96 Portal Hypertension
 Anatomy and Physiology of the Portal System
Anatomy and Physiology of the Portal System
The term portal system refers to a venous system that begins and ends in capillaries. The portal venous system commences in the capillaries of the intestine and ends in the hepatic sinusoids. The portal venous system drains blood from the gastrointestinal (GI) tract, pancreas, gallbladder, and spleen. The portal vein originates from the confluence of the splenic vein and the superior mesenteric vein. The inferior mesenteric vein and short gastric veins drain into the splenic vein. The superior mesenteric vein drains all the blood from the small bowel and the right colon, while the inferior mesenteric vein drains the blood from the remainder of the colon and most of the rectum. Flow in the portal vein is normally about 1 L/min (approximately 20% cardiac output) with a mean pressure of 7 mm Hg. Although the blood in the portal vein is the outflow from capillary beds and therefore has relatively low oxygen content, 70% of hepatic oxygenation is derived from portal flow. The blood flowing through the hepatic artery supplies the remainder of hepatic oxygen consumption and is the primary blood supply to the biliary tree. The portal vein carries a high concentration of nutrients and hormones, facilitating the liver’s central role in fat, carbohydrate, drug, and protein metabolism. Toxic substances are removed by hepatocytes, and bacteria (and bacterial products) are removed by Kupffer cells. Portal venous blood and hepatic arterial blood mix at the sinusoidal level, and there exists an adenosine-mediated local hepatic arterial autoregulatory “buffer response” that increases arterial inflow in response to low portal flow; however, total hepatic flow is not preserved when hepatic arterial flow is decreased. This buffer response is also dysregulated in sepsis.1
 Pathophysiology
Pathophysiology
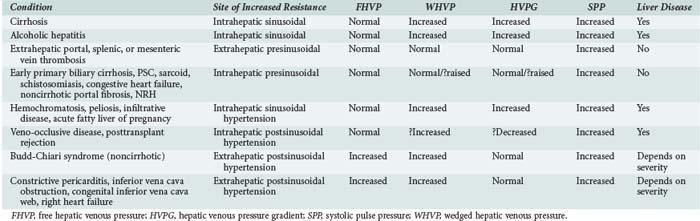
According to Ohm’s law (as applied to the cardiovascular system rather than an electrical circuit), the pressure within a vessel is determined by the flow of the blood in that vessel divided by the resistance. Apparent resistance depends upon a number of factors, including the length of the vessel, the radius of the vessel, and the viscosity of the blood. Since length and blood viscosity remain relatively constant, changes in radius are of paramount importance for determining changes in apparent resistance. An increase in blood flow in the portal vein and hepatic artery are important to the development of portal hypertension in some cases, but the increase in resistance seems to be the most important factor and is used to classify portal hypertension (PHT). The origin of PHT can be divided into cirrhotic and noncirrhotic and presinusoidal, sinusoidal, and postsinusoidal (Table 96-1). In response to PHT, vascular collaterals develop, and vascular resistance drops in the splanchnic bed, leading to the development of a hyperdynamic circulation. As a consequence, splanchnic and portal venous inflow increases, and PHT persists even with the development of vascular collaterals. As the pressure within the portal system continues to rise, portal blood flow decreases and hepatic perfusion deteriorates. The liver is deprived of portal blood, and this tends to accelerate the progression of liver disease. The hyperdynamic circulation and PHT also contribute to the development of portopulmonary syndrome (pulmonary hypertension and PHT), hepatopulmonary syndrome (hypoxia and intrapulmonary shunting in association with PHT), cirrhotic cardiomyopathy, ascites, and hepatorenal syndrome.
 Diagnosis of Portal Hypertension
Diagnosis of Portal Hypertension
PHT is defined as a portal pressure that is 5 mm Hg greater than the pressure measured in the inferior vena cava or a pressure of more than 15 mm Hg in the splenic vein or portal pressure measured at surgery. If the gradient is greater than 10 mm Hg, then clinically significant PHT is present. The direct consequences of PHT are formation of portosystemic collaterals and splenomegaly. Portosystemic collaterals can become clinically apparent as gastric or esophageal varices, umbilical vein recanalization, retroperitoneal collaterals, and/or rectal or ileostomy varices. The complications of PHT are variceal bleeding, ascites, spontaneous bacterial peritonitis, hepatic encephalopathy, hyperdynamic circulation, and hypersplenism. Varices are rarely (maybe never) seen if the gradient is less than 10 mm Hg.2 Variceal bleeding is not observed if the pressure gradient is less than 12 mm Hg, and protection from variceal bleeding is gained if the pressure gradient can be manipulated to less than 12 mm Hg or a 20% reduction in pressure is achieved.3
Indirect measurements also can be used to assess the portal pressure gradient. This procedure involves measurement of the free and wedged hepatic venous pressure using catheterization of the right hepatic vein. Wedged hepatic venous pressure (measured using a balloon-tipped catheter) reflects the pressure in a static column of blood from the hepatic vein to the sinusoid. It is an assessment of sinusoidal pressure rather than portal venous pressure and therefore may underestimate the portal pressure gradient in disease states characterized by pre-sinusoidal hypertension (see Table 96-1). The free hepatic venous pressure is obtained with the catheter in the hepatic vein and gives an assessment of caval pressure. Free hepatic venous pressure is not elevated in patients with diseases characterized by pre-sinusoidal and sinusoidal PHT, but it is characteristically raised in post-hepatic (or extrahepatic postsinusoidal) etiologies. The gradient between the two measurements is called the hepatic venous pressure gradient and is the most commonly quoted parameter in the medical literature regarding management of PHT. Both the absolute value of hepatic venous pressure gradient and the change in hepatic venous pressure gradient with pharmacotherapy have prognostic significance related to the risk of variceal bleeding.4
 Complications of Portal Hypertension
Complications of Portal Hypertension
Varices
Two main mechanisms have been implicated in the pathogenesis of variceal hemorrhage in patients with established varices and PHT: erosions secondary to acid reflux and spontaneous rupture. Effects related to ascites and changes in plasma volume also have been implicated in the genesis of bleeding. Ascites may be a factor in variceal hemorrhage, because ascites can transmit intraabdominal pressure and thereby increase the pressure inside the esophageal varices. Some studies have shown a decrease up to 10% in the hepatic vein pressure gradient (HVPG) and a decrease in portal flow with paracentesis. Drainage of large volume of ascites can decrease intraabdominal pressure, leading to splanchnic dilation and increased blood flow against a fixed resistance in the liver.5 This circumstance would increase portal pressure and, hence, the risk of bleeding. The pressure inside the varices does seem to be affected by intraabdominal pressure, but whether this affects the risk of bleeding is not known. Erosions secondary to esophagitis also have been suggested as an important factor for the bleeding process.6,7 However, there was no evidence of acid reflux in patients studied with a pH electrode, and treatment with cimetidine did not affect the rates of rebleeding from varices.8
The pressure inside the varices is directly dependent on the portal pressure and also on the radius of the varix. The pressure within the varix is inversely proportional to wall thickness. Therefore, varices are more likely to bleed when they are larger and have a thinner wall. Large, thin-walled varices are generally located near the gastroesophageal junction where the veins are more superficial and less surrounded by other tissues. There is a relationship between the risk of bleeding and portal pressure. If the HVPG is greater than 20 mm Hg after an initial bleed, it is a poor prognostic sign, and there is a substantial risk of rebleeding and mortality.9 In a recent study, patients underwent portal pressure measurement after initial endoscopy and control of bleeding. Those whose pressure was above 20 mm Hg were randomized to early transjugular intrahepatic portosystemic shunt (TIPS) or conventional treatment. The group who underwent TIPS shunt insertion had an improved outcome compared to the standard-of-care high-risk group and similar to that of the low-risk group. Recent work has also suggested that portal pressure may be equally predicted by Child-Pugh score, and thus in this group of patients, consideration should be given to early TIPS shunt insertion.
