[level-membership-for-internal-medicine-category]
UNSTABLE HEMOGLOBINS
Amino acid substitutions that reduce solubility or increase susceptibility to oxidation result in unstable hemoglobins that precipitate, forming inclusion bodies injurious to the RBC membrane. Representative mutations are those that interfere with contact points between the α and β subunits (e.g., Hb Philly [β35Tyr→Phe]), alter the helical segments (e.g., Hb Genova [β28Leu→Pro]), or disrupt interactions of the hydrophobic pockets of the globin subunits with heme (e.g., Hb Köln [β98Val→Met]) (Table 127-3). The inclusions, called Heinz bodies, are clinically detectable by staining with supravital dyes such as crystal violet. Removal of these inclusions by the spleen generates pitted, rigid cells that have shortened life spans, producing hemolytic anemia of variable severity, sometimes requiring chronic transfusion support. Splenectomy may be needed to correct the anemia. Leg ulcers and premature gallbladder disease due to bilirubin loading are frequent stigmata.
|
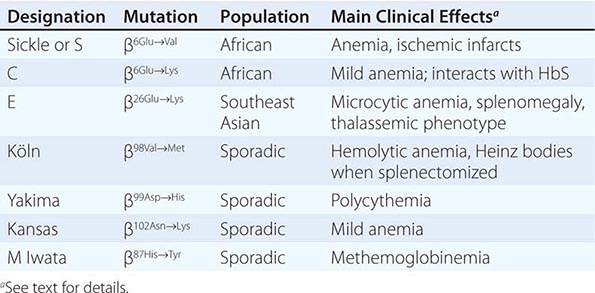
REPRESENTATIVE ABNORMAL HEMOGLOBINS WITH ALTERED SYNTHESIS OR FUNCTION |
Unstable hemoglobins occur sporadically, often by spontaneous new mutations. Heterozygotes are often symptomatic because a significant Heinz body burden can develop even when the unstable variant accounts for only a portion of the total hemoglobin. Symptomatic unstable hemoglobins tend to be β-globin variants, because sporadic mutations affecting only one of the four α globins alleles would generate only 20–30% abnormal hemoglobin.
HEMOGLOBINS WITH ALTERED OXYGEN AFFINITY
High-affinity hemoglobins (e.g., Hb Yakima [β99Asp→His]) bind oxygen more readily but deliver less O2 to tissues at normal capillary Po2 levels (Fig. 127-2). Mild tissue hypoxia ensues, stimulating RBC production and erythrocytosis (Table 127-3). In extreme cases, the hematocrits can rise to 60–65%, increasing blood viscosity and producing typical symptoms (headache, somnolence, or dizziness). Phlebotomy may be required. Typical mutations alter interactions within the heme pocket or disrupt the Bohr effect or salt-bond site. Mutations that impair the interaction of HbA with 2,3-BPG can increase O2 affinity because 2,3-BPG binding lowers O2 affinity.
Low-affinity hemoglobins (e.g., Hb Kansas [β102Asn→Lys]) bind sufficient oxygen in the lungs, despite their lower oxygen affinity, to achieve nearly full saturation. At capillary oxygen tensions, they lose sufficient amounts of oxygen to maintain homeostasis at a low hematocrit (Fig. 127-2) (pseudoanemia). Capillary hemoglobin desaturation can also be sufficient to produce clinically apparent cyanosis. Despite these findings, patients usually require no specific treatment.
METHEMOGLOBINEMIAS
Methemoglobin is generated by oxidation of the heme iron moieties to the ferric state, causing a characteristic bluish-brown muddy color resembling cyanosis. Methemoglobin has such high oxygen affinity that virtually no oxygen is delivered. Levels >50–60% are often fatal.
Congenital methemoglobinemia arises from globin mutations that stabilize iron in the ferric state (e.g., HbM Iwata [α87His→Tyr], Table 127-3) or from mutations that impair the enzymes that reduce methemoglobin to hemoglobin (e.g., methemoglobin reductase, NADP diaphorase). Acquired methemoglobinemia is caused by toxins that oxidize heme iron, notably nitrate and nitrite-containing compounds, including drugs commonly used in cardiology and anesthesiology.
DIAGNOSIS AND MANAGEMENT OF PATIENTS WITH UNSTABLE HEMOGLOBINS, HIGH-AFFINITY HEMOGLOBINS, AND METHEMOGLOBINEMIA
Unstable hemoglobin variants should be suspected in patients with nonimmune hemolytic anemia, jaundice, splenomegaly, or premature biliary tract disease. Severe hemolysis usually presents during infancy as neonatal jaundice or anemia. Milder cases may present in adult life with anemia or only as unexplained reticulocytosis, hepatosplenomegaly, premature biliary tract disease, or leg ulcers. Because spontaneous mutation is common, family history of anemia may be absent. The peripheral blood smear often shows anisocytosis, abundant cells with punctate inclusions, and irregular shapes (i.e., poikilocytosis).
The two best tests for diagnosing unstable hemoglobins are the Heinz body preparation and the isopropanol or heat stability test. Many unstable Hb variants are electrophoretically silent. A normal electrophoresis does not rule out the diagnosis. Mass spectroscopy or direct gene analysis will provide a definitive diagnosis.
Severely affected patients may require transfusion support for the first 3 years of life, because splenectomy before age 3 is associated with a significantly higher immune deficit. Splenectomy is usually effective thereafter, but occasional patients may require lifelong transfusion support. After splenectomy, patients can develop cholelithiasis and leg ulcers, hypercoagulable states, and susceptibility to overwhelming sepsis. Splenectomy should thus be avoided or delayed unless it is the only alternative. Precipitation of unstable hemoglobins is aggravated by oxidative stress, e.g., infection and antimalarial drugs, which should be avoided where possible.
High-O2 affinity hemoglobin variants should be suspected in patients with erythrocytosis. The best test for confirmation is measurement of the P50. A high-O2 affinity hemoglobin causes a significant left shift (i.e., lower numeric value of the P50); confounding conditions, e.g., tobacco smoking or carbon monoxide exposure, can also lower the P50.
High-affinity hemoglobins are often asymptomatic; rubor or plethora may be telltale signs. When the hematocrit approaches 60%, symptoms of high blood viscosity and sluggish flow (headache, lethargy, dizziness, etc.) may be present. These persons may benefit from judicious phlebotomy. Erythrocytosis represents an appropriate attempt to compensate for the impaired oxygen delivery by the abnormal variant. Overzealous phlebotomy may stimulate increased erythropoiesis or aggravate symptoms by thwarting this compensatory mechanism. The guiding principle of phlebotomy should be to improve oxygen delivery by reducing blood viscosity and increasing blood flow rather than restoration of a normal hematocrit. Phlebotomy-induced modest iron deficiency may aid in control.
Low-affinity hemoglobins should be considered in patients with cyanosis or a low hematocrit with no other reason apparent after thorough evaluation. The P50 test confirms the diagnosis. Counseling and reassurance are the interventions of choice.
Methemoglobin should be suspected in patients with hypoxic symptoms who appear cyanotic but have a Pao2 sufficiently high that hemoglobin should be fully saturated with oxygen. A history of nitrite or other oxidant ingestions may not always be available; some exposures may be inapparent to the patient, and others may result from suicide attempts. The characteristic muddy appearance of freshly drawn blood can be a critical clue. The best diagnostic test is methemoglobin assay, which is usually available on an emergency basis.
Methemoglobinemia often causes symptoms of cerebral ischemia at levels >15%; levels >60% are usually lethal. Intravenous injection of 1 mg/kg of methylene blue is effective emergency therapy. Milder cases and follow-up of severe cases can be treated orally with methylene blue (60 mg three to four times each day) or ascorbic acid (300–600 mg/d).
THALASSEMIA SYNDROMES
The thalassemia syndromes are inherited disorders of α- or β-globin biosynthesis. The reduced supply of globin diminishes production of hemoglobin tetramers, causing hypochromia and microcytosis. Unbalanced accumulation of α and β subunits occurs because the synthesis of the unaffected globins proceeds at a normal rate. Unbalanced chain accumulation dominates the clinical phenotype. Clinical severity varies widely, depending on the degree to which the synthesis of the affected globin is impaired, altered synthesis of other globin chains, and coinheritance of other abnormal globin alleles.
CLINICAL MANIFESTATIONS OF β THALASSEMIA SYNDROMES
Mutations causing thalassemia can affect any step in the pathway of globin gene expression: transcription, processing of the mRNA precursor, translation, and posttranslational metabolism of the β-globin polypeptide chain. The most common forms arise from mutations that derange splicing of the mRNA precursor or prematurely terminate translation of the mRNA.
Hypochromia and microcytosis characterize all forms of β thalassemia because of the reduced amounts of hemoglobin tetramers (Fig. 127-5). In heterozygotes (β thalassemia trait), this is the only abnormality seen. Anemia is minimal. In more severe homozygous states, unbalanced α- and β-globin accumulation causes accumulation of highly insoluble unpaired α chains. They form toxic inclusion bodies that kill developing erythroblasts in the marrow. Few of the proerythroblasts beginning erythroid maturation survive. The surviving RBCs bear a burden of inclusion bodies that are detected in the spleen, shortening the RBC life span and producing severe hemolytic anemia. The resulting profound anemia stimulates erythropoietin release and compensatory erythroid hyperplasia, but the marrow response is sabotaged by the ineffective erythropoiesis. Anemia persists. Erythroid hyperplasia can become exuberant and produce masses of extramedullary erythropoietic tissue in the liver and spleen.
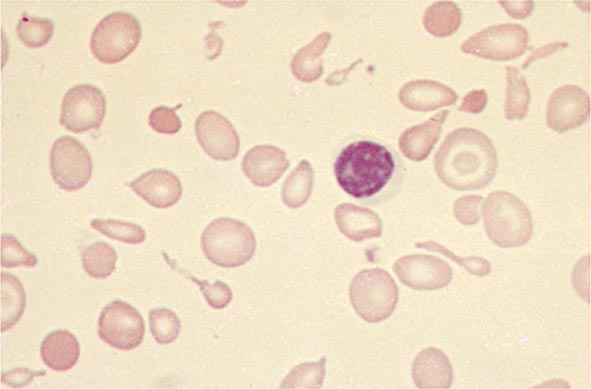
FIGURE 127-5 β Thalassemia intermedia. Microcytic and hypochromic red blood cells are seen that resemble the red blood cells of severe iron-deficiency anemia. Many elliptical and teardrop-shaped red blood cells are noted.
Massive bone marrow expansion deranges growth and development. Children develop characteristic “chipmunk” facies due to maxillary marrow hyperplasia and frontal bossing. Thinning and pathologic fracture of long bones and vertebrae may occur due to cortical invasion by erythroid elements and profound growth retardation. Hemolytic anemia causes hepatosplenomegaly, leg ulcers, gallstones, and high-output congestive heart failure. The conscription of caloric resources to support erythropoiesis leads to inanition, susceptibility to infection, endocrine dysfunction, and in the most severe cases, death during the first decade of life. Chronic transfusions with RBCs improve oxygen delivery, suppress the excessive ineffective erythropoiesis, and prolong life, but the inevitable side effects, notably iron overload, often prove fatal by age 30 years.
Severity is highly variable. Known modulating factors are those that ameliorate the burden of unpaired α-globin inclusions. Alleles associated with milder synthetic defects and coinheritance of α thalassemia trait reduce clinical severity by reducing accumulation of excess α globin. HbF persists to various degrees in β thalassemias. γ-Globin gene chains can substitute for β chains, generating more hemoglobin and reducing the burden of α-globin inclusions. The terms β thalassemia major and β thalassemia intermedia are used to reflect the clinical heterogeneity. Patients with β thalassemia major require intensive transfusion support to survive. Patients with β thalassemia intermedia have a somewhat milder phenotype and can survive without transfusion. The terms β thalassemia minor and β thalassemia trait describe asymptomatic heterozygotes for β thalassemia.
THALASSEMIA SYNDROMES
The four classic α thalassemias, most common in Asians, are α thalassemia-2 trait, in which one of the four α-globin loci is deleted; α thalassemia-1 trait, with two deleted loci; HbH disease, with three loci deleted; and hydrops fetalis with Hb Barts, with all four loci deleted (Table 127-4). Nondeletion forms of α thalassemia also exist.
|
THE α THALASSEMIAS |
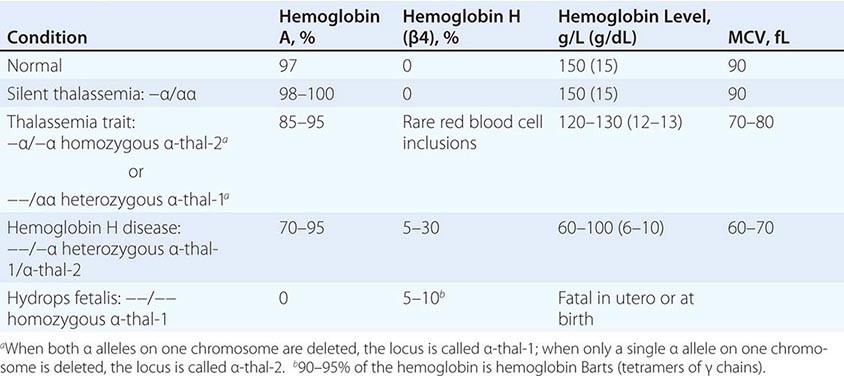
α Thalassemia-2 trait is an asymptomatic, silent carrier state. α Thalassemia-1 trait resembles β thalassemia minor. Offspring doubly heterozygous for α thalassemia-2 and α thalassemia-1 exhibit a more severe phenotype called HbH disease. Heterozygosity for a deletion that removes both genes from the same chromosome (cis deletion) is common in Asians and in those from the Mediterranean region, as is homozygosity for α thalassemia-2 (trans deletion). Both produce asymptomatic hypochromia and microcytosis.
In HbH disease, HbA production is only 25–30% normal. Fetuses accumulate some unpaired γ chains (Hb Barts; γ-chain tetramers). In adults, unpaired β chains accumulate and are soluble enough to form β4 tetramers called HbH. HbH forms few inclusions in erythroblasts and precipitates in circulating RBC. Patients with HbH disease have thalassemia intermedia characterized by moderately severe hemolytic anemia but milder ineffective erythropoiesis. Survival into midadult life without transfusions is common.
The homozygous state for the α thalassemia-1 cis deletion (hydrops fetalis) causes total absence of α-globin synthesis. No physiologically useful hemoglobin is produced beyond the embryonic stage. Excess γ globin forms tetramers called Hb Barts (γ4), which has a very high oxygen affinity. It delivers almost no O2 to fetal tissues, causing tissue asphyxia, edema (hydrops fetalis), congestive heart failure, and death in utero. α Thalassemia-2 trait is common (15–20%) among people of African descent. The cis α thalassemia-1 deletion is almost never seen, however. Thus, α thalassemia-2 and the trans form of α thalassemia-1 are very common, but HbH disease and hydrops fetalis are rare.
It has been known for some time that some patients with myelodysplasia or erythroleukemia produce RBC clones containing HbH. This phenomenon is due to mutations in the ATRX pathway that affect the LCR of the α-globin gene cluster.
DIAGNOSIS AND MANAGEMENT OF THALASSEMIAS
The diagnosis of β-thalassemia major is readily made during childhood on the basis of severe anemia accompanied by the characteristic signs of massive ineffective erythropoiesis: hepatosplenomegaly, profound microcytosis, a characteristic blood smear (Fig. 127-5), and elevated levels of HbF, HbA2, or both. Many patients require chronic hypertransfusion therapy designed to maintain a hematocrit of at least 27–30% so that erythropoiesis is suppressed. Splenectomy is required if the annual transfusion requirement (volume of RBCs per kilogram of body weight per year) increases by >50%. Folic acid supplements may be useful. Vaccination with Pneumovax in anticipation of eventual splenectomy is advised, as is close monitoring for infection, leg ulcers, and biliary tract disease. Many patients develop endocrine deficiencies as a result of iron overload. Early endocrine evaluation is required for glucose intolerance, thyroid dysfunction, and delayed onset of puberty or secondary sexual characteristics.
Patients with β thalassemia intermedia exhibit similar stigmata but can survive without chronic hypertransfusion. Management is particularly challenging because a number of factors can aggravate the anemia, including infection, onset of puberty, and development of splenomegaly and hypersplenism. Some patients may eventually benefit from splenectomy. The expanded erythron can cause absorption of excessive dietary iron and hemosiderosis, even without transfusion. Some patients eventually become transfusion dependent.
β Thalassemia minor (i.e., thalassemia trait) usually presents as profound microcytosis and hypochromia with target cells, but only minimal or mild anemia. The mean corpuscular volume is rarely >75 fL; the hematocrit is rarely <30–33%. Hemoglobin analysis classically reveals an elevated HbA2 (3.5–7.5%), but some forms are associated with normal HbA2 and/or elevated HbF. Genetic counseling and patient education are essential. Patients with β thalassemia trait should be warned that their blood picture resembles iron deficiency and can be misdiagnosed. They should eschew empirical use of iron, yet iron deficiency requiring replacement therapy can develop during pregnancy or from chronic bleeding.
Persons with α thalassemia trait may exhibit mild hypochromia and microcytosis usually without anemia. HbA2 and HbF levels are normal. Affected individuals usually require only genetic counseling. HbH disease resembles β thalassemia intermedia, with the added complication that the HbH molecule behaves like moderately unstable hemoglobin. Patients with HbH disease should undergo splenectomy if excessive anemia or a transfusion requirement develops. Oxidative drugs should be avoided. Iron overload leading to death can occur in more severely affected patients.
PREVENTION
Antenatal diagnosis of thalassemia syndromes is now widely available. DNA diagnosis is based on polymerase chain reaction (PCR) amplification of fetal DNA, obtained by amniocentesis or chorionic villus biopsy followed by hybridization to allele-specific oligonucleotide probes or direct DNA sequencing.
THALASSEMIC STRUCTURAL VARIANTS
Thalassemic structural variants are characterized by both defective synthesis and abnormal structure.
HEMOGLOBIN LEPORE
Hb Lepore [α2(δβ)2] arises by an unequal crossover and recombination event that fuses the proximal end of the δ-gene with the distal end of the closely linked β-gene. It is common in the Mediterranean basin. The resulting chromosome contains only the fused δβ gene. The Lepore (δβ) globin is synthesized poorly because the fused gene is under the control of the weak δ-globin promoter. Hb Lepore alleles have a phenotype like β thalassemia, except for the added presence of 2–20% Hb Lepore. Compound heterozygotes for Hb Lepore and a classic β thalassemia allele may also have severe thalassemia.
HEMOGLOBIN E
![]() HbE (i.e., α2β226Glu→Lys) is extremely common in Cambodia, Thailand, and Vietnam. The gene has become far more prevalent in the United States as a result of immigration of Asian persons, especially in California, where HbE is the most common variant detected. HbE is mildly unstable but not enough to affect RBC life span significantly. Heterozygotes resemble individuals with a mild β-thalassemia trait. Homozygotes have somewhat more marked abnormalities but are asymptomatic. Compound heterozygotes for HbE and a β thalassemia gene can have β thalassemia intermedia or β thalassemia major, depending on the severity of the coinherited thalassemic gene.
HbE (i.e., α2β226Glu→Lys) is extremely common in Cambodia, Thailand, and Vietnam. The gene has become far more prevalent in the United States as a result of immigration of Asian persons, especially in California, where HbE is the most common variant detected. HbE is mildly unstable but not enough to affect RBC life span significantly. Heterozygotes resemble individuals with a mild β-thalassemia trait. Homozygotes have somewhat more marked abnormalities but are asymptomatic. Compound heterozygotes for HbE and a β thalassemia gene can have β thalassemia intermedia or β thalassemia major, depending on the severity of the coinherited thalassemic gene.
The βE allele contains a single base change in codon 26 that causes the amino acid substitution. This mutation also activates a cryptic RNA splice site, generating a structurally abnormal globin mRNA that cannot be translated, from about 50% of the initial pre-mRNA molecules. The remaining 40–50% are normally spliced and generate functional mRNA that is translated into βE-globin because the mature mRNA carries the base change that alters codon 26.
Genetic counseling of the persons at risk for HbE should focus especially on the interaction of HbE with β thalassemia, because HbE homozygosity is a condition associated with mildly asymptomatic microcytosis, hypochromia, and hemoglobin levels rarely <100 g/L (<10 g/dL).
HEREDITARY PERSISTENCE OF FETAL HEMOGLOBIN
HPFH is characterized by continued synthesis of high levels of HbF in adult life. No deleterious effects are apparent, even when all of the hemoglobin produced is HbF. These rare patients demonstrate convincingly that prevention or reversal of the fetal to adult hemoglobin switch would provide effective therapy for sickle cell anemia and β thalassemia.
ACQUIRED HEMOGLOBINOPATHIES
The two most important acquired hemoglobinopathies are carbon monoxide poisoning and methemoglobinemia (see above). Carbon monoxide has a higher affinity for hemoglobin than does oxygen; it can replace oxygen and diminish O2 delivery. Chronic elevation of carboxyhemoglobin levels to 10 or 15%, as occurs in smokers, can lead to secondary polycythemia. Carboxyhemoglobin is cherry red in color and masks the development of cyanosis usually associated with poor O2 delivery to tissues.
Abnormalities of hemoglobin biosynthesis have also been described in blood dyscrasias. In some patients with myelodysplasia, erythroleukemia, or myeloproliferative disorders, elevated HbF or a mild form of HbH disease may also be seen. The abnormalities are not severe enough to alter the course of the underlying disease.
EXPERIMENTAL THERAPIES
BONE MARROW TRANSPLANTATION, GENE THERAPY, AND MANIPULATION OF HbF
Bone marrow transplantation provides stem cells able to express normal hemoglobin; it has been used in a large number of patients with β thalassemia and a smaller number of patients with sickle cell anemia. Early in the course of disease, before end-organ damage occurs, transplantation is curative in 80–90% of patients. In highly experienced centers, the treatment-related mortality is <10%. Because survival into adult life is possible with conventional therapy, the decision to transplant is best made in consultation with specialized centers.
Gene therapy of thalassemia and sickle cell disease has proved to be an elusive goal, but experimental advances are raising expectations.
Reestablishing high levels of fetal hemoglobin synthesis should ameliorate the symptoms of β-chain hemoglobinopathies. Cytotoxic agents such as hydroxyurea and cytarabine promote high levels of HbF synthesis, probably by stimulating proliferation of the primitive HbF-producing progenitor cell population (i.e., F cell progenitors). Unfortunately, this regimen has not yet been effective in β thalassemia. Butyrates stimulate HbF production, but only transiently. Pulsed or intermittent administration has been found to sustain HbF induction in the majority of patients with sickle cell disease. It is unclear whether butyrates will have similar activity in patients with β thalassemia.
APLASTIC AND HYPOPLASTIC CRISIS IN PATIENTS WITH HEMOGLOBINOPATHIES
Patients with hemolytic anemias sometimes exhibit an alarming decline in hematocrit during and immediately after acute illnesses. Bone marrow suppression occurs in almost everyone during acute and chronic inflammatory illnesses. In patients with short RBC life spans, suppression can affect RBC counts more dramatically. These hypoplastic crises are usually transient and self-correcting before intervention is required.
Aplastic crisis refers to a profound cessation of erythroid activity in patients with chronic hemolytic anemias. It is associated with a rapidly falling hematocrit. Episodes are usually self-limited. Aplastic crises are caused by infection with a particular strain of parvovirus, B19A. Children infected with this virus usually develop permanent immunity. Aplastic crises do not often recur and are rarely seen in adults. Management requires close monitoring of the hematocrit and reticulocyte count. If anemia becomes symptomatic, transfusion support is indicated. Most crises resolve spontaneously within 1–2 weeks.
128 |
Megaloblastic Anemias |
The megaloblastic anemias are a group of disorders characterized by the presence of distinctive morphologic appearances of the developing red cells in the bone marrow. The marrow is usually hypercellular and the anemia is based on ineffective erythropoiesis. The cause is usually a deficiency of either cobalamin (vitamin B12) or folate, but megaloblastic anemia may occur because of genetic or acquired abnormalities that affect the metabolism of these vitamins or because of defects in DNA synthesis not related to cobalamin or folate (Table 128-1). Cobalamin and folate absorption and metabolism are described next, followed by the biochemical basis, clinical and laboratory features, causes, and treatment of megaloblastic anemia.
|
CAUSES OF MEGALOBLASTIC ANEMIA |
COBALAMIN
Cobalamin (vitamin B12) exists in a number of different chemical forms. All have a cobalt atom at the center of a corrin ring. In nature, the vitamin is mainly in the 2-deoxyadenosyl (ado) form, which is located in mitochondria. It is the cofactor for the enzyme methylmalonyl coenzyme A (CoA) mutase. The other major natural cobalamin is methylcobalamin, the form in human plasma and in cell cytoplasm. It is the cofactor for methionine synthase. There are also minor amounts of hydroxocobalamin to which methyl- and adocobalamin are converted rapidly by exposure to light.
DIETARY SOURCES AND REQUIREMENTS
Cobalamin is synthesized solely by microorganisms. Ruminants obtain cobalamin from the foregut, but the only source for humans is food of animal origin, e.g., meat, fish, and dairy products. Vegetables, fruits, and other foods of nonanimal origin are free from cobalamin unless they are contaminated by bacteria. A normal Western diet contains 5–30 μg of cobalamin daily. Adult daily losses (mainly in the urine and feces) are 1–3 μg (~0.1% of body stores), and because the body does not have the ability to degrade cobalamin, daily requirements are also about 1–3 μg. Body stores are of the order of 2–3 mg, sufficient for 3–4 years if supplies are completely cut off.
ABSORPTION
Two mechanisms exist for cobalamin absorption. One is passive, occurring equally through buccal, duodenal, and ileal mucosa; it is rapid but extremely inefficient, with <1% of an oral dose being absorbed by this process. The normal physiologic mechanism is active; it occurs through the ileum and is efficient for small (a few micrograms) oral doses of cobalamin, and it is mediated by gastric intrinsic factor (IF). Dietary cobalamin is released from protein complexes by enzymes in the stomach, duodenum, and jejunum; it combines rapidly with a salivary glycoprotein that belongs to the family of cobalamin-binding proteins known as haptocorrins (HCs). In the intestine, the haptocorrin is digested by pancreatic trypsin and the cobalamin is transferred to IF.
IF (gene at chromosome 11q13) is produced in the gastric parietal cells of the fundus and body of the stomach, and its secretion parallels that of hydrochloric acid. Normally, there is a vast excess of IF. The IF-cobalamin complex passes to the ileum, where IF attaches to a specific receptor (cubilin) on the microvillus membrane of the enterocytes. Cubilin also is present in yolk sac and renal proximal tubular epithelium. Cubilin appears to traffic by means of amnionless (AMN), an endocytic receptor protein that directs sublocalization and endocytosis of cubilin with its ligand IF-cobalamin complex. The cobalamin-IF complex enters the ileal cell, where IF is destroyed. After a delay of about 6 h, the cobalamin appears in portal blood attached to transcobalamin (TC) II.
Between 0.5 and 5 μg of cobalamin enter the bile each day. This binds to IF, and a major portion of biliary cobalamin normally is reabsorbed together with cobalamin derived from sloughed intestinal cells. Because of the appreciable amount of cobalamin undergoing enterohepatic circulation, cobalamin deficiency develops more rapidly in individuals who malabsorb cobalamin than it does in vegans, in whom reabsorption of biliary cobalamin is intact.
TRANSPORT
Two main cobalamin transport proteins exist in human plasma; they both bind cobalamin—one molecule for one molecule. One HC, also known as TC I, is closely related to other cobalamin-binding HCs in milk, gastric juice, bile, saliva, and other fluids. The gene TCNL is at chromosome 11q11-q12.3. These HCs differ from each other only in the carbohydrate moiety of the molecule. TC I is derived primarily from the specific granules in neutrophils. Normally, it is about two-thirds saturated with cobalamin, which it binds tightly. TC I does not enhance cobalamin entry into tissues. Glycoprotein receptors on liver cells are involved in the removal of TC I from plasma, and TC I may play a role in the transport of cobalamin analogues (which it binds more effectively than IF) to the liver for excretion in bile.
The other major cobalamin transport protein in plasma is transcobalamin, also known as TC II. The gene is on chromosome 22q11-q13.1. As for IF and HC, there are nine exons. The three proteins are likely to have a common ancestral origin. TC II is synthesized by liver and by other tissues, including macrophages, ileum, and vascular endothelium. It normally carries only 20–60 ng of cobalamin per liter of plasma and readily gives up cobalamin to marrow, placenta, and other tissues, which it enters by receptor-mediated endocytosis involving the TC II receptor and megalin (encoded by the LRP-2 gene). The TC II cobalamin is internalized by endocytosis via clathrin-coated pits; the complex is degraded, but the receptor probably is recycled to the cell membrane as is the case for transferrin. Export of “free” cobalamin is via the ATP-binding cassette drug transporter alias multidrug resistance protein 1.
FOLATE
DIETARY FOLATE
Folic (pteroylglutamic) acid is a yellow, crystalline, water-soluble substance. It is the parent compound of a large family of natural folate compounds, which differ from it in three respects: (1) they are partly or completely reduced to di- or tetrahydrofolate (THF) derivatives, (2) they usually contain a single carbon unit (Table 128-2), and (3) 70–90% of natural folates are folate-polyglutamates.
|
BIOCHEMICAL REACTIONS OF FOLATE COENZYMES |
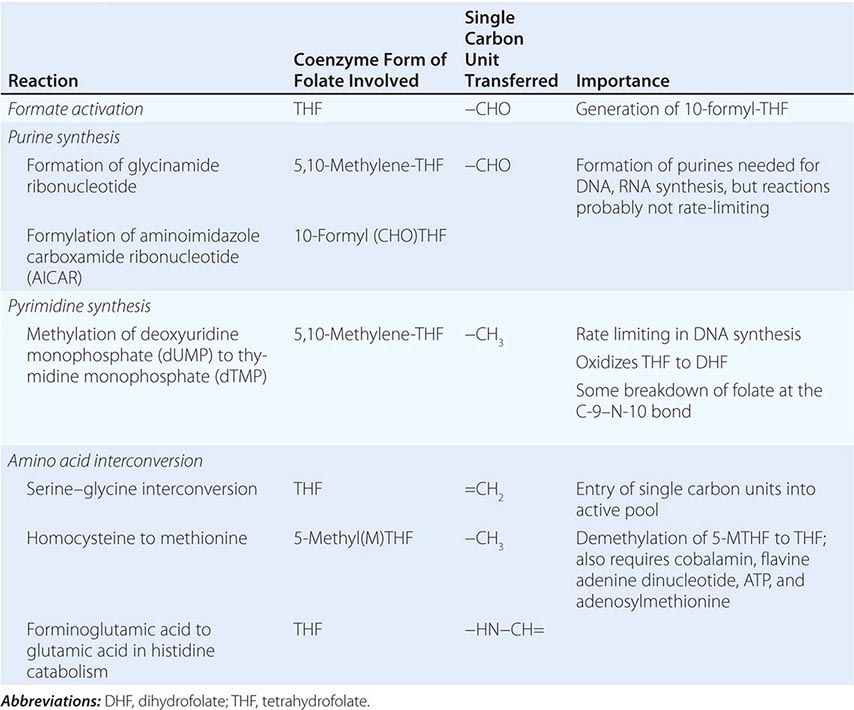
Most foods contain some folate. The highest concentrations are found in liver, yeast, spinach, other greens, and nuts (>100 μg/100 g). The total folate content of an average Western diet is ~250 μg daily, but the amount varies widely according to the type of food eaten and the method of cooking. Folate is easily destroyed by heating, particularly in large volumes of water. Total body folate in the adult is ~10 mg, with the liver containing the largest store. Daily adult requirements are ~100 μg, and so stores are sufficient for only 3–4 months in normal adults and severe folate deficiency may develop rapidly.
ABSORPTION
Folates are absorbed rapidly from the upper small intestine. The absorption of folate polyglutamates is less efficient than that of monoglutamates; on average, ~50% of food folate is absorbed. Polyglutamate forms are hydrolyzed to the monoglutamate derivatives either in the lumen of the intestine or within the mucosa. All dietary folates are converted to 5-methylTHF (5-MTHF) within the small intestinal mucosa before entering portal plasma. The monoglutamates are actively transported across the enterocyte by a proton-coupled folate transporter (PCFT, SCL46A1). This is situated at the apical brush border and is most active at pH 5.5, which is about the pH of the duodenal and jejunal surface. Genetic mutations of this protein underlie hereditary malabsorption of folate (see below). Pteroylglutamic acid at doses >400 μg is absorbed largely unchanged and converted to natural folates in the liver. Lower doses are converted to 5-MTHF during absorption through the intestine.
