chimeric antibodies have been successfully used in the treatment of children with high-risk neuroblastoma.49
Local Tumor Control
Radiation Oncology
Radiation therapy may be combined with operation in a strategic manner to deliver the highest effective dose to a well-defined site while minimizing the dose to surrounding normal structures.50 Preoperative radiation may permit a smaller treatment area because the operative bed has not been manipulated. In larger tumors, preoperative radiation may reduce the lesion volume sufficiently to allow a subsequent resection. In addition, potential tumor seeding during operative removal may be reduced because cells that may be surgically disseminated have been rendered incapable of reproducing. On the other hand, preoperative radiation may delay the surgical procedure and alter the staging information obtained at operation.
For these reasons, many combined strategies use postoperative radiation, which allows the treatment fields and doses to be determined after operative resection and histologic assessment.51 Higher doses can be delivered postoperatively once the target volumes have been more accurately defined. Doses to the periphery of the tumor can be fine-tuned, depending on the presence of gross, microscopic, or no residual disease. However, postoperative delivery may require a wider treatment area after extensive surgical manipulation.
Soft tissue sarcoma provides a model for the adjunctive role of radiation therapy. Resection is the primary method of obtaining local control; however, radiation therapy can be effective when clear margins are not possible.52 Combined therapy has resulted in dramatically improved survival.53 In extremity lesions, radiation also allows more conservative resection with limb sparing. Although local tumor control rates of 75–98% have been achieved with limb salvage rates greater than 80%, wound complications occur in as many as 40% of patients. Neoadjuvant radiation at more modest doses (30 Gy total) has decreased the complication rate while maintaining excellent (>95%) five-year local control, and ultimately, limb salvage.52 Postoperative adjuvant radiation therapy also may be advantageous.54
Several aspects of radiation treatment in pediatric patients warrant special consideration. Attention should be paid to immobilizing or sedating children so that ionizing doses can be targeted to the desired area without inappropriate exposure of surrounding tissues. Pediatric radiation oncologists may use lower treatment doses and accept a higher recurrence rate to ensure lower toxicity, especially in important developing organs such as the brain. The normal tolerance of organs or tissues may be adversely affected when chemotherapeutic agents are also used. The long-term effects of combined-modality therapy must be considered in regard to musculoskeletal and dental tissues, central nervous system (CNS) and neuropsychological sequelae, and endocrine and gonadal dysfunction, as well as direct effects on the heart, lungs, or kidneys.55 The following sections describe techniques that allow safe, efficacious doses of radiation to be delivered, often in combination with surgical excision, to produce the maximal therapeutic benefit.
Brachytherapy
Children with soft tissue sarcomas can benefit from specialized radiation treatment strategies. For children who have microscopic residual disease after operation, radiation provides excellent local tumor control.56 High-dose-rate brachytherapy can be delivered in only a few minutes, which is particularly helpful in young children. In contrast, low-dose-rate brachytherapy provided by low energy source techniques requires sedation, immobilization, long exposure times, and hospitalization. The short therapy duration associated with high-dose-rate therapy also allows rapid reinstitution of chemotherapy. Morbidity is usually related to skin or mucosal reactions, which may progress as a ‘recall’ phenomenon in patients treated with radiosensitizing agents such as anthracyclines.57 Brachytherapy, alone or in combination with external-beam irradiation, has been shown to provide a high rate of local tumor control in pediatric soft tissue sarcomas.58–60
Intraoperative Radiation
Intraoperative radiation therapy (IORT) allows the radiation dose to be directly applied to the target area while shielding adjacent structures. Whenever disease remains in surgically inaccessible areas, IORT may be an effective adjunct. Phase I and II studies have demonstrated that IORT can be performed safely in children.61 It is used for patients in whom unresectable disease is present at diagnosis or for delayed primary or second-look procedures, residual lesions, or local tumor recurrence.
In children with retroperitoneal tumors, IORT may, however, lead to urologic complications. In a study reported in 1990, 3 of 6 patients treated with IORT and external-beam therapy required intervention for fibrotic ureteral strictures or renal artery stenosis; in two cases, the injured structures were within the supplemental external-beam treatment field. Also, neuropathies developed in two other patients. Nevertheless, all patients were survivors for up to 42 months’ follow-up.62 In a later study, the complications were minimized through more extensive dissection of normal structures and avoidance of overlapping radiation fields.63
Intensity-Modulated Radiation Therapy
Techniques continue to evolve to improve the impact of radiation therapy on tumor response while minimizing the dose of radiation imparted to surrounding normal tissues. Stereotactic radiation therapy is sometimes used for CNS tumors.64–67 Imaging systems, treatment-planning software, and delivery systems have undergone dramatic advancements that allow sophisticated delivery of more precise courses of radiation. Intensity-modulated radiation therapy (IMRT) is an advanced form of three-dimensional conformal therapy that uses non-uniform radiation beam intensities determined by using various computer-based optimization techniques. Experience with IMRT in children is growing. Reports of mixed tumor cases, including pediatric patients, suggest that IMRT will be effective in reducing treatment-related morbidity and allow dose escalation to the target volume.68,69 Significant reductions in radiation exposure to critical structures has been shown for intracranial, cervical, and abdominopelvic lesions.70
Proton Therapy
