135e |
Less Common Hematologic Malignancies |
The most common lymphoid malignancies are discussed in Chap. 134, myeloid leukemias in Chaps. 132 and 133, myelodysplastic syndromes in Chap. 130, and myeloproliferative syndromes in Chap. 131. This chapter will focus on the more unusual forms of hematologic malignancy. The diseases discussed here are listed in Table 135e-1. Each of these entities accounts for less than 1% of hematologic neoplasms.
|
UNUSUAL LYMPHOID AND MYELOID MALIGNANCIES |
LYMPHOID MALIGNANCIES
Precursor B-cell and precursor T-cell neoplasms are discussed in Chap. 134. All the lymphoid tumors discussed here are mature B cell or T cell, natural killer (NK) cell neoplasms.
MATURE B-CELL NEOPLASMS
B-Cell Prolymphocytic Leukemia (B-PLL) This is a malignancy of medium-sized (about twice the size of a normal small lymphocyte), round lymphocytes with a prominent nucleolus and light blue cytoplasm on Wright’s stain. It dominantly affects the blood, bone marrow, and spleen and usually does not cause adenopathy. The median age of affected patients is 70 years, and men are more often affected than women (male-to-female ratio is 1.6). This entity is distinct from chronic lymphoid leukemia (CLL) and does not develop as a consequence of that disease.
Clinical presentation is generally from symptoms of splenomegaly or incidental detection of an elevated white blood cell (WBC) count. The clinical course can be rapid. The cells express surface IgM (with or without IgD) and typical B-cell markers (CD19, CD20, CD22). CD23 is absent, and about one-third of cases express CD5. The CD5 expression along with the presence of the t(11;14) translocation in 20% of cases leads to confusion in distinguishing B-PLL from the leukemic form of mantle cell lymphoma. No reliable criteria for the distinction have emerged. About half of patients have mutation or loss of p53, and deletions have been noted in 11q23 and 13q14. Nucleoside analogues like fludarabine and cladribine and combination chemotherapy (cyclophosphamide, doxorubicin, vincristine, and prednisone [CHOP]) have produced responses. CHOP plus rituximab may be more effective than CHOP alone, but the disease is sufficiently rare that large series have not been reported. Splenectomy can produce palliation of symptoms but appears to have little or no impact on the course of the disease.
Splenic Marginal Zone Lymphoma (SMZL) This tumor of mainly small lymphocytes originates in the marginal zone of the spleen white pulp, grows to efface the germinal centers and mantle, and invades the red pulp. Splenic hilar nodes, bone marrow, and peripheral blood may be involved. The circulating tumor cells have short surface villi and are called villous lymphocytes. Table 135e-2 shows differences in tumor cells of a number of neoplasms of small lymphocytes that aid in the differential diagnosis. SMZL cells express surface immunoglobulin and CD20, but are negative for CD5, CD10, CD43, and CD103. Lack of CD5 distinguishes SMZL from CLL, and lack of CD103 separates SMZL from hairy cell leukemia.
|
IMMUNOPHENOTYPE OF TUMORS OF SMALL LYMPHOCYTES |
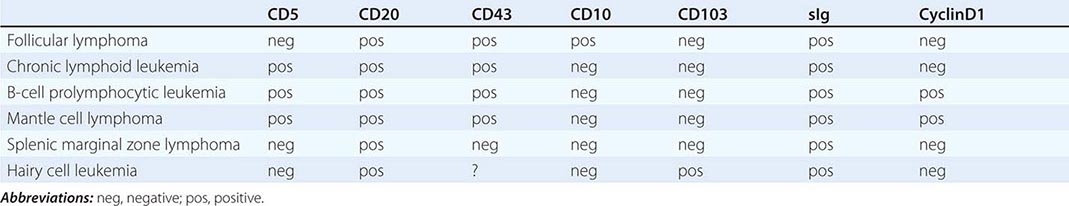
The median age of patients with SMZL is mid-fifties, and men and women are equally represented. Patients present with incidental or symptomatic splenomegaly or incidental detection of lymphocytosis in the peripheral blood with villous lymphocytes. Autoimmune anemia or thrombocytopenia may be present. The immunoglobulin produced by these cells contains somatic mutations that reflect transit through a germinal center, and ongoing mutations suggest that the mutation machinery has remained active. About 40% of patients have either deletions or translocations involving 7q21, the site of the FLNC gene (filamin Cγ, involved in cross-linking actin filaments in the cytoplasm). NOTCH2 mutations are seen in 25% of patients. Chromosome 8p deletions may also be noted. The genetic lesions typically found in extranodal marginal zone lymphomas [e.g., trisomy 3 and t(11;18)] are uncommon in SMZL.
The clinical course of disease is generally indolent with median survivals exceeding 10 years. Patients with elevated lactate dehydrogenase (LDH) levels, anemia, and hypoalbuminemia generally have a poorer prognosis. Long remissions can be seen after splenectomy. Rituximab is also active. A small fraction of patients undergo histologic progression to diffuse large B-cell lymphoma with a concomitant change to a more aggressive natural history. Experience with combination chemotherapy in SMZL is limited.
Hairy Cell Leukemia Hairy cell leukemia is a tumor of small lymphocytes with oval nuclei, abundant cytoplasm, and distinctive membrane projections (hairy cells). Patients have splenomegaly and diffuse bone marrow involvement. While some circulating cells are noted, the clinical picture is dominated by symptoms from the enlarged spleen and pancytopenia. The mechanism of the pancytopenia is not completely clear and may be mediated by both inhibitory cytokines and marrow replacement. The marrow has an increased level of reticulin fibers; indeed, hairy cell leukemia is a common cause of inability to aspirate bone marrow or so-called “dry tap” (Table 135e-3). Monocytopenia is profound and may explain a predisposition to atypical mycobacterial infection that is observed clinically. The tumor cells have strong expression of CD22, CD25, and CD103; soluble CD25 level in serum is an excellent tumor marker for disease activity. The cells also express tartrate-resistant acid phosphatase. The immunoglobulin genes are rearranged and mutated, indicating the influence of a germinal center. No specific cytogenetic abnormality has been found, but most cases contain the activating BRAF mutation V600E.
|
DIFFERENTIAL DIAGNOSIS OF “DRY TAP”–INABILITY TO ASPIRATE BONE MARROW |

The median age of affected patients is mid-fifties, and the male-to-female ratio is 5:1. Treatment options are numerous. Splenectomy is often associated with prolonged remission. Nucleosides including cladribine and deoxycoformycin are highly active but are also associated with further immunosuppression and can increase the risk of certain opportunistic infections. However, after brief courses of these agents, patients usually obtain very durable remissions during which immune function spontaneously recovers. Interferon α is also an effective therapy but is not as effective as nucleosides. Chemotherapy-refractory patients have responded to vemurafenib, a BRAF inhibitor.