About 60–90 μg of folate enters the bile each day and is excreted into the small intestine. Loss of this folate, together with the folate of sloughed intestinal cells, accelerates the speed with which folate deficiency develops in malabsorption conditions.
TRANSPORT
Folate is transported in plasma; about one-third is loosely bound to albumin, and two-thirds is unbound. In all body fluids (plasma, cerebrospinal fluid, milk, bile), folate is largely, if not entirely, 5-MTHF in the monoglutamate form. Three types of folate-binding protein are involved. A reduced folate transporter (RFC, SLC19A1) is the major route of delivery of plasma folate (5-MTHF) to cells. Two folate receptors, FR2 and FR3 embedded in the cell membrane by a glycosyl phosphatidylinositol anchor, transport folate into the cell via receptor-mediated endocytosis. The third protein, PCFT, transports folate at low pH from the vesicle to the cell cytoplasm. The reduced folate transporter also mediates uptake of methotrexate by cells.
BIOCHEMICAL FUNCTIONS
Folates (as the intracellular polyglutamate derivatives) act as coenzymes in the transfer of single-carbon units (Fig. 128-1 and Table 128-2). Two of these reactions are involved in purine synthesis and one in pyrimidine synthesis necessary for DNA and RNA replication. Folate is also a coenzyme for methionine synthesis, in which methylcobalamin is also involved and in which THF is regenerated. THF is the acceptor of single carbon units newly entering the active pool via conversion of serine to glycine. Methionine, the other product of the methionine synthase reaction, is the precursor for S-adenosylmethionine (SAM), the universal methyl donor involved in >100 methyltransferase reactions (Fig. 128-1).
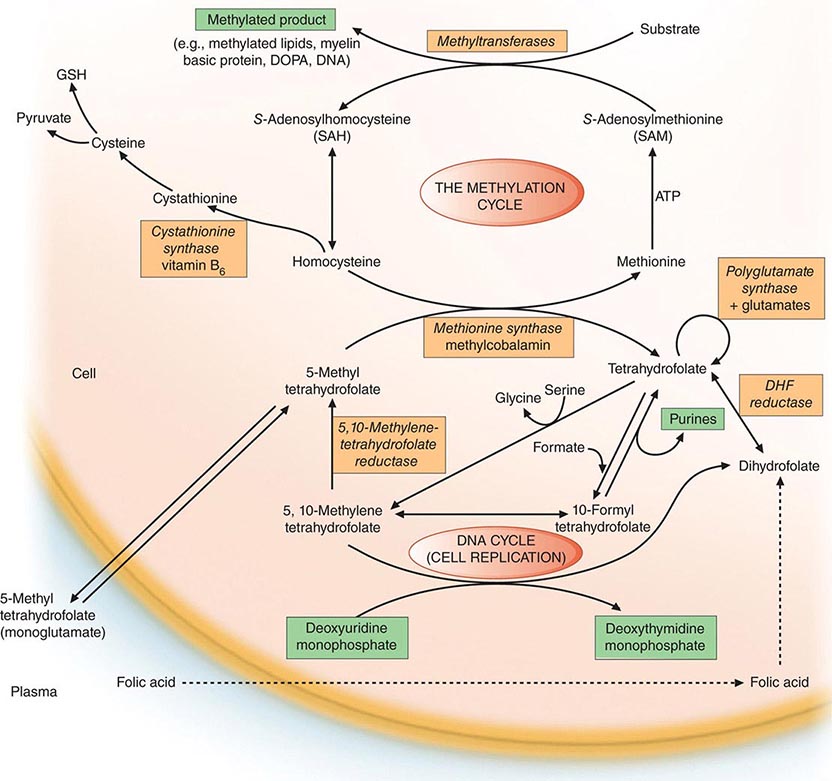
FIGURE 128-1 The role of folates in DNA synthesis and in formation of S-adenosylmethionine (SAM), which is involved in numerous methylation reactions. DHF, dihydrofolate; GSH, glutathione. (Reprinted from AV Hoffbrand et al [eds]: Postgraduate Haematology, 5th ed. Oxford, UK, Blackwell Publishing, 2005; with permission.)
During thymidylate synthesis, 5,10-methylene-THF is oxidized to DHF (dihydrofolate). The enzyme DHF reductase converts this to THF. The drugs methotrexate, pyrimethamine, and (mainly in bacteria) trimethoprim inhibit DHF reductase and so prevent formation of active THF coenzymes from DHF. A small fraction of the folate coenzyme is not recycled during thymidylate synthesis but is degraded at the C9-N10 bond.
BIOCHEMICAL BASIS OF MEGALOBLASTIC ANEMIA
The common feature of all megaloblastic anemias is a defect in DNA synthesis that affects rapidly dividing cells in the bone marrow. All conditions that give rise to megaloblastic changes have in common a disparity in the rate of synthesis or availability of the four immediate precursors of DNA: the deoxyribonucleoside triphosphates (dNTPs)—dA(adenine)TP and dG(guanine)TP (purines), dT(thymine)TP and dC(cytosine)TP (pyrimidines). In deficiencies of either folate or cobalamin, there is failure to convert deoxyuridine monophosphate (dUMP) to deoxythymidine monophosphate (dTMP), the precursor of dTTP (Fig 128-1). This is the case because folate is needed as the coenzyme 5,10-methylene-THF polyglutamate for conversion of dUMP to dTMP; the availability of 5,10-methylene-THF is reduced in either cobalamin or folate deficiency. An alternative theory for megaloblastic anemia in cobalamin or folate deficiency is misincorporation of uracil into DNA because of a buildup of deoxyuridine triphosphate (dUTP) at the DNA replication fork as a consequence of the block in conversion of dUMP to dTMP.
COBALAMIN-FOLATE RELATIONS
Folate is required for many reactions in mammalian tissues. Only two reactions in the body are known to require cobalamin. Methylmalonyl CoA isomerization requires adocobalamin, and the methylation of homocysteine to methionine requires both methylcobalamin and 5-MTHF (Fig. 128-1). This reaction is the first step in the pathway by which 5-MTHF, which enters bone marrow and other cells from plasma, is converted into all the intracellular folate coenzymes. The coenzymes are all polyglutamated (the larger size aiding retention in the cell), but the enzyme folate polyglutamate synthase can use only THF, not MTHF, as substrate. In cobalamin deficiency, MTHF accumulates in plasma, and intracellular folate concentrations fall due to failure of formation of THF, the substrate on which folate polyglutamates are built. This has been termed THF starvation, or the methylfolate trap.
This theory explains the abnormalities of folate metabolism that occur in cobalamin deficiency (high serum folate, low cell folate, positive purine precursor aminoimidazole carboxamide ribonucleotide [AICAR] excretion; Table 128-2) and also why the anemia of cobalamin deficiency responds to folic acid in large doses.
CLINICAL FEATURES
Many symptomless patients are detected through the finding of a raised mean corpuscular volume (MCV) on a routine blood count. The main clinical features in more severe cases are those of anemia. Anorexia is usually marked, and there may be weight loss, diarrhea, or constipation. Glossitis, angular cheilosis, a mild fever in more severely anemic patients, jaundice (unconjugated), and reversible melanin skin hyperpigmentation also may occur with a deficiency of either folate or cobalamin. Thrombocytopenia sometimes leads to bruising, and this may be aggravated by vitamin C deficiency or alcohol in malnourished patients. The anemia and low leukocyte count may predispose to infections, particularly of the respiratory and urinary tracts. Cobalamin deficiency has also been associated with impaired bactericidal function of phagocytes.
GENERAL TISSUE EFFECTS OF COBALAMIN AND FOLATE DEFICIENCIES
Epithelial Surfaces After the marrow, the next most frequently affected tissues are the epithelial cell surfaces of the mouth, stomach, and small intestine and the respiratory, urinary, and female genital tracts. The cells show macrocytosis, with increased numbers of multinucleate and dying cells. The deficiencies may cause cervical smear abnormalities.
Complications of Pregnancy The gonads are also affected, and infertility is common in both men and women with either deficiency. Maternal folate deficiency has been implicated as a cause of prematurity, and both folate deficiency and cobalamin deficiency have been implicated in recurrent fetal loss and neural tube defects, as discussed below.
Neural Tube Defects Folic acid supplements at the time of conception and in the first 12 weeks of pregnancy reduce by ~70% the incidence of neural tube defects (NTDs) (anencephaly, meningomyelocele, encephalocele, and spina bifida) in the fetus. Most of this protective effect can be achieved by taking folic acid, 0.4 mg daily, at the time of conception.
The incidence of cleft palate and harelip also can be reduced by prophylactic folic acid. There is no clear simple relationship between maternal folate status and these fetal abnormalities, although overall the lower the maternal folate, the greater the risk to the fetus. NTDs also can be caused by antifolate and antiepileptic drugs.
An underlying maternal folate metabolic abnormality has also been postulated. One abnormality has been identified: reduced activity of the enzyme 5,10-methylene-THF reductase (MTHFR) (Fig. 128-1) caused by a common C677T polymorphism in the MTHFR gene. In one study, the prevalence of this polymorphism was found to be higher than in controls in the parents of NTD fetuses and in the fetuses themselves: homozygosity for the TT mutation was found in 13% of cases compared with 5% of control subjects. The polymorphism codes for a thermolabile form of MTHFR. The homozygous state results in a lower mean serum and red cell folate level compared with control subjects, as well as significantly higher serum homocysteine levels. Tests for mutations in other enzymes possibly associated with NTDs, e.g., methionine synthase and serine–glycine hydroxymethylase, have been negative. Serum vitamin B12 levels are also lower in the sera of mothers of NTD infants than in controls. In addition, maternal TC II receptor polymorphisms are associated with increased risk of NTD births. There are, however, no studies showing dietary fortification with vitamin B12 reduces the incidence of NTDs.
Cardiovascular Disease Children with severe homocystinuria (blood levels ≥100 μmol/L) due to deficiency of one of three enzymes, methionine synthase, MTHFR, or cystathionine synthase (Fig. 128-1), have vascular disease, e.g., ischemic heart disease, cerebrovascular disease, or pulmonary embolus, as teenagers or in young adulthood. Lesser degrees of raised serum homocysteine and low levels of serum folate and homozygous inherited mutations of MTHFR have been found to be associated with cerebrovascular, peripheral vascular, and coronary heart disease and with deep vein thrombosis. Prospective randomized trials of lowering homocysteine levels with supplements of folic acid, vitamin B12, and vitamin B6 against placebo over a 5-year period in patients with vascular disease or diabetes have not, however, shown a reduction of first event fatal or nonfatal myocardial infarction, nor have these supplements reduced the risk of recurrent cardiovascular disease after an acute myocardial infarct. Meta-analysis showed an 18% reduction in strokes but no significant prevention of death from any cause. Venous thrombosis has been reported to be more frequent in vitamin B12–deficient subjects than in controls. This was ascribed to raised plasma homocysteine levels in vitamin B12 deficiency.
Malignancy Prophylactic folic acid in pregnancy has been found in some but not all studies to reduce the subsequent incidence of acute lymphoblastic leukemia (ALL) in childhood. A significant negative association has also been found with the MTHFR C677T polymorphism and leukemias with mixed lineage leukemia (MLL) translocations, but a positive association with hyperdiploidy in infants with ALL or acute myeloid leukemia or with childhood ALL. A second polymorphism in the MTHFR gene, A1298C, is also strongly associated with hyperdiploid leukemia. There are various positive and negative associations between polymorphisms in folate-dependent enzymes and the incidence of adult ALL. The C677T polymorphism is thought to lead to increased thymidine pools and “better quality” of DNA synthesis by shunting one-carbon groups toward thymidine and purine synthesis. This may explain its reported association with a lower risk for colorectal cancer. Most but not all studies suggest that prophylactic folic acid also protects against colon adenomas. Other tumors that have been associated with folate polymorphisms or status include follicular lymphoma, breast cancer, and gastric cancer. A meta-analysis of 50,000 individuals given folic acid or placebo in cardiovascular or colon adenoma prevention trials found that folic acid supplementation did not substantially increase or decrease the incidence of site-specific cancer during the first 5 years of treatment. Because folic acid may “feed” tumors, it probably should be avoided in those with established tumors unless there is severe megaloblastic anemia due to folate deficiency.
Neurologic Manifestations Cobalamin deficiency may cause a bilateral peripheral neuropathy or degeneration (demyelination) of the posterior and pyramidal tracts of the spinal cord and, less frequently, optic atrophy or cerebral symptoms.
The patient, more frequently male, presents with paresthesias, muscle weakness, or difficulty in walking and sometimes dementia, psychotic disturbances, or visual impairment. Long-term nutritional cobalamin deficiency in infancy leads to poor brain development and impaired intellectual development. Folate deficiency has been suggested to cause organic nervous disease, but this is uncertain, although methotrexate injected into the cerebrospinal fluid may cause brain or spinal cord damage.
An important clinical problem is the nonanemic patient with neurologic or psychiatric abnormalities and a low or borderline serum cobalamin level. In such patients, it is necessary to try to establish whether there is significant cobalamin deficiency, e.g., by careful examination of the blood film, tests for serum gastrin level and for antibodies to IF or parietal cells, along with serum methylmalonic acid (MMA) measurement if available. A trial of cobalamin therapy for at least 3 months will usually also be needed to determine whether the symptoms improve.
The biochemical basis for cobalamin neuropathy remains obscure. Its occurrence in the absence of methylmalonic aciduria in TC II deficiency suggests that the neuropathy is related to the defect in homocysteine-methionine conversion. Accumulation of S-adenosylhomocysteine in the brain, resulting in inhibition of transmethylation reactions, has been suggested.
Psychiatric disturbance is common in both folate and cobalamin deficiencies. This, like the neuropathy, has been attributed to a failure of the synthesis of SAM, which is needed in methylation of biogenic amines (e.g., dopamine) as well as that of proteins, phospholipids, and neurotransmitters in the brain (Fig. 128-1). Associations between lower serum folate or cobalamin levels and higher homocysteine levels and the development of decreased cognitive function and dementia in Alzheimer’s disease have been reported. A meta-analysis of randomized, placebo-controlled trials of homocysteine-lowering B-vitamin supplementation of individuals with and without cognitive impairment, however, showed that supplementation with vitamin B12, vitamin B6, and folic acid alone or in combination did not improve cognitive function. It is unknown whether prolonged treatment with these B vitamins can reduce the risk of dementia in later life.
HEMATOLOGIC FINDINGS
PERIPHERAL BLOOD
Oval macrocytes, usually with considerable anisocytosis and poikilocytosis, are the main feature (Fig. 128-2A). The MCV is usually >100 fL unless a cause of microcytosis (e.g., iron deficiency or thalassemia trait) is present. Some of the neutrophils are hypersegmented (more than five nuclear lobes). There may be leukopenia due to a reduction in granulocytes and lymphocytes, but this is usually >1.5 × 109/L; the platelet count may be moderately reduced, rarely to <40 × 109/L. The severity of all these changes parallels the degree of anemia. In a nonanemic patient, the presence of a few macrocytes and hypersegmented neutrophils in the peripheral blood may be the only indication of the underlying disorder.
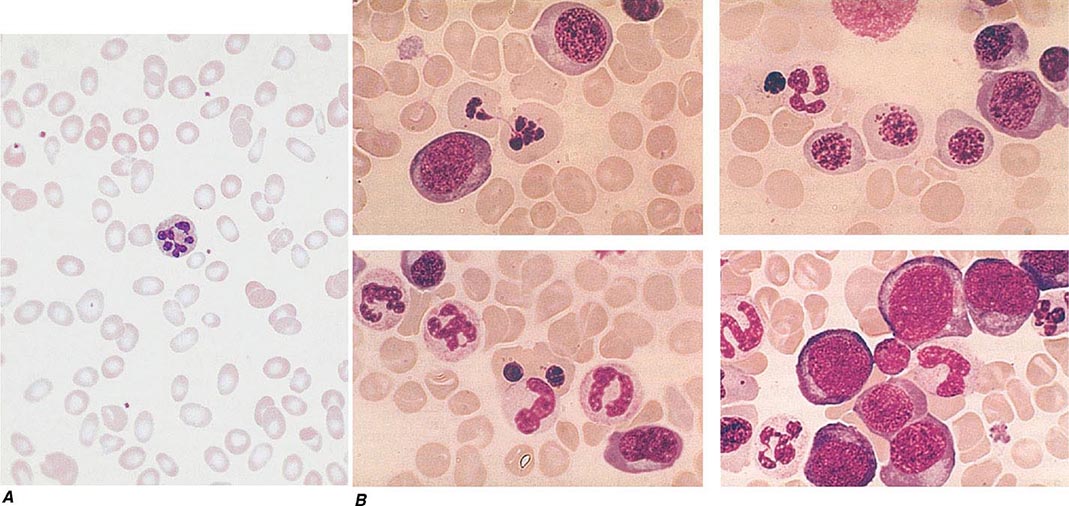
FIGURE 128-2 A. The peripheral blood in severe megaloblastic anemia. B. The bone marrow in severe megaloblastic anemia. (Reprinted from AV Hoffbrand et al [eds]: Postgraduate Haematology, 5th ed. Oxford, UK, Blackwell Publishing, 2005; with permission.)
BONE MARROW
In a severely anemic patient, the marrow is hypercellular with an accumulation of primitive cells due to selective death by apoptosis of more mature forms. The erythroblast nucleus maintains a primitive appearance despite maturation and hemoglobinization of the cytoplasm. The cells are larger than normoblasts, and an increased number of cells with eccentric lobulated nuclei or nuclear fragments may be present (Fig. 128-2B). Giant and abnormally shaped metamyelocytes and enlarged hyperpolyploid megakaryocytes are characteristic. In severe cases, the accumulation of primitive cells may mimic acute myeloid leukemia, whereas in less anemic patients, the changes in the marrow may be difficult to recognize. The terms intermediate, mild, and early have been used. The term megaloblastoid does not mean mildly megaloblastic. It is used to describe cells with both immature-appearing nuclei and defective hemoglobinization and is usually seen in myelodysplasia.
CHROMOSOMES
Bone marrow cells, transformed lymphocytes, and other proliferating cells in the body show a variety of changes, including random breaks, reduced contraction, spreading of the centromere, and exaggeration of secondary chromosomal constrictions and overprominent satellites. Similar abnormalities may be produced by antimetabolite drugs (e.g., cytosine arabinoside, hydroxyurea, and methotrexate) that interfere with either DNA replication or folate metabolism and that also cause megaloblastic appearances.
INEFFECTIVE HEMATOPOIESIS
There is an accumulation of unconjugated bilirubin in plasma due to the death of nucleated red cells in the marrow (ineffective erythropoiesis). Other evidence for this includes raised urine urobilinogen, reduced haptoglobins and positive urine hemosiderin, and a raised serum lactate dehydrogenase. A weakly positive direct antiglobulin test due to complement can lead to a false diagnosis of autoimmune hemolytic anemia.
CAUSES OF COBALAMIN DEFICIENCY
Cobalamin deficiency is usually due to malabsorption. The only other cause is inadequate dietary intake.
INADEQUATE DIETARY INTAKE
Adults Dietary cobalamin deficiency arises in vegans who omit meat, fish, eggs, cheese, and other animal products from their diet. The largest group in the world consists of Hindus, and it is likely that many millions of Indians are at risk of deficiency of cobalamin on a nutritional basis. Subnormal serum cobalamin levels are found in up to 50% of randomly selected, young, adult Indian vegans, but the deficiency usually does not progress to megaloblastic anemia since the diet of most vegans is not totally lacking in cobalamin and the enterohepatic circulation of cobalamin is intact. Dietary cobalamin deficiency may also arise rarely in nonvegetarian individuals who exist on grossly inadequate diets because of poverty or psychiatric disturbance.
Infants Cobalamin deficiency has been described in infants born to severely cobalamin-deficient mothers. These infants develop megaloblastic anemia at about 3–6 months of age, presumably because they are born with low stores of cobalamin and because they are fed breast milk with low cobalamin content. The babies have also shown growth retardation, impaired psychomotor development, and other neurologic sequelae.
GASTRIC CAUSES OF COBALAMIN MALABSORPTION
See Tables 128-3 and 128-4.
|
CAUSES OF COBALAMIN DEFICIENCY SUFFICIENTLY SEVERE TO CAUSE MEGALOBLASTIC ANEMIA |
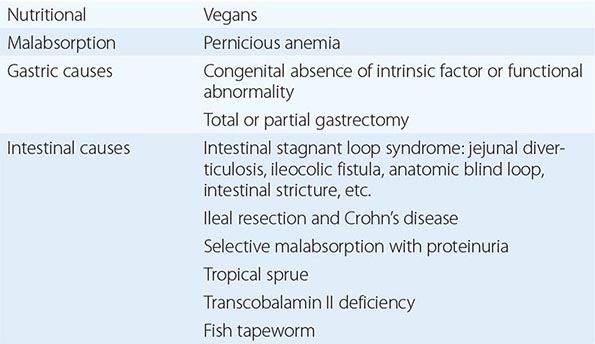
|
MALABSORPTION OF COBALAMIN MAY OCCUR IN THE FOLLOWING CONDITIONS BUT IS NOT USUALLY SUFFICIENTLY SEVERE AND PROLONGED TO CAUSE MEGALOBLASTIC ANEMIA |
Pernicious Anemia Pernicious anemia (PA) may be defined as a severe lack of IF due to gastric atrophy. It is a common disease in north Europeans but occurs in all countries and ethnic groups. The overall incidence is about 120 per 100,000 population in the United Kingdom (UK). The ratio of incidence in men and women among whites is ~1:1.6, and the peak age of onset is 60 years, with only 10% of patients being <40 years of age. However, in some ethnic groups, notably black individuals and Latin Americans, the age at onset of PA is generally lower. The disease occurs more commonly than by chance in close relatives and in persons with other organ-specific autoimmune diseases, e.g., thyroid diseases, vitiligo, hypoparathyroidism, and Addison’s disease. It is also associated with hypogammaglobulinemia, with premature graying or blue eyes, and persons of blood group A. An association with human leukocyte antigen (HLA) 3 has been reported in some but not all series and, in those with endocrine disease, with HLA-B8, -B12, and -BW15. Life expectancy is normal in women once regular treatment has begun. Men have a slightly subnormal life expectancy as a result of a higher incidence of carcinoma of the stomach than in control subjects. Gastric output of hydrochloric acid, pepsin, and IF is severely reduced. The serum gastrin level is raised, and serum pepsinogen I levels are low.
Gastric Biopsy A single endoscopic examination is recommended if PA is diagnosed. Gastric biopsy usually shows atrophy of all layers of the body and fundus, with loss of glandular elements, an absence of parietal and chief cells and replacement by mucous cells, a mixed inflammatory cell infiltrate, and perhaps intestinal metaplasia. The infiltrate of plasma cells and lymphocytes contains an excess of CD4 cells. These are directed against gastric H/K-ATPase. The antral mucosa is usually well preserved. Helicobacter pylori infection occurs infrequently in PA, but it has been suggested that H. pylori gastritis occurs at an early phase of atrophic gastritis and presents in younger patients as iron-deficiency anemia but in older patients as PA. H. pylori is suggested to stimulate an autoimmune process directed against parietal cells, with the H. pylori infection then being gradually replaced, in some individuals, by an autoimmune process.
Serum Antibodies Two types of IF immunoglobulin G antibody may be found in the sera of patients with PA. One, the “blocking,” or type I, antibody, prevents the combination of IF and cobalamin, whereas the “binding,” or type II, antibody prevents attachment of IF to ileal mucosa. Type I occurs in the sera of ~55% of patients, and type II in 35%. IF antibodies cross the placenta and may cause temporary IF deficiency in a newborn infant. Patients with PA also show cell-mediated immunity to IF. Type I antibody has been detected rarely in the sera of patients without PA but with thyrotoxicosis, myxedema, Hashimoto’s disease, or diabetes mellitus and in relatives of PA patients. IF antibodies also have been detected in gastric juice in ~80% of PA patients. These gastric antibodies may reduce absorption of dietary cobalamin by combining with small amounts of remaining IF.
Parietal cell antibody is present in the sera of almost 90% of adult patients with PA but is frequently present in other subjects. Thus, it occurs in as many as 16% of randomly selected female subjects age >60 years. The parietal cell antibody is directed against the α and β subunits of the gastric proton pump (H+,K+-ATPase).
JUVENILE PERNICIOUS ANEMIA
This usually occurs in older children and resembles PA of adults. Gastric atrophy, achlorhydria, and serum IF antibodies are all present, although parietal cell antibodies are usually absent. About one-half of these patients show an associated endocrinopathy such as autoimmune thyroiditis, Addison’s disease, or hypoparathyroidism; in some, mucocutaneous candidiasis occurs.
CONGENITAL INTRINSIC FACTOR DEFICIENCY OR FUNCTIONAL ABNORMALITY
An affected child usually presents with megaloblastic anemia in the first to third year of life; a few have presented as late as the second decade. The child usually has no demonstrable IF but has a normal gastric mucosa and normal secretion of acid. The inheritance is autosomal recessive. Parietal cell and IF antibodies are absent. Variants have been described in which the child is born with IF that can be detected immunologically but is unstable or functionally inactive, unable to bind cobalamin or to facilitate its uptake by ileal receptors.
GASTRECTOMY
After total gastrectomy, cobalamin deficiency is inevitable, and prophylactic cobalamin therapy should be commenced immediately after the operation. After partial gastrectomy, 10–15% of patients also develop this deficiency. The exact incidence and time of onset are most influenced by the size of the resection and the preexisting size of cobalamin body stores.
FOOD COBALAMIN MALABSORPTION
Failure of release of cobalamin from binding proteins in food is believed to be responsible for this condition, which is more common in the elderly. It is associated with low serum cobalamin levels, with or without raised serum levels of MMA and homocysteine. Typically, these patients have normal cobalamin absorption, as measured with crystalline cobalamin, but show malabsorption when a modified test using food-bound cobalamin is used. The frequency of progression to severe cobalamin deficiency and the reasons for this progression are not clear.
INTESTINAL CAUSES OF COBALAMIN MALABSORPTION
Intestinal Stagnant Loop Syndrome Malabsorption of cobalamin occurs in a variety of intestinal lesions in which there is colonization of the upper small intestine by fecal organisms. This may occur in patients with jejunal diverticulosis, enteroanastomosis, or an intestinal stricture or fistula or with an anatomic blind loop due to Crohn’s disease, tuberculosis, or an operative procedure.
Ileal Resection Removal of ≥1.2 m of terminal ileum causes malabsorption of cobalamin. In some patients after ileal resection, particularly if the ileocecal valve is incompetent, colonic bacteria may contribute further to the onset of cobalamin deficiency.
Selective Malabsorption of Cobalamin with Proteinuria (Imerslund’s Syndrome; Imerslund-Gräsbeck Syndrome; Congenital Cobalamin Malabsorption; Autosomal Recessive Megaloblastic Anemia; MGA1) This autosomally recessive disease is the most common cause of megaloblastic anemia due to cobalamin deficiency in infancy in Western countries. More than 200 cases have been reported, with familial clusters in Finland, Norway, the Middle East, and North Africa. The patients secrete normal amounts of IF and gastric acid but are unable to absorb cobalamin. In Finland, impaired synthesis, processing, or ligand binding of cubilin due to inherited mutations is found. In Norway, mutation of the gene for AMN has been reported. Other tests of intestinal absorption are normal. Over 90% of these patients show nonspecific proteinuria, but renal function is otherwise normal and renal biopsy has not shown any consistent renal defect. A few have shown aminoaciduria and congenital renal abnormalities, such as duplication of the renal pelvis.
Tropical Sprue Nearly all patients with acute and subacute tropical sprue show malabsorption of cobalamin; this may persist as the principal abnormality in the chronic form of the disease, when the patient may present with megaloblastic anemia or neuropathy due to cobalamin deficiency. Absorption of cobalamin usually improves after antibiotic therapy and, in the early stages, folic acid therapy.
Fish Tapeworm Infestation The fish tapeworm (Diphyllobothrium latum) lives in the small intestine of humans and accumulates cobalamin from food, rendering the cobalamin unavailable for absorption. Individuals acquire the worm by eating raw or partly cooked fish. Infestation is common around the lakes of Scandinavia, Germany, Japan, North America, and Russia. Megaloblastic anemia or cobalamin neuropathy occurs only in those with a heavy infestation.
Gluten-Induced Enteropathy Malabsorption of cobalamin occurs in ~30% of untreated patients (presumably those in whom the disease extends to the ileum). Cobalamin deficiency is not severe in these patients and is corrected with a gluten-free diet.
Severe Chronic Pancreatitis In this condition, lack of trypsin is thought to cause dietary cobalamin attached to gastric non-IF (R) binder to be unavailable for absorption. It also has been proposed that in pancreatitis, the concentration of calcium ions in the ileum falls below the level needed to maintain normal cobalamin absorption.
HIV Infection Serum cobalamin levels tend to fall in patients with HIV infection and are subnormal in 10–35% of those with AIDS. Malabsorption of cobalamin not corrected by IF has been shown in some, but not all, patients with subnormal serum cobalamin levels. Cobalamin deficiency sufficiently severe to cause megaloblastic anemia or neuropathy is rare.
Zollinger-Ellison Syndrome Malabsorption of cobalamin has been reported in the Zollinger-Ellison syndrome. It is thought that there is a failure to release cobalamin from R-binding protein due to inactivation of pancreatic trypsin by high acidity, as well as interference with IF binding of cobalamin.
Radiotherapy Both total-body irradiation and local radiotherapy to the ileum (e.g., as a complication of radiotherapy for carcinoma of the cervix) may cause malabsorption of cobalamin.
Graft-versus-Host Disease This commonly affects the small intestine. Malabsorption of cobalamin due to abnormal gut flora, as well as damage to ileal mucosa, is common.
Drugs The drugs that have been reported to cause malabsorption of cobalamin are listed in Table 105-4. However, megaloblastic anemia due to these drugs is rare.
ABNORMALITIES OF COBALAMIN METABOLISM
Congenital Transcobalamin II Deficiency or Abnormality Infants with TC II deficiency usually present with megaloblastic anemia within a few weeks of birth. Serum cobalamin and folate levels are normal, but the anemia responds to massive (e.g., 1 mg three times weekly) injections of cobalamin. Some cases show neurologic complications. The protein may be present but functionally inert. Genetic abnormalities found include mutations of an intra-exonic cryptic splice site, extensive deletion, single nucleotide deletion, nonsense mutation, and an RNA editing defect. Malabsorption of cobalamin occurs in all cases, and serum immunoglobulins are usually reduced. Failure to institute adequate cobalamin therapy or treatment with folic acid may lead to neurologic damage.
Congenital Methylmalonic Acidemia and Aciduria Infants with this abnormality are ill from birth with vomiting, failure to thrive, severe metabolic acidosis, ketosis, and mental retardation. Anemia, if present, is normocytic and normoblastic. The condition may be due to a functional defect in either mitochondrial methylmalonyl CoA mutase or its cofactor adocobalamin. Mutations in the methylmalonyl CoA mutase are not responsive, or only poorly responsive, to treatment with cobalamin. A proportion of infants with failure of adocobalamin synthesis respond to cobalamin in large doses. Some children have combined methylmalonic aciduria and homocystinuria due to defective formation of both cobalamin coenzymes. This usually presents in the first year of life with feeding difficulties, developmental delay, microcephaly, seizures, hypotonia, and megaloblastic anemia.
Acquired Abnormality of Cobalamin Metabolism: Nitrous Oxide Inhalation Nitrous oxide (N2O) irreversibly oxidizes methylcobalamin to an inactive precursor; this inactivates methionine synthase. Megaloblastic anemia has occurred in patients undergoing prolonged N2O anesthesia (e.g., in intensive care units). A neuropathy resembling cobalamin neuropathy has been described in dentists and anesthetists who are exposed repeatedly to N2O. Methylmalonic aciduria does not occur as adocobalamin is not inactivated by N2O.
CAUSES OF FOLATE DEFICIENCY
|
CAUSES OF FOLATE DEFICIENCY |
aIn severely folate-deficient patients with causes other than those listed under Dietary, poor dietary intake is often present. bDrugs inhibiting dihydrofolate reductase are discussed in the text.
NUTRITIONAL
Dietary folate deficiency is common. Indeed, in most patients with folate deficiency a nutritional element is present. Certain individuals are particularly prone to have diets containing inadequate amounts of folate (Table 128-5). In the United States and other countries where fortification of the diet with folic acid has been adopted, the prevalence of folate deficiency has dropped dramatically and is now almost restricted to high-risk groups with increased folate needs. Nutritional folate deficiency occurs in kwashiorkor and scurvy and in infants with repeated infections or those who are fed solely on goats’ milk, which has a low folate content.
