Chapter 7 Primary Mediastinal Neoplasms
Introduction
Neoplasms of the mediastinum are a diverse group of benign and malignant pathologic entities that include both primary and secondary malignancies. Most neoplasms of the mediastinum are metastases, typically from lung cancer, although extrathoracic neoplasms such as breast cancer and melanoma have a predilection for spread to the mediastinum. Primary neoplasms of the mediastinum are uncommon, and whereas the majority in adults are benign, those in children are malignant.1 In terms of primary neoplasms, the most common anterior mediastinal neoplasms include thymomas, teratomas, and lymphomas. Neoplasms of the middle mediastinum are typically congenital cysts, including foregut and pericardial cysts, whereas those that arise in the posterior mediastinum are often neurogenic neoplasms.
Epidemiology and Risk Factors
Thymic, germ cell, and neurogenic neoplasms are a heterogeneous group of primary mediastinal neoplasms. Thymic neoplasms account for 17%, lymphomas 16%, and neurogenic neoplasms 14% of all cases of primary mediastinal neoplasms.2 Germ cell tumors (teratomas, seminomas, embryonal carcinomas, endodermal sinus tumors, and choriocarcinomas) account for approximately 15% of all mediastinal tumors in adults and 24% in children.3 The remainder of the neoplasms in the mediastinum represents a large group of miscellaneous entities that includes mediastinal cysts (bronchogenic, esophageal, pericardial, thymic, and neurenteric) that account for 15% to 20% of all mediastinal masses.
Thymomas are the most common of the thymic neoplasms and characteristically are located in the anterior mediastinum. Thymomas typically occur in patients older than 40 years, are rare in children, and affect men and women equally.4–6 Thymic neuroendocrine tumors (carcinoid, small cell carcinoma, and large cell carcinoma) are uncommon. Thymic carcinoid tumor is the most common of this group of tumors. Affected patients are typically in the fourth and fifth decades of life and there is a male predominance.
Germ cell tumors usually occur in young adults (mean age 27 yr).7 Most malignant germ cell neoplasms (>90%) occur in men, whereas benign neoplasms (mature teratomas) occur with equal incidence in men and women.7 Teratomas are the most common germ cell neoplasm, representing 70% of all germ cell neoplasms in childhood and 60% in adults.7 Men and woman are equally affected. Malignant germ cell neoplasms are divided into seminomas and nonseminomatous neoplasms. Seminomas are the most common pure histologic type, accounting for 40% of such neoplasms, and usually occur in men between the third and fourth decades of life.7 The nonseminomatous germ cell tumors of the mediastinum include embryonal cell carcinoma, endodermal sinus tumor, choriocarcinoma, and mixed germ cell tumors. Teratoma with embryonal cell carcinoma (teratocarcinoma) is the most common subtype, whereas pure endodermal sinus tumors, choriocarcinomas, and embryonal carcinomas are much less common.
Neurogenic neoplasms represent 75% of all primary posterior mediastinal masses.8 These neoplasms are classified as tumors of peripheral nerves (neurofibromas, schwannomas, and malignant tumors of nerve sheath origin), sympathetic ganglia (ganglioneuromas, ganglioneuroblastomas, and neuroblastomas), or parasympathetic ganglia (paraganglioma and pheochromocytoma). Peripheral nerves are more commonly involved in adults and schwannomas constitute 75% of this group, whereas sympathetic ganglia neoplasms are more common in children. Schwannomas and neurofibromas typically occur with equal frequency in men and women, most commonly in the third and fourth decades of life. Thirty percent to 45% of neurofibromas9 occur in patients with neurofibromatosis (NF), and multiple neurogenic tumors or a single plexiform neurofibroma is considered pathognomonic of the disease.10 Malignant tumors of nerve sheath origin (also termed malignant neurofibromas, malignant schwannomas, or neurofibrosarcomas) are rare and typically develop from solitary or plexiform neurofibromas in the third to fifth decades of life. Up to 50% occur in patients with NF-1, and in these patients, tumors occur at an earlier age (typically adolescents) and with a higher incidence than in the general population.4,11 Neuroblastomas are the most common extracranial solid neoplasms in children, accounting for 10% of all childhood neoplasms. Neuroblastomas are typically diagnosed at a median age of younger than 2 years, ganglioneuroblastomas at 5.5 years of age, and ganglioneuromas at 10 years.12
Anatomy and Pathology
The anterior mediastinum is bounded anteriorly by the sternum; posteriorly by the pericardium, aorta, and brachiocephalic vessels; superiorly by the thoracic inlet; and inferiorly by the diaphragm. Its contents include the thymus, lymph nodes, and fat. The middle mediastinum is bounded by the pericardium, and its contents include the heart and pericardium, aorta, vena cava, brachiocephalic vessels, pulmonary vessels, trachea and main bronchi, lymph nodes, and phrenic, vagus, and left recurrent laryngeal nerves. The posterior mediastinum is bounded anteriorly by the trachea and pericardium, anteroinferiorly by the diaphragm, posteriorly by the vertebral column, and superiorly by the thoracic inlet. Although the true anatomic posterior boundary is the vertebral column, with respect to mediastinal disease, masses in the paravertebral regions are usually included in the posterior mediastinum. The contents of the posterior mediastinum include the esophagus, descending aorta, azygos and hemiazygos veins, thoracic duct, vagus and splanchnic nerves, lymph nodes, and fat.13
In the anterior mediastinum, significant proportions of the neoplasms arise from the thymus. Accordingly, an understanding of the normal appearance and location of the thymus is important in the detection and diagnosis of these neoplasms. The thymus consists of two lobes placed in close contact along the midline extending from the fourth costal cartilage upward, as high as the lower border of the thyroid gland. However, ectopic thymic tissue can occur at any level in the pathway of normal thymic descent, from the angle of the mandible to the upper anterior mediastinum. The two lobes generally differ in size; the left lobe is larger than the right and extends more inferiorly. Thymic hyperplasia, an increase in weight and size, has two distinct histologic forms: true and lymphoid hyperplasia. True hyperplasia occurs in children and young adults recovering from severe illness or trauma or after chemotherapy (Figure 7-1). Lymphoid hyperplasia occurs most commonly in patients with myasthenia gravis but also in association with other diseases such as hyperthyroidism, Graves’ disease, rheumatoid arthritis, and scleroderma.14
Thymic neoplasms are usually epithelial in origin and include thymomas and carcinomas. Most thymomas are solid neoplasms that are encapsulated and localized to the thymus. However, one third have necrosis, hemorrhage, or cystic components; invasion of the capsule and involvement of the surrounding structures occurs in approximately one third of cases.15 Thymomas have a wide variety of histologic features, and there is a strong association between the histologic findings and invasiveness as well as prognosis.16 Thymic carcinomas usually manifest histologically as large, solid, and infiltrating masses with cystic and necrotic areas. They are histologically classified as low or high grade, with squamous cell–like or lymphoepithelioma-like variants being the most common cell types.4 Thymic neuroendocrine neoplasms are uncommon. These tumors typically manifest as a large, lobulated, and usually invasive anterior mediastinal mass that can exhibit areas of hemorrhage and necrosis.4,17 Malignant potential ranges from relatively benign (thymic carcinoid) to highly malignant (small cell/large cell carcinoma of the thymus). Typical carcinoid has low mitotic activity (<2 mitoses/2 mm2) without necrosis, whereas atypical carcinoid has a higher degree of mitotic rate (2-10 mitoses/2 mm2) and/or necrosis. Small cell and large cell neuroendocrine carcinomas have a higher rate of mitotic activity (>10 mitosis/2 mm2) and associated necrosis.
In the posterior mediastinum, most of the neoplasms are neurogenic in origin. Nerve sheath tumors are either schwannomas, neurofibromas, or malignant tumors of nerve sheath origin (malignant neurofibroma, malignant schwannoma, and neurogenic fibrosarcoma). Histologically, schwannomas are encapsulated tumors which arise from Schwann cells located in the nerve sheath and grow along the nerve, causing extrinsic compression.11 They are heterogeneous in composition and can have low cellularity, areas of cystic degeneration, hemorrhage, and small calcifications. Neurofibromas differ from schwannomas in that they are unencapsulated and result from proliferation of all nerve elements, including Schwann cells, nerve fibers, and fibroblasts. They grow by diffusely expanding the nerve. Plexiform neurofibromas are variants of neurofibromas that infiltrate along nerve trunks or plexuses.18 Ganglion cell tumors arise from the autonomic nervous system rather than nerve sheaths and range from benign encapsulated neoplasms (ganglioneuromas) to moderately aggressive neoplasms (ganglioneuroblastomas) to malignant unencapsulated masses (neuroblastomas). They are derived from cells of embryologic origin or sympathetic ganglia. After the abdomen, the thorax is the second most common location of neuroblastomas,19 whereas ganglioneuromas and ganglioneuroblastomas are more common in the sympathetic chain of the posterior mediastinum. Ganglioneuromas are benign tumors composed of one or more mature ganglionic cells. Ganglioneuroblastomas have histologic features of both ganglioneuromas and neuroblastomas. They are the least common type of neurogenic tumors. Neuroblastomas are the most aggressive type and are composed of small round cells arranged in sheets or pseudorosettes.20,21
Key Points Anatomy: Anterior mediastinal masses
• Thymic origin: Thymic hyperplasia, thymic epithelial tumor (thymoma, thymic carcinoma, thymic carcinoid, thymolipoma), and cyst.