Nodal Marginal Zone B-Cell Lymphoma This rare node-based disease bears an uncertain relationship with extranodal marginal zone lymphomas, which are often mucosa-associated and are called mucosa-associated lymphoid tissue (MALT) lymphomas, and SMZLs. Patients may have localized or generalized adenopathy. The neoplastic cell is a marginal zone B cell with monocytoid features and has been called monocytoid B-cell lymphoma in the past. Up to one-third of the patients may have extranodal involvement, and involvement of the lymph nodes can be secondary to the spread of a mucosal primary lesion. In authentic nodal primaries, the cytogenetic abnormalities associated with MALT lymphomas [trisomy 3 and t(11;18)] are very rare. The clinical course is indolent. Patients often respond to combination chemotherapy, although remissions have not been durable. Few patients have received CHOP plus rituximab, which is likely to be an effective approach to management.
Mediastinal (Thymic) Large B-Cell Lymphoma This entity was originally considered a subset of diffuse large B-cell lymphoma; however, additional study has identified it as a distinct entity with its own characteristic clinical, genetic, and immunophenotypic features. This is a disease that can be bulky in size but usually remains confined to the mediastinum. It can be locally aggressive, including progressing to produce a superior vena cava obstruction syndrome or pericardial effusion. About one-third of patients develop pleural effusions, and 5–10% can disseminate widely to kidney, adrenal, liver, skin, and even brain. The disease affects women more often than men (male-to-female ratio is 1:2–3), and the median age is 35–40 years.
The tumor is composed of sheets of large cells with abundant cytoplasm accompanied by variable, but often abundant, fibrosis. It is distinguished from nodular sclerosing Hodgkin’s disease by the paucity of normal lymphoid cells and the absence of lacunar variants of Reed-Sternberg cells. However, more than one-third of the genes that are expressed to a greater extent in primary mediastinal large B-cell lymphoma than in usual diffuse large B-cell lymphoma are also overexpressed in Hodgkin’s disease, suggesting a possible pathogenetic relationship between the two entities that affect the same anatomic site. Tumor cells may overexpress MAL. The genome of tumor cells is characterized by frequent chromosomal gains and losses. The tumor cells in mediastinal large B-cell lymphoma express CD20, but surface immunoglobulin and HLA class I and class II molecules may be absent or incompletely expressed. Expression of lower levels of class II HLA identifies a subset with poorer prognosis. The cells are CD5 and CD10 negative but may show light staining with anti-CD30. The cells are CD45 positive, unlike cells of classical Hodgkin’s disease.
Methotrexate, leucovorin, doxorubicin, cyclophosphamide, vincristine, prednisone, and bleomycin (MACOP-B) and rituximab plus CHOP are effective treatments, achieving 5-year survival of 75–87%. Dose-adjusted therapy with prednisone, etoposide, vincristine, cyclophosphamide, and doxorubicin (EPOCH) plus rituximab has produced 5-year survival of 97%. A role for mediastinal radiation therapy has not been definitively demonstrated, but it is frequently used, especially in patients whose mediastinal area remains positron emission tomography–avid after four to six cycles of chemotherapy.
Intravascular Large B-Cell Lymphoma This is an extremely rare form of diffuse large B-cell lymphoma characterized by the presence of lymphoma in the lumen of small vessels, particularly capillaries. It is also known as malignant angioendotheliomatosis or angiotropic large cell lymphoma. It is sufficiently rare that no consistent picture has emerged to define a clinical syndrome or its epidemiologic and genetic features. It is thought to remain inside vessels because of a defect in adhesion molecules and homing mechanisms, an idea supported by scant data suggesting absence of expression of β-1 integrin and ICAM-1. Patients commonly present with symptoms of small-vessel occlusion, skin lesions, or neurologic symptoms. The tumor cell clusters can promote thrombus formation. In general, the clinical course is aggressive and the disease is poorly responsive to therapy. Often a diagnosis is not made until very late in the course of the disease.
Primary Effusion Lymphoma This entity is another variant of diffuse large B-cell lymphoma that presents with pleural effusions, usually without apparent tumor mass lesions. It is most common in the setting of immune deficiency disease, especially AIDS, and is caused by human herpes virus 8 (HHV-8)/Kaposi’s sarcoma herpes virus (KSHV). It is also known as body cavity–based lymphoma. Some patients have been previously diagnosed with Kaposi’s sarcoma. It can also occur in the absence of immunodeficiency in elderly men of Mediterranean heritage, similar to Kaposi’s sarcoma but even less common.
The malignant effusions contain cells positive for HHV-8/KSHV, and many are also co-infected with Epstein-Barr virus. The cells are large with large nuclei and prominent nucleoli that can be confused with Reed-Sternberg cells. The cells express CD20 and CD79a (immunoglobulin-signaling molecule), although they often do not express immunoglobulin. Some cases aberrantly express T-cell markers such as CD3 or rearranged T-cell receptor genes. No characteristic genetic lesions have been reported, but gains in chromosome 12 and × material has been seen, similar to other HIV-associated lymphomas. The clinical course is generally characterized by rapid progression and death within 6 months.
Lymphomatoid Granulomatosis This is an angiocentric, angiodestructive lymphoproliferative disease comprised by neoplastic Epstein-Barr virus–infected monoclonal B cells accompanied and outnumbered by a polyclonal reactive T-cell infiltrate. The disease is graded based on histologic features such as cell number and atypia in the B cells. It is most often confused with extranodal NK–T cell lymphoma, nasal type, which can also be angiodestructive and is Epstein-Barr virus–related. The disease usually presents in adults (males > females) as a pulmonary infiltrate. Involvement is often entirely extranodal and can include kidney (32%), liver (29%), skin (25%), and brain (25%). The disease often but not always occurs in the setting of immune deficiency.
The disease can be remitting and relapsing in nature or can be rapidly progressive. The course is usually predicted by the histologic grade. The disease is highly responsive to combination chemotherapy and is curable in most cases. Some investigators have claimed that low-grade disease (grade I and II) can be treated with interferon α.