MALABSORPTION
Malabsorption of dietary folate occurs in tropical sprue and in gluten-induced enteropathy. In the rare congenital recessive syndrome of selective malabsorption of folate due to mutation of the proton-coupled folate transporter (PCFT), there is an associated defect of folate transport into the cerebrospinal fluid, and these patients show megaloblastic anemia, which responds to physiologic doses of folic acid given parenterally but not orally. They also show mental retardation, convulsions, and other central nervous system abnormalities. Minor degrees of malabsorption may also occur after jejunal resection or partial gastrectomy, in Crohn’s disease, and in systemic infections, but in these conditions, if severe deficiency occurs, it is usually largely due to poor nutrition. Malabsorption of folate has been described in patients receiving sulfasalazine (Salazopyrin), cholestyramine, and triamterene.
EXCESS UTILIZATION OR LOSS
Pregnancy Folate requirements are increased by 200–300 μg to ~400 μg daily in a normal pregnancy, partly because of transfer of the vitamin to the fetus but mainly because of increased folate catabolism due to cleavage of folate coenzymes in rapidly proliferating tissues. Megaloblastic anemia due to this deficiency is prevented by prophylactic folic acid therapy. It occurred in 0.5% of pregnancies in the UK and other Western countries before prophylaxis with folic acid, but the incidence is much higher in countries where the general nutritional status is poor.
Prematurity A newborn infant, whether full term or premature, has higher serum and red cell folate concentrations than does an adult. However, a newborn infant’s demand for folate has been estimated to be up to 10 times that of adults on a weight basis, and the neonatal folate level falls rapidly to the lowest values at about 6 weeks of age. The falls are steepest and are liable to reach subnormal levels in premature babies, a number of whom develop megaloblastic anemia responsive to folic acid at about 4–6 weeks of age. This occurs particularly in the smallest babies (<1500 g birth weight) and those who have feeding difficulties or infections or have undergone multiple exchange transfusions. In these babies, prophylactic folic acid should be given.
Hematologic Disorders Folate deficiency frequently occurs in chronic hemolytic anemia, particularly in sickle cell disease, autoimmune hemolytic anemia, and congenital spherocytosis. In these and other conditions of increased cell turnover (e.g., myelofibrosis, malignancies), folate deficiency arises because it is not completely reutilized after performing coenzyme functions.
Inflammatory Conditions Chronic inflammatory diseases such as tuberculosis, rheumatoid arthritis, Crohn’s disease, psoriasis, exfoliative dermatitis, bacterial endocarditis, and chronic bacterial infections cause deficiency by reducing the appetite and increasing the demand for folate. Systemic infections also may cause malabsorption of folate. Severe deficiency is virtually confined to the patients with the most active disease and the poorest diet.
Homocystinuria This is a rare metabolic defect in the conversion of homocysteine to cystathionine. Folate deficiency occurring in most of these patients may be due to excessive utilization because of compensatory increased conversion of homocysteine to methionine.
Long-Term Dialysis Because folate is only loosely bound to plasma proteins, it is easily removed from plasma by dialysis. In patients with anorexia, vomiting, infections, and hemolysis, folate stores are particularly likely to become depleted. Routine folate prophylaxis is now given.
Congestive Heart Failure, Liver Disease Excess urinary folate losses of >100 μg per day may occur in some of these patients. The explanation appears to be release of folate from damaged liver cells.
ANTIFOLATE DRUGS
A large number of epileptics who are receiving long-term therapy with phenytoin or primidone, with or without barbiturates, develop low serum and red cell folate levels. The exact mechanism is unclear. Alcohol may also be a folate antagonist, as patients who are drinking spirits may develop megaloblastic anemia that will respond to normal quantities of dietary folate or to physiologic doses of folic acid only if alcohol is withdrawn. Macrocytosis of red cells is associated with chronic alcohol intake even when folate levels are normal. Inadequate folate intake is the major factor in the development of deficiency in spirit-drinking alcoholics. Beer is relatively folate-rich in some countries, depending on the technique used for brewing.
The drugs that inhibit DHF reductase include methotrexate, pyrimethamine, and trimethoprim. Methotrexate has the most powerful action against the human enzyme, whereas trimethoprim is most active against the bacterial enzyme and is likely to cause megaloblastic anemia only when used in conjunction with sulfamethoxazole in patients with preexisting folate or cobalamin deficiency. The activity of pyrimethamine is intermediate. The antidote to these drugs is folinic acid (5-formyl-THF).
CONGENITAL ABNORMALITIES OF FOLATE METABOLISM
Some infants with congenital defects of folate enzymes (e.g., cyclohydrolase or methionine synthase) have had megaloblastic anemia.
DIAGNOSIS OF COBALAMIN AND FOLATE DEFICIENCIES
The diagnosis of cobalamin or folate deficiency has traditionally depended on the recognition of the relevant abnormalities in the peripheral blood and analysis of the blood levels of the vitamins.
COBALAMIN DEFICIENCY
Serum Cobalamin This is measured by an automated enzyme-linked immunosorbent assay (ELISA) or competitive-binding luminescence assay (CBLA). Normal serum levels range from 118–148 pmol/L (160–200 ng/L) to ~738 pmol/L (1000 ng/L). In patients with megaloblastic anemia due to cobalamin deficiency, the level is usually <74 pmol/L (100 ng/L). In general, the more severe the deficiency, the lower is the serum cobalamin level. In patients with spinal cord damage due to the deficiency, levels are very low even in the absence of anemia. Values between 74 and 148 pmol/L (100 and 200 ng/L) are regarded as borderline. They may occur, for instance, in pregnancy, in patients with megaloblastic anemia due to folate deficiency. They may also be due to heterozygous, homozygous, or compound heterozygous mutations of the gene TCN1 that codes for haptocorrin (transcobalamin I). There is no clinical or hematologic abnormality. The serum cobalamin level is sufficiently robust, cost-effective, and most convenient to rule out cobalamin deficiency in the vast majority of patients suspected of having this problem. However, problems have arisen with commercial CBLA assays involving intrinsic factor in PA patients with intrinsic antibodies in serum. These antibodies may cause false normal serum vitamin B12 levels in up to 50% of cases tested. Where clinical indications of PA are strong, a normal serum vitamin B12 does not rule out the diagnosis. Serum MMA levels will be elevated in PA (see below).
Serum Methylmalonate and Homocysteine In patients with cobalamin deficiency sufficient to cause anemia or neuropathy, the serum MMA level is raised. Sensitive methods for measuring MMA and homocysteine in serum have been introduced and recommended for the early diagnosis of cobalamin deficiency, even in the absence of hematologic abnormalities or subnormal levels of serum cobalamin. Serum MMA levels fluctuate, however, in patients with renal failure. Mildly elevated serum MMA and/or homocysteine levels occur in up to 30% of apparently healthy volunteers, with serum cobalamin levels up to 258 pmol/L (350 ng/L) and normal serum folate levels; 15% of elderly subjects, even with cobalamin levels >258 pmol/L (>350 ng/L), have this pattern of raised metabolite levels. These findings bring into question the exact cutoff points for normal MMA and homocysteine levels. It is also unclear at present whether these mildly raised metabolite levels have clinical consequences.
Serum homocysteine is raised in both early cobalamin and folate deficiency but may be raised in other conditions, e.g., chronic renal disease, alcoholism, smoking, pyridoxine deficiency, hypothyroidism, and therapy with steroids, cyclosporine, and other drugs. Levels are also higher in serum than in plasma, in men than in premenopausal women, in women taking hormone replacement therapy or in oral contraceptive users, and in elderly persons and patients with several inborn errors of metabolism affecting enzymes in trans-sulfuration pathways of homocysteine metabolism. Thus, homocysteine levels must be carefully interpreted for diagnosis of cobalamin or folate deficiency.
Tests for the Cause of Cobalamin Deficiency Only vegans, strict vegetarians, or people living on a totally inadequate diet will become vitamin B12 deficient because of inadequate intake. Studies of cobalamin absorption once were widely used, but difficulty in obtaining radioactive cobalamin and ensuring that IF preparations are free of viruses has made these tests obsolete. Tests to diagnose PA include serum gastrin, which is raised; serum pepsinogen I, which is low in PA (90–92%) but also in other conditions; and gastric endoscopy. Tests for IF and parietal cell antibodies are also used, as well as tests for individual intestinal diseases.
FOLATE DEFICIENCY
Serum Folate This is also measured by an ELISA technique. In most laboratories, the normal range is from 11 nmol/L (2 μg/L) to ~82 nmol/L (15 μg/L). The serum folate level is low in all folate-deficient patients. It also reflects recent diet. Because of this, serum folate may be low before there is hematologic or biochemical evidence of deficiency. Serum folate rises in severe cobalamin deficiency because of the block in conversion of MTHF to THF inside cells; raised levels have also been reported in the intestinal stagnant loop syndrome due to absorption of bacterially synthesized folate.
Red Cell Folate The red cell folate assay is a valuable test of body folate stores. It is less affected than the serum assay by recent diet and traces of hemolysis. In normal adults, concentrations range from 880–3520 μmol/L (160–640 μg/L) of packed red cells. Subnormal levels occur in patients with megaloblastic anemia due to folate deficiency but also in nearly two-thirds of patients with severe cobalamin deficiency. False-normal results may occur if a folate-deficient patient has received a recent blood transfusion or if a patient has a raised reticulocyte count. Serum homocysteine assay is discussed earlier.
Tests for the Cause of Folate Deficiency The diet history is important. Tests for transglutaminase antibodies are performed to confirm or exclude celiac disease. If positive, duodenal biopsy is needed. An underlying disease causing increased folate breakdown should also be excluded.
MEGALOBLASTIC ANEMIA NOT DUE TO COBALAMIN OR FOLATE DEFICIENCY OR ALTERED METABOLISM
This may occur with many antimetabolic drugs (e.g., hydroxyurea, cytosine arabinoside, 6-mercaptopurine) that inhibit DNA replication. Antiviral nucleoside analogues used in treatment of HIV infection may also cause macrocytosis and megaloblastic marrow changes. In the rare disease orotic aciduria, two consecutive enzymes in purine synthesis are defective. The condition responds to therapy with uridine, which bypasses the block. In thiamine-responsive megaloblastic anemia, there is a genetic defect in the high-affinity thiamine transport (SLC19A2) gene. This causes defective RNA ribose synthesis through impaired activity of transketolase, a thiamine-dependent enzyme in the pentose cycle. This leads to reduced nucleic acid production. It may be associated with diabetes mellitus and deafness and the presence of many ringed sideroblasts in the marrow. The explanation is unclear for megaloblastic changes in the marrow in some patients with acute myeloid leukemia and myelodysplasia.
129 |
Hemolytic Anemias and Anemia Due to Acute Blood Loss |
DEFINITIONS
A finite life span is a distinct characteristic of red cells. Hence, a logical, time-honored classification of anemias is in three groups: (1) decreased production of red cells, (2) increased destruction of red cells, and (3) acute blood loss. Decreased production is covered in Chaps. 126, 128, and 130; increased destruction and acute blood loss are covered in this chapter.
All patients who are anemic as a result of either increased destruction of red cells or acute blood loss have one important element in common: the anemia results from overconsumption of red cells from the peripheral blood, whereas the supply of cells from the bone marrow is normal (indeed, it is usually increased). On the other hand, these two groups differ in that physical loss of red cells from the bloodstream or from the body itself, as in acute hemorrhage, is fundamentally different from destruction of red cells within the body, as in hemolytic anemias. Therefore, the clinical aspects and pathophysiology of anemia in these two groups of patients are quite different, and they will be considered separately.
HEMOLYTIC ANEMIAS
With respect to primary etiology, anemias due to increased destruction of red cells, which we know as hemolytic anemias (HAs), may be inherited or acquired; from a clinical point of view, they may be more acute or more chronic, and they may vary from mild to very severe; the site of hemolysis may be predominantly intravascular or extravascular. With respect to mechanisms, HAs may be due to intracorpuscular causes or to extracorpuscular causes (Table 129-1). But before reviewing the individual types of HA, it is appropriate to consider what they have in common.
|
CLASSIFICATION OF HEMOLYTIC ANEMIASa |

GENERAL CLINICAL AND LABORATORY FEATURES
The clinical presentation of a patient with anemia is greatly influenced in the first place by whether the onset is abrupt or gradual, and HAs are no exception. A patient with autoimmune HA or with favism may be a medical emergency, whereas a patient with mild hereditary spherocytosis or with cold agglutinin disease may be diagnosed after years. This is due in large measure to the remarkable ability of the body to adapt to anemia when it is slowly progressing (Chap. 77).
What differentiates HAs from other anemias is that the patient has signs and symptoms arising directly from hemolysis (Table 129-2). At the clinical level, the main sign is jaundice; in addition, the patient may report discoloration of the urine. In many cases of HA, the spleen is enlarged, because it is a preferential site of hemolysis; and in some cases, the liver may be enlarged as well. In all severe congenital forms of HA, there may also be skeletal changes due to overactivity of the bone marrow (although they are never as severe as they are in thalassemia).
|
FEATURES COMMON TO MOST PATIENTS WITH A HEMOLYTIC DISORDER |
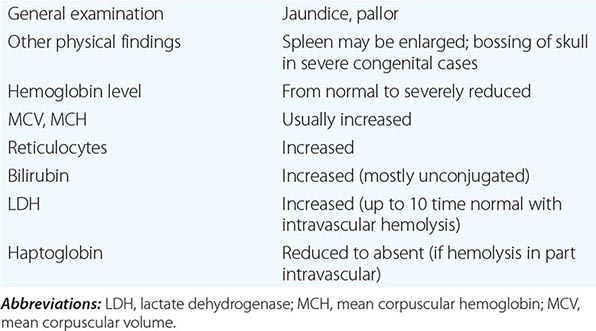
The laboratory features of HA are related to hemolysis per se and the erythropoietic response of the bone marrow. Hemolysis regularly produces an increase in unconjugated bilirubin and aspartate aminotransferase (AST) in the serum; urobilinogen will be increased in both urine and stool. If hemolysis is mainly intravascular, the telltale sign is hemoglobinuria (often associated with hemosiderinuria); in the serum, there is hemoglobin, lactate dehydrogenase (LDH) is increased, and haptoglobin is reduced. In contrast, the bilirubin level may be normal or only mildly elevated. The main sign of the erythropoietic response by the bone marrow is an increase in reticulocytes (a test all too often neglected in the initial workup of a patient with anemia). Usually the increase will be reflected in both the percentage of reticulocytes (the more commonly quoted figure) and the absolute reticulocyte count (the more definitive parameter). The increased number of reticulocytes is associated with an increased mean corpuscular volume (MCV) in the blood count. On the blood smear, this is reflected in the presence of macrocytes; there is also polychromasia, and sometimes one sees nucleated red cells. In most cases, a bone marrow aspirate is not necessary in the diagnostic workup; if it is done, it will show erythroid hyperplasia. In practice, once an HA is suspected, specific tests will usually be required for a definitive diagnosis of a specific type of HA.
GENERAL PATHOPHYSIOLOGY
The mature red cell is the product of a developmental pathway that brings the phenomenon of differentiation to an extreme. An orderly sequence of events produces synchronous changes, whereby the gradual accumulation of a huge amount of hemoglobin in the cytoplasm (to a final level of 340 g/L, i.e., about 5 mM) goes hand in hand with the gradual loss of cellular organelles and of biosynthetic abilities. In the end, the erythroid cell undergoes a process that has features of apoptosis, including nuclear pyknosis and actual loss of the nucleus. However, the final result is more altruistic than suicidal; the cytoplasmic body, instead of disintegrating, is now able to provide oxygen to all cells in the human organism for some remaining 120 days of the red cell life span.
As a result of this unique process of differentiation and maturation, intermediary metabolism is drastically curtailed in mature red cells (Fig. 129-1); for instance, cytochrome-mediated oxidative phosphorylation has been lost with the loss of mitochondria (through a process of physiologic autophagy); therefore, there is no backup to anaerobic glycolysis, which in the red cell is the only provider of adenosine triphosphate (ATP). Also the capacity of making protein has been lost with the loss of ribosomes. This places the cell’s limited metabolic apparatus at risk, because if any protein component deteriorates, it cannot be replaced, as it would be in most other cells; and in fact the activity of most enzymes gradually decreases as red cells age. At the same time, during their long time in circulation, various red cell components inevitably accumulate damage; in senescent red cells, the membrane protein band 3 molecules (see below and Fig. 129-1), having bound hemichromes on their intracellular domains, tend to cluster. Now they bind anti–band 3 IgG antibodies (present in most people) and C3 complement fragments; thus they become opsonized and are eventually removed by phagocytosis in the reticuloendothelial system.
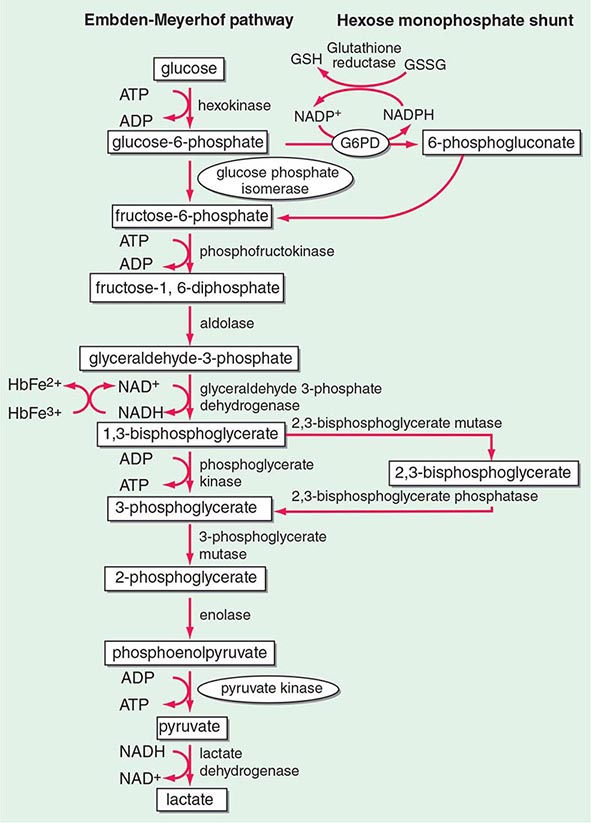
FIGURE 129-1 Red blood cell (RBC) metabolism. The Embden-Meyerhof pathway (glycolysis) generates ATP for energy and membrane maintenance. The generation of NADPH maintains hemoglobin in a reduced state. The hexose monophosphate shunt generates NADPH that is used to reduce glutathione, which protects the red cell against oxidant stress. Regulation of 2,3-bisphosphoglycerate levels is a critical determinant of oxygen affinity of hemoglobin. Enzyme deficiency states in order of prevalence: glucose 6-phosphate dehydrogenase (G6PD) > pyruvate kinase > glucose-6-phosphate isomerase > rare deficiencies of other enzymes in the pathway. The more common enzyme deficiencies are encircled.
Another consequence of the relative simplicity of red cells is that they have a very limited range of ways to manifest distress under hardship; in essence, any sort of metabolic failure will eventually lead either to structural damage to the membrane or to failure of the cation pump. In either case, the life span of the red cell is reduced, which is the definition of a hemolytic disorder. If the rate of red cell destruction exceeds the capacity of the bone marrow to produce more red cells, the hemolytic disorder will manifest as HA.
Thus, the essential pathophysiologic process common to all HAs is an increased red cell turnover; and in many HAs, this is due at least in part to an acceleration of the senescence process described above. The gold standard for proving that the life span of red cells is reduced (compared to the normal value of about 120 days) is a red cell survival study, which can be carried out by labeling the red cells with 51Cr and measuring residual radioactivity over several days or weeks: however, this classic test is now available in very few centers, and it is rarely necessary. If the hemolytic event is transient, it does not usually cause any long-term consequences, except for an increased requirement for erythropoietic factors, particularly folic acid. However, if hemolysis is recurrent or persistent, the increased bilirubin production favors the formation of gallstones. If a considerable proportion of hemolysis takes place in the spleen, as is often the case, splenomegaly may become increasingly a feature, and hypersplenism may develop, with consequent neutropenia and/or thrombocytopenia.
The increased red cell turnover also has metabolic consequences. In normal subjects, the iron from effete red cells is very efficiently recycled by the body; however, with chronic intravascular hemolysis, the persistent hemoglobinuria will cause considerable iron loss, needing replacement. With chronic extravascular hemolysis, the opposite problem, iron overload, is more common, especially if the patient needs frequent blood transfusions. Chronic iron overload will cause secondary hemochromatosis; this will cause damage particularly to the liver, eventually leading to cirrhosis, and to the heart muscle, eventually causing heart failure.
Compensated Hemolysis Versus Hemolytic Anemia Red cell destruction is a potent stimulus for erythropoiesis, which is mediated by erythropoietin (EPO) produced by the kidney. This mechanism is so effective that in many cases the increased output of red cells from the bone marrow can fully balance an increased destruction of red cells. In such cases, we say that hemolysis is compensated. The pathophysiology of compensated hemolysis is similar to what we have just described, except there is no anemia. This notion is important from the diagnostic point of view, because a patient with a hemolytic condition, even an inherited one, may present without anemia; and it is also important from the point of view of management, because compensated hemolysis may become “decompensated,” i.e., anemia may suddenly appear, in certain circumstances, for instance in pregnancy, folate deficiency, or renal failure interfering with adequate EPO production. Another general feature of chronic HAs is seen when any intercurrent condition, such as an acute infection, depresses erythropoiesis. When this happens, in view of the increased rate of red cell turnover, the effect will be predictably much more marked than in a person who does not have hemolysis. The most dramatic example is infection by parvovirus B19, which may cause a rather precipitous fall in hemoglobin—an occurrence sometimes referred to as aplastic crisis.
INHERITED HEMOLYTIC ANEMIAS
There are three essential components in the red cell: (1) hemoglobin, (2) the membrane-cytoskeleton complex, and (3) the metabolic machinery necessary to keep hemoglobin and the membrane-cytoskeleton complex in working order. Diseases caused by abnormalities of hemoglobin, or hemoglobinopathies, are covered in Chap. 127. Here we will deal with diseases of the other two components.
Hemolytic Anemias due to Abnormalities of the Membrane-Cytoskeleton Complex The detailed architecture of the red cell membrane is complex, but its basic design is relatively simple (Fig. 129-2). The lipid bilayer incorporates phospholipids and cholesterol, and it is spanned by a number of proteins that have their hydrophobic transmembrane domain(s) embedded in the membrane; most of these proteins also extend to both the outside (extracellular domains) and the inside of the cell (cytoplasmic domains). Other proteins are tethered to the membrane through a glycosylphosphatidylinositol (GPI) anchor; these have only an extracellular domain, and they include ion channels, receptors for complement components, and receptors for other ligands. The most abundant red cell membrane proteins are glycophorins and the so-called band 3, an anion transporter. The extracellular domains of many of these proteins are heavily glycosylated, and they carry antigenic determinants that correspond to blood groups. Underneath the membrane, and tangential to it, is a network of other proteins that make up the cytoskeleton. The main cytoskeletal protein is spectrin, the basic unit of which is a dimer of α-spectrin and β-spectrin. The membrane is physically linked to the cytoskeleton by a third set of proteins (including ankyrin and the so-called band 4.1 and band 4.2), which thus make these two structures intimately connected to each other.
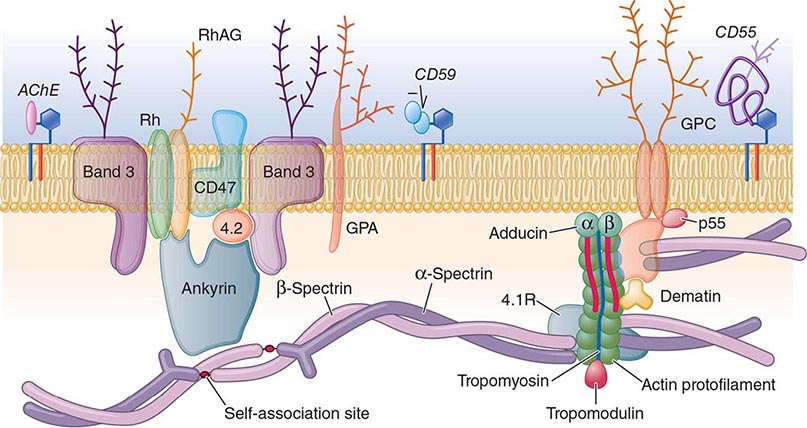
FIGURE 129-2 The red cell membrane. In this figure, one sees, within the lipid bilayer, several membrane proteins, of which band 3 (anion exchanger 1 [AE1]) is the most abundant; the α-β spectrin dimers that associate to form most of the cytoskeleton; and several proteins (e.g., ankyrin) that connect the membrane to the cytoskeleton. In addition, as examples of glycosylphosphatidylinositol (GPI)-linked proteins, one sees acetylcholinesterase (AChE) and the two complement-regulatory proteins CD59 and CD55. The (nonrealistic) shapes of the protein moieties of the GPI-linked proteins are meant to indicate that they can be very different from each other and that, unlike with the other membrane proteins shown, the entire polypeptide chain is extracellular. Branched lines symbolize carbohydrate moiety of proteins. The molecules are obviously not drawn to the same scale. Additional explanations can be found in the text. (From N Young et al: Clinical Hematology. Copyright Elsevier, 2006; with permission.)
The membrane-cytoskeleton complex is so integrated that, not surprisingly, an abnormality of almost any of its components will be disturbing or disruptive, causing structural failure, which results ultimately in hemolysis. These abnormalities are almost invariably inherited mutations; thus, diseases of the membrane-cytoskeleton complex belong to the category of inherited HAs. Before the red cells lyse, they often exhibit more or less specific morphologic changes that alter the normal biconcave disk shape. Thus, the majority of the diseases in this group have been known for over a century as hereditary spherocytosis and hereditary elliptocytosis. Over the past 20 years, their molecular basis has been elucidated; it has emerged that both conditions can arise from mutations in several genes with considerable overlap (Fig. 129-3).
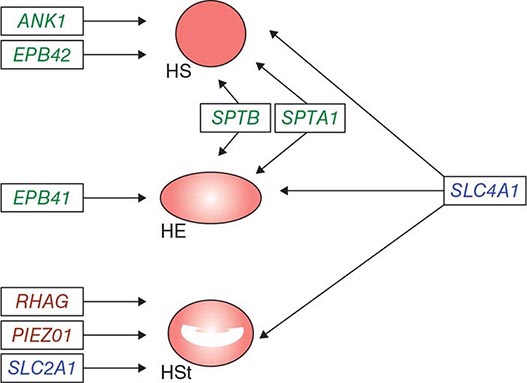
FIGURE 129-3 Hereditary spherocytosis (HS), hereditary elliptocytosis (HE), and hereditary stomatocytosis (HSt) are three morphologically distinct forms of congenital hemolytic anemia. It has emerged that each one can arise from mutation of one of several genes and that different mutations of the same gene can give one or another form. (See also Table 129-3.)
HEREDITARY SPHEROCYTOSIS (HS) This is a relatively common type of genetically determined HA, with an estimated frequency of at least 1 in 5000. Its identification is credited to Minkowksy and Chauffard, who, at the end of the nineteenth century, reported families who had the presence of numerous spherocytes in the peripheral blood (Fig 129-4A). In vitro studies revealed that the red cells were abnormally susceptible to lysis in hypotonic media; indeed, the presence of osmotic fragility became the main diagnostic test for HS. Today we know that HS, thus defined, is genetically heterogeneous; i.e., it can arise from a variety of mutations in one of several genes (Table 129-3). It has been also recognized that the inheritance of HS is not always autosomal dominant (with the patient being heterozygous); indeed, some of the most severe forms are instead autosomal recessive (with the patient being homozygous).
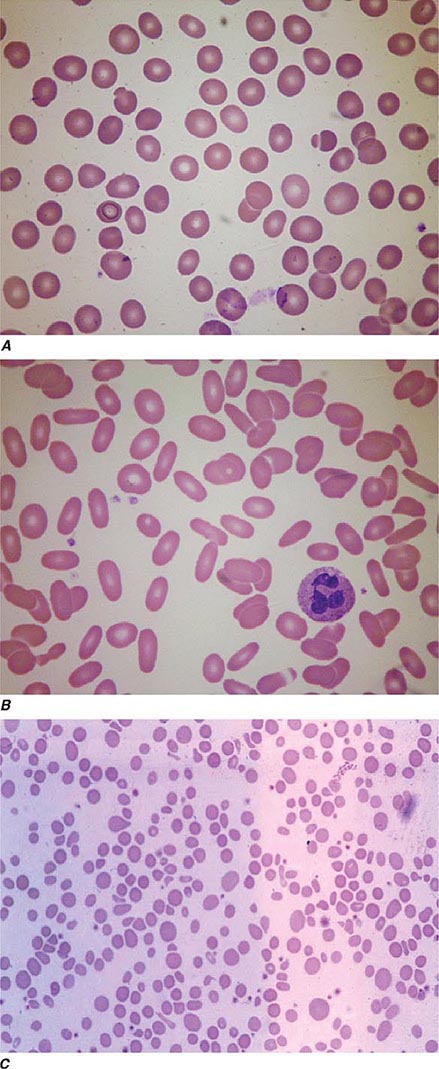
FIGURE 129-4 Peripheral blood smear from patients with membrane-cytoskeleton abnormalities. A. Hereditary spherocytosis. B. Hereditary elliptocytosis, heterozygote. C. Elliptocytosis, with both alleles of the α-spectrin gene mutated.
|
INHERITED DISEASES OF THE RED CELL MEMBRANE-CYTOSKELETON COMPLEX |
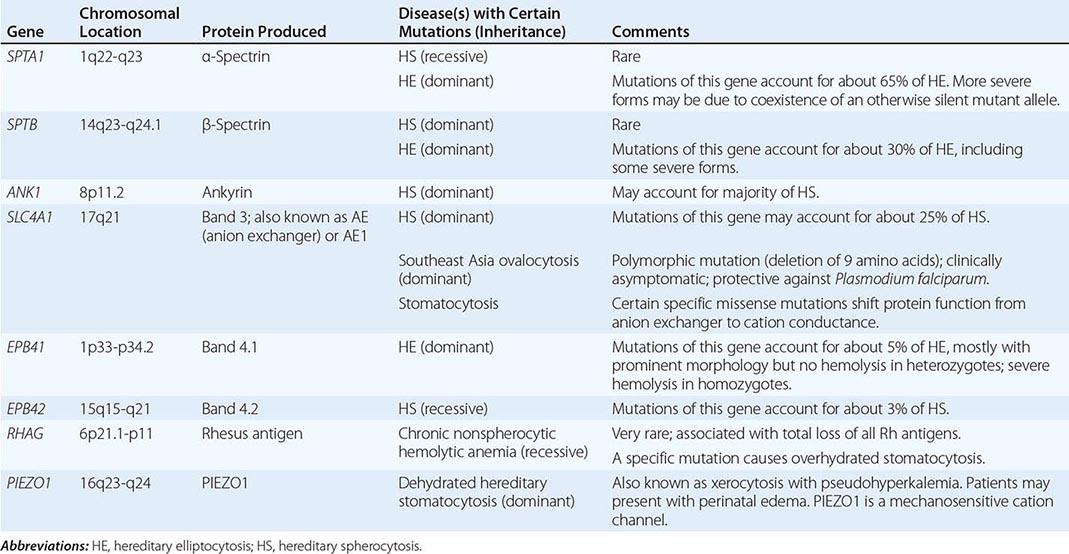
Clinical presentation and diagnosis The spectrum of clinical severity of HS is broad. Severe cases may present in infancy with severe anemia, whereas mild cases may present in young adults or even later in life. The main clinical findings are jaundice, an enlarged spleen, and often gallstones; indeed, it may be the finding of gallstones in a young person that triggers diagnostic investigations.
The variability in clinical manifestations that is observed among patients with HS is largely due to the different underlying molecular lesions (Table 129-3). Not only are mutations of several genes involved, but also individual mutations of the same gene can also give very different clinical manifestations. In milder cases, hemolysis is often compensated (see above), and this may cause variation in time even in the same patient, due to the fact that intercurrent conditions (e.g., pregnancy, infection) may cause decompensation. The anemia is usually normocytic, with the characteristic morphology that gives the disease its name. An increased mean corpuscular hemoglobin concentration (MCHC) on an ordinary blood count report should raise the suspicion of HS, because HS is almost the only condition in which this abnormality occurs. It has been apparent for a long time that the spleen plays a special role in HS through a dual mechanism. On one hand, like in many other HAs, the spleen itself is a major site of destruction; on the other hand, transit through the splenic circulation makes the defective red cells more spherocytic and, therefore, accelerates their demise, even though that may take place elsewhere.
When there is a family history, it is usually easy to make a diagnosis based on features of HA and typical red cell morphology. However, there may be no family history for at least two reasons. First, the patient may have a de novo mutation, i.e., a mutation that has taken place in a germ cell of one of his parents or early after zygote formation. Second, the patient may have a recessive form of HS (Table 129-3). In such cases, more extensive laboratory investigations are required, including osmotic fragility, the acid glycerol lysis test, the eosin-5′-maleimide (EMA)–binding test, and SDS-gel electrophoresis of membrane proteins; these tests are usually carried out in laboratories with special expertise in this area. Sometimes a definitive diagnosis can be obtained only by molecular studies demonstrating a mutation in one of the genes underlying HS (Table 129-3).
