33 Plastic and Reconstructive Surgery
PEDIATRIC PLASTIC SURGERY is performed in children of all ages, even in utero.1 However, the majority of children who undergo plastic surgical and reconstructive procedures are between 2 and 9 years of age, with a median age of 5 years.2 A wide spectrum of associated craniofacial abnormalities, underlying medical conditions, and surgical procedures characterize this pediatric population. Consequently, a thorough preoperative assessment, consultation with medical and surgical teams, and anticipation and preparation for potential complications are of paramount importance to ensure a successful perioperative outcome. The incidence of major morbidity and mortality has been reduced over the last 30 years from 16.5% and 1.6% to less than 0.1% and 0.1%, respectively, in children undergoing major craniofacial surgeries.3
Cleft Lip and Palate
Cleft lip and palate are among the more common congenital malformations, occurring with an estimated incidence of approximately 1 in 700 births worldwide.4 More common in males than in females, this malformation likely results from both environmental and genetic causes. Parental occupation, in particular paternal farming, increases the risk of cleft lip or palate in the offspring, whereas the maternal occupation presents no additional risk.5 Folate metabolism disturbances and increased maternal homocysteine levels also may be contributory.6 Cleft lip with or without cleft palate has been linked to several loci on chromosomes 1, 2, 4, 6, 14, 17, and 19, suggesting a genetic basis for some of these anomalies.7–9
These disorders are associated with more than 400 syndromes; the more common syndromes are presented in Table 33-1. Cleft lip and palate begin as a defect in palatal growth in the first trimester of pregnancy. Fetal magnetic resonance imaging (MRI) provides a greater degree of resolution of defects in the posterior palate and lateral extent of cleft with greater diagnostic accuracy than ultrasound. MRI also enables early detection of potential syndromic conditions by providing a complete study of the fetal head and biometric development of the facial bones.10
TABLE 33-1 Syndromes Commonly Associated with Cleft Lip and Palate
Anesthetic Considerations
Surgical correction of cleft lip defect is usually performed at 3 months of age to allow sufficient time for maturation and associated abnormalities to become apparent. Preoperative assessment may reveal abnormalities such as mandibular hypoplasia in Pierre Robin sequence (Fig. 33-1 and E-Fig. 33-1) or restricted neck movement as in Klippel-Feil syndrome (E-Fig. 33-2).4 Pierre Robin sequence is defined as the triad of micrognathia, glossoptosis (caudally displaced insertion of the tongue), and respiratory distress in the first 24 to 48 hours after birth. The presence of other anomalies might warrant additional clinical or laboratory investigations. Cleft lip repair usually involves minimal blood loss, so for children with hematocrit values greater than 30%, homologous blood donation is unnecessary. Type and screen of donated blood is usually sufficient for a hematocrit value less than 30%.11


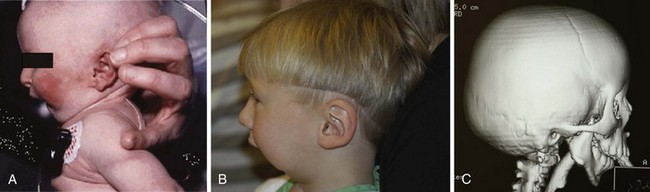
The frequency of difficult airways in children with cleft lip and palate varies from 4.7% to 8.4%.12–14 The incidence of difficult intubation in children with bilateral cleft is greater than that with unilateral cleft.14 Furthermore, micrognathia is an independent predictor of a difficult airway. The incidence of difficult laryngoscopy is approximately 50% in children with micrognathia but only about 4% in those without. In infants and young children, micrognathia may be subtle and not always easily detected. However, the presence of microtia, particularly bilateral microtia, which has a 42% incidence of difficult intubation with bilateral compared with 2% with unilateral microtia, should prompt a closer examination of the mandible for hypoplastic growth and raise the possibility of a hemifacial microsomia or Treacher Collins syndrome.15 Intubation difficulty with isolated micrognathia decreases with increasing age, with the greatest difficulty presenting in infants younger than 6 months. Tracheal intubation becomes easier with age in isolated micrognathia. This has been attributed to rapid growth of the mandible, which catches up to the maxilla, thereby aligning the two bones, by 2 years of age in most cases. A careful review of previous anesthetic records may forewarn of a difficult airway. In our experience, tracheal intubation is not particularly difficult in most infants and children with cleft lip or palate, unless concomitant defects such as micrognathia are present.
Induction of anesthesia via facemask is usually uncomplicated in infants with cleft lip and palate. Laryngoscopy should be performed using a straight blade via a right paraglossal approach (blade inserted into the pharyngeal gutter with tongue displaced to the opposite side)16 (or a left approach if a left-handed blade is available), taking care to avoid inserting the blade into the cleft (see also Fig. 12-28, A-C). If the mandible is hypoplastic, external laryngeal manipulation may be required to visualize the larynx. In some centers, the tongue is sutured to either the mandible or lower lip to preclude airway obstruction in infants with Pierre Robin sequence in the postnatal period. In such instances, the tongue cannot be displaced to the left to expose the larynx. To facilitate laryngoscopy in such cases, the tongue is first released from the lower lip using ketamine sedation. Direct laryngoscopy follows. If direct laryngoscopy fails to expose the larynx, then alternative airway maneuvers such as fiberoptic intubation through a laryngeal mask airway (LMA) may be used after an inhalational induction of anesthesia and release of the tongue (see Chapter 12 and Fig. 12-22, A-C).
A variety of tracheal tubes can be used to secure the airway for cleft lip and palate surgery, although the ideal tracheal tube is perhaps the oral Ring-Adair-Elwyn (RAE) tube, which can be fixed centrally to the chin to facilitate optimal surgical access. Reinforced tracheal tubes are suitable alternatives, but in both cases care must be taken to fix the tube at the correct depth to avoid endobronchial intubation. Throat packs usually impinge on the surgical field and are not normally required for cleft palate repair. The lungs are ventilated for the duration of the procedure, usually 1 to 2 hours. Inhalational or intravenous anesthetics combined with a short-acting opioid such as fentanyl (1 to 2 µg/kg) can be used for maintenance of anesthesia. Bilateral infraorbital nerve blocks may be used to provide postoperative analgesia for cleft lip repairs (see Fig. 41-11, A-C, and Chapter 41 videos) . Such blocks reduce the need for opioids and antiemetics, improve the ability to feed,17 and increase parental satisfaction.18 A combination of infraorbital and external nasal nerve blocks for pain control after cleft lip repair is an alternative.19
. Such blocks reduce the need for opioids and antiemetics, improve the ability to feed,17 and increase parental satisfaction.18 A combination of infraorbital and external nasal nerve blocks for pain control after cleft lip repair is an alternative.19
During cleft palate surgery, the pharyngeal space is reduced dramatically (Fig. 33-2 and E-Fig. 33-3). The trachea is extubated after the upper airway reflexes have returned and the child is completely awake. These children are at particular risk for acute upper airway obstruction in the immediate postextubation period as a result of upper airway narrowing, edema, blood, and residual anesthetic effects.20–25 Accordingly, it is very important to extubate the trachea only when the child is completely awake. Late postoperative edema26 and severe subcutaneous emphysema are additional complications. Upper respiratory tract infections are common in this age group, and if these infections are present, they should weigh heavily in favor of delaying surgery until they are resolved. Antibiotics may reduce the incidence of postoperative respiratory complications.27

A nasopharyngeal airway may be inserted by the surgeon before extubation to ensure a patent airway after extubation and permit suctioning the airway without damaging the palatal repair (see Fig. 33-2). Arm restraints are used in many centers to prevent suture disruption. These children are monitored for signs of upper airway obstruction during the recovery period for approximately 48 hours.20 As soon as the child is awake, feeding with clear fluids is allowed. Postoperative pain is managed with a combination of opioids and acetaminophen. Sphenopalatine and infraorbital nerve blocks with a long-acting local anesthetic can be placed at the end of the procedure to prevent pain after cleft lip and palate repair.17 Palatal nerve block (nasopalatine, greater and lesser palatine)28 or a bilateral suprazygomatic maxillary nerve block reduce postoperative pain and favor early feeding.29
Craniosynostosis
Craniosynostosis, a congenital anomaly in which one or more cranial sutures closes prematurely, occurs in approximately 1 in 2000 births, and affects males more frequently than females. Embryologically, the cranial vault starts to ossify at 8 weeks after conception. Fusion of the parietal and frontal bones is usually completed by 7 months after conception. Postnatally, the anterolateral fontanelle closes by 3 months, the posterior fontanelle by 3 to 6 months, the anterior fontanelle by 9 to 18 months, and the posterolateral fontanelle by 2 years. Premature osseous obliteration of a bony suture might result from the absence of osteoinhibitory signals from the suture. Craniosynostosis may be categorized as simple (or nonsyndromic) (65% to 80% of cases), involving closure of one suture, or complex (or syndromic) (20% to 30%), involving closure of two or more sutures and often associated with a variety of clinical features and metabolic diseases (Table 33-2).30,31
TABLE 33-2 Classification of Craniosynostosis
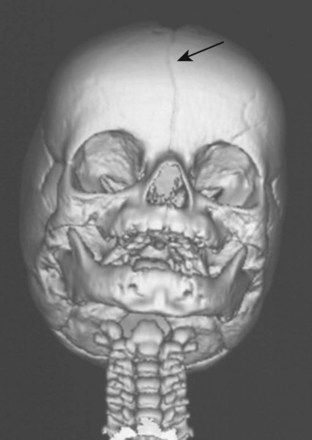
In the child with craniosynostosis, the head may assume various shapes depending on the sutures involved (Fig. 33-3 and E-Fig. 33-4). The frequency of single suture closures varies with the specific suture: sagittal (50%), coronal (20%), and metopic (10%).30 The coronal suture is more commonly associated with syndromic craniosynostoses.

Although approximately 80% of premature suture closures are isolated defects, the remaining 20% involve multiple suture closures associated with more than 150 syndromes that present with a myriad of clinical features (see Table 33-2).30 Apert syndrome occurs in approximately 1 in 100,000 live births, usually as a sporadic mutation, although autosomal dominant inheritance patterns can occur. This syndrome phenotypically manifests as cloverleaf skull (craniosynostosis), hypertelorism, proptosis, midface hypoplasia, and syndactyly (upper or lower extremity). Development is often complicated by increased intracranial pressure (ICP) and obstructive sleep apnea (OSA).32,33 Whether children with Apert syndrome develop normal intelligence quotients (IQs) is unclear. One study reported that 32% of children with Apert syndrome had IQs greater than 70.34 Timing of cranial surgery may affect the child’s IQ; surgery in the first year of life was associated with an IQ greater than 70 in more than 50% of children in one study, whereas surgery after the first year of life was associated with an IQ greater than 70 in only 7%. Two other factors predicted improved IQ indices: absence of a defect in the septum pellucidum and noninstitutional residence (i.e., family home residence). Crouzon syndrome is phenotypically similar to Apert syndrome but has different ophthalmologic defects, specifically, optic atrophy occurring in up to 20%, and the absence of digital involvement.35 Fifty percent of Crouzon syndrome defects are sporadic mutations, and the remainder are familial. Pfeiffer syndrome occurs in approximately 1 in 25,000 live births. Most cases are familial with an autosomal dominant inheritance pattern, although many remain sporadic. The phenotype of Pfeiffer syndrome is similar to that of Apert syndrome but includes broad thumb, large first toe, and polydactyly. Children with Pfeiffer syndrome have normal intelligence. Carpenter syndrome is associated with craniosynostosis, syndactyly, cardiac defects, and obesity.36 Cognitive impairment is common.36 A relatively new but rare syndrome, Shprintzen-Goldberg, is characterized by craniosynostosis and a phenotype that resembles that of Marfan syndrome.
Indications for cranial vault reconstruction include increased ICP, severe exophthalmos, OSA, craniofacial deformity, and psychosocial reasons. If uncorrected, the deformed cranium may cause severe neurologic sequelae, including visual loss and developmental delay (see Chapter 23).37–49 Because rapid brain growth during infancy determines skull shape, surgical correction is undertaken within the first months of life to achieve the best cosmetic results.
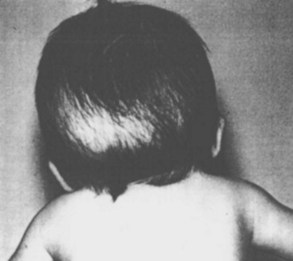
Cranial vault reconstruction may involve the anterior or posterior aspect of the skull or both (total cranial vault reconstruction). Less invasive approaches to correct craniosynostosis are available and may be associated with reduced morbidity. Surgical correction may employ an extended strip craniectomy in which the cranial vault is split in multiple segments, allowing the skull to grow with the brain (Fig. 33-4 and E-Fig. 33-5). This technique is used in children younger than 6 months of age and is believed to be less invasive than total cranial vault reconstruction, although children are required to wear a protective helmet after surgery.50,51 Endoscopic strip craniectomy is increasingly being used in early infancy and has various benefits compared with the open procedures. Spring-assisted cranioplasty, a technique preferentially used in infants, involves performing a midline osteotomy along the fused sagittal suture and placing springs across the osteotomy to increase the biparietal dimension. Spring-assisted cranioplasty may be associated with reduced intraoperative blood loss, reduced transfusion requirement, and shorter duration of hospital stay.52 In a single-center study of 100 children with spring-assisted craniosynostosis, no child was transfused and none was admitted to the intensive care unit.53
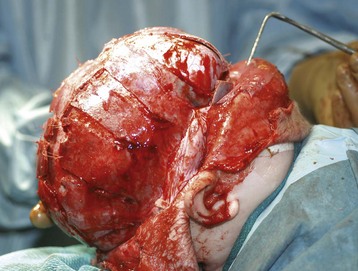