• Germ cell tumors: Teratoma (mature, immature, malignant), seminoma, nonseminomatous.
• Lymphoma: Hodgkin’s and non-Hodgkin’s.
• Thyroid mass: Goiter, thyroid cancer.
• Miscellaneous: Adenopathy, parathyroid adenoma, mesenchymal tumors (lymphangioma, hemangioma).
Key Points Pathology
• Thymic neoplasms: Epithelial (thymomas and carcinoma); neuroendocrine tumors (carcinoid-typical and atypical, small and large cell neuroendocrine carcinoma).
• Germ cell tumors: Teratoma (mature, immature, and malignant); nonteratomatous tumors (seminomas and nonseminomas). Seminomas (germinomas); nonseminomatous germ cell tumors (embryonal carcinoma, endodermal sinus tumor, choriocarcinoma, and mixed type).
• Neurogenic sheath tumors: Schwannomas or neurofibromas.
• Ganglion cell tumors: Ganglioneuromas, ganglioneuroblastomas, and neuroblastomas arise from the autonomic nervous system rather than the nerve sheath.
Clinical Presentation
Thymomas usually are an incidental finding, but they can manifest as chest pain, cough, or dyspnea in up to one third of patients.22 Myasthenia gravis, characteristically associated with thymomas, occurs most frequently in women. Thirty percent to 50% of patients with thymomas have myasthenia gravis, whereas 10% to 15% of patients with myasthenia gravis have a thymoma.23 Patients with myasthenia gravis usually have diplopia, ptosis, dysphagia, and fatigue. Ten percent of patients with a thymoma have hypogammaglobulinemia and 5% of patients have pure red cell aplasia. Thymomas are also associated with various autoimmune disorders such as systemic lupus erythematosus, polymyositis, or myocarditis.2 Thymic carcinomas are frequently symptomatic at presentation owing to marked local invasion and involvement of mediastinal structures. Symptoms include chest pain, dyspnea, cough, SVC syndrome, usually due to compression or invasion of the SVC; unlike thymomas, paraneoplastic syndromes are rare. Thymic neuroendocrine tumors are also associated with ectopic secretion of hormones. Up to 50% of affected patients with thymic carcinoid tumors have hormonal abnormalities and up to 35% have Cushing’s syndrome due to tumoral production of adrenocorticotropic hormone (ACTH). Nonfunctioning thymic carcinoids may be seen in association with the multiple endocrine neoplasia syndrome type 1.
Patients with germ cell tumors are often asymptomatic. Large tumors can manifest clinically as cough, dyspnea, chest pain, or pulmonary infection. Rarely, teratomas may erode and rupture into adjacent airway, resulting in expectoration of oily material or hair (tricophtysis). Seminomas can manifest as SVC syndrome in 10% of cases. Beta-human chorionic gonadotropin (β-hCG) and alpha-fetoprotein (AFP) levels are usually normal. However, 7% to 8% of patients with pure seminomas are reported to have elevated serum levels of β-hCG and elevation of serum lactate dehydrogenase (LDH) levels occurs in 80% of patients with advanced seminomas. Importantly, elevation of AFP indicates a nonseminomatous component of the tumor. Most (90%) of patients with nonseminomatous germ cell tumors of the mediastinum are symptomatic at presentation with chest pain, cough, dyspnea, weight loss, and fever. Serologic tumor markers are important in the diagnostic evaluation and 71% of affected patients have elevated levels of AFP and 54% have elevated levels of β-hCG.24 There is an association between malignant nonseminomatous germ cell tumors of the mediastinum and hematologic malignancies. In addition, approximately 20% of cases are associated with Klinefelter’s syndrome (gynecomastia, testicular atrophy, and 47 XXY karyotype).
Nerve sheath tumors are usually asymptomatic. Because most patients with a benign neurogenic tumor are asymptomatic, the development of pain often indicates malignant transformation (malignant neurofibromas, malignant schwannomas, or neurofibrosarcomas). Mediastinal neuroblastomas can manifest clinically due to local mass effect leading to respiratory distress or spinal cord compression. Neuroblastomas and, less frequently, ganglioneuroblastoma and ganglioneuroma can produce metabolically active catecholamines responsible for hypertension, flushing, and watery diarrhea syndrome.25,26 Catecholamine derivatives, such as vanilmandelic acid and homovanilic acid, can be secreted and associated with elevated catecholamine plasma levels and are helpful in establishing the diagnosis.
Key Points Middle and posterior mediastinal masses
• Vascular: Aorta (aneurysm, dissection, and congenital abnormalities), pulmonary artery (aneurysm and pulmonary hypertension), and venous abnormalities (left SVC and azygos/hemiazygos system abnormalities).
• Adenopathy. Infectious (tuberculosis, histoplasmosis, and coccidioidomycosis), sarcoidosis, lymphoma, metastatic disease (head and neck, melanoma, breast, and genitourinary), Castleman’s disease.
• Cysts: Pericardial, esophageal, bronchogenic, meningocele, pancreatic pseudocyst, neurenteric, cystic tumors.
• Esophageal: Megaesophagus, esophageal varices, neoplasms.
• Neurogenic tumors: Nerve sheath (neurofibroma, schwannoma, and malignant tumors of nerve sheath origin), ganglion cell (neuroblastoma, ganglioneuroma, and ganglioneuroblastoma), paraganglia cell (paraganglioma).
• Miscellaneous: Hematoma, abscess, hiatal hernia, congenital hernia.
Staging
In 1999, the World Health Organization (WHO) Consensus Committee published a histologic classification of tumors of the thymus. In this scheme, thymomas are classified on the combined basis of the morphology of the neoplastic epithelial cells and the lymphocyte–to–epithelial cell ratio. This classification has resulted in six separate histologic subtypes of thymomas (types A, AB, B1, B2, B3, and C) (Table 7-1). An update on the WHO classification was published in 2004.27 It retained its classifications of A through B3 thymomas but relegated type C to a separate category of thymic carcinoma and acknowledged that certain types of thymoma do not fall into these categories. The applicability and clinical significance of the WHO histologic classification of thymomas is still being evaluated. It appears that most thymomas can be classified using the WHO criteria, although the clinical and prognostic relevance of this classification has not been conclusively determined. Most thymomas of types A and AB tend to have no local invasion, are completely resectable, and have no recurrence or tumor-related deaths. There is an increasing tendency to local invasion, incomplete resection, and recurrence after resection in the order of types of B1 and B2, type B3, and type C thymomas. In this regard, most type C thymomas are locally invasive, many are incompletely resected and there is a high early relapse rate and poor prognosis. The WHO histologic subtype classification of thymomas has been reported to correlate with the staging classification devised by Masaoka and coworkers. The Masaoka staging system is a pathologic staging system that is based on the presence of capsular invasion and is currently the most widely used to determine therapy.28 In this staging system, the stages are defined as stage I, macroscopically encapsulated and microscopically no capsular invasion; stage II, macroscopic invasion into surrounding fatty tissue of mediastinal pleura or microscopic invasion into capsule; stage III, macroscopic invasion into a neighboring organ; stage IVa, pleural or pericardial dissemination; and stage IVb, lymphogenous or hematogenous metastasis.