HEREDITARY ELLIPTOCYTOSIS (HE) HE is at least as heterogeneous as HS, both from the genetic point of view (Table 129-3, Fig. 129-3) and from the clinical point of view. Again, it is the shape of the red cells (Fig. 129-4B) that gives the name to the condition, but there is no direct correlation between the elliptocytic morphology and clinical severity. In fact, some mild or even asymptomatic cases may have nearly 100% elliptocytes, whereas in severe cases, all kinds of bizarre poikilocytes can predominate. Clinical features and recommended management are similar to those outlined above for HS. Although the spleen may not have the specific role it has in HS, in severe cases, splenectomy may be beneficial. The prevalence of HE causing clinical disease is similar to that of HS. However, an in-frame deletion of nine amino acids in the SLC4A1 gene encoding band 3, causing the so-called Southeast Asia ovalocytosis, has a frequency of up to 7% in certain populations, presumably as a result of malaria selection; it is asymptomatic in heterozygotes and probably lethal in homozygotes.
Disorders of Cation Transport These rare conditions with autosomal dominant inheritance are characterized by increased intracellular sodium in red cells, with concomitant loss of potassium; indeed, they are sometimes discovered through the incidental finding, in a blood test, of a high serum K+ (pseudohyperkalemia). In patients from some families, the cation transport disturbance is associated with gain of water; as a result, the red cells are overhydrated (low MCHC), and on a blood smear, the normally round-shaped central pallor is replaced by a linear-shaped central pallor, which has earned this disorder the name stomatocytosis (Fig. 129-3). In patients from other families, instead, the red cells are dehydrated (high MCHC), and their consequent rigidity has earned this disorder the name xerocytosis. One would surmise that in these disorders the primary defect may be in a cation transporter; indeed, xerocytosis results from mutations in PIEZO1. In other patients with stomatocytosis, mutations are found in other genes also related to solute transport (Table 129-3), including SLC4A1 (encoding band 3), the Rhesus gene RHAG, and the glucose transporter gene SLC2A1 responsible for a special form called cryohydrocytosis. Hemolysis can vary from relatively mild to quite severe. From the practical point of view, it is important to know that in stomatocytosis, splenectomy is strongly contraindicated because it has been followed in a significant proportion of cases by severe thromboembolic complications.
Enzyme Abnormalities When there is an important defect in the membrane or in the cytoskeleton, hemolysis is a direct consequence of the fact that the very structure of the red cell is abnormal. Instead, when one of the enzymes is defective, the consequences will depend on the precise role of that enzyme in the metabolic machinery of the red cell, which, in first approximation, has two important functions: (1) to provide energy in the form of ATP and (2) to prevent oxidative damage to hemoglobin and to other proteins by providing sufficient reductive potential; the key molecule for this is NADPH.
ABNORMALITIES OF THE GLYCOLYTIC PATHWAY Because red cells, in the course of their differentiation, have sacrificed not only their nucleus and their ribosomes, but also their mitochondria, they rely exclusively on the anaerobic portion of the glycolytic pathway for producing energy in the form of ATP. Most of the ATP is required by the red cell for cation transport against a concentration gradient across the membrane. If this fails, due to a defect of any of the enzymes of the glycolytic pathway (Table 129-4), the result will be hemolytic disease.
|
RED CELL ENZYME ABNORMALITIES CAUSING HEMOLYSIS |
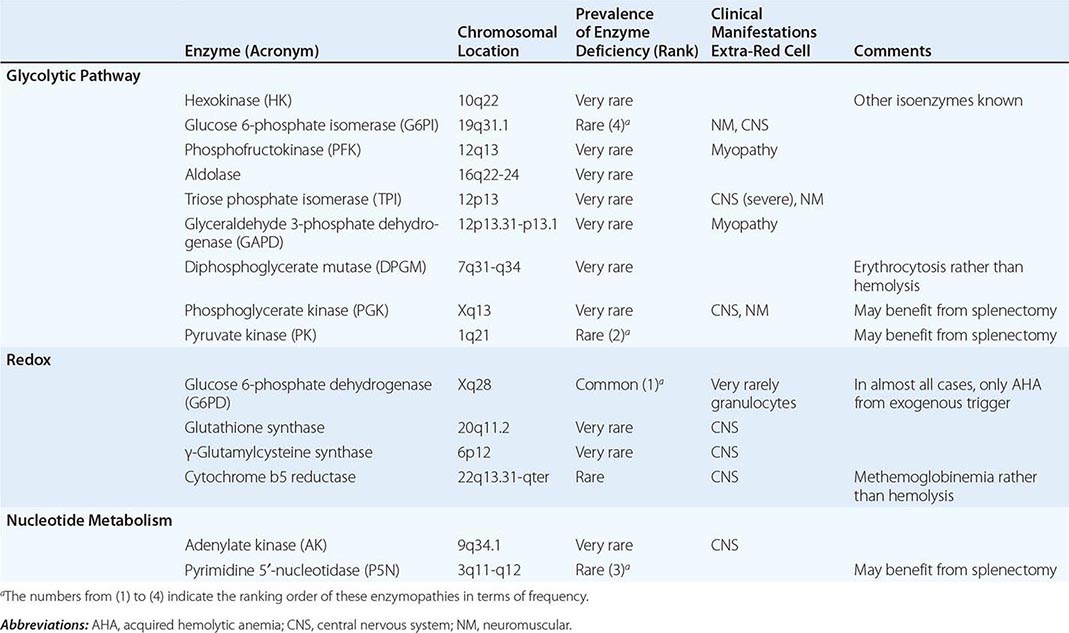
Pyruvate kinase deficiency Abnormalities of the glycolytic pathway are all inherited and all rare. Among them, deficiency of pyruvate kinase (PK) is the least rare, with an estimated prevalence in most populations of the order of 1:10,000. However, very recently, a polymorphic PK mutation (E277K) was found in some African populations, with heterozygote frequencies of 1–7%, suggesting that this may be another malaria-related polymorphism. The clinical picture of homozygous (or compound biallelic) PK deficiency is that of an HA that often presents in the newborn with neonatal jaundice; the jaundice persists, and it is usually associated with a very high reticulocytosis. The anemia is of variable severity; sometimes it is so severe as to require regular blood transfusion treatment, whereas sometimes it is mild, bordering on a nearly compensated hemolytic disorder. As a result, the diagnosis may be delayed, and in some cases, it is made, for instance, in a young woman during her first pregnancy, when the anemia may get worse. The delay in diagnosis may be also helped by the fact that the anemia is remarkably well tolerated, because the metabolic block at the last step in glycolysis causes an increase in bisphosphoglycerate (or DPG; Fig. 129-1), a major effector of the hemoglobin-oxygen dissociation curve; thus, the oxygen delivery to the tissues is enhanced, a remarkable compensatory feat.
Other glycolytic enzyme abnormalities All of these defects are rare to very rare (Table 129-4), and all cause hemolytic anemia with varying degrees of severity. It is not unusual for the presentation to be in the guise of severe neonatal jaundice, which may require exchange transfusion; if the anemia is less severe, it may present later in life, or it may even remain asymptomatic and be detected incidentally when a blood count is done for unrelated reasons. The spleen is often enlarged. When other systemic manifestations occur, they can involve the central nervous system (sometimes entailing severe mental retardation, particularly in the case of triose phosphate isomerase deficiency), the neuromuscular system, or both. This is not altogether surprising, if we consider that these are housekeeping genes. The diagnosis of hemolytic anemia is usually not difficult, thanks to the triad of normomacrocytic anemia, reticulocytosis, and hyperbilirubinemia. Enzymopathies should be considered in the differential diagnosis of any chronic Coombs-negative hemolytic anemia. Unlike with membrane disorders where the red cells show characteristic morphologic abnormalities, in most cases of glycolytic enzymopathies, these are conspicuous by their absence. A definitive diagnosis can be made only by demonstrating the deficiency of an individual enzyme by quantitative assays; these are carried out in only a few specialized laboratories. If a particular molecular abnormality is already known in the family, then one could test directly for that defect at the DNA level, thus bypassing the need for enzyme assays. Of course the time may be getting nearer when a patient will present with her or his exome already sequenced, and we will need to concentrate on which genes to look up within the file. The principles for the management of these conditions are similar as for PK deficiency. In one case of phosphoglycerate kinase deficiency, allogeneic bone marrow transplantation (BMT) effectively controlled the hematologic manifestations but did not reverse neurologic damage.
ABNORMALITIES OF REDOX METABOLISM
Glucose 6-phosphate dehydrogenase (G6PD) deficiency G6PD is a housekeeping enzyme critical in the redox metabolism of all aerobic cells (Fig. 129-1). In red cells, its role is even more critical, because it is the only source of NADPH, which directly and via glutathione (GSH) defends these cells against oxidative stress (Fig. 129-5). G6PD deficiency is a prime example of an HA due to interaction between an intracorpuscular cause and an extracorpuscular cause, because in the majority of cases hemolysis is triggered by an exogenous agent. Although a decrease in G6PD activity is present in most tissues of G6PD-deficient subjects, in other cells, the decrease is much less marked than in red cells, and it does not seem to impact on clinical expression.
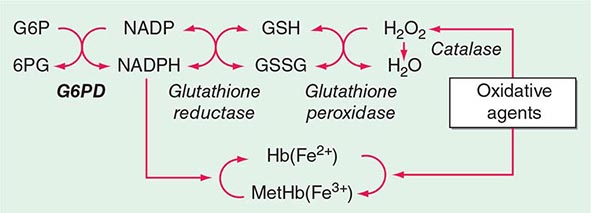
FIGURE 129-5 Diagram of redox metabolism in the red cell. 6PG, 6-phosphogluconate; G6P, glucose 6-phosphate; G6PD, glucose 6-phosphate dehydrogenase; GSH, reduced glutathione; GSSG, oxidized glutathione; Hb, hemoglobin; MetHb, methemoglobin; NADP, nicotinamide adenine dinucleotide phosphate; NADPH, reduced nicotinamide adenine dinucleotide phosphate.
GENETIC CONSIDERATIONS
![]() The G6PD gene is X-linked, and this has important implications. First, because males have only one G6PD gene (i.e., they are hemizygous for this gene), they must be either normal or G6PD deficient. By contrast, females, who have two G6PD genes, can be either normal or deficient (homozygous) or intermediate (heterozygous). As a result of the phenomenon of × chromosome inactivation, heterozygous females are genetic mosaics, with a highly variable ratio of G6PD-normal to G6PD-deficient cells and an equally variable degree of clinical expression; some heterozygotes can be just as affected as hemizygous males. The enzymatically active form of G6PD is either a dimer or a tetramer of a single protein subunit of 514 amino acids. G6PD-deficient subjects have been found invariably to have mutations in the coding region of the G6PD gene (Fig. 129-5). Almost all of the approximately 180 different mutations known are single missense point mutations, entailing single amino acid replacements in the G6PD protein. In most cases, these mutations cause G6PD deficiency by decreasing the in vivo stability of the protein; thus, the physiologic decrease in G6PD activity that takes place with red cell aging is greatly accelerated. In some cases, an amino acid replacement can also affect the catalytic function of the enzyme.
The G6PD gene is X-linked, and this has important implications. First, because males have only one G6PD gene (i.e., they are hemizygous for this gene), they must be either normal or G6PD deficient. By contrast, females, who have two G6PD genes, can be either normal or deficient (homozygous) or intermediate (heterozygous). As a result of the phenomenon of × chromosome inactivation, heterozygous females are genetic mosaics, with a highly variable ratio of G6PD-normal to G6PD-deficient cells and an equally variable degree of clinical expression; some heterozygotes can be just as affected as hemizygous males. The enzymatically active form of G6PD is either a dimer or a tetramer of a single protein subunit of 514 amino acids. G6PD-deficient subjects have been found invariably to have mutations in the coding region of the G6PD gene (Fig. 129-5). Almost all of the approximately 180 different mutations known are single missense point mutations, entailing single amino acid replacements in the G6PD protein. In most cases, these mutations cause G6PD deficiency by decreasing the in vivo stability of the protein; thus, the physiologic decrease in G6PD activity that takes place with red cell aging is greatly accelerated. In some cases, an amino acid replacement can also affect the catalytic function of the enzyme.
Among these mutations, those underlying chronic nonspherocytic hemolytic anemia (CNSHA; see below) are a discrete subset. This much more severe clinical phenotype can be ascribed in some cases to adverse qualitative changes (for instance, a decreased affinity for the substrate, glucose 6-phosphate) or simply to the fact that the enzyme deficit is more extreme, because of a more severe instability of the enzyme. For instance, a cluster of mutations map at or near the dimer interface, and clearly they compromise severely the formation of the dimer.
![]() Epidemiology G6PD deficiency is widely distributed in tropical and subtropical parts of the world (Africa, Southern Europe, the Middle East, Southeast Asia, and Oceania) (Fig. 129-6) and wherever people from those areas have migrated. A conservative estimate is that at least 400 million people have a G6PD deficiency gene. In several of these areas, the frequency of a G6PD deficiency gene may be as high as 20% or more. It would be quite extraordinary for a trait that causes significant pathology to spread widely and reach high frequencies in many populations without conferring some biologic advantage. Indeed, G6PD is one of the best-characterized examples of genetic polymorphisms in the human species. Clinical field studies and in vitro experiments strongly support the view that G6PD deficiency has been selected by Plasmodium falciparum malaria, by virtue of the fact that it confers a relative resistance against this highly lethal infection. Different G6PD variants underlie G6PD deficiency in different parts of the world. Some of the more widespread variants are G6PD Mediterranean on the shores of that sea, in the Middle East, and in India; G6PD A– in Africa and in Southern Europe; G6PD Vianchan and G6PD Mahidol in Southeast Asia; G6PD Canton in China; and G6PD Union worldwide. The heterogeneity of polymorphic G6PD variants is proof of their independent origin, and it supports the notion that they have been selected by a common environmental agent, in keeping with the concept of convergent evolution (Fig. 129-6).
Epidemiology G6PD deficiency is widely distributed in tropical and subtropical parts of the world (Africa, Southern Europe, the Middle East, Southeast Asia, and Oceania) (Fig. 129-6) and wherever people from those areas have migrated. A conservative estimate is that at least 400 million people have a G6PD deficiency gene. In several of these areas, the frequency of a G6PD deficiency gene may be as high as 20% or more. It would be quite extraordinary for a trait that causes significant pathology to spread widely and reach high frequencies in many populations without conferring some biologic advantage. Indeed, G6PD is one of the best-characterized examples of genetic polymorphisms in the human species. Clinical field studies and in vitro experiments strongly support the view that G6PD deficiency has been selected by Plasmodium falciparum malaria, by virtue of the fact that it confers a relative resistance against this highly lethal infection. Different G6PD variants underlie G6PD deficiency in different parts of the world. Some of the more widespread variants are G6PD Mediterranean on the shores of that sea, in the Middle East, and in India; G6PD A– in Africa and in Southern Europe; G6PD Vianchan and G6PD Mahidol in Southeast Asia; G6PD Canton in China; and G6PD Union worldwide. The heterogeneity of polymorphic G6PD variants is proof of their independent origin, and it supports the notion that they have been selected by a common environmental agent, in keeping with the concept of convergent evolution (Fig. 129-6).
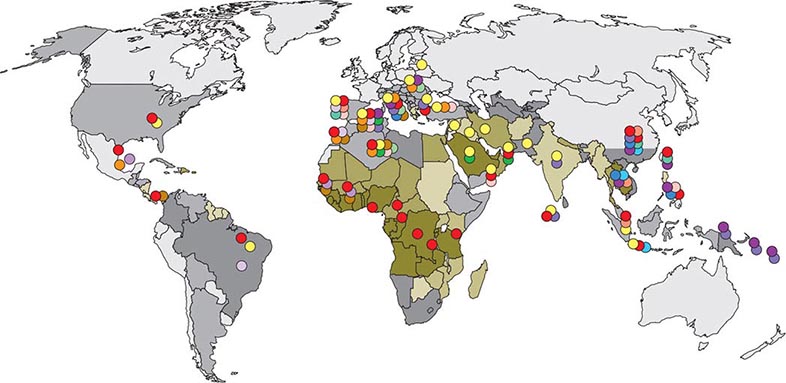
FIGURE 129-6 Epidemiology of glucose 6-phosphate dehydrogenase (G6PD) deficiency throughout the world. The different shadings indicate increasingly high levels of prevalence, up to about 20%; the different colored symbols indicate individual genetic variants of G6PD, each one having a different mutation. (From L Luzzatto et al, in C Scriver et al [eds]: The Metabolic & Molecular Bases of Inherited Disease, 8th ed. New York, McGraw-Hill, 2001.)
Clinical manifestations The vast majority of people with G6PD deficiency remain clinically asymptomatic throughout their lifetime; however, all of them have an increased risk of developing neonatal jaundice (NNJ) and a risk of developing acute HA (AHA) when challenged by a number of oxidative agents. NNJ related to G6PD deficiency is very rarely present at birth; the peak incidence of clinical onset is between day 2 and day 3, and in most cases, the anemia is not severe. However, NNJ can be very severe in some G6PD-deficient babies, especially in association with prematurity, infection, and/or environmental factors (such as naphthalene-camphor balls, which are used in babies’ bedding and clothing), and the risk of severe NNJ is also increased by the coexistence of a monoallelic or biallelic mutation in the uridyl transferase gene (UGT1A1; the same mutations are associated with Gilbert’s syndrome). If inadequately managed, NNJ associated with G6PD deficiency can produce kernicterus and permanent neurologic damage.
AHA can develop as a result of three types of triggers: (1) fava beans, (2) infections, and (3) drugs (Table 129-5). Typically, a hemolytic attack starts with malaise, weakness, and abdominal or lumbar pain. After an interval of several hours to 2–3 days, the patient develops jaundice and often dark urine. The onset can be extremely abrupt, especially with favism in children. The anemia is moderate to extremely severe, usually normocytic and normochromic, and due partly to intravascular hemolysis; hence, it is associated with hemoglobinemia, hemoglobinuria, high LDH, and low or absent plasma haptoglobin. The blood film shows anisocytosis, polychromasia, and spherocytes typical of hemolytic anemias. The most typical feature of G6PD deficiency is the presence of bizarre poikilocytes, with red cells that appear to have unevenly distributed hemoglobin (“hemighosts”) and red cells that appear to have had parts of them bitten away (“bite cells” or “blister cells”) (Fig. 129-7). A classical test, now rarely carried out, is supravital staining with methyl violet, which, if done promptly, reveals the presence of Heinz bodies (consisting of precipitates of denatured hemoglobin and hemichromes), which are regarded as a signature of oxidative damage to red cells (they are also seen with unstable hemoglobins). LDH is high, and so is the unconjugated bilirubin, indicating that there is also extravascular hemolysis. The most serious threat from AHA in adults is the development of acute renal failure (this is exceedingly rare in children). Once the threat of acute anemia is over and in the absence of comorbidity, full recovery from AHA associated with G6PD deficiency is the rule.
|
DRUGS THAT CARRY RISK OF CLINICAL HEMOLYSIS IN PERSONS WITH GLUCOSE 6-PHOSPHATE DEHYDROGENASE DEFICIENCY |
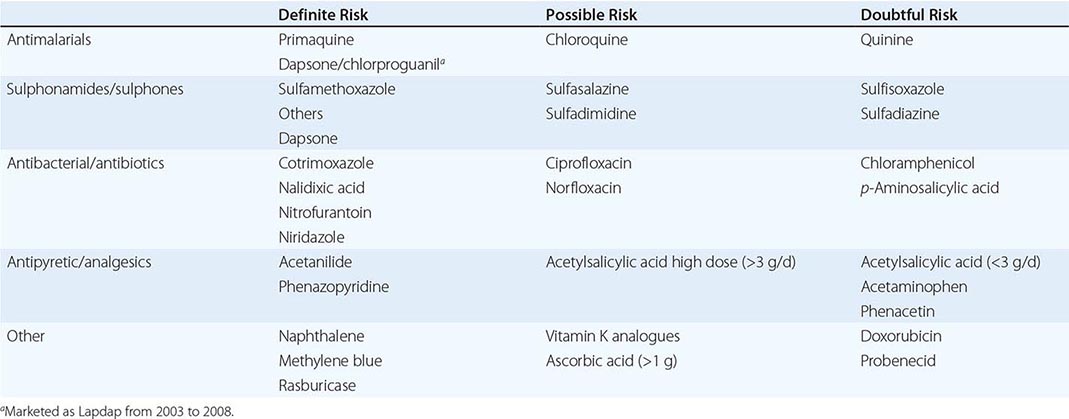
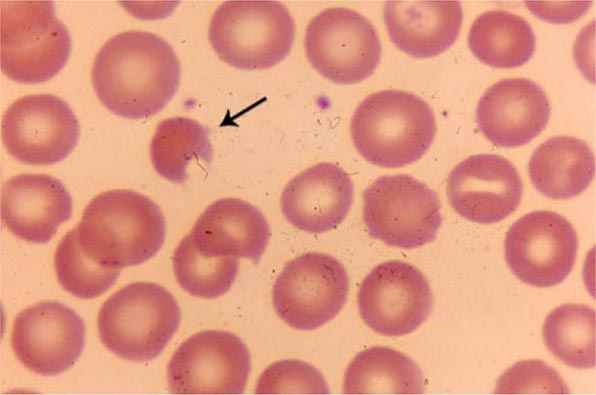
FIGURE 129-7 Peripheral blood smear from a glucose 6-phosphate dehydrogenase (G6PD)-deficient boy experiencing hemolysis. Note the red cells that are misshapen and called “bite” cells. (From MA Lichtman et al: Lichtman’s Atlas of Hematology: http://www.accessmedicine.com. Copyright © The McGraw-Hill Companies, Inc. All rights reserved.)
Although it was primaquine (PQ) that led to the discovery of G6PD deficiency, this drug has not been very prominent subsequently, because it is not necessary for the treatment of life-threatening P. falciparum malaria. Today there is a revival of interest in PQ because it is the only effective agent for eliminating the gametocytes of P. falciparum (thus preventing further transmission) and eliminating the hypnozoites of Plasmodium vivax (thus preventing endogenous relapse). In countries aiming to eliminate malaria, there may be a call for mass administration of PQ; this ought to be associated with G6PD testing. At the other end of the historic spectrum, the latest addition to the list of potentially hemolytic drugs (Table 129-5) is rasburicase; again G6PD testing ought to be made mandatory before giving this drug because fatal cases have been reported in newborns with kidney injury and in adults with tumor lysis syndrome.
A very small minority of subjects with G6PD deficiency have chronic nonspherocytic hemolytic anemia (CNSHA) of variable severity. The patient is nearly always a male, usually with a history of NNJ, who may present with anemia, unexplained jaundice, or gallstones later in life. The spleen may be enlarged. The severity of anemia ranges in different patients from borderline to transfusion dependent. The anemia is usually normomacrocytic, with reticulocytosis. Bilirubin and LDH are increased. Although hemolysis is, by definition, chronic in these patients, they are also vulnerable to acute oxidative damage, and therefore the same agents that can cause AHA in people with the ordinary type of G6PD deficiency will cause severe exacerbations in people with CNSHA associated with G6PD deficiency. In some cases of CNSHA, the deficiency of G6PD is so severe in granulocytes that it becomes rate-limiting for their oxidative burst, with consequent increased susceptibility to some bacterial infections.
Laboratory diagnosis The suspicion of G6PD deficiency can be confirmed by semiquantitative methods often referred to as screening tests, which are suitable for population studies and can correctly classify male subjects, in the steady state, as G6PD normal or G6PD deficient. However, in clinical practice, a diagnostic test is usually needed when the patient has had a hemolytic attack; this implies that the oldest, most G6PD-deficient red cells have been selectively destroyed, and young red cells, having higher G6PD activity, are being released into the circulation. Under these conditions, only a quantitative test can give a definitive result. In males, this test will identify normal hemizygotes and G6PD-deficient hemizygotes; among females, some heterozygotes will be missed, but those who are at most risk of hemolysis will be identified. Of course, G6PD deficiency also can be diagnosed by DNA testing.
Other abnormalities of the redox system As mentioned above, GSH is a key player in the defense against oxidative stress. Inherited defects of GSH metabolism are exceedingly rare, but each one can give rise to chronic HA (Table 129-4). A rare, peculiar, usually self-limited severe HA of the first month of life, called infantile poikilocytosis, may be associated with deficiency of glutathione peroxidase (GSHPX) due not to an inherited abnormality, but to transient nutritional deficiency of selenium, an element essential for the activity of GSHPX.
PYRIMIDINE 5′-NUCLEOTIDASE (P5N) DEFICIENCY P5N is a key enzyme in the catabolism of nucleotides arising from the degradation of nucleic acids that takes place in the final stages of erythroid cell maturation. How exactly its deficiency causes HA is not well understood, but a highly distinctive feature of this condition is a morphologic abnormality of the red cells known as basophilic stippling. The condition is rare, but it probably ranks third in frequency among red cell enzyme defects (after G6PD deficiency and PK deficiency). The anemia is lifelong, of variable severity, and may benefit from splenectomy.
Familial (Atypical) Hemolytic-Uremic Syndrome The term familial (atypical) hemolytic-uremic syndrome is used to designate a group of rare disorders, mostly affecting children, characterized by microangiopathic HA with presence of fragmented erythrocytes in the peripheral blood smear, thrombocytopenia (usually mild), and acute renal failure. (The word atypical is part of the phrase for historical reasons; it was hemolytic-uremic syndrome [HUS] caused by infection with Escherichia coli producing the Shiga toxin that was regarded as typical). The genetic basis of atypical HUS (aHUS) has been elucidated. Studies of >100 families have revealed that those family members who developed HUS had mutations in any one of several genes encoding complement regulatory proteins: complement factor H (CFH), CD46 or membrane cofactor protein (MCP), complement factor I (CFI), complement component C3, complement factor B (CFB), and thrombomodulin. Thus, whereas all other inherited HAs are due to intrinsic red cell abnormalities, this group is unique in that hemolysis results from an inherited defect external to red cells (Table 129-1). Because the regulation of the complement cascade has considerable redundancy, in the steady state, any of the above abnormalities can be tolerated. However, when an intercurrent infection or some other trigger activates complement through the alternative pathway, the deficiency of one of the complement regulators becomes critical. Endothelial cells get damaged, especially in the kidney; at the same time, and partly as a result of this, there will be brisk hemolysis (thus, the more common Shiga toxin–related HUS [Chap. 149] can be regarded as a phenocopy of aHUS). aHUS is a severe disease, with up to 15% mortality in the acute phase and up to 50% of cases progressing to end-stage renal disease. Not infrequently, aHUS undergoes spontaneous remission; but because its basis is an inherited abnormality, it is not surprising that, given renewed exposure to a trigger, the syndrome will tend to recur; when it does, the prognosis is always serious. The standard treatment has been plasma exchange, which will supply the deficient complement regulator. The anti-C5 complement inhibitor eculizumab (see below) was found to greatly ameliorate the microangiopathic picture, with improvement in platelet counts and in renal function, thus abrogating the need for plasma exchange. It remains to be seen for how long eculizumab treatment will have to be continued in individual patients and whether it will influence the controversial issue of kidney (and liver) transplantation.
ACQUIRED HEMOLYTIC ANEMIA
Mechanical Destruction of Red Cells Although red cells are characterized by the remarkable deformability that enables them to squeeze through capillaries narrower than themselves for thousands of times in their lifetime, there are at least two situations in which they succumb to shear, if not to wear and tear; the result is intravascular hemolysis, resulting in hemoglobinuria (Table 129-6). One situation is acute and self-inflicted, march hemoglobinuria. Why sometimes a marathon runner may develop this complication, whereas on another occasion, this does not happen, we do not know (perhaps her or his footwear needs attention). A similar syndrome may develop after prolonged barefoot ritual dancing or intense playing of bongo drums. The other situation is chronic and iatrogenic (it has been called microangiopathic hemolytic anemia). It takes place in patients with prosthetic heart valves, especially when paraprosthetic regurgitation is present. If the hemolysis consequent on mechanical trauma to the red cells is mild, and if the supply of iron is adequate, the loss may be largely compensated; if more than mild anemia develops, reintervention to correct regurgitation may be required.
|
DISEASES AND CLINICAL SITUATIONS WITH PREDOMINANTLY INTRAVASCULAR HEMOLYSIS |
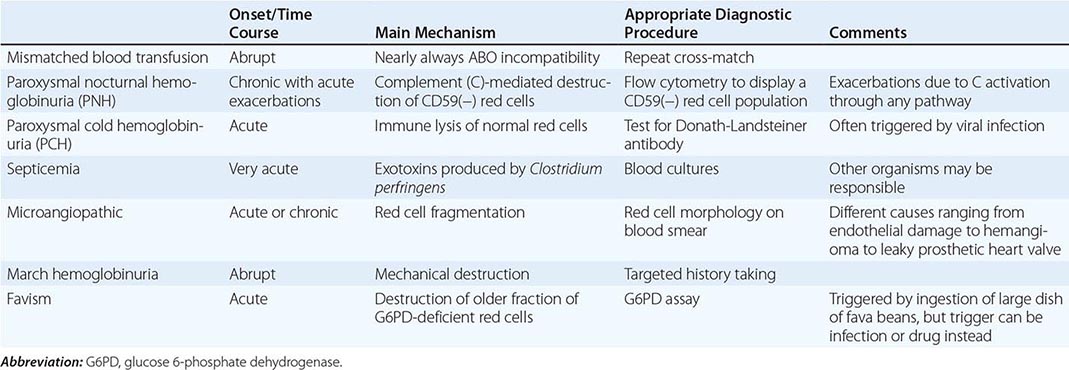
Infection By far the most frequent infectious cause of HA, in endemic areas, is malaria (Chap. 248). In other parts of the world, the most frequent direct cause is probably Shiga toxin–producing E. coli O157:H7, now recognized as the main etiologic agent of HUS, which is more common in children than in adults (Chap. 149). Life-threatening intravascular hemolysis, due to a toxin with lecithinase activity, occurs with Clostridium perfringens sepsis, particularly following open wounds, septic abortion, or as a disastrous accident due to a contaminated blood unit. Rarely, and if at all in children, HA is seen with sepsis or endocarditis from a variety of organisms. In addition, bacterial and viral infections can cause HA by indirect mechanisms (see above section on G6PD deficiency and Table 129-6).
Immune Hemolytic Anemias These can arise through at least two distinct mechanisms. (1) There is a true autoantibody directed against a red cell antigen, i.e., a molecule present on the surface of red cells. (2) When an antibody directed against a certain molecule (e.g., a drug) reacts with that molecule, red cells may get caught in the reaction, whereby they are damaged or destroyed. Because the antibodies involved differ in optimum reactivity temperatures, they are classified in the time-honored categories of “cold” and “warm” (Table 129-7). Autoantibody-mediated HAs may be seen in isolation (when they are called idiopathic) or as part of a systemic autoimmune disorder such as systemic lupus erythematosus. Here we discuss the most distinctive clinical pictures.
|
CLASSIFICATION OF ACQUIRED IMMUNE HEMOLYTIC ANEMIAS |
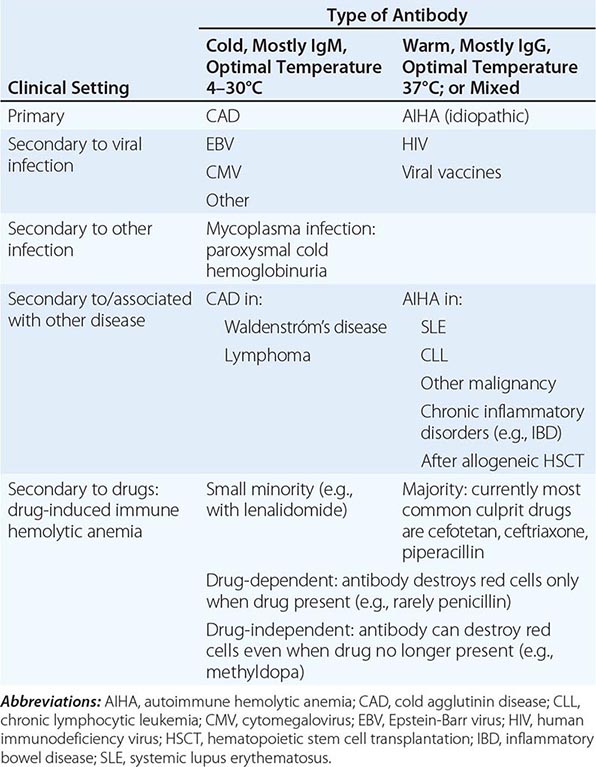
AUTOIMMUNE HEMOLYTIC ANEMIA (AIHA) Once a red cell is coated by an autoantibody (see [1] above), it will be destroyed by one or more mechanisms. In most cases, the Fc portion of the antibody will be recognized by the Fc receptor of macrophages, and this will trigger erythrophagocytosis. Thus, destruction of red cells will take place wherever macrophages are abundant, i.e., in the spleen, liver, or bone marrow; this is called extravascular hemolysis (Fig. 129-8). Because of the special anatomy of the spleen, this organ is particularly efficient in trapping antibody-coated red cells, and often this is the predominant site of red cell destruction. In some cases, the nature of the antibody is such (usually an IgM antibody) that the antigen-antibody complex on the surface of red cells is able to activate complement (C); as a result, a large amount of membrane attack complex will form, and the red cells may be destroyed directly; this is known as intravascular hemolysis.
