Portal hypertension
Aetiology and pathophysiology of portal hypertension
Traditionally, portal hypertension has been classified as prehepatic, intrahepatic or posthepatic, with the intrahepatic causes subdivided into presinusoidal, sinusoidal and postsinusoidal (Table 8.1). Prehepatic causes are usually due to portal vein thrombosis, which is discussed later in this chapter. The main cause of portal hypertension in the West is cirrhosis. This is a sinusoidal obstruction to portal flow with varying causes. Viral hepatitis and alcoholic liver disease are the most common causes, but others include primary biliary cirrhosis, primary sclerosing cholangitis and haemochromatosis. Presinusoidal obstruction due to hepatic fibrosis occurs in schistosomiasis. Worldwide, this is one of the commonest causes of portal hypertension and, as it is usually associated with normal liver function, has a better prognosis. The main causes of postsinusoidal portal hypertension are hepatic venous thrombosis (Budd–Chiari syndrome) and veno-occlusive disease.
Table 8.1
| Presinusoidal | Sinusoidal | Postsinusoidal |
| Extrahepatic | Cirrhotic | Budd–Chiari syndrome |
| Portal vein thrombosis | Postviral (B, C) | Veno-occlusive disease |
| Splenic vein thrombosis | Alcoholic | Caval web |
| Increased splenic flow (tropical splenomegaly, myelofibrosis) | Cryptogenic Primary biliary cirrhosis Primary sclerosing cholangitis |
Constrictive pericarditis |
| Intrahepatic | Chronic active hepatitis | |
| Schistosomiasis | Haemochromotosis | |
| Congenital hepatic fibrosis | Wilson’s disease | |
| Sarcoidosis | Non-cirrhotic | |
| Acute alcoholic hepatitis | ||
| Cytotoxic drugs |
Experimental studies have demonstrated that the initial factor in the pathophysiology of portal hypertension is the increase in vascular resistance to portal blood flow. In cirrhosis, this increase in resistance occurs in the hepatic microcirculation (sinusoidal portal hypertension), and is a consequence of both a ‘passive’ and an ‘active’ component. The ‘passive’ component is the mechanical consequence of the hepatic architectural disorder resulting from histological cirrhosis, and the ‘active’ component is the active contraction of portal/septal myofibroblasts, activated stellate cells and portal venules. The increase in intrahepatic tone is probably a consequence of an imbalance between an increase in the endogenous vasoconstrictor substances, such as endothelin, noradrenaline, leukotrienes and angiotensin II, and a relative decrease in the endogenous vasodilator nitric oxide.1 Vasodilatory drugs (for example, calcium channel blockers) may restore the equilibrium in intrahepatic tone, although they are not used for this indication in clinical practice.
The natural history of portal hypertension
The prevalence of oesophageal varices in patients with cirrhosis and portal hypertension is high. When cirrhosis is diagnosed, varices are present in 40% of compensated and 60% of decompensated cirrhotics.2 After the initial diagnosis of cirrhosis, varices develop with an incidence of 5% per year; subsequently, they may progress from small to large at an incidence of 10–15% per year.3 Rapid progression of hepatic decompensation is associated with a rapid increase in size, whilst improvement in liver function, particularly when associated with removal of the injurious agent (e.g. abstinence from alcohol), may result in a decrease in size or disappearance of the varices.4,5
The overall incidence of variceal bleeding following diagnosis is of the order of 25% in unselected patients. The most important predictive factors of variceal bleeding are severity of liver dysfunction, size of varices and intravariceal wall pressure (which although difficult to measure may correlate at endoscopy with the presence of red spots or red weals).6 Traditionally, liver dysfunction has been classified using the Child–Pugh score7 (Table 8.2), but a more recent scoring system, the MELD (Model for End-stage Liver Disease), may be a better prognostic indicator (Box 8.1).8 Variceal size may be the best single predictor of variceal bleeding and generally it is used to decide whether a patient should be given prophylactic therapy or not. Whether a patient dies from a variceal bleed depends on the severity of the accompanying liver failure; those with a high Child–Pugh or MELD score have been reported to have as high a risk of mortality as 30–50% within 6 weeks of the index bleed.9 However, a more realistic figure would be 20% at 6 weeks with an immediate mortality from uncontrolled bleeding as low as 5–8%. Indeed, in 40–50% of patients who bleed and develop hypotension, variceal bleeding stops spontaneously, probably as a result of reflex splanchnic vasoconstriction with associated reduction in portal pressure and blood flow; this beneficial response is nullified by over-transfusing the patient.
Box 8.1 Model for End-stage Liver Disease (MELD)
MELD is calculated for patients over the age of 12 based on the following variables:
The formula incorporates these variables as:
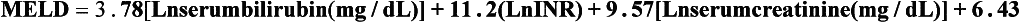
The following rules must be observed when using this formula:
• 1 is the minimum acceptable value for any of the three variables.
• The maximum acceptable value for serum creatinine is 4.
• The maximum value for the MELD score is 40. All values higher than 40 are given a score of 40.
• If the patient has been dialysed twice within the last 7 days, then the value for serum creatinine used should be 4.0.
Table 8.2
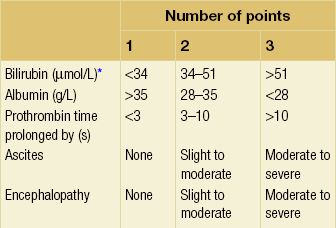
Grade A 5–6 points; Grade B 7–9 points; Grade C 10–15 points.
*In primary biliary cirrhosis, the point scoring for bilirubin level is adjusted as follows: 1, < 68; 2, 68–170; 3, > 170.
The incidence of re-bleeding ranges between 30% and 40% within the first 6 weeks; this risk peaks in the first 5 days following the index bleed. Bleeding gastric varices, active bleeding at emergency endoscopy, low serum albumin levels, renal failure and a hepatic venous pressure gradient > 20 mmHg have all been reported as significant indicators of an early risk of re-bleeding.10–12 Patients surviving a first episode of variceal bleeding have a very high risk of re-bleeding (63%) and death (33%), and this is the basis for treating all patients to prevent further bleeding.9
Presentation
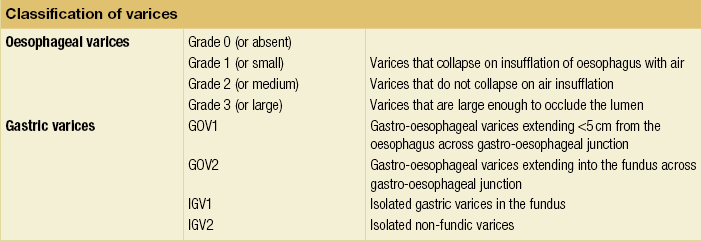
Portal hypertension may present acutely with variceal bleeding or be discovered during the investigation of a patient with liver disease. Varices are usually easily diagnosed at endoscopy and patients will then be investigated systematically. A classification of the grading of varices is given in Table 8.3. Presentation of patients with liver disease is variable and ranges from non-specific tiredness to advanced encephalopthy with decompensation. External features of advanced liver disease such as spider naevi, palmar erythema and ascites are easy to detect, although these signs will be lacking in many patients. Splenomegaly is probably the most useful physical sign, although some patients will have the classic sign of dilated umbilical vein collaterals (caput medusae).