It may be equally possible to measure variceal pressure at the time of endoscopic band ligation. This appears to be feasible and safe, although it remains at present an experimental method, and more studies are required.10
Portal hypertensive gastropathy is a complication of PHT that causes flow and pressure changes in the gastric mucosa. Mild or chronic bleeding is observed in 35% of the patients with mild gastropathy and 90% of those with severe gastropathy. Overt bleeding happens in 30% of those with mild and in 60% of those with severe gastropathy.11 The administration of propranolol decreases gastric mucosal blood flow and is effective in reducing bleeding in portal hypertensive gastropathy.12,13 TIPS and transplant are effective modes of treatment.
Diagnosis of Variceal Bleeding
When a patient with a possible hepatic disorder presents with hematemesis or melena, the most common cause of bleeding is from varices.14 Sometimes it is useful to insert a nasogastric tube followed by lavage of the stomach, both as a diagnostic tool and also to clear the gastric cavity before endoscopy. Since patients with PHT also can bleed from gastritis, esophagitis, Mallory-Weiss tears, and peptic ulcers, the most accurate method for the diagnosis of bleeding varices is upper endoscopy; accuracy exceeds 90%. Frequently one or two doses of 250 mg of erythromycin are administered before the procedure to help clear the stomach before the procedure.
Acute Variceal Hemorrhage
Because of the high risk of infection in this population of patients, it is necessary to give prophylactic antibiotics. In a recent meta-analysis, treatment with antibiotics was associated with an increase in hospital survival and decrease in infection. The risk of rebleeding also was lower in patients receiving antibiotics.14,15 Oral norfloxacin, 400 mg twice a day; intravenous (IV) ciprofloxacin, 400 mg twice a day; or levofloxacin, 500 mg twice a day are the recommended antibiotics in 5-day courses.
The lack of tachycardia or hypotension in these patients is not indicative of stability, because up to 25% of blood volume may be lost without any hemodynamic changes.16 Blood volume should be restored, and coagulation factor support in the form of fresh frozen plasma and platelets may be required.
 Treatment
Treatment
Pharmacotherapy
Terlipressin (Glypressin) is a prodrug of vasopressin that has some intrinsic activity. It acts on vasopressin-1 receptors within arteriolar smooth muscle and induces vasoconstriction via phospholipase C–dependent signaling.17 Treatment with terlipressin results in splanchnic vasoconstriction and decreases splanchnic inflow, thereby reducing portal pressure. Terlipressin also reduces collateral blood flow and variceal pressure.18
Compared with vasopressin, terlipressin is associated with a lower incidence of systemic ischemic events, and unlike vasopressin, terlipressin can be used safely without coadministration of nitroglycerin or other organic nitrates. Terlipressin has a longer half-life than vasopressin and can be administered intermittently. A dose of 2 mg IV given four times daily is as effective as endoscopic sclerotherapy for achieving initial control of variceal bleeding and preventing early rebleeding.19 Terlipressin has been shown to decrease mortality and length of stay when administered to a high-risk population of patients presenting with acute upper GI hemorrhage.20
Terlipressin is well tolerated and has few side effects and may represent first-line treatment in acute hemorrhage until endoscopy can be performed in a controlled environment. In a recent meta-analysis, terlipressin was more effective than placebo but less effective than octreotide for controlling bleeding.21 Despite these findings, a recent study showed equivalence between terlipressin and octreotide.22,23
The duration of treatment should be governed by the clinical situation. After 48 hours of therapy, however, the dose should be tapered (initially halved), seeking to achieve a course of therapy lasting 6 to 7 days. A recent study compared endoscopic banding therapy and banding in addition to 5 days of terlipressin.24 Outcome was improved in the cohort that received combined therapy.
Treatment with somatostatin also may be considered. The dose of somatostatin is 250 µg as a bolus followed by 250 µg every hour as an infusion. The efficacy of somatostatin in the control of bleeding is not totally clear. In a recent meta-analysis of studies of somatostatin compared to control or no treatment, the use of somatostatin was associated with initial hemostasis but not with a decrease in mortality or rebleeding.25 The treatment effect of somatostatin amounted to lowering the transfusion requirement by 0.5 units of blood per patient. In view of these findings, somatostatin cannot be recommended as the first-line agent for the control of variceal bleeding.
The somatostatin analogue, octreotide, may act by blocking the acute rise in portal pressure associated with fluid resuscitation in the face of GI hemorrhage. Its use is associated with improved outcome after therapeutic endoscopy. Octreotide is a somatostatin analog that has much longer half-life and therefore can be given as a bolus or infusion. Octreotide acts by blocking the vasodilatory effects of glucagon and vasoactive intestinal peptide. The side-effect profile for octreotide is more favorable than the side-effect profiles for terlipressin or vasopressin. In a recent meta-analysis, octreotide was more effective than no treatment or vasopressin/terlipressin.18
In a recent study, vapreotide was given for 5 days to patients acutely bleeding from varices.26,27 A decrease in the number of bleedings during the index endoscopy and in the next 5 days was observed.