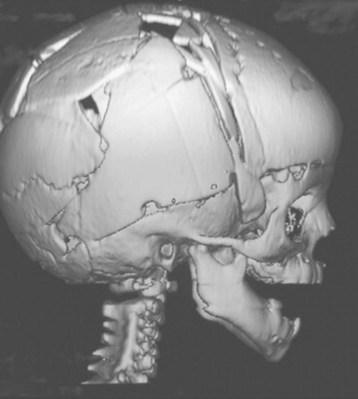
E-FIGURE 33-5 Three-dimensional MRI of a child after extended strip craniectomy (see Fig. 33-4). This approach is commonly used in infants younger than 6 months of age. To protect the fragile skull bones, a protective helmet must be worn after surgery until cranial remodeling is complete.
Preoperative assessment of children with craniosynostosis should focus on airway management, eye protection, and ICP. An important consideration in children with midfacial hypoplasia and large tonsils is the presence of OSA. OSA may be present in 50% to 70% of children with syndromic craniosynostosis.33,54,55 Although some recommend preoperative adenotonsillectomy to treat OSA in children with craniosynostosis, neither airway dimensions nor airway collapse is improved. Midfacial advancement may be required to resolve OSA, and even then, residual airway obstruction may persist.33,54 Preoperative endoscopy has been recommended to assess the severity of midfacial hypoplasia and whether OSA is likely to persist after midface advancement. Careful titration of opioids in the perioperative period is indicated if the child exhibits severe nocturnal desaturation (i.e., if the Sao2 nadir is less than 85%) (see Chapter 31). Upper airway obstruction may also occur postoperatively in children who received opioids as a direct effect of opioids on the hypoglossal nucleus.56
Postoperative pain is generally not severe and is managed effectively with a combination of acetaminophen, nonsteroidal antiinflammatory drugs (NSAIDs), and intravenous opioids. Opioids remain the mainstay of pain management, but careful titration of reduced doses (approximately 50% less) must be considered if OSA is present. Given the small incidence of craniosynostosis and the large variability in the management of these patients, multicenter trials are required to determine the optimal management strategy for these children.57
Airway Management
Meticulous preoperative planning and evaluation of the airway is essential, particularly for OSA.54 A facemask may prove to be a challenge to seal on their flat faces and the nasal passages may be obstructed, together creating a difficult mask ventilation.58 External fixator devices on the face may also present challenges in managing the airway (E-Fig. 33-6).

Blood Loss, Coagulopathy, and Hyponatremia
Crystalloid solutions are commonly administered for minimal to moderate surgical blood loss and fluid shifts during craniosynostosis surgeries. Although lactated Ringer’s solution is most commonly used in North America, some advocate using normal saline because it may be less likely to induce hyponatremia and an acid-base disturbance than lactated Ringer’s solution. However, a recent study comparing the two solutions suggests that normal saline is more likely to induce (metabolic) acidosis than lactated Ringer’s solution in infants undergoing craniosynostosis.59 The explanation for this finding has been elusive, but it has been attributed to hyperchloremia, a dilutional acidosis, or both.
Surgery for craniosynostosis is associated with an increased incidence of cardiac arrest as a result of sudden massive blood loss.3,30,60,61 Although these procedures are extradural, significant blood loss from the scalp and cranium can occur. The blood loss can be so rapid during the surgery that the expression “trauma in progress” is applicable. The risk of massive blood loss and the need for invasive monitoring are determined in part by the specific suture involved, number of sutures scheduled for repair, type of surgery, and expertise of the surgeon.61 In children undergoing endoscopic strip craniectomy, weight less than 5 kg, those undergoing sagittal endoscopic craniectomy, those with syndromic craniosynostosis, and earlier date of surgery in the series were associated with blood transfusion.62 Some centers advocate commencing blood transfusions at the time of skin incision (particularly in infants) to prevent hemodynamic instability and the need for a rapid transfusion, but this must be tempered by whether the surgery is open or endoscopic (see later discussion).
To manage the large volume and rapidity of the blood loss, it is essential to establish large-bore peripheral venous access. Central venous access (see also Fig. 48-2), usually via the femoral route, can provide a useful estimate of the cardiac filling pressures but should not be used for rapid transfusion in small infants, because hyperkalemic cardiac arrests may occur with old blood infused rapidly into the right atrium and the lumens may limit the rate of blood infusion. Estimation of ongoing blood loss can be difficult because of the use of large volumes of irrigation fluid and blood loss onto surgical drapes and gowns.63 Invasive arterial blood pressure (BP) monitoring and serial blood gas sampling are indicated in this type of surgery (see Fig. 48-10, A-D). A urinary catheter should be inserted to monitor urine output.
Several blood conservation strategies have been proposed for this type of surgery, including preoperative recombinant human erythropoietin, directed blood donation (from the parents or siblings), acute normovolemic hemodilution, antifibrinolytics, and induced hypotension (see Chapter 10 for a more detailed discussion). These strategies should be combined with meticulous surgical technique and attention to hemostasis. Furthermore, bleeding from the scalp incision may be reduced by infiltration with a dilute (1 : 400,000) epinephrine-containing solution. The use of the reverse Trendelenburg position may help to decrease venous pressure and blood loss from osteotomy sites but also increases the risk of venous air embolism (which has been reported to be 5% to 80%; see later discussion). For this reason, the horizontal position is preferred.
Blood-conserving dual therapy with recombinant human erythropoietin (to optimize preoperative hematocrit) and use of a cell saver reduces transfusion in children undergoing craniosynostosis repair.64 Administration of preoperative recombinant human erythropoietin, in combination with elemental iron (6 mg/kg/day to a maximum of 200 mg/day for 6 weeks) increases the preoperative hematocrit value and decreases the need for autologous blood transfusion.65,66 If iron stores are at all compromised, iron therapy combined with oral vitamin C (to increase gastrointestinal absorption) should begin 3 weeks before erythropoietin therapy.67
Little evidence exists to suggest that autologous or directed blood donation decreases perioperative morbidity in craniosynostosis surgery, although it will decrease the number of blood unit exposures.68,69 Infants as young as 3 months have predonated, although the limited volume of predonated blood is unlikely to preclude all blood transfusions during craniosynostosis or repeat craniosynostosis surgery.68,69
Acute normovolemic hemodilution is a labor-intensive technique in which blood is collected from the child after induction of anesthesia but immediately before surgery and replaced with an equal volume of crystalloid or colloid, such as 5% albumin. Although acute normovolemic hemodilution may be beneficial in children with rare blood types, no evidence exists that it reduces either the incidence of homologous transfusion or the amount of homologous blood transfused in children undergoing craniosynostosis repair.70
The coagulation profile and clotting factors after fresh frozen plasma (FFP) or 5% albumin during craniofacial surgery have been compared in a nonrandomized study in infants younger than 12 months of age.71 The increases in activated partial thromboplastin time (aPTT) and decreases in the plasma concentration of factors XI and XIII and antithrombin 3 were less after intraoperative FFP than after 5% albumin. Fibrinogen concentrations remained stable in the FFP-treated group but decreased in the albumin-treated group. However, it should be emphasized that no clinical indication exists to administer FFP when the blood loss is less than 1 blood volume (Chapter 10). Recombinant factor VIIa has been used successfully for intractable hemorrhage during cranial vault reconstruction in an infant, although this is an isolated report.72
Antifibrinolytic therapy may decrease blood loss during craniosynostosis repair in children. Tranexamic acid (TXA) is a widely used agent, and several dosing regimens have been described: (1) A loading dose of 15 mg/kg TXA after induction of anesthesia, followed by an infusion of 10 mg/kg/hr until skin closure73; (2) 50 mg/kg TXA loading dose, followed by an infusion of 5 mg/kg/hr74 until skin closure; and (3) 15 mg/kg at induction of anesthesia, every 4 hours during surgery, and every 8 hours after surgery for 24 hours after surgery. Although dose-response data from a single study are lacking, two studies73,74 report that TXA reduces intraoperative and postoperative bleeding and transfusion requirements.75 Another antifibrinolytic, ε-aminocaproic acid (EACA), is effective in reducing bleeding in cardiac and spinal procedures, although its effectiveness in reducing blood loss during craniosynostosis has not been established. The dose that has been used for spinal surgery is 75 to 100 mg/kg loading dose intravenously over 15 to 20 minutes before skin incision, followed by 10 to 15 mg/kg/hr until skin closure.76
Induced hypotension defined as a 10% to 20% reduction in the mean arterial BP decreases intraoperative surgical blood loss and operating time,77 although studies demonstrating its efficacy during craniosynostosis surgery are lacking. A variety of pharmacologic agents have been used to induce hypotension, including inhalational agents, vasodilators, β-blockers, and remifentanil.77,78 Invasive arterial pressure monitoring is essential whenever hypotensive anesthesia is used. Induced hypotension should be used with caution in the presence of increased ICP because of the risk of compromising cerebral perfusion pressure (i.e., the difference between mean arterial pressure [MAP] and either ICP or central venous pressure, whichever is greater). Increases in ICP mandate invasive monitoring of MAP to ensure an adequate cerebral perfusion pressure. It is considered prudent to maintain normovolemia and normocapnia when induced hypotension is used (see Chapter 10 for a more detailed description of these techniques).
Whether used alone or in combination,79–81 the preceding techniques seldom obviate the need for all blood transfusions during craniofacial surgery. Therefore intravenous access with large-bore catheters remains essential and at least 2 units of packed red blood cells (PRBCs) should be crossmatched and available in the operating room at all times. All intravenous fluids should be administered via a fluid warmer to prevent hypothermia. Maintaining normothermia preserves normal coagulation indices and theoretically reduces bleeding and transfusion-related complications.82 A coagulopathy should always be anticipated once the blood loss exceeds 1 blood volume. Serial determinations of the international normalized ratio, partial thromboplastin time, platelet count, and fibrinogen concentration will identify an evolving coagulopathy and indicate which blood products, FFP, platelets, or cryoprecipitate, will best correct the abnormalities (see Chapter 10).
Endoscopic repair of craniosynostosis has become a rapidly growing surgical approach to reduce bleeding and decrease morbidity.83,84 Independent risk factors for bleeding during endoscopic strip craniectomy include low body weight (<5 kg), sagittal suture surgery (related to proximity to the sagittal vein), syndromic craniosynostosis, and earlier date of surgery.62
Hyponatremia and cerebral salt wasting syndrome are associated with craniosynostosis repair.85–89 Both intraoperative and postoperative hyponatremia have been described, with the latter occurring in approximately 30% of children. In a retrospective review of a cleft palate and craniofacial database, postoperative hyponatremia was significantly associated with preoperative increased ICP, blood loss, and female gender with normal preoperative ICP.89 The average reduction in sodium concentration was more pronounced in children who received hyponatremic (hypotonic) (5% dextrose and 0.2% or 0.5% NaCl) compared with normonatremic (isotonic) postoperative intravenous fluids.89 The perioperative use of balanced salt solutions is recommended to prevent hyponatremia (see Chapter 8 for a more in-depth discussion of new perioperative fluid management recommendations). In infants, the addition of 5% dextrose to the balanced salt solution may be required to avoid intraoperative or postoperative hypoglycemia.
Increased Intracranial Pressure
Early surgery for craniosynostosis is often indicated to prevent increases in ICP.31,32 One third of children with craniofacial dysostosis syndrome and 15% to 20% of children with single-suture craniosynostosis have increased ICP (>15 mm Hg).90 Approximately 40% to 50% of children with syndromic craniosynostosis have associated hydrocephalus, although differentiation from nonprogressive ventriculomegaly may be difficult.90–92 Timing of surgery may affect neurocognitive development and intelligence because these are adversely affected by sustained increased ICP. Associated OSA resulting in hypoxemia and hypercapnia may lead to an increase in cerebral blood volume and thereby exacerbate intracranial hypertension.93 Untreated intracranial hypertension may lead to optic atrophy and visual impairment.35,94 As a consequence, when increased ICP has been identified either preoperatively or postoperatively, placement of a ventriculoperitoneal shunt should be considered.92 This is more common an occurrence in Crouzon and Pfeiffer syndromes.92
For children who present with signs of intracranial hypertension, it is important to follow basic principles of neuroanesthesia to prevent further increases in ICP and decreases in cerebral perfusion pressure (see Chapter 24 for a more in-depth discussion). It may be prudent to use protective measures to attenuate the hypertensive response to laryngoscopy and intubation, including the administration of a short-acting opioid, a β-blocker, or topical local anesthesia to the upper airway. Intraoperatively, the anesthesiologist faces numerous challenges to control ICP. Mild to moderate hyperventilation (to an end-tidal carbon dioxide of 30 to 35 mm Hg), especially when signs of herniation are evident, avoidance of hypervolemia, and, where indicated, appropriate use of mannitol, furosemide, and dexamethasone may be employed to reduce ICP, reduce brain volume, and facilitate brain retraction. Although cranial vault reconstruction increases intracranial volume and reduces ICP,95 children remain at risk of increased ICP after surgery and require close ophthalmologic and clinical follow-up, even after a cosmetically successful cranial expansion.96,97
Venous Air Embolism
Venous air embolism (VAE) may occur during any operative procedure in which the operative site is above the level of the heart and noncollapsible veins are exposed to atmospheric pressure.98–105 The incidence of VAE in children undergoing craniectomy for craniosynostosis repair has been reported to be as great as 83%,105 although hemodynamically significant VAE is rare. The incidence associated with endoscopic craniectomy may be as small as 2%.62 Significant hypovolemia resulting from surgical blood loss can lead to a decrease in both systemic and central venous pressures and the development of a pressure gradient between the right atrium and the surgical site. This gradient increases the potential to entrain air via open dural sinuses or bony venous sinusoids.105,106 If the entrained volume of air is sufficiently large, right ventricular outflow obstruction may ensue, causing acute right-sided heart failure and cardiovascular collapse. Smaller volumes of air may cause a reduction in cardiac output, hypotension, and myocardial or cerebral ischemia.103 Transesophageal echocardiography (documenting the presence of air in the right ventricular outflow tract), precordial Doppler ultrasonography (continuous machinery mill murmur), end-tidal carbon dioxide (precipitous decrease in carbon dioxide tension), and nitrogen monitoring (sudden increase in nitrogen concentration in the exhaled breath) have been used to identify VAE with different sensitivities, well before cardiovascular collapse occurs (see Figure 24-6).103,107–110 Applying bone wax to the open edges of cut bone, reducing the degree of or avoiding the reverse Trendelenburg position, introducing positive-pressure ventilation with 5 cm of positive end-expiratory pressure, and maintaining normovolemia help to prevent VAE. Fluid resuscitation, vasopressors, and aspiration of air from the right side of the heart may prevent episodes of VAE from progressing to cardiovascular collapse.99,105,110
Prolonged Surgery
As with all surgeries that last several hours, preventing the complications associated with prolonged anesthesia is paramount.111 Nerve palsies, pressure necrosis of the skin, ophthalmic complications, hypothermia, and acidosis may occur. Careful positioning of the extremities, use of an egg-crate type mattress, and avoiding pressure to the eyes, particularly when the surgical procedure requires the prone position, will prevent the majority of these adverse outcomes. In children with proptosis, such as Crouzon syndrome, it is particularly essential to suture the eyelids closed after applying lubrication to prevent corneal abrasions. Anterior ischemic optic neuropathy that can cause transient or permanent postoperative blindness is a rare complication that occurs in the absence of external pressure to the eyes.112
Hypothermia is another major concern. Factors that predispose to hypothermia include the relatively large surface area exposed during surgery and the transfusion of large volumes of relatively cold intravenous fluids. Effective measures to prevent hypothermia include warming the operating room, insulating the child, and the use of forced air warmers and warming devices for blood and intravenous fluids. Preventing hypothermia and limiting blood loss and transfusion requirements are key factors in preventing the development of perioperative metabolic disturbance.113
Orbital Hypertelorism
The term orbital hypertelorism describes abnormally separated orbits. This deformity may occur in isolation or in association with other congenital abnormalities, such as facial clefts and Apert syndrome (Fig. 33-5). Surgical repair involves mobilization and repositioning of the orbit through either a subcranial approach, which leaves the roof of the orbit intact, or an intracranial approach via a frontal craniectomy. This procedure is performed in children older than 5 years who may have already undergone extensive surgical reconstruction. Surgical manipulation of the globe may elicit the oculocardiac reflex, resulting in bradyarrhythmias or asystole. Oculocardiac reflex may also occur during midface and orthognathic procedures (see also Fig. 32-3, A and B).114 These arrhythmias are prevented by administering a prophylactic anticholinergic such as atropine (10-20 µg/kg) or glycopyrrolate (5-10 µg/kg). Discontinuing the surgical stimulus almost always increases the heart rate.
Midface Procedures
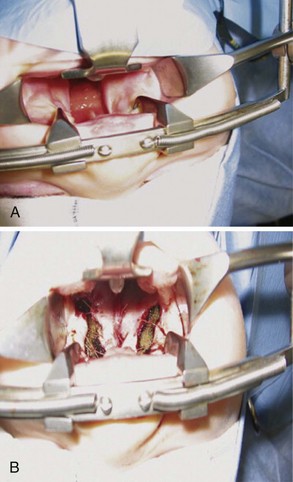
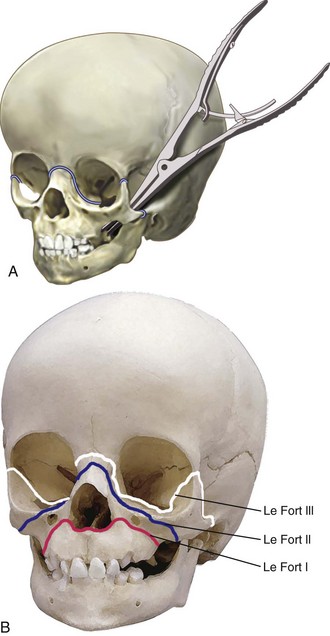
Midface advancement to improve facial appearance is commonly required for children with maxillary hypoplasia, such as those with Crouzon, Apert (see Fig. 33-5), and Pfeiffer syndromes (E-Fig. 33-7).115–117 This procedure is typically performed in preschool children, although complications such as proptosis, corneal ulceration, ocular dislocations, and airway obstruction may necessitate earlier intervention.117–120 A LeFort II procedure is similar to a LeFort III, with the difference that the osteotomy is oriented vertically through the infraorbital rim. Thus the nasal pyramid and the maxilla move forward as a single unit. Le Fort III osteotomy and monobloc procedures (Fig. 33-6) have the potential for significant complications, including massive blood loss, airway difficulties, cerebrospinal fluid leak, and infection.111,121–123

