Potential advances in treatment
Targeting the microenvironment in haematological malignancy
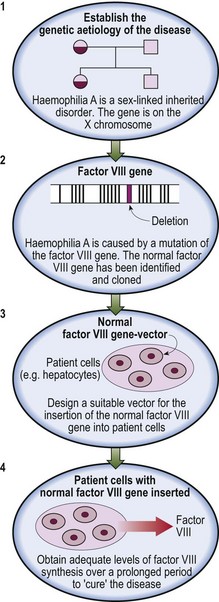
The tumour microenvironment is currently thought to play a crucial role in cancer biology. A better understanding of the complex interactions between tumour and non-malignant cells is likely to translate into better characterization of tumour pathophysiology and novel approaches to therapy. In haematological malignancy, follicular lymphoma (see also p. 61) provides an excellent model for the study of the microenvironment. This disease accounts for approximately a quarter of all cases of non-Hodgkin’s lymphoma. Modern treatment with a combination of chemotherapy agents and the anti-CD20 monoclonal antibody rituximab has improved overall survival but cure is elusive. Better therapeutic strategies are needed. Technologies such as immunocytochemistry, functional cytotoxicity assays and 3-D confocal imaging have improved our understanding of the role of the microenvironment in this disease and offered the prospect of new treatments which tip the balance against the tumour cells. The various interactions are complex (Fig 51.1) but it is clear that CD8+ cytotoxic lymphocytes play an active role and have the potential to be employed in the treatment of follicular lymphoma. Most simplistically, a higher content of T-cells appears to confer a better prognosis while pro-tumour macrophages may worsen the outlook. Follicular lymphoma cells can manipulate the microenvironment to their own advantage. Stimulation of CD4+ T-cell cytokine release promotes tumour cell growth and survival. The tumour cells also employ mechanisms to block entry of cytotoxic T-cells into the tumour site and to inhibit recruitment and trafficking of previously healthy T-cells. The challenge is to find ways of supplementing current immunotherapy regimens (e.g. rituximab and chemotherapy) by tipping the balance in the tumour microenvironment towards enhanced T-cell anti-tumour activity and away from immunosuppression. Possible agents include immunomodulating drugs such as lenalidomide. Such considerations may also apply in other haematological malignancies. For instance, encouraging clinical results have been obtained in chronic lymphocytic leukaemia following the targeting of the tumour cells with genetically modified autologous T-cells.
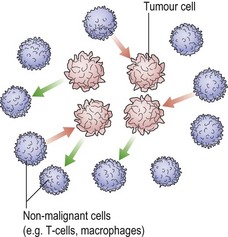
Fig 51.1 A schematic view of the microenvironment in follicular lymphoma.
Cytotoxic CD8+ lymphocytes exert anti-tumour effects (red arrows) while tumour cells seek self-preservation (green arrows) by stimulating CD4+ T-cells to secrete favourable cytokines, inhibiting cytotoxic T-cells, and erecting a ‘tumour barrier’.
New oral anticoagulant drugs
Anticoagulant drugs are used both in the treatment and prevention of thromboembolic disease (see pp. 80–81). Conventional heparin and warfarin were discovered over 60 years ago while low molecular weight heparin is a more recent addition. All of these drugs have disadvantages, none fulfilling all the criteria for an ‘ideal’ anticoagulant, namely a high efficacy to safety index, a predictable dose response with no need for laboratory monitoring, administration by parenteral and oral routes, a rapid onset of action, a safe antidote, freedom from non-anticoagulant side-effects and minimal interaction with other drugs. Any new agent must aspire to meet as many of these requirements as possible without being prohibitively expensive. Attempts to develop better anticoagulants have focused on specific steps or enzymes in the coagulation pathway. Agents under evaluation include inhibitors of the factor VIIIc/tissue factor pathway, factor Xa inhibitors, activated protein C and soluble thrombomodulin and direct thrombin inhibitors (Fig 51.2).
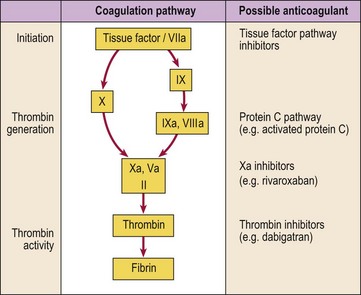
Gene therapy
Gene therapy is potentially a very powerful therapeutic tool applicable to a wide range of diseases including several blood disorders. After years of failure, there have been recent successes in the treatment of infants with immunodeficiency and in selected patients with malignancy. The basis of the technique is the insertion of a new functional gene into a cell (Fig 51.3). The gene is introduced into the target cell by use of a ‘vector’. Disabled viruses are commonly used as vectors because they can undertake tasks necessary for successful gene transfer such as binding to the target cell and delivering the viral genome to the nucleus for transcription. Non-viral vectors based on plasmid DNA produced in bacteria and combined with lipids are also used. The optimum vector and delivery method is likely to vary depending on the disease under treatment. Early protocols mainly involved an ex vivo approach where the gene was inserted into cells taken from the patient. However, more recently the vector is normally given directly to the patient (in vivo approach). The problems have been numerous and have included difficulties in characterising and accessing the target cells, poor efficacy of gene transfer, short-lived expression of the newly introduced gene, and safety issues. The latter especially relate to viral vectors which have caused clinical symptoms of infection and induced massive immunological responses. Two children cured of their immunodeficiency by gene therapy subsequently developed leukaemia as a result of insertional mutagenesis.