Therapeutic aims for pharmacological therapy in portal hypertension

Varices do not develop until the HVPG increases to 10–12 mmHg and the HVPG must be greater than 12 mmHg for the appearance of complications such as variceal bleeding and ascites.13 Longitudinal studies of patients with complications of portal hypertension have demonstrated that when an HVPG decreases to less than 12 mmHg with pharmacological therapy, TIPS or an improvement in liver function, variceal bleeding is prevented and varices may decrease in size or disappear altogether.14 When this target is not reached, a substantial reduction in portal pressure by more than 20% still offers protection against variceal bleeding15 and thus these two parameters are regarded as the end-points to therapeutic strategies to lower portal pressure.
Recent evidence suggests that these therapeutic end-points may also reduce the risk of other complications of portal hypertension, including ascites, spontaneous bacterial peritonitis and hepatorenal syndrome.16,17
Oesophageal varices
Primary prophylaxis for the prevention of variceal haemorrhage

A meta-analysis has indicated that indefinite treatment with propanolol or nadolol significantly reduces the bleeding risk from 25% with non-active treatment or placebo to 15% with beta-blockers over a median follow-up period of 24 months; there was no significant reduction in mortality.3 The benefit of therapy was only proven in those patients with grade II (or larger) varices; there was no evidence to support the use of primary prophylactic therapy in patients with grade I varices.

Withdrawal of therapy was associated with a return to the same bleeding risk (25%) as the untreated subpopulation; indeed, there may also be an increased risk of mortality over untreated patients in those individuals who stop therapy.18,19
Assessment of the success of primary prophylactic therapy is ideally undertaken by measurement of the HVPG before and after initiating therapy, with the aim being to reduce the HVPG to < 12 mmHg or to reduce it by > 20% from its baseline value.14 In practice, however, measurement of HVPG does require specific training and is probably not cost-effective for assessing primary prophylactic therapy. Thus, the clinician faces the question of how to adjust the dose of beta-blocker to maximise its beneficial effects. Traditional practice has recommended a stepwise increase in dose until the heart rate decreases by 25%, is < 55 beats per minute, or there is arterial hypotension or clinical intolerance. This means that the dose of the beta-blocker is titrated against its β1 effects (cardiac) and clinical tolerance; however, a fall in portal pressure results from blockade of both β1 and β2 receptors, and the fall in portal pressure does not readily correlate with the fall in heart rate or blood pressure. Therefore, titration solely against clinical tolerance may be the most useful surrogate marker of the maximal dose of beta-blocker in the absence of HVPG measurement.
When compared with propanolol, carvedilol significantly increased the number of patients achieving a target reduction of HVPG (< 12 mmHg or > 20% reduction from baseline HVPG).19 There is considerable controversy about how to give the carvedilol because of its hypotensive side-effects; however, the above study demonstrated that lower doses (12.5 mg/day) result in good tolerance. In practice, the usual starting dose is 6.25 mg/day and the usual maintenance dose 12.5 mg/day.
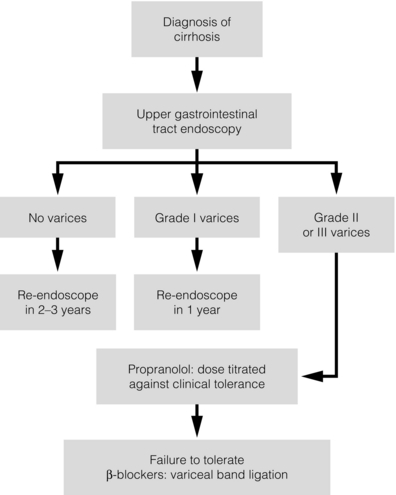
In patients who are unable to tolerate beta-blockers (15–20%) because of side-effects or relative/absolute contraindications (for example, asthma), treatment with nitrates is ineffective, despite its portal pressure-lowering properties.20 Therefore, variceal band ligation (VBL) therapy is the only option for patients with high-risk varices (grade II or above) and contraindications to beta-blockers. More controversially, a meta-analysis has suggested that VBL is a more effective mode of treatment than beta-blockers for primary prophylaxis.21 However, this analysis included four trials, only two of which have been published in full; therefore, it seems reasonable to recommend that, for the time being, beta-blockers remain the primary prophylactic therapy of choice in terms of cost and convenience. Of course, VBL does not reduce portal pressure (and therefore measurement of HVPG following endoscopic monotherapy is of no value) and this may leave the patient at risk of developing other complications of portal hypertension. An algorithm for the primary prevention of variceal bleeding is given in Fig. 8.1.
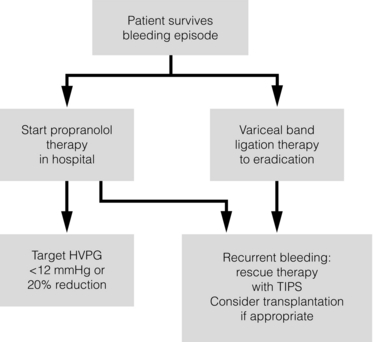
Prevention of re-bleeding from oesophageal varices (secondary prophylaxis)
Following a variceal bleed, patients with cirrhosis should be managed in two ways: firstly, they should receive urgent and active treatment for the prevention of re-bleeding; secondly, they should be examined for signs of physiological stress following their bleed, which might indicate a need for an elective liver transplant assessment (Fig. 8.2). Management of non-cirrhotic patients is discussed later in this chapter.