MATURE T-CELL AND NK CELL NEOPLASMS
T-Cell Prolymphocytic Leukemia This is an aggressive leukemia of medium-sized prolymphocytes involving the blood, marrow, nodes, liver, spleen, and skin. It accounts for 1–2% of all small lymphocytic leukemias. Most patients present with elevated WBC count (often >100,000/μL), hepatosplenomegaly, and adenopathy. Skin involvement occurs in 20%. The diagnosis is made from peripheral blood smear, which shows cells about 25% larger than those in small lymphocytes, with cytoplasmic blebs and nuclei that may be indented. The cells express T-cell markers like CD2, CD3, and CD7; two-thirds of patients have cells that are CD4+ and CD8–, and 25% have cells that are CD4+ and CD8+. T-cell receptor β chains are clonally rearranged. In 80% of patients, inversion of chromosome 14 occurs between q11 and q32. Ten percent have t(14;14) translocations that bring the T-cell receptor alpha/beta gene locus into juxtaposition with oncogenes TCL1 and TCL1b at 14q32.1. Chromosome 8 abnormalities are also common. Deletions in the ATM gene are also noted. Activating JAK3 mutations have also been reported.
The course of the disease is generally rapid, with median survival of about 12 months. Responses have been seen with the anti-CD52 antibody, nucleoside analogs, and CHOP chemotherapy. Small numbers of patients with T-cell prolymphocytic leukemia have also been treated with high-dose therapy and allogeneic bone marrow transplantation after remission has been achieved with conventional-dose therapy.
T-Cell Large Granular Lymphocytic Leukemia T-cell large granular lymphocytic leukemia (LGL leukemia) is characterized by increases in the number of LGLs in the peripheral blood (2000–20,000/μL) often accompanied by severe neutropenia, with or without concomitant anemia. Patients may have splenomegaly and frequently have evidence of systemic autoimmune disease, including rheumatoid arthritis, hypergammaglobulinemia, autoantibodies, and circulating immune complexes. Bone marrow involvement is mainly interstitial in pattern, with fewer than 50% lymphocytes on differential count. Usually the cells express CD3, T-cell receptors, and CD8; NK-like variants may be CD3–. The leukemic cells often express Fas and Fas ligand.
The course of the disease is generally indolent and dominated by the neutropenia. Paradoxically, immunosuppressive therapy with cyclosporine, methotrexate, or cyclophosphamide plus glucocorticoids can produce an increase in granulocyte counts. Nucleosides have been used anecdotally. Occasionally the disease can accelerate to a more aggressive clinical course.
Aggressive NK Cell Leukemia NK neoplasms are very rare, and they may follow a range of clinical courses from very indolent to highly aggressive. They are more common in Asians than whites, and the cells frequently harbor a clonal Epstein-Barr virus episome. The peripheral blood white count is usually not greatly elevated, but abnormal large lymphoid cells with granular cytoplasm are noted. The aggressive form is characterized by symptoms of fever and laboratory abnormalities of pancytopenia. Hepatosplenomegaly is common; node involvement is less common. Patients may have hemophagocytosis, coagulopathy, or multiorgan failure. Serum levels of Fas ligand are elevated.
The cells express CD2 and CD56 and do not have rearranged T-cell receptor genes. Deletions involving chromosome 6 are common. The disease can be rapidly progressive. Some forms of NK neoplasms are more indolent. They tend to be discovered incidentally with LGL lymphocytosis and do not manifest the fever and hepatosplenomegaly characteristic of the aggressive leukemia. The cells are also CD2 and CD56 positive, but they do not contain clonal forms of Epstein-Barr virus and are not accompanied by pancytopenia or autoimmune disease.
Extranodal NK/T-Cell Lymphoma, Nasal Type Like lymphomatoid granulomatosis, extranodal NK/T-cell lymphoma tends to be an angiocentric and angiodestructive lesion, but the malignant cells are not B cells. In most cases, they are CD56+ Epstein-Barr virus–infected cells; occasionally they are CD56– Epstein-Barr virus–infected cytotoxic T cells. They are most commonly found in the nasal cavity. Historically, this illness was called lethal midline granuloma, polymorphic reticulosis, and angiocentric immunoproliferative lesion. This form of lymphoma is prevalent in Asia, Mexico, and Central and South America; it affects males more commonly than females. When it spreads beyond the nasal cavity, it may affect soft tissue, the gastrointestinal tract, or the testis. In some cases, hemophagocytic syndrome may influence the clinical picture. Patients may have B symptoms. Many of the systemic manifestations of disease are related to the production of cytokines by the tumor cells and the cells responding to their signals. Deletions and inversions of chromosome 6 are common.
Many patients with extranodal NK/T-cell lymphoma, nasal type have excellent antitumor responses with combination chemotherapy regimens, particularly those with localized disease. Radiation therapy is often used after completion of chemotherapy. Four risk factors have been defined, including B symptoms, advanced stage, elevated LDH, and regional lymph node involvement. Patient survival is linked to the number of risk factors: 5-year survival is 81% for zero risk factors, 64% for one risk factor, 32% for two risk factors, and 7% for three or four risk factors. Combination regimens without anthracyclines have been touted as superior to CHOP, but data are sparse. High-dose therapy with stem cell transplantation has been used, but its role is unclear.
Enteropathy-Type T-Cell Lymphoma Enteropathy-type T-cell lymphoma is a rare complication of longstanding celiac disease. It most commonly occurs in the jejunum or the ileum. In adults, the lymphoma may be diagnosed at the same time as celiac disease, but the suspicion is that the celiac disease was a longstanding precursor to the development of lymphoma. The tumor usually presents as multiple ulcerating mucosal masses, but may also produce a dominant exophytic mass or multiple ulcerations. The tumor expresses CD3 and CD7 nearly always and may or may not express CD8. The normal-appearing lymphocytes in the adjacent mucosa often have a similar phenotype to the tumor. Most patients have the HLA genotype associated with celiac disease, HLA DQA1*0501 or DQB1*0201.
The prognosis of this form of lymphoma is typically (median survival is 7 months) poor, but some patients have a good response to CHOP chemotherapy. Patients who respond can develop bowel perforation from responding tumor. If the tumor responds to treatment, recurrence may develop elsewhere in the celiac disease–affected small bowel.
Hepatosplenic T-Cell Lymphoma Hepatosplenic T-cell lymphoma is a malignancy derived from T cells expressing the gamma/delta T-cell antigen receptor that affects mainly the liver and fills the sinusoids with medium-size lymphoid cells. When the spleen is involved, dominantly the red pulp is infiltrated. It is a disease of young people, especially young people with an underlying immunodeficiency or with an autoimmune disease that demands immunosuppressive therapy. The use of thiopurine and infliximab is particularly common in the history of patients with this disease. The cells are CD3+ and usually CD4– and CD8–. The cells may contain isochromosome 7q, often together with trisomy 8. The lymphoma has an aggressive natural history. Combination chemotherapy may induce remissions, but most patients relapse. Median survival is about 2 years. The tumor does not appear to respond to reversal of immunosuppressive therapy.