Therapeutic Endoscopy
The timing of endoscopy has recently been addressed; one study suggested that the determinants of outcome following acute variceal hemorrhage were “door-to-needle time” (threshold value: 15 hours) and Model for End-stage Liver Disease (MELD) score.28 A similar study from a Canadian group did not show any effect of door-to-needle time.29 However, in this study, hemodynamically unstable patients were excluded and underwent endoscopy within 4 hours. Determinants of outcome were infection on admission, albumin level, and MELD score.
Sclerotherapy
In the sclerotherapy approach, a sclerosant is injected directly into the varix. A variety of sclerosants are in use, but ethanolamine and sodium tetradecyl sulfate are the most common. The immediate effect of controlling bleeding is probably due to edema caused by the injection of the sclerosant; thrombosis occurs later. Injection sclerotherapy can be accompanied by complications (Table 96-2). The rate of mortality related to severe complications is approximately 15%. The most common long-term complication is esophageal stricture.
| Site | Complication |
|---|---|
| Local | Ulcers |
| Bleeding | |
| Stricture | |
| Esophageal dysmotility | |
| Regional | Perforation |
| Mediastinitis | |
| Pleural effusion | |
| Systemic | Sepsis |
| Aspiration |
Band Ligation Therapy
In band ligation therapy, a rubber band is placed on a variceal column that has been aspirated into a cylinder attached to the endoscope. The initial hemostatic effect is caused by strangulation of the vessel that is the source of variceal hemorrhage; later, thrombosis and ischemia result, leaving a shallow mucosal ulcer. Endoscopic band ligation is associated with fewer complications than endoscopic sclerotherapy, and systemic complications are rare.30
Although superficial ulceration is a side effect of endoscopic band ligation, stricture formation is rare. The most hazardous complication is rebleeding associated with early shedding of the band. In a recent meta-analysis, band ligation was as effective as sclerotherapy in the control of bleeding.31 But because it is associated with fewer side effects and better long-term control of the varices, band ligation is preferred. However, results from a recent study suggest that there is an increased risk of infections as the number of band ligations is greater.32
A recently published meta-analysis demonstrated that combined pharmacologic and endoscopic treatments is better than each of these treatments alone.33,34
Glue (Butyl Cyanoacrylate)
Results from a recent study of a small number of patients with decompensated liver disease and severe esophageal variceal hemorrhage suggest that injection of tissue glue rather than a sclerosant may result in improved initial hemostasis, reduced rebleeding, and improved survival.35,36 However, this approach requires further study and comparison with endoscopic band ligation and other therapies before it is universally adopted.
Esophageal Varices Versus Gastric Varices
Gastric varices can be subclassified according to their anatomic position, relationship to esophageal varices, and whether they are primary in origin or whether they develop as a result of obliteration of esophageal varices. Endoscopic management of bleeding gastric varices can be technically demanding. Recent evidence shows that cyanoacrylate is the treatment of choice for most gastric varices.37 Cyanoacrylate injection can cause complications such as embolic phenomena.
If treatment fails to control the bleeding, early TIPS should be considered. If the bleeding is not controlled acutely, temporary hemostasis may be achieved with placement of a tamponade-inducing device such as a Sengstaken-Blakemore tube or, more safely, a Minnesota balloon. Prior to placing this sort of device, the vast majority of patients should be endotracheally intubated and mechanically ventilated to protect the airway and prevent aspiration.8
 Failure of Therapy/Salvage
Failure of Therapy/Salvage
Therapy failure is defined as:
In 10% to 20% of patients, initial methods fail to control variceal bleeding.38 This group of patients is at high risk for having a poor outcome, as discussed later. Salvage therapy relies on other modalities for halting ongoing bleeding.
Mechanical Salvage Methods
The use of balloon tamponade to control variceal hemorrhage has decreased dramatically with the widespread use of vasoactive agents and therapeutic endoscopy. Nonetheless, balloon tamponade still has a role in the emergency management of uncontrollable bleeding from varices. Inflation of the gastric balloon results in tamponade of the varices, reduces blood flow into the plexus, and controls bleeding. The use of balloon tamponade effectively controls bleeding in 90% of patients.39
In the vast majority of cases, adequate control is achieved by inflation of the gastric balloon plus adequate traction without inflation of the esophageal balloon.40,41 It is rarely necessary to inflate the esophageal balloon, and it is important to appreciate that this maneuver contributes significantly to the incidence of potentially life-threatening complications. Constant pressure on the gastroesophageal junction is achieved with skin traction or fixed traction using a helmet.
In approximately 50% patients, bleeding recurs upon deflation of the gastric balloon.42,43 Potential complications associated with the use of compression devices include pulmonary aspiration, esophageal mucosal ischemia and ulceration, and misplacement of the device with gastric balloon inflation in the esophagus, leading to esophageal perforation.
A recent study proposes an alternative in the form of a self-expandable metal stent to compress the esophageal varices. Placement of this device does not require endoscopy. Experience with this device is limited, and therefore more studies are needed on this topic.44 In addition, it should be recognized that this option would not be effective if bleeding is originating from gastric varices.