Meta-analyses of studies using beta-blocker therapy to prevent re-bleeding have demonstrated both a significantly decreased mortality (27% in controls to 20% in beta-blocker-treated individuals) and a decreased incidence of re-bleeding (63% to 42%).3
VBL also both improves survival and significantly decreases re-bleeding rates; it is superior to endoscopic sclerotherapy since it is associated with significantly fewer complications.22,23 Currently, it is unclear whether pharmacological therapy is better than VBL or vice versa; studies have demonstrated a variety of outcomes with reference to re-bleeding rates, but none have indicated any clear difference in survival.15,24,25 A combination of pharmacological therapy and endoscopic therapy is commonly used, but evidence suggesting a better outcome with this combination compared with monotherapy is hard to find. Likewise, combination therapy of nitrates and beta-blockers has not been consistently shown to be more effective than beta-blockers alone or VBL.15,26
Treatment for bleeding oesophageal varices

Antibiotics should be instituted from admission, since these increase the survival of bleeding patients; norfloxacin 400 mg/12 hours or ciprofloxacin 250 mg/12 hours are the antibiotics of choice.27,28
Finally, early therapy should also involve starting a vasoactive drug from admission (usually terlipressin or octreotide); a number of randomised controlled trials demonstrate that early administration of vasoactive drugs facilitates endoscopy, improves control of bleeding and reduces the 5 day re-bleeding rate.29,30 Initiation of these measures in association with endoscopic therapy at the time of diagnostic endoscopy will control bleeding in approximately 75% of patients. However, as in most trials, in acute variceal bleeding this combined approach failed to improve overall mortality compared with drug or endoscopic therapy alone. The optimal duration of vasoactive drug therapy is not well established and requires evaluation; current recommendations are to continue the drug for 5 days, since this covers the period of maximum risk of re-bleeding.
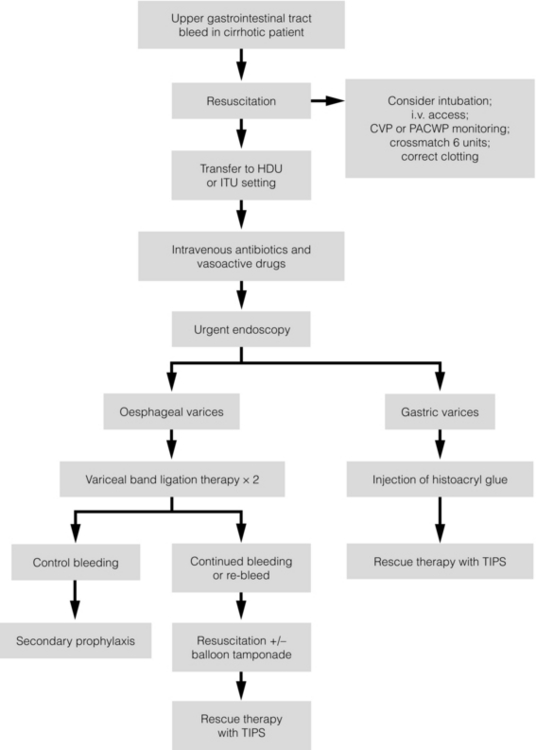
Endoscopic therapy should be performed at the time of diagnostic endoscopy, within 12 hours of admission in a resuscitated patient. However, if the patient is stable, endoscopic therapy can probably be postponed until within normal working hours. There are multiple randomised controlled trials examining modes of endoscopic therapy in acute variceal bleeding. These have compared: endoscopic therapy with no therapy; endoscopic therapy with vasoactive drug therapy; endoscopic sclerotherapy with variceal band ligation therapy; combined endoscopic therapy with variceal band ligation therapy; and endoscopic therapy with TIPS. Endoscopic therapy is certainly superior to no therapy;31 of the two endoscopic therapies, variceal band ligation therapy should be considered the treatment of choice since it is associated with significantly fewer complications (oesophageal stricturing or oesophageal ulcer formation) and significantly fewer sessions of therapy to eradicate the varices. However, there is probably no difference in re-bleeding or mortality rates between the two therapies. Likewise, there is little evidence to support combined endoscopic therapy for the treatment of bleeding varices.32 In practice, however, it is sometimes beneficial for the endoscopist to use a small volume of sclerosant initially to improve vision in order to place some variceal bands to achieve eventual haemostasis. If endoscopic therapy fails to control bleeding, balloon tamponade should be used as a ‘bridge’ until definitive therapy can be offered. In practice, this usually means a further attempt at endoscopic band ligation therapy followed by second-line therapies. An algorithm for the management of variceal bleeding in cirrhotics is given in Fig. 8.3.
Gastric varices
Treatment of acute gastric variceal bleeding is very challenging. Medical management is similar to the treatment of oesophageal varices. Terlipressin and octreotide are useful for control of acute bleeding, while beta-blockers may also be as effective as secondary prophylactic therapy. The Sengstaken–Blakemore tube may have some utility for controlling bleeding from junctional (GOV1 or GOV2) varices but has little effect on controlling bleeding varices in the fundus or further down the stomach. Some endoscopic therapies are promising, but quality data are scarce; sclerotherapy, glue injection, thrombin and variceal band ligation therapy have all been reported. Control of bleeding using sclerotherapy with cyanoacrylate has been reported as efficacious in 62–100% of cases, with successful obliteration of varices in 0–94%.33,34

A randomised controlled trial from Taiwan has confirmed that endoscopic sclerotherapy with cyanoacrylate was more effective and also safer than band ligation in the management of bleeding gastric varices.35

Recently, the first prospective, randomised controlled trial comparing TIPS with cyanoacrylate injection in the prevention of gastric re-bleeding was published. This concluded that TIPS was more effective than glue injection in preventing re-bleeding from gastric varices, although the two modalities shared a similar mortality rate and frequency of complications.36
Portal hypertensive gastropathy
The presence of portal hypertensive gastropathy (PHG) is strongly correlated with the severity of cirrhosis, its overall prevalence in cirrhosis being about 80%.37 However, the incidence of acute bleeding is low, occurring in about 2.5% of patients over an 18-month follow-up period, with an associated mortality of 12.5%; the incidence of chronic bleeding is significantly higher at 12%. Propanolol, octreotide and terlipressin have all been proposed for the treatment of acute bleeding from PHG based on their ability to decrease portal blood flow. In a randomised controlled trial, propanolol was found to reduce recurrent bleeding from PHG.38 Once again, TIPS is considered as the rescue therapy of choice in patients who have repeated bleeding from PHG despite propanolol therapy.