Table 7-1 World Health Organization Classification Scheme for Thymic Epithelial Tumors
| TUMOR TYPE | DESCRIPTION |
|---|---|
| A | Medullary |
| AB | Mixed |
| B1 | Lymphocyte rich, predominantly cortical |
| B2 | Cortical |
| B3 | Epithelial (well-differentiated thymic carcinoma) |
| C | Thymic carcinoma |
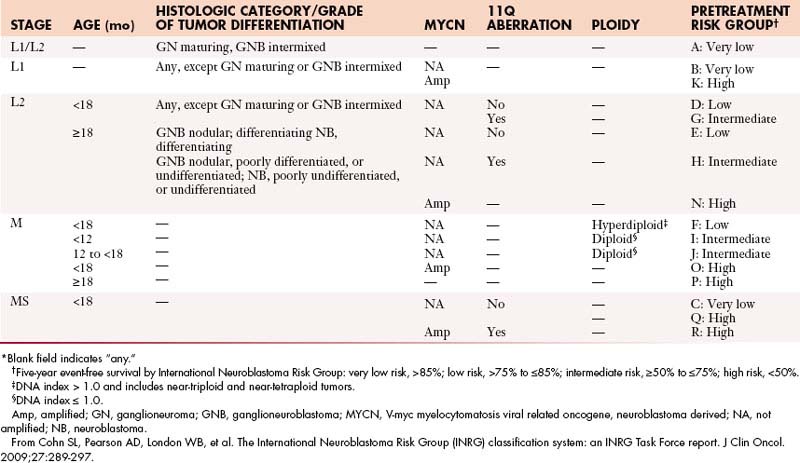
Neuroblastomas are currently staged according to the International Neuroblastoma Staging System (INSS), which is based on surgical findings as well as lymph node and metastatic involvement. However, new guidelines for a pretreatment staging system have been developed by the International Neuroblastoma Risk Group (INRG); this staging system is being implemented clinically.29 This staging model is based on clinical and imaging features rather than surgical findings. Currently, both of these staging systems have an important role in the determination of appropriate treatment and prediction of outcome in patients with neuroblastoma (Table 7-2).
Table 7-2 Comparison between International Neuroblastoma Staging System and International Neuroblastoma Risk Group Staging System
| INSS | INRGSS |
|---|---|
| Stage 1: Localized tumor with complete gross excision; ± microscopic residual disease; representative ipsilateral lymph node negative for tumor microscopically. | Stage L1: Localized tumor not involving vital structures as defined by IDRFs and confined to one body compartment. |
| Stage 2A: Localized tumor with incomplete gross excision; representative ipsilateral lymph node negative for tumor microscopically. | Stage L2: Locoregional tumor with presence of one or more IDRFs. |
| Stage 2B: Localized tumor with or without complete gross excision; ipsilateral lymph node positive for tumor microscopically; enlarged contralateral lymph nodes should be negative microscopically. | Equals stage L2. |
| Stage 3: Unresectable unilateral tumor infiltrating across the midline; ± regional lymph node involvement; or localized unilateral tumor with contralateral regional lymph node involvement or midline tumor with bilateral extension by infiltration (unresectable) or by lymph node involvement. | Equals stage L2. |
| Stage 4: Any primary tumor with dissemination to distant lymph nodes, bone, bone marrow, liver, skin, or other organs. | Stage M: Distant metastatic disease (except stage MS). Distant lymph node involvement is metastatic disease. Ascites and pleural effusion, even if malignant cells are present, do not constitute metastatic disease unless they are remote from the primary tumor. |
| Stage 4S: Localized primary tumor in infants < 1 yr (localized as in stage 1, 2A, or 2B) with dissemination limited to skin, liver, or bone marrow (<10% malignant cells). | Stage MS: Metastatic disease in children < 547 days (18 mo) of age with metastases confined to skin, liver, and/or bone marrow (<10% malignant cells); MIBG scan must be negative in bone and bone marrow. Primary tumor can be L1 or L2 with no limitations in terms of crossing or infiltration of the midline. |
IDRFs, image-defined risk factors; INRGSS, International Neuroblastoma Risk Group Staging System; INSS, International Neuroblastoma Staging System; MIBG, iodine-123-metaiodobenzylguanidine.
Adapted from Monclair T, Brodeur GM, Ambros PF, et al. The International Neuroblastoma Risk Group (INRG) staging system: an INRG Task Force report. J Clin Oncol. 2009;27:298-303.
Key Points Staging thymic neoplasms
• The Masaoka classification is based on surgical and pathologic findings.
• The WHO classification is based on histologic findings.
• The Masaoka staging system is currently the most widely used to determine therapy.
• Neuroblastoma is staged according to the INSS or INRG systems.
• The INSS is based on surgical findings and tumor spread, and the INRG is based on imaging and clinical features.
Patterns of Spread
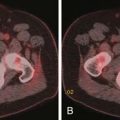
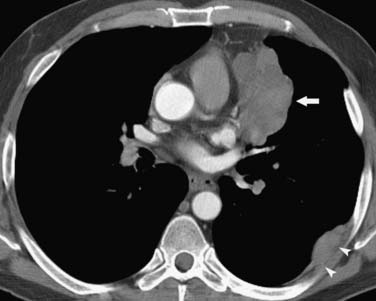
Thymomas can remain localized or spread through the mediastinum in a contiguous fashion. Characteristically, if invasion of the adjacent pleura occurs, dissemination in the pleural space can result in solitary, multiple, or diffuse metastases distant to the primary mass. Historically, this dissemination of metastases in the pleural space has been referred to as drop metastases (Figure 7-2). Pleural effusion is uncommon, despite often extensive pleural metastatic disease. Transdiaphragmatic spread has been reported in up to one third of patients.30 Pericardial involvement due to contiguous spread is common and can manifest as nodular or diffuse thickening and a pericardial effusion. Systemic dissemination is rare, although lung metastases can occur. Thymic carcinomas are aggressive malignancies that often exhibit marked local invasion and early dissemination. In 40% of cases, there is invasion of adjacent organs, 40% present with nodal metastatic disease, and 10% have pleural or pericardial involvement. Distant metastases to lung, liver, adrenal glands, brain, and bone occur in 40% of patients.
Although mature teratomas are benign, rare cases of tumor rupture into the lung, pleural space, and pericardium have been reported. Seminomas usually have lymphatic or systemic dissemination, and local invasion of adjacent structures is rare. Metastases to regional nodes as well as metastatic involvement of cervical (25%) and abdominal lymph nodes (8%) are reported.31 In cases of nonseminomatous tumors, invasion of the adjacent structures such as lung and mediastinal pleural is frequent.32 Pleural and pericardial effusion are common owing to local and direct invasion. Chest wall invasion is more frequently associated with larger masses. Hematogenous disseminated metastases to lungs, liver, brain, and bones are common and occur in up to 50% of patients. Metastatic spread to lymph nodes is less frequent.
Because neurogenic tumors usually arise in a paravertebral location, intraspinal extension is common. Ten percent of paravertebral neurofibromas and schwannomas extend into the neural foramina and spinal canal.11 Compression and destruction of adjacent structures can occur as a result of aggressive local tumor invasion. Ganglioneuromas are encapsulated benign tumors without evidence of local or distant dissemination. Ganglioneuroblastomas and neuroblastomas are more aggressive, with evidence of local and intraspinal invasion. Neuroblastomas have a tendency to grow across the midline and lymph node involvement can occur. Lymphatic and hematogenous dissemination are common and sites of metastatic involvement include bone (60%), regional lymph nodes (45%), orbit (20%), liver (15%), brain (14%), and lung (10%).
Key Points Patterns of spread
• Thymoma. Most slow growth and remained localized. In one third of cases contiguous invasion of adjacent structures occurs. Noncontiguous pleural involvement (“drop metastasis”) is reported.
• Thymic carcinoma: Local invasion and hematogenous dissemination to brain, lung, liver, adrenal glands, and bones are common.
• Germ cell tumor. Teratomas are typically benign although rarely malignant transformation and local invasion occurs. With seminomas, local invasion does not occur and hematogenous dissemination has been reported. Nonseminomatous tumors commonly invade adjacent structures and distant metastases are common.
• Neurogenic tumor. Neurofibroma and schwannoma are benign and rarely undergo malignant degeneration. Malignant tumors can cause local invasion. Ganglioneuroma is benign, but neuroblastoma can cause distant metastases.