Proton therapy is very costly and there are currently fewer than 50 centers in the world capable of providing this treatment. Although most patients who have undergone therapy to date are adults, limited experience with pediatric CNS tumors and sarcomas has been reported.71,72 Researchers at the Massachusetts General Hospital have described 30 patients with Ewing sarcoma who received proton therapy as a component of multimodal management. Three-year local control in 86% of patients was achieved with few adverse events.71 The utility of proton therapy in other childhood tumors, including neuroblastoma and rhabdomyosarcoma, is also being explored.72
Innovative Adjunctive Techniques
Radiofrequency Ablation
Radiofrequency ablation (RFA) is a technique that applies thermal energy via a probe that results in coagulation necrosis of the target tissue. The technique involves image-guided application of the probe, primarily using ultrasound. The probe can be introduced percutaneously, laparoscopically, or via an open exposure. In adults, the most common applications are for primary or metastatic hepatic lesions, renal tumors, and pulmonary lesions.73–77 Treatment of pediatric tumors with RFA is largely anecdotal. A small series reported the use of percutaneous RFA on fetuses with sacrococcygeal teratoma, but found a 50% fetal mortality rate.78 Another report described severe soft tissue injury and sciatic nerve destruction at birth in an infant treated prenatally.79
Transcatheter Arterial Chemoembolization
Transcatheter arterial chemoembolization (TACE) is a technique used to directly instill chemotherapeutic agents into a tumor, thereby minimizing systemic toxicity. TACE is most commonly used in the management of liver lesions, taking advantage of the preferential arterial blood supply to these tumors.80,81
Although the pediatric experience with TACE is limited, there is some evidence to support its efficacy.82,83 In one series, a suspension of cisplatin, doxorubicin, and mitomycin-C mixed with bovine collagen and radiopaque contrast material was used in 11 children with unresectable or recurrent lesions.82 Six hepatoblastoma patients had an initial partial response, as measured by imaging and α-fetoprotein levels. Of these patients, three underwent subsequent surgical resection; one had progressive disease and died, and two survived for more than 15 months. The other three patients eventually also died of known progressive disease. In three children with hepatocellular carcinoma, one underwent surgical resection and was a long-term survivor for more than 65 months, one was alive with disease for more than 36 months, and one died of progressive liver failure with no evidence of malignancy. In another relatively recent report documenting the outcomes of 16 infants and children with unresectable hepatoblastoma, Li et al reported that TACE facilitated safe and complete surgical resection in 12 cases.83 Three patients underwent partial resection. One patient underwent successful orthotopic liver transplantation after receiving TACE therapy.
Overall, chemoembolization is feasible in young patients and is associated with tolerable toxicity. This option represents a possible therapeutic alternative in persistent, unresectable, or recurrent hepatoblastoma, or in nonmetastatic hepatocellular carcinoma. Its use has also been proposed in unresectable Wilms tumor.84
Sentinel Lymph Node Biopsy
The utility of sentinel lymph node biopsy in children is evolving. Although it is standard practice for patients with intermediate thickness melanomas,85 its value in soft tissue sarcomas remains unclear.86,87
The initial draining lymph node (sentinel node) is predictive of regional nodal metastases in a variety of tumors.88 It is most commonly used to predict nodal status of patients with melanoma or breast cancer. In most cases, a combination of technetium-labeled sulfur colloid with either Lymphazurin or methylene blue dye is used to localize the sentinel node. Preoperative lymphoscintigraphy provides information regarding the location of the primary lymph node draining the tumor site. Lymphatic mapping is accomplished by injecting the technetium-labeled sulfur colloid at the primary tumor site. The affected region is then imaged to identify the sentinel lymph node. Just before incision, the blue dye is injected at the primary tumor site and a gamma probe is used to identify areas of high counts. The underlying tissue is then examined for lymph nodes containing the blue dye. The lymph nodes are removed and sent for histopathologic examination. If the sentinel node shows no evidence of metastases, the related regional lymphatic bed is highly likely to be tumor free. In these patients, the morbidity of lymphadenectomy can be avoided.
Late Effects of Cancer Therapy
According to the most recent National Cancer Institute statistics: (1) there are more than 328,000 survivors of childhood cancer in the USA and (2) 1 in 900 adults is now a cancer survivor.89 The remarkable increase in survival has created a new and growing population who are at increased risk for late adverse effects of treatment and a diminished quality of life. All aspects of combined-modality therapy can contribute to the late effects. A number of chemotherapy agents have been associated with late toxicities. In addition, radiation therapy can significantly inhibit further growth of bone, muscle, heart, and kidney within the radiation field, and also can affect fertility.
Growth retardation is a late effect unique to children. The degree of impairment depends on the dose of chemotherapy or radiation and the age of the child at the time of therapy. The younger the child is at the time of the insult, the more severe the sequelae. More than 50% of childhood brain tumor patients treated with 3000 cGy or more to the whole brain will have severe growth retardation, with adult height being lower than the 5th percentile.90 Cranial irradiation can lead to growth hormone deficiency, which results in poor linear growth, unless growth hormone replacement is given. In addition, patients who have received total-body irradiation or spinal radiation may not be able to achieve their full height potential because the irradiated bones have limited growth potential, even with growth hormone stimulation.91
Adjuvant therapy can also cause musculoskeletal problems, including scoliosis, avascular necrosis, osteoporosis, and atrophy or hypoplasia of tissues. Radiation to the head and neck results in hypoplasia of the jaw, orbit, or neck, with associated atrophy of the soft tissues. Associated endocrine, dental, and psychological consequences can occur.92 Osteoporosis occurs as a result of corticosteroid treatment and from high-dose irradiation, as used for sarcomas.