Second-line therapies
TIPS (transjugular intrahepatic portosystemic shunt)

The degree of shunting can be tailored to some extent by adjusting the diameter of the balloon-dilated shunt against the resulting pressure gradient, directly measured through the catheter.39

An early disadvantage of TIPS was the poor primary patency rate, but this can be significantly improved by the use of covered stents, as demonstrated in a recent randomised trial.40
TIPS has been compared unfavourably with surgery (see later section on surgical shunts) because of the high rate of reintervention, yet overall survival has been similar in randomised trials of both H-graft portocaval shunts and distal splenorenal shunts versus TIPS.41,42 However, TIPS is usually preferred in patients with more advanced liver disease and in those likely to need future transplantation. Patients with more severe liver disease may be candidates for liver transplantation, but TIPS can stabilise some to enable survival long enough to receive a successful transplant. Moreover, the MELD score can be used to predict likely survival following TIPS.43
TIPS for variceal bleeding
Uncontrolled bleeding from oesophagogastric or ectopic varices in the presence of a patent portal vein can usually be controlled by TIPS. The procedure can be performed on patients considered too sick for surgery. The mortality of these patients is due more to their general condition rather than the TIPS procedure. The 30-day mortality after TIPS in the UK National Confidential Enquiry into Perioperative Death study was 17%.44 In this study, 80% of patients dying after TIPS had the procedure performed urgently or as an emergency for bleeding varices.
Meta-analyses of several trials compare TIPS with endoscopic sclerotherapy and/or banding for prevention of recurrent variceal bleeding.45,46 Additional medical therapy was included in some. TIPS was more successful at preventing re-bleeding but with no overall improvement in mortality. Encephalopathy was overall more frequent in the TIPS patients, but not in every trial. In some studies patients in the endoscopic groups were rescued by TIPS because of significant recurrent bleeding. General consensus is that endoscopic and medical therapy should be the primary treatment and TIPS reserved for those cases where control is not achieved. TIPS may be combined effectively with medical treatment or endoscopic variceal eradication after bleeding has been controlled. Long-term TIPS surveillance and reintervention may then be less necessary.47,48
Other procedures have been used to control bleeding from varices when venous anatomy permits catheter access. Retrograde balloon occlusion of gastric varices has been mainly used in Asia as an alternative to TIPS when there is a patent gastrorenal venous connection.49,50
Surgical options
Portal systemic shunts
The variety of surgical shunts described for portal hypertension is perhaps a testament to the ingenuity of surgeons (Fig. 8.4). With the passing of the ‘shunt era’ most surgical trainees will not have seen a shunt, which now has a limited application for a selected group of patients. These are mainly those with non-cirrhotic portal hypertension and patients living in areas where newer therapies are not available. However, in some units where an active interest in shunt surgery has been maintained, a combination of very experienced surgeons and an excellent organisation has allowed for good results with emergency shunt surgery.51
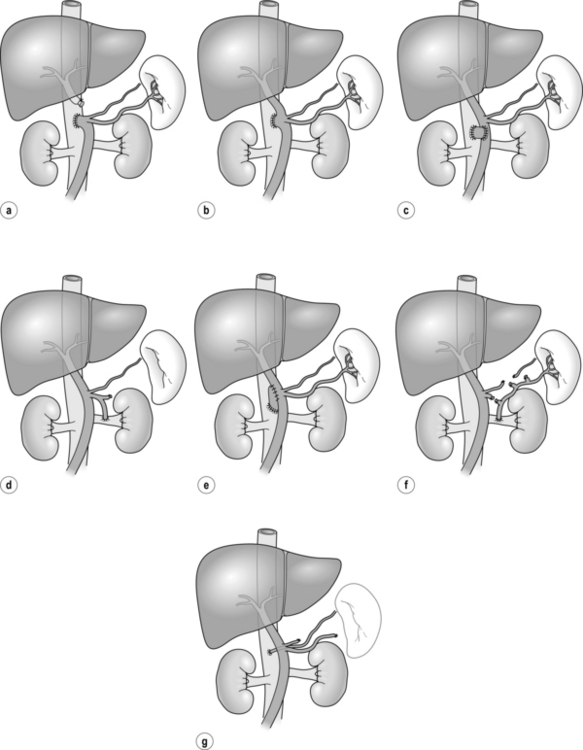
Figure 8.4 Non-selective (a–d) and selective (e–g) portosystemic shunts. (a) End-to-side portocaval. (b) Side-to-side portocaval. (c) Mesocaval (jugular vein graft or prosthesis). (d) Proximal splenorenal. (e) Small-diameter PTFE H-graft portocaval. (f) Distal splenorenal (Warren). (g) Left gastric-to-IVC (Inokuchi).

A controlled crossover trial comparing distal splenorenal (Warren) shunt (DSRS) with endoscopic sclerotherapy showed that shunting produced better control of bleeding but did not produce any survival advantage.52
Shunt operations can be classified into selective or non-selective shunts. The former carry lower rates of hepatic encephalopathy but are less successful in controlling acute bleeding. The two main procedures that have achieved popularity are the DSRS and the interposition portocaval or mesocaval shunt utilising a small-diameter prosthetic H-graft (see Fig. 8.4e,f). Direct primary portocaval anastomosis produces the most effective lowering of portal pressure but with the highest encephalopathy rates, and the advantage of the small-diameter portocaval H-graft is that it is selective and maintains some portal flow. This shunt has been compared to TIPS in a single-centre randomised trial in which the entry criterion was variceal bleeding in patients who had failed or ‘were not amenable to’ sclerotherapy or banding; recruitment was rapid, which suggests a low threshold to proceed with second-line therapies.53 There was a higher 30-day mortality in shunt patients but a better long-term control of bleeding than that seen in the TIPS patients. It should be recognised that the expertise needed for TIPS insertion and the protocols for subsequent surveillance will vary between centres, such that results should be interpreted with caution as they may reflect local interest and expertise. It had previously been established that shunt surgery for cirrhotics carries significant postoperative mortality rates, being as high as 26.1% for Child C patients even in specialised centres.54 Furthermore 5-year survival rates in patients with advanced liver disease are poor and shunt surgery carries an additional burden due to the risks of hepatic encephalopathy.