Anesthetic concerns are similar to those for orbital hypertelorism and craniosynostosis. Children with Apert and Crouzon syndromes often present with incomplete or complete nasal obstruction that results from choanal atresia or midface hypoplasia. As a consequence, mask anesthesia can be difficult, even with an oral airway in situ. However, laryngoscopy and tracheal intubation in these children is usually uncomplicated. The diameter of the tracheal tube requires careful consideration because these children may require prolonged postoperative ventilation until postoperative facial and laryngeal edema has resolved. The decision to intubate the trachea via the oral or nasal route must be discussed with the surgeon before induction of anesthesia. A nasotracheal tube can be used throughout the procedure, or the surgeon may request an intraoperative change from the oral to the nasal tracheal position after completing the midfacial osteotomies.124 To perform the latter maneuver, the anesthesiologist wears a sterile surgical gown and gloves and uses sterile equipment, including laryngoscope, Magill forceps, and tube exchange catheter (see E-Fig. 12-3). Visualizing the glottis during surgery may be difficult because of the presence of airway edema and blood in the hypopharynx. A tube exchange catheter is passed through a naris and into the trachea alongside the orotracheal tube. The nasal tube is then passed over the exchange catheter, and its tip is positioned at the glottic opening. The oral tube is then removed, and the nasal tube is advanced (rotating the bevel 90 degrees clockwise or counterclockwise as needed to pass the vocal cords and arytenoids)125,126 and visualized as it passes through the glottic aperture. Once the tube position is confirmed, the catheter is removed and the nasal tube is sutured securely to the nasal septum. Given the proximity of the tracheal tube to the surgical site, damage to the tracheal tube can occur during surgery.127,128 Vigilance is required at all times to detect an accidental disconnection or damage to the tracheal tube. The anesthesiologist must be prepared to respond immediately to an unexpected interruption in ventilation and replace the tracheal tube. A nasogastric tube is placed after surgery to prevent gastric distention and reduce the likelihood of postoperative nausea and vomiting. A wire cutter must be available at the bedside at all times if intermaxillary fixation is used to stabilize the facial bones and mandible. In the intensive care unit, the presence of an audible leak around the tracheal tube is an important criterion to determine absence of laryngeal or periglottic edema and therefore readiness for tracheal extubation.127 Intraoperative blood loss is not typically as great as for craniosynostosis surgery. Hypotensive anesthesia may reduce or prevent the need for blood transfusions during maxillary orthognathic surgery.129

Hemifacial Microsomia, Treacher Collins Syndrome, and Goldenhar Syndrome
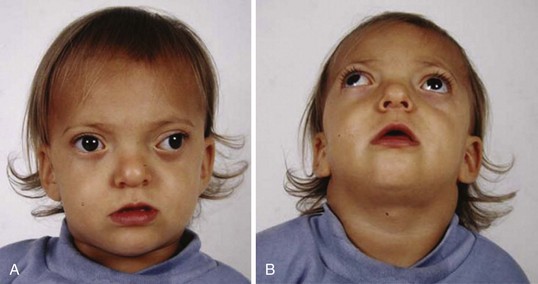
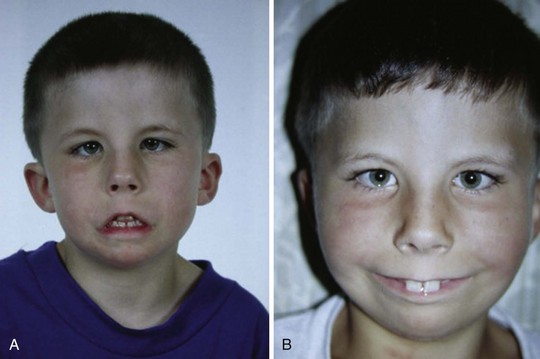
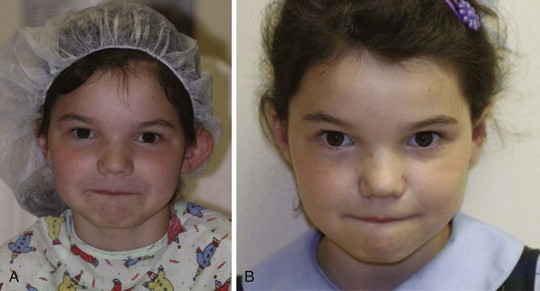
Hemifacial microsomias, also known as otomandibular dysostosis (Fig. 33-7), result from a malformation of the first and second branchial (or pharyngeal) arches. This is the second most common facial defect after clefts. These disorders are classified according to the classification orbital distortion, mandibular hypoplasia, ear anomaly, nerve involvement, and soft tissue deficiency (OMENS).4,130–133 Piezosurgery is a relatively new technique used to perform osteotomies using ultrasonic frequencies during mandibular distraction in children with hemifacial microsomia.134 Airway difficulty increases with the complexity of the defect from unilateral to bilateral mandibular or temporomandibular involvement. The disorder may include mandibular hypoplasia, temporomandibular joint dysostosis, cleft palate, and auricular, ophthalmologic, and facial nerve defects. Goldenhar syndrome (Fig. 33-8 and E-Fig. 33-8) is the most common form of this disorder. Vertebral anomalies are present in 40% and congenital heart defects occur in 35% of children with this syndrome. Airway management is complicated by midfacial hypoplasia, asymmetry of mouth opening, and mandibular retrognathia. Overall, tracheal intubation in children with unilateral hemifacial microsomia is easy in 70% and very difficult in 9%.130 In contrast, tracheal intubation in children with bilateral mandibular hypoplasia is evenly distributed among easy, difficult, and very difficult.130 The airway anomalies associated with this syndrome predispose to OSA.
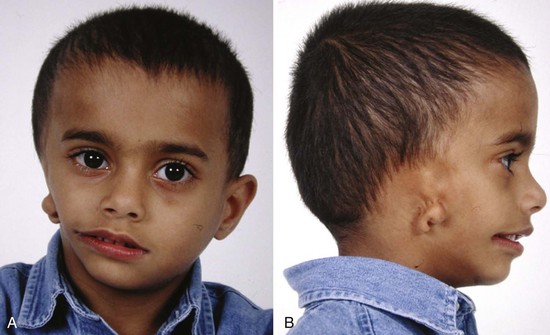
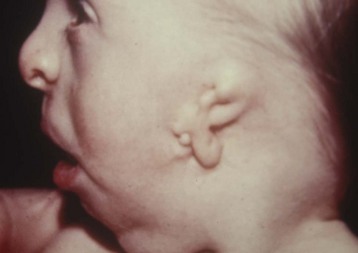

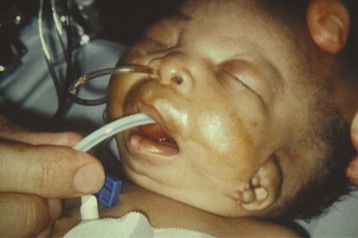
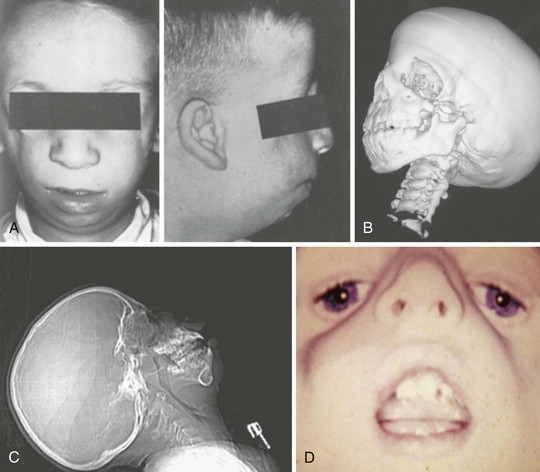
The craniofacial abnormalities of mandibular hypoplasia, macrostomia, and cleft palate in Treacher Collins syndrome (Fig. 33-9 and E-Fig. 33-9) often present difficulties for airway management that increase with increasing age.4,130 Other clinical features of the syndrome include hypoplastic zygomatic arches, ophthalmic features (including sloping palpebral fissures, coloboma of the eyes, and notched lower eyelids) and microtia, choanal atresia, cardiovascular defects, and renal anomalies. Mandibular distraction osteogenesis is a surgical option considered when upper airway obstruction is due to mandibular deficiency. This avoids tracheostomy or other surgical intervention and allows for future growth of the mandible. Children with hemifacial microsomia have a poor psychosocial outcome.135
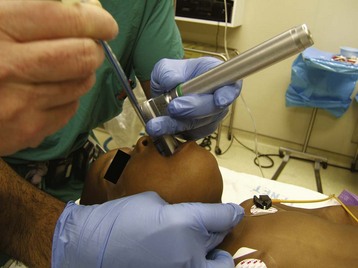
FIGURE 33-9 Two-person intubation technique. The intubator (first person) performs laryngoscopy and manipulates the larynx with external laryngeal manipulation (gloved hands). With the larynx in view, the intubator cocks his or her head to the left while holding position and the assistant (second person, nongloved hand) who is standing on the intubator’s right, then passes the tracheal tube through the larynx.137