Shunt Surgery/Interventional Radiology
Side-to-side portacaval shunt is an example of a total shunt that is achieved either by direct anastomosis of the portal vein to the inferior vena cava or anastomosis using a short interpositional graft. Traditionally, the graft diameter is greater than 12 mm, producing total portal decompression. This procedure controls variceal bleeding in 95% to 98% of patients and controls ascites in more than 90% of patients. The encephalopathy rate is 30% to 40%. If the diameter of the graft is reduced to 8 mm, this type of shunt is known as a partial shunt. It does not provide total portal decompression, thus the risk of rebleeding is higher, but rates of both encephalopathy and ascites/liver failure are lower.45,46
“Selective” shunts, such as the distal splenorenal shunt, aim to address the issue of portal flow diversion. The aim of this shunt is to decompress the gastroesophageal junction and the spleen through the splenic vein to the renal vein. PHT is thus maintained in the superior mesenteric and portal vein to maintain blood flow to the liver.47,48
TIPS achieves the same effect in terms of decompression of the portal system without the operative risk. Depending on the diameter of the intrahepatic shunt, TIPS can be viewed as either a total or a partial shunt. It can be used in the setting of refractory acute hemorrhage when both endoscopic and pharmacologic strategies have failed. Use of TIPS, however, is not clearly associated with a survival benefit. TIPS carries a higher risk of precipitating encephalopathy and is significantly more expensive than either endoscopic or pharmacologic strategies.49,50 The exact subgroup of patients for whom salvage TIPS leads to a favorable outcome has not been characterized.51,52
New radiologic methods have been developed that can be performed with fluoroscopy or even at the bedside of the patient in an intensive care environment. One such method is percutaneous transhepatic variceal embolization (PTVE) with 2-octyl cyanoacrylate (2-OCA). The effectiveness of PTVE with 2-OCA for controlling bleeding from esophageal varices is dependent upon the site and range of embolization. If the lower-esophageal and periesophageal varices and/or the cardial submucosal and perforating vessels are sufficiently obliterated, PTVE with 2-OCA can preventing variceal recurrence and rebleeding.53
Devascularization procedures combine components of splenectomy and gastric and esophageal devascularization. The aim of these procedures is to reduce inflow to variceal beds and thereby reduce the risk of bleeding. Because portal flow is maintained, the risk of encephalopathy is low (10%-15%). In patients with extensive portomesenteric venous occlusion or previous splenectomy, devascularization may offer an alternative decompressive strategy in selected cases when anatomic considerations make surgical or radiologic shunting impossible. Generally, it is felt that Child-Pugh B and C patients are likely to do less well with a surgical operative procedure than a TIPS shunt, owing to the risks of hepatic decompensation. Recent studies compared surgical and medical shunts and showed that outcomes were similar, although proper selection of patients was important.54,55
 Prognosis
Prognosis
The mortality rate following a variceal bleed is often quoted as in the range of 30% to 60%.56 Several reports of improved outcome since the introduction of therapeutic endoscopy may alter this estimate.57,58
The overall improvement in survival over the last 20 years is attributed to decreased early mortality, largely due to effective control of bleeding and prevention of rebleeding (due to treatment of the initial bleed, use of antibiotics, and secondary prophylaxis), rather than modification of the natural history of the disease. Survival increased both at 30 days and 6 years after hemorrhage in historical cohorts compared to those treated contemporaneously.59 A recent publication predicted 6-week outcome looking at the values of creatinine, Child-Pugh score, and number and type of infections.60
 Complications
Complications
Sepsis, Renal Failure, Multiple Organ Dysfunction Syndrome
Significantly, bacterial infection is associated with both an increase in failure to control bleeding and early rebleeding. Bacterial infection is associated with poor short-term prognosis. The use of broad-spectrum antibiotics after variceal hemorrhage has been shown to reduce the infection rate, decrease the rebleeding rate, and more importantly, improve early survival.61
Primary Prophylaxis
All cirrhotic patients should be screened for esophageal varices with endoscopy. If moderate to large varices are found, it is necessary to start prophylactic treatment with a nonselective β-adrenergic blocker such as propranolol or nadolol. The dose of the drug should be titrated to achieve a heart rate that is 25% lower than baseline, or 55 bpm. If the patient bleeds while on β-adrenergic blocker prophylaxis, band ligation should be performed while the patient continues to be on β-adrenergic blocker therapy to avoid bleeding from portal hypertensive gastropathy and to prolong the durability of the endoscopic treatment. If a patient is intolerant to β-adrenergic blocker therapy, band ligation should be performed as primary prophylaxis.62 There is marked up-regulation of the hepatic and mesenteric expression of β3-adrenergic receptors (ARs) in human cirrhosis and in two different animal models of cirrhosis. β3-AR-agonists should be further evaluated for therapy of PHT.63 A recent study has shown carvedilol to be as effective as propranolol in primary prophylaxis.64,65
Secondary Prophylaxis
After an initial variceal bleed, as many as 60% of untreated patients will bleed again. Rebleeding is most frequent in the 6 weeks following an index variceal bleed and is seen in up to 40% of patients.66 Risk factors for early rebleeding include age older than 60 years, high severity of initial bleed, renal failure, ascites, active bleeding on endoscopy, red signs, clot on varix, hypoalbuminemia, and hepatic venous pressure gradient greater than 20 mm Hg. The risk of late rebleeding is related to the severity of liver disease, endoscopic findings indicative of high risk of rebleeding, and continued alcohol intake, along with the poor prognostic indicators mentioned earlier.
Ascites
Accumulation of fluid in the peritoneal cavity is called ascites. The most common cause is cirrhosis.67 Ascites is present in 20% to 60% of cirrhotic patients at the time of presentation. Leakage of sinusoidal fluid in cirrhosis happens as a result of sinusoidal hypertension due to the regenerative nodules and surrounding fibrosis. The other factor related to the pathogenesis of ascites in cirrhosis is expansion of plasma volume as a consequence of excessive renal retention of salt and water. The symptoms are increased abdominal girth, weight gain, and frequently edema. Tense ascites can result in respiratory compromise due to diaphragmatic splinting and or hydrothorax. The best method for diagnosing ascites is abdominal ultrasound, which can detect as little as 100 mL of ascites. Ascites total protein and serum ascites albumin gradient (SAAG) are important in determining the etiology of the ascites. If total protein concentration in ascitic fluid is greater than 25 g/dL, the diagnosis is likely malignancy, tuberculosis, or a post-sinusoidal form of PHT such as Budd-Chiari syndrome. In these cases, the SAAG will be less than 1 : 1. If the SAAG is more than 1 : 1 is and the total protein concentration in ascetic fluid is less than 2.5 g/dL, then the most likely diagnosis is cirrhosis.