Specific organs are often affected by chemotherapy. Heart, liver, lung, thyroid, and gonadal function are often impaired. Gonadal dysfunction (azoospermia, amenorrhea) frequently results from alkylator treatment. Therapy with mechlorethamine, vincristine, prednisone, and procarbazine has resulted in azoospermia in 80–100% of all male patients.93 Combination chemotherapy programs for childhood Hodgkin disease have been adjusted to replace mechlorethamine with cyclophosphamide and eliminate dacarbazine from standard treatments in an attempt to decrease the infertility risk. It should be noted that the children of childhood cancer survivors are not at an increased risk for congenital anomalies.94
Cardiotoxicity from anthracycline antibiotics has been a problem in the treatment of Ewing sarcoma, osteosarcoma, and lymphomas. However, the use of continuous infusion anthracycline can decrease the risk of cardiac muscle damage and subsequent congestive heart failure.95 Another strategy is the use of dexrazoxane to prevent anthracycline-induced cardiotoxicity.28 Survivors of childhood brain tumor therapy treated with a combination of chemotherapy, irradiation, and surgery had a significantly increased risk of stroke, blood clots, and angina-like symptoms compared with their siblings.96
Other significant organ-related late effects include chronic renal insufficiency from cisplatin therapy, chronic cystitis from cyclophosphamide or ifosfamide treatment, and prolonged hypogammaglobulinemia and T-lymphocyte dysfunction after multiple high-dose alkylators for bone marrow transplant.97
The most significant late effect of cancer therapy is the risk of secondary malignancy. This risk is highest in patients who have received both chemotherapy and radiation therapy. In 2001, Neglia and colleagues reported that the estimated cumulative incidence of all subsequent malignant neoplasms in a cohort of 13,581 childhood cancer survivors was 3.2% at 20 years after the primary diagnosis.98 Hodgkin disease survivors have the highest secondary malignancy rates. Breast cancer is the most common solid tumor, with an estimated actuarial incidence in women of 35% by age 40 years. These patients are also at risk of developing leukemia, non-Hodgkin lymphoma, and thyroid carcinoma.99 In survivors of childhood Hodgkin disease, the cumulative estimated incidence of second malignancies at 30 years ranges from 18% to 31%.100,101 Patients who have received additional multimodality therapy for recurrent Hodgkin disease have the highest risk of second tumors. Patients with soft tissue sarcomas, retinoblastoma, and Ewing sarcoma who receive high-dose radiation to the primary lesion are at increased risk for secondary osteosarcoma within the radiation field.102 Etoposide has been recognized as causing secondary AML.
References
1. Farber, S, Diamond, LK. Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl-glutamic acid (aminopterin). N Engl J Med. 1948; 238:787–793.
2. Li, MC, Hertz, R, Spencer, D, et al. Effect of methotrexate upon choriocarcinoma and chorioadenoma. Proc Soc Exp Biol Med. 1956; 93:361–366.
3. Frei, E, III., Freireich, EJ, Gehan, E, et al. Studies of sequential and combination antimetabolite therapy in acute leukemia: 6-Mercaptopurine and methotrexate. Blood. 1961; 18:431–454.
4. Green, DM, Jaffe, N. Wilms’ tumor: Model of a curable pediatric malignant solid tumor. Cancer Treat Rev. 1978; 5:143–172.
5. D’Angio, GJ, Evans, AE, Breslow, N, et al. The treatment of Wilms’ tumor: Results of the National Wilms’ Tumor Study. Cancer. 1976; 38:647.
6. Link, MP, Goorin, AM, Miser, AW, et al. The effect of adjuvant chemotherapy on relapse-free survival in patients with osteosarcoma of the extremity. N Engl J Med. 1986; 314:1600–1606.
7. Schwenn, MR, Blattner, SR, Lynch, E, et al. HiC-COM: A 2-month intensive chemotherapy regimen for children with stage III and IV Burkitt’s lymphoma and B-cell acute lymphoblastic leukemia. J Clin Oncol. 1991; 9:133–138.
8. Cheung, NK, Heller, G. Chemotherapy dose intensity correlates strongly with response, median survival and median progression-free survival in metastatic neuroblastoma. J Clin Oncol. 1991; 9:1050–1058.
9. Antman, K, Ayash, L, Elias, A, et al. A phase II study of high dose cyclophosphamide, thiotepa, and carboplatin with autologous marrow support in women with measurable advanced breast cancer responding to standard therapy. J Clin Oncol. 1992; 10:102–110.
10. Grier, H, Krailo, M, Link, M, et al. Improved outcome in non-metastatic Ewing’s sarcoma and PNET of bone with the addition of ifosfamide and etoposide to vincristine, Adriamycin, cyclophosphamide, and actinomycin: A Children’s Cancer Group and Pediatric Oncology Group report. J Clin Oncol. 1994; 13(Suppl):421.
11. Clark, PI, Slevin, ML, Joel, SP, et al. A randomized trial of two etoposide schedules in small-cell lung cancer: The influence of pharmacokinetics on efficacy and toxicity. J Clin Oncol. 1994; 12:1427–1435.
12. Worth, L, Jeha, S, Kleinerman, E. Biologic response modifiers in pediatric cancer. Hematol Oncol Clin North Am. 2001; 5:723–740.
13. Parker, SL, Tong, T, Bolden, S, et al. Cancer statistics, 1997. CA Cancer J Clin. 1997; 47:5–27.
14. National Cancer Institute fact sheet [Internet]. Available at http://www.cancer.gov/cancertopics/factsheet/Sites-Types/childhood.
15. Gloeckler Ries, LA, Percy, CL, Bunin, GR. SEER Pediatric Monograph. Available at http://seer.cancer.gov/publications/childhood/introduction.
16. Kreissman, SG. Molecular genetics: Toward an understanding of childhood cancer. Semin Pediatr Surg. 1993; 2:2–10.
17. Brodeur, GM. Neuroblastoma: Clinical applications of molecular parameters. Brain Pathol. 1990; 1:47–54.
18. MacGregor, PF. Gene expression in cancer: The application of microarrays. Expert Rev Mol Diagn. 2003; 3:185–200.
19. Goldie, JH, Coldman, AJ. A mathematic model for relating the drug sensitivity of tumors to the spontaneous mutation rate. Cancer Treat Rep. 1979; 63:1727–1731.
20. Balis, FM, Holcenberg, JS, Poplack, DG. General principles of chemotherapy. In Pizzo P, Poplack D, eds.: Principles and practice of pediatric oncology, 3rd ed, Philadelphia: Lippincott-Raven, 1997.
21. D’Angio, GJ, Evans, AE, Breslow, N, et al. The treatment of Wilms’ tumor: Results of the National Wilms’ Tumor Study. Cancer. 1976; 38:633–646.
22. Green, DM, Breslow, NE, Evans, I, et al. The effect of chemotherapy dose intensity on the hematological toxicity of treatment for Wilms’ tumor: A report from the National Wilms’ Tumor Study. Am J Pediatr Hematol Oncol. 1994; 16:207–212.