Imaging
Importantly, in the assessment of thymic neoplasms, the normal thymus in young children and thymic hyperplasia can mimic a mediastinal mass on chest radiographic assessment. CT and MRI are useful in differentiating normal thymic tissue and the hyperplastic thymus from tumors. Thymic hyperplasia manifesting as a mass on CT or MRI is differentiated from a primary mediastinal neoplasm based on diffuse, symmetrical enlargement with preservation of normal shape of the thymus (see Figure 7-1). Similar findings distinguish rebound hyperplasia, which occurs 3 to 8 months after cessation of chemotherapy in approximately 25% of patients, from recurrence of the initial neoplasm.33 When CT and conventional MRI sequences are not able to differentiate between a normal thymus or thymic hyperplasia and a thymoma, chemical shift MRI sequences (in-phase and out-of-phase gradient echo sequences) can be helpful in the diagnosis—that is, homogeneous signal decrease occurs in normal and hyperplastic tissue compared with absence of signal decrease in tumors. PET/CT imaging is being increasing performed in oncologic patients. The normal thymus can have mildly increased fluoro-2-deoxy-d-glucose (FDG), especially in children. In addition, increased FDG uptake in the thymus, mainly related to hyperplasia after chemotherapy, has been reported to occur in 28% of patients and is higher in younger patients (in 80% of patients < 10 yr of age, compared with 8% of patients in the 31- to 40-yr age group).34
Thymic Neoplasms
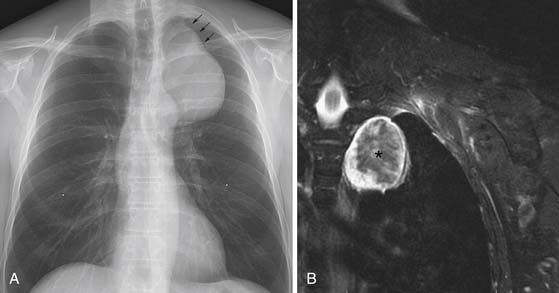
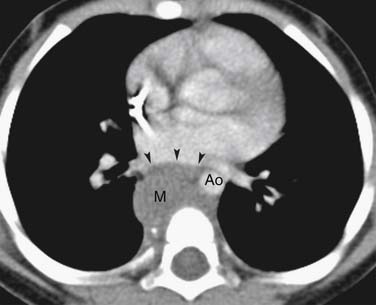
Thymomas are usually located anterior to the aortic arch but can occur in the cardiophrenic recesses.4,35 They typically manifest as a 1- to 10-cm (mean 5 cm) smooth or lobulated well-marginated mass that characteristically arises from one lobe of the thymus. Although thymomas typically manifest as a unilateral mass, bilateral mediastinal involvement can occur.36 On CT, thymomas are usually of homogeneous soft tissue attenuation (see Figure 7-2). Calcification, seen in up to 7% of cases, is usually thin, linear, and located in the capsule. Enhancement after intravenous administration of contrast is usually homogeneous except in the one third of thymomas that are necrotic or cystic or contain hemorrhage. Tomiyama and colleagues37 suggested that a combination of lobulated or irregular contour, cystic or necrotic portion within the tumor and multifocal calcification is more suggestive of invasive thymoma (Figure 7-3). On MRI, thymomas manifest with low to intermediate signal intensity (similar to skeletal muscle) on T1-weighted images and high signal intensity on T2-weighted images. Heterogeneous signal intensity is present in those tumors that have focal areas of necrosis, hemorrhage, and cystic change. MRI can occasionally reveal fibrous septa within the masses. Pleural metastases manifest on CT and MRI as an isolated pleural nodule, multifocal masses, or contiguous pleural involvement, which can be smooth, nodular, or diffuse, mimicking malignant pleural mesothelioma. Pleural effusion is uncommon, despite often extensive pleural metastatic disease.4,38 Pericardial thickening and/or effusion typically are associated with invasive thymomas. The differentiation of benign from invasive thymomas is usually not possible on CT and MRI.
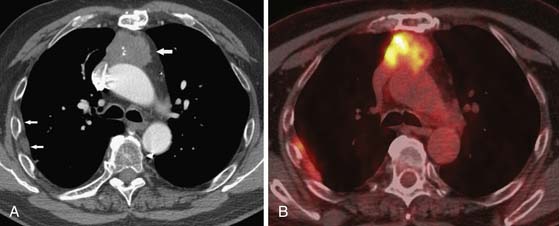
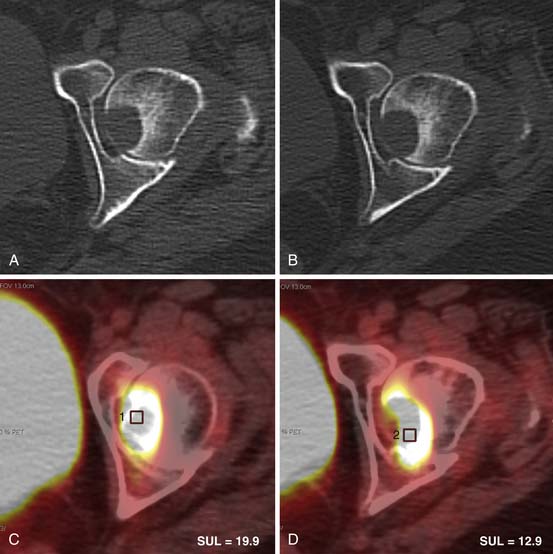
The precise role of PET/CT in the evaluation of thymomas is unclear. One difficulty is that FDG uptake in the normal thymus is variable and increased FDG uptake by the thymus is common, especially in young patients.34 Although it has been reported that PET/CT can be useful in distinguishing thymic epithelial tumors according to the WHO classification as well as for staging the extent of the disease,39–41 in our experience, PET/CT is unreliable in distinguishing noninvasive from invasive thymomas or thymic carcinomas. Thymic carcinomas commonly manifest as large, poorly marginated anterior mediastinal masses that are calcified in 10% to 40% of cases.37,42,43 Intrathoracic lymphadenopathy is common. On CT, thymic carcinomas are usually of heterogeneous attenuation and have poorly defined margins. On MRI, they typically have intermediate signal intensity (slightly higher than skeletal muscle) on T1-weighted images and high signal intensity on T2-weighted images. Signal intensity can be heterogeneous because of hemorrhage and necrosis within the mass. MRI can be helpful for revealing local soft tissue and vascular invasion. Similar to the evaluation of thymomas, the role of PET/CT in the diagnosis and staging of thymic carcinomas has not been clearly established. It has been reported that thymic carcinomas typically exhibit increased FDG uptake on PET/CT that is usually higher and more homogeneous than in thymomas and thymic hyperplasia.39,41,44 However, clinically, the role of FDG-PET imaging in differentiating thymomas from thymic carcinomas is limited, although the detection of unexpected nodal and distant metastases is useful in patient management.
Thymic neuroendocrine tumors usually manifest as a large anterior mediastinal mass, with a propensity for local invasion and metastasis (Figures 7-4 and 7-5). Focal areas of necrosis or punctuate and dystrophic calcification may be present.17 On CT or MRI, the masses are usually of heterogeneous attenuation or signal intensity, respectively. Differentiation between thymic neuroendocrine tumors and invasive thymic epithelial tumors may be difficult on the basis of imaging findings alone. Thymic neuroendocrine tumors have a poor prognosis owing to a high prevalence of recurrence and metastasis.