Airway Management
Airway management of children with hemifacial dysostoses is traditionally known to be difficult. It is essential that all equipment for management of the difficult airway be present in the operating room before induction of anesthesia (see Table 12-10). For infants and children with difficult airways, an inhalational induction is the most commonly used technique. In contrast, in older children and adolescents, either an inhalational induction or intravenous sedation (using dexmedetomidine or propofol) with topical local anesthesia applied to the upper airway may be used to facilitate tracheal intubation. In all cases, it is essential to maintain spontaneous respirations until the airway is secured so that oxygenation and ventilation will be maintained if the airway cannot be secured.
A variety of techniques may be used to control the difficult airway, including a flexible fiberoptic bronchoscope, Glidescope (Verathon Inc., Bothell, Wash.), the Airtraq (Prodol Meditec S.A., Vizcaya, Spain) disposable optical laryngoscope, Truview Infant laryngoscope (Truphatek International Truview Infant laryngoscope Limited, Netanya, Israel), LMA,136 and others (see Chapter 12). We have used the two-person intubation technique, in which the first anesthesiologist applies external posterior laryngeal pressure while performing laryngoscopy to optimize the view of the glottis, while the second anesthesiologist inserts the tracheal tube into the trachea when the view is adequate (see Fig. 33-9).137 The second anesthesiologist also may assist with more advanced airway management both in terms of helping to observe the child and assisting with the use of advanced airway devices.
For children with a history of upper airway obstruction or midfacial hypoplasia who present with a tracheostomy, the tracheostomy tube can be replaced with a cuffed tracheostomy tube or an armored tube that is sutured in place for the duration of the surgery. Changes in the position of the head and neck can cause displacement of the tracheal tube, so care should be taken to confirm tracheal tube position after the child is positioned for surgery.138 This is especially important for cranial vault procedures that involve extremes of neck extension, such as might occur during reconstruction of the supraorbital bar. Airway equipment and additional tracheal tubes must be available in the operating room at all times. It is essential to document an audible leak around the tracheal tube at the time of tracheal intubation (with the cuff on the tracheal tube deflated), because the presence of a leak is often used postoperatively to determine suitability for extubation when significant airway edema has developed. OSA associated with midfacial anomalies may result in supraglottic obstruction during induction and emergence.54,58
Orthognathic Surgery
Airway management is a major concern, particularly in children with a hypoplastic mandible or temporomandibular dysfunction.4 A high index of suspicion for atlantoaxial instability is required if juvenile rheumatoid arthritis is the underlying disease process. The anticipated difficult airway can be managed using fiberoptic intubation, with sedation or topical local anesthesia or an inhalation induction and maintenance of spontaneous ventilation until the trachea is intubated, as discussed earlier. Nasotracheal intubation using a preformed tracheal tube is the preferred method of tracheal intubation. Careful stabilization and fixation of the tube using transseptal suturing to prevent unintended extubation is often employed. Excessive pressure on the ala nasi (causing ischemia) can be avoided by fixing the nasal RAE tube to the forehead with the nasal curve positioned away from the ala. LeFort I advancements require close communication between the surgery and anesthesia teams because the nasotracheal tube has been dislodged once the maxilla is fully mobilized. If intermaxillary fixation is used postoperatively, wire cutters must be immediately available at all times while the child is monitored in an intensive care setting.
To reduce intraoperative blood loss, controlled hypotension is commonly employed using any of a range of pharmacologic agents, including inhalational anesthetic agents, β-blockers, and remifentanil. The literature is extensive on the salutary effect of induced hypotension in reducing intraoperative blood loss and improving the quality of the surgical field during orthognathic surgery.129,139–148 Invasive BP monitoring is indicated to monitor BP continuously during controlled hypotension and to facilitate intraoperative evaluation of blood gases and hematocrit. In some cases, a mild degree of hypotension is sufficient for optimal surgical conditions (systolic BP 85 to 90 mm Hg). Dexamethasone (0.2 mg/kg) may reduce postoperative airway edema.149 After the return of protective airway reflexes, the trachea is extubated and the child is monitored overnight in a high-dependency setting with the ability to establish an airway should acute airway obstruction develop.
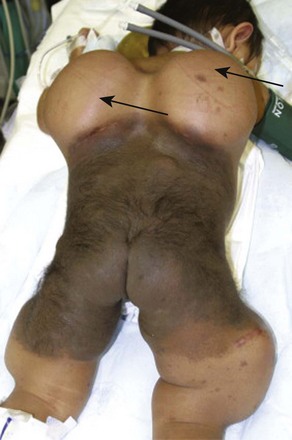
Cystic Hygromas and Hemangiomas
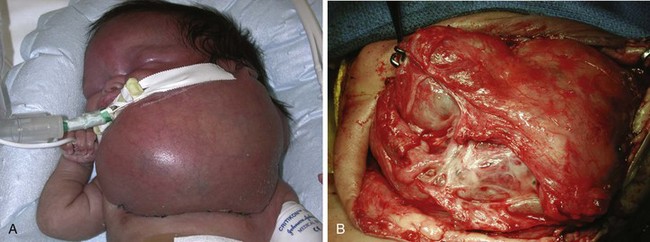
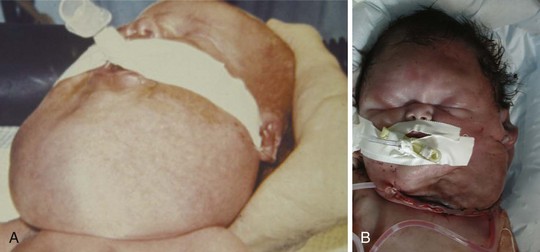
Cystic hygroma is a rare congenital malformation of the lymphatic system occurring with an incidence of 1 in 16,000 births, most frequently involving the axilla and neck (Fig. 33-10 and E-Fig 33-10). The pathology consists of multiple loculated cysts that contain lymph fluid or blood (see Fig. 33-10, B). In most cases, cystic hygromas are present at birth, although 80% to 90% are diagnosed within the first 2 years of life. The natural history is spontaneous resolution, although most require repeated aspirations, sclerotherapy, or surgical excision to debulk the mass (see E-Fig. 33-10, B).150–152 Cystic hygroma can be associated with other chromosomal abnormalities such as Noonan and Turner syndromes, in which case the anesthetic management is guided by the underlying syndrome. Some children require an urgent tracheostomy in the first hours of life to relieve an obstructed airway (Chapter 37). During the preoperative assessment, the airway should be examined and evaluated by radiographs for involvement of supraglottic and infraglottic structures. Acute airway obstruction can occur during induction if cystic lesions are present in the upper airway. Fiberoptic intubation may be required if the larynx is distorted by the lesions, in which case spontaneous ventilation should be maintained until the airway is secured.153 Postoperative complications of surgical excision include laryngeal edema, airway obstruction, pneumonia, facial palsy, and infection.150,151,154

Hemangiomas, also known as juvenile or infantile hemangiomas, are the most common benign tumors in infancy, affecting up to 10% of infants.155 The majority of hemangiomas are uncomplicated and require no treatment. The natural course begins with a proliferation phase that starts within the first few months of life, followed by an involution phase of variable length. It is estimated that involution occurs at a rate of 10% per year. Hemangiomas can affect all organs, and intervention is required when the lesion affects the function of vital organs such as the eyes, airway, or liver.156 Hemangiomas that occur in the subglottic region must be considered in the differential diagnosis of a noninfectious cause of croup in infants younger than 3 months of age. When present around the eyes and on the face (Fig. 33-11), hemangiomas are often associated with lesions in the airway.157,158 They can occur in any part of the airway and can cause airway and feeding difficulties. Airway procedures to resect or remove hemangiomas are generally undertaken between 1 and 11 months of age.159 Limb hemangiomas generally present with cosmetic concerns and bleeding. Rarely, children with large hemangiomas develop high-output heart failure.

Treatment options for hemangiomas include nonintervention, systemic corticosteroid treatment, corticosteroid injection, surgical excision, and laser ablation. Propranolol is an emerging treatment option for hemangiomas.160 It may be used in children without contraindications either as an oral monotherapy at a dose of 2 mg/kg/day divided into three doses or in combination with oral prednisone 3 mg/kg/day tapered after 4 to 6 weeks of therapy. Propranolol reduces the need for surgical intervention but requires 6 months to 1 year of therapy.161 Surgical treatment is reserved for superficial hemangiomas in locations that are surgically accessible.156 Laser treatment for superficial lesions is commonly performed as an outpatient procedure,162 and routine anesthetic precautions should be taken. Children with hemangiomas who had airway procedures received more steroids and had increased admissions and mortality compared with those without airway procedures.159
Arteriovenous malformations are present at birth but can go unrecognized for years, especially when they are intracranial. Large arteriovenous malformations may cause high-output cardiac failure, necessitating therapeutic intervention. Treatment options may include chemotherapy, corticosteroid therapy, embolization, and surgical excision. Children who undergo excision of the hemangiomas often require blood products during surgery, but platelets should be transfused with care because an accumulation within the malformation may increase its size.163 NSAIDs should be avoided because of their effects on platelet function.
Möbius Syndrome
Möbius sequence is a rare neurologic disorder (2 to 20 in 1,000,000 births) characterized by congenital palsy of the facial (VII) and abducens (VI) cranial nerves, resulting in unilateral or bilateral facial weakness and defective extraocular eye movement (E-Fig. 33-11). These classic features may be associated with other cranial nerves palsies, ophthalmic abnormalities, developmental delay, and various craniofacial, limb, and musculoskeletal malformations, resulting in a variable pattern of clinical expression.164–168 Involvement of cranial nerves IX and X is associated with pharyngeal dysfunction, dysphagia, feeding difficulties, retention of oral secretions, and recurrent aspiration pneumonia. Associated micrognathia, microstomia, limited mouth opening, and other orofacial abnormalities may make tracheal intubation difficult.169 Other associations include gastroesophageal reflux, hypotonia of skeletal muscles, congenital cardiac abnormalities, spinal abnormalities, and peripheral neuropathies. Central alveolar hypoventilation has been described in association with Möbius sequence and may be secondary to hypoplasia of midbrain respiratory centers.170 Central alveolar hypoventilation, compounded by upper airway hypotonia and the effects of sedatives, opioids, and anesthetic agents, can predispose to postoperative respiratory compromise. The absence of facial expression secondary to paresis of the facial nerve can make it difficult to assess and evaluate postoperative pain.171 The anesthetic plan for the child with Möbius sequence must be tailored to the individual based on the clinical expression of the syndrome.

The most common surgical procedure performed in children with Möbius sequence is a segmental gracilis muscle transplantation, in which the muscle is transplanted to the face and revascularized to the facial artery and vein.172 Motor innervation of the gracilis requires a functioning cranial nerve such as the masseter branch of the trigeminal nerve. The aim of this facial reanimation is to facilitate facial expression and provide lower lip support to reduce drooling and improve speech.172 Anesthetic considerations include those for prolonged surgery, avoidance of neuromuscular block to facilitate intraoperative nerve stimulation, and avoidance of hypocapnia, hypothermia, and hypotension to ensure graft perfusion. The latter considerations are also applicable to the postoperative period. Other surgical procedures commonly performed in children with Möbius sequence include strabismus surgery and orthopedic procedures to improve limb function.
Congenital Intraoral Fibrous Bands
Congenital intraoral fibrous bands (e.g., pterygium syndrome and syngnathia) can present an almost impossible airway to secure even with advanced pediatric fiberoptic skills (E-Fig. 33-12). Syngnathia reduces mouth opening as a result of fusion of maxilla and mandible and often presents as a part of Van der Woude and popliteal pterygium syndromes.173 These children often present with airway and feeding difficulties. Depending on the severity of the bands, these children may present formidable anesthetic challenges. Intravenous ketamine anesthesia in the spontaneously breathing neonate may allow division of the adhesions in the first few days of life, precluding the need for a surgical airway or facilitating tracheal intubation if other surgery is required.173

Intraoral Tumors
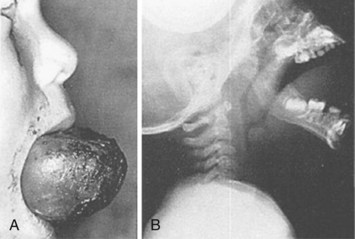
Intraoral tumors are rare in children (E-Fig. 33-13) but, if massive, may present great challenges in securing the airway. Those that present the greatest difficulties preclude visualizing the larynx (E-Fig. 33-14) and present an increased risk of intraoperative bleeding. Preoperative radiographic studies are required to delineate the extent of involvement of the upper airway and whether the supraglottic region or the nasopharynx is clear for passage of a bronchoscope. If the tumors are sufficiently large that laryngoscopy is precluded, fiberoptic nasal intubation must be considered. If the tumor is resectable, a tracheostomy could be a back-up plan. The risk of bleeding depends on the vascularity of the tumor and whether the tongue is involved. If the vascular supply of the tumor can be isolated, bleeding should be easily controlled. If, however, the tumor cannot be separated from the tongue, bleeding can be controlled only by clamping the tongue before resecting the tumor. This should prevent excessive blood loss and permit a hemostatic closure of the resection surface. The trachea should remain intubated postoperatively until both airway and lingual edema have abated.