Subcutaneous Panniculitis-Like T-Cell Lymphoma Subcutaneous panniculitis-like T-cell lymphoma involves multiple subcutaneous collections of neoplastic T cells that are usually cytotoxic cells in phenotype (i.e., contain perforin and granzyme B and express CD3 and CD8). The rearranged T-cell receptor is usually alpha/beta-derived, but occasionally the gamma/delta receptors are involved, particularly in the setting of immunosuppression. The cells are negative for Epstein-Barr virus. Patients may have a hemophagocytic syndrome in addition to the skin infiltration; fever and hepatosplenomegaly may also be present. Nodes are generally not involved. Patients frequently respond to combination chemotherapy, including CHOP. When the disease is progressive, the hemophagocytic syndrome can be a component of a fulminant downhill course. Effective therapy can reverse the hemophagocytic syndrome.
Blastic NK Cell Lymphoma The neoplastic cells express NK cell markers, especially CD56, and are CD3 negative. They are large blastic-appearing cells and may produce a leukemia picture, but the dominant site of involvement is the skin. Morphologically, the cells are similar to the neoplastic cells in acute lymphoid and myeloid leukemia. No characteristic chromosomal abnormalities have been described. The clinical course is rapid, and the disease is largely unresponsive to typical lymphoma treatments.
Primary Cutaneous CD30+ T-Cell Lymphoma This tumor involves the skin and is composed of cells that appear similar to the cells of anaplastic T-cell lymphoma. Among cutaneous T-cell tumors, about 25% are CD30+ anaplastic lymphomas. If dissemination to lymph nodes occurs, it is difficult to distinguish between the cutaneous and systemic forms of the disease. The tumor cells are often CD4+, and the cells contain granules that are positive for granzyme B and perforin in 70% of cases. The typical t(2;5) of anaplastic T-cell lymphoma is absent; indeed, its presence should prompt a closer look for systemic involvement and a switch to a diagnosis of anaplastic T-cell lymphoma. This form of lymphoma has sporadically been noted as a rare complication of silicone on saline breast implants. Cutaneous CD30+ T-cell lymphoma often responds to therapy. Radiation therapy can be effective, and surgery can also produce long-term disease control. Five-year survival exceeds 90%.
Angioimmunoblastic T-Cell Lymphoma Angioimmunoblastic T-cell lymphoma is a systemic disease that accounts for about 15% of all T-cell lymphomas. Patients frequently have fever, advanced stage, diffuse adenopathy, hepatosplenomegaly, skin rash, polyclonal hypergammaglobulinemia, and a wide range of autoantibodies including cold agglutinins, rheumatoid factor, and circulating immune complexes. Patients may have edema, arthritis, pleural effusions, and ascites. The nodes contain a polymorphous infiltrate of neoplastic T cells and nonneoplastic inflammatory cells together with proliferation of high endothelial venules and follicular dendritic cells. The most common chromosomal abnormalities are trisomy 3, trisomy 5, and an extra × chromosome. Aggressive combination chemotherapy can induce regressions. The underlying immune defects make conventional lymphoma treatments more likely to produce infectious complications.
MYELOID MALIGNANCIES
The World Health Organization (WHO) system uses peripheral blood counts and smear analysis, bone marrow morphology, and cytogenetic and molecular genetic tests in order to classify myeloid malignancies into five major categories (Table 135e-4). In this chapter, we focus on chronic neutrophilic leukemia; atypical chronic myeloid leukemia, BCR-ABL1 negative; chronic myelomonocytic leukemia; juvenile myelomonocytic leukemia; chronic eosinophilic leukemia, not otherwise specified; mastocytosis; myeloproliferative neoplasm (MPN), unclassifiable (MPN-U); myelodysplastic syndrome (MDS)/MPN, unclassifiable (MDS/MPN-U); refractory anemia with ring sideroblasts associated with marked thrombocytosis (RARS-T); and myeloid and lymphoid neoplasms with eosinophilia and abnormalities of PDGFRA, PDGFRB, or FGFR1. This chapter also includes histiocytic and dendritic cell neoplasms, transient myeloproliferative disorders, and a broader discussion on primary eosinophilic disorders including hypereosinophilic syndrome (HES).
|
WORLD HEALTH ORGANIZATION CLASSIFICATION OF MYELOID MALIGNANCIES |
aAML-related precursor neoplasms include therapy-related MDS and myeloid sarcoma.
bEither monocytopenia or bicytopenia: hemoglobin level <10 g/dL, absolute neutrophil count <1.8 × 109/L, or platelet count <100 × 109/L. However, higher blood counts do not exclude the diagnosis in the presence of unequivocal histologic/cytogenetic evidence for MDS. cGenetic rearrangements involving platelet-derived growth factor receptor α/β (PDGFRA/PDGFRB) or fibroblast growth factor receptor 1 (FGFR1).
CHRONIC NEUTROPHILIC LEUKEMIA
Chronic neutrophilic leukemia (CNL) is characterized by mature neutrophilic leukocytosis with few or no circulating immature granulocytes. CNL is associated with activating mutations of the gene (CSF3R) encoding for the receptor for granulocyte colony-stimulating factor (G-CSF), also known as colony-stimulating factor 3 (CSF3). Patients with CNL might be asymptomatic at presentation but also display constitutional symptoms, splenomegaly, anemia, and thrombocytopenia. Median survival is approximately 2 years, and causes of death include leukemic transformation, progressive disease associated with severe cytopenias, and marked treatment-refractory leukocytosis. CNL is rare, with less than 200 reported cases. Median age at diagnosis is approximately 67 years, and the disease is equally prevalent in both genders.
Pathogenesis CSF3 is the main growth factor for granulocyte proliferation and differentiation. Accordingly, recombinant CSF3 is used for the treatment of severe neutropenia, including severe congenital neutropenia (SCN). Some patients with SCN acquire CSF3R mutations, and the frequency of such mutations is significantly higher (~80%) in patients who experience leukemic transformation. SCN-associated CSF3R mutations occur in the region of the gene coding for the cytoplasmic domain of CSF3R and result in truncation of the C-terminal-negative regulatory domain. A different class of CSF3R mutations is noted in ~90% of patients with CNL; these are mostly membrane proximal, with the most frequent being a C-to-T substitution at nucleotide 1853 (T618I). About 40% of the T618I-mutated cases also harbored SETBP1 mutations. CSF3R T618I induces a lethal myeloproliferative disorder in a mouse model and is associated with in vitro sensitivity to JAK inhibition.