A recently reported multicentre randomised trial of TIPS versus DSRS showed no overall difference in survival and a tendency for TIPS to be more cost-effective in terms of lives saved.42,55
On current evidence there is no role for routine shunting in cirrhotic patients. Shunts should be avoided in patients in whom transplantation is an option as they significantly increase the risk of surgery. If endoscopic and radiological approaches fail, surgery away from the liver hilum is recommended either as a splenorenal or interposition mesocaval shunt.56
Liver transplantation
In 1997, minimal selection criteria, based on studies of the natural history of compensated chronic liver diseases, were developed to aid such a process.57 The minimal listing criteria were: an estimated 1-year survival ≤ 90%; Child–Pugh score ≥ 7 (Class B or C); or portal hypertensive bleeding; or an episode of spontaneous bacterial peritonitis regardless of the Child–Pugh score. The basis of these criteria is that the expected outcome of the untreated patient would be significantly worse than that of the outcomes from liver transplantation. This recognises the significantly worse prognosis of decompensated cirrhosis, which in those with hepatitis C, for example, dramatically reduces from a 91% 5-year survival to 50%.58 Spontaneous bacterial peritonitis carries an adverse outcome in these patients, with 1-year survival falling from 66% to 38% in one report,59 and despite the many therapeutic modalities available for treatment, the only definitive therapy for recurrent variceal bleeding is liver transplantation.60
Broadly, liver transplantation should be considered in any patient who is able to cooperate with the treatment and in whom an anticipated survival rate of at least 50% at 5 years postgrafting is likely to be achieved. The decisions to proceed to liver replacement should be made by a multidisciplinary team including an experienced hepatologist. Today, MELD scoring is widely used to list patients for liver transplant. Transplanting a patient with a MELD of < 15 is associated with a poorer outcome than would be expected on the waiting list and transplanting these patients may not be the best use of the limited organ pool (see Box 8.1).
Management of ascites
Ascites is a common feature of portal hypertension, although the exact mechanisms remain under debate.61 Ascites in chronic liver disease can be effectively treated by a number of medical, surgical or radiological techniques. The new development of ascites should be investigated for bacterial peritonitis, portal vein thrombosis or hepatic malignancy. Initial treatment involves dietary sodium restriction and diuretic therapy. Unresponsive patients may benefit from regular large-volume paracentesis with concurrent intravenous administration of 20% human albumin.62 Though peritoneovenous shunting is effective in controlling ascites, potential risks include disseminated intravascular coagulation, sepsis and cardiac failure. It has few advantages over large-volume paracentesis and is not recommended for patients who are transplant candidates.63 Refractory ascites is an indication for transplant assessment.
TIPS can also be very effective in controlling ascites refractory to medical treatment, but many such patients have very advanced liver disease with poor prognosis. The immediate risk is worsening liver failure and hepatic encephalopathy, and advice from an experienced hepatologist should be sought before TIPS. Older patients and those with renal dysfunction fare worse, and if patients with severe ascites are liver transplant candidates this may be a better option than TIPS. Patients with better liver function and disproportionate ascites, especially those with liver disease that can improve, e.g. by withdrawal from alcohol, respond well to TIPS. Some trials have shown TIPS to be more effective than medical treatment plus paracentesis, but patient selection is most important. Some studies have shown an improved survival and quality of life in patients having TIPS for ascites, whereas others have not.64–66 Surgical shunts are no longer recommended for resistant ascites due to high perioperative mortality and encephalopathy rates.
Budd–Chiari syndrome
Acute Budd–Chiari syndrome
In the acute presentation there will usually be abdominal pain and swelling. If there is a short stenosis or occlusion of the hepatic vein(s) and/or inferior vena cava (IVC), balloon dilatation or stenting is very effective. Transjugular, transfemoral or transhepatic access may be required. Occlusion or stenosis of the IVC (sometimes a web) may similarly respond to balloon dilatation. If the dilated or recanalised segment of hepatic vein is not satisfactorily maintained after balloon inflation, a metal stent can be inserted to maintain the patent lumen.67 These approaches have the benefit of restoring physiological hepatic vein outflow in at least one of the main hepatic veins. Adjunctive pharmacological or mechanical thrombolysis may assist these procedures in selected cases, especially when acute thrombosis complicates an otherwise successful vein recanalisation. There are individual case reports of systemic thrombolysis producing improvement but these are rare.68

TIPS can be used in both acute and chronic Budd–Chiari, and would now be regarded as the treatment of choice for those not responding to medical therapy and/or hepatic vein recanalisation.69,70
A recent study has shown that TIPS was the most frequent treatment modality applied in a 2-year multicentre European study of new Budd–Chiari presentations.71 The advantage of TIPS is decompression of the portal vein above the compressed part of the IVC within the caudate lobe and avoidance of laparotomy. With their tendency to thrombosis, Budd–Chiari patients have a greater need for reintervention than other TIPS patients but covered stents have shown improved patency.72
Extended TIPS can be a successful treatment for patients with Budd–Chiari syndrome complicated by portal and mesenteric vein thrombosis. A few cases have been described in which TIPS was a stabilising factor before liver transplantation, but most patients improve sufficiently after TIPS to avoid the need for transplantation. If TIPS cannot be achieved then surgical shunt can be performed or liver transplant when there is significant liver failure. In summary, there is a progressive hierarchy of radiological procedures that can manage the majority of Budd–Chiari patients according to their individual venous anatomy. These procedures are effective in combination with appropriate medical therapy.73
Chronic Budd–Chiari syndrome
Many patients present with significant ascites and marked changes on liver biopsy, which include significant fibrosis or even cirrhosis. It is likely that for some patients the hepatic venous obstruction is sequential and that the condition is asymptomatic until a second or final (third) hepatic vein is occluded. Either hepatic vein dilatation or TIPS should be performed based on the presence of identifiable hepatic veins within the liver.70,73 Shunt procedures should be reserved for radiological approach failures. Our local experience is that significant jaundice is an adverse prognostic sign and in these cases liver transplantation may be required.
Non-cirrhotic portal hypertension
Portal vein thrombosis
Portal vein thrombosis is rare in the West but is seen more frequently in Third World countries and is thought to be the result of umbilical sepsis in the neonatal period. Presentation can be in early childhood but is usually delayed to the early teenage years. The symptoms are usually that of a sudden variceal bleed, although some patients may be picked up by the presence of significant splenomegaly with or without haematological features of hypersplenism. The management of the acute bleed is similar to patients with cirrhosis. Re-bleeding or the presence of large gastric varices should be considered as a clear indication for a surgical shunt. Given the risks of splenectomy in the young, a spleen-preserving procedure is recommended. In a small child, splenorenal shunts are less practical because of the small size of the vessels and interposition mesocaval shunts using autologous jugular vein have high success rates with good long-term patency.74 In larger children, the distal splenorenal (Warren) shunt is usually favoured, although side-to-side splenorenal shunts have been reported in significant numbers from centres with a high prevalence of portal vein thrombosis.75 The natural history of these patients is interesting in that as they grow the varices become less symptomatic, and certainly shunting is not indicated unless bleeding episodes occur.