23. Picci, P, Rougraff, BT, Bacci, G, et al. Prognostic significance of histopathologic response to chemotherapy in nonmetastatic Ewing’s sarcoma of the extremities. J Clin Oncol. 1993; 11:1763–1769.
24. Provisor, AJ, Ettinger, LJ, Nachman, JB, et al. Treatment of nonmetastatic osteosarcoma of the extremity with preoperative and postoperative chemotherapy: A report from the Children’s Cancer Group. J Clin Oncol. 1997; 5:76–84.
25. Shamberger, RC, Allarde-Segundo, A, Kozakewich, HP, et al. Surgical management of stage III and IV neuroblastoma: Resection before or after chemotherapy? J Pediatr Surg. 1991; 26:1113–1118.
26. Householder, SE, Rackoff, W, Goldman, J, et al. A case-control retrospective study of the efficacy of granulocyte colony stimulating factor in children with neuroblastoma. Am J Pediatr Hematol Oncol. 1994; 16:132–137.
27. Tepler, I, Elias, L, Smith, JW, et al. A randomized placebo-controlled trial of recombinant human interleukin-11 in cancer patients with severe thrombocytopenia due to chemotherapy. Blood. 1996; 87:3607–3614.
28. Lipshultz, SE. Dexrazoxane for protection against cardiotoxic effects of anthracyclines in children. J Clin Oncol. 1996; 14:328–331.
29. Strother, D, Ashley, D, Kellie, SJ, et al. Feasibility of four consecutive high-dose chemotherapy cycles with stem-cell rescue for patients with newly diagnosed medulloblastoma or supratentorial primitive neuroectodermal tumor after craniospinal radiotherapy: Results of a collaborative study. J Clin Oncol. 2001; 19:2696–2704.
30. Welte, K, Reiter, A, Mempel, K, et al. A randomized phase-III study of the efficacy of granulocyte colony-stimulating factor in children with high-risk acute lymphoblastic leukemia. Berlin-Frankfurt-Munster Study Group. Blood. 1996; 87:3143–3150.
31. Burdach, SE, Müschenich, M, Josephs, W, et al. Granulocyte-macrophage-colony stimulating factor for prevention of neutropenia and infections in children and adolescents with solid tumors. Results of a prospective randomized study. Cancer. 1995; 76:510–516.
32. Hawkins, DS, Felgenhauer, J, Park, J, et al. Peripheral blood stem cell support reduces the toxicity of intensive chemotherapy for children and adolescents with metastatic sarcomas. Cancer. 2002; 95:1354–1365.
33. Frei, E, Canellos, GP. Dose: A critical factor in cancer chemotherapy. Am J Med. 1980; 69:585–594.
34. Broun, R, Nichols, CR, Kneebone, P, et al. Long-term outcome of patients with relapsed and refractory germ cell tumors treated with high-dose chemotherapy and autologous bone marrow rescue. Ann Intern Med. 1992; 117:124–128.
35. Smith, MA, Ungerleider, RS, Horowitz, ME, et al. Influence of doxorubicin dose intensity on response and outcome for patients with osteogenic sarcoma and Ewing’s sarcoma. J Natl Cancer Inst. 1991; 83:1460–1470.
36. Kaye, SB, Lewis, CR, Dave, J, et al. A randomized study of two doses of cisplatin and cyclophosphamide in epithelial ovarian cancer. Lancet. 1992; 340:329–333.
37. Kreissman, SG, Rackoff, W, Lee, M, et al. High dose cyclophosphamide with carboplatin: A tolerable regimen suitable for dose intensification in children with solid tumors. J Pediatr Hematol Oncol. 1997; 19:309–312.
38. Gordon, MS, McCaskill-Stevens, WJ, Battiato, LA, et al. A phase I trial of recombinant human interleukin 11 (Neumega rgIL-11 growth factor) in women with breast cancer receiving chemotherapy. Blood. 1996; 87:3615–3624.
39. Ali-Nazir, A, Davenport, G, Reaman, G, et al. A phase I/II trial of rhIL-11 following ifosfamide, carboplatin, and etoposide (ICE) chemotherapy in pediatric patients with solid tumors or lymphoma. J Clin Oncol. 1996; 15(Suppl):274.
40. Shaw, IC, Graham, MI. Mesna: A short review. Cancer Treat Rev. 1987; 14:67–86.
41. Moritz, T, MacKay, W, Glassner, B, et al. Retrovirus-mediated expression of DNA repair protein in bone marrow protects hematopoietic cells from nitrosourea-induced toxicity in vitro and in vivo. Cancer Res. 1995; 55:2608–2614.
42. Sonis, S, Muska, A, O’Brien, J, et al. Alterations in the frequency, severity, and duration of chemotherapy induced mucositis in hamsters by interleukin-11. Eur J Cancer. 1995; 31:261–266.
43. Fouladi, M, Chintagumpala, M, Ashley, D, et al. Amifostine protects against cisplatin-induced ototoxicity in children with average-risk medulloblastoma. J Clin Oncol. 2008; 26:3749–3755.
44. Corey, SJ. Targeted therapy: for kids, too. Review. Pediatr Blood Cancer. 2005; 45:623–634.
45. Levitzki, A, Klein, S. Signal transduction therapy of cancer. Mol Aspects Med. 2010; 31:287–329.
46. Restifo, NP, Snzol, M. Cancer vaccines. In: DeVita VT, Hellman S, Rosenberg SA, eds. Cancer: Principles and Practice of Oncology. 5th ed. Philadelphia: Lippincott-Raven; 1997:3033.
47. Rosenberg, SA. Principles of cancer management: Biologic therapy. In: DeVita VT, Hellman S, Rosenberg SA, eds. Cancer: Principles and Practice of Oncology. 5th ed. Philadelphia: Lippincott-Raven; 1997:364.