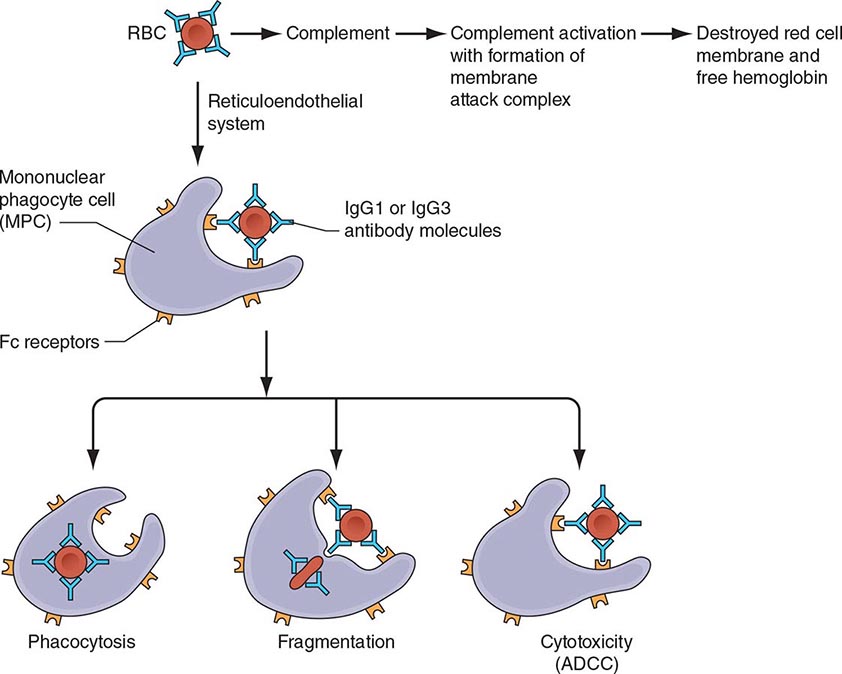
FIGURE 129-8 Mechanism of antibody-mediated immune destruction of red blood cells (RBCs). ADCC, antibody-dependent cell-mediated cytotoxicity. (From N Young et al: Clinical Hematology. Philadelphia, Elsevier, 2006; with permission.)
Clinical features AIHA is a serious condition; without appropriate treatment, it may have a mortality of approximately 10%. The onset is often abrupt and can be dramatic. The hemoglobin level can drop, within days, to as low as 4 g/dL; the massive red cell removal will produce jaundice; and sometimes the spleen is enlarged. When this triad is present, the suspicion of AIHA must be high. When hemolysis is (in part) intravascular, the telltale sign will be hemoglobinuria, which the patient may report or about which we must enquire or test for. The diagnostic test for AIHA is the direct antiglobulin test developed in 1945 by R. R. A. Coombs and known since by this name. The beauty of this test is that it detects directly the pathogenetic mediator of the disease, i.e., the presence of antibody on the red cells themselves. When the test is positive, it clinches the diagnosis; when it is negative, the diagnosis is unlikely. However, the sensitivity of the Coombs test varies depending on the technique that is used, and in doubtful cases, a repeat in a specialized lab is advisable; the term Coombs-negative AIHA is a last resort. In some cases, the autoantibody has a defined identity; it may be specific for an antigen belonging to the Rhesus system (it is often anti-e). In many cases, it is regarded as “nonspecific” because it reacts with virtually all types of red cells.
When AIHA develops in a person who is already known to have, for instance, systemic lupus or chronic lymphocytic leukemia (Table 129-7), we call it a complication; conversely, when AIHA presents on its own, it may be a pointer to an underlying condition that we ought to seek out. In both cases, what brings about AIHA remains, as in other autoimmune disorders, obscure. In some cases, AIHA can be associated, on first presentation or subsequently, with autoimmune thrombocytopenia (Evans’ syndrome).
PAROXYSMAL COLD HEMOGLOBINURIA (PCH) PCH is a rather rare form of AIHA occurring mostly in children, usually triggered by a viral infection, usually self-limited, and characterized by the involvement of the so-called Donath-Landsteiner antibody. In vitro, this antibody has unique serologic features; it has anti-P specificity and binds to red cells only at a low temperature (optimally at 4°C), but when the temperature is shifted to 37°C, lysis of red cells takes place in the presence of complement. Consequently, in vivo there is intravascular hemolysis, resulting in hemoglobinuria. Clinically the differential diagnosis must include other causes of hemoglobinuria (Table 129-6), but the presence of the Donath-Landsteiner antibody will prove PCH. Active supportive treatment, including blood transfusion, is needed to control the anemia; subsequently, recovery is the rule.
COLD AGGLUTININ DISEASE (CAD) This designation is used for a form of chronic AIHA that usually affects the elderly and has special clinical and pathologic features. First, the term cold refers to the fact that the autoantibody involved reacts with red cells poorly or not at all at 37°C, whereas it reacts strongly at lower temperatures. As a result, hemolysis is more prominent the more the body is exposed to the cold. The antibody is usually IgM; usually it has an anti-I specificity (the I antigen is present on the red cells of almost everybody), and it may have a very high titer (1:100,000 or more has been observed). Second, the antibody is produced by an expanded clone of B lymphocytes, and sometimes its concentration in the plasma is high enough to show up as a spike in plasma protein electrophoresis, i.e., as a monoclonal gammopathy. Third, because the antibody is IgM, CAD is related to Waldenström’s macroglobulinemia (WM) (Chap. 136), although in most cases, the other clinical features of this disease are not present. Thus, CAD must be regarded as a form of WM (i.e., as a low-grade mature B cell lymphoma) that manifests at an earlier stage precisely because the unique biologic properties of the IgM that it produces give the clinical picture of chronic HA.
In mild forms of CAD, avoidance of exposure to cold may be all that is needed to enable the patient to have a reasonably comfortable quality of life; but in more severe forms, the management of CAD is not easy. Blood transfusion is not very effective because donor red cells are I positive and will be rapidly removed. Immunosuppressive/cytotoxic treatment with azathioprine or cyclophosphamide can reduce the antibody titer, but clinical efficacy is limited, and in view of the chronic nature of the disease, the side effects may prove unacceptable. Unlike in AIHA, prednisone and splenectomy are ineffective. Plasma exchange will remove antibody and is, therefore, in theory, a rational approach, but it is laborious and must be carried out at frequent intervals if it is to be beneficial. The management of CAD has changed significantly with the advent of rituximab; although its impact on CAD is not as great as on AIHA, up to 60% of patients respond, and remissions may be more durable with a rituximab-fludarabine combination. Given the long clinical course of CAD, it remains to be seen with what schedule or periodicity these agents will need to be administered.
Toxic Agents and Drugs A number of chemicals with oxidative potential, whether medicinal or not, can cause hemolysis even in people who are not G6PD deficient (see above). Examples are hyperbaric oxygen (or 100% oxygen), nitrates, chlorates, methylene blue, dapsone, cisplatin, and numerous aromatic (cyclic) compounds. Other chemicals may be hemolytic through nonoxidative, largely unknown mechanisms; examples include arsine, stibine, copper, and lead. The HA caused by lead poisoning is characterized by basophilic stippling; it is in fact a phenocopy of that seen in P5N deficiency (see above), suggesting it is mediated at least in part by lead inhibiting this enzyme.
In these cases, hemolysis appears to be mediated by a direct chemical action on red cells. But drugs can cause hemolysis through at least two other mechanisms. (1) A drug can behave as a hapten and induce antibody production; in rare subjects, this happens, for instance, with penicillin. Upon a subsequent exposure, red cells are caught, as innocent bystanders, in the reaction between penicillin and antipenicillin antibodies. Hemolysis will subside as soon as penicillin administration is stopped. (2) A drug can trigger, perhaps through mimicry, the production of an antibody against a red cell antigen. The best known example is methyldopa, an antihypertensive agent no longer in use, which in a small fraction of patients stimulated the production of the Rhesus antibody anti-e. In patients who have this antigen, the anti-e is a true autoantibody, which then causes an autoimmune HA (see below). Usually this will gradually subside once methyldopa is discontinued.
Severe intravascular hemolysis can be caused by the venom of certain snakes (cobras and vipers), and HA can also follow spider bites.
Paroxysmal Nocturnal Hemoglobinuria (PNH) PNH is an acquired chronic HA characterized by persistent intravascular hemolysis subject to recurrent exacerbations. In addition to hemolysis, there is often pancytopenia and a distinct tendency to venous thrombosis. This triad makes PNH a truly unique clinical condition; however, when not all of these three features are manifest on presentation, the diagnosis is often delayed, although it can always be made by appropriate laboratory investigations (see below).
![]() PNH has about the same frequency in men and women and is encountered in all populations throughout the world, but it is a rare disease; its prevalence is estimated to be approximately 5 per million (it may be somewhat less rare in Southeast Asia and in the Far East). There is no evidence of inherited susceptibility. PNH has never been reported as a congenital disease, but it can present in small children or as late as in the seventies, although most patients are young adults.
PNH has about the same frequency in men and women and is encountered in all populations throughout the world, but it is a rare disease; its prevalence is estimated to be approximately 5 per million (it may be somewhat less rare in Southeast Asia and in the Far East). There is no evidence of inherited susceptibility. PNH has never been reported as a congenital disease, but it can present in small children or as late as in the seventies, although most patients are young adults.
CLINICAL FEATURES The patient may seek medical attention because, one morning, she or he passed blood instead of urine (Fig. 129-9). This distressing or frightening event may be regarded as the classical presentation; however, more frequently, this symptom is not noticed or is suppressed. Indeed, the patient often presents simply as a problem in the differential diagnosis of anemia, whether symptomatic or discovered incidentally. Sometimes, the anemia is associated from the outset with neutropenia, thrombocytopenia, or both, thus signaling an element of bone marrow failure (see below). Some patients may present with recurrent attacks of severe abdominal pain defying a specific diagnosis and eventually found to be related to thrombosis. When thrombosis affects the hepatic veins, it may produce acute hepatomegaly and ascites, i.e., a full-fledged Budd-Chiari syndrome, which, in the absence of liver disease, ought to raise the suspicion of PNH.

FIGURE 129-9 Consecutive urine samples from a patient with paroxysmal nocturnal hemoglobinuria (PNH). The variation in the severity of hemoglobinuria within hours is probably unique to this condition.
The natural history of PNH can extend over decades. Without treatment, the median survival is estimated to be about 8–10 years; in the past, the most common cause of death has been venous thrombosis, followed by infection secondary to severe neutropenia and hemorrhage secondary to severe thrombocytopenia. Rarely (estimated 1–2% of all cases), PNH may terminate in acute myeloid leukemia. On the other hand, full spontaneous recovery from PNH has been documented, albeit rarely.
LABORATORY INVESTIGATIONS AND DIAGNOSIS The most consistent blood finding is anemia, which may range from mild to moderate to very severe. The anemia is usually normomacrocytic, with unremarkable red cell morphology. If the MCV is high, it is usually largely accounted for by reticulocytosis, which may be quite marked (up to 20%, or up to 400,000/μL). The anemia may become microcytic if the patient is allowed to become iron deficient as a result of chronic urinary blood loss through hemoglobinuria. Unconjugated bilirubin is mildly or moderately elevated; LDH is typically markedly elevated (values in the thousands are common); and haptoglobin is usually undetectable. All of these findings make the diagnosis of hemolytic anemia compelling. Hemoglobinuria may be overt in a random urine sample; if it is not, it may be helpful to obtain serial urine samples, because hemoglobinuria can vary dramatically from day to day and even from hour to hour. The bone marrow is usually cellular, with marked to massive erythroid hyperplasia, often with mild to moderate dyserythropoietic features (not to be confused with myelodysplastic syndrome). At some stage of the disease, the marrow may become hypocellular or even frankly aplastic (see below).
The definitive diagnosis of PNH must be based on the demonstration that a substantial proportion of the patient’s red cells have an increased susceptibility to complement (C), due to the deficiency on their surface of proteins (particularly CD59 and CD55) that normally protect the red cells from activated C. The sucrose hemolysis test is unreliable; in contrast, the acidified serum (Ham) test is highly reliable but is carried out only in a few labs. The gold standard today is flow cytometry, which can be carried out on granulocytes as well as on red cells. A bimodal distribution of cells, with a discrete population that is CD59 and CD55 negative, is diagnostic of PNH. In PNH patients, this population is at least 5% of the total red cells and at least 20% of the total granulocytes.
PATHOPHYSIOLOGY Hemolysis in PNH is mainly intravascular and is due to an intrinsic abnormality of the red cell, which makes it exquisitely sensitive to activated C, whether it is activated through the alternative pathway or through an antigen-antibody reaction. The former mechanism is mainly responsible for chronic hemolysis in PNH; the latter explains why the hemolysis can be dramatically exacerbated in the course of a viral or bacterial infection. Hypersusceptibility to C is due to deficiency of several protective membrane proteins (Fig. 129-10), of which CD59 is the most important, because it hinders the insertion into the membrane of C9 polymers. The molecular basis for the deficiency of these proteins has been pinpointed not to a defect in any of the respective genes, but rather to the shortage of a unique glycolipid molecule, GPI (Fig. 129-2), which, through a peptide bond, anchors these proteins to the surface membrane of cells. The shortage of GPI is due in turn to a mutation in an X-linked gene, called PIG-A, required for an early step in GPI biosynthesis. In virtually each patient, the PIG-A mutation is different. This is not surprising, because these mutations are not inherited; rather, each one takes place de novo in a hemopoietic stem cell (i.e., they are somatic mutations). As a result, the patient’s marrow is a mosaic of mutant and nonmutant cells, and the peripheral blood always contains both PNH cells and normal (non-PNH) cells. Thrombosis is one of the most immediately life-threatening complications of PNH and yet one of the least understood in its pathogenesis. It could be that deficiency of CD59 on the PNH platelet causes inappropriate platelet activation; however, other mechanisms are possible.
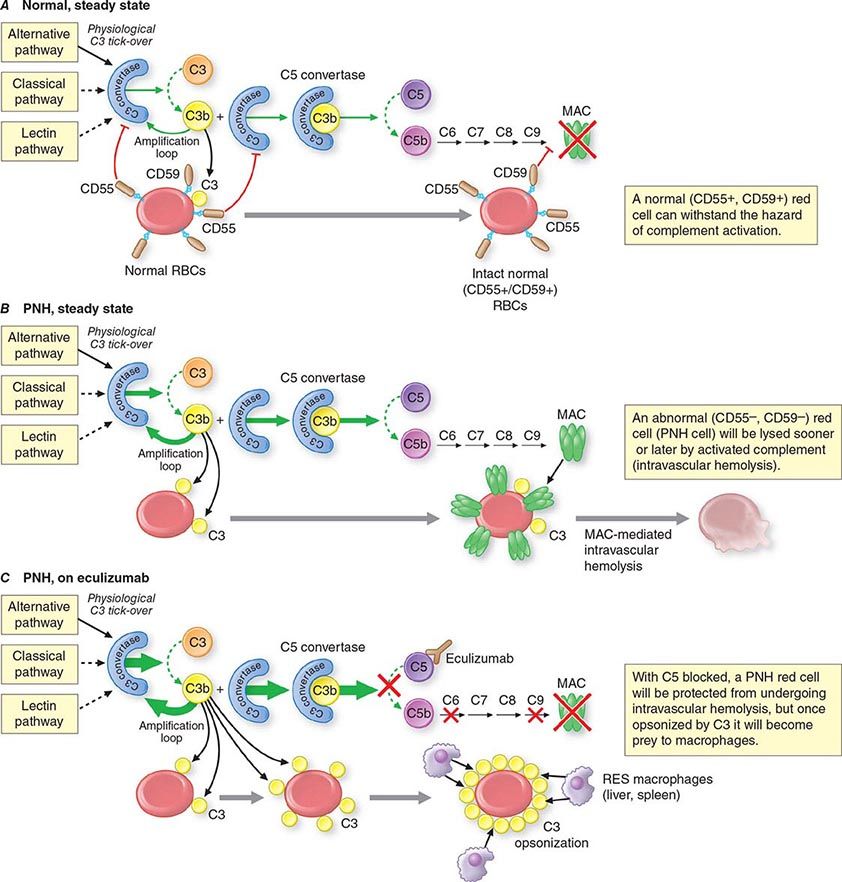
FIGURE 129-10 The complement cascade and the fate of red cells. A. Normal red cells are protected from complement activation and subsequent hemolysis by CD55 and CD59. These two proteins, being GPI-linked, are missing from the surface of PNH red cells as a result of a somatic mutation of the X-linked PIG-A gene that encodes a protein required for an early step of the GPI molecule biosynthesis. B. In the steady state, PNH erythrocytes suffer from spontaneous (tick-over) complement activation, with consequent intravascular hemolysis through formation of the membrane attack complex (MAC); when extra complement is activated through the classical pathway, an exacerbation of hemolysis will result. C. On eculizumab, PNH erythrocytes are protected from hemolysis from the inhibition of C5 cleavage; however, upstream complement activation may lead to C3 opsonization and possible extravascular hemolysis. GPI, glycosylphosphatidylinositol; PNH, paroxysmal nocturnal hemoglobinuria. (From L Luzzatto et al: Haematologica 95:523, 2010.)
BONE MARROW FAILURE (BMF) AND RELATIONSHIP BETWEEN PNH AND APLASTIC ANEMIA (AA) It is not unusual that patients with firmly established PNH have a previous history of well-documented AA; indeed, BMF preceding overt PNH is probably the rule rather than the exception. On the other hand, sometimes a patient with PNH becomes less hemolytic and more pancytopenic and ultimately has the clinical picture of AA. Because AA is probably an organ-specific autoimmune disease, in which T cells cause damage to hematopoietic stem cells, the same may be true of PNH, with the specific proviso that the damage spares PNH stem cells. PIG-A mutations can be demonstrated in normal people, and there is evidence from mouse models that PNH stem cells do not expand when the rest of the bone marrow is normal. Thus, we can visualize PNH as always having two components: failure of normal hematopoiesis and massive expansion of a PNH clone. Findings supporting this notion include skewing of the T cell repertoire and the demonstration of GPI-reactive T cells in patients with PNH.
ANEMIA DUE TO ACUTE BLOOD LOSS
Blood loss causes anemia by two main mechanisms: (1) by the direct loss of red cells; and (2) if the loss of blood is protracted, it will gradually deplete iron stores, eventually resulting in iron deficiency. The latter type of anemia is covered in Chap. 126; here we are concerned with the former type, i.e., posthemorrhagic anemia, which follows acute blood loss. This can be external (e.g., after trauma or obstetric hemorrhage) or internal (e.g., from bleeding in the gastrointestinal tract, rupture of the spleen, rupture of an ectopic pregnancy, subarachnoid hemorrhage). In any of these cases, after the sudden loss of a large amount of blood, there are three clinical/pathophysiologic stages. (1) At first, the dominant feature is hypovolemia, which poses a threat particularly to organs that normally have a high blood supply, like the brain and the kidneys; therefore, loss of consciousness and acute renal failure are major threats. It is important to note that at this stage an ordinary blood count will not show anemia, because the hemoglobin concentration is not affected. (2) Next, as an emergency response, baroreceptors and stretch receptors will cause release of vasopressin and other peptides, and the body will shift fluid from the extravascular to the intravascular compartment, producing hemodilution; thus, the hypovolemia gradually converts to anemia. The degree of anemia will reflect the amount of blood lost. If after 3 days the hemoglobin is, for example, 7 g/dL, it means that about half of the entire blood has been lost. (3) Provided bleeding does not continue, the bone marrow response will gradually ameliorate the anemia.
The diagnosis of acute posthemorrhagic anemia (APHA) is usually straightforward, although sometimes internal bleeding episodes (e.g., after a traumatic injury), even when large, may not be immediately obvious. Whenever an abrupt fall in hemoglobin has taken place, whatever history is given by the patient, APHA should be suspected. Supplementary history may have to be obtained by asking the appropriate questions, and appropriate investigations (e.g., a sonogram or an endoscopy) may have to be carried out.
130 |
Bone Marrow Failure Syndromes Including Aplastic Anemia and Myelodysplasia |
The hypoproliferative anemias are normochromic, normocytic, or macrocytic and are characterized by a low reticulocyte count. Hypoproliferative anemia is also a prominent feature of hematologic diseases that are described as bone marrow failure states; these include aplastic anemia, myelodysplastic syndrome (MDS), pure red cell aplasia (PRCA), and myelophthisis. Anemia in these disorders is often not a solitary or even the major hematologic finding. More frequent in bone marrow failure is pancytopenia: anemia, leukopenia, and thrombocytopenia. Low blood counts in the marrow failure diseases result from deficient hematopoiesis, as distinguished from blood count depression due to peripheral destruction of red cells (hemolytic anemias), platelets (idiopathic thrombocytopenic purpura [ITP] or due to splenomegaly), and granulocytes (as in the immune leukopenias). Marrow damage and dysfunction also may be secondary to infection, inflammation, or cancer.
Hematopoietic failure syndromes are classified by dominant morphologic features of the bone marrow (Table 130-1). Although practical distinction among these syndromes usually is clear, some processes are so closely related that the diagnosis may be complex. Patients may seem to suffer from two or three related diseases simultaneously, or one diagnosis may appear to evolve into another. Many of these syndromes share an immune-mediated mechanism of marrow destruction and some element of genomic instability resulting in a higher rate of malignant transformation.
|
DIFFERENTIAL DIAGNOSIS OF PANCYTOPENIA |
It is important that the internist and general practitioner recognize the marrow failure syndromes, as their prognosis may be poor if the patient is untreated; effective therapies are often available but sufficiently complicated in their choice and delivery so as to warrant the care of a hematologist or oncologist.
APLASTIC ANEMIA
DEFINITION
Aplastic anemia is pancytopenia with bone marrow hypocellularity. Acquired aplastic anemia is distinguished from iatrogenic aplasia, marrow hypocellularity after intensive cytotoxic chemotherapy for cancer. Aplastic anemia can also be constitutional: the genetic diseases Fanconi anemia and dyskeratosis congenita, although frequently associated with typical physical anomalies and the development of pancytopenia early in life, can also present as marrow failure in normal-appearing adults. Acquired aplastic anemia is often stereotypical in its manifestations, with the abrupt onset of low blood counts in a previously well young adult; seronegative hepatitis or a course of an incriminated medical drug may precede the onset. The diagnosis in these instances is uncomplicated. Sometimes blood count depression is moderate or incomplete, resulting in anemia, leukopenia, and thrombocytopenia in some combination. Aplastic anemia is related to both paroxysmal nocturnal hemoglobinuria (PNH; Chap. 129) and to MDS, and in some cases, a clear distinction among these disorders may not be possible.
EPIDEMIOLOGY
![]() The incidence of acquired aplastic anemia in Europe and Israel is two cases per million persons annually. In Thailand and China, rates of five to seven per million have been established. In general, men and women are affected with equal frequency, but the age distribution is biphasic, with the major peak in the teens and twenties and a second rise in older adults.
The incidence of acquired aplastic anemia in Europe and Israel is two cases per million persons annually. In Thailand and China, rates of five to seven per million have been established. In general, men and women are affected with equal frequency, but the age distribution is biphasic, with the major peak in the teens and twenties and a second rise in older adults.
ETIOLOGY
The origins of aplastic anemia have been inferred from several recurring clinical associations (Table 130-2); unfortunately, these relationships are not reliable in an individual patient and may not be etiologic. In addition, although most cases of aplastic anemia are idiopathic, little other than history separates these cases from those with a presumed etiology such as a drug exposure.
|
CLASSIFICATION OF APLASTIC ANEMIA AND SINGLE CYTOPENIAS |

Radiation Marrow aplasia is a major acute sequela of radiation. Radiation damages DNA; tissues dependent on active mitosis are particularly susceptible. Nuclear accidents involve not only power plant workers but also employees of hospitals, laboratories, and industry (food sterilization, metal radiography, etc.), as well as innocents exposed to stolen, misplaced, or misused sources. Whereas the radiation dose can be approximated from the rate and degree of decline in blood counts, dosimetry by reconstruction of the exposure can help to estimate the patient’s prognosis and also to protect medical personnel from contact with radioactive tissue and excreta. MDS and leukemia, but probably not aplastic anemia, are late effects of radiation.
Chemicals Benzene is a notorious cause of bone marrow failure: epidemiologic, clinical, and laboratory data link benzene to aplastic anemia, acute leukemia, and blood and marrow abnormalities. For leukemia, incidence is correlated with cumulative exposure, but susceptibility must also be important, because only a minority of even heavily exposed workers develop myelotoxicity. The employment history is important, especially in industries where benzene is used for a secondary purpose, usually as a solvent. Benzene-related blood diseases have declined with regulation of industrial exposure. Although benzene is no longer generally available as a household solvent, exposure to its metabolites occurs in the normal diet and in the environment. The association between marrow failure and other chemicals is much less well substantiated.
Drugs (Table 130-3) Many chemotherapeutic drugs have marrow suppression as a major toxicity; effects are dose dependent and will occur in all recipients. In contrast, idiosyncratic reactions to a large and diverse group of drugs may lead to aplastic anemia without a clear dose-response relationship. These associations rest largely on accumulated case reports until a large international study in Europe in the 1980s quantitated drug relationships, especially for nonsteroidal analgesics, sulfonamides, thyrostatic drugs, some psychotropics, penicillamine, allopurinol, and gold. Association does not equal causation: a drug may have been used to treat the first symptoms of bone marrow failure (antibiotics for fever or the preceding viral illness) or provoked the first symptom of a preexisting disease (petechiae by nonsteroidal anti-inflammatory agents administered to the thrombocytopenic patient). In the context of total drug use, idiosyncratic reactions, although individually devastating, are rare events. Risk estimates are usually lower when determined in population-based studies. Furthermore, the low absolute risk is also made more obvious: even a 10- or 20-fold increase in risk translates, in a rare disease, to just a handful of drug-induced aplastic anemia cases among hundreds of thousands of exposed persons.
|
SOME DRUGS AND CHEMICALS ASSOCIATED WITH APLASTIC ANEMIA |
Note: Terms set in italics show the most consistent association with aplastic anemia.
Infections Hepatitis is the most common preceding infection, and posthepatitis marrow failure accounts for approximately 5% of etiologies in most series. Patients are usually young men who have recovered from a bout of liver inflammation 1 to 2 months earlier; the subsequent pancytopenia is very severe. The hepatitis is seronegative (non-A, non-B, non-C) and possibly due to an as yet undiscovered infectious agent. Fulminant liver failure in childhood also follows seronegative hepatitis, and marrow failure occurs at a high rate in these patients. Aplastic anemia can rarely follow infectious mononucleosis. Parvovirus B19, the cause of transient aplastic crisis in hemolytic anemias and of some PRCAs (see below), does not usually cause generalized bone marrow failure. Mild blood count depression is frequent in the course of many viral and bacterial infections but resolves with the infection.
Immunologic Diseases Aplasia is a major consequence and the inevitable cause of death in transfusion-associated graft-versus-host disease (GVHD) that can occur after infusion of nonirradiated blood products to an immunodeficient recipient. Aplastic anemia is strongly associated with the rare collagen vascular syndrome eosinophilic fasciitis that is characterized by painful induration of subcutaneous tissues (Chap. 382). Thymoma and hypoimmunoglobulinemia are occasional associations with aplastic anemia. Pancytopenia with marrow hypoplasia can also occur in systemic lupus erythematosus (SLE).
Pregnancy Aplastic anemia very rarely may occur and recur during pregnancy and resolve with delivery or with spontaneous or induced abortion.
Paroxysmal Nocturnal Hemoglobinuria An acquired mutation in the PIG-A gene in a hematopoietic stem cell is required for the development of PNH, but PIG-A mutations probably occur commonly in normal individuals. If the PIG-A mutant stem cell proliferates, the result is a clone of progeny deficient in glycosylphosphatidylinositol-linked cell surface membrane proteins (Chap. 129). Small clones of deficient cells can be detected by sensitive flow cytometry tests in one-half or more of patients with aplastic anemia at the time of presentation. Functional studies of bone marrow from PNH patients, even those with mainly hemolytic manifestations, show evidence of defective hematopoiesis. Patients with an initial clinical diagnosis of PNH, especially younger individuals, may later develop frank marrow aplasia and pancytopenia; patients with an initial diagnosis of aplastic anemia may suffer from hemolytic PNH years after recovery of blood counts.
Constitutional Disorders Fanconi anemia, an autosomal recessive disorder, manifests as congenital developmental anomalies, progressive pancytopenia, and an increased risk of malignancy. Chromosomes in Fanconi anemia are peculiarly susceptible to DNA cross-linking agents, the basis for a diagnostic assay. Patients with Fanconi anemia typically have short stature, café au lait spots, and anomalies involving the thumb, radius, and genitourinary tract. At least 16 different genetic defects (all but one with an identified gene) have been defined; the most common, type A Fanconi anemia, is due to a mutation in FANCA. Most of the Fanconi anemia gene products form a protein complex that activates FANCD2 by monoubiquitination to play a role in the cellular response to DNA damage and especially interstrand cross-linking.
Dyskeratosis congenita is characterized by the triad of mucous membrane leukoplasia, dystrophic nails, reticular hyperpigmentation, and with the development of aplastic anemia in childhood. Dyskeratosis is due to mutations in genes of the telomere repair complex, which acts to maintain telomere length in replicating cells: the X-linked variety is due to mutations in the DKC1 (dyskerin) gene; the more unusual autosomal dominant type is due to mutation in TERC, which encodes an RNA template, and TERT, which encodes the catalytic reverse transcriptase, telomerase. Mutations in TNF2, a component of the shelterin complex, proteins that bind the telomere DNA, also occur.
In Shwachman-Diamond syndrome, presentation is early in life with neutropenia with pancreatic insufficiency and malabsorption; most patients have compound heterozygous mutations in SBDS that may affect both ribosomal biogenesis (as in Diamond-Blackfan anemia; see below) and marrow stroma function.
While these constitutional syndromes can on occasion present in adults, genetic mutations are also risk factors for bone marrow failure. In the recently recognized telomeropathies, mutations in TERT and TERC have subtle effects on hematopoietic function. Typical presentations include not only severe but also moderate aplastic anemia, which can be chronic and not progressive, and isolated macrocytic anemia or thrombocytopenia. Physical anomalies are usually not found in the patient, although early hair graying is a clue to the diagnosis. A careful family history may disclose pulmonary fibrosis and hepatic cirrhosis. Specific involvement of the bone marrow, liver, and lung is highly variable, as is penetrance of clinical phenotype, both within families and among kindreds. Variable penetrance means that TERT and TERC mutations represent risk factors for marrow failure, as family members with the same mutations may have normal or only slight hematologic abnormalities but more subtle evidence of (compensated) hematopoietic insufficiency.
PATHOPHYSIOLOGY
Bone marrow failure results from severe damage to the hematopoietic cell compartment. In aplastic anemia, replacement of the bone marrow by fat is apparent in the morphology of the biopsy specimen (Fig. 130-1) and magnetic resonance imaging (MRI) of the spine. Cells bearing the CD34 antigen, a marker of early hematopoietic cells, are greatly diminished, and in functional studies, committed and primitive progenitor cells are virtually absent; in vitro assays have suggested that the stem cell pool is reduced to ≤1% of normal in severe disease at the time of presentation.