TIPS and portal vein thrombosis
Interventions have extended into the portal venous system by percutaneous transhepatic, transjugular and even the trans-splenic routes in selected cases.76 Limited acute portal vein thrombus is relatively easily treated by TIPS combined with thrombolysis, including clot disruption by balloon or other devices.77 Patients with normal liver may only require transhepatic portal vein procedures for success, but those with hepatic portal hypertension benefit from TIPS improved outflow as well. Chronic portal vein thrombosis is often associated with extensive portal collaterals forming a portal vein cavernoma. If there is an appropriate clinical indication, then these can be traversed and the main portal vein flow can be restored by balloon dilatation and/or stent insertion.78 More extensive occlusion involving the splenic and superior mesenteric veins may respond but with more difficulty, and those with underlying liver disease fare worse.
References
1. Rodríguez-Vilarrupla, A., Fernández, M., Bosch, J., et al, Current concepts on the pathophysiology of portal hypertension. Ann Hepatol 2007; 6:28–36. 17297426
2. Schepis, F., Camma, C., Nicefero, D., et al, Which patients should undergo endoscopic screening for esophageal varices detection? Hepatology 2001; 33:333–338. 11172334
3. D’Amico, G., Pagliaro, L., Bosch, J., Pharmacological treatment of portal hypertension: an evidence based approach. Semin Liver Dis 1999; 19:475–505. 10643630 Meta-analysis illustrating the benefits of treating patients with beta-blockers after an episode of bleeding oesophageal varices both in terms of decreasing risk of re-bleeding and decreasing mortality.
4. Zoli, M., Merkel, C., Magalotti, D., et al, Natural history of cirrhotic patients with small esophageal varices: a prospective study. Am J Gastroenterol 2000; 95:503–508. 10685758
5. Vorobioff, J., Grozmann, R.J., Picabea, E., et al, Prognostic value of hepatic venous pressure gradient measurements in alcoholic cirrhosis: a 10 year prospective study. Gastroenterology 1996; 111:701–709. 8780575
6. The North Italian Endoscopic Club for the Study and Treatment of Esophageal Varices, Prediction of the first variceal haemorrhage in patients with cirrhosis of the liver and esophageal varices: a prospective multicenter study. N Engl J Med 1988; 319:983–989. 3262200
7. Albers, I., Hartmann, H., Bircher, J., et al, Superiority of the Child–Pugh classification to quantitative liver function tests for assessing prognosis of liver cirrhosis. Scand J Gastroenterol 1989; 24:269–276. 2734585
8. Kamath, P.S., Wiesner, R.H., Malinchoc, M., et al, A model to predict survival in patients with end-stage liver disease. Hepatology 2001; 33:464–470. 11172350
9. D’Amico, G., Luca, A., Natural history. Clinical–haemodynamic correlations. Prediction of the risk of bleeding. Baillière’s Best Pract Res Clin Gastroenterol 1997; 11:243–256. 9395746
10. Ben Ari, Z., Cardin, F., McCormik, A.P., et al, A predictive model for failure to control bleeding during acute variceal haemorrhage. J Hepatol 1999; 31:443–450. 10488702
11. Goulis, J., Armonis, A., Patch, D., et al, Bacterial infection is independently associated with failure to control bleeding in cirrhotic patients with gastrointestinal haemorrhage. Hepatology 1998; 27:1207–1212. 9581672
12. Moitinho, E., Escorsell, A., Bandi, J.C., et al, Prognostic value of early measurements of portal pressure in acute variceal bleeding. Gastroenterology 1999; 117:626–631. 10464138
13. Garcia-Tsao, G., Grozsmann, R.J., Fisher, R.L., et al, Portal pressure, presence of gastroesophageal varices and variceal bleeding. Hepatology 1985; 5:419–424. 3873388
14. Feu, F., Garcia-Pagan, J.C., Bosch, J., et al, Relation between portal pressure response to pharmacotherapy and risk of recurrent variceal bleeding in patients with cirrhosis. Lancet 1995; 346:1056–1059. 7564785
15. Villaneuva, C., Minana, J., Ortiz, J., et al, Endoscopic ligation compared with combined treatment with nadolol and isosorbide mononitrate to prevent variceal bleeding. N Engl J Med 2001; 345:647–655. 11547718 References 13–15 provide the rationale for the therapeutic aim of pharmacological therapy to decrease the HVPG to < 12 mmHg or reduce it to 20% of its baseline value.
16. Tarantino, I., Abraldes, J.G., Turnes, J., et al. The HVPG response to pharmacological treatment of portal hypertension predicts prognosis and the risk of developing complications of cirrhosis. J Hepatol. 2002; 36(Suppl. 1):15A.
17. Bosch, J., Abraldes, J.G., Groszmann, R., Current management of portal hypertension. J Hepatol 2003; 38:S54–S68. 12591186
18. Abraczinskas, D.R., Ookubo, R., Grace, N.D., et al, Propanolol for the prevention of first esophageal haemorrhage: a lifetime commitment? Hepatology 2001; 34:1096–1102. 11731997
19. Banares, R., Moitinho, E., Matilla, A., et al, Randomised comparison of long-term carvedilol and propanolol administration in the treatment of portal hypertension in cirrhosis. Hepatology 2002; 36:1367–1373. 12447861 References 18 and 19 provide the rationale for the use of long-term beta-blockade to reduce the risk of bleeding in grade II or III varices.
20. Borroni, G., Salerno, F., Cazzaniga, M., et al, Nadolol is superior to isosorbide mononitrate for the prevention of the first variceal bleeding in cirrhotic patients with ascites. J Hepatol 2002; 37:315–321. 12175626
21. Imperiale, T.F., Chalasani, N., A meta-analysis of endoscopic variceal ligation for primary prophylaxis of esophageal variceal bleeding. Hepatology 2001; 33:908–914. 12460123
22. De Franchis, R., Primignami, M., Endoscopic treatments for portal hypertension. Semin Liver Dis 1999; 19:439–455. 10643628
23. Laine, L., El Newihi, H.M., Migikovsky, B., et al, Endoscopic ligation compared with sclerotherapy for the treatment of bleeding esophageal varices. Ann Intern Med 1993; 119:1–7. 8498757
24. Patch, D., Sabin, C.A., Goulis, J., et al, A randomised, controlled trial of medical therapy versus endoscopic ligation for the prevention of variceal rebleeding in patients with cirrhosis. Gastroenterology 2002; 123:1013–1019. 12360462
25. Lo, G.H., Chen, W.C., Chen, M.H., et al, Banding ligation versus nadolol and isosorbide mononitrate for the prevention of esophageal rebleeding. Gastroenterology 2002; 123:728–734. 12198699
26. Gournay, J., Masliah, C., Martin, T., et al, Isosorbide mononitrate and propanolol compared with propanolol alone for the prevention of variceal re-bleeding. Hepatology 2000; 31:1239–1245. 10827148
27. Bernard, B., Grange, J.D., Khac, E.N., et al, Antibiotic prophylaxis for the prevention of bacterial infections in cirrhotic patients with gastrointestinal bleeding: a meta-analysis. Hepatology 1999; 29:1655–1661. 10347104
28. Rimola, A., Garcia-Tsao, G., Navasa, M., et al, Diagnosis, treatment and prophylaxis of spontaneous bacterial peritonitis: a consensus document. International Ascites Club. J Hepatol 2000; 32:142–153. 10673079 References 27 and 28 illustrate the benefit to cirrhotic patients of prophylactic antibiotics following a variceal bleed by decreasing mortality.