48. Frost, JD, Hank, JA, Reahman, GH, et al. A phase I/IB trial of murine monoclonal anti-GD2 antibody 14. G2a plus interleukin-2 in children with refractory neuroblastoma: A report of the Children’s Cancer Group. Cancer. 1997; 80:317–333.
49. Yu, AL, Gilman, AL, Ozkaynak, MF, et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med. 2010; 363:1324–1334.
50. Eisbruch, A, Lichter, AS. What a surgeon needs to know about radiation. Ann Surg Oncol. 1997; 4:516–522.
51. Liu, L, Glicksman, AS. The role of radiation in the management of soft tissue sarcoma. Med Health RI. 1997; 80:32–36.
52. Temple, WJ, Temple, CLF, Arthur, K, et al. Prospective cohort study of neoadjuvant treatment in conservative surgery of soft tissue sarcomas. Ann Surg Oncol. 1997; 4:586–590.
53. Marcus, KC, Grier, HE, Shamberger, RC, et al. Childhood soft tissue sarcoma: A 20-year experience. J Pediatr. 1997; 131:603–607.
54. Lindberg, RD, Martin, RG, Romsdahl, MM, et al. Conservative surgery and postoperative radiotherapy in 300 adults with soft-tissue sarcomas. Cancer. 1981; 47:2391–2397.
55. Fryer, CJH. Principles of pediatric radiation oncology. In: Holland JF, Frei E, Bast RC, eds. Cancer Medicine. 4th ed. Baltimore: Williams & Wilkins; 1996:2899–2905.
56. Donaldson, S, Breneman, J, Asmar, L, et al. Hyperfractionated radiation in children with rhabdomyosarcoma. Results of an intergroup rhabdomyosarcoma pilot study. Int J Radiat Oncol Biol Phys. 1995; 32:903–911.
57. Nag, S, Grecula, J, Ruymann, FB. Aggressive chemotherapy, organ-preserving surgery, and high-dose-rate remote brachytherapy in the treatment of rhabdomyosarcoma in infants and young children. Cancer. 1993; 72:2769–2776.
58. Nag, S, Olson, T, Ruymann, F, et al. High-dose-rate brachytherapy in childhood sarcomas: A local control strategy preserving bone growth and function. Med Pediatr Oncol. 1995; 25:463–469.
59. Nag, S, Martinez-Monge, R, Ruyman, F, et al. Innovation in the management of soft tissue sarcomas in infants and young children: High dose brachytherapy. J Clin Oncol. 1997; 15:3075–3084.
60. Merchant, TE, Parsh, N, del Valle, PL, et al. Brachytherapy for pediatric soft-tissue sarcoma. Int J Radiat Oncol Biol Phys. 2000; 46:427–432.
61. Zelefsky, MJ, LaQuaglia, MP, Ghavimi, F, et al. Preliminary results of phase I/II study of high-dose-rate intraoperative radiation therapy for pediatric tumors. J Surg Oncol. 1996; 62:267–272.
62. Ritchey, ML, Gunderson, LL, Smithson, WA, et al. Pediatric urologic complications with intraoperative radiation therapy. J Urol. 1990; 143:89–91.
63. Haase, GM, Meagher, D, Jr., McNeely, LK, et al. Electron beam intraoperative radiation therapy for pediatric neoplasms. Cancer. 1994; 74:740–747.
64. Mehta, MP. The physical, biologic, and clinical basis of radiosurgery. Curr Probl Cancer. 1995; 19:265–329.
65. Shaw, E, Scott, C, Souhami, L, et al. Radiosurgery for the treatment of previously irradiated recurrent primary brain tumors and brain metastases: Initial analysis of Radiation Therapy Oncology Group protocol (RTOG) 9005. Int J Radiat Oncol Biol Phys. 1994; 30:166.
66. Smyth, MD, Sneed, PK, Ciricillo, SF, et al. Stereotactic radiosurgery for pediatric intracranial arteriovenous malformations: The University of California at San Francisco experience. J Neurosurg. 2002; 97:48–55.
67. Shrieve, DC, Alexander, E, Wen, PY, et al. Comparison of stereotactic radiosurgery and brachytherapy in the treatment of recurrent glioblastoma multiforme. Neurosurg. 1995; 36:275–284.
68. Teh, BS, Mai, WY, Grant, WH, et al. Intensity modulated radiotherapy (IMRT) decreases treatment-related morbidity and potentially enhances tumor control. Cancer Invest. 2002; 20:437–451.
69. Swift, P. Novel techniques in the delivery of radiation in pediatric oncology. Pediatr Clin North Am. 2002; 9:1107–1129.
70. Bhatnagar, A, Deutsch, M. The role for intensity modulated radiation therapy (IMRT) in pediatric population. Technol Cancer Res Treat. 2006; 5:591–595.
71. Merchant, TE. Proton beam therapy in pediatric oncology. Cancer J. 2009; 15:298–305.
72. Cotter, SE, McBride, SM, Yock, TI. Proton radiation for solid tumors of childhood. Technol Cancer ResTreat. 2012; 11:267–277.
73. Curley, SA. Radiofrequency ablation of malignant liver tumors. Ann Surg Oncol. 2003; 10:338–347.
74. Scaife, CL, Curley, SA. Complication, local recurrence, and survival rates after radiofrequency ablation for hepatic malignancies. Surg Oncol Clin North Am. 2003; 12:243–255.
75. Mayo-Smith, WW, Dupuy, DE, Parikh, PM, et al. Image-guided percutaneous radiofrequency ablation of solid renal masses: Techniques and outcomes of 38 treatment sessions in 32 consecutive patients. AJR Am J Roentgenol. 2003; 180:1503–1508.
76. Farrell, MA, Charboneau, WJ, DiMarco, DS, et al. Image-guided radiofrequency ablation of solid renal tumors. Am J Roentgenol. 2003; 180:1509–1513.
77. Herrera, LJ, Fernando, HC, Perry, Y, et al. Radiofrequency ablation of pulmonary malignant tumors in non-surgical candidates. J Thorac Cardiovasc Surg. 2003; 125:929–937.