Brachial Plexus Surgery
Brachial plexus injury occurs in 0.5 to 5 infants per 1000 live births as a result of birth trauma.174,175 Erb palsy involves damage to nerve roots C5, C6, and C7, whereas Klumpke palsy involves roots C8 and T1.175 Complete plexus palsies are the most devastating injuries, resulting in a flail and insensate arm.175,176 Although 75% of brachial plexus injuries resolve spontaneously and completely within the first month after birth, 25% result in permanent disability and impairment.175,177,178 Surgical intervention is indicated if the motor function does not improve after 3 months of age.177,178 Clinically significant diaphragmatic palsy is associated with 2.4% of neonates with brachial plexus injury. In the very young, diaphragmatic palsy requires aggressive intervention before brachial plexus repair. For brachial plexus surgery to be successful, the nerve root cannot be completely avulsed from the spinal cord. Therefore detailed imaging is required to characterize the nature of the injury: avulsion of the nerve root from the spinal cord, disruption of the nerve within the nerve sheath, or disruption of the nerve and the nerve sheath. Because irreversible loss of the neuromotor end plate may occur, surgery is often undertaken before 12 months of age.174,177–181 Microsurgical intervention is performed in infants with global lesions and Horner syndrome by 3 months of age. The aim is to improve function with no expectation of complete recovery; without intervention these children have severe functional deficits. Conversely, if recovery of biceps occurs by 3 months, treatment is performed without microsurgical intervention.182 The treatment of choice includes resection of neuromas with interpositional nerve grafting.183 Nerve grafting is being increasingly performed for treating neonatal brachial plexus injury. Donor nerves include motor branches of C4, intercostal nerves, inferior branches of the 11th cranial nerve, pectoral nerves, and sural nerves.184 Synthetic collagen nerve conduits have been approved as nerve guidance channels in microsurgery and may be an option for the future.
Preoperative MRI for assessing bone and joint deformities may require administration of general anesthesia. Repair of brachial plexus may be challenging because the only extremity for intravenous access and monitoring (BP and pulse oximetry) is the contralateral upper extremity. Both lower extremities are usually prepped and draped for harvesting the sural nerves or other donor nerves for the repair. Because these infants are usually 9 to 12 months of age, they are chubby, making intravenous access more difficult. This surgery often takes up to 12 hours, so the considerations for prolonged anesthesia must be invoked, such as protecting pressure points during positioning. Muscle relaxants are avoided to facilitate intraoperative electrophysiologic testing.185 An indwelling urinary catheter is essential to decompress the bladder. Analgesic requirement is minimal except during brief periods of surgical stimulation. Remifentanil provides excellent intraoperative analgesia and permits rapid adjustment of the depth of anesthesia. Maintenance of normothermia and prevention of fluid overload are important during this prolonged surgery. Blood loss is minimal, and maintenance fluids usually suffice.174 Prolonged administration of propofol is not recommended because of the risk of propofol infusion syndrome and delayed emergence. In its stead, remifentanil combined with inhalation agent may be a more appropriate regimen. Postoperative analgesia requirements are minimal, and acetaminophen and NSAIDs usually provide adequate pain relief. Shoulder spica casts may be applied to avoid sudden neck movements postoperatively if the lower branches of the accessory nerve are used for reconstruction.185
Otoplasty
Protruding ears (commonly known as “bat ears”) are common in the Caucasian population, occurring with an incidence of up to 5% (E-Fig. 33-15).186 Children with protruding ears are generally healthy, with about two thirds undergoing surgical correction before the age of 8 years or as soon as the child expresses concern about the deformity.187 Younger children are more likely to require general anesthesia, whereas those older than age 8 years may tolerate the procedure using local anesthetic infiltration or nerve blocks.188 Laser techniques are now employed increasingly to perform cartilage reshaping.189 The main complication of general anesthesia is postoperative nausea and vomiting, which can last up to 2 days after surgery in approximately 80% of children.190 However, postoperative nausea and vomiting may be reduced by surgical and anesthetic techniques, including the use of propofol and avoidance of packing of the external auditory meatus and concha.190,191 To provide optimal surgical access and positioning of the child, a preformed, low-profile tracheal tube, such as the RAE tube, may be required, but flexible LMAs provide equally satisfactory conditions in the ventilated or spontaneously breathing child.192 Infiltration with local anesthetic (usually less than 10 mL of 1% lidocaine with 1 : 100,000 epinephrine) attenuates the surgical stimulus and reduces the intraoperative opioid requirements. The use of a long-acting local anesthetic combined with a nerve block, acetaminophen, and NSAIDs provides adequate postoperative analgesia in most children. This multimodal approach may obviate the need for opioids and thereby reduce the incidence of postoperative nausea and vomiting.193 Multimodal therapy (ondansetron and dexamethasone) provides specific antiemetic prophylaxis or therapy.

Congenital Hand Anomalies
Congenital limb malformations exhibit a wide spectrum of phenotypic manifestations. Syndactyly may occur as an isolated malformation (Fig. 33-12 and E-Fig. 33-16) or part of a syndrome, the most common being Apert syndrome but also with Poland syndrome, in association with skeletal abnormalities and gastrointestinal and cardiac malformations. Limb malformations are more frequent in males than females, and they affect both upper and lower limbs in approximately 50% of children with a deformity. Early separation of digits is favored if the ring and little fingers or index finger and thumb are involved, because the differing longitudinal growth rates will lead to greater deformities.194 Surgery is usually performed between 6 and 18 months of age.195,196 An association has been reported between syndactyly and prolonged QT interval, with life-threatening arrhythmias reported in one child during anesthesia.197 Timothy syndrome is a multisystem disorder with cardiac, facial, limb, and neurodevelopmental features.198
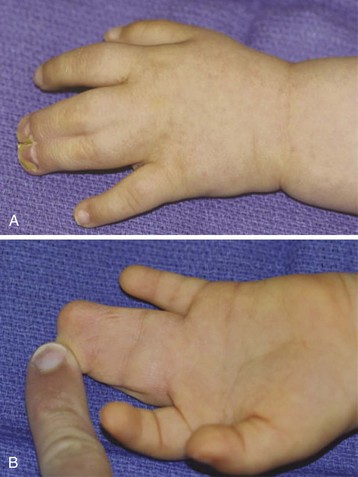
FIGURE 33-12 Syndactyly of the first and second digit of an infant: dorsal aspect (A) and volar aspect (B).

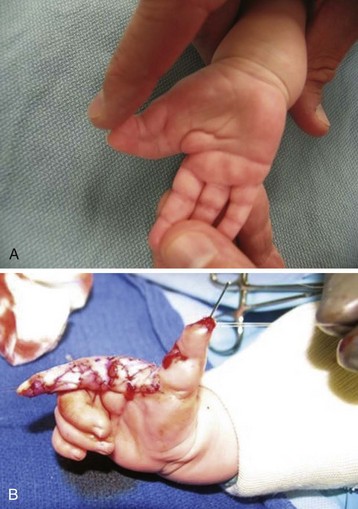
Duplicated thumb can be present as an isolated anomaly and is present in approximately 1 in 3000 births. Hypoplastic thumbs are associated with systemic syndromes such as Holt-Oram syndrome; vertebral, anal, cardiac, tracheal, esophageal, renal, limb (VACTERL) anomalies; Fanconi anemia; Nager syndrome; and thrombocytopenia-absent radius.199 A complete evaluation of the child is generally warranted because abnormalities can occur in the cardiovascular, neurologic, and hematopoietic systems.199 Genetic testing is not generally needed in isolated thumb duplications.
Tissue Expanders
Tissue expansion has become a major treatment modality in the management of giant congenital hairy pigmented nevi (see E-Fig. 33-17), hemangiomas, meningomyelocele, abdominal wall defects, and secondary reconstruction of extensive burn scars.200–208 Tissue expanders effectively allow removal of the affected area and preserve sensation in a durable flap with minimal donor site morbidity.209 These devices consist of a silicone shell that stretches to accommodate serial injections of saline when placed subcutaneously or, in the case of the scalp, under the galea, through an incision made in normal tissue adjacent to the lesion or defect (Fig. 33-13).204,210 Osmotic tissue expanders have been used in burn scars, congenital nevi, alopecia, or foot deformities with reduced infection rates and low cost. Tissue expansion requires at least two surgical procedures, one to insert the expander and a second to remove it when expansion is complete; some children may require serial insertions or multiple expanders.204 Reconstruction of areas of the head and neck constitute a particular challenge because expansion without oral, visual, or airway compromise is required.211 Complications of tissue expansion include infection, skin erosion, leakage, migration, and flap necrosis.207,212–215 Perioperative antibiotics are given at insertion and removal, although their effectiveness in preventing infection has not been established.202,207,209,213,214

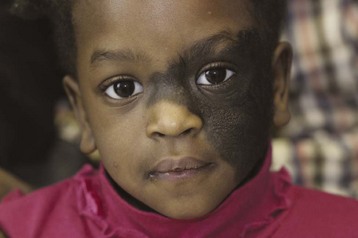
Hairy Pigmented Nevi
Congenital melanocytic nevi characteristically vary in size, shape, surface texture, and hairiness. They are frequently excised because they are disfiguring and have the potential to become malignant. Serial surgical excision is common, but skin grafting and tissue expanders are also used (E-Fig. 33-17).209 The position and size determine the frequency of excision and anesthetic technique. If the face, head, or neck is involved, airway management should be discussed with the surgeon to allow optimal surgical access (Fig. 33-14).