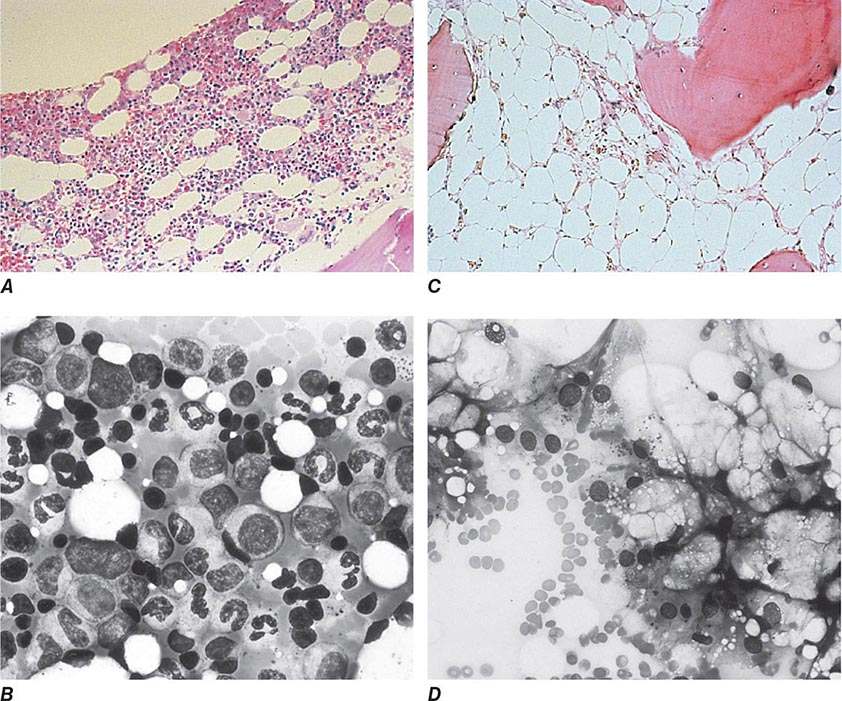
FIGURE 130-1 Normal and aplastic bone marrow. A. Normal bone marrow biopsy. B. Normal bone marrow aspirate smear. The marrow is normally 30–70% cellular, and there is a heterogeneous mix of myeloid, erythroid, and lymphoid cells. C. Aplastic anemia biopsy. D. Marrow smear in aplastic anemia. The marrow shows replacement of hematopoietic tissue by fat and only residual stromal and lymphoid cells.
An intrinsic stem cell defect exists for the constitutional aplastic anemias: cells from patients with Fanconi anemia exhibit chromosome damage and death on exposure to certain chemical agents. Telomeres are short in some patients with aplastic anemia, due to heterozygous mutations in genes of the telomere repair complex. Telomeres may also shorten physiologically in acquired marrow failure due to replicative demands on a limited stem cell pool.
Drug Injury Extrinsic damage to the marrow follows massive physical or chemical insults such as high doses of radiation and toxic chemicals. For the more common idiosyncratic reaction to modest doses of medical drugs, altered drug metabolism has been invoked as a likely mechanism. The metabolic pathways of many drugs and chemicals, especially if they are polar and have limited water solubility, involve enzymatic degradation to highly reactive electrophilic compounds; these intermediates are toxic because of their propensity to bind to cellular macromolecules. For example, derivative hydroquinones and quinolones are responsible for benzene-induced tissue injury. Excessive generation of toxic intermediates or failure to detoxify the intermediates may be genetically determined and apparent only on specific drug challenge; the complexity and specificity of the pathways imply multiple susceptibility loci and would provide an explanation for the rarity of idiosyncratic drug reactions.
Immune-Mediated Injury The recovery of marrow function in some patients prepared for bone marrow transplantation with antilymphocyte globulin first suggested that aplastic anemia might be immune mediated. Consistent with this hypothesis was the frequent failure of simple bone marrow transplantation from a syngeneic twin, without conditioning cytotoxic chemotherapy, which also argued both against simple stem cell absence as the cause and for the presence of a host factor producing marrow failure. Laboratory data support an important role for the immune system in aplastic anemia. Blood and bone marrow cells of patients can suppress normal hematopoietic progenitor cell growth, and removal of T cells from aplastic anemia bone marrow improves colony formation in vitro. Increased numbers of activated cytotoxic T cell clones are observed in aplastic anemia patients and usually decline with successful immunosuppressive therapy; type 1 cytokines are implicated; and interferon γ (IFN-γ) induces Fas expression on CD34 cells, leading to apoptotic cell death. The early immune system events in aplastic anemia are not well understood, but an oligoclonal, T cell response implies antigenic stimulus. The rarity of aplastic anemia despite common exposures (medicines, seronegative hepatitis) suggests that genetically determined features of the immune response can convert a normal physiologic response into a sustained abnormal autoimmune process, including polymorphisms in histocompatibility antigens, cytokine genes, and genes that regulate T cell polarization and effector function.
CLINICAL FEATURES
History Aplastic anemia can appear abruptly or insidiously. Bleeding is the most common early symptom; a complaint of days to weeks of easy bruising, oozing from the gums, nose bleeds, heavy menstrual flow, and sometimes petechiae will have been noticed. With thrombocytopenia, massive hemorrhage is unusual, but small amounts of bleeding in the central nervous system can result in catastrophic intracranial or retinal hemorrhage. Symptoms of anemia are also frequent, including lassitude, weakness, shortness of breath, and a pounding sensation in the ears. Infection is an unusual first symptom in aplastic anemia (unlike in agranulocytosis, where pharyngitis, anorectal infection, or frank sepsis occurs early). A striking feature of aplastic anemia is the restriction of symptoms to the hematologic system, and patients often feel and look remarkably well despite drastically reduced blood counts. Systemic complaints and weight loss should point to other etiologies of pancytopenia. Prior drug use, chemical exposure, and preceding viral illnesses must often be elicited with repeated questioning. A family history of hematologic diseases or blood abnormalities, of pulmonary or liver fibrosis, or of early hair graying points to a telomeropathy.
Physical Examination Petechiae and ecchymoses are typical, and retinal hemorrhages may be present. Pelvic and rectal examinations can often be deferred but, when performed, should be undertaken with great gentleness to avoid trauma; these will often show bleeding from the cervical os and blood in the stool. Pallor of the skin and mucous membranes is common except in the most acute cases or those already transfused. Infection on presentation is unusual but may occur if the patient has been symptomatic for a few weeks. Lymphadenopathy and splenomegaly are highly atypical of aplastic anemia. Café au lait spots and short stature suggest Fanconi anemia; peculiar nails and leukoplakia suggest dyskeratosis congenita; early graying (and use of hair dyes to mask it!) suggests a telomerase defect.
LABORATORY STUDIES
Blood The smear shows large erythrocytes and a paucity of platelets and granulocytes. Mean corpuscular volume (MCV) is commonly increased. Reticulocytes are absent or few, and lymphocyte numbers may be normal or reduced. The presence of immature myeloid forms suggests leukemia or MDS; nucleated red blood cells (RBCs) suggest marrow fibrosis or tumor invasion; abnormal platelets suggest either peripheral destruction or MDS.
Bone Marrow The bone marrow is usually readily aspirated but dilute on smear, and the fatty biopsy specimen may be grossly pale on withdrawal; a “dry tap” instead suggests fibrosis or myelophthisis. In severe aplasia, the smear of the aspirated specimen shows only red cells, residual lymphocytes, and stromal cells; the biopsy (which should be >1 cm in length) is superior for determination of cellularity and shows mainly fat under the microscope, with hematopoietic cells occupying <25% of the marrow space; in the most serious cases, the biopsy is virtually all fat. The correlation between marrow cellularity and disease severity is imperfect, in part because marrow cellularity declines physiologically with aging. Additionally, some patients with moderate disease by blood counts will have empty iliac crest biopsies, whereas “hot spots” of hematopoiesis may be seen in severe cases. If an iliac crest specimen is inadequate, cells may also be obtained by aspiration from the sternum. Residual hematopoietic cells should have normal morphology, except for mildly megaloblastic erythropoiesis; megakaryocytes are invariably greatly reduced and usually absent. Granulomas may indicate an infectious etiology of the marrow failure.
Ancillary Studies Chromosome breakage studies of peripheral blood using diepoxybutane or mitomycin C should be performed on children and younger adults to exclude Fanconi anemia. Very short telomere length (available commercially) strongly suggests the presence of a telomerase or shelterin mutation, which can be pursued by family studies and nucleotide sequencing. Chromosome studies of bone marrow cells are often revealing in MDS and should be negative in typical aplastic anemia. Flow cytometry offers a sensitive diagnostic test for PNH. Serologic studies may show evidence of viral infection, such as Epstein-Barr virus and HIV. Posthepatitis aplastic anemia is seronegative. The spleen size should be determined by computed tomography (CT) scanning or ultrasound if the physical examination of the abdomen is unsatisfactory. Occasionally MRI may be helpful to assess the fat content of vertebrae in order to distinguish aplasia from MDS.
DIAGNOSIS
The diagnosis of aplastic anemia is usually straightforward, based on the combination of pancytopenia with a fatty bone marrow. Aplastic anemia is a disease of the young and should be a leading diagnosis in the pancytopenic adolescent or young adult. When pancytopenia is secondary, the primary diagnosis is usually obvious from either history or physical examination: the massive spleen of alcoholic cirrhosis, the history of metastatic cancer or SLE, or miliary tuberculosis on chest radiograph (Table 130-1).
Diagnostic problems can occur with atypical presentations and among related hematologic diseases. Although pancytopenia is most common, some patients with bone marrow hypocellularity have depression of only one or two of three blood lines, with later progression to pancytopenia. The bone marrow in constitutional aplastic anemia is morphologically indistinguishable from the aspirate in acquired disease. The diagnosis can be suggested by family history, abnormal blood counts since childhood, or the presence of associated physical anomalies. Aplastic anemia may be difficult to distinguish from the hypocellular variety of MDS: MDS is favored by finding morphologic abnormalities, particularly of megakaryocytes and myeloid precursor cells, and typical cytogenetic abnormalities (see below).
PROGNOSIS
The natural history of severe aplastic anemia is rapid deterioration and death. Historically, provision first of RBC and later of platelet transfusions and effective antibiotics were of some benefit, but few patients show spontaneous recovery. The major prognostic determinant is the blood count. Severe disease has been defined by the presence of two of three parameters: absolute neutrophil count <500/μL, platelet count <20,000/μL, and corrected reticulocyte count <1% (or absolute reticulocyte count <60,000/μL). In the era of effective immunosuppressive therapies, absolute numbers of reticulocytes (>25,000/μL) and lymphocytes (>1000/μL) may be better predictors of response to treatment and long-term outcome.
PURE RED CELL APLASIA
Other, more restricted forms of marrow failure occur, in which only a single circulating cell type is affected and the marrow shows corresponding absence or decreased numbers of specific precursor cells: aregenerative anemia as in PRCA (see below), thrombocytopenia with amegakaryocytosis (Chap. 140), and neutropenia without marrow myeloid cells in agranulocytosis (Chap. 80). In general, and in contrast to aplastic anemia and MDS, the unaffected lineages appear quantitatively and qualitatively normal. Agranulocytosis, the most frequent of these syndromes, is usually a complication of medical drug use (with agents similar to those related to aplastic anemia), either by a mechanism of direct chemical toxicity or by immune destruction. Agranulocytosis has an incidence similar to aplastic anemia but is especially frequent among older adults and in women. The syndrome should resolve with discontinuation of exposure, but significant mortality is attached to neutropenia in the older and often previously unwell patient. Both pure white cell aplasia (agranulocytosis without incriminating drug exposure) and amegakaryocytic thrombocytopenia are exceedingly rare and, like PRCA, appear to be due to destructive antibodies or lymphocytes and can respond to immunosuppressive therapies. In all of the single-lineage failure syndromes, progression to pancytopenia or leukemia is unusual.
DEFINITION AND DIFFERENTIAL DIAGNOSIS
PRCA is characterized by anemia, reticulocytopenia, and absent or rare erythroid precursor cells in the bone marrow. The classification of PRCA is shown in Table 130-4. In adults, PRCA is acquired. An identical syndrome can occur constitutionally: Diamond-Blackfan anemia, or congenital PRCA, is diagnosed at birth or in early childhood and often responds to glucocorticoid treatment; mutations in ribosome protein genes are etiologic. Temporary red cell failure occurs in transient aplastic crisis of hemolytic anemias due to acute parvovirus infection (Chap. 221) and in transient erythroblastopenia of childhood, which occurs in normal children.
|
CLASSIFICATION OF PURE RED CELL APLASIA |
CLINICAL ASSOCIATIONS AND ETIOLOGY
PRCA has important associations with immune system diseases. A small minority of cases occur with a thymoma. More frequently, red cell aplasia can be the major manifestation of large granular lymphocytosis or complicate chronic lymphocytic leukemia. Some patients may be hypogammaglobulinemic. Infrequently (compared to agranulocytosis), PRCA can be due to an idiosyncratic drug reaction. Subcutaneous administration of erythropoietin (EPO) has provoked PRCA mediated by neutralizing antibodies.
Like aplastic anemia, PRCA results from diverse mechanisms. Antibodies to RBC precursors are frequently present in the blood, but T cell inhibition is probably the more common immune mechanism. Cytotoxic lymphocyte activity restricted by histocompatibility locus or specific for human T cell leukemia/lymphoma virus I–infected cells and natural killer cell activity inhibitory of erythropoiesis have been demonstrated in particularly well-studied individual cases.
PERSISTENT PARVOVIRUS B19 INFECTION
Chronic parvovirus infection is an important, treatable cause of PRCA. This common virus causes a benign exanthem of childhood (fifth disease) and a polyarthralgia/arthritis syndrome in adults. In patients with underlying hemolysis (or any condition that increases demand for RBC production), parvovirus infection can cause a transient aplastic crisis and an abrupt but temporary worsening of the anemia due to failed erythropoiesis. In normal individuals, acute infection is resolved by production of neutralizing antibodies to the virus, but in the setting of congenital, acquired, or iatrogenic immunodeficiency, persistent viral infection may occur. The bone marrow shows red cell aplasia and the presence of giant pronormoblasts (Fig. 130-2), which is the cytopathic sign of B19 parvovirus infection. Viral tropism for human erythroid progenitor cells is due to its use of erythrocyte P antigen as a cellular receptor for entry. Direct cytotoxicity of virus causes anemia if demands on erythrocyte production are high; in normal individuals, the temporary cessation of red cell production is not clinically apparent, and skin and joint symptoms are mediated by immune complex deposition.
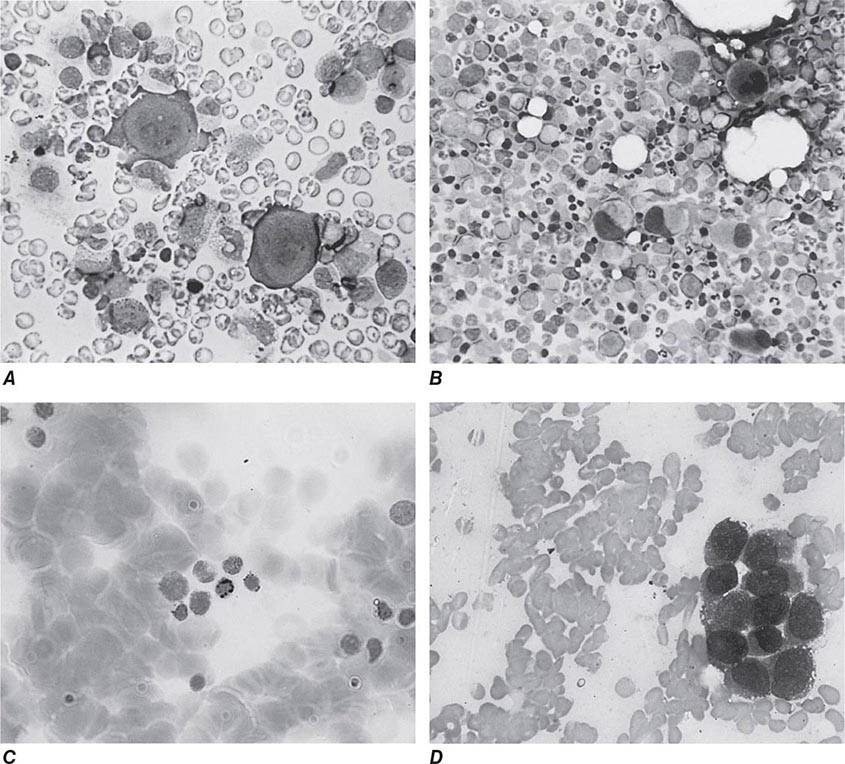
FIGURE 130-2 Pathognomonic cells in marrow failure syndromes. A. Giant pronormoblast, the cytopathic effect of B19 parvovirus infection of the erythroid progenitor cell. B. Uninuclear megakaryocyte and microblastic erythroid precursors typical of the 5q–myelodysplasia syndrome. C. Ringed sideroblast showing perinuclear iron granules. D. Tumor cells present on a touch preparation made from the marrow biopsy of a patient with metastatic carcinoma.
MYELODYSPLASIA
DEFINITION
The myelodysplastic syndromes (MDS) are a heterogeneous group of hematologic disorders broadly characterized by both (1) cytopenias due to bone marrow failure and (2) a high risk of development of acute myeloid leukemia (AML). Anemia, often with thrombocytopenia and neutropenia, occurs with dysmorphic (abnormal appearing) and usually cellular bone marrow, which is evidence of ineffective blood cell production. In patients with “low-risk” MDS, marrow failure dominates the clinical course. In other patients, myeloblasts are present at diagnosis, chromosomes are abnormal, and the “high risk” is due to leukemic progression. MDS may be fatal due to the complications of pancytopenia or the incurability of leukemia, but a large proportion of patients will die of concurrent disease, the comorbidities typical in an elderly population. A clinically useful nosology of these often confusing entities was first developed by the French-American-British Cooperative Group in 1983. Five entities were defined: refractory anemia (RA), refractory anemia with ringed sideroblasts (RARS), refractory anemia with excess blasts (RAEB), refractory anemia with excess blasts in transformation (RAEB-t), and chronic myelomonocytic leukemia (CMML). The World Health Organization (WHO) classification (2002) recognized that the distinction between RAEB-t and AML is arbitrary and grouped them together as acute leukemia and that CMML behaves as a myeloproliferative disease; the WHO classification also separated refractory anemias with dysmorphic change restricted to erythroid lineage from those with multilineage changes. In a 2008 revision, specific categories for unilineage dysplasias were added (Table 130-5).
|
WORLD HEALTH ORGANIZATION (WHO) CLASSIFICATION OF MYELODYSPLASTIC SYNDROMES/NEOPLASMS |
The diagnosis of MDS may be a challenge, because sometimes subtle clinical and pathologic features must be distinguished and precise diagnostic categorization requires a hematopathologist knowledgeable in the latest classification scheme. Nonetheless, it is important that the internist and primary care physician be sufficiently familiar with MDS to expedite referral to a hematologist, both because many new therapies are now available to improve hematopoietic function and the judicious use of supportive care can improve the patient’s quality of life.
EPIDEMIOLOGY
Idiopathic MDS is a disease of the elderly; the mean age at onset is older than 70 years. There is a slight male preponderance. MDS is a relatively common form of bone marrow failure, with reported incidence rates of 35 to >100 per million persons in the general population and 120 to >500 per million in the older adult. MDS is rare in children, but monocytic leukemia can be seen. Secondary or therapy-related MDS is not age related. Rates of MDS have increased over time, due to better recognition of the syndrome by physicians and an aging population.
ETIOLOGY AND PATHOPHYSIOLOGY
MDS is associated with environmental exposures such as radiation and benzene; other risk factors have been reported inconsistently. Secondary MDS occurs as a late toxicity of cancer treatment, usually a combination of radiation and the radiomimetic alkylating agents such as busulfan, nitrosourea, or procarbazine (with a latent period of 5–7 years) or the DNA topoisomerase inhibitors (2-year latency). Acquired aplastic anemia, Fanconi anemia, and other constitutional marrow failure diseases can evolve into MDS. However, the typical MDS patient does not have a suggestive environmental exposure history or a preceding hematologic disease. MDS is a disease of aging, suggesting random cumulative intrinsic and environmental damage to marrow cells.
MDS is a clonal hematopoietic stem cell disorder characterized by disordered cell proliferation and impaired differentiation, resulting in cytopenias and risk of progression to leukemia. Both chromosomal and genetic instability have been implicated, and both are likely aging-related. Cytogenetic abnormalities are found in approximately one-half of patients, and some of the same specific lesions are also seen in frank leukemia; aneuploidy (chromosome loss or gain) is more frequent than translocations. More sensitive assays, such as comparative genomic hybridization and single nucleotide polymorphism arrays, reveal chromosomal abnormalities in a large proportion of patients with normal conventional cytogenetics. Accelerated telomere attrition may destabilize the genome in marrow failure and predispose to acquisition of chromosomal lesions. Cytogenetic abnormalities are not random (loss of all or part of 5, 7, and 20, trisomy of 8) and may be related to etiology (11q23 following topoisomerase II inhibitors). The type and number of cytogenetic abnormalities strongly correlate with the probability of leukemic transformation and survival.
Genomics has illuminated the role of point mutations in the pathophysiology of MDS. Recurrent somatic mutations, acquired in the abnormal marrow cells and absent in the germline, have been identified in almost 100 genes. Many of the same genes are also mutated in AML without MDS, whereas others are distinctive in subtypes of MDS. A prominent example of the latter is the discovery of mutations in genes of the RNA splicing machinery, especially SF3B1, which strongly associate with sideroblastic anemia. Some mutations correlate with prognosis: spliceosome defects with favorable outcome, and mutations in EZH2, TP53, RUNX1, and ASXL1 with poor outcome. Mutations and cytogenetic abnormalities are not independent: TP53 mutations associate with complex cytogenetic abnormalities and TET2 mutations with normal cytogenetics. Correlation and exclusion in the pattern of mutations indicate a functional genomic architecture. Analysis of deep sequencing results in patients whose MDS evolved to AML has shown evidence of clonal succession, with founder clones acquiring further mutations that allow clonal dominance. Furthermore, the prevalence of abnormal cells by morphology underestimates bone marrow involvement by MDS clones, as cells normal in appearance are apparently derived from the abnormal clones. Both presenting and evolving hematologic manifestations result from the accumulation of multiple genetic lesions: loss of tumor-suppressor genes, activating oncogene mutations, epigenetic pathways that affect mRNA processing and methylation status, or other harmful alterations. Pathophysiology has been linked to mutations and chromosome abnormalities in some specific MDS syndromes. The 5q– deletion leads to heterozygous loss of a ribosomal protein gene that is also mutant in Diamond-Blackfan anemia, and both are characterized by deficient erythropoiesis. An immune pathophysiology may underlie trisomy 8 MDS, in which patients often experience improved blood counts after immunosuppressive therapy; there is T cell activity directed to hematopoietic progenitors, which the cytogenetically aberrant clone resists. However, in general for MDS, the role of the immune system and its cells and cytokines; the role of the hematopoietic stem cell niche, the microenvironment, and cell-cell interactions; the fate of normal cells in the Darwinian competitive environment of the dysplastic marrow; and how mutant cells produce marrow failure in MDS are not well understood.
CLINICAL FEATURES
Anemia dominates the early course. Most symptomatic patients complain of the gradual onset of fatigue and weakness, dyspnea, and pallor, but at least one-half the patients are asymptomatic, and their MDS is discovered only incidentally on routine blood counts. Previous chemotherapy or radiation exposure is an important historic fact. Fever and weight loss should point to a myeloproliferative rather than myelodysplastic process. MDS in childhood is rare and, when diagnosed, increases the likelihood of an underlying genetic disease. Children with Down syndrome are susceptible to MDS, and a family history may indicate a hereditary form of sideroblastic anemia, Fanconi anemia, or a telomeropathy. Inherited GATA2 mutations, as in the MonoMAC syndrome (with increased susceptibility to viral, mycobacteria, and fungal infections, as well as deficient numbers of monocytes, natural killer cells, and B lymphocytes), also cause MDS in young patients.
The physical examination is remarkable for signs of anemia; approximately 20% of patients have splenomegaly. Some unusual skin lesions, including Sweet syndrome (febrile neutrophilic dermatosis), occur with MDS. Accompanying autoimmune syndromes are not infrequent. In the younger patient, stereotypical anomalies point to a constitutional syndrome (short stature, abnormal thumbs in Fanconi anemia; early graying in the telomeropathies; cutaneous warts in GATA2 deficiency).
LABORATORY STUDIES
Blood Anemia is present in most cases, either alone or as part of bi- or pancytopenia; isolated neutropenia or thrombocytopenia is more unusual. Macrocytosis is common, and the smear may be dimorphic with a distinctive population of large red blood cells. Platelets are also large and lack granules. In functional studies, they may show marked abnormalities, and patients may have bleeding symptoms despite seemingly adequate numbers. Neutrophils are hypogranulated; have hyposegmented, ringed, or abnormally segmented nuclei; contain Döhle bodies; and may be functionally deficient. Circulating myeloblasts usually correlate with marrow blast numbers, and their quantity is important for classification and prognosis. The total white blood cell count (WBC) is usually normal or low, except in chronic myelomonocytic leukemia. As in aplastic anemia, MDS can be associated with a clonal population of PNH cells. Genetic testing is commercially available for constitutional syndromes.
Bone Marrow The bone marrow is usually normal or hypercellular, but in about 20% of cases, it is sufficiently hypocellular to be confused with aplasia. No single characteristic feature of marrow morphology distinguishes MDS, but the following are commonly observed: dyserythropoietic changes (especially nuclear abnormalities) and ringed sideroblasts in the erythroid lineage; hypogranulation and hyposegmentation in granulocytic precursors, with an increase in myeloblasts; and megakaryocytes showing reduced numbers of or disorganized nuclei. Megaloblastic nuclei associated with defective hemoglobinization in the erythroid lineage are common. Prognosis strongly correlates with the proportion of marrow blasts. Cytogenetic analysis and fluorescent in situ hybridization can identify chromosomal abnormalities.
DIFFERENTIAL DIAGNOSIS
Deficiencies of vitamin B12 or folate should be excluded by appropriate blood tests; vitamin B6 deficiency can be assessed by a therapeutic trial of pyridoxine if the bone marrow shows ringed sideroblasts. Marrow dysplasia can be observed in acute viral infections, drug reactions, or chemical toxicity but should be transient. More difficult are the distinctions between hypocellular MDS and aplasia or between refractory anemia with excess blasts and early acute leukemia. The WHO considers the presence of 20% blasts in the marrow as the criterion that separates AML from MDS. In young patients, underlying, predisposing genetic diseases should be considered (see above).
PROGNOSIS
The median survival varies greatly from years for patients with 5q– or sideroblastic anemia to a few months in refractory anemia with excess blasts or severe pancytopenia associated with monosomy 7; an International Prognostic Scoring System (IPSS; Table 130-6) assists in making predictions. Even “low-risk” MDS has significant morbidity and mortality. Most patients die as a result of complications of pancytopenia and not due to leukemic transformation; perhaps one-third will succumb to other diseases unrelated to their MDS. Precipitous worsening of pancytopenia, acquisition of new chromosomal abnormalities on serial cytogenetic determination, increase in the number of blasts, and marrow fibrosis are all poor prognostic indicators. The outlook in therapy-related MDS, regardless of type, is extremely poor, and most patients will progress within a few months to refractory AML.
|
INTERNATIONAL PROGNOSTIC SCORING SYSTEM (IPSS) |
MYELOPHTHISIC ANEMIAS
Fibrosis of the bone marrow (see Fig. 129-2), usually accompanied by a characteristic blood smear picture called leukoerythroblastosis, can occur as a primary hematologic disease, called myelofibrosis or myeloid metaplasia (Chap. 131), and as a secondary process, called myelophthisis. Myelophthisis, or secondary myelofibrosis, is reactive. Fibrosis can be a response to invading tumor cells, usually an epithelial cancer of breast, lung, or prostate origin or neuroblastoma. Marrow fibrosis may occur with infection of mycobacteria (both Mycobacterium tuberculosis and Mycobacterium avium), fungi, or HIV and in sarcoidosis. Intracellular lipid deposition in Gaucher’s disease and obliteration of the marrow space related to absence of osteoclast remodeling in congenital osteopetrosis also can produce fibrosis. Secondary myelofibrosis is a late consequence of radiation therapy or treatment with radiomimetic drugs. Usually the infectious or malignant underlying processes are obvious. Marrow fibrosis can also be a feature of a variety of hematologic syndromes, especially chronic myeloid leukemia, multiple myeloma, lymphomas, myeloma, and hairy cell leukemia.
The pathophysiology has three distinct features: proliferation of fibroblasts in the marrow space (myelofibrosis); the extension of hematopoiesis into the long bones and into extramedullary sites, usually the spleen, liver, and lymph nodes (myeloid metaplasia); and ineffective erythropoiesis. The etiology of the fibrosis is unknown but most likely involves dysregulated production of growth factors: platelet-derived growth factor and transforming growth factor β have been implicated. Abnormal regulation of other hematopoietins would lead to localization of blood-producing cells in nonhematopoietic tissues and uncoupling of the usually balanced processes of stem cell proliferation and differentiation. Myelofibrosis is remarkable for pancytopenia despite very large numbers of circulating hematopoietic progenitor cells.
Anemia is dominant in secondary myelofibrosis, usually normocytic and normochromic. The diagnosis is suggested by the characteristic leukoerythroblastic smear (see Fig. 129-1). Erythrocyte morphology is highly abnormal, with circulating nucleated RBCs, teardrops, and shape distortions. WBC numbers are often elevated, sometimes mimicking a leukemoid reaction, with circulating myelocytes, promyelocytes, and myeloblasts. Platelets may be abundant and are often of giant size. Inability to aspirate the bone marrow, the characteristic “dry tap,” can allow a presumptive diagnosis in the appropriate setting before the biopsy is decalcified.
The course of secondary myelofibrosis is determined by its etiology, usually a metastatic tumor or an advanced hematologic malignancy. Treatable causes must be excluded, especially tuberculosis and fungus. Transfusion support can relieve symptoms.
131 |
Polycythemia Vera and Other Myeloproliferative Neoplasms |
The World Health Organization (WHO) classification of the chronic myeloproliferative neoplasms (MPNs) includes eight disorders, some of which are rare or poorly characterized (Table 131-1) but all of which share an origin in a multipotent hematopoietic progenitor cell; overproduction of one or more of the formed elements of the blood without significant dysplasia; and a predilection to extramedullary hematopoiesis, myelofibrosis, and transformation at varying rates to acute leukemia. Within this broad classification, however, significant phenotypic heterogeneity exists. Some diseases such as chronic myelogenous leukemia (CML), chronic neutrophilic leukemia (CNL), and chronic eosinophilic leukemia (CEL) express primarily a myeloid phenotype, whereas in other diseases, such as polycythemia vera (PV), primary myelofibrosis (PMF), and essential thrombocytosis (ET), erythroid or megakaryocytic hyperplasia predominates. The latter three disorders, in contrast to the former three, also appear capable of transforming into each other.
|
WORLD HEALTH ORGANIZATION CLASSIFICATION OF CHRONIC MYELOPROLIFERATIVE NEOPLASMS |
Such phenotypic heterogeneity has a genetic basis; CML is the consequence of the balanced translocation between chromosomes 9 and 22 [t(9;22)(q34;11)]; CNL has been associated with a t(15;19) translocation; and CEL occurs with a deletion or balanced translocations involving the PDGFRα gene. By contrast, to a greater or lesser extent, PV, PMF, and ET are characterized by a mutation, V617F, that causes constitutive activation of JAK2, a tyrosine kinase essential for the function of the erythropoietin and thrombopoietin receptors but not the granulocyte colony-stimulating factor receptor. This important distinction is also reflected in the natural histories of CML, CNL, and CEL, which are usually measured in years, and their high rate of leukemic transformation. By contrast, the natural history of PV, PMF, and ET is usually measured in decades, and transformation to acute leukemia is uncommon in PV and ET in the absence of exposure to mutagenic drugs. This chapter, therefore, will focus only on PV, PMF, and ET, because their clinical and genetic overlap is substantial even though their clinical courses are distinctly different.
The other chronic myeloproliferative neoplasms will be discussed in Chaps. 133 and 135e.
POLYCYTHEMIA VERA
PV is a clonal disorder involving a multipotent hematopoietic progenitor cell in which phenotypically normal red cells, granulocytes, and platelets accumulate in the absence of a recognizable physiologic stimulus. The most common of the chronic MPNs, PV occurs in 2.5 per 100,000 persons, sparing no adult age group and increasing with age to rates over 10/100,000. Familial transmission is infrequent, and women predominate among sporadic cases.
ETIOLOGY
![]() The etiology of PV is unknown. Although nonrandom chromosome abnormalities such as deletion 20q and trisomy 8 and 9 have been documented in up to 30% of untreated PV patients, unlike CML, no consistent cytogenetic abnormality has been associated with the disorder. However, a mutation in the autoinhibitory pseudokinase domain of the tyrosine kinase JAK2—that replaces valine with phenylalanine (V617F), causing constitutive kinase activation—appears to have a central role in the pathogenesis of PV.
The etiology of PV is unknown. Although nonrandom chromosome abnormalities such as deletion 20q and trisomy 8 and 9 have been documented in up to 30% of untreated PV patients, unlike CML, no consistent cytogenetic abnormality has been associated with the disorder. However, a mutation in the autoinhibitory pseudokinase domain of the tyrosine kinase JAK2—that replaces valine with phenylalanine (V617F), causing constitutive kinase activation—appears to have a central role in the pathogenesis of PV.