Abdominal Compartment Syndrome
Large-volume paracentesis puts patients at risk of further central volume depletion and, hence, cardiovascular and renal dysfunction. It is normal to administer 20% albumin IV to prevent these complications. Most papers suggest that removing less than 5 L of ascites will not put the patient at risk of a post paracentesis cardiovascular disfunction syndrome; however, most physicians administer a vial of 100 mL of 20% albumin for every 2 L evacuated.68
Spontaneous Bacterial Peritonitis
Spontaneous bacterial peritonitis (SBE) is the development of infection in ascites in the absence of an obvious source of infection (intestinal perforation or abdominal abscess) or another site of inflammation, such as cholecystitis or pancreatitis. The most common pathogens are Escherichia coli, Streptococcus pneumonia, and Klebsiella spp., although there has been an increase in other gram-positive organisms in the last decade, possibly related to the increased use of norfloxacin in the community setting as primary prophylaxis to decrease the incidence of bacterial peritonitis.69 Recent data suggest that some genetic variants of NOD2 are related not only to the development of SBP but also with risk of death.70
SBE is the most common type of infection in cirrhotic patients and accounts for about 25% of all infections.71 Patients may present with signs of generalized peritonitis (diffuse pain, abdominal tenderness, fever, decreased bowel sounds), however, the clinical picture may be very mild, and a high level of suspicion is needed in any patient with cirrhosis who presents unwell with ascites. A diagnostic paracentesis should be performed in all patients with suspected SBP and also in those admitted to the hospital for the first time with ascites and in those presenting with encephalopathy or renal failure.
 Other Complications of Portal Hypertension Syndrome
Other Complications of Portal Hypertension Syndrome
Hepatorenal Syndrome
Hepatorenal syndrome (HRS) is a clinical condition that appears in patients with advanced chronic liver disease, impaired renal function, and abnormalities in the renal circulation. HRS is characterized by renal vasoconstriction and decreased glomerular filtration rate. At the same time, marked arterial vasodilatation is apparent in the systemic (extrarenal) circulation.72
HRS usually occurs in patients with advanced cirrhosis, although it can also be seen in other situations such as acute liver failure or severe alcoholic hepatitis. There are two types of HRS: type I, associated with a worse prognosis and generally rapidly progressive; and type II, defined as impairment in renal function that does not meet criteria for type I and probably involves several different types of renal injury. HRS is characterized by oliguria, a rapid and progressive rise in serum creatinine concentration in less than 2 weeks, and urinary sodium of less than 10 mEq/L. It is also necessary to differentiate HRS from the more common prerenal azotemia. Whereas prerenal kidney dysfunction responds to intravascular volume expansion, HRS does not respond to IV fluid administration or the removal of diuretics. In addition, renal causes of renal dysfunction should be excluded by urinalysis, imaging, blood tests, and if necessary, renal biopsy (usually via the transjugular route). Venovenous hemofiltration as well as terlipressin or noradrenaline are beneficial in some cases.73 In a recent meta-analysis, administration of terlipressin and albumin increased short-term survival in HRS type I, but there is a lack of data to provide clear recommendations for HRS type 2.74 Recently, a study has shown that a positive response to midodrine, octreotide, and albumin can select a population of patients whose renal function can respond completely to TIPS as a second-line treatment.75
Hepatic Encephalopathy
Hepatic encephalopathy (HE) is a neuropsychiatric syndrome in patients with liver disease and/or major portosystemic shunting. The classic definition of Adams and Foley76,77 led to several different definitions of HE. Currently, three different types of HE are recognized. Type A (A is for acute) refers to HE seen in acute liver failure. In Type A HE, cerebral edema is almost always present. Cerebral edema can lead to intracranial hypertension and its associated complications. Type B (B is for bypass) appears in patients with significant portosystemic shunts without intrinsic liver disease and is very rare. Finally, type C (C is for chronic or cirrhosis) is seen in patients with chronic liver disease and PHT. In these patients, many of the products that normally are filtered and eliminated by the liver are delivered to the systemic circulation and, hence, the brain. There are several hypotheses for the pathogenesis of HE, including excessive production of ammonia, systemic inflammation, high levels of the neurotransmitter, gamma-aminobutyric acid (GABA), false neurotransmitters, and endogenous benzodiazepines.78,79 In patients presenting with HE, possible triggering factors should be identified and treated. These potential triggering factors include infection, bleeding, constipation, and electrolyte and acid-base abnormalities. There are five grades of HE. Grade 0, or subclinical, can only be detected with psychometric tests. In grade 1, the patient is euphoric or depressed and has sleep pattern alterations, frequently associated with vivid nightmares. In grade 2, the patient tends to sleep but is easily arousable, while in grade 3, calling vigorously or inflicting pain are needed to wake the patient. In grade 4, the patient is in a coma, and diagnosis is based on the previous medical history of the patient, physical examination that can show extrapyramidal signs such as rigidity of the limbs or clonus, and the electroencephalogram, which will show triphasic delta waves in the frontal lobe. Treatment is based on avoidance and prevention of precipitating factors and in improving protein intake by feeding dairy products and vegetable-based diets. Laxatives in the form of lactulose or other disaccharides may be used, aiming for two to three soft bowel movements per day. Antibiotics are reserved for patients who respond poorly to disaccharides. Rifaximin recently has been proposed in this context, and it appears to be an effective and safe treatment option for HE.80 Artificial liver support devices, specifically the MARS device, have been shown to result in more rapid resolution of HE.81 L-Ornithine L-aspartate (LOLA) also may ameliorate encephalopathy,82 although further studies are required. A recent randomized controlled study in acute liver failure was not able to demonstrate any improvement in HE or survival in the LOLA-treated population.83 The definitive treatment for HE is liver transplantation.84,85
Garcia-Tsao G, Groszmann RJ, Fisher RL, et al. Portal pressure, presence of gastroesophageal varices and variceal bleeding. Hepatology. 1985;5:419-424.
Gonzalez R, Zamora J, Gomez-Camarero J, Molinero LM, Bañares R, Albillos A. Meta-analysis: combination endoscopic and drug therapy to prevent variceal rebleeding in cirrhosis. Ann Intern Med. 2008;149:109-122.
Boyer TD, ZJ Haskal. American Association for the Study of Liver Diseases. The role of transjugular intrahepatic portosystemic shunt (TIPS) in the management of portal hypertension: update 2009. Hepatology. 2010;51:306.
Lee SW, Lee TY, Chang CS. Independent factors associated with recurrent bleeding in cirrhotic patients with esophageal variceal hemorrhage. Dig Dis Sci. 2009;54:1128-1134.
Bernard B, Grangé JD, Khac EN, Amiot X, Opolon P, Poynard T. Antibiotic prophylaxis for the prevention of bacterial infection in cirrhotic patients with gastrointestinal bleeding: a meta-analysis. Hepatology. 1999;29:1655-1661.
Davenport A. Management of acute kidney injury in liver disease. Contrib Nephrol. 2010;165:197-205.
Blei AT, J Córdoba. Practice Parameters Committee of the American College of Gastroenterology. Hepatic encephalopathy. Am J Gastroenterol. 2001;96:1968-1976.