In general, these children are healthy. In the cooperative and motivated child, subcutaneous infusion of a very dilute local anesthetic (e.g., ropivacaine 0.08% to 0.3%) mixed with epinephrine 1 : 1,000,000 can be used to provide painless tumescent anesthesia.216 The local anesthetic is infused through a 30-gauge needle at an initial rate of 120 mL/hr. Blanching of skin identifies the area that is anesthetized. This method has been used successfully in children 7 years of age and older.217 To avoid toxicity, local anesthetic volume and dosing guidelines should be followed (see Chapter 41). The addition of longer-acting local anesthetics to the infiltrate will enhance postoperative analgesia. Repeated reconstructive procedures are often required, and attention should be paid to providing appropriate premedication where necessary (see Chapter 4).
Cosmetic Procedures
According to American Society of Plastic Surgeons, the most common cosmetic surgeries performed in adolescents are nose reshaping, male breast reduction, ear surgery, laser hair removal, laser treatment of leg veins, and laser skin resurfacing.218 Breast implants and liposuction are the most controversial cosmetic procedures performed in this age group, although combined they only comprise 5% of cosmetic surgery in this age group. In 2011, 73% of male breast reduction and 28% of otoplasty occurred in this age group.218 The breast augmentation procedures were performed on an outpatient basis.219 These patients are generally healthy. Routine anesthetic induction with endotracheal intubation with no additional invasive monitoring is generally all that is required. This group of patients may be at increased risk of postoperative nausea and vomiting and will benefit from multimodal prophylaxis. Postoperative discomfort is common, requiring administration of systemic opioids or nerve blocks.
Liposuction is usually scheduled as outpatient surgery, although extensive liposuction may require an overnight admission to the hospital. This latter procedure is associated with acute complications, including itching, bruising, and swelling, damage to nerves or vital organs, blood loss, and embolization of fat or blood. Lidocaine toxicity and fluid accumulation at the surgical site are directly proportional to the volume of aspirated fat and number of treated sites.220
Trauma
A considerable proportion of plastic surgical procedures in children are performed for trauma and emergency surgery. Procedures include treatment of simple lacerations, animal bites, tendon, nerve and vascular repair, reimplantation of digits and limbs, and treatment of burns (see Chapter 34). The cooperative and fasted child with minor trauma can often undergo minor surgical procedures using local anesthesia, commonly administered by the plastic surgeon in the emergency department. This can be supplemented with inhalation of a 50 : 50 mixture of nitrous oxide and oxygen (Entonox),221 intravenous administration of small doses of a benzodiazepine and opioid (e.g., midazolam 50 µg/kg, and fentanyl 0.5 µg/kg) or ketamine according to locally established sedation protocols (see Chapters 45 and 47). Alternatively, a single-injection digital nerve block is safe and effective for minor surgical procedures in children.222
Surgery for extensive injuries to digits and limbs usually requires general anesthesia because children do not tolerate prolonged application of a tourniquet. The severity and urgency of the injury dictate the timing of the surgery. The general principles of care for the child with trauma should be followed (see Chapter 38), with particular attention directed to identifying more life-threatening injuries. For urgent surgery in the presence of a full stomach, precautions against aspiration of gastric contents should be considered. For postoperative pain control, a combination of regional anesthesia or systemic analgesia is usually adequate. Continuous brachial plexus or other nerve blocks may improve tissue perfusion223 and facilitate cooperation during postoperative physiotherapy for procedures such as digital reimplantation. Continuous nerve block may attenuate the signs associated with compartment syndrome, and frequent and meticulous attention must be paid to perfusion of the extremity.
Antony AK, Sloan GM. Airway obstruction following palatoplasty: analysis of 247 consecutive operations. Cleft Palate Craniofac J. 2002;39:145–148.
Faberowski LW, Black S, Mickle JP. Incidence of venous air embolism during craniectomy for craniosynostosis repair. Anesthesiology. 2000;92:20–23.
Goobie SM, Meier PM, Pereira LM, et al. Efficacy of tranexamic acid in pediatric craniosynostosis surgery: a double-blind, placebo-controlled trial. Anesthesiology. 2011;114:862–871.
Lavoie J. Blood transfusion risks and alternative strategies in pediatric patients. Pediatr Anesth. 2011;21:14–24.
Meier PM, Goobie SM, DiNardo JA, et al. Endoscopic strip craniectomy in early infancy: the initial five years of anesthesia experience. Anesth Analg. 2011;112:407–414.
Nargozian C. The airway in patients with craniofacial abnormalities. Pediatr Anesth. 2004;14:53–59.
1 Harling TR, Stelnicki EJ, Hedrick MH, Longaker MT. In utero models of craniofacial surgery. World J Surg. 2003;27:108–116.
2 Fisher QA, Nichols D, Stewart FC, et al. Assessing pediatric anesthesia practices for volunteer medical services abroad. Anesthesiology. 2001;95:1315–1322.
3 Czerwinski M, Hopper RA, Gruss J, Fearon JA. Major morbidity and mortality rates in craniofacial surgery: an analysis of 8101 major procedures. Plast Reconstr Surg. 2010;126:181–186.
4 Nargozian C. The airway in patients with craniofacial abnormalities. Paediatric Anaesthesia. 2004;14:53–59.
5 Mirilas P, Mentessidou A, Kontis E, et al. Parental exposures and risk of nonsyndromic orofacial clefts in offspring: a case-control study in Greece. Int J Pediatr Otorhinolaryngol. 2011;75:695–699.
6 Blanton SH, Henry RR, Yuan Q, et al. Folate pathway and nonsyndromic cleft lip and palate. Birth Defects Res A Clin Mol Teratol. 2011;91:50–60.
7 Prescott NJ, Lees MM, Winter RM, Malcolm S. Identification of susceptibility loci for nonyndromic cleft lip with or without cleft palate in a two stage genome scan of affected sib-pairs. Hum Genet. 2000;106:345–350.
8 Zeiger JS, Hetmanski JB, Beaty TH, et al. Evidence for linkage of nonsyndromic cleft lip with or without cleft palate to a region on chromosome 2. Eur J Hum Genet. 2003;11:835–839.
9 Mossey PA, Little J, Munger RG, Dixon MJ, Shaw WC. Cleft lip and palate. Lancet. 2009;374:1773–1785.
10 Manganaro L, Tomei A, Fierro F, et al. Fetal MRI as a complement to US in the evaluation of cleft lip and palate. Radiol Med. 2011;116:1134–1148.
11 Adeyemo WL, Adeyemo TA, Ogunlewe MO, et al. Blood transfusion requirements in cleft lip surgery. Int J Pediatr Otorhinolaryngol. 2011.
12 Rinaldi PA, Dogra S, Sellman GL. Difficult intubation in paediatric palatoplasty. Anaesthesia. 1993;48:358–359.
13 Gunawardana RH. Difficult laryngoscopy in cleft lip and palate surgery. Br J Anaesth. 1996;76:757–759.
14 Xue FS, Zhang GH, Li P, et al. The clinical observation of difficult laryngoscopy and difficult intubation in infants with cleft lip and palate. Paediatr Anaesth. 2006;16:283–289.
15 Uezono S, Holzman RS, Goto T, et al. Prediction of difficult airway in school-aged patients with microtia. Paediatri Anaesth. 2001;11:409–413.
16 Sen I, Kumar S, Bhardwaj N, Wig J. A left paraglossal approach for oral intubation in children scheduled for bilateral orofacial cleft reconstruction surgery: a prospective observational study. Paediatr Anaesth. 2009;19:159–163.
17 Rajamani A, Kamat V, Rajavel VP, Murthy J, Hussain SA. A comparison of bilateral infraorbital nerve block with intravenous fentanyl for analgesia following cleft lip repair in children. Paediatr Anaesth. 2007;17:133–139.
18 Takmaz SA, Uysal HY, Uysal A, et al. Bilateral extraoral, infraorbital nerve block for postoperative pain relief after cleft lip repair in pediatric patients: a randomized, double-blind controlled study. Ann Plast Surg. 2009;63:59–62.
19 Salloum ML, Eberlin KR, Sethna N, Hamdan US. Combined use of infraorbital and external nasal nerve blocks for effective perioperative pain control during and after cleft lip repair. Cleft Palate Craniofac J. 2009;46:629–635.
20 Antony AK, Sloan GM. Airway obstruction following palatoplasty: analysis of 247 consecutive operations. Cleft Palate Craniofac J. 2002;39:145–148.
21 Chan MT, Chan MS, Mui KS, Ho BP. Massive lingual swelling following palatoplasty: an unusual cause of upper airway obstruction. Anaesthesia. 1995;50:30–34.
22 Lee JT, Kingston HG. Airway obstruction due to massive lingual oedema following cleft palate surgery. Can Anaesth Soc J. 1985;32:265–267.
23 Patane PS, White SE. Macroglossia causing airway obstruction following cleft palate repair. Anesthesiology. 1989;71:995–996.
24 Saldien V, Coppejans HC, Vercauteren MP, et al. Acute respiratory distress after upper airway obstruction following palatoplasty. Int J Pediatr Otorhinolaryngol. 2003;67:403–407.
25 Dell’Oste C, Savron F, Pelizzo G, Sarti A. Acute airway obstruction in an infant with Pierre Robin syndrome after palatoplasty. Acta Anaesthesiol Scand. 2004;48:787–789.
26 Mukozawa M, Kono T, Fujiwara S, Takakura K. Late onset tongue edema after palatoplasty. Acta Anaesthesiol Taiwan. 2011;49:29–31.
27 Takemura H, Yasumoto K, Toi T, Hosoyamada A. Correlation of cleft type with incidence of perioperative respiratory complications in infants with cleft lip and palate. Paediatr Anaesth. 2002;12:585–588.
28 Jonnavithula N, Durga P, Madduri V, et al. Efficacy of palatal block for analgesia following palatoplasty in children with cleft palate. Paediatr Anaesth. 2010;20:727–733.
29 Mesnil M, Dadure C, Captier G, et al. A new approach for peri-operative analgesia of cleft palate repair in infants: the bilateral suprazygomatic maxillary nerve block. Paediatr Anaesth. 2010;20:343–349.
30 Koh JL, Gries H. Perioperative management of pediatric patients with craniosynostosis. Anesthesiol Clin. 2007;25:465–481. viii
31 Tuncbilek G, Alanay Y, Uzun H, et al. Intracranial and extracranial malformations in patients with craniofacial anomalies. J Craniofac Surg. 2010;21:1460–1464.
32 Marucci DD, Dunaway DJ, Jones BM, Hayward RD. Raised intracranial pressure in Apert syndrome. Plast Reconstr Surg. 2008;122:1162–1168.
33 Al-Saleh S, Riekstins A, Forrest CR, et al. Sleep-related disordered breathing in children with syndromic craniosynostosis. J Craniomaxillofac Surg. 2011;39:153–157.
34 Renier D, Arnaud E, Cinalli G, et al. Prognosis for mental function in Apert’s syndrome. J Neurosurg. 1996;85:66–72.
35 Kreiborg S, Cohen MM, Jr. Ocular manifestations of Apert and Crouzon syndromes: qualitative and quantitative findings. J Craniofac Surg. 2010;21:1354–1357.
36 Batra YK, Rajeev S, Nishtala S, Grover G. Anesthetic implications of Carpenter syndrome (acrocephalopolysyndactyly type II). Paediatr Anaesth. 2008;18:1235–1237.
37 Bellew M, Chumas P, Mueller R, Liddington M, Russell J. Pre- and postoperative developmental attainment in sagittal synostosis. Arch Dis Child. 2005;90:346–350.
38 Cohen SR, Cho DC, Nichols SL, et al. American society of maxillofacial surgeons outcome study: preoperative and postoperative neurodevelopmental findings in single-suture craniosynostosis. Plast Reconstr Surg. 2004;114:841–847.
39 Dollfus H, Verloes A. Dysmorphology and the orbital region: a practical clinical approach. Surv Ophthalmol. 2004;49:547–561.
40 Gupta S, Ghose S, Rohatgi M, Kumar A, Das A. The optic nerve in children with craniosynostosis: a pre and post surgical evaluation. Doc Ophthalmol. 1993;83:271–278.
41 Hertle RW, Quinn GE, Minguini N, Katowitz JA. Visual loss in patients with craniofacial synostosis. J Pediatr Ophthalmol Strabismus. 1991;28:344–349.
42 Kapp-Simon KA, Leroux B, Cunningham M, Speltz ML. Multisite study of infants with single-suture craniosynostosis: preliminary report of presurgery development. Cleft Palate Craniofac J. 2005;42:377–384.
43 Kelleher MO, Murray DJ, McGillivary A, et al. Behavioral, developmental, and educational problems in children with nonsyndromic trigonocephaly. J Neurosurg. 2006;105:382–384.
44 Liasis A, Nischal KK, Walters B, et al. Monitoring visual function in children with syndromic craniosynostosis: a comparison of 3 methods. Arch Ophthalmol. 2006;124:1119–1126.
45 Panchal J, Amirsheybani H, Gurwitch R, et al. Neurodevelopment in children with single-suture craniosynostosis and plagiocephaly without synostosis. Plast Reconstr Surg. 2001;108:1492–1498.
46 Speltz ML, Endriga MC, Mouradian WE. Presurgical and postsurgical mental and psychomotor development of infants with sagittal synostosis. Cleft Palate Craniofac J. 1997;34:374–379.
47 Speltz ML, Kapp-Simon K, Collett B, et al. Cunningham ML: Neurodevelopment of infants with single-suture craniosynostosis: presurgery comparisons with case-matched controls. Plast Reconstr Surg. 2007;119:1874–1881.
48 Warschausky S, Angobaldo J, Kewman D, et al. Early development of infants with untreated metopic craniosynostosis. Plast Reconstr Surg. 2005;115:1518–1523.
49 Bartels MC, Vaandrager JM, de Jong TH, Simonsz HJ. Visual loss in syndromic craniosynostosis with papilledema but without other symptoms of intracranial hypertension. J Craniofac Surg. 2004;15:1019–1022.
50 Cartwright CC, Jimenez DF, Barone CM, Baker L. Endoscopic strip craniectomy: a minimally invasive treatment for early correction of craniosynostosis. J Neurosci Nurs. 2003;35:130–138.
51 Maugans TA, McComb JG, Levy ML. Surgical management of sagittal synostosis: a comparative analysis of strip craniectomy and calvarial vault remodeling. Pediatr Neurosurg. 1997;27:137–148.