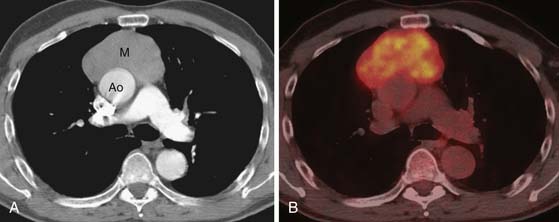
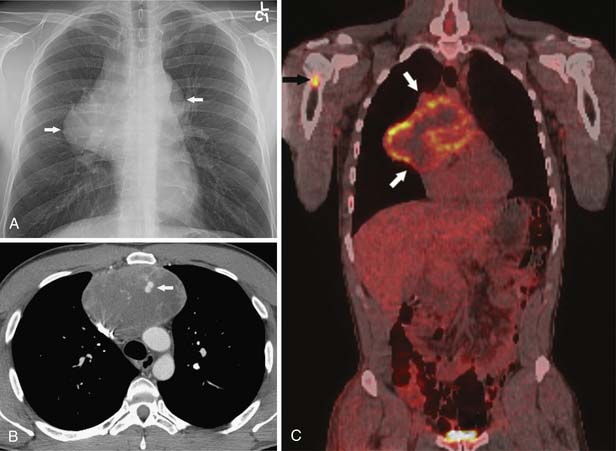
Germ Cell Tumors
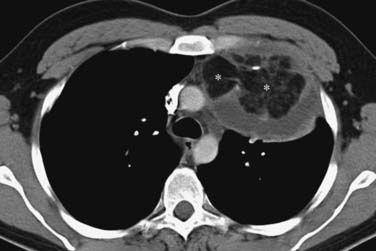
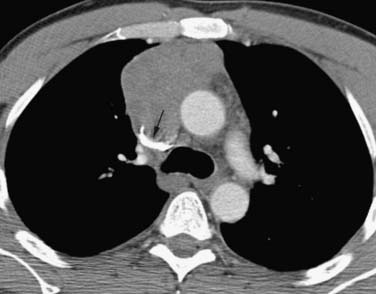
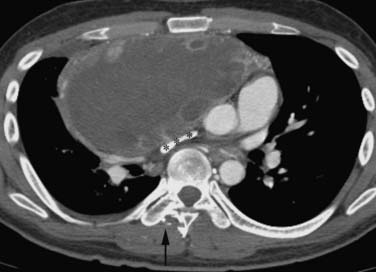
These neoplasms have a range of manifestations in the mediastinum. Mature teratomas manifest on CT or MRI as smooth or lobulated mediastinal masses that typically have cystic and solid components, whereas malignant teratomas are usually poorly marginated masses containing areas of necrosis. The combination of fluid, soft tissue, calcium, and/or fat is diagnostic of teratomas (Figure 7-6). The finding of a fat-fluid level within a mass on CT or MRI is also diagnostic of a teratoma. Fat occurs in up to 75% of mature teratomas and up to 40% of malignant teratomas. However, only 17% to 39% of mature teratomas will have all tissue components and approximately 15% of mature teratomas manifest only a unilocular or multilocular cystic component. In this regard, these masses can mimic mediastinal cysts. In terms of differentiation, benign mediastinal cysts manifest as well-defined thin-walled masses with low attenuation (0-20 HU) on CT and no enhancement after contrast administration45 whereas teratomas show rim enhancement and enhancement of tissue septa. Because simple cysts can contain proteinaceous fluid and have high attenuation on CT, MRI can be useful in differentiating these lesions. Simple cysts characteristically have low signal intensity on T1-weighted image sequences and high signal intensity on T2-weighted sequences (high signal intensity occurs on T1-weighted image sequences when proteinaceous material or hemorrhage is present). Teratomas containing fat typically manifest as high signal intensity on T1-weighted images. A fat-saturation MRI technique can be used to detect and distinguish fat from hemorrhage. Importantly, if a cystic neoplasm is suspected because the CT or MRI findings are atypical or because the mass has increased in size, aspiration may be required for diagnosis. Mature teratomas can rupture into lung, pleural space, and pericardium, and CT or MRI can be useful in detecting fat within these regions. In addition, ancillary changes such as adjacent parenchymal consolidation or atelectasis, pleural effusion, and pericardial effusion are associated with rupture into the pleural or pericardial space. PET/CT is not useful in the evaluation of mature teratomas owing to the lack of FDG avidity. Seminomas manifest as large/bulky, well-marginated masses with lobulated contours and typically have homogeneous attenuation or signal intensity on CT and MRI, respectively. However, cysts or areas of necrosis can occur and calcification is rare. Seminomas enhance only slightly after administration of intravenous contrast material. Compression of adjacent mediastinal structures is common but invasion is uncommon (Figure 7-7). Extension into the middle and posterior compartments and obliteration of fat planes can occur. In contradistinction, nonseminomatous tumors are usually large, unencapsulated, heterogeneous soft tissue masses that tend to invade and infiltrate adjacent structures, including the lung and chest wall. Fat planes are typically obliterated, and the interface between the tumor and the adjacent lung may be irregular and spiculated owing to lung invasion. These tumors can contain large areas of hemorrhage, necrosis, and cyst formation (Figure 7-8). Associated pleural and pericardial effusions can occur. Metastases to the regional lymph nodes and distant sites are common. Multiple pulmonary nodules representing metastases can be present. The role of PET/CT has not been clearly elucidated in patients with primary mediastinal seminomas and nonseminomatous tumors.
Neurogenic Tumors
The benign peripheral nerve tumors (schwannomas and neurofibromas) are slowly growing neoplasms that often are radiologically indistinguishable. Schwannomas and neurofibromas are usually sharply marginated, spherical, and lobulated paravertebral masses. On CT imaging, punctate calcification and low-attenuation areas caused by the presence of fat, cystic change, or hemorrhage can be seen. Enhancement after intravenous contrast administration is variable and can be homogeneous, heterogeneous,46 or peripheral.18 Enlargement of neural foramina with or without extension into the spinal canal and osseous abnormalities, such as rib erosion and splaying of the ribs, can occur. MRI is the preferred modality for demonstrating the intraspinal extension of the tumor or the presence of an associated spinal cord abnormality (Figure 7-9). On MRI, neurofibromas and schwannomas have variable signal intensity on T1-weighted images but typically have similar signal intensity to the spinal cord. On T2-weighted images, these neoplasms characteristically have high signal intensity peripherally and low signal intensity centrally (target sign) due to collagen deposition (Figure 7-10). This feature, when present, helps distinguish neurofibromas from other mediastinal tumors. Also, areas of cystic degeneration within the mass can result in foci of increased signal intensity on T2-weighted images. Although the high signal intensity of schwannomas and neurofibromas on T2-weighted images can facilitate differentiation of tumors from spinal cord, the tumors can be obscured by the high signal intensity of cerebrospinal fluid. Schwannomas and neurofibromas, however, enhance with gadolinium; this feature can be useful in detecting and determining intradural extension of these tumors. Paravertebral neurofibromas and schwannomas that extend into the spinal canal manifest as dumbbell-shaped masses with widening of the affected neural foramen. In several small series, neurofibromas and schwannomas have been reported to be FDG-avid on PET/CT. On CT, plexiform neurofibromas manifest as low-attenuation, poorly marginated masses located along the mediastinal nerves and sympathetic chains. MRI of plexiform neurofibromas shows the infiltrative nature of the tumors, and the masses have low signal on both T1-weighted and T2-weighted images owing to the fibrous nature of the tumors.
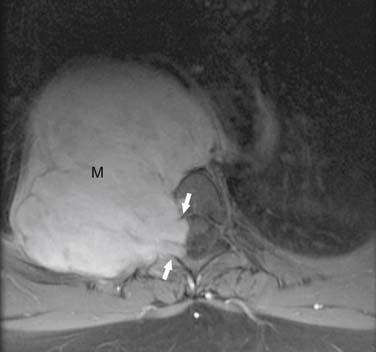
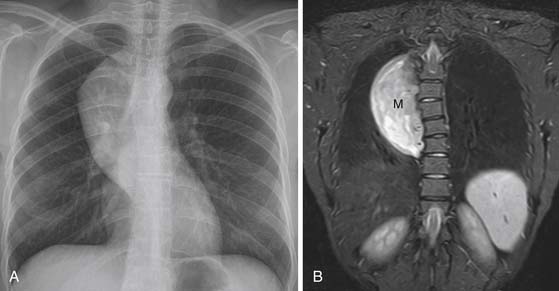
Malignant tumors of nerve sheath origin are rare but typically develop from solitary or plexiform neurofibromas. On CT or MRI, malignant tumors of nerve sheath origin typically manifest as posterior mediastinal masses larger than 5 cm in diameter. Although benign and malignant neurogenic tumors cannot be differentiated with certainty, findings that suggest malignancy include a sudden change in size of a preexisting mass or development of heterogeneous signal intensity on MRI (caused by necrosis and hemorrhage). The presence of multiple target signs throughout the lesion on MRI favors the diagnosis of a plexiform neurofibroma rather than a malignant tumor of nerve sheath origin. Neurofibrosarcomas are often FDG-avid and when the standardized uptake value (SUV) is greater than 3, aggressive behavior has been reported. The sympathetic ganglia tumors, ganglioneuromas and ganglioneuroblastomas, usually manifest as well-marginated, elliptical, posterior mediastinal masses that extend vertically over three to five vertebral bodies (Figure 7-11). They are usually located lateral to the spine and can cause pressure erosion on adjacent vertebral bodies. On CT, they are typically heterogeneous and contain stippled or punctate calcification in up to 30% of masses.47 On T1- and T2-weighted MRI, they are usually homogeneous and of intermediate signal intensity. Occasionally, these lesions are heterogeneous and of high signal intensity on T2-weighted images.48 Ganglioneuroblastomas are typically larger and more aggressive with evidence of local and intraspinal invasion than ganglioneuromas. Neuroblastomas manifest as a paraspinal mass of heterogeneous, predominantly soft tissue attenuation (Figure 7-12). The masses usually contain areas of hemorrhage, necrosis, and cystic generation. Calcification occurs in up to 80% of cases and can be coarse, mottled, solid, or ring-shaped.49 Neuroblastomas often show widespread local invasion and have irregular margins, although many of these lesions are well-marginated on CT or MRI. The primary tumor can also spread through the neural foramina, resulting in a classic dumbbell-shaped tumor.12 On CT, discrete lytic or mixed lytic and sclerotic areas or metaphyseal lucencies are typical of metastatic osseous involvement. On MRI, neuroblastomas show homogeneous or heterogeneous signal intensity on all sequences and variable enhancement after contrast administration.48 Iodine-123-metaiodobenzylguanidine (123I-MIBG) is an essential part of the evaluation for patients with neuroblastomas and improves the detection of the primary malignancy as well as metastases and is also useful in the assessment of response to therapy. The role of PET/CT in evaluation of patients with neuroblastoma is unclear. PET/CT can be useful in 123I-MIBG-negative neuroblastoma patients and is better in the evaluation of disease extent in the chest, abdomen, and pelvis. PET/CT is superior in depicting stage 1 and 2 neuroblastoma, although 123I-MIBG is overall superior in the evaluation of stage 4 neuroblastoma, mainly because of the better detection of bone or marrow metastases.50
Key Points Imaging
• Thymic neoplasms. Thymoma, a lobulated mass within the anterior mediastinum. Mediastinal fat, great vessel invasion as well as pleural seeding are findings suggestive of local invasion. Thymic carcinoma, a large anterior mediastinal mass with local and distant metastasis.