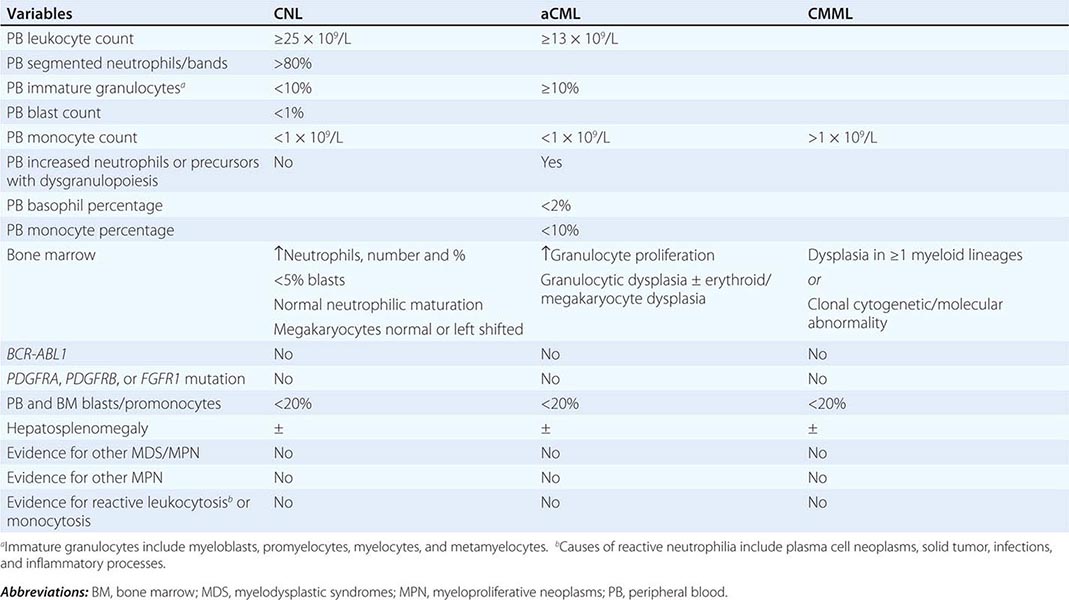
Diagnosis Diagnosis of CNL requires exclusion of the more common causes of neutrophilia including infections and inflammatory processes. In addition, one should be mindful of the association between some forms of metastatic cancer or plasma cell neoplasms with secondary neutrophilia. Neoplastic neutrophilia also occurs in other myeloid malignancies including atypical chronic myeloid leukemia and chronic myelomonocytic leukemia. Accordingly, the WHO diagnostic criteria for CNL are designed to exclude the possibilities of both secondary/reactive neutrophilia and leukocytosis associated with myeloid malignancies other than CNL (Table 135e-5): leukocytosis (≥25 × 109/L), >80% segmented/band neutrophils, <10% immature myeloid cells, <1% circulating blasts, and absence of dysgranulopoiesis or monocytosis. Bone marrow in CNL is hypercellular and displays increased number and percentage of neutrophils with a very high myeloid-to-erythroid ratio and minimal left shift, myeloid dysplasia, or reticulin fibrosis.
|
WORLD HEALTH ORGANIZATION DIAGNOSTIC CRITERIA FOR CHRONIC NEUTROPHILIC LEUKEMIA (CNL); ATYPICAL CHRONIC MYELOID LEUKEMIA, BCRABL1 NEGATIVE (ACML); AND CHRONIC MYELOMONOCYTIC LEUKEMIA (CMML) |
Treatment Current treatment in CNL is largely palliative and suboptimal in its efficacy. Several drugs alone or in combination have been tried, and none have shown remarkable efficacy. As such, allogeneic stem cell transplantation (ASCT) is reasonable to consider in the presence of symptomatic disease, especially in younger patients. Otherwise, cytoreductive therapy with hydroxyurea is probably as good as any treatment, and a more intensive combination chemotherapy may not have additional value. However, response to hydroxyurea therapy is often transient, and some have successfully used interferon α as an alternative drug. Response to treatment with ruxolitinib (a JAK1 and JAK2 inhibitor) has been reported but has not been confirmed.
ATYPICAL CHRONIC MYELOID LEUKEMIA
Atypical chronic myeloid leukemia, BCR-ABL1 negative (aCML) is formally classified under the MDS/MPN category of myeloid malignancies and is characterized by left shifted granulocytosis and dysgranulopoiesis. The differential diagnosis of aCML includes chronic myeloid leukemia (CML), which is distinguished by the presence of BCR-ABL1; CNL, which is distinguished by the absence of dysgranulopoiesis and presence of CSF3R mutations; and chronic myelomonocytic leukemia, which is distinguished by the presence of monocytosis (absolute monocyte count >1 × 109/L). The WHO diagnostic criteria for aCML are listed in Table 135e-5 and include granulocytosis (WBC ≥13 × 109/L), neutrophilia with dysgranulopoiesis, ≥10% immature granulocytes, <20% peripheral blood myeloblasts, <10% peripheral blood monocytes, <2% basophils, and absence of otherwise specific mutations such as BCR-ABL1. The bone marrow is hypercellular with granulocyte proliferation and dysplasia with or without erythroid or megakaryocytic dysplasia.
The molecular pathogenesis of aCML is incompletely understood; about one-fourth of the patients express SETBP1 mutations, which are, however, also found in several other myeloid malignancies, including CNL and chronic myelomonocytic leukemia. SETBP1 mutations in aCML were prognostically detrimental and mostly located between codons 858 and 871; similar mutations are seen with Schinzel-Giedion syndrome (a congenital disease with severe developmental delay and various physical stigmata including midface retraction, large forehead, and macroglossia).
In a series of 55 patients with WHO-defined aCML, median age at diagnosis was 62 years with female preponderance (57%); splenomegaly was reported in 54% of the patients, red cell transfusion requirement in 65%, abnormal karyotype in 20% (20q– and trisomy 8 being the most frequent), and leukemic transformation in 40%. Median survival was 25 months. Outcome was worse in patients with marked leukocytosis, transfusion requirement, and increased immature cells in the peripheral blood. Conventional chemotherapy is largely ineffective in the treatment of aCML. However, a favorable experience with ASCT was reported in nine patients; after a median follow-up of 55 months, the majority of the patients remained in complete remission.
CHRONIC MYELOMONOCYTIC LEUKEMIA
Chronic myelomonocytic leukemia (CMML) is classified under the WHO category of MDS/MPN and is defined by an absolute monocyte count (AMC) of >1 × 109/L in the peripheral blood. Median age at diagnosis ranges between 65 and 75 years, and there is a 2:1 male predominance. Clinical presentation is variable and depends on whether the disease presents with MDS-like or MPN-like phenotype; the former is associated with cytopenias and the latter with splenomegaly and features of myeloproliferation such as fatigue, night sweats, weight loss, and cachexia. About 20% of patients with CMML experience serositis involving the joints (arthritis), pericardium (pericarditis and pericardial effusion), pleura (pleural effusion), or peritoneum (ascites).