52 Mackenzie KA, Davis C, Yang A, MacFarlane MR. Evolution of surgery for sagittal synostosis: the role of new technologies. J Craniofac Surg. 2009;20:129–133.
53 Ririe DG, Smith TE, Wood BC, et al. Time-dependent perioperative anesthetic management and outcomes of the first 100 consecutive cases of spring-assisted surgery for sagittal craniosynostosis. Paediatr Anaesth. 2011;21:1015–1019.
54 Bannink N, Nout E, Wolvius EB, et al. Obstructive sleep apnea in children with syndromic craniosynostosis: long-term respiratory outcome of midface advancement. Int J Oral Maxillofac Surg. 2010;39:115–121.
55 Gonsalez S, Hayward R, Jones B, Lane R. Upper airway obstruction and raised intracranial pressure in children with craniosynostosis. Eur Respir J. 1997;10:367–375.
56 Hajiha M, DuBord MA, Liu H, Horner RL. Opioid receptor mechanisms at the hypoglossal motor pool and effects on tongue muscle activity in vivo. J Physiol. 2009;587:2677–2692.
57 Stricker PA, Cladis FP, Fiadjoe JE, McCloskey JJ, Maxwell LG. Perioperative management of cildren undergoing craniofacial reconstruction surgery: a practice survey. Paediatr Anaesth. 2011;21:1026–1035.
58 Barnett S, Moloney C, Bingham R. Perioperative complications in children with Apert syndrome: a review of 509 anesthetics. Paediatric Anaesthesia. 2011;21:72–77.
59 Zunini GS, Rando KA, Cox RG. Fluid replacement in craniofacial pediatric surgery: normal saline or Ringer’s lactate? J Craniofac Surg. 2011;22:1370–1374.
60 Bhananker SM, Ramamoorthy C, Geiduschek JM, et al. Anesthesia-related cardiac arrest in children: update from the Pediatric Perioperative Cardiac Arrest Registry. Anesth Analg. 2007;105:344–350.
61 van Uitert A, Megens JHAM, Breugem CC, et al. Factors influencing blood loss and allogeneic blood transfusion practice in craniosynostosis surgery. Paediatr Anaesth. 2011;21:1192–1197.
62 Meier PM, Goobie SM, DiNardo JA, et al. Endoscopic strip craniectomy in early infancy: the initial five years of anesthesia experience. Anesth Analg. 2011;112:407–414.
63 Seruya M, Oh AK, Boyajian MJ, et al. Unreliability of intraoperative estimated blood loss in extended sagittal synostectomies. J Neurosurg Pediatr. 2011;8:443–449.
64 Krajewski K, Ashley RK, Pung N, et al. Successful blood conservation during craniosynostotic correction with dual therapy using procrit and cell saver. J Craniofac Surg. 2008;19:101–105.
65 Fearon JA, Weinthal J. The use of recombinant erythropoietin in the reduction of blood transfusion rates in craniosynostosis repair in infants and children. Plast Reconstr Surg. 2002;109:2190–2196.
66 Helfaer MA, Carson BS, James CS, et al. Increased hematocrit and decreased transfusion requirements in children given erythropoietin before undergoing craniofacial surgery. J Neurosurg. 1998;88:704–708.
67 Cooper MJ, Cockell KA, L’Abbe MR. The iron status of Canadian adolescents and adults: current knowledge and practical implications. Can J Diet Pract Res. 2006;67:130–138.
68 Kemmotsu H, Joe K, Nakamura H, Yamashita M. Predeposited autologous blood transfusion for surgery in infants and children. J Pediatr Surg. 1995;30:659–661.
69 Thompson HW, Luban NL. Autologous blood transfusion in the pediatric patient. J Pediatr Surg. 1995;30:1406–1411.
70 Hans P, Collin V, Bonhomme V, et al. Evaluation of acute normovolemic hemodilution for surgical repair of craniosynostosis. J Neurosurg Anesthesiol. 2000;12:33–36.
71 Hildebrandt B, Machotta A, Riess H, et al. Intraoperative fresh-frozen plasma versus human albumin in craniofacial surgery: a pilot study comparing coagulation profiles in infants younger than 12 months. Thromb Haemost. 2007;98:172–177.
72 Stricker PA, Petersen C, Fiadjoe JE, McCloskey JJ. Successful treatment of intractable hemorrhage with recombinant factor VIIa during cranial vault reconstruction in an infant. Paediatr Anaesth. 2009;19:806–807.
73 Dadure C, Sauter M, Bringuier S, et al. Intraoperative tranexamic acid reduces blood transfusion in children undergoing craniosynostosis surgery: a randomized double-blind study. Anesthesiology. 2011;114:856–861.
74 Goobie SM, Meier PM, Pereira LM, et al. Efficacy of tranexamic acid in pediatric craniosynostosis surgery: a double-blind, placebo-controlled trial. Anesthesiology. 2011;114:862–871.
75 Duran de la FP, Garcia-Fernandez J, Perez-Lopez C, Carceller F, Gilsanz RF. [Usefulness of tranexamic acid in cranial remodeling surgery]. Rev Esp Anestesiol Reanim. 2003;50:388–394.
76 Dhawale AA, Shah SA, Sponseller PD, et al. Are antifibrinolytics helpful in decreasing blood loss and transfusions during spinal fusion surgery in children with cerebral palsy scoliosis? Spine (Phila Pa 1976.). 2012;37:E549–E555.
77 Diaz JH, Lockhart CH. Hypotensive anaesthesia for craniectomy in infancy. Br J Anaesth. 1979;51:233–235.
78 Caverni V, Rosa G, Pinto G, Tordiglione P, Favaro R. Hypotensive anesthesia and recovery of cognitive function in long-term craniofacial surgery. J Craniofac Surg. 2005;16:531–536.
79 Velardi F, Di CA, Di RC, et al. “No allogeneic blood transfusion” protocol for the surgical correction of craniosynostoses. I. Rationale. Childs Nerv Syst. 1998;14:722–731.
80 Velardi F, Di Chirico A, Di Rocco C, et al. “No allogeneic blood transfusion” protocol for the surgical correction of craniosynostoses. II. Clinical application. Childs Nerv Syst. 1998;14:732–739.
81 Meara JG, Smith EM, Harshbarger RJ, et al. Blood-conservation techniques in craniofacial surgery. Ann Plast Surg. 2005;54:525–529.
82 Cavallini M, Baruffaldi Preis FW, Casati A. Effects of mild hypothermia on blood coagulation in patients undergoing elective plastic surgery. Plast Reconstr Surg. 2005;116:316–321.
83 Jimenez DF, Barone CM, McGee ME, Cartwright CC, Baker CL. Endoscopy-assisted wide-vertex craniectomy, barrel stave osteotomies, and postoperative helmet molding therapy in the management of sagittal suture craniosynostosis. J Neurosurg. 2004;100:407–417.
84 Murad GJ, Clayman M, Seagle MB, et al. Endoscopic-assisted repair of craniosynostosis. Neurosurg Focus. 2005;19:E6.
85 Byeon JH, Yoo G. Cerebral salt wasting syndrome after calvarial remodeling in craniosynostosis. J Korean Med Sci. 2005;20:866–869.
86 Levine JP, Stelnicki E, Weiner HL, Bradley JP, McCarthy JG. Hyponatremia in the postoperative craniofacial pediatric patient population: a connection to cerebral salt wasting syndrome and management of the disorder. Plast Reconstr Surg. 2001;108:1501–1508.
87 Lee SJ, Huh EJ, Byeon JH. Two cases of cerebral salt wasting syndrome developing after cranial vault remodeling in craniosynostosis children. J Korean Med Sci. 2004;19:627–630.
88 Rando K, Zunini G, Negroto A. Intraoperative hyponatremia during craniofacial surgery. Paediatr Anaesth. 2009;19:358–363.
89 Cladis FP, Bykowski M, Schmitt E, et al. Postoperative hyponatremia following calvarial vault remodeling in craniosynostosis. Paediatr Anaesth. 2011;21:1020–1025.
90 Tamburrini G, Caldarelli M, Massimi L, Santini P, DI RC. Intracranial pressure monitoring in children with single suture and complex craniosynostosis: a review. Childs Nerv Syst. 2005;21:913–921.
91 Noetzel MJ, Marsh JL, Palkes H, Gado M. Hydrocephalus and mental retardation in craniosynostosis. J Pediatr. 1985;107:885–892.
92 Collmann H, Sorensen N, Krauss J. Hydrocephalus in craniosynostosis: a review. Childs Nerv Syst. 2005;21:902–912.
93 Hayward R, Gonsalez S. How low can you go? Intracranial pressure, cerebral perfusion pressure, and respiratory obstruction in children with complex craniosynostosis. J Neurosurg. 2005;102:16–22.
94 Liasis A, Walters B, Thompson D, et al. Visual field loss in children with craniosynostosis. Childs Nerv Syst. 2011;27:1289–1296.
95 Renier D, Sainte-Rose C, Marchac D, Hirsch JF. Intracranial pressure in craniostenosis. J Neurosurg. 1982;57:370–377.
96 Hudgins RJ, Cohen SR, Burstein FD, Boydston WR. Multiple suture synostosis and increased intracranial pressure following repair of single suture, nonsyndromal craniosynostosis. Cleft Palate Craniofac J. 1998;35:167–172.
97 Pollack IF, Losken HW, Biglan AW. Incidence of increased intracranial pressure after early surgical treatment of syndromic craniosynostosis. Pediatr Neurosurg. 1996;24:202–209.
98 Cucchiara RF, Bowers B. Air embolism in children undergoing suboccipital craniotomy. Anesthesiology. 1982;57:338–339.
99 Bithal PK, Pandia MP, Dash HH, et al. Comparative incidence of venous air embolism and associated hypotension in adults and children operated for neurosurgery in the sitting position. Eur J Anaesthesiol. 2004;21:517–522.
100 Harrison EA, Mackersie A, McEwan A, Facer E. The sitting position for neurosurgery in children: a review of 16 years’ experience. Br J Anaesth. 2002;88:12–17.
101 Harris MM, Strafford MA, Rowe RW, et al. Venous air embolism and cardiac arrest during craniectomy in a supine infant. Anesthesiology. 1986;65:547–550.
102 Gronert GA, Messick JM, Jr., Cucchiara RF, Michenfelder JD. Paradoxical air embolism from a patent foramen ovale. Anesthesiology. 1979;50:548–549.
103 Mirski MA, Lele AV, Fitzsimmons L, Toung TJ. Diagnosis and treatment of vascular air embolism. Anesthesiology. 2007;106:164–177.
104 Tobias JD, Johnson JO, Jimenez DF, Barone CM, McBride DS, Jr. Venous air embolism during endoscopic strip craniectomy for repair of craniosynostosis in infants. Anesthesiology. 2001;95:340–342.
105 Faberowski LW, Black S, Mickle JP. Incidence of venous air embolism during craniectomy for craniosynostosis repair. Anesthesiology. 2000;92:20–23.
106 Palmon SC, Moore LE, Lundberg J, Toung T. Venous air embolism: a review. J Clin Anesth. 1997;9:251–257.
107 Furuya H, Suzuki T, Okumura F, Kishi Y, Uefuji T. Detection of air embolism by transesophageal echocardiography. Anesthesiology. 1983;58:124–129.
108 Hurter D, Sebel PS. Detection of venous air embolism: a clinical report using end-tidal carbon dioxide monitoring during neurosurgery. Anaesthesia. 1979;34:578–582.
109 Jaffe RA, Siegel LC, Schnittger I, Propst JW, Brock-Utne JG. Epidural air injection assessed by transesophageal echocardiography. Reg Anesth. 1995;20:152–155.
110 Sato S, Toya S, Ohira T, Mine T, Greig NH. Echocardiographic detection and treatment of intraoperative air embolism. J Neurosurg. 1986;64:440–444.
111 Jones BM, Jani P, Bingham RM, Mackersie AM, Hayward R. Complications in paediatric craniofacial surgery: an initial four year experience. Br J Plast Surg. 1992;45:225–231.
112 Lee J, Crawford MW, Drake J, Buncic JR, Forrest C. Anterior ischemic optic neuropathy complicating cranial vault reconstruction for sagittal synostosis in a child. J Craniofac Surg. 2005;16:559–562.
113 Choi AY, Ahmad NS, de Beer DA. Metabolic changes during major craniofacial surgery. Paediatr Anaesth. 2010;20:851–855.
114 Lubbers HT, Zweifel D, Gratz KW, Kruse A. Classification of potential risk factors for trigeminocardiac reflex in craniomaxillofacial surgery. J Oral Maxillofac Surg. 2010;68:1317–1321.
115 Cedars MG, Linck DL, Chin M, Toth BA. Advancement of the midface using distraction techniques. Plast Reconstr Surg. 1999;103:429–441.
116 Meazzini MC, Mazzoleni F, Caronni E, Bozzetti A. Le Fort III advancement osteotomy in the growing child affected by Crouzo’s and Apert’s syndromes: presurgical and postsurgical growth. J Craniofac Surg. 2005;16:369–377.
117 Meling TR, Tveten S, Due-Tonnessen BJ, Skjelbred P, Helseth E. Monobloc and midface distraction osteogenesis in pediatric patients with severe syndromal craniosynostosis. Pediatr Neurosurg. 2000;33:89–94.
118 Britto JA, Evans RD, Hayward RD, Jones BM. Maxillary distraction osteogenesis in Pfeiffer’s syndrome: urgent ocular protection by gradual midfacial skeletal advancement. Br J Plast Surg. 1998;51:343–349.
119 Posnick JC. Craniofacial dysostosis: staging of reconstruction and management of the midface deformity. Neurosurg Clin N Am. 1991;2:683–702.
120 Uemura T, Hayashi T, Satoh K, et al. A case of improved obstructive sleep apnea by distraction osteogenesis for midface hypoplasia of an infantile Crouzon’s syndrome. J Craniofac Surg. 2001;12:73–77.
121 Israele V, Siegel JD. Infectious complications of craniofacial surgery in children. Rev Infect Dis. 1989;11:9–15.
122 Matsumoto K, Nakanishi H, Seike T, Koizumi Y, Hirabayashi S. Intracranial hemorrhage resulting from skull base fracture as a complication of Le Fort III osteotomy. J Craniofac Surg. 2003;14:545–548.
123 Newhouse RF, Schow SR, Kraut RA, Price JC. Life-threatening hemorrhage from a Le Fort I osteotomy. J Oral Maxillofac Surg. 1982;40:117–119.
124 Posnick JC, al-Qattan MM, Armstrong D. Monobloc and facial bipartition osteotomies for reconstruction of craniofacial malformations: a study of extradural dead space and morbidity. Plast Reconstr Surg. 1996;97:1118–1128.
125 Johnson DM, From AM, Smith RB, From RP, Maktabi MA. Endoscopic study of mechanisms of failure of endotracheal tube advancement into the trachea during awake fiberoptic orotracheal intubation. Anesthesiology. 2005;102:910–914.
126 Asai T, Shingu K. Difficulty in advancing a tracheal tube over a fibreoptic bronchoscope: incidence, causes and solutions. Br J Anaesth. 2004;92:870–881.
127 Crysdale WS, Kohli-Dang N, Mullins GC, et al. Airway management in craniofacial surgery: experience in 542 patients. J Otolaryngol. 1987;16:207–215.
128 Wolfe SA, Morrison G, Page LK, Berkowitz S. The monobloc frontofacial advancement: do the pluses outweigh the minuses? Plast Reconstr Surg. 1993;91:977–987.
129 Varol A, Basa S, Ozturk S. The role of controlled hypotension upon transfusion requirement during maxillary downfracture in double-jaw surgery. J Craniomaxillofac Surg. 2010;38:345–349.
