34 Primary pulmonary hypertension
Salient features
History
• Syncope, near syncope or dizziness (13–88%)
• Determine whether the patient is on oral contraceptives, fenfluramine or aminorex (N Engl J Med 1996;335:609–16).
• Determine whether the patient has habitually consumed plant products from Crotalaria species (particularly if from the Caribbean).
• Determine whether there is a family history: the chromosome locus 2q31-q32 has been identified in one familial cohort of primary pulmonary hypertension (Circulation 1997;95:2603–6).
• Determine whether the patient is positive for the human immunodeficiency virus (HIV) (HIV-associated pulmonary hypertension is associated with poor prognosis).
Advanced-level questions
What is clinical classification of pulmonary hypertension that guides therapy?
• Pulmonary arterial hypertension: disease-targeted therapies (but caution in venoocclusive disease)
• Pulmonary hypertension with left heart disease: medical, interventional and surgical therapies for chronic heart failure, coronary artery disease, valve disease and pericardial disease
• Pulmonary hypertension associated with lung diseases and/or hypoxaemia: therapy to treat the primary lung disorder, oxygen, disease-targeted therapies when pulmonary hypertension out of proportion to lung disease
• Pulmonary hypertension caused by chronic thrombotic and/or embolic disease: pulmonary endarterectomy (PEA) for proximal disease; disease-targeted therapies for distal disease, significant residual post-PEA pulmonary hypertension or late redevelopment of symptomatic pulmonary hypertension post-PEA
How would you investigate such a patient?
• Blood investigations include routine haematology and biochemistry, thyroid function, thrombophilia screen in chronic thromboembolic pulmonary hypertension; autoimmune screen (for anti-centromere antibody, anti-SCL70, U1-ribonucleoprotein (U1RNP), anti-phospholipid antibodies, hepatitis serology, serum ACE, HIV.
• Urine for human chorionic gonadotrophin.
• Chest radiograph shows enlarged main pulmonary arteries with reduced peripheral branches, enlargement of the RV.
• Pulmonary function testing includes arterial blood-gas study, 6 min walk test and nocturnal oxygen saturation monitoring.
• ECG will show right ventricular and right atrial hypertrophy.
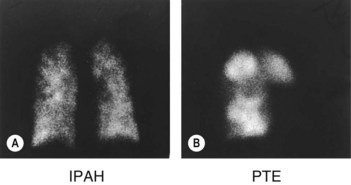
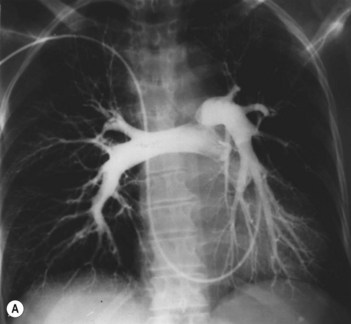
• Ventilation–perfusion scan can exclude pulmonary emboli (Fig. 34.1).
• Echocardiography with right heart catheterization and pulmonary angiography.
What are the measurements made during right heart catheterization in such a patient?
What are the imaging procedures to be considered in such a patient?
• Chest radiography to looking for increase in cardiac chambers, increased pulmonary artery size, hypoperfused areas of lung and evidence of parenchymal lung disease.
• High-resolution CT of lungs to look for parenchymal lung disease, mosaic perfusion (a sign of pulmonary vascular embolism or thrombosis but for which there are other causes such as air trapping) and features of pulmonary venous hypertension.
• CT pulmonary angiography to look for enlargement of pulmonary arteries, filling defects and webs in the arteries. Detects enlarged bronchial circulation.
• Ventilation–perfusion scanning is more sensitive for chronic pulmonary thromboembolism than CT pulmonary angiography but is not helpful when there is underlying parenchymal lung disease.
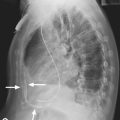
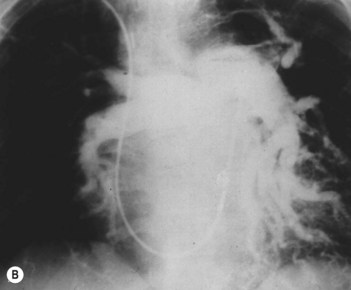
• Selective pulmonary angiography by direct injection of the pulmonary arteries (Fig. 34.2): gold standard for delineating chronic pulmonary thromboembolism to define the location and extent of disease. It may be superseded by MR angiography or multislice CT.
• Echocardiography is the screening tool of choice as it detects cardiac disease (congenital, myocardial, valvular, intracavity clot or tumour, pericardial). Use of contrast may be helpful to identify shunts.
• Cardiac MRI is a good tool for imaging the RV and is helpful in delineating congenital heart defects, and the pulmonary circulation by angiography.
• Abdominal ultrasound is used for investigation of liver disease and suspected portal hypertension.
What are the theories for the cause of primary pulmonary hypertension?
• Excess endothelial production of the vasoconstrictor thromboxane relative to dilator prostaglandins such as prostaglandin I2 (prostacyclin)
• Excess endothelin-1 levels relative to nitric oxide. Inhaled nitric oxide and endothelin-1 antagonists reduce pulmonary hypertension
• Excessive thrombosis in situ caused by increased platelet activation, plasminogen activator inhibitor levels and decreased thrombomodulin
• Inhibition or down regulation of potassium (Kv) channels in pulmonary artery smooth muscle cells and platelets
• Activation of elastase and matrix metalloprotease, which enhances production of mitogens
What treatment is available for primary pulmonary hypertension?
• Diuretics: useful in reducing excessive preload in patients with right heart failure, particularly when hepatic congestion and ascites are present.
• Oral anticoagulants: warfarin is the anticoagulant of choice, in doses adjusted to achieve an international normalized ratio (INR) of approximately 2.0. Anticoagulants nearly double the 3-year survival rate (Circulation 1984;70:580–7).
• Calcium channel blockers (nifedipine, diltiazem): patients who respond to calcium channel blockers have a 5-year survival rate of 95% (N Engl J Med 1992;327:76–81).
• Intravenous epoprostenol (formerly prostacyclin): potent short-acting vasodilator and inhibitor of platelet aggregation that is natually produced by the vascular endothelium (N Engl J Med 1996;334:296–301, N Engl J Med 1998;338:273–7).
• Atrial septostomy: the creation of a right-to-left shunt by blade–balloon atrial septostomy has been reported to improve forward output and alleviate right-sided heart failure by providing blood with a low-resistance channel, thereby decompressing the right atrium and improving filling of the left side of the heart (Circulation 1995;91:2028–35).
• Lung transplantation and combined heart–lung transplantation: survival rates after the two procedures are similar. Even markedly depressed right ventricular function improves considerably with single- or double-lung transplantation.
• PPARγ-activating agents: data suggest that the genes involved in development of pulmonary hypertension are targets of the insulin-sensitizing transcription factor peroxisome proliferator-activated receptor γ (PPARγ), and that PPARγ activation could lead to their beneficial induction or repression and subsequent antiproliferative, anti-inflammatory, proapoptotic and direct vasodilatory effects in the vasculature. PPARγ acts downstream of bone morphogenetic protein receptor II (BMP-RII), which is the cell surface receptor that is mutated or dysfunctional in many forms of pulmonary hypertension. Insulin resistance may be an environmental risk factor or disease modifier (’second hit’); it has, therefore, been suggested that PPARγ-activating agents might be beneficial in the future treatment of both insulin-resistant and insulin-sensitive PAH patients with or without BMP-RII mutations (Sci Transl Med 2009;1:12–14).