• Germ cell tumors: Teratoma, a multiloculated cystic mass, can contain adipose tissue. Seminoma is a homogeneous and well-marginated mass. Nonseminomatous germ cell tumor is a large and irregular mass due to hemorrhage, necrosis, or cystic change.
• Neurogenic tumors: Neurofibroma and schwannoma are posterior mediastinal with enlargement of neural foramina. The MRI target appearance is high signal intensity in the periphery and intermediate signal intensity in the central zone. Ganglion cell is a paraspinal soft tissue mass with elongated vertical direction. Neuroblastomas are heterogeneous, predominantly soft tissue attenuation paraspinal masses, with calcification in up to 80%.
Treatment
Thymic Tumors
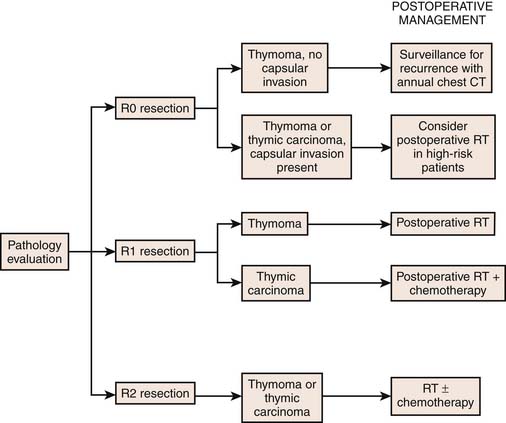
Surgery resection is the cornerstone of treatment for patients with thymomas.51 Generally, postoperative radiotherapy is recommended in patients with incompletely resected thymomas and chemoradiation should be considered in nonsurgical patients. Treatment recommendations are dependent on stage. In this regard, patients with stage I tumors are treated with surgical resection alone. In patients with Masaoka stage II who undergo complete resection, adjuvant radiation therapy is controversial. However, in Masaoka stage II tumors of a high-risk WHO category such as B2 or B3, adjuvant radiotherapy should be considered. Adjuvant radiotherapy is generally considered an effective treatment in patients with advanced thymomas (Masaoka stages III and IVa). Chemotherapy is not considered as a treatment of choice in localized, surgically resectable thymomas. However, cisplatin-based chemotherapy is advocated in patients with inoperable or gross residual disease after resection, mainly for Masaoka stage III or IV thymomas.52 Multimodality therapy has been used to manage patients with unresectable tumors (usually Masaoka III, IVa, and IVb thymomas). In this regard, induction chemoradiotherapy can be used to downstage thymomas to improve surgical respectability.53–55 In addition, Lucchi and associates have reported reasonable long-term survival in Masaoka stage III and IVa thymomas using neoadjuvant chemotherapy, surgery, and postoperative radiotherapy or primary surgical resection followed by adjuvant chemotherapy or radiation therapy or both.56 In view of the complexity of management of thymic tumors, a schematic approach as outlined by the National Comprehensive Cancer Network (NCCN) is included to clarify the management of resectable and advanced disease (Figures 7-13 and 7-14).
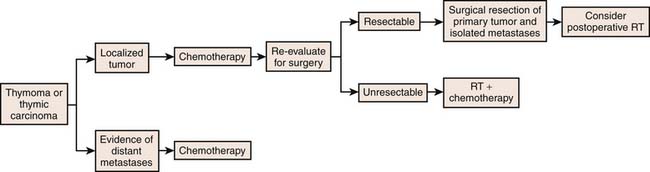
Germ Cell Tumors
The treatment of mature teratomas consists of complete surgical resection. Complete resection is important because residual disease is associated with a risk of malignant transformation. The treatment approach for mediastinal seminomas and nonseminomas is based on recommendations of the International Germ Cell Cancer Collaborative Group.57 This approach is based on a risk assignment algorithm that is used in clinical practice that includes the histology of the primary tumor, serum tumor marker levels, and presence of nonpulmonary visceral metastases. The therapeutic management of seminomas is typically chemoradiation. Surgical resection can also be used in patients with bulky or residual tumors. The therapy of choice for nonseminomatous tumor is a cisplatin-based chemotherapy regimen followed by resection of residual tumor.
Neurogenic Tumors
Neurofibromas and schwannomas can be either observed or resected. For malignant transformation, adjuvant chemotherapy and radiation overall has only a limited effect on survival and, accordingly, when possible, resection should be performed. The treatment of patients with neuroblastomas is complex and evolving. Importantly, neuroblastomas detected in infants by screening or incidentally on sonography before birth can be observed without obtaining a definitive histologic diagnosis and without surgical intervention owing to the likelihood of spontaneous regression. Otherwise, treatment strategies for patients with neuroblastomas depend on a risk stratification that includes multiple factors such as age at diagnosis, staging system, histopathology, and genetic abnormalities such as MYCN (V-myc myelocytomatosis vital related oncogene, neuroblastoma derived) oncogene amplification, aberration of chromosome 1p or 11q, and DNA index. Generally, patients are stratified into low-, intermediate-, or high-risk groups (Table 7-3). Patients in the low-risk group are managed by surgery alone, although surgery can be combined with chemotherapy in cases of symptomatic patients or in stage IVS. Management of the intermediate-risk patients is surgical resection and chemotherapy. Patients in the high-risk group receive an intensive combination of induction chemotherapy, radiation therapy, surgery, high-dose chemotherapy with autologous stem cell rescue, and differentiation therapy with retinoic acid. Radiation therapy can be used to treat the residual tumor and sites of metastases.
Key Points Treatment
• Thymic neoplasms: Complete tumor resection is the preferred treatment for localized disease; advanced disease is treated with multimodality therapy.
• Germ cell tumors: Teratoma is treated with surgical excision. Seminoma and nonseminomatous germ cell tumors are treated with cisplatin-based chemotherapy and radiotherapy. Surgical resection is helpful in cases of residual mass.
• Neurogenic tumors: Neurofibroma and schwannoma are treated surgically. Neuroblastoma treatment depends on risk stratification. Low-risk groups are treated with surgery. Intermediate-risk groups are treated with surgery followed by chemotherapy. High-risk groups are treated with chemotherapy, which can be followed by surgery, radiation therapy, and stem cell transplant.
Surveillance
Each specific neoplasm discussed has a different approach for evaluation after definitive treatment. A complete explanation is beyond the scope of this chapter and the reader is referred to the NCCN. However, in all cases, patients should be managed by an experienced multidisciplinary team. In patients with thymic tumors, annual chest CT is performed to evaluate for disease recurrence. CT of the abdomen and pelvis, brain MRI, and PET/CT scans are performed based on sites of metastases and symptomatology. Surveillance for germ cell tumors depends on the type and stage of the tumor. Usually, patients with seminoma are followed after treatment with serum markers (AFP, β-hCG, and LDH) and chest CT. In cases of nonseminomatous tumors, surveillance is also done with serum markers (LDH, β-hCG, and AFP) and chest and abdominal/pelvic CT. PET/CT, brain MRI, and bone scan are obtained when clinically indicated. Although the incidence is higher in patients with NF-1, malignant nerve sheath tumors occur in patients with or without NF, and evaluation with CT or MRI is required if the patient presents with chest pain or other symptomatology suspicious for malignant degeneration of a preexisting neurogenic tumor. Evaluation by chest CT is done routinely and MRI can also be useful in cases with neural foramina or spinal cord invasion. In cases of neuroblastoma, CT is performed routinely to detect the relapse that occurs in a substantial portion of patients. The most reliable test to detect disease progression or recurrence is 123I-MIBG imaging.