130 Nargozian C, Ririe DG, Bennun RD, Mulliken JB. Hemifacial microsomia: anatomical prediction of difficult intubation. Paediatric Anaesthesia. 1999;9:393–398.
131 Vento AR, LaBrie RA, Mulliken JB. The O.M.E.N.S. classification of hemifacial microsomia. Cleft Palate Craniofac J. 1991;28:68–76.
132 Rodgers SF, Eppley BL, Nelson CL, Sadove AM. Hemifacial microsomia: assessment of classification systems. J Craniofac Surg. 1991;2:114–126.
133 Horgan JE, Padwa BL, LaBrie RA, Mulliken JB. OMENS-Plus: analysis of craniofacial and extracraniofacial anomalies in hemifacial microsomia. Cleft Palate Craniofac J. 1995;32:405–412.
134 Robiony M, Polini F. Piezosurgery: a safe method to perform osteotomies in young children affected by hemifacial microsomia. J Craniofac Surg. 2010;21:1813–1815.
135 Dufton LM, Speltz ML, Kelly JP, et al. Psychosocial outcomes in children with hemifacial microsomia. J Pediatr Psychol. 2011;36:794–805.
136 Inada T, Fujise K, Tachibana K, Shingu K. Orotracheal intubation through the laryngeal mask airway in paediatric patients with Treacher-Collins syndrome. Paediatr Anaesth. 1995;5:129–132.
137 Lerman J, Creighton RE. Two hands, three sites: show me the vocal cords. Paediatr Anaeth. 2006;16:96.
138 Sugiyama K, Yokoyama K. Displacement of the endotracheal tube caused by change of head position in pediatric anesthesia: evaluation by fiberoptic bronchoscopy. Anesth Analg. 1996;82:251–253.
139 Blau WS, Kafer ER, Anderson JA. Esmolol is more effective than sodium nitroprusside in reducing blood loss during orthognathic surgery. Anesth Analg. 1992;75:172–178.
140 Fromme GA, MacKenzie RA, Gould AB, Jr., Lund BA, Offord KP. Controlled hypotension for orthognathic surgery. Anesth Analg. 1986;65:683–686.
141 Golia JK, Woo R, Farole A, Seltzer JL. Nitroglycerin-controlled circulation in orthognathic surgery. J Oral Maxillofac Surg. 1985;43:342–345.
142 Lessard MR, Trepanier CA, Baribault JP, et al. Isoflurane-induced hypotension in orthognathic surgery. Anesth Analg. 1989;69:379–383.
143 McNulty S, Sharifi-Azad S, Farole A. Induced hypotension with labetalol for orthognathic surgery. J Oral Maxillofac Surg. 1987;45:309–311.
144 Praveen K, Narayanan V, Muthusekhar MR, Baig MF. Hypotensive anaesthesia and blood loss in orthognathic surgery: a clinical study. Br J Oral Maxillofac Surg. 2001;39:138–140.
145 Precious DS, Splinter W, Bosco D. Induced hypotensive anesthesia for adolescent orthognathic surgery patients. J Oral Maxillofac Surg. 1996;54:680–683.
146 Rodrigo C. Induced hypotension during anesthesia with special reference to orthognathic surgery. Anesth Prog. 1995;42:41–58.
147 Tobias JD. Nicardipine for controlled hypotension during orthognathic surgery. Plast Reconstr Surg. 1997;99:1539–1543.
148 Zellin G, Rasmusson L, Palsson J, Kahnberg KE. Evaluation of hemorrhage depressors on blood loss during orthognathic surgery: a retrospective study. J Oral Maxillofac Surg. 2004;62:662–666.
149 Weber CR, Griffin JM. Evaluation of dexamethasone for reducing postoperative edema and inflammatory response after orthognathic surgery. J Oral Maxillofac Surg. 1994;52:35–39.
150 Burezq H, Williams B, Chitte SA. Management of cystic hygromas: 30 year experience. J Craniofac Surg. 2006;17:815–818.
151 Tran NN, Tran XN. Cystic hygroma in children: a report of 126 cases. J Pediatr Surg. 1974;9:191–195.
152 Wu MP, Wu RC, Lee JS, Yao WJ, Kuo PL. Spontaneous resolution of fetal mediastinal cystic hygroma. Int J Gynaecol Obstet. 1995;48:295–298.
153 Bryan Y, Chwals W, Ovassapian A. Sedation and fiberoptic intubation of a neonate with a cystic hygroma. Acta Anaesthesiol Scand. 2005;49:122–123.
154 Ameh EA, Nmadu PT. Cervical cystic hygroma: pre-, intra-, and post-operative morbidity and mortality in Zaria, Nigeria. Pediatr Surg Int. 2001;17:342–343.
155 Alper JC, Holmes LB. The incidence and significance of birthmarks in a cohort of 4,641 newborns. Pediatr Dermatol. 1983;1:58–68.
156 Higuera S, Gordley K, Metry DW, Stal S. Management of hemangiomas and pediatric vascular malformations. J Craniofac Surg. 2006;17:783–789.
157 Orlow SJ, Isakoff MS, Blei F. Increased risk of symptomatic hemangiomas of the airway in association with cutaneous hemangiomas in a “beard” distribution. J Pediatr. 1997;131:643–646.
158 Leikensohn JR, Benton C, Cotton R. Subglottic hemangioma. J Otolaryngol. 1976;5:487–492.
159 Perkins JA, Oliaei S, Garrison MM, Manning SC, Christakis DA. Airway procedures and hemangiomas: treatment patterns and outcome in U.S. pediatric hospitals. Int J Pediatr Otorhinolaryngol. 2009;73:1302–1307.
160 Leaute-Labreze C, Dumas DLR, Hubiche T, et al. Propranolol for severe hemangiomas of infancy. N Engl J Med. 2008;358:2649–2651.
161 Truong MT, Perkins JA, Messner AH, Chang KW. Propranolol for the treatment of airway hemangiomas: a case series and treatment algorithm. Int J Pediatr Otorhinolaryngol. 2010;74:1043–1048.
162 Isago T, Kono T, Nozaki M, et al. Ambulatory anesthesia for children undergoing laser treatment. Surg Today. 2006;36:765–768.
163 Kloiber R, Poon MC, Molnar CP. Platelet sequestration in a vascular malformation of Klippel-Trenaunay syndrome. AJR Am J Roentgenol. 1987;149:1275–1277.
164 Abramson DL, Cohen MM, Jr., Mulliken JB. Möbius syndrome: classification and grading system. Plast Reconstr Surg. 1998;102:961–967.
165 Carr MM, Ross DA, Zuker RM. Cranial nerve defects in congenital facial palsy. J Otolaryngol. 1997;26:80–87.
166 Cronemberger MF, de Castro Moreira JB, Brunoni D, et al. Ocular and clinical manifestations of Möbius’ syndrome. J Pediatr Ophthalmol Strabismus. 2001;38:156–162.
167 Kumar D. Moebius syndrome. J Med Genet. 1990;27:122–126.
168 Terzis JK, Noah EM. Möbius and Möbius-like patients: etiology, diagnosis, and treatment options. Clin Plast Surg. 2002;29:497–514.
169 Ferguson S. Moebius syndrome: a review of the anaesthetic implications. Paediatr Anaesth. 1996;6:51–56.
170 Igarashi M, Rose DF, Storgion SA. Moebius syndrome and central respiratory dysfunction. Pediatr Neurol. 1997;16:237–240.
171 Gondipalli P, Tobias JD. Anesthetic implications of Möbius syndrome. J Clin Anesth. 2006;18:55–59.
172 Zuker RM, Goldberg CS, Manktelow RT. Facial animation in children with Möbius syndrome after segmental gracilis muscle transplant. Plast Reconstr Surg. 2000;106:1–8.
173 Valnicek SM, Clarke HM. Syngnathia: a report of two cases. Cleft Palate Craniofac J. 1993;30:582–585.
174 La Scala GC, Rice SB, Clarke HM. Complications of microsurgical reconstruction of obstetrical brachial plexus palsy. Plast Reconstr Surg. 2003;111:1383–1388.
175 Andersen J, Watt J, Olson J, Van AJ. Perinatal brachial plexus palsy. Paediatr Child Health. 2006;11:93–100.
176 Shenaq SM, Berzin E, Lee R, et al. Brachial plexus birth injuries and current management. Clin Plast Surg. 1998;25:527–536.
177 Chuang DC, Mardini S, Ma HS. Surgical strategy for infant obstetrical brachial plexus palsy: experiences at Chang Gung Memorial Hospital. Plast Reconstr Surg. 2005;116:132–142.
178 Waters PM. Comparison of the natural history, the outcome of microsurgical repair, and the outcome of operative reconstruction in brachial plexus birth palsy. J Bone Joint Surg Am. 1999;81:649–659.
179 Kawabata H, Masada K, Tsuyuguchi Y, et al. Early microsurgical reconstruction in birth palsy. Clin Orthop Relat Res. 1987:233–242.
180 Laurent JP, Lee RT. Birth-related upper brachial plexus injuries in infants: operative and nonoperative approaches. J Child Neurol. 1994;9:111–117.
181 Laurent JP, Lee R, Shenaq S, et al. Neurosurgical correction of upper brachial plexus birth injuries. J Neurosurg. 1993;79:197–203.
182 Hale HB, Bae DS, Waters PM. Current concepts in the management of brachial plexus birth palsy. J Hand Surg Am. 2010;35:322–331.
183 Capek L, Clarke HM, Curtis CG. Neuroma-in-continuity resection: early outcome in obstetrical brachial plexus palsy. Plast Reconstr Surg. 1998;102:1555–1562.
184 Dodds SD, Wolfe SW. Perinatal brachial plexus palsy. Curr Opin Pediatr. 2000;12:40–47.
185 Hazama A, Kinouchi K, Kitamura S, Fukumitsu K. Brachial plexus birth injuries: anaesthesia for surgical nerve reconstruction and preoperative myelography and computed tomographic myelography. Paediatr Anaesth. 1999;9:403–407.
186 Adamson PA, Strecker HD. Otoplasty techniques. Facial Plast Surg. 1995;11:284–300.
187 Songu M, Adibelli H. Otoplasty in children younger than 5 years of age. Int J Pediatr Otorhinolaryngol. 2010;74:292–296.
188 Caouette-Laberge L, Guay N, Bortoluzzi P, Belleville C. Otoplasty: anterior scoring technique and results in 500 cases. Plast Reconstr Surg. 2000;105:504–515.
189 Ragab A. Carbon dioxide laser-assisted cartilage reshaping otoplasty: a new technique for prominent ears. Laryngoscope. 2010;120:1312–1318.
190 Ridings P, Gault D, Khan L. Reduction in postoperative vomiting after surgical correction of prominent ears. Br J Anaesth. 1994;72:592–593.
191 Woodward WM, Barker I, John RE, Peacock JE. Propofol infusion vs thiopentone/isoflurane anaesthesia for prominent ear correction in children. Pediatr Anaesth. 1997;7:379–383.
192 Engelhardt T, Johnston G, Kumar MM. Comparison of cuffed, uncuffed tracheal tubes and laryngeal mask airways in low flow pressure controlled ventilation in children. Paediatr Anaesth. 2006;16:140–143.
193 Cregg N, Conway F, Casey W. Analgesia after otoplasty: regional nerve blockade vs local anaesthetic infiltration of the ear. Can J Anaesth. 1996;43:141–147.
194 Chong AK. Common congenital hand conditions. Singapore Med J. 2010;51:965–971.
195 Dao KD, Shin AY, Billings A, Oberg KC, Wood VE. Surgical treatment of congenital syndactyly of the hand. J Am Acad Orthop Surg. 2004;12:39–48.
196 Tonkin MA. Failure of differentiation. I. Syndactyly. Hand Clin. 2009;25:171–193.
197 Joseph-Reynolds AM, Auden SM, Sobczyzk WL. Perioperative considerations in a newly described subtype of congenital long QT syndrome. Paediatr Anaesth. 1997;7:237–241.
198 U.S. National Library of Medicine. Genetics Home Reference. Timothy syndrome. http://ghr.nlm.nih.gov/condition/timothy-syndrome, 2012. Available at (accessed Oct. 20, 2012)
199 Ashbaugh H, Gellman H. Congenital thumb deformities and associated syndromes. J Craniofac Surg. 2009;20:1039–1044.
200 Arnell K. Primary and secondary tissue expansion gives high quality skin and subcutaneous coverage in children with a large myelomeningocele and kyphosis. Acta Neurochir (Wien). 2006;148:293–297.
201 Bax NM, van DZ, Pull ter Gunne AJ, Rovekamp MH. Treatment of giant omphalocele by enlargement of the abdominal cavity with a tissue expander. J Pediatr Surg. 1993;28:1181–1184.
202 Farzaneh FC, Kaldari S, Becker M, Wikstrom SO. Tissue expansion 1984-1999: a 15-year review. Scand J Plast Reconstr Surg Hand Surg. 2006;40:89–92.
203 Foglia R, Kane A, Becker D, Asz-Sigall J, Mychaliska G. Management of giant omphalocele with rapid creation of abdominal domain. J Pediatr Surg. 2006;41:704–709.
204 Hudson DA, Arasteh E. Serial tissue expansion for reconstruction of burns of the head and neck. Burns. 2001;27:481–487.
205 Neale HW, Kurtzman LC, Goh KB, et al. Tissue expanders in the lower face and anterior neck in pediatric burn patients: limitations and pitfalls. Plast Reconstr Surg. 1993;91:624–631.
206 Paletta CE, Huang DB, Dehghan K, Kelly C. The use of tissue expanders in staged abdominal wall reconstruction. Ann Plast Surg. 1999;42:259–265.
207 Pisarski GP, Mertens D, Warden GD, Neale HW. Tissue expander complications in the pediatric burn patient. Plast Reconstr Surg. 1998;102:1008–1012.
208 Spence RJ. Experience with novel uses of tissue expanders in burn reconstruction of the face and neck. Ann Plast Surg. 1992;28:453–464.
209 Lindzon G, Zuker RM. Congenital melanocytic nevi. J Craniofac Surg. 2003;14:876–879.
210 MacLennan SE, Corcoran JF, Neale HW. Tissue expansion in head and neck burn reconstruction. Clin Plast Surg. 2000;27:121–132.
211 Chadha IA, Multani ST, Chadha A. Tissue expander causing iatrogenic difficult intubation. Anaesthesia. 1996;51:1160–1161.
212 Casanova D, Bali D, Bardot J, Legre R, Magalon G. Tissue expansion of the lower limb: complications in a cohort of 103 cases. Br J Plast Surg. 2001;54:310–316.
213 Gibstein LA, Abramson DL, Bartlett RA, et al. Tissue expansion in children: a retrospective study of complications. Ann Plast Surg. 1997;38:358–364.
214 Hurvitz KA, Rosen H, Meara JG. Pediatric cervicofacial tissue expansion. Int J Pediatr Otorhinolaryngol. 2005;69:1509–1513.
215 Iconomou TG, Michelow BJ, Zuker RM. Tissue expansion in the pediatric patient. Ann Plast Surg. 1993;31:134–140.
216 Breuninger H, Wehner-Caroli J. Slow infusion tumescent anesthesia. Dermatol Surg. 1998;24:759–763.
217 Moehrle M, Breuninger H. Dermatosurgery using subcutaneous infusion anesthesia with prilocaine and ropivacaine in children. Pediatr Dermatol. 2001;18:469–472.
218 American Society of Plastic Surgeons, 2011 plastic surgery statistics report. Available at http://www.plasticsurgery.org/News-and-Resources/2011-Statistics-.html accessed Oct. 20, 2012
219 Zuckerman D, Abraham A. Teenagers and cosmetic surgery: focus on breast augmentation and liposuction. J Adolesc Health. 2008;43:318–324.
220 Iverson RE, Lynch DJ. Practice advisory on liposuction. Plast Reconstr Surg. 2004;113:1478–1490.
221 Bar-Meir E, Zaslansky R, Regev E, et al. Nitrous oxide administered by the plastic surgeon for repair of facial lacerations in children in the emergency room. Plast Reconstr Surg. 2006;117:1571–1575.
222 Antevy PM, Zuckerbraun NS, Saladino RA, Pitetti RD. Evaluation of a transthecal digital nerve block in the injured pediatric patient. Pediatr Emerg Care. 2010;26:177–180.
223 Kurt E, Ozturk S, Isik S, Zor F. Continuous brachial plexus blockade for digital replantations and toe-to-hand transfers. Ann Plast Surg. 2005;54:24–27.