JAK2 is a member of an evolutionarily well-conserved, nonreceptor tyrosine kinase family and serves as the cognate tyrosine kinase for the erythropoietin and thrombopoietin receptors. It also functions as an obligate chaperone for these receptors in the Golgi apparatus and is responsible for their cell-surface expression. The conformational change induced in the erythropoietin and thrombopoietin receptors following binding to their respective cognate ligands, erythropoietin or thrombopoietin, leads to JAK2 autophosphorylation, receptor phosphorylation, and phosphorylation of proteins involved in cell proliferation, differentiation, and resistance to apoptosis. Transgenic animals lacking JAK2 die as embryos from severe anemia. Constitutive activation of JAK2, on the other hand, explains the erythropoietin hypersensitivity, erythropoietin-independent erythroid colony formation, rapid terminal differentiation, increase in Bcl-XL expression, and apoptosis resistance in the absence of erythropoietin that characterize the in vitro behavior of PV erythroid progenitor cells.
Importantly, the JAK2 gene is located on the short arm of chromosome 9, and loss of heterozygosity on chromosome 9p due to mitotic recombination is the most common cytogenetic abnormality in PV. The segment of 9p involved contains the JAK2 locus, and loss of heterozygosity in this region leads to homozygosity for JAK2 V617F. More than 95% of PV patients express this mutation, as do approximately 50% of PMF and ET patients. Homozygosity for the mutation occurs in approximately 30% of PV patients and 60% of PMF patients but is rare in ET. Over time, a portion of PV JAK2 V617F heterozygotes become homozygotes due to mitotic recombination, but usually not after 10 years of the disease. Most PV patients who do not express JAK2 V617F express a mutation in exon 12 of the kinase and are not clinically different from those who do, nor do JAK2 V617F heterozygotes differ clinically from homozygotes. Interestingly, the predisposition to acquire mutations in JAK2 appears to be associated with a specific JAK2 gene haplotype, GGCC. JAK2 V617F is the basis for many of the phenotypic and biochemical characteristics of PV such as elevation of the leukocyte alkaline phosphatase (LAP) score; however, it cannot solely account for the entire PV phenotype and is probably not the initiating lesion in the three MPNs. First, PV patients with the same phenotype and documented clonal disease lack any mutation of JAK2. Second, ET and PMF patients have the same mutation but different clinical phenotypes. Third, familial PV can occur without the mutation, even when other members of the same family express it. Fourth, not all the cells of the malignant clone express JAK2 V617F. Fifth, JAK2 V617F has been observed in patients with long-standing idiopathic erythrocytosis. Sixth, in some patients, JAK2 V617F appears to be acquired after another mutation. Finally, in some JAK2 V617F–positive PV or ET patients, acute leukemia can occur in a JAK2 V617F–negative progenitor cell. However, although JAK2 V617F alone may not be sufficient to cause PV, it appears essential for the transformation of ET to PV, although not for its transformation to PMF.
[/level-membership-for-internal-medicine-category][not-level-membership-for-internal-medicine-category]
UNSTABLE HEMOGLOBINS
Amino acid substitutions that reduce solubility or increase susceptibility to oxidation result in unstable hemoglobins that precipitate, forming inclusion bodies injurious to the RBC membrane. Representative mutations are those that interfere with contact points between the α and β subunits (e.g., Hb Philly [β35Tyr→Phe]), alter the helical segments (e.g., Hb Genova [β28Leu→Pro]), or disrupt interactions of the hydrophobic pockets of the globin subunits with heme (e.g., Hb Köln [β98Val→Met]) (Table 127-3). The inclusions, called Heinz bodies, are clinically detectable by staining with supravital dyes such as crystal violet. Removal of these inclusions by the spleen generates pitted, rigid cells that have shortened life spans, producing hemolytic anemia of variable severity, sometimes requiring chronic transfusion support. Splenectomy may be needed to correct the anemia. Leg ulcers and premature gallbladder disease due to bilirubin loading are frequent stigmata.
|
REPRESENTATIVE ABNORMAL HEMOGLOBINS WITH ALTERED SYNTHESIS OR FUNCTION |
Unstable hemoglobins occur sporadically, often by spontaneous new mutations. Heterozygotes are often symptomatic because a significant Heinz body burden can develop even when the unstable variant accounts for only a portion of the total hemoglobin. Symptomatic unstable hemoglobins tend to be β-globin variants, because sporadic mutations affecting only one of the four α globins alleles would generate only 20–30% abnormal hemoglobin.
HEMOGLOBINS WITH ALTERED OXYGEN AFFINITY
High-affinity hemoglobins (e.g., Hb Yakima [β99Asp→His]) bind oxygen more readily but deliver less O2 to tissues at normal capillary Po2 levels (Fig. 127-2). Mild tissue hypoxia ensues, stimulating RBC production and erythrocytosis (Table 127-3). In extreme cases, the hematocrits can rise to 60–65%, increasing blood viscosity and producing typical symptoms (headache, somnolence, or dizziness). Phlebotomy may be required. Typical mutations alter interactions within the heme pocket or disrupt the Bohr effect or salt-bond site. Mutations that impair the interaction of HbA with 2,3-BPG can increase O2 affinity because 2,3-BPG binding lowers O2 affinity.
Low-affinity hemoglobins (e.g., Hb Kansas [β102Asn→Lys]) bind sufficient oxygen in the lungs, despite their lower oxygen affinity, to achieve nearly full saturation. At capillary oxygen tensions, they lose sufficient amounts of oxygen to maintain homeostasis at a low hematocrit (Fig. 127-2) (pseudoanemia). Capillary hemoglobin desaturation can also be sufficient to produce clinically apparent cyanosis. Despite these findings, patients usually require no specific treatment.
METHEMOGLOBINEMIAS
Methemoglobin is generated by oxidation of the heme iron moieties to the ferric state, causing a characteristic bluish-brown muddy color resembling cyanosis. Methemoglobin has such high oxygen affinity that virtually no oxygen is delivered. Levels >50–60% are often fatal.
Congenital methemoglobinemia arises from globin mutations that stabilize iron in the ferric state (e.g., HbM Iwata [α87His→Tyr], Table 127-3) or from mutations that impair the enzymes that reduce methemoglobin to hemoglobin (e.g., methemoglobin reductase, NADP diaphorase). Acquired methemoglobinemia is caused by toxins that oxidize heme iron, notably nitrate and nitrite-containing compounds, including drugs commonly used in cardiology and anesthesiology.
DIAGNOSIS AND MANAGEMENT OF PATIENTS WITH UNSTABLE HEMOGLOBINS, HIGH-AFFINITY HEMOGLOBINS, AND METHEMOGLOBINEMIA
Unstable hemoglobin variants should be suspected in patients with nonimmune hemolytic anemia, jaundice, splenomegaly, or premature biliary tract disease. Severe hemolysis usually presents during infancy as neonatal jaundice or anemia. Milder cases may present in adult life with anemia or only as unexplained reticulocytosis, hepatosplenomegaly, premature biliary tract disease, or leg ulcers. Because spontaneous mutation is common, family history of anemia may be absent. The peripheral blood smear often shows anisocytosis, abundant cells with punctate inclusions, and irregular shapes (i.e., poikilocytosis).
The two best tests for diagnosing unstable hemoglobins are the Heinz body preparation and the isopropanol or heat stability test. Many unstable Hb variants are electrophoretically silent. A normal electrophoresis does not rule out the diagnosis. Mass spectroscopy or direct gene analysis will provide a definitive diagnosis.
Severely affected patients may require transfusion support for the first 3 years of life, because splenectomy before age 3 is associated with a significantly higher immune deficit. Splenectomy is usually effective thereafter, but occasional patients may require lifelong transfusion support. After splenectomy, patients can develop cholelithiasis and leg ulcers, hypercoagulable states, and susceptibility to overwhelming sepsis. Splenectomy should thus be avoided or delayed unless it is the only alternative. Precipitation of unstable hemoglobins is aggravated by oxidative stress, e.g., infection and antimalarial drugs, which should be avoided where possible.
High-O2 affinity hemoglobin variants should be suspected in patients with erythrocytosis. The best test for confirmation is measurement of the P50. A high-O2 affinity hemoglobin causes a significant left shift (i.e., lower numeric value of the P50); confounding conditions, e.g., tobacco smoking or carbon monoxide exposure, can also lower the P50.
High-affinity hemoglobins are often asymptomatic; rubor or plethora may be telltale signs. When the hematocrit approaches 60%, symptoms of high blood viscosity and sluggish flow (headache, lethargy, dizziness, etc.) may be present. These persons may benefit from judicious phlebotomy. Erythrocytosis represents an appropriate attempt to compensate for the impaired oxygen delivery by the abnormal variant. Overzealous phlebotomy may stimulate increased erythropoiesis or aggravate symptoms by thwarting this compensatory mechanism. The guiding principle of phlebotomy should be to improve oxygen delivery by reducing blood viscosity and increasing blood flow rather than restoration of a normal hematocrit. Phlebotomy-induced modest iron deficiency may aid in control.
Low-affinity hemoglobins should be considered in patients with cyanosis or a low hematocrit with no other reason apparent after thorough evaluation. The P50 test confirms the diagnosis. Counseling and reassurance are the interventions of choice.
Methemoglobin should be suspected in patients with hypoxic symptoms who appear cyanotic but have a Pao2 sufficiently high that hemoglobin should be fully saturated with oxygen. A history of nitrite or other oxidant ingestions may not always be available; some exposures may be inapparent to the patient, and others may result from suicide attempts. The characteristic muddy appearance of freshly drawn blood can be a critical clue. The best diagnostic test is methemoglobin assay, which is usually available on an emergency basis.
Methemoglobinemia often causes symptoms of cerebral ischemia at levels >15%; levels >60% are usually lethal. Intravenous injection of 1 mg/kg of methylene blue is effective emergency therapy. Milder cases and follow-up of severe cases can be treated orally with methylene blue (60 mg three to four times each day) or ascorbic acid (300–600 mg/d).
THALASSEMIA SYNDROMES
The thalassemia syndromes are inherited disorders of α- or β-globin biosynthesis. The reduced supply of globin diminishes production of hemoglobin tetramers, causing hypochromia and microcytosis. Unbalanced accumulation of α and β subunits occurs because the synthesis of the unaffected globins proceeds at a normal rate. Unbalanced chain accumulation dominates the clinical phenotype. Clinical severity varies widely, depending on the degree to which the synthesis of the affected globin is impaired, altered synthesis of other globin chains, and coinheritance of other abnormal globin alleles.
CLINICAL MANIFESTATIONS OF β THALASSEMIA SYNDROMES
Mutations causing thalassemia can affect any step in the pathway of globin gene expression: transcription, processing of the mRNA precursor, translation, and posttranslational metabolism of the β-globin polypeptide chain. The most common forms arise from mutations that derange splicing of the mRNA precursor or prematurely terminate translation of the mRNA.
Hypochromia and microcytosis characterize all forms of β thalassemia because of the reduced amounts of hemoglobin tetramers (Fig. 127-5). In heterozygotes (β thalassemia trait), this is the only abnormality seen. Anemia is minimal. In more severe homozygous states, unbalanced α- and β-globin accumulation causes accumulation of highly insoluble unpaired α chains. They form toxic inclusion bodies that kill developing erythroblasts in the marrow. Few of the proerythroblasts beginning erythroid maturation survive. The surviving RBCs bear a burden of inclusion bodies that are detected in the spleen, shortening the RBC life span and producing severe hemolytic anemia. The resulting profound anemia stimulates erythropoietin release and compensatory erythroid hyperplasia, but the marrow response is sabotaged by the ineffective erythropoiesis. Anemia persists. Erythroid hyperplasia can become exuberant and produce masses of extramedullary erythropoietic tissue in the liver and spleen.
FIGURE 127-5 β Thalassemia intermedia. Microcytic and hypochromic red blood cells are seen that resemble the red blood cells of severe iron-deficiency anemia. Many elliptical and teardrop-shaped red blood cells are noted.
Massive bone marrow expansion deranges growth and development. Children develop characteristic “chipmunk” facies due to maxillary marrow hyperplasia and frontal bossing. Thinning and pathologic fracture of long bones and vertebrae may occur due to cortical invasion by erythroid elements and profound growth retardation. Hemolytic anemia causes hepatosplenomegaly, leg ulcers, gallstones, and high-output congestive heart failure. The conscription of caloric resources to support erythropoiesis leads to inanition, susceptibility to infection, endocrine dysfunction, and in the most severe cases, death during the first decade of life. Chronic transfusions with RBCs improve oxygen delivery, suppress the excessive ineffective erythropoiesis, and prolong life, but the inevitable side effects, notably iron overload, often prove fatal by age 30 years.
Severity is highly variable. Known modulating factors are those that ameliorate the burden of unpaired α-globin inclusions. Alleles associated with milder synthetic defects and coinheritance of α thalassemia trait reduce clinical severity by reducing accumulation of excess α globin. HbF persists to various degrees in β thalassemias. γ-Globin gene chains can substitute for β chains, generating more hemoglobin and reducing the burden of α-globin inclusions. The terms β thalassemia major and β thalassemia intermedia are used to reflect the clinical heterogeneity. Patients with β thalassemia major require intensive transfusion support to survive. Patients with β thalassemia intermedia have a somewhat milder phenotype and can survive without transfusion. The terms β thalassemia minor and β thalassemia trait describe asymptomatic heterozygotes for β thalassemia.
THALASSEMIA SYNDROMES
The four classic α thalassemias, most common in Asians, are α thalassemia-2 trait, in which one of the four α-globin loci is deleted; α thalassemia-1 trait, with two deleted loci; HbH disease, with three loci deleted; and hydrops fetalis with Hb Barts, with all four loci deleted (Table 127-4). Nondeletion forms of α thalassemia also exist.
|
THE α THALASSEMIAS |
α Thalassemia-2 trait is an asymptomatic, silent carrier state. α Thalassemia-1 trait resembles β thalassemia minor. Offspring doubly heterozygous for α thalassemia-2 and α thalassemia-1 exhibit a more severe phenotype called HbH disease. Heterozygosity for a deletion that removes both genes from the same chromosome (cis deletion) is common in Asians and in those from the Mediterranean region, as is homozygosity for α thalassemia-2 (trans deletion). Both produce asymptomatic hypochromia and microcytosis.
In HbH disease, HbA production is only 25–30% normal. Fetuses accumulate some unpaired γ chains (Hb Barts; γ-chain tetramers). In adults, unpaired β chains accumulate and are soluble enough to form β4 tetramers called HbH. HbH forms few inclusions in erythroblasts and precipitates in circulating RBC. Patients with HbH disease have thalassemia intermedia characterized by moderately severe hemolytic anemia but milder ineffective erythropoiesis. Survival into midadult life without transfusions is common.
The homozygous state for the α thalassemia-1 cis deletion (hydrops fetalis) causes total absence of α-globin synthesis. No physiologically useful hemoglobin is produced beyond the embryonic stage. Excess γ globin forms tetramers called Hb Barts (γ4), which has a very high oxygen affinity. It delivers almost no O2 to fetal tissues, causing tissue asphyxia, edema (hydrops fetalis), congestive heart failure, and death in utero. α Thalassemia-2 trait is common (15–20%) among people of African descent. The cis α thalassemia-1 deletion is almost never seen, however. Thus, α thalassemia-2 and the trans form of α thalassemia-1 are very common, but HbH disease and hydrops fetalis are rare.
It has been known for some time that some patients with myelodysplasia or erythroleukemia produce RBC clones containing HbH. This phenomenon is due to mutations in the ATRX pathway that affect the LCR of the α-globin gene cluster.
DIAGNOSIS AND MANAGEMENT OF THALASSEMIAS
The diagnosis of β-thalassemia major is readily made during childhood on the basis of severe anemia accompanied by the characteristic signs of massive ineffective erythropoiesis: hepatosplenomegaly, profound microcytosis, a characteristic blood smear (Fig. 127-5), and elevated levels of HbF, HbA2, or both. Many patients require chronic hypertransfusion therapy designed to maintain a hematocrit of at least 27–30% so that erythropoiesis is suppressed. Splenectomy is required if the annual transfusion requirement (volume of RBCs per kilogram of body weight per year) increases by >50%. Folic acid supplements may be useful. Vaccination with Pneumovax in anticipation of eventual splenectomy is advised, as is close monitoring for infection, leg ulcers, and biliary tract disease. Many patients develop endocrine deficiencies as a result of iron overload. Early endocrine evaluation is required for glucose intolerance, thyroid dysfunction, and delayed onset of puberty or secondary sexual characteristics.
Patients with β thalassemia intermedia exhibit similar stigmata but can survive without chronic hypertransfusion. Management is particularly challenging because a number of factors can aggravate the anemia, including infection, onset of puberty, and development of splenomegaly and hypersplenism. Some patients may eventually benefit from splenectomy. The expanded erythron can cause absorption of excessive dietary iron and hemosiderosis, even without transfusion. Some patients eventually become transfusion dependent.
β Thalassemia minor (i.e., thalassemia trait) usually presents as profound microcytosis and hypochromia with target cells, but only minimal or mild anemia. The mean corpuscular volume is rarely >75 fL; the hematocrit is rarely <30–33%. Hemoglobin analysis classically reveals an elevated HbA2 (3.5–7.5%), but some forms are associated with normal HbA2 and/or elevated HbF. Genetic counseling and patient education are essential. Patients with β thalassemia trait should be warned that their blood picture resembles iron deficiency and can be misdiagnosed. They should eschew empirical use of iron, yet iron deficiency requiring replacement therapy can develop during pregnancy or from chronic bleeding.
Persons with α thalassemia trait may exhibit mild hypochromia and microcytosis usually without anemia. HbA2 and HbF levels are normal. Affected individuals usually require only genetic counseling. HbH disease resembles β thalassemia intermedia, with the added complication that the HbH molecule behaves like moderately unstable hemoglobin. Patients with HbH disease should undergo splenectomy if excessive anemia or a transfusion requirement develops. Oxidative drugs should be avoided. Iron overload leading to death can occur in more severely affected patients.
PREVENTION
Antenatal diagnosis of thalassemia syndromes is now widely available. DNA diagnosis is based on polymerase chain reaction (PCR) amplification of fetal DNA, obtained by amniocentesis or chorionic villus biopsy followed by hybridization to allele-specific oligonucleotide probes or direct DNA sequencing.
THALASSEMIC STRUCTURAL VARIANTS
Thalassemic structural variants are characterized by both defective synthesis and abnormal structure.
HEMOGLOBIN LEPORE
Hb Lepore [α2(δβ)2] arises by an unequal crossover and recombination event that fuses the proximal end of the δ-gene with the distal end of the closely linked β-gene. It is common in the Mediterranean basin. The resulting chromosome contains only the fused δβ gene. The Lepore (δβ) globin is synthesized poorly because the fused gene is under the control of the weak δ-globin promoter. Hb Lepore alleles have a phenotype like β thalassemia, except for the added presence of 2–20% Hb Lepore. Compound heterozygotes for Hb Lepore and a classic β thalassemia allele may also have severe thalassemia.
HEMOGLOBIN E
![]() HbE (i.e., α2β226Glu→Lys) is extremely common in Cambodia, Thailand, and Vietnam. The gene has become far more prevalent in the United States as a result of immigration of Asian persons, especially in California, where HbE is the most common variant detected. HbE is mildly unstable but not enough to affect RBC life span significantly. Heterozygotes resemble individuals with a mild β-thalassemia trait. Homozygotes have somewhat more marked abnormalities but are asymptomatic. Compound heterozygotes for HbE and a β thalassemia gene can have β thalassemia intermedia or β thalassemia major, depending on the severity of the coinherited thalassemic gene.
HbE (i.e., α2β226Glu→Lys) is extremely common in Cambodia, Thailand, and Vietnam. The gene has become far more prevalent in the United States as a result of immigration of Asian persons, especially in California, where HbE is the most common variant detected. HbE is mildly unstable but not enough to affect RBC life span significantly. Heterozygotes resemble individuals with a mild β-thalassemia trait. Homozygotes have somewhat more marked abnormalities but are asymptomatic. Compound heterozygotes for HbE and a β thalassemia gene can have β thalassemia intermedia or β thalassemia major, depending on the severity of the coinherited thalassemic gene.
The βE allele contains a single base change in codon 26 that causes the amino acid substitution. This mutation also activates a cryptic RNA splice site, generating a structurally abnormal globin mRNA that cannot be translated, from about 50% of the initial pre-mRNA molecules. The remaining 40–50% are normally spliced and generate functional mRNA that is translated into βE-globin because the mature mRNA carries the base change that alters codon 26.
Genetic counseling of the persons at risk for HbE should focus especially on the interaction of HbE with β thalassemia, because HbE homozygosity is a condition associated with mildly asymptomatic microcytosis, hypochromia, and hemoglobin levels rarely <100 g/L (<10 g/dL).
HEREDITARY PERSISTENCE OF FETAL HEMOGLOBIN
HPFH is characterized by continued synthesis of high levels of HbF in adult life. No deleterious effects are apparent, even when all of the hemoglobin produced is HbF. These rare patients demonstrate convincingly that prevention or reversal of the fetal to adult hemoglobin switch would provide effective therapy for sickle cell anemia and β thalassemia.
ACQUIRED HEMOGLOBINOPATHIES
The two most important acquired hemoglobinopathies are carbon monoxide poisoning and methemoglobinemia (see above). Carbon monoxide has a higher affinity for hemoglobin than does oxygen; it can replace oxygen and diminish O2 delivery. Chronic elevation of carboxyhemoglobin levels to 10 or 15%, as occurs in smokers, can lead to secondary polycythemia. Carboxyhemoglobin is cherry red in color and masks the development of cyanosis usually associated with poor O2 delivery to tissues.
Abnormalities of hemoglobin biosynthesis have also been described in blood dyscrasias. In some patients with myelodysplasia, erythroleukemia, or myeloproliferative disorders, elevated HbF or a mild form of HbH disease may also be seen. The abnormalities are not severe enough to alter the course of the underlying disease.
EXPERIMENTAL THERAPIES
BONE MARROW TRANSPLANTATION, GENE THERAPY, AND MANIPULATION OF HbF
Bone marrow transplantation provides stem cells able to express normal hemoglobin; it has been used in a large number of patients with β thalassemia and a smaller number of patients with sickle cell anemia. Early in the course of disease, before end-organ damage occurs, transplantation is curative in 80–90% of patients. In highly experienced centers, the treatment-related mortality is <10%. Because survival into adult life is possible with conventional therapy, the decision to transplant is best made in consultation with specialized centers.
Gene therapy of thalassemia and sickle cell disease has proved to be an elusive goal, but experimental advances are raising expectations.
Reestablishing high levels of fetal hemoglobin synthesis should ameliorate the symptoms of β-chain hemoglobinopathies. Cytotoxic agents such as hydroxyurea and cytarabine promote high levels of HbF synthesis, probably by stimulating proliferation of the primitive HbF-producing progenitor cell population (i.e., F cell progenitors). Unfortunately, this regimen has not yet been effective in β thalassemia. Butyrates stimulate HbF production, but only transiently. Pulsed or intermittent administration has been found to sustain HbF induction in the majority of patients with sickle cell disease. It is unclear whether butyrates will have similar activity in patients with β thalassemia.
APLASTIC AND HYPOPLASTIC CRISIS IN PATIENTS WITH HEMOGLOBINOPATHIES
Patients with hemolytic anemias sometimes exhibit an alarming decline in hematocrit during and immediately after acute illnesses. Bone marrow suppression occurs in almost everyone during acute and chronic inflammatory illnesses. In patients with short RBC life spans, suppression can affect RBC counts more dramatically. These hypoplastic crises are usually transient and self-correcting before intervention is required.
Aplastic crisis refers to a profound cessation of erythroid activity in patients with chronic hemolytic anemias. It is associated with a rapidly falling hematocrit. Episodes are usually self-limited. Aplastic crises are caused by infection with a particular strain of parvovirus, B19A. Children infected with this virus usually develop permanent immunity. Aplastic crises do not often recur and are rarely seen in adults. Management requires close monitoring of the hematocrit and reticulocyte count. If anemia becomes symptomatic, transfusion support is indicated. Most crises resolve spontaneously within 1–2 weeks.
128 |
Megaloblastic Anemias |
The megaloblastic anemias are a group of disorders characterized by the presence of distinctive morphologic appearances of the developing red cells in the bone marrow. The marrow is usually hypercellular and the anemia is based on ineffective erythropoiesis. The cause is usually a deficiency of either cobalamin (vitamin B12) or folate, but megaloblastic anemia may occur because of genetic or acquired abnormalities that affect the metabolism of these vitamins or because of defects in DNA synthesis not related to cobalamin or folate (Table 128-1). Cobalamin and folate absorption and metabolism are described next, followed by the biochemical basis, clinical and laboratory features, causes, and treatment of megaloblastic anemia.
|
CAUSES OF MEGALOBLASTIC ANEMIA |
COBALAMIN
Cobalamin (vitamin B12) exists in a number of different chemical forms. All have a cobalt atom at the center of a corrin ring. In nature, the vitamin is mainly in the 2-deoxyadenosyl (ado) form, which is located in mitochondria. It is the cofactor for the enzyme methylmalonyl coenzyme A (CoA) mutase. The other major natural cobalamin is methylcobalamin, the form in human plasma and in cell cytoplasm. It is the cofactor for methionine synthase. There are also minor amounts of hydroxocobalamin to which methyl- and adocobalamin are converted rapidly by exposure to light.
DIETARY SOURCES AND REQUIREMENTS
Cobalamin is synthesized solely by microorganisms. Ruminants obtain cobalamin from the foregut, but the only source for humans is food of animal origin, e.g., meat, fish, and dairy products. Vegetables, fruits, and other foods of nonanimal origin are free from cobalamin unless they are contaminated by bacteria. A normal Western diet contains 5–30 μg of cobalamin daily. Adult daily losses (mainly in the urine and feces) are 1–3 μg (~0.1% of body stores), and because the body does not have the ability to degrade cobalamin, daily requirements are also about 1–3 μg. Body stores are of the order of 2–3 mg, sufficient for 3–4 years if supplies are completely cut off.
ABSORPTION
Two mechanisms exist for cobalamin absorption. One is passive, occurring equally through buccal, duodenal, and ileal mucosa; it is rapid but extremely inefficient, with <1% of an oral dose being absorbed by this process. The normal physiologic mechanism is active; it occurs through the ileum and is efficient for small (a few micrograms) oral doses of cobalamin, and it is mediated by gastric intrinsic factor (IF). Dietary cobalamin is released from protein complexes by enzymes in the stomach, duodenum, and jejunum; it combines rapidly with a salivary glycoprotein that belongs to the family of cobalamin-binding proteins known as haptocorrins (HCs). In the intestine, the haptocorrin is digested by pancreatic trypsin and the cobalamin is transferred to IF.
IF (gene at chromosome 11q13) is produced in the gastric parietal cells of the fundus and body of the stomach, and its secretion parallels that of hydrochloric acid. Normally, there is a vast excess of IF. The IF-cobalamin complex passes to the ileum, where IF attaches to a specific receptor (cubilin) on the microvillus membrane of the enterocytes. Cubilin also is present in yolk sac and renal proximal tubular epithelium. Cubilin appears to traffic by means of amnionless (AMN), an endocytic receptor protein that directs sublocalization and endocytosis of cubilin with its ligand IF-cobalamin complex. The cobalamin-IF complex enters the ileal cell, where IF is destroyed. After a delay of about 6 h, the cobalamin appears in portal blood attached to transcobalamin (TC) II.
Between 0.5 and 5 μg of cobalamin enter the bile each day. This binds to IF, and a major portion of biliary cobalamin normally is reabsorbed together with cobalamin derived from sloughed intestinal cells. Because of the appreciable amount of cobalamin undergoing enterohepatic circulation, cobalamin deficiency develops more rapidly in individuals who malabsorb cobalamin than it does in vegans, in whom reabsorption of biliary cobalamin is intact.
TRANSPORT
Two main cobalamin transport proteins exist in human plasma; they both bind cobalamin—one molecule for one molecule. One HC, also known as TC I, is closely related to other cobalamin-binding HCs in milk, gastric juice, bile, saliva, and other fluids. The gene TCNL is at chromosome 11q11-q12.3. These HCs differ from each other only in the carbohydrate moiety of the molecule. TC I is derived primarily from the specific granules in neutrophils. Normally, it is about two-thirds saturated with cobalamin, which it binds tightly. TC I does not enhance cobalamin entry into tissues. Glycoprotein receptors on liver cells are involved in the removal of TC I from plasma, and TC I may play a role in the transport of cobalamin analogues (which it binds more effectively than IF) to the liver for excretion in bile.
The other major cobalamin transport protein in plasma is transcobalamin, also known as TC II. The gene is on chromosome 22q11-q13.1. As for IF and HC, there are nine exons. The three proteins are likely to have a common ancestral origin. TC II is synthesized by liver and by other tissues, including macrophages, ileum, and vascular endothelium. It normally carries only 20–60 ng of cobalamin per liter of plasma and readily gives up cobalamin to marrow, placenta, and other tissues, which it enters by receptor-mediated endocytosis involving the TC II receptor and megalin (encoded by the LRP-2 gene). The TC II cobalamin is internalized by endocytosis via clathrin-coated pits; the complex is degraded, but the receptor probably is recycled to the cell membrane as is the case for transferrin. Export of “free” cobalamin is via the ATP-binding cassette drug transporter alias multidrug resistance protein 1.
FOLATE
DIETARY FOLATE
Folic (pteroylglutamic) acid is a yellow, crystalline, water-soluble substance. It is the parent compound of a large family of natural folate compounds, which differ from it in three respects: (1) they are partly or completely reduced to di- or tetrahydrofolate (THF) derivatives, (2) they usually contain a single carbon unit (Table 128-2), and (3) 70–90% of natural folates are folate-polyglutamates.
|
BIOCHEMICAL REACTIONS OF FOLATE COENZYMES |
Most foods contain some folate. The highest concentrations are found in liver, yeast, spinach, other greens, and nuts (>100 μg/100 g). The total folate content of an average Western diet is ~250 μg daily, but the amount varies widely according to the type of food eaten and the method of cooking. Folate is easily destroyed by heating, particularly in large volumes of water. Total body folate in the adult is ~10 mg, with the liver containing the largest store. Daily adult requirements are ~100 μg, and so stores are sufficient for only 3–4 months in normal adults and severe folate deficiency may develop rapidly.
ABSORPTION
Folates are absorbed rapidly from the upper small intestine. The absorption of folate polyglutamates is less efficient than that of monoglutamates; on average, ~50% of food folate is absorbed. Polyglutamate forms are hydrolyzed to the monoglutamate derivatives either in the lumen of the intestine or within the mucosa. All dietary folates are converted to 5-methylTHF (5-MTHF) within the small intestinal mucosa before entering portal plasma. The monoglutamates are actively transported across the enterocyte by a proton-coupled folate transporter (PCFT, SCL46A1). This is situated at the apical brush border and is most active at pH 5.5, which is about the pH of the duodenal and jejunal surface. Genetic mutations of this protein underlie hereditary malabsorption of folate (see below). Pteroylglutamic acid at doses >400 μg is absorbed largely unchanged and converted to natural folates in the liver. Lower doses are converted to 5-MTHF during absorption through the intestine.
About 60–90 μg of folate enters the bile each day and is excreted into the small intestine. Loss of this folate, together with the folate of sloughed intestinal cells, accelerates the speed with which folate deficiency develops in malabsorption conditions.
TRANSPORT
Folate is transported in plasma; about one-third is loosely bound to albumin, and two-thirds is unbound. In all body fluids (plasma, cerebrospinal fluid, milk, bile), folate is largely, if not entirely, 5-MTHF in the monoglutamate form. Three types of folate-binding protein are involved. A reduced folate transporter (RFC, SLC19A1) is the major route of delivery of plasma folate (5-MTHF) to cells. Two folate receptors, FR2 and FR3 embedded in the cell membrane by a glycosyl phosphatidylinositol anchor, transport folate into the cell via receptor-mediated endocytosis. The third protein, PCFT, transports folate at low pH from the vesicle to the cell cytoplasm. The reduced folate transporter also mediates uptake of methotrexate by cells.
BIOCHEMICAL FUNCTIONS
Folates (as the intracellular polyglutamate derivatives) act as coenzymes in the transfer of single-carbon units (Fig. 128-1 and Table 128-2). Two of these reactions are involved in purine synthesis and one in pyrimidine synthesis necessary for DNA and RNA replication. Folate is also a coenzyme for methionine synthesis, in which methylcobalamin is also involved and in which THF is regenerated. THF is the acceptor of single carbon units newly entering the active pool via conversion of serine to glycine. Methionine, the other product of the methionine synthase reaction, is the precursor for S-adenosylmethionine (SAM), the universal methyl donor involved in >100 methyltransferase reactions (Fig. 128-1).