Pathogenesis Clonal cytogenetic abnormalities are seen in about one-third of patients with CMML and include trisomy 8 and abnormalities of chromosome 7. Almost all patients with CMML harbor somatic mutations involving epigenetic regulator genes (e.g., ASXL1, TET2), spliceosome pathway genes (e.g., SRSF2), DNA damage response genes (e.g., TP53), and tyrosine kinases/transcription factors (e.g., KRAS, NRAS, CBL, and RUNX1). However, none of these mutations are specific to CMML, and their precise pathogenetic contribution is unclear.
Diagnosis Reactive monocytosis is uncommon but has been reported in association with certain infections and inflammatory conditions. Clonal (i.e., neoplastic) monocytosis defines CMML but is also seen with juvenile myelomonocytic leukemia and acute myeloid leukemia with monocytic differentiation. The WHO diagnostic criteria for CMML are listed in Table 135e-5 and include persistent AMC >1 × 109/L, absence of BCR-ABL1, absence of the PDGFRA or PDGFRB mutations, <20% blasts and promonocytes in the peripheral blood and bone marrow, and dysplasia involving one or more myeloid lineages.
The bone marrow in CMML is hypercellular with granulocytic and monocytic proliferation. Dysplasia is often present and may involve one, two, or all myeloid lineages. On immunophenotyping, the abnormal cells often express myelomonocytic antigens such as CD13 and CD33, with variable expression of CD14, CD68, CD64, and CD163. Monocytic-derived cells are almost always positive for the cytochemical nonspecific esterases (e.g., butyrate esterase), whereas normal granulocytic precursors are positive for lysozyme and chloroacetate esterase. In CMML, it is common to have a hybrid cytochemical staining pattern with cells expressing both chloroacetate and butyrate esterases simultaneously (dual esterase staining).
Prognosis A meta-analysis showed median survival of 1.5 years in CMML. Numerous prognostic systems have attempted to better define and stratify the natural history of CMML. One of these, the Mayo prognostic model, assigns one point each to the following four independent prognostic variables: AMC >10 × 109/L, presence of circulating immature cells, hemoglobin <10 g/dL, and platelet count <100,000/mL. This model stratified patients into three risk groups: low (0 points), intermediate (1 point), and high (≥2 points), translating to median survival times of 32, 18, and 10 months, respectively.
A French study incorporated ASXL1 mutational status in 312 CMML patients. In a multivariable model, independent predictors of poor survival were WBC >15 × 109/L (3 points), ASXL1 mutations (2 points), age >65 years (2 points), platelet count <100,000/mL (2 points), and hemoglobin <10 g/dL in females and <11 g/dL in males (2 points). This model stratified patients into three groups: low (0–4 points), intermediate (5–7 points), and high risk (8–12 points), with median survival times of not reached, 38.5 months, and 14.4 months, respectively.
Treatment Current treatment consists of hydroxyurea and supportive care, including red cell transfusions and use of erythropoiesis-stimulating agents (ESAs). The value of hydroxyurea was reinforced by a randomized trial against oral etoposide. No other single or combination chemotherapy has been shown to be superior to hydroxyurea. ASCT is a viable treatment option for transplant-eligible patients with poor prognostic features. Given the MDS/MPN overlap phenotype and the presence of MDS-like genetic/methylation abnormalities in CMML, hypomethylating agents such as 5-azacitidine and decitabine have been used with limited efficacy.
JUVENILE MYELOMONOCYTIC LEUKEMIA
Juvenile myelomonocytic leukemia (JMML) is primarily a disease of early childhood and is included, along with CMML, in the MDS/MPN WHO category. Both CMML and JMML feature leukocytosis, monocytosis, and hepatosplenomegaly. Additional characteristic features in JMML include thrombocytopenia and elevated fetal hemoglobin. Myeloid progenitors in JMML display granulocyte-macrophage colony-stimulating factor (GM-CSF) hypersensitivity that has been attributed to dysregulated RAS/MAPK signaling. The latter is believed to result from mutually exclusive mutations involving RAS, PTPN11, and NF1. A third of patients with JMML that is not associated with Noonan’s syndrome carry PTPN11 mutations, whereas the incidence of NF1 in patients without neurofibromatosis type 1 and RAS mutations is approximately 15% each. Drug therapy is relatively ineffective in JMML, and the treatment of choice is ASCT, which results in a 5-year survival of approximately 50%.
MDS/MPN-U
The WHO classifies patients with morphologic and laboratory features that resemble both MDS and MPN as MDS/MPN overlap. This category includes CMML, aCML, and JMML, which have been described above. In addition, MDS/MPN includes a fourth category referred to as MDS/MPN, unclassifiable (MDS/MPN-U). Diagnosis of MDS/MPN-U requires the presence of both MDS and MPN features that are not adequate to classify patients as CMML, aCML, or JMML. MDS/MPN includes the provisional category of RARS-T.
RARS-T is classified in the MDS/MPN category because it shares dysplastic features with RARS and myeloproliferative features with essential thrombocythemia (ET). In one study, 111 patients with RARS-T were compared with 33 patients with RARS. The frequency of SF3B1 mutations in RARS-T (87%) was similar to that in RARS (85%). JAK2 V617F mutation was detected in 49% of RARS-T patients (including 48% of those mutated for SF3B1) but none of those with RARS. In RARS-T, SF3B1 mutations were more frequent in females (95%) than in males (77%), and mean ring sideroblast counts were higher in SF3B1-mutated patients. Median overall survival was 6.9 years in SF3B1-mutated patients versus 3.3 years in unmutated patients. Six-year survival was 67% in JAK2-mutated patients versus 32% in unmutated patients. Multivariable analysis identified younger age and JAK2 and SF3B1 mutations as favorable factors.
In one series, 85 patients with non-RARS-T MDS/MPN, median age was 70 years, and 72% were males. Splenomegaly at presentation was present in 33%, thrombocytosis in 13%, leukocytosis in 18%, JAK2 mutations in 30%, and abnormal karyotype in 51%; the most frequent cytogenetic abnormality was trisomy 8. Median survival was 12.4 months and favorably affected by thrombocytosis. Treatment with hypomethylating agents, immunomodulators, or ASCT did not appear to favorably affect survival.