New Therapies
The increasing understanding of tumor-associated genes/cancer immunotherapy and molecular-targeted therapeutic options has clinical potential in the future treatment of patients with thymic, germ cell, and neurogenic tumors of the mediastinum. Because the response rates of currently used standard therapy for advanced stage thymomas and thymic carcinomas are not optimal, there has been an interest in new therapeutic targets. A novel target that has the potential to optimize the response rate in thymomas and thymic carcinomas is cyclooxygenase-2 (COX-2), which is overexpressed in several thymomas and thymic carcinomas.58 Another promising target is the epidermal growth factor receptor (EGFR), which has a high frequency of overexpression in thymomas and, to a lesser extent, in thymic carcinomas. In a phase II study of gefitinib treatment in 26 patients with advanced thymic malignancies, 1 patient had a partial response and 14 patients (54%) had stable disease.58 Furthermore, cetuximab, a monoclonal antibody with EGFR inhibitor activity, given with traditional chemotherapy regimens in patients with advanced-stage thymoma (stages III and IVA) before surgery, is being evaluated in a phase II trial. In addition, experimental drugs that block the insulin-like growth factor-1 receptor (IGF-1R) and sunitinib, a multitargeted receptor tyrosine kinase (RTK) inhibitor with antiangiogenic effect, are being evaluated in patients with invasive thymomas. In patients with germ cell tumors, new antitumoral agents such as bevacizumab combined with oxaliplatin are being assessed in patients with refractory or relapsed germ cell tumors. Pegfilgrastim, an immunostimulator that functions as a granulocyte colony–stimulating factor, in combination with chemotherapy is being studied in patients with untreated germ cell tumors.
Patients with NF-1 and multiple neurofibromas and large plexiform neurofibromas have provided the impetus for the development of new agents for targeted molecular therapy in patients with neurogenic neoplasms. In this regard, the principal therapeutic option, surgical resection, may not be feasible in patients with multiple neurofibromas or plexiform neurofibromas. In addition, there is a high recurrence rate after resection and a risk of malignant transformation in patients with large plexiform neurofibromas.59–61 Accordingly, innovative therapeutic options are being investigated in preclinical and clinical trials. Proposed treatments that target mast cell functioning, the Ras signaling pathways, the downstream effector of the Ras signaling pathway, and inhibition of PI3K and KIT all have the potential to be effective in the management of patients with neurofibromas.59,62–65 An additional therapeutic option is to target the mTOR pathway with the mTOR inhibitor rapamycin.66–68 Some of these studies have shown encouraging preclinical results in the treatment of malignant peripheral nerve sheath tumors.59,69
The development of new therapeutic strategies is important in patients with neuroblastoma and advanced tumor stages because the prognosis with current therapy is poor. Monoclonal antibody therapy may have a role in the future treatment of patients with advanced neuroblastoma. Currently, the Children Oncologic Group is studying the use of monoclonal antibody therapy with granulocyte-macrophage colony–stimulating factor and interleukin-2 combined with cis-retinoic acid after chemotherapy.70,71 In addition, the New Approaches to Neuroblastoma Therapy consortium is evaluating the inclusion of myeloablative doses of 131I-MIBG with chemotherapy prior to stem cell transplantation in patients with an incomplete response to induction chemotherapy. The use of new therapies is evolving and beyond the scope of this article. The interested reader is referred to a recent article by Wagner and coworkers72 that reviews the new treatment options for high-risk neuroblastoma.
The Radiology Report
1. Hoffman O.A., Gillespie D.J., Aughenbaugh G.L., Brown L.R. Primary mediastinal neoplasms (other than thymoma). Mayo Clin Proc. 1993;68:880-891.
2. Davis R.D.Jr., Oldham H.N.Jr., Sabiston D.C.Jr. Primary cysts and neoplasms of the mediastinum: recent changes in clinical presentation, methods of diagnosis, management, and results. Ann Thorac Surg. 1987;44:229-237.
3. Strollo D.C., Rosado de Christenson M.L., Jett J.R. Primary mediastinal tumors. Part 1: tumors of the anterior mediastinum. Chest. 1997;112:511-522.
4. Rosai J., Levine G.D. Tumors of the thymus. Washington, DC: Armed Forces Institute of Pathology; 1976.
5. O’Gara R.W., Horn R.C.Jr., Enterline H.T. Tumors of the anterior mediastinum. Cancer. 1958;11:562-590.
6. Morgenthaler T.I., Brown L.R., Colby T.V., et al. Thymoma. Mayo Clin Proc. 1993;68:1110-1123.
7. Nichols C.R. Mediastinal germ cell tumors. Clinical features and biologic correlates. Chest. 1991;99:472-479.
8. Davidson K.G., Walbaum P.R., McCormack R.J. Intrathoracic neural tumours. Thorax. 1978;33:359-367.
9. Reed J.C., Hallet K.K., Feigin D.S. Neural tumors of the thorax: subject review from the AFIP. Radiology. 1978;126:9-17.
10. Ribet M.E., Cardot G.R. Neurogenic tumors of the thorax. Ann Thorac Surg. 1994;58:1091-1095.
11. Aughenbaugh G.L. Thoracic manifestations of neurocutaneous diseases. Radiol Clin North Am. 1984;22:741-756.
12. Lonergan G.J., Schwab C.M., Suarez E.S., Carlson C.L. Neuroblastoma, ganglioneuroblastoma, and ganglioneuroma: radiologic-pathologic correlation. Radiographics. 2002;22:911-934.
13. Aquino S.L., Duncan G., Taber K.H., et al. Reconciliation of the anatomic, surgical, and radiographic classifications of the mediastinum. J Comput Assist Tomogr. 2001;25:489-492.
14. Lattes R., Pachter M.R. Benign lymphoid masses of probable hamartomatous nature. Analysis of 12 cases. Cancer. 1962;15:197-214.
15. Lee J.K.T., Sagel S.S., Stanley R.J. Computed body tomography with MRI correlation. New York: Raven Press; 1989.
16. Okumura M., Ohta M., Tateyama H., et al. The World Health Organization histologic classification system reflects the oncologic behavior of thymoma: a clinical study of 273 patients. Cancer. 2002;94:624-632.
17. Wick M.R., Carney J.A., Bernatz P.E., Brown L.R. Primary mediastinal carcinoid tumors. Am J Surg Pathol. 1982;6:195-205.
18. Kumar A.J., Kuhajda F.P., Martinez C.R., et al. Computed tomography of extracranial nerve sheath tumors with pathological correlation. J Comput Assist Tomogr. 1983;7:857-865.
19. Hiorns M.P., Owens C.M. Radiology of neuroblastoma in children. Eur Radiol. 2001;11:2071-2081.
20. Shields T.W., Reynolds M. Neurogenic tumors of the thorax. Surg Clin North Am. 1988;68:645-668.
21. Page D.L., DeLellis R.A., Hough A.J. Atlas of Tumor Pathology: Tumors of the Adrenal. Washington, DC: Armed Forces Institute of Pathology; 1986. 219-260
22. Lewis J.E., Wick M.R., Scheithauer B.W., et al. Thymoma. A clinicopathologic review. Cancer. 1987;60:2727-2743.
23. Osserman K.E., Genkins G. Studies in myasthenia gravis: review of a twenty-year experience in over 1200 patients. Mt Sinai J Med. 1971;38:497-537.
24. Bukowski R.M., Wolf M., Kulander B.G., et al. Alternating combination chemotherapy in patients with extragonadal germ cell tumors. A Southwest Oncology Group study. Cancer. 1993;71:2631-2638.
25. Gale A.W., Jelihovsky T., Grant A.F., et al. Neurogenic tumors of the mediastinum. Ann Thorac Surg. 1974;17:434-443.
26. Caty M.G., Shamberger R.C. Abdominal tumors in infancy and childhood. Pediatr Clin North Am. 1993;40:1253-1271.
27. Travis W.D., Brambilla E., Müller-Hermelink H.K., Harris C.C. Pathology and genetics of tumours of the lung, pleura, thymus and heart. World Health Organization Classification of Tumors. France: IARC Press; 2004.
28. Masaoka A., Monden Y., Nakahara K., Tanioka T. Follow-up study of thymomas with special reference to their clinical stages. Cancer. 1981;48:2485-2492.