A review of the current recommendations for management of hepatic encephalopathy.
1 Heller J, Sogni P, Tazi KA, Chagneau C, Poirel O, Moreau R, et al. Abnormal regulation of aortic NOS2 and NOS3 activity and expression from portal vein-stenosed rats after lipopolysaccharide administration. Hepatology. 1999;30(3):698.
2 Escorsell A, Ginès A, Llach J, García-Pagán JC, Bordas JM, Bosch J, et al. Increasing intra-abdominal pressure increases pressure, volume, and wall tension in esophageal varices. Hepatology. 2002;36(4 Pt 1):936.
3 Garcia-Tsao G, Groszmann RJ, Fisher RL, et al. Portal pressure, presence of gastroesophageal varices and variceal bleeding. Hepatology. 1985;5:419.
4 Feu F, Garcia-Pagan JC, Bosch J, et al. Relationship between portal pressure response and risk of recurrent variceal haemorrhage in patients with cirrhosis. Lancet. 1995;346:1056.
5 Ruiz-del-Arbol L, Monescillo A, Jimenéz W, Garcia-Plaza A, Arroyo V, Rodés J. Paracentesis-induced circulatory dysfunction: mechanism and effect on hepatic hemodynamics in cirrhosis. Gastroenterology. 1997;113(2):579.
6 Eckardt VF, Grace ND, Kantrowitz PA. Does lower esophageal sphincter incompetency contribute to esophageal bleeding? Gastroenterology. 1976;71(2):185.
7 Eckardt VF, Grace ND. Gastroesophageal reflux and bleeding esophageal varices. Gastroenterology. 1979;76(1):39.
8 Macdougall BR, Williams R. A controlled clinical trial of cimetidine in the recurrence of variceal hemorrhage: implications about the pathogenesis of hemorrhage. Hepatology. 1983;3(1):69.
9 D’Amico G, De Franchis R, Cooperative Study Group. Upper digestive bleeding in cirrhosis. Post-therapeutic outcome and prognostic indicators. Hepatology. 2003;38(3):599.
10 Liao WC, Hou MC, Lin HC, Tan PC, Liao TM, Li HS. Feasibility of needle puncture measurement of variceal pressure in patients undergoing endoscopic variceal ligation. Hepatogastroenterology. 2009;56(90):398.
11 Primignani M, Carpinelli L, Preatoni P, Battaglia G, Carta A, Prada A, et al. Natural history of portal hypertensive gastropathy in patients with liver cirrhosis. The New Italian Endoscopic Club for the study and treatment of esophageal varices (NIEC). Gastroenterology. 2000;119(1):181.
12 Pérez-Ayuso RM, Piqué JM, Bosch J, Panés J, González A, Pérez R, et al. Propranolol in prevention of recurrent bleeding from severe portal hypertensive gastropathy in cirrhosis. Lancet. 1991;337(8755):1431.
13 Panés J, Bordas JM, Piqué JM, García-Pagán JC, Feu F, Terés J, et al. Effects of propranolol on gastric mucosal perfusion in cirrhotic patients with portal hypertensive gastropathy. Hepatology. 1993;17(2):213.
14 Chalasani N, Imperiale TF, Ismail A, Sood G, Carey M, Wilcox CM, et al. Predictors of large esophageal varices in patients with cirrhosis. Am J Gastroenterol. 1999;94(11):3285.
15 Soares-Weiser K, Brezis M, Tur-Kaspa R, Paul M, Yahav J, et al. Antibiotic prophylaxis of bacterial infections in cirrhotic inpatients: a meta-analysis of randomized controlled trials. Scand J Gastroenterol. 2003;38(2):193.
16 Boyer TD. Portal hypertensive hemorrhage: pathogenesis and risk factors. Semin Gastrointest Dis. 1995;6(3):125.
17 Grillone LR, Clark MA, Godfrey RW, Stassen F, Crooke ST. Vasopressin induces V1 receptors to activate phosphatidylinositol- and phosphatidylcholine-specific phospholipase C and stimulates the release of arachidonic acid by at least two pathways in the smooth muscle cell line, A-10. J Biol Chem. 1988;263(6):2658.
18 Nevens F. Review article: a critical comparison of drug therapies in currently used therapeutic strategies for variceal haemorrhage. Aliment Pharmacol Ther. 2004;20(Suppl 3):18.
19 Escorsell A, Ruiz Del Arbol L, Planas R, et al. Multicenter randomized controlled trial of terlipressin versus sclerotherapy in the treatment of acute variceal bleeding: The TEST Study. Hepatology. 2000;32:471.
20 Ioannou GN, Doust J, Rockey DC. Systematic review: terlipressin in acute oesophageal variceal haemorrhage. Aliment Pharmacol Ther. 2003;17(1):53.
21 Abid S, Jafri W, Hamid S, Salih M, Azam Z, Mumtaz K, et al. Terlipressin vs. octreotide in bleeding esophageal varices as an adjuvant therapy with endoscopic band ligation: a randomized double-blind placebo-controlled trial. Am J Gastroenterol. 2009;104(3):617.
22 Pedretti G, Elia G, Calzetti C, Magnani G, Fiaccadori F. Octreotide versus terlipressin in acute variceal hemorrhage in liver cirrhosis. Emergency control and prevention of early rebleeding. Clin Invest. 1994;72(9):653.
23 Lo GH, Chen WC, Wang HM, Lin CK, Chan HH, Tsai WL, et al. Low-dose terlipressin plus banding ligation versus low-dose terlipressin alone in the prevention of very early rebleeding of oesophageal varices. Gut. 2009;58(9):1275.
24 Gøtzsche PC, Hróbjartsson A. Somatostatin analogues for acute bleeding oesophageal varices. Cochrane Database Syst Rev. 2005;(1):CD000193.
25 Corley DA, Cello JP, Adkisson W, Ko WF, Kerlikowske K. Octreotide for acute esophageal variceal bleeding: a meta-analysis. Gastroenterology. 2001;120(4):946.
26 Calès P, Masliah C, Bernard B, Garnier PP, Silvain C, Szostak-Talbodec N, et al. Early administration of vapreotide for variceal bleeding in patients with cirrhosis. N Engl J Med. 2001;344(1):23.
27 Fortune BE, Jackson J, Leonard J, Trotter JF. Vapreotide: a somatostatin analog for the treatment of acute variceal bleeding. Expert Opin Pharmacother. 2009;10(14):2337.