78. Paek, BW, Jennings, RW, Harrison, MR, et al. Radiofrequency ablation of human fetal sacrococcygeal teratoma. Am J Obstet Gynecol. 2001; 184:503–507.
79. Ibrahim, D, Ho, E, Scherl, LA, et al. Newborn with an open posterior hip dislocation and sciatic nerve injury after intrauterine radiofrequency ablation of a sacrococcygeal teratoma. J Pediatr Surg. 2003; 38:248–250.
80. Gunvén, P. Liver embolizations in oncology: A review. Part I. (chemo)embolizations. Med Oncol. 2008; 25:1–11.
81. Bittles, MA, Hoffer, FA. Interventional radiology and the care of the pediatric oncology patient. Surg Oncol. 2007; 16:229–233.
82. Malogolowkin, MH, Stanley, P, Steele, DA, et al. Feasibility and toxicity of chemoembolization for children with liver tumors. J Clin Oncol. 2000; 18:1279–1284.
83. Li, J, Chu, J, Yang, J, et al. Preoperative transcatheter selective arterial chemoembolization in treatment of unresectable hepatoblastoma in infants and children. Cardiovasc Intervent Radiol. 2008; 31:1117–1123.
84. Li, M, Zhou, Y, Huang, Y, et al. A retrospective study of the preoperative treatment of advanced Wilms tumor in children with chemotherapy versus transcatheter arterial chemoembolization alone or combined with short-term chemotherapy. J Vasc Interv Radiol. 2011; 22:279–286.
85. Topar, G, Zelger, B. Assessment of value of the sentinel lymph node biopsy in melanoma in children and adolescents and applicability of subcutaneous infusion anesthesia. J Pediatr Surg. 2007; 42:1716–1720.
86. DeCorti, F, Dall’Igna, P, Bisogno, G, et al. Sentinel node biopsy in pediatric soft tissue sarcomas of extremities. Pediatr Blood Cancer. 2009; 52:51–54.
87. Pacella, SJ, Lowe, L, Bradford, C, et al. The utility of sentinel lymph node biopsy in head and neck melanoma in the pediatric population. Plast Reconstr Surg. 2003; 112:1257–1265.
88. Morton, DL, Wend, D, Wong, JH, et al. Technical details of intraoperative lymphatic mapping for early stage melanoma. Arch Surg. 1992; 127:392–399.
89. Mariotto, AB, Rowland, JH, Yabroff, KR, et al. Long-term survivors of childhood cancers in the United States. Cancer Epidemiol Biomarkers Prev. 2009; 18:1033–1040.
90. Oberfield, SE, Allen, JC, Pollack, J, et al. A long-term endocrine sequelae after treatment of medulloblastoma: Prospective study of growth and thyroid function. J Pediatr. 1986; 108:219–223.
91. Huma, Z, Boulad, F, Black, P, et al. Growth in children after bone marrow transplantation for acute leukemia. Blood. 1995; 86:819–824.
92. Larson, DL, Kroll, S, Jaffe, N, et al. Long-term effects of radiotherapy in childhood and adolescence. Am J Surg. 1990; 160:348–350.
93. Whitehead, E, Shalet, SM, Morris-Jones, PH, et al. Gonadal function after combination chemotherapy for Hodgkin’s disease in childhood. Arch Dis Child. 1982; 57:287–291.
94. Green, DM, Zevon, MA, Lowrie, G, et al. Congenital anomalies in children of patients who received chemotherapy for cancer in childhood or adolescence. N Engl J Med. 1991; 325:141–146.
95. Legha, SS, Benjamin, RS, Mackay, B, et al. Reduction of doxorubicin cardiotoxicity by prolonged continuous infusion. Ann Intern Med. 1982; 96:133–139.
96. Gurney, J, Kaden-Lottick, N, Packer, R, et al. Endocrine and cardiovascular late effects among adult survivors of childhood brain tumors: Childhood Cancer Survivors Study. Cancer. 2003; 97:663–673.
97. Blatt, J, Copeland, DR, Bleyer, WA. Late effects of childhood cancer and its treatment. In: Pizzo PA, Poplack DG, eds. Principles and Practice of Pediatric Oncology. 3rd ed. Philadelphia: Lippincott-Raven; 1997:1303.
98. Neglia, JP, Friedman, DL, Yasui, Y, et al. Second malignant neoplasms in five-year survivors of childhood cancer; childhood cancer survivor study. J Natl Cancer Inst. 2001; 93:618–629.
99. Bhatia, S, Robison, LL, Oberlin, O, et al. Breast cancer and other second neoplasm after childhood Hodgkin’s disease. N Engl J Med. 1996; 334:745–751.
100. Jenkin, D, Greenberg, M, Fitzgerald, A. Second malignant tumours in childhood Hodgkin’s disease. Med Pediatr Oncol. 1996; 26:373–379.
101. Sankila, R, Garwicz, S, Olsen, JH, et al. Risk of subsequent malignant neoplasms among 1,641 Hodgkin’s disease patients diagnosed in childhood and adolescence: A population-based cohort study in the five Nordic countries. J Clin Oncol. 1996; 14:1442–1446.