29. Mueller S., Matthay K.K. Neuroblastoma: biology and staging. Curr Oncol Rep. 2009;11:431-438.
30. Scatarige J.C., Fishman E.K., Zerhouni E.A., Siegelman S.S. Transdiaphragmatic extension of invasive thymoma. AJR Am J Roentgenol. 1985;144:31-35.
31. Bokemeyer C., Droz J.P., Horwich A., et al. Extragonadal seminoma: an international multicenter analysis of prognostic factors and long term treatment outcome. Cancer. 2001;91:1394-1401.
32. Takeda S., Miyoshi S., Ohta M., et al. Primary germ cell tumors in the mediastinum: a 50-year experience at a single Japanese institution. Cancer. 2003;97:367-376.
33. Choyke P.L., Zeman R.K., Gootenberg J.E., et al. Thymic atrophy and regrowth in response to chemotherapy: CT evaluation. AJR Am J Roentgenol. 1987;149:269-272.
34. Jerushalmi J., Frenkel A., Bar-Shalom R., et al. Physiologic thymic uptake of 18F-FDG in children and young adults: a PET/CT evaluation of incidence, patterns, and relationship to treatment. J Nucl Med. 2009;50:849-853.
35. Verstandig A.G., Epstein D.M., Miller W.T.Jr., et al. Thymoma—report of 71 cases and a review. Crit Rev Diagn Imaging. 1992;33:201-230.
36. Rosado-de-Christenson M.L., Galobardes J., Moran C.A. Thymoma: radiologic-pathologic correlation. Radiographics. 1992;12:151-168.
37. Tomiyama N., Johkoh T., Mihara N., et al. Using the World Health Organization Classification of thymic epithelial neoplasms to describe CT findings. AJR Am J Roentgenol. 2002;179:881-886.
38. Moran C.A., Travis W.D., Rosado-de-Christenson M., et al. Thymomas presenting as pleural tumors. Report of eight cases. Am J Surg Pathol. 1992;16:138-144.
39. Sung Y.M., Lee K.S., Kim B.T., et al. 18F-FDG PET/CT of thymic epithelial tumors: usefulness for distinguishing and staging tumor subgroups. J Nucl Med. 2006;47:1628-1634.
40. Luzzi L., Campione A., Gorla A., et al. Role of fluorine-fluorodeoxyglucose positron emission tomography/computed tomography in preoperative assessment of anterior mediastinal masses. Eur J Cardiothorac Surg. 2009;36:475-479.
41. Endo M., Nakagawa K., Ohde Y., et al. Utility of 18FDG-PET for differentiating the grade of malignancy in thymic epithelial tumors. Lung Cancer. 2008;61:350-355.
42. Sadohara J., Fujimoto K., Muller N.L., et al. Thymic epithelial tumors: comparison of CT and MR imaging findings of low-risk thymomas, high-risk thymomas, and thymic carcinomas. Eur J Radiol. 2006;60:70-79.
43. Jeong Y.J., Lee K.S., Kim J., et al. Does CT of thymic epithelial tumors enable us to differentiate histologic subtypes and predict prognosis? AJR Am J Roentgenol. 2004;183:283-289.
44. Kumar A., Regmi S.K., Dutta R., et al. Characterization of thymic masses using (18)F-FDG PET-CT. Ann Nucl Med. 2009;23:569-577.
45. Kuhlman J.E., Fishman E.K., Wang K.P., et al. Mediastinal cysts: diagnosis by CT and needle aspiration. AJR Am J Roentgenol. 1988;150:75-78.
46. Coleman B.G., Arger P.H., Dalinka M.K., et al. CT of sarcomatous degeneration in neurofibromatosis. AJR Am J Roentgenol. 1983;140:383-387.
47. Sofka C.M., Semelka R.C., Kelekis N.L., et al. Magnetic resonance imaging of neuroblastoma using current techniques. Magn Reson Imaging. 1999;17:193-198.
48. Sakai F., Sone S., Kiyono K., et al. Intrathoracic neurogenic tumors: MR-pathologic correlation. AJR Am J Roentgenol. 1992;159:279-283.
49. Stark D.D., Moss A.A., Brasch R.C., et al. Neuroblastoma: diagnostic imaging and staging. Radiology. 1983;148:101-105.
50. Sharp S.E., Shulkin B.L., Gelfand M.J., et al. 123I-MIBG scintigraphy and 18F-FDG PET in neuroblastoma. J Nucl Med. 2009;50:1237-1243.
51. Girard N., Mornex F., Van Houtte P., et al. Thymoma: a focus on current therapeutic management. J Thorac Oncol. 2009;4:119-126.
52. Falkson C.B., Bezjak A., Darling G., et al. The management of thymoma: a systematic review and practice guideline. J Thorac Oncol. 2009;4:911-919.
53. Bretti S., Berruti A., Loddo C., et al. Multimodal management of stages III-IVa malignant thymoma. Lung Cancer. 2004;44:69-77.
54. Jacot W., Quantin X., Valette S., et al. Multimodality treatment program in invasive thymic epithelial tumor. Am J Clin Oncol. 2005;28:5-7.
55. Tomaszek S., Wigle D.A., Keshavjee S., Fischer S. Thymomas: review of current clinical practice. Ann Thorac Surg. 2009;87:1973-1980.
56. Lucchi M., Ambrogi M.C., Duranti L., et al. Advanced stage thymomas and thymic carcinomas: results of multimodality treatments. Ann Thorac Surg. 2005;79:1840-1844.
57. International Germ Cell Consensus Classification: a prognostic factor-based staging system for metastatic germ cell cancers. J Clin Oncol. 1997;15:594-603.
58. Meister M., Schirmacher P., Dienemann H., et al. Mutational status of the epidermal growth factor receptor (EGFR) gene in thymomas and thymic carcinomas. Cancer Lett. 2007;248:186-191.
59. Gottfried O.N., Viskochil D.H., Couldwell W.T. Neurofibromatosis Type 1 and tumorigenesis: molecular mechanisms and therapeutic implications. Neurosurg Focus. 2010;28:E8.
60. Katz D., Lazar A., Lev D. Malignant peripheral nerve sheath tumour (MPNST): the clinical implications of cellular signalling pathways. Expert Rev Mol Med. 2009;11:e30.
61. Tucker T., Friedman J.M., Friedrich R.E., et al. Longitudinal study of neurofibromatosis 1 associated plexiform neurofibromas. J Med Genet. 2009;46:81-85.
62. Weiss B., Bollag G., Shannon K. Hyperactive Ras as a therapeutic target in neurofibromatosis type 1. Am J Med Genet. 1999;89:14-22.
63. Yang F.C., Ingram D.A., Chen S., et al. Neurofibromin-deficient Schwann cells secrete a potent migratory stimulus for Nf1± mast cells. J Clin Invest. 2003;112:1851-1861.
64. Yang F.C., Ingram D.A., Chen S., et al. Nf1-dependent tumors require a microenvironment containing Nf1±- and c-kit-dependent bone marrow. Cell. 2008;135:437-448.
65. Yan N., Ricca C., Fletcher J., et al. Farnesyltransferase inhibitors block the neurofibromatosis type I (NF1) malignant phenotype. Cancer Res. 1995;55:3569-3575.
66. Brems H., Beert E., de Ravel T., Legius E. Mechanisms in the pathogenesis of malignant tumours in neurofibromatosis type 1. Lancet Oncol. 2009;10:508-515.
67. Johannessen C.M., Johnson B.W., Williams S.M., et al. TORC1 is essential for NF1-associated malignancies. Curr Biol. 2008;18:56-62.
68. Johannessen C.M., Reczek E.E., James M.F., et al. The NF1 tumor suppressor critically regulates TSC2 and mTOR. Proc Natl Acad Sci U S A. 2005;102:8573-8578.
69. Bhola P., Banerjee S., Mukherjee J., et al. Preclinical in vivo evaluation of rapamycin in human malignant peripheral nerve sheath explant xenograft. Int J Cancer. 2010;126:563-571.
70. Cheung N.K., Kushner B.H., Cheung I.Y., et al. Anti-G(D2) antibody treatment of minimal residual stage 4 neuroblastoma diagnosed at more than 1 year of age. J Clin Oncol. 1998;16:3053-3060.
71. Simon T., Hero B., Faldum A., et al. Consolidation treatment with chimeric anti-GD2-antibody ch14.18 in children older than 1 year with metastatic neuroblastoma. J Clin Oncol. 2004;22:3549-3557.
72. Wagner L.M., Danks M.K., et al. New therapeutic targets for the treatment of high-risk neuroblastoma. J Cell Biochem. 2009;107:46-57.