28 Hsu YC, Chung CS, Tseng CH, Lin TL, Liou JM, Wu MS, et al. Delayed endoscopy as a risk factor for in-hospital mortality in cirrhotic patients with acute variceal hemorrhage. J Gastroenterol Hepatol. 2009;24(7):1294.
29 Cheung J, Soo I, Bastiampillai R, Zhu Q, Ma M. Urgent vs. non-urgent endoscopy in stable acute variceal bleeding. Am J Gastroenterol. 2009;104(5):1125.
30 Laine L, Cook D. Endoscopic ligation compared with sclerotherapy for treatment of esophageal variceal bleeding. A meta-analysis. Ann Intern Med. 1995;123(4):280.
31 Nevens F. Review article: a critical comparison of drug therapies in currently used therapeutic strategies for variceal haemorrhage. Aliment Pharmacol Ther. 2004;20(Suppl 3):18.
32 Lee SW, Lee TY, Chang CS. Independent factors associated with recurrent bleeding in cirrhotic patients with esophageal variceal hemorrhage. Dig Dis Sci. 2009;54(5):1128.
33 Gonzalez R, Zamora J, Gomez-Camarero J, Molinero LM, Bañares R, Albillos A. Meta-analysis: Combination endoscopic and drug therapy to prevent variceal rebleeding in cirrhosis. Ann Intern Med. 2008;149(2):109.
34 GH Lo. Combination of endoscopic band ligation with terlipressin is better than that with octreotide in shortening hospital stay? Am J Gastroenterol. 2009;104(11):2855.
35 Maluf-Filho F, Sakai P, Ishioka S, Matuguma SE. Endoscopic sclerosis versus cyanoacrylate endoscopic injection for the first episode of variceal bleeding: a prospective, controlled, and randomized study in Child-Pugh class C patients. Endoscopy. 2001;33(5):421.
36 Cipolletta L, Zambelli A, Bianco MA, De Grazia F, Meucci C, Lupinacci G, et al. Acrylate glue injection for acutely bleeding oesophageal varices: A prospective cohort study. Dig Liver Dis. 2009;41(10):729.
37 Tripathi D. Therapies for bleeding gastric varices: is the fog starting to clear? Gastrointest Endosc. 2009;70(5):888.
38 Dagradi AE. The natural history of esophageal varices in patients with alcoholic liver disease: An endoscopic and clinical study. Am J Gastroenterol. 1972;57:520.
39 Panés J, Terés J, Bosch J, Rodés J. Efficacy of balloon tamponade in treatment of bleeding gastric and esophageal varices. Results in 151 consecutive episodes. Dig Dis Sci. 1988;33(4):454.
40 Sarin SK, Nundy S. Balloon tamponade in the management of bleeding oesophageal varices. Ann R Coll Surg Engl. 1984;66(1):30.
41 Agger P, Andersen JR, Burcharth F. Does the oesophageal balloon compress oesophageal varices? Scand J Gastroenterol. 1978;13(2):225.
42 Panés J, Terés J, Bosch J, Rodés J. Efficacy of balloon tamponade in treatment of bleeding gastric and esophageal varices. Results in 151 consecutive episodes. Dig Dis Sci. 1988;33(4):454.
43 Paquet KJ, Feussner H. Endoscopic sclerosis and esophageal balloon tamponade in acute hemorrhage from esophagogastric varices: a prospective controlled randomized trial. Hepatology. 1985;5(4):580.
44 Wright G, Lewis H, Hogan B, Burroughs A, Patch D, O’Beirne J. A self-expanding metal stent for complicated variceal hemorrhage: experience at a single center. Gastrointest Endosc. 2010;71(1):71.
45 Stipa S, Balducci G, Ziparo V, Stipa F. Lucandri GTotal shunting and elective management of variceal bleeding. World J Surg. 1994;18(2):200.
46 Busuttil RW. Selective and nonselective shunts for variceal bleeding. A prospective study of 103 patients. Am J Surg. 1984;148(1):27.
47 Warren WD, Zeppa R, Fomon JJ. Selective trans-splenic decompression of gastroesophageal varices by distal splenorenal shunt. Ann Surg. 1992;216(3):248.
48 Henderson JM, Gilmore GT, Hooks MA, Galloway JR, Dodson TF, Hood MM, et al. Selective shunt in the management of variceal bleeding in the era of liver transplantation. Ann Surg. 1967;166(3):437.
49 Meddi P, Merli M, Lionetti R, De Santis A, Valeriano V, Masini A, et al. Cost analysis for the prevention of variceal rebleeding: a comparison between transjugular intrahepatic portosystemic shunt and endoscopic sclerotherapy in a selected group of Italian cirrhotic patients. Hepatology. 1999;29(4):1074.
50 Papatheodoridis GV, Goulis J, Leandro G, Patch D, Burroughs AK. Transjugular intrahepatic portosystemic shunt compared with endoscopic treatment for prevention of variceal rebleeding: A meta-analysis. Hepatology. 1999;30(3):612.
51 Riggio O, Ridola L, Lucidi C, Angeloni S. Emerging issues in the use of transjugular intrahepatic portosystemic shunt (TIPS) for management of portal hypertension: Time to update the guidelines? Dig Liver Dis. 2010;42(7):462.
52 Boyer TD, ZJ Haskal. American Association for the Study of Liver Diseases. The role of transjugular intrahepatic portosystemic shunt (TIPS) in the management of portal hypertension: update 2009. Hepatology. 2010;51(1):306.
53 Zhang CQ, Liu FL, Liang B, Xu HW, Xu L, Feng K, et al. A modified percutaneous transhepatic varices embolization with 2-octyl cyanoacrylate in the treatment of bleeding esophageal varices. J Clin Gastroenterol. 2009;43(5):463.
54 Bajaj JS, Ananthakrishnan A, Saeian K. Survey of attitudes of AASLD members toward balloon tamponade. Hepatology. 2005;41(6):1435.
55 Henderson JM, Boyer TD, Kutner MH, Galloway JR, Rikkers LF, Jeffers LJ, et al. Distal splenorenal shunt versus transjugular intrahepatic portal systematic shunt for variceal bleeding: a randomized trial. Gastroenterology. 2006;130(6):1643.