102. Smith, MB, Xue, H, Strong, L, et al. Forty year experience with second malignancies after treatment of childhood cancer: Analysis of outcome following the development of the second malignancy. J Pediatr Surg. 1993; 28:1342–1348.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]Chapter 64
Principles of Adjuvant Therapy in Childhood Cancer
Historical Overview
Early strides in improving oncologic outcomes through the use of single chemotherapeutic agents were first reported by Farber in 19481 and Li in 1956.2 As additional chemotherapeutic agents were developed, they were combined in multidrug regimens that demonstrated both significantly improved response rates and response duration.3,4 By the late 1970s, multimodal therapy was shown to improve cure rates in children with Wilms tumor5 and was being adopted for the treatment of rhabdomyosarcoma, Ewing sarcoma, lymphoma, and other solid tumors.6 The close collaboration of multidisciplinary cooperative groups and the development of improved supportive care measures added impetus to progress. During the 1990s, dose-intensive chemotherapy programs were shown to be successful in improving outcome for patients with Burkitt lymphoma, neuroblastoma, and other advanced-stage solid tumors.7–9 In addition, improvements in outcome were achieved either by alternating effective groups of chemotherapeutic agents to overcome or prevent resistance,10 or by administering agents by continuous infusion rather than bolus.11
By 2001, noncytotoxic biologic therapies (e.g., signal transduction inhibitors, various tissue growth factor receptor inhibitors, antiangiogenesis agents, tumor-targeted antibodies, and adoptive immunotherapy techniques) were developed to target specific biologic pathways.12 In addition, improvements in radiation therapy have led to the development of intraoperative radiation therapy and radiosurgery techniques. Collectively, these advancements have resulted in the continually increasing survival of children with solid tumors and a profound improvement in their quality of life.
Incidence and Survival Rates
Although childhood cancers account for only 2% of all reported cancer cases in the general population, they account for 10% of all deaths in children.13 On average, 1 to 2 of every 10,000 children in the USA develop cancer each year.14 The distribution of the types of cancer in childhood is different than in adults. Whereas most cancers in adults have an epithelial cell origin, <10% of childhood cancers fall into this category.
The incidence of specific cancers varies by age, gender, and race. Overall, however, it rose from 11.5 cases per 100,000 children in 1975 to 14.8 per 100,000 in 2004.14 The peak incidence (>200 cases per million) is in children younger than 2 years of age. The incidence then decreases to a low of 82.5 cases per million by age 9 years, at which point it begins to rise again through adolescence. In children younger than age 2, central nervous system malignancies, neuroblastoma, acute myeloid leukemia (AML), Wilms tumor, and retinoblastoma account for the majority of diagnoses. In children 2 to 4 years of age, acute lymphoblastic leukemia (ALL) is the most common childhood cancer. After age 9 years, the incidence of Hodgkin disease, osteosarcoma, and Ewing sarcoma begins to increase sharply.13
Over the past several decades, the mortality from childhood cancers has declined dramatically (40%),15 while the 5-year survival rate has risen from 58% in 1975–1977 to close to 80% in 1996–2003.14
Tumor Biology
Understanding normal cell growth and regulation is a prerequisite to understanding both the genetic basis for the development of childhood cancer and the mechanisms of action of chemotherapeutic agents designed to kill rapidly proliferating cancer cells (Fig. 64-1). Normal cell growth occurs by the regulated progression of the cell through the cell cycle of DNA replication and mitosis. This cycle is separated by two intervening growth phases, referred to as G1 and G2. Cells can temporarily leave the cell cycle and enter a resting state referred to as G0. They are programmed to proceed through the cell cycle by a series of external and internal stimuli. Binding of proteins (growth factors) to cell surface receptors stimulates a cascade of cytoplasmic signaling proteins (membrane kinases and signal transducers) that carries the stimuli to the nucleus. Other proteins (transcription factors) then bind to the DNA, resulting in the expression of growth-regulating genes. When functioning normally, these genes promote or prevent cell division, direct the cell to differentiate, or initiate apoptosis, the process of programmed cell death.
FIGURE 64-1 The cell cycle. Normal cell growth proceeds through DNA replication (S) and mitosis (M), separated by two growth phases (G1 and G2). Cells leave the cell cycle to enter a resting phase (G0) to differentiate or to die. Chemotherapy agents act at specific sites along the cell cycle, as indicated. (Adapted from Balis FRM, Holcenberg JS, Poplack DG. General principles of chemotherapy. In Pizzo P, Poplack D, editors. Principles and Practice of Pediatric Oncology. 3rd ed. Philadelphia: Lippincott-Raven; 1997. p. 219.)
Cancer Cytogenetics
The presence of consistent cytogenetic abnormalities associated with a specific childhood leukemia or solid tumor is useful in both cancer diagnosis and prognosis. Specific cytogenetic aberrations have been identified in rhabdomyosarcoma, Ewing sarcoma, synovial sarcoma, germ cell tumors, medulloblastoma, neuroblastoma, retinoblastoma, and Wilms tumor.16 Chromosomal aberrations can also be helpful in predicting prognosis. For example, the finding of a chromosome 1q deletion, the presence of double-minute chromatin bodies, or the presence of homogeneous staining regions in neuroblastoma confer a poor prognosis.17
The treatment of Wilms tumor based on risk stratification determined by multiple tumor characteristics that impact prognosis (including loss of heterozygosity at 1p and 16q) is a topic of ongoing study. In the future, specific tumors may be identified by a specific ‘fingerprint’ determined by microarray analysis that can simultaneously analyze expression of thousands of genes on a single chip.18 The ability to tailor therapy to individual patients based on the genetic characteristics of their particular tumor is quickly becoming a reality.
Clincial Trials
Combination chemotherapy remains the mainstay of treatment. The likelihood of cure is maximized when all available active agents are administered simultaneously after local control measures have been undertaken and the tumor burden is as low as possible.19 This approach has led to increased survival rates in children with neuroblastoma, Ewing sarcoma, anaplastic Wilms tumor, and osteosarcoma.
Adjuvant and Neoadjuvant Chemotherapy
The use of adjuvant chemotherapy is supported by the finding that less than 20% of sarcoma and lymphoma patients with initially nonmetastatic solid tumors can be cured by operative or radiation therapy alone or combined.20 Tumor recurrence is generally at a distant site, lending support to the hypothesis that micrometastatic disease exists at the time of presentation for most patients with clinically nonmetastatic disease. As many as 40% of patients with Wilms tumor can be cured with resection or radiation therapy alone. However, survival increases to 90% with the addition of combination chemotherapy.21
As the goal of adjuvant chemotherapy is to prevent the growth of metastatic disease, it is vital that chemotherapy begins as soon as possible after local control measures are employed. For this reason, most current chemotherapy protocols for childhood solid tumors advise that chemotherapy is initiated within 2 weeks of operation.22 In children with Wilms tumor, rapid assessment of tumor biology and risk stratification is important for determining the appropriate chemotherapy regimen.