MYELOPROLIFERATIVE NEOPLASM, UNCLASSIFIABLE (MPN-U)
The category of MPN-U includes MPN-like neoplasms that cannot be clearly classified as one of the other seven subcategories of MPN (Table 135e-4). Examples include patients presenting with unusual thrombosis or unexplained organomegaly with normal blood counts but found to carry MPN-characteristic mutations such as JAK2 and CALR or display bone marrow morphology that is consistent with MPN. It is possible that some cases of MPN-U represent earlier disease stages in polycythemia vera (PV) or ET that fail to meet the threshold hemoglobin levels (18.5 g/dL in men or 16.5 g/dL in women) or platelet counts (450 × 109/L) that are required by the WHO diagnostic criteria. Specific treatment interventions might not be necessary in asymptomatic patients with MPN-U, whereas patients with arterial thrombotic complications might require cytoreductive and aspirin therapy and those with venous thrombosis might require systemic anticoagulation.
TRANSIENT MYELOPROLIFERATIVE DISORDER (TMD)
TMD constitutes an often but not always transient phenomenon of abnormal megakaryoblast proliferation, which occurs in approximately 10% of infants with Down’s syndrome. TMD is usually recognized at birth and either undergoes spontaneous regression (75% of cases) or progresses into acute megakaryoblastic leukemia (AMKL) (25% of cases). Almost all patients with TMD and TMD-derived AMKL display somatic GATA1 mutations. TMD-associated GATA1 mutations constitute exon 2 insertions, deletions, or missense mutations, affecting the N-terminal transactivation domain of GATA-1, and result in loss of full-length (50-kDa) GATA-1 and its replacement with a shorter isoform (40-kDa) that retains friend of GATA-1 (FOG-1) binding. In contrast, inherited forms of exon 2 GATA1 mutations produce a phenotype with anemia, whereas exon 4 mutations that affect the N-terminal, FOG-1-interactive domain produce familial dyserythropoietic anemia with thrombocytopenia or X-linked macrothrombocytopenia.
EOSINOPHILIC DISORDERS
Eosinophilia refers to a peripheral blood absolute eosinophil count (AEC) that is above the upper normal limit of the reference range. The term hypereosinophilia is used when the AEC is >1500 × 109/L. Eosinophilia is operationally classified as secondary (nonneoplastic proliferation of eosinophils) and primary (proliferation of eosinophils that is either neoplastic or otherwise unexplained) (Table 135e-6). Secondary eosinophilia is by far the most frequent cause of eosinophilia and is often associated with infections, especially those related to tissue-invasive helminths; allergic/vasculitic diseases; drugs; and metastatic cancer. Primary eosinophilia is the focus of this chapter and is considered when a cause for secondary eosinophilia is not readily apparent.
|
DIAGNOSIS OF CHRONIC EOSINOPHILIC LEUKEMIA AND HYPEREOSINOPHILIC SYNDROME |
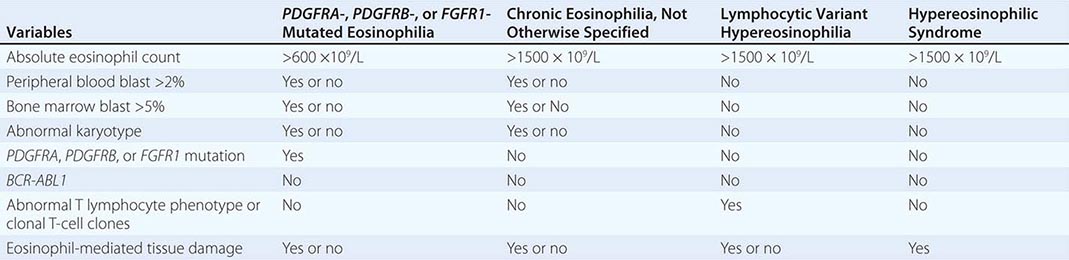
Primary Eosinophilia Primary eosinophilia is classified as clonal or idiopathic. Diagnosis of clonal eosinophilia requires morphologic, cytogenetic, or molecular evidence of a myeloid neoplasm. Idiopathic eosinophilia is considered when both secondary and clonal eosinophilias have been ruled out as a possibility. HES is a subcategory of idiopathic eosinophilia with persistent AEC of ≥1.5 × 109/L and associated with eosinophil-mediated organ damage (Table 135e-7). An HES-like disorder that is associated with clonal or phenotypically abnormal T cells is referred to as lymphocytic variant hypereosinophilia (Table 135e-7).
|
PRIMARY EOSINOPHILIA CLASSIFICATION |
Clonal Eosinophilia Examples of clonal eosinophilia include eosinophilia associated with acute myeloid leukemia (AML), MDS, CML, mastocytosis, and MDS/MPN overlap. Myeloid neoplasm-associated eosinophilia also includes the WHO MPN subcategory of chronic eosinophilic leukemia, not otherwise specified (CEL-NOS) and the WHO myeloid malignancy subcategory referred to as myeloid/lymphoid neoplasms with eosinophilia and mutations involving platelet-derived growth factor receptor (PDGFR) α/β or fibroblast growth factor receptor 1 (FGFR1).
The diagnostic workup for clonal eosinophilia that is not associated with morphologically overt myeloid malignancy should start with peripheral blood mutation screening for FIP1L1-PDGFRA and PDGFRB mutations using fluorescence in situ hybridization (FISH) or reverse transcription polymerase chain reaction. This is crucial because such eosinophilia is easily treated with imatinib. If mutation screening is negative, a bone marrow examination with cytogenetic studies is indicated. In this regard, one must first pay attention to the presence or absence of 5q33, 4q12, or 8p11.2 translocations, which, if present, would suggest PDGFRB-, PDGFRA-, or FGFR1-rearranged clonal eosinophilia, respectively. The presence of 5q33 or 4q12 translocations predicts favorable response to treatment with imatinib mesylate, whereas 8p11.2 translocations are associated with aggressive myeloid malignancies that are refractory to current drug therapy.
CEL-NOS is considered in the presence of cytogenetic/morphologic evidence of a myeloid malignancy that is otherwise not classifiable. Specifically, CEL-NOS is distinguished from HES by the presence of either a cytogenetic abnormality or greater than 2% peripheral blood blasts or greater than 5% bone marrow blasts (Table 135e-7). HES or idiopathic eosinophilia is considered in the absence of both morphologic and molecular evidence of clonal eosinophilia. However, before making a working diagnosis of HES, one has to exclude lymphocytic variant hypereosinophilia by excluding the presence of phenotypically abnormal T lymphocytes (by flow cytometry) and clonal T-cell gene rearrangements.