56 Altamirano J, Zapata L, Agustin S, Muntaner L, González-Angulo A, Ortiz AL, et al. Predicting 6-week mortality after acute variceal bleeding: role of classification and regression tree analysis. Ann Hepatol. 2009;8(4):308.
57 Chalasani N, Kahi C, Francois F, Pinto A, Marathe A, Bini EJ. Improved patient survival after acute variceal bleeding: a multicenter, cohort study. Am J Gastroenterol. 2003;98(3):653.
58 Carbonell N, Pauwels A, Serfaty L, Fourdan O, Lévy VG, Poupon R. Improved survival after variceal bleeding in patients with cirrhosis over the past two decades. Hepatology. 2004;40(3):652.
59 El-Serag HB, Everhart JE. Improved survival after variceal hemorrhage over an 11-year period in the Department of Veterans Affairs. Am J Gastroenterol. 2000;95(12):3566.
60 Augustin S, Muntaner L, Altamirano JT, González A, Saperas E, Dot J, et al. Predicting early mortality after acute variceal hemorrhage based on classification and regression tree analysis. Clin Gastroenterol Hepatol. 2009;7(12):1347.
61 Bernard B, Grange JD, Khac EN, et al. Antibiotic prophylaxis for the prevention of bacterial infection in cirrhotic patients with GI bleeding: A meta-analysis. Hepatology. 1999;29:1655-1661.
62 Köklü S, Yuksel O, Coban S, Basar O. Is endoscopic band ligation necessary for primary prophylaxis of esophageal varices without high bleeding risks? Hepatology. 2009;50(6):2052.
63 Trebicka J, Hennenberg M, Schulze Pröbsting A, Laleman W, Klein S, Granzow M. Role of beta3-adrenoceptors for intrahepatic resistance and portal hypertension in liver cirrhosis. Hepatology. 2009;50(6):1924-1935.
64 Hemstreet BA. Evaluation of carvedilol for the treatment of portal hypertension. Pharmacotherapy. 2004;24(1):94-104.
65 Tsochatzis EA, Triantos CK, Burroughs AK. Gastrointestinal bleeding: Carvedilol-the best beta-blocker for primary prophylaxis? Nat Rev Gastroenterol Hepatol. 2009;6(12):692-694.
66 D’Amico G, Pagliaro L, Bosch J. Pharmacological treatment of portal hypertension: An evidence based approach. Semin Liver Dis. 1999;19:475-505.
67 Khan J, Pikkarainen P, Karvonen AL, Mäkelä T, Peräaho M, Pehkonen E, Collin P. Ascites: aetiology, mortality and the prevalence of spontaneous bacterial peritonitis. Scand J Gastroenterol. 2009;44(8):970-974.
68 BA Runyon. Practice Guidelines Committee, American Association for the Study of Liver Diseases (AASLD). Management of adult patients with ascites due to cirrhosis. Hepatology. 2004;39(3):841-856.
69 Rogers GB, Russell LE, Preston PG, Marsh P, Collins JE, Saunders J. Characterisation of bacteria in ascites–reporting the potential of culture-independent, molecular analysis. Eur J Clin Microbiol Infect Dis. 2010;29(5):533.
70 Appenrodt B, Grünhage F, Gentemann MG, Thyssen L, Sauerbruch T, Lammert F. Nucleotide-binding oligomerization domain containing 2 (NOD2) variants are genetic risk factors for death and spontaneous bacterial peritonitis in liver cirrhosis. Hepatology. 2010;51(4):1327.
71 Fernández J, Navasa M, Gómez J, Colmenero J, Vila J, Arroyo V, Rodés J. Bacterial infections in cirrhosis: epidemiological changes with invasive procedures and norfloxacin prophylaxis. Hepatology. 2002;35(1):140.
72 Carl DE, Sanyal A. The management of hepatorenal syndrome. Minerva Gastroenterol Dietol. 2009;55(2):207.
73 Davenport A. Management of acute kidney injury in liver disease. Contrib Nephrol. 2010;165:197.
74 Gluud LL, Christensen K, Christensen E, Krag A. Systematic review of randomized trials on vasoconstrictor drugs for hepatorenal syndrome. Hepatology. 2010;51(2):576.
75 Wong F, Pantea L, Sniderman K. Midodrine, octreotide, albumin, and TIPS in selected patients with cirrhosis and type 1 hepatorenal syndrome. Hepatology. 2004;40(1):55.
76 Adams RD, Foley JM. The neurological disorder associated with liver disease. Res Publ Assoc Res Nerv Ment Dis. 1953;32:198.
77 Foley JM, Watson CW, Adams RD. Significance of the electroencephalographic changes in hepatic coma. Trans Am Neurol Assoc. 1950;51:161-165.
78 Quero Guillén JC, Carmona Soria I, García Montes JM, Jiménez Sáenz M, Herrerías Gutiérrez JM. Hepatic encephalopathy: nomenclature, pathogenesis and treatment. Rev Esp Enferm Dig. 2003;95(2):135-142. 127
79 Sundaram V, Shaikh OS. Hepatic encephalopathy: pathophysiology and emerging therapies. Med Clin North Am. 2009;93(4):819.
80 Maclayton DO, Eaton-Maxwell A. Rifaximin for treatment of hepatic encephalopathy. Ann Pharmacother. 2009;43(1):77.
81 Hassanein TI, Tofteng F, Brown RSJr, McGuire B, Lynch P, Mehta R, et al. Randomized controlled study of extracorporeal albumin dialysis for hepatic encephalopathy in advanced cirrhosis. Hepatology. 2007;46(6):1853.
82 Schmid M, Peck-Radosavljevic M, König F, Mittermaier C, Gangl A, Ferenci P. A double-blind, randomized, placebo-controlled trial of intravenous L-ornithine-L-aspartate on postural control in patients with cirrhosis. Liver Int. 2010;30(4):574.
83 Acharya SK, Bhatia V, Sreenivas V, Khanal S, Panda SK. Efficacy of L-ornithine-L-aspartate in acute liver failure: a double-blind, randomized, placebo-controlled study. Gastroenterology. 2009;136(7):2159.
84 Romero-Gómez M. Pharmacotherapy of hepatic encephalopathy in cirrhosis. Expert Opin Pharmacother. 2010;11(8):1317.
85 Blei AT, Córdoba J. Practice Parameters Committee of the American College of Gastroenterology. Hepatic encephalopathy. Am J Gastroenterol. 2001;96(7):1968.