29. Avgerinos, A., Nevens, F., Raptis, S., et al, Early administration of somatostatin and efficacy of sclerotherapy in acute variceal bleeds: the European acute bleeding oesophageal variceal episodes (ABOVE) randomised trial. Lancet 1997; 350:1495–1499. 9388396
30. Cales, P., Masliah, C., Bernard, B., et al, Early administration of vapreotide for variceal bleeding in patients with cirrhosis. French Club for the Study of Portal Hypertension. N Engl J Med 2001; 344:23–28. 11136956
31. Infante-Rivard, C., Esnaola, S., Villeneuve, J.P., Role of endoscopic variceal sclerotherapy in the long-term management of variceal bleeding: a meta-analysis. Gastroenterology 1989; 96:1087–1092. 2784398
32. Singh, P., Pooran, N., Indaram, A., et al, Combined ligation and sclerotherapy versus ligation alone for secondary prophylaxis of esophageal variceal bleeding: a meta-analysis. Am J Gastroenterol 2002; 97:623–629. 11922557
33. Huang, Y.H., Yeh, H.Z., Chen, G.H., et al, Endoscopic treatment of bleeding gastric varices by N-butyl-2-cyanoacrylate (Histoacryl) injection: long-term efficacy and safety. Gastrointest Endosc 2000; 52:160–167. 10922085
34. Kind, R., Guglielmi, A., Rodella, L., et al, Bucrylate treatment of bleeding gastric varices: 12 years’ experience. Endoscopy 2000; 32:512–519. 10917182
35. Lo, G.H., Lai, K.H., Cheng, J.S., et al, A prospective randomised trial of butyl cyanoacrylate injection versus band ligation in the management of bleeding gastric varices. Hepatology 2001; 33:1060–1064. 11343232 This was the first study to compare banding with the injection of ‘glue’ in the treatment of gastric varices as a prospective randomised trial.
36. Lo, G.H., Liang, H.L., Chen, W.C., et al, A prospective, randomised controlled trial of transjugular intrahepatic portosystemic shunt versus cyanoacrylate injection in the prevention of gastric variceal rebleeding. Endoscopy 2007; 39:679–685. 17661241 A further rare, randomised controlled trial in gastric variceal bleeding from the same institution. This study compared the efficacy and complications of TIPS with glue injections in preventing re-bleeding from gastric varices. TIPS was more effective than glue injection in preventing re-bleeding, but was associated with a similar risk of mortality and frequency of complications.
37. Primignani, M., Carpinelli, L., Preatoni, P., et al, Natural history of portal hypertensive gastropathy in patients with liver cirrhosis. The New Italian Endoscopic Club for the study and treatment of esophageal varices (NIEC). Gastroenterology 2000; 119:181–187. 10889167
38. Perez-Ayuso, R.M., Pique, J.M., Bosch, J., et al, Propanolol in the prevention of recurrent bleeding from severe portal hypertensive gastropathy in cirrhosis. Lancet 1991; 337:1431–1434. 1675316
39. Rossle, M., Siegerstetter, V., Olchewski, M., et al, How much reduction in portal pressure is necessary to prevent variceal rebleeding? A longitudinal study in 225 patients with transjugular intrahepatic portosystemic shunts. Am J Gastroenterol 2001; 96:3379–3383. 11774952 Observations on 225 patients having TIPS follow-up. Reduction of pressure gradient by 25–50% of original may be sufficient to prevent re-bleeding rather than the target of 12 mmHg.
40. Bureau, C., Pagan, J.C., Layrargues, G.P., et al, Patency of stents covered with polytetrafluoroethylene in patients treated by transjugular intrahepatic portosystemic shunts: long-term results of a randomized multicentre study. Liver Int 2007; 27:742–747. 17617116 PTFE-covered stents require less reintervention with no disadvantage at 2 years.
41. Rosemurgy, A.S., Zervos, E.E., Blooston, M., et al, Post shunt resource consumption favors small-diameter prosthetic H-graft portocaval shunt over TIPS for patients with poor hepatic reserve. Ann Surg 2003; 237:820–825. 12796578
42. Henderson, J.M., Boyer, T.D., Kutner, M.H., et al, Distal splenorenal shunt versus transjugular intrahepatic portal systematic shunt for variceal bleeding: a randomized trial. Gastroenterology 2006; 130:1643–1651. 16697728
43. Ferral, H., Gamboa, P., Postoak, D.W., et al, Survival after elective transjugular intrahepatic portosystemic shunt creation: prediction with model for end-stage liver disease score. Radiology 2004; 231:231–236. 14990811
44. , NCEPOD report on Vascular Interventional Radiology. www.ncepod.org.uk;, 2000.
45. Burroughs, A.K., Vangeli, M., Transjugular intrahepatic portosystemic shunt versus endoscopic therapy: randomized trials for secondary prophylaxis of variceal bleeding: an updated meta-analysis. Scand J Gastroenterol 2002; 37:249–252. 11916185
46. Khan, S., Tudur Smith, C., Williamson, P., et al, Portosystemic shunts versus endoscopic therapy for variceal rebleeding in patients with cirrhosis. Cochrane Database Syst Rev 2006; CD000553. 17054131
47. Brensing, K.A., Horsch, M., Textor, J., et al, Hemodynamic effects of propranolol and nitrates in cirrhotic patients with transjugular intrahepatic portosystemic stent-shunt. Scand J Gastorenterol 2002; 37:1070–1076. 12374234
48. Tripathi, D., Lui, H.F., Helmy, A., et al, Randomised controlled trial of long term portographic follow up versus variceal band ligation following transjugular intrahepatic portosystemic stent shunt for preventing oesophageal variceal rebleeding. Gut 2004; 53:431–437. 14960530
49. Hiraga, N., Aikata, H., Takaki, S., et al, The long-term outcome of patients with bleeding gastric varices after balloon-occluded retrograde transvenous obliteration. J Gastroenterol 2007; 42:663–672. 17701130
50. Cho, S.K., Shin, S.W., Lee, I.H., et al, Balloon-occluded retrograde transvenous obliteration of gastric varices: outcomes and complications in 49 patients. AJR Am J Roentgenol 2007; 189:W365–W372. 18029851
51. Orloff, M.J., Isenberg, J.I., Wheeler, H.O., et al, A randomised trial of liver transplantation in a randomized controlled trial of emergency treatment of acutely bleeding esophageal varices in cirrhosis. Transplant Proc. 2010;42(10):4101–4108. 21168637
52. Henderson, J.M., Kutner, M.H., Millikan, W.J., Jr., et al, Endoscopic variceal sclerosis compared with distal splenorenal shunt to prevent recurrent variceal bleeding in cirrhosis. A prospective, randomized trial. Ann Intern Med 1990; 112:262–269. 2404448 Shunting produced better control of bleeding but did not produce any survival advantage.