Neoadjuvant chemotherapy has become standard in the treatment of Ewing sarcoma and osteosarcoma, and has the theoretical advantage of minimizing resistance to chemotherapy.10,23,24 Delayed surgical intervention may allow a more complete or less morbid resection, as well as histologic assessment of tumor responsiveness to the chemotherapy agents. Neoadjuvant chemotherapy is beneficial only in tumors for which a known highly effective combination chemotherapy program limits the risk of tumor progression at the primary site. For example, diagnostic biopsy followed by neoadjuvant chemotherapy and delayed resection of the primary tumor for complex neuroblastoma reduces the operative complication rate without compromising survival.25
Dose Intensity and Duration of Chemotherapy
Advances in supportive care have enabled increased dose intensity of active chemotherapy agents in pediatric clinical trials. These advances decrease or minimize the toxic effects of higher-dose chemotherapy on normal tissues. The use of cytokines (granulocyte colony-stimulating factor [G-CSF] and interleukin-11 [IL-11]) to speed recovery of white blood cells and platelets,26,27 and the use of cardioprotectant agents to allow use of a higher cumulative dose of doxorubicin have helped in the development of new dose-intensive therapy for solid tumors.28 Similarly, progress in bone marrow and stem cell transplantation have allowed dose intensities to be pushed to the upper limits.29–32
The positive impact of increasing the dose intensity on improving response rate and survival has been demonstrated for Burkitt lymphoma, osteosarcoma, Ewing sarcoma, testicular cancer, and advanced ovarian cancer.7–9,33–36 The duration of chemotherapy programs for most pediatric solid tumors has been 1 year. However, as the dose intensity of chemotherapy programs has increased, the duration of therapy has concomitantly decreased. This downward trend is likely to continue.
Chemotherapeutic Agents
Chemotherapy drugs are divided into classes by their mechanism of action. These classes include alkylating agents (cisplatin and its analogs), antimetabolites, topoisomerase inhibitors, antimicrotubule agents, differentiation agents, miscellaneous nonclassified agents, and biologic agents. Understanding the mechanism of action for each of these agents helps in establishing combination therapies with synergistic antitumor effects. The most common agents from each class, their mechanism of action, common side effects, and tumors in which they are active are listed in Table 64-1.
TABLE 64-1
Data from Balis FM, Holcenberg JS, Poplack DG. General principles of chemotherapy. In: Pizzo P, Poplack D, editors. Principles and Practice of Pediatric Oncology. 3rd ed. Philadelphia: Lippincott-Raven; 1997. p. 215–72; Ratain M, Teicher B, O’Dwyer P, et al. Pharmacology of cancer chemotherapy. In: DeVita V, Hellman S, Rosenberg S, editors. Cancer: Principles and Practice of Oncology. Philadelphia: Lippincott-Raven; 1997. p. 375–85; Dorr R, Von Hoff D. Drug monographs. In: Dorr R, Von Hoff D, editors. Cancer Chemotherapy Handbook. Norwalk, CT: Appleton & Lange; 1994. p. 129.
Acute Chemotherapy Toxicity and Supportive Care
Myelosuppression is an expected side effect of almost all treatment protocols for solid tumors. Transfusions of packed cells and platelets are frequently needed. Of greatest concern is the risk of severe life-threatening bacterial or fungal infections that occur during episodes of neutropenia. In dose-intensive regimens, more than 75% of chemotherapy courses result in hospitalization for fever, with the incidence of bacteremia ranging from 10– 20% per course.37
Success in improving treatment outcomes is partially attributed to advances in supportive care. The routine use of hematopoietic growth factors, specifically G-CSF, results in more rapid granulocyte recovery and shorter hospitalizations for fever and neutropenia.26 In addition, IL-11 enhances platelet recovery, decreases the depth of the platelet nadir, and decreases platelet transfusion requirements.27,38,39 It is well tolerated and is beneficial in combination regimens that induce severe thrombocytopenia.40,41
The gastrointestinal tract is injured by cytarabine, anthracyclines, and high-dose methotrexate; however, the folate derivative Leucovorin can be given to rescue normal mucosal and bone marrow cells from the effects of the latter. No rescue is known for the mucositis and diarrhea which occur with other agents. In addition to enhancing platelet production, IL-11 may help speed recovery from gastrointestinal injury after chemotherapy.42
Renal toxicity can occur from the use of cisplatin, ifosfamide, and high-dose methotrexate. Cisplatin causes renal tubular damage, leading to elevation of levels of blood urea nitrogen and creatinine; this effect is generally reversible. Both ifosfamide and cisplatin cause renal electrolyte wasting in which hypokalemia, hypocalcemia, hypophosphatemia, and hypomagnesemia can occur. Renal injury from these agents can be improved by hyperhydration and forced diuresis. Mesna can prevent hemorrhagic cystitis resulting from cyclophosphamide and ifosfamide by binding to the bladder-toxic acrolein metabolites.40
Additionally, a recent study demonstrates that amifostine administered prior to and during cisplatin infusion significantly reduces the risk of severe ototoxicity in children with average risk medulloblastoma who are undergoing treatment with dose-intense chemotherapy.43
Targeting Biologic Pathways
Signal Transduction Inhibitors
Cellular signaling is a basic biologic function of all normal cells. Signaling can be extracellular (e.g., growth factor receptor tyrosine kinase) or through multiple intracellular effector and survival pathways (e.g., RAS, RAF, TP53, BCL-2). Cancer cells differ from normal cells as they are associated with chromosomal mutations. These mutations result in cancer cells being dependent on several hyperactive signaling pathways. The rationale behind signal transduction therapy is that blocking the hyperactive pathways induces cancer cell apoptosis. By contrast, normal cells have redundant signaling pathways that protect them against cell death. Multiple new signal transduction inhibitors are in development.44,45