FIGURE 128-1 The role of folates in DNA synthesis and in formation of S-adenosylmethionine (SAM), which is involved in numerous methylation reactions. DHF, dihydrofolate; GSH, glutathione. (Reprinted from AV Hoffbrand et al [eds]: Postgraduate Haematology, 5th ed. Oxford, UK, Blackwell Publishing, 2005; with permission.)
During thymidylate synthesis, 5,10-methylene-THF is oxidized to DHF (dihydrofolate). The enzyme DHF reductase converts this to THF. The drugs methotrexate, pyrimethamine, and (mainly in bacteria) trimethoprim inhibit DHF reductase and so prevent formation of active THF coenzymes from DHF. A small fraction of the folate coenzyme is not recycled during thymidylate synthesis but is degraded at the C9-N10 bond.
BIOCHEMICAL BASIS OF MEGALOBLASTIC ANEMIA
The common feature of all megaloblastic anemias is a defect in DNA synthesis that affects rapidly dividing cells in the bone marrow. All conditions that give rise to megaloblastic changes have in common a disparity in the rate of synthesis or availability of the four immediate precursors of DNA: the deoxyribonucleoside triphosphates (dNTPs)—dA(adenine)TP and dG(guanine)TP (purines), dT(thymine)TP and dC(cytosine)TP (pyrimidines). In deficiencies of either folate or cobalamin, there is failure to convert deoxyuridine monophosphate (dUMP) to deoxythymidine monophosphate (dTMP), the precursor of dTTP (Fig 128-1). This is the case because folate is needed as the coenzyme 5,10-methylene-THF polyglutamate for conversion of dUMP to dTMP; the availability of 5,10-methylene-THF is reduced in either cobalamin or folate deficiency. An alternative theory for megaloblastic anemia in cobalamin or folate deficiency is misincorporation of uracil into DNA because of a buildup of deoxyuridine triphosphate (dUTP) at the DNA replication fork as a consequence of the block in conversion of dUMP to dTMP.
COBALAMIN-FOLATE RELATIONS
Folate is required for many reactions in mammalian tissues. Only two reactions in the body are known to require cobalamin. Methylmalonyl CoA isomerization requires adocobalamin, and the methylation of homocysteine to methionine requires both methylcobalamin and 5-MTHF (Fig. 128-1). This reaction is the first step in the pathway by which 5-MTHF, which enters bone marrow and other cells from plasma, is converted into all the intracellular folate coenzymes. The coenzymes are all polyglutamated (the larger size aiding retention in the cell), but the enzyme folate polyglutamate synthase can use only THF, not MTHF, as substrate. In cobalamin deficiency, MTHF accumulates in plasma, and intracellular folate concentrations fall due to failure of formation of THF, the substrate on which folate polyglutamates are built. This has been termed THF starvation, or the methylfolate trap.
This theory explains the abnormalities of folate metabolism that occur in cobalamin deficiency (high serum folate, low cell folate, positive purine precursor aminoimidazole carboxamide ribonucleotide [AICAR] excretion; Table 128-2) and also why the anemia of cobalamin deficiency responds to folic acid in large doses.
CLINICAL FEATURES
Many symptomless patients are detected through the finding of a raised mean corpuscular volume (MCV) on a routine blood count. The main clinical features in more severe cases are those of anemia. Anorexia is usually marked, and there may be weight loss, diarrhea, or constipation. Glossitis, angular cheilosis, a mild fever in more severely anemic patients, jaundice (unconjugated), and reversible melanin skin hyperpigmentation also may occur with a deficiency of either folate or cobalamin. Thrombocytopenia sometimes leads to bruising, and this may be aggravated by vitamin C deficiency or alcohol in malnourished patients. The anemia and low leukocyte count may predispose to infections, particularly of the respiratory and urinary tracts. Cobalamin deficiency has also been associated with impaired bactericidal function of phagocytes.
GENERAL TISSUE EFFECTS OF COBALAMIN AND FOLATE DEFICIENCIES
Epithelial Surfaces After the marrow, the next most frequently affected tissues are the epithelial cell surfaces of the mouth, stomach, and small intestine and the respiratory, urinary, and female genital tracts. The cells show macrocytosis, with increased numbers of multinucleate and dying cells. The deficiencies may cause cervical smear abnormalities.
Complications of Pregnancy The gonads are also affected, and infertility is common in both men and women with either deficiency. Maternal folate deficiency has been implicated as a cause of prematurity, and both folate deficiency and cobalamin deficiency have been implicated in recurrent fetal loss and neural tube defects, as discussed below.
Neural Tube Defects Folic acid supplements at the time of conception and in the first 12 weeks of pregnancy reduce by ~70% the incidence of neural tube defects (NTDs) (anencephaly, meningomyelocele, encephalocele, and spina bifida) in the fetus. Most of this protective effect can be achieved by taking folic acid, 0.4 mg daily, at the time of conception.
The incidence of cleft palate and harelip also can be reduced by prophylactic folic acid. There is no clear simple relationship between maternal folate status and these fetal abnormalities, although overall the lower the maternal folate, the greater the risk to the fetus. NTDs also can be caused by antifolate and antiepileptic drugs.
An underlying maternal folate metabolic abnormality has also been postulated. One abnormality has been identified: reduced activity of the enzyme 5,10-methylene-THF reductase (MTHFR) (Fig. 128-1) caused by a common C677T polymorphism in the MTHFR gene. In one study, the prevalence of this polymorphism was found to be higher than in controls in the parents of NTD fetuses and in the fetuses themselves: homozygosity for the TT mutation was found in 13% of cases compared with 5% of control subjects. The polymorphism codes for a thermolabile form of MTHFR. The homozygous state results in a lower mean serum and red cell folate level compared with control subjects, as well as significantly higher serum homocysteine levels. Tests for mutations in other enzymes possibly associated with NTDs, e.g., methionine synthase and serine–glycine hydroxymethylase, have been negative. Serum vitamin B12 levels are also lower in the sera of mothers of NTD infants than in controls. In addition, maternal TC II receptor polymorphisms are associated with increased risk of NTD births. There are, however, no studies showing dietary fortification with vitamin B12 reduces the incidence of NTDs.
Cardiovascular Disease Children with severe homocystinuria (blood levels ≥100 μmol/L) due to deficiency of one of three enzymes, methionine synthase, MTHFR, or cystathionine synthase (Fig. 128-1), have vascular disease, e.g., ischemic heart disease, cerebrovascular disease, or pulmonary embolus, as teenagers or in young adulthood. Lesser degrees of raised serum homocysteine and low levels of serum folate and homozygous inherited mutations of MTHFR have been found to be associated with cerebrovascular, peripheral vascular, and coronary heart disease and with deep vein thrombosis. Prospective randomized trials of lowering homocysteine levels with supplements of folic acid, vitamin B12, and vitamin B6 against placebo over a 5-year period in patients with vascular disease or diabetes have not, however, shown a reduction of first event fatal or nonfatal myocardial infarction, nor have these supplements reduced the risk of recurrent cardiovascular disease after an acute myocardial infarct. Meta-analysis showed an 18% reduction in strokes but no significant prevention of death from any cause. Venous thrombosis has been reported to be more frequent in vitamin B12–deficient subjects than in controls. This was ascribed to raised plasma homocysteine levels in vitamin B12 deficiency.
Malignancy Prophylactic folic acid in pregnancy has been found in some but not all studies to reduce the subsequent incidence of acute lymphoblastic leukemia (ALL) in childhood. A significant negative association has also been found with the MTHFR C677T polymorphism and leukemias with mixed lineage leukemia (MLL) translocations, but a positive association with hyperdiploidy in infants with ALL or acute myeloid leukemia or with childhood ALL. A second polymorphism in the MTHFR gene, A1298C, is also strongly associated with hyperdiploid leukemia. There are various positive and negative associations between polymorphisms in folate-dependent enzymes and the incidence of adult ALL. The C677T polymorphism is thought to lead to increased thymidine pools and “better quality” of DNA synthesis by shunting one-carbon groups toward thymidine and purine synthesis. This may explain its reported association with a lower risk for colorectal cancer. Most but not all studies suggest that prophylactic folic acid also protects against colon adenomas. Other tumors that have been associated with folate polymorphisms or status include follicular lymphoma, breast cancer, and gastric cancer. A meta-analysis of 50,000 individuals given folic acid or placebo in cardiovascular or colon adenoma prevention trials found that folic acid supplementation did not substantially increase or decrease the incidence of site-specific cancer during the first 5 years of treatment. Because folic acid may “feed” tumors, it probably should be avoided in those with established tumors unless there is severe megaloblastic anemia due to folate deficiency.
Neurologic Manifestations Cobalamin deficiency may cause a bilateral peripheral neuropathy or degeneration (demyelination) of the posterior and pyramidal tracts of the spinal cord and, less frequently, optic atrophy or cerebral symptoms.
The patient, more frequently male, presents with paresthesias, muscle weakness, or difficulty in walking and sometimes dementia, psychotic disturbances, or visual impairment. Long-term nutritional cobalamin deficiency in infancy leads to poor brain development and impaired intellectual development. Folate deficiency has been suggested to cause organic nervous disease, but this is uncertain, although methotrexate injected into the cerebrospinal fluid may cause brain or spinal cord damage.
An important clinical problem is the nonanemic patient with neurologic or psychiatric abnormalities and a low or borderline serum cobalamin level. In such patients, it is necessary to try to establish whether there is significant cobalamin deficiency, e.g., by careful examination of the blood film, tests for serum gastrin level and for antibodies to IF or parietal cells, along with serum methylmalonic acid (MMA) measurement if available. A trial of cobalamin therapy for at least 3 months will usually also be needed to determine whether the symptoms improve.
The biochemical basis for cobalamin neuropathy remains obscure. Its occurrence in the absence of methylmalonic aciduria in TC II deficiency suggests that the neuropathy is related to the defect in homocysteine-methionine conversion. Accumulation of S-adenosylhomocysteine in the brain, resulting in inhibition of transmethylation reactions, has been suggested.
Psychiatric disturbance is common in both folate and cobalamin deficiencies. This, like the neuropathy, has been attributed to a failure of the synthesis of SAM, which is needed in methylation of biogenic amines (e.g., dopamine) as well as that of proteins, phospholipids, and neurotransmitters in the brain (Fig. 128-1). Associations between lower serum folate or cobalamin levels and higher homocysteine levels and the development of decreased cognitive function and dementia in Alzheimer’s disease have been reported. A meta-analysis of randomized, placebo-controlled trials of homocysteine-lowering B-vitamin supplementation of individuals with and without cognitive impairment, however, showed that supplementation with vitamin B12, vitamin B6, and folic acid alone or in combination did not improve cognitive function. It is unknown whether prolonged treatment with these B vitamins can reduce the risk of dementia in later life.
HEMATOLOGIC FINDINGS
PERIPHERAL BLOOD
Oval macrocytes, usually with considerable anisocytosis and poikilocytosis, are the main feature (Fig. 128-2A). The MCV is usually >100 fL unless a cause of microcytosis (e.g., iron deficiency or thalassemia trait) is present. Some of the neutrophils are hypersegmented (more than five nuclear lobes). There may be leukopenia due to a reduction in granulocytes and lymphocytes, but this is usually >1.5 × 109/L; the platelet count may be moderately reduced, rarely to <40 × 109/L. The severity of all these changes parallels the degree of anemia. In a nonanemic patient, the presence of a few macrocytes and hypersegmented neutrophils in the peripheral blood may be the only indication of the underlying disorder.
FIGURE 128-2 A. The peripheral blood in severe megaloblastic anemia. B. The bone marrow in severe megaloblastic anemia. (Reprinted from AV Hoffbrand et al [eds]: Postgraduate Haematology, 5th ed. Oxford, UK, Blackwell Publishing, 2005; with permission.)
BONE MARROW
In a severely anemic patient, the marrow is hypercellular with an accumulation of primitive cells due to selective death by apoptosis of more mature forms. The erythroblast nucleus maintains a primitive appearance despite maturation and hemoglobinization of the cytoplasm. The cells are larger than normoblasts, and an increased number of cells with eccentric lobulated nuclei or nuclear fragments may be present (Fig. 128-2B). Giant and abnormally shaped metamyelocytes and enlarged hyperpolyploid megakaryocytes are characteristic. In severe cases, the accumulation of primitive cells may mimic acute myeloid leukemia, whereas in less anemic patients, the changes in the marrow may be difficult to recognize. The terms intermediate, mild, and early have been used. The term megaloblastoid does not mean mildly megaloblastic. It is used to describe cells with both immature-appearing nuclei and defective hemoglobinization and is usually seen in myelodysplasia.
CHROMOSOMES
Bone marrow cells, transformed lymphocytes, and other proliferating cells in the body show a variety of changes, including random breaks, reduced contraction, spreading of the centromere, and exaggeration of secondary chromosomal constrictions and overprominent satellites. Similar abnormalities may be produced by antimetabolite drugs (e.g., cytosine arabinoside, hydroxyurea, and methotrexate) that interfere with either DNA replication or folate metabolism and that also cause megaloblastic appearances.
INEFFECTIVE HEMATOPOIESIS
There is an accumulation of unconjugated bilirubin in plasma due to the death of nucleated red cells in the marrow (ineffective erythropoiesis). Other evidence for this includes raised urine urobilinogen, reduced haptoglobins and positive urine hemosiderin, and a raised serum lactate dehydrogenase. A weakly positive direct antiglobulin test due to complement can lead to a false diagnosis of autoimmune hemolytic anemia.
CAUSES OF COBALAMIN DEFICIENCY
Cobalamin deficiency is usually due to malabsorption. The only other cause is inadequate dietary intake.
INADEQUATE DIETARY INTAKE
Adults Dietary cobalamin deficiency arises in vegans who omit meat, fish, eggs, cheese, and other animal products from their diet. The largest group in the world consists of Hindus, and it is likely that many millions of Indians are at risk of deficiency of cobalamin on a nutritional basis. Subnormal serum cobalamin levels are found in up to 50% of randomly selected, young, adult Indian vegans, but the deficiency usually does not progress to megaloblastic anemia since the diet of most vegans is not totally lacking in cobalamin and the enterohepatic circulation of cobalamin is intact. Dietary cobalamin deficiency may also arise rarely in nonvegetarian individuals who exist on grossly inadequate diets because of poverty or psychiatric disturbance.
Infants Cobalamin deficiency has been described in infants born to severely cobalamin-deficient mothers. These infants develop megaloblastic anemia at about 3–6 months of age, presumably because they are born with low stores of cobalamin and because they are fed breast milk with low cobalamin content. The babies have also shown growth retardation, impaired psychomotor development, and other neurologic sequelae.
GASTRIC CAUSES OF COBALAMIN MALABSORPTION
See Tables 128-3 and 128-4.
|
CAUSES OF COBALAMIN DEFICIENCY SUFFICIENTLY SEVERE TO CAUSE MEGALOBLASTIC ANEMIA |
|
MALABSORPTION OF COBALAMIN MAY OCCUR IN THE FOLLOWING CONDITIONS BUT IS NOT USUALLY SUFFICIENTLY SEVERE AND PROLONGED TO CAUSE MEGALOBLASTIC ANEMIA |
Pernicious Anemia Pernicious anemia (PA) may be defined as a severe lack of IF due to gastric atrophy. It is a common disease in north Europeans but occurs in all countries and ethnic groups. The overall incidence is about 120 per 100,000 population in the United Kingdom (UK). The ratio of incidence in men and women among whites is ~1:1.6, and the peak age of onset is 60 years, with only 10% of patients being <40 years of age. However, in some ethnic groups, notably black individuals and Latin Americans, the age at onset of PA is generally lower. The disease occurs more commonly than by chance in close relatives and in persons with other organ-specific autoimmune diseases, e.g., thyroid diseases, vitiligo, hypoparathyroidism, and Addison’s disease. It is also associated with hypogammaglobulinemia, with premature graying or blue eyes, and persons of blood group A. An association with human leukocyte antigen (HLA) 3 has been reported in some but not all series and, in those with endocrine disease, with HLA-B8, -B12, and -BW15. Life expectancy is normal in women once regular treatment has begun. Men have a slightly subnormal life expectancy as a result of a higher incidence of carcinoma of the stomach than in control subjects. Gastric output of hydrochloric acid, pepsin, and IF is severely reduced. The serum gastrin level is raised, and serum pepsinogen I levels are low.
Gastric Biopsy A single endoscopic examination is recommended if PA is diagnosed. Gastric biopsy usually shows atrophy of all layers of the body and fundus, with loss of glandular elements, an absence of parietal and chief cells and replacement by mucous cells, a mixed inflammatory cell infiltrate, and perhaps intestinal metaplasia. The infiltrate of plasma cells and lymphocytes contains an excess of CD4 cells. These are directed against gastric H/K-ATPase. The antral mucosa is usually well preserved. Helicobacter pylori infection occurs infrequently in PA, but it has been suggested that H. pylori gastritis occurs at an early phase of atrophic gastritis and presents in younger patients as iron-deficiency anemia but in older patients as PA. H. pylori is suggested to stimulate an autoimmune process directed against parietal cells, with the H. pylori infection then being gradually replaced, in some individuals, by an autoimmune process.
Serum Antibodies Two types of IF immunoglobulin G antibody may be found in the sera of patients with PA. One, the “blocking,” or type I, antibody, prevents the combination of IF and cobalamin, whereas the “binding,” or type II, antibody prevents attachment of IF to ileal mucosa. Type I occurs in the sera of ~55% of patients, and type II in 35%. IF antibodies cross the placenta and may cause temporary IF deficiency in a newborn infant. Patients with PA also show cell-mediated immunity to IF. Type I antibody has been detected rarely in the sera of patients without PA but with thyrotoxicosis, myxedema, Hashimoto’s disease, or diabetes mellitus and in relatives of PA patients. IF antibodies also have been detected in gastric juice in ~80% of PA patients. These gastric antibodies may reduce absorption of dietary cobalamin by combining with small amounts of remaining IF.
Parietal cell antibody is present in the sera of almost 90% of adult patients with PA but is frequently present in other subjects. Thus, it occurs in as many as 16% of randomly selected female subjects age >60 years. The parietal cell antibody is directed against the α and β subunits of the gastric proton pump (H+,K+-ATPase).
JUVENILE PERNICIOUS ANEMIA
This usually occurs in older children and resembles PA of adults. Gastric atrophy, achlorhydria, and serum IF antibodies are all present, although parietal cell antibodies are usually absent. About one-half of these patients show an associated endocrinopathy such as autoimmune thyroiditis, Addison’s disease, or hypoparathyroidism; in some, mucocutaneous candidiasis occurs.
CONGENITAL INTRINSIC FACTOR DEFICIENCY OR FUNCTIONAL ABNORMALITY
An affected child usually presents with megaloblastic anemia in the first to third year of life; a few have presented as late as the second decade. The child usually has no demonstrable IF but has a normal gastric mucosa and normal secretion of acid. The inheritance is autosomal recessive. Parietal cell and IF antibodies are absent. Variants have been described in which the child is born with IF that can be detected immunologically but is unstable or functionally inactive, unable to bind cobalamin or to facilitate its uptake by ileal receptors.
GASTRECTOMY
After total gastrectomy, cobalamin deficiency is inevitable, and prophylactic cobalamin therapy should be commenced immediately after the operation. After partial gastrectomy, 10–15% of patients also develop this deficiency. The exact incidence and time of onset are most influenced by the size of the resection and the preexisting size of cobalamin body stores.
FOOD COBALAMIN MALABSORPTION
Failure of release of cobalamin from binding proteins in food is believed to be responsible for this condition, which is more common in the elderly. It is associated with low serum cobalamin levels, with or without raised serum levels of MMA and homocysteine. Typically, these patients have normal cobalamin absorption, as measured with crystalline cobalamin, but show malabsorption when a modified test using food-bound cobalamin is used. The frequency of progression to severe cobalamin deficiency and the reasons for this progression are not clear.
INTESTINAL CAUSES OF COBALAMIN MALABSORPTION
Intestinal Stagnant Loop Syndrome Malabsorption of cobalamin occurs in a variety of intestinal lesions in which there is colonization of the upper small intestine by fecal organisms. This may occur in patients with jejunal diverticulosis, enteroanastomosis, or an intestinal stricture or fistula or with an anatomic blind loop due to Crohn’s disease, tuberculosis, or an operative procedure.
Ileal Resection Removal of ≥1.2 m of terminal ileum causes malabsorption of cobalamin. In some patients after ileal resection, particularly if the ileocecal valve is incompetent, colonic bacteria may contribute further to the onset of cobalamin deficiency.
Selective Malabsorption of Cobalamin with Proteinuria (Imerslund’s Syndrome; Imerslund-Gräsbeck Syndrome; Congenital Cobalamin Malabsorption; Autosomal Recessive Megaloblastic Anemia; MGA1) This autosomally recessive disease is the most common cause of megaloblastic anemia due to cobalamin deficiency in infancy in Western countries. More than 200 cases have been reported, with familial clusters in Finland, Norway, the Middle East, and North Africa. The patients secrete normal amounts of IF and gastric acid but are unable to absorb cobalamin. In Finland, impaired synthesis, processing, or ligand binding of cubilin due to inherited mutations is found. In Norway, mutation of the gene for AMN has been reported. Other tests of intestinal absorption are normal. Over 90% of these patients show nonspecific proteinuria, but renal function is otherwise normal and renal biopsy has not shown any consistent renal defect. A few have shown aminoaciduria and congenital renal abnormalities, such as duplication of the renal pelvis.
Tropical Sprue Nearly all patients with acute and subacute tropical sprue show malabsorption of cobalamin; this may persist as the principal abnormality in the chronic form of the disease, when the patient may present with megaloblastic anemia or neuropathy due to cobalamin deficiency. Absorption of cobalamin usually improves after antibiotic therapy and, in the early stages, folic acid therapy.
Fish Tapeworm Infestation The fish tapeworm (Diphyllobothrium latum) lives in the small intestine of humans and accumulates cobalamin from food, rendering the cobalamin unavailable for absorption. Individuals acquire the worm by eating raw or partly cooked fish. Infestation is common around the lakes of Scandinavia, Germany, Japan, North America, and Russia. Megaloblastic anemia or cobalamin neuropathy occurs only in those with a heavy infestation.
Gluten-Induced Enteropathy Malabsorption of cobalamin occurs in ~30% of untreated patients (presumably those in whom the disease extends to the ileum). Cobalamin deficiency is not severe in these patients and is corrected with a gluten-free diet.
Severe Chronic Pancreatitis In this condition, lack of trypsin is thought to cause dietary cobalamin attached to gastric non-IF (R) binder to be unavailable for absorption. It also has been proposed that in pancreatitis, the concentration of calcium ions in the ileum falls below the level needed to maintain normal cobalamin absorption.
HIV Infection Serum cobalamin levels tend to fall in patients with HIV infection and are subnormal in 10–35% of those with AIDS. Malabsorption of cobalamin not corrected by IF has been shown in some, but not all, patients with subnormal serum cobalamin levels. Cobalamin deficiency sufficiently severe to cause megaloblastic anemia or neuropathy is rare.
Zollinger-Ellison Syndrome Malabsorption of cobalamin has been reported in the Zollinger-Ellison syndrome. It is thought that there is a failure to release cobalamin from R-binding protein due to inactivation of pancreatic trypsin by high acidity, as well as interference with IF binding of cobalamin.
Radiotherapy Both total-body irradiation and local radiotherapy to the ileum (e.g., as a complication of radiotherapy for carcinoma of the cervix) may cause malabsorption of cobalamin.
Graft-versus-Host Disease This commonly affects the small intestine. Malabsorption of cobalamin due to abnormal gut flora, as well as damage to ileal mucosa, is common.
Drugs The drugs that have been reported to cause malabsorption of cobalamin are listed in Table 105-4. However, megaloblastic anemia due to these drugs is rare.
ABNORMALITIES OF COBALAMIN METABOLISM
Congenital Transcobalamin II Deficiency or Abnormality Infants with TC II deficiency usually present with megaloblastic anemia within a few weeks of birth. Serum cobalamin and folate levels are normal, but the anemia responds to massive (e.g., 1 mg three times weekly) injections of cobalamin. Some cases show neurologic complications. The protein may be present but functionally inert. Genetic abnormalities found include mutations of an intra-exonic cryptic splice site, extensive deletion, single nucleotide deletion, nonsense mutation, and an RNA editing defect. Malabsorption of cobalamin occurs in all cases, and serum immunoglobulins are usually reduced. Failure to institute adequate cobalamin therapy or treatment with folic acid may lead to neurologic damage.
Congenital Methylmalonic Acidemia and Aciduria Infants with this abnormality are ill from birth with vomiting, failure to thrive, severe metabolic acidosis, ketosis, and mental retardation. Anemia, if present, is normocytic and normoblastic. The condition may be due to a functional defect in either mitochondrial methylmalonyl CoA mutase or its cofactor adocobalamin. Mutations in the methylmalonyl CoA mutase are not responsive, or only poorly responsive, to treatment with cobalamin. A proportion of infants with failure of adocobalamin synthesis respond to cobalamin in large doses. Some children have combined methylmalonic aciduria and homocystinuria due to defective formation of both cobalamin coenzymes. This usually presents in the first year of life with feeding difficulties, developmental delay, microcephaly, seizures, hypotonia, and megaloblastic anemia.
Acquired Abnormality of Cobalamin Metabolism: Nitrous Oxide Inhalation Nitrous oxide (N2O) irreversibly oxidizes methylcobalamin to an inactive precursor; this inactivates methionine synthase. Megaloblastic anemia has occurred in patients undergoing prolonged N2O anesthesia (e.g., in intensive care units). A neuropathy resembling cobalamin neuropathy has been described in dentists and anesthetists who are exposed repeatedly to N2O. Methylmalonic aciduria does not occur as adocobalamin is not inactivated by N2O.
CAUSES OF FOLATE DEFICIENCY
|
CAUSES OF FOLATE DEFICIENCY |
aIn severely folate-deficient patients with causes other than those listed under Dietary, poor dietary intake is often present. bDrugs inhibiting dihydrofolate reductase are discussed in the text.
NUTRITIONAL
Dietary folate deficiency is common. Indeed, in most patients with folate deficiency a nutritional element is present. Certain individuals are particularly prone to have diets containing inadequate amounts of folate (Table 128-5). In the United States and other countries where fortification of the diet with folic acid has been adopted, the prevalence of folate deficiency has dropped dramatically and is now almost restricted to high-risk groups with increased folate needs. Nutritional folate deficiency occurs in kwashiorkor and scurvy and in infants with repeated infections or those who are fed solely on goats’ milk, which has a low folate content.
MALABSORPTION
Malabsorption of dietary folate occurs in tropical sprue and in gluten-induced enteropathy. In the rare congenital recessive syndrome of selective malabsorption of folate due to mutation of the proton-coupled folate transporter (PCFT), there is an associated defect of folate transport into the cerebrospinal fluid, and these patients show megaloblastic anemia, which responds to physiologic doses of folic acid given parenterally but not orally. They also show mental retardation, convulsions, and other central nervous system abnormalities. Minor degrees of malabsorption may also occur after jejunal resection or partial gastrectomy, in Crohn’s disease, and in systemic infections, but in these conditions, if severe deficiency occurs, it is usually largely due to poor nutrition. Malabsorption of folate has been described in patients receiving sulfasalazine (Salazopyrin), cholestyramine, and triamterene.
EXCESS UTILIZATION OR LOSS
Pregnancy Folate requirements are increased by 200–300 μg to ~400 μg daily in a normal pregnancy, partly because of transfer of the vitamin to the fetus but mainly because of increased folate catabolism due to cleavage of folate coenzymes in rapidly proliferating tissues. Megaloblastic anemia due to this deficiency is prevented by prophylactic folic acid therapy. It occurred in 0.5% of pregnancies in the UK and other Western countries before prophylaxis with folic acid, but the incidence is much higher in countries where the general nutritional status is poor.
Prematurity A newborn infant, whether full term or premature, has higher serum and red cell folate concentrations than does an adult. However, a newborn infant’s demand for folate has been estimated to be up to 10 times that of adults on a weight basis, and the neonatal folate level falls rapidly to the lowest values at about 6 weeks of age. The falls are steepest and are liable to reach subnormal levels in premature babies, a number of whom develop megaloblastic anemia responsive to folic acid at about 4–6 weeks of age. This occurs particularly in the smallest babies (<1500 g birth weight) and those who have feeding difficulties or infections or have undergone multiple exchange transfusions. In these babies, prophylactic folic acid should be given.
Hematologic Disorders Folate deficiency frequently occurs in chronic hemolytic anemia, particularly in sickle cell disease, autoimmune hemolytic anemia, and congenital spherocytosis. In these and other conditions of increased cell turnover (e.g., myelofibrosis, malignancies), folate deficiency arises because it is not completely reutilized after performing coenzyme functions.
Inflammatory Conditions Chronic inflammatory diseases such as tuberculosis, rheumatoid arthritis, Crohn’s disease, psoriasis, exfoliative dermatitis, bacterial endocarditis, and chronic bacterial infections cause deficiency by reducing the appetite and increasing the demand for folate. Systemic infections also may cause malabsorption of folate. Severe deficiency is virtually confined to the patients with the most active disease and the poorest diet.
Homocystinuria This is a rare metabolic defect in the conversion of homocysteine to cystathionine. Folate deficiency occurring in most of these patients may be due to excessive utilization because of compensatory increased conversion of homocysteine to methionine.
Long-Term Dialysis Because folate is only loosely bound to plasma proteins, it is easily removed from plasma by dialysis. In patients with anorexia, vomiting, infections, and hemolysis, folate stores are particularly likely to become depleted. Routine folate prophylaxis is now given.
Congestive Heart Failure, Liver Disease Excess urinary folate losses of >100 μg per day may occur in some of these patients. The explanation appears to be release of folate from damaged liver cells.
ANTIFOLATE DRUGS
A large number of epileptics who are receiving long-term therapy with phenytoin or primidone, with or without barbiturates, develop low serum and red cell folate levels. The exact mechanism is unclear. Alcohol may also be a folate antagonist, as patients who are drinking spirits may develop megaloblastic anemia that will respond to normal quantities of dietary folate or to physiologic doses of folic acid only if alcohol is withdrawn. Macrocytosis of red cells is associated with chronic alcohol intake even when folate levels are normal. Inadequate folate intake is the major factor in the development of deficiency in spirit-drinking alcoholics. Beer is relatively folate-rich in some countries, depending on the technique used for brewing.
The drugs that inhibit DHF reductase include methotrexate, pyrimethamine, and trimethoprim. Methotrexate has the most powerful action against the human enzyme, whereas trimethoprim is most active against the bacterial enzyme and is likely to cause megaloblastic anemia only when used in conjunction with sulfamethoxazole in patients with preexisting folate or cobalamin deficiency. The activity of pyrimethamine is intermediate. The antidote to these drugs is folinic acid (5-formyl-THF).
CONGENITAL ABNORMALITIES OF FOLATE METABOLISM
Some infants with congenital defects of folate enzymes (e.g., cyclohydrolase or methionine synthase) have had megaloblastic anemia.
DIAGNOSIS OF COBALAMIN AND FOLATE DEFICIENCIES
The diagnosis of cobalamin or folate deficiency has traditionally depended on the recognition of the relevant abnormalities in the peripheral blood and analysis of the blood levels of the vitamins.
COBALAMIN DEFICIENCY
Serum Cobalamin This is measured by an automated enzyme-linked immunosorbent assay (ELISA) or competitive-binding luminescence assay (CBLA). Normal serum levels range from 118–148 pmol/L (160–200 ng/L) to ~738 pmol/L (1000 ng/L). In patients with megaloblastic anemia due to cobalamin deficiency, the level is usually <74 pmol/L (100 ng/L). In general, the more severe the deficiency, the lower is the serum cobalamin level. In patients with spinal cord damage due to the deficiency, levels are very low even in the absence of anemia. Values between 74 and 148 pmol/L (100 and 200 ng/L) are regarded as borderline. They may occur, for instance, in pregnancy, in patients with megaloblastic anemia due to folate deficiency. They may also be due to heterozygous, homozygous, or compound heterozygous mutations of the gene TCN1 that codes for haptocorrin (transcobalamin I). There is no clinical or hematologic abnormality. The serum cobalamin level is sufficiently robust, cost-effective, and most convenient to rule out cobalamin deficiency in the vast majority of patients suspected of having this problem. However, problems have arisen with commercial CBLA assays involving intrinsic factor in PA patients with intrinsic antibodies in serum. These antibodies may cause false normal serum vitamin B12 levels in up to 50% of cases tested. Where clinical indications of PA are strong, a normal serum vitamin B12 does not rule out the diagnosis. Serum MMA levels will be elevated in PA (see below).
Serum Methylmalonate and Homocysteine
[/not-level-membership-for-internal-medicine-category]