53. Rosemurgy, A., Serafini, F., Zweibel, B., et al, Transjugular intrahepatic portosystemic shunt vs. small-diameter prosthetic H-graft portacaval shunt: extended follow-up of an expanded randomized prospective trial. J Gastrointest Surg 2000; 4:589–597. 11307093
54. Rikkers, L.F., The changing spectrum of treatment for variceal bleeding. Ann Surg 1998; 228:536–546. 9790343
55. Boyer, T.D., Henderson, J.M., Heerey, A.M., et al, Cost of preventing variceal rebleeding with transjugular intrahepatic portal systemic shunt and distal splenorenal shunt. J Hepatol 2008; 48:407–414. 18045724
56. Dell’Era, A., Grande, L., Barros-Schelotto, P., et al, Impact of prior portosystemic shunt procedures on outcome of liver transplantation. Surgery 2005; 137:620–625. 15933628
57. Lucey, M.R., Brown, K.A., Everson, G.T., et al, Minimal criteria for placement of adults on liver transplant waiting list: a report of a national conference organized by the American Association for the Study of Liver Diseases. Liver Transpl Surg 1997; 3:628–637. 9404965
58. Fattovich, G., Giustina, G., Degos, F., et al, Morbidity and mortality in compensated cirrhosis type C: a retrospective follow up study of 384 patients. Gastroenterology 1997; 112:463–472. 9024300
59. Andreu, M., Sola, R., Sitges-Serra, A., et al, Risks for spontaneous bacterial peritonitis in cirrhotic patients with ascites. Gastroenterology 1993; 104:1133–1138. 8462803
60. D’Amico, G., Pagliaro, L., Bosch, J., The treatment of portal hypertension: a meta-analytic review. Hepatology 1995; 22:332–354. 7601427
61. Jalan, R., Hayes, P.C., Hepatic encephalopathy and ascites. Lancet 1997; 350:1309–1315. 9357420
62. Gines, A., Fernandez-Esparrach, G., Monescillo, A., et al, Randomized trial comparing albumin, dextran 70, and polygeline in cirrhotic patients with ascites treated by paracentesis. Gastroenterology 1996; 111:1002–1010. 8831595
63. Gines, P., Arroyo, V., Vargas, V., et al, Paracentesis with intravenous infusion of albumin as compared with peritoneovenous shunting in cirrhosis with refractory ascites. N Engl J Med 1991; 325:829–835. 1875966
64. Salerno, F., Camma, C., Enea, M., et al, Transjugular intrahepatic portosystemic shunt for refractory ascites: a meta-analysis of individual patient data. Gastroenterology 2007; 133:825–834. 17678653
65. Deltenre, P., Mathurin, P., Dharancy, S., et al, Transjugular intrahepatic portosystemic shunt in refractory ascites: a meta-analysis. Liver Int 2005; 25:349–356. 15780061
66. Saab, S., Nieto, J.M., Lewis, S.K., Runyon, B.A., TIPS versus paracentesis for cirrhotic patients with refractory ascites. Cochrane Database Syst Rev 2006; 18 CD004889. 17054221
67. Beckett, D., Olliff, S., Interventional radiology in the management of Budd Chiari syndrome. Cardiovasc Intervent Radiol 2008; 31:839–847. 18214592
68. Sharma, S., Texeira, A., Texeira, P., et al, Pharmacological thrombolysis in Budd Chiari syndrome: a single centre experience and review of the literature. J Hepatol 2004; 40:172–180. 14672630
69. Perello, A., Garcia Pagan, J.C., et al, TIPS is a useful long-term derivative therapy for patients with Budd Chiari syndrome uncontrolled by medical therapy. Hepatology 2002; 35:132–139. 11786969
70. Eapen, C.E., Velissaris, D., Heydtmann, M., et al, Favourable medium term outcome following hepatic vein recanalisation and/or transjugular intrahepatic portosystemic shunt for Budd Chiari syndrome. Gut 2006; 55:878–884. 16174658
71. Heydtmann, M., Raffa, S., Olliff, S., et al. One year survival in Budd–Chiari syndrome treated with TIPS: an international study. Gut. 2007; 56(Suppl. II):A2.
72. Hernández-Guerra, M., Turnes, J., Rubinstein, P., et al, PTFE-covered stents improve TIPS patency in Budd–Chiari syndrome. Hepatology 2004; 40:1197–1202. 15486923
73. Olliff, S.P., Transjugular intrahepatic portosystemic shunt in the management of Budd Chiari syndrome. Eur J Gastroenterol Hepatol 2006; 18:1151–1154. 17033433
74. Gauthier, F., De Dreuzy, O., Valayer, J., et al, H-type shunt with an autologous venous graft for treatment of portal hypertension in children. J Pediatr Surg 1989; 24:1041–1043. 2809948
75. Mitra, S.K., Rao, K.L., Narasimhan, K.L., et al, Side-to-side lienorenal shunt without splenectomy in non-cirrhotic portal hypertension in children. J Pediatr Surg 1993; 28:398–401. 8468654
76. Tuite, D.J., Rehman, J., Davies, M.H., et al, Percutaneous transsplenic access in the management of bleeding varices from chronic portal vein thrombosis. J Vasc Interv Radiol 2007; 18:1571–1575. 18057293
77. Uflacker, R., Applications of percutaneous mechanical thrombectomy in transjugular intrahepatic portosystemic shunt and portal vein thrombosis. Tech Vasc Interv Radiol 2003; 6:59–69. 12772131
78. Bilbao, J.I., Elorz, M., Vivas, I., et al, Transjugular intra-hepatic portosystemic shunt (TIPS) in the treatment of venous symptomatic chronic portal thrombosis in non-cirrhotic patients. Cardiovasc Intervent Radiol 2004; 27:474–480. 15383850