Chronic Eosinophilic Leukemia, Not Otherwise Specified (CEL-NOS) CEL-NOS is a subset of clonal eosinophilia that is neither molecularly defined nor classified as an alternative clinicopathologically assigned myeloid malignancy. We prefer to use the term strictly in patients with an HES phenotype who also display either a clonal cytogenetic/molecular abnormality or excess blasts in the bone marrow or peripheral blood. The WHO defines CEL-NOS in the presence of an AEC ≥1.5 × 109/L that is accompanied by either the presence of myeloblast excess (either >2% in the peripheral blood or 5–19% in the bone marrow) or evidence of myeloid clonality. Cytogenetic abnormalities in CEL, other than those that are associated with molecularly defined eosinophilic disorders, include trisomy 8 (the most frequent), t(10;11)(p14;q21), and t(7;12)(q11;p11). CEL-NOS does not respond to imatinib, and treatment strategies are often not different from those used in other similar MPNs and include ASCT for transplant-eligible patients with poor risk factors and participation in experimental treatment protocols otherwise.
PDGFR-Mutated Eosinophilia Both platelet-derived growth factor receptors α (PDGFRA located on chromosome 4q12) and β (PDGFRB located on chromosome 5q31-q32) are involved in MPN-relevant activating mutations. Clinical phenotype in both instances includes prominent blood eosinophilia and excellent response to imatinib therapy. In regard to PDGFRA mutations, the most popular is FIP1L1-PDGFRA, a karyotypically occult del(4)(q12) that was described in 2003 as an imatinib-sensitive activating mutation. Functional studies have demonstrated transforming properties in cell lines and the induction of MPN in mice. Cloning of the FIP1L1-PDGFRA fusion gene identified a novel molecular mechanism for generating this constitutively active fusion tyrosine kinase, wherein a ~800-kb interstitial deletion within 4q12 fuses the 5′ portion of FIP1L1 to the 3′ portion of PDGFRA. FIP1L1-PDGFRA occurs in a very small subset of patients who present with the phenotypic features of either systemic mastocytosis or HES, but the presence of the mutation reliably predicts complete hematologic and molecular response to imatinib therapy.
The association between eosinophilic myeloid malignancies and PDGFRB rearrangement was first characterized and published in 1994 when fusion of the tyrosine kinase–encoding region of PDGFRB to the ets-like gene, ETV6 [ETV6-PDGFRB, t(5;12)(q33;p13)] was demonstrated. The fusion protein was transforming to cell lines and resulted in constitutive activation of PDGFRB signaling. Since then, several other PDGFRB fusion transcripts with similar disease phenotypes have been described, cell line transformation and myeloproliferative disease (MPD) induction in mice has been demonstrated, and imatinib therapy was proven effective when used.
FGFR1-Mutated Eosinophilia The 8p11 myeloproliferative syndrome (EMS) (also known as human stem cell leukemic/lymphoma syndrome) constitutes a clinical phenotype with features of both lymphoma and eosinophilic MPN and characterized by a fusion mutation that involves the gene for fibroblast growth factor receptor 1 (FGFR1), which is located on chromosome 8p11. In EMS, both myeloid and lymphoid lineage cells exhibit the 8p11 translocation, thus demonstrating the stem cell origin of the disease. The disease features several 8p11-linked chromosome translocations, and some of the corresponding fusion FGFR1 mutants have been shown to transform cell lines and induce EMS- or CML-like disease in mice depending on the specific FGFR1 partner gene (ZNF198 or BCR, respectively). Consistent with this laboratory observation, some patients with BCR-FGFR1 mutation manifest a more indolent CML-like disease. The mechanism of FGFR1 activation in EMS is similar to that seen with PDGFRB-associated MPD; the tyrosine kinase domain of FGFR1 is juxtaposed to a dimerization domain from the partner gene. EMS is aggressive and requires combination chemotherapy followed by ASCT.
Hypereosinophilic Syndrome (HES) Blood eosinophilia that is neither secondary nor clonal is operationally labeled as being idiopathic. HES is a subcategory of idiopathic eosinophilia with persistent increase of the AEC to ≥1.5 × 109/L and presence of eosinophil-mediated organ damage, including cardiomyopathy, gastroenteritis, cutaneous lesions, sinusitis, pneumonitis, neuritis, and vasculitis. In addition, some patients manifest thromboembolic complications, hepatosplenomegaly, and either cytopenia or cytosis.
Bone marrow histologic and cytogenetic/molecular studies should be examined before a working diagnosis of HES is made. Additional blood studies that are currently recommended during the evaluation of HES include serum tryptase (an increased level suggests systemic mastocytosis and warrants molecular studies to detect FIP1L1-PDGFRA), T-cell immunophenotyping, and T-cell receptor antigen gene rearrangement analysis (a positive test suggests an underlying clonal or phenotypically abnormal T-cell disorder). In addition, initial evaluation in HES should include echocardiogram and measurement of serum troponin levels to screen for myocardial involvement by the disease.
Initial evaluation of the patient with eosinophilia should include tests that facilitate assessment of target organ damage, including complete blood count, chest x-ray, echocardiogram, and serum troponin level. An increased level of serum cardiac troponin has been shown to correlate with the presence of cardiomyopathy in HES. Typical echocardiographic findings in HES include ventricular apical thrombus, posterior mitral leaflet or tricuspid valve abnormality, endocardial thickening, dilated left ventricle, and pericardial effusion.
Glucocorticoids are the cornerstone of therapy in HES. Treatment with oral prednisone is usually started at 1 mg/kg per day and continued for 1–2 weeks before the dose is tapered slowly over the ensuing 2–3 months. If symptoms recur at a prednisone dose level of >10 mg/d, either hydroxyurea or interferon α is used as steroid-sparing agent. In patients who do not respond to usual therapy as outlined above, mepolizumab or alemtuzumab might be considered. Mepolizumab targets interleukin 5 (IL-5), a well-recognized survival factor for eosinophils. Alemtuzumab targets the CD52 antigen, which has been shown to be expressed by eosinophils but not by neutrophils.
MASTOCYTOSIS
Mast cell disease (MCD) is defined as tissue infiltration by morphologically and immunophenotypically abnormal mast cells. MCD is classified into two broad categories: cutaneous mastocytosis and systemic mastocytosis (SM). MCD in adults is usually systemic, and the clinical course can be either indolent or aggressive, depending on the respective absence or presence of impaired organ function. Symptoms and signs of MCD include urticaria pigmentosa, mast cell mediator release symptoms (e.g., headache, flushing, lightheadedness, syncope, anaphylaxis, pruritus, urticaria, angioedema, nausea, diarrhea, abdominal cramps), and organ damage (lytic bone lesions, osteoporosis, hepatosplenomegaly, cytopenia). Aggressive SM can be associated with another myeloid malignancy, including MPN, MDS, or MDS/MPN overlap (e.g., CMML), or present as overt mast cell leukemia. In general, life expectancy is near normal in indolent SM but significantly shortened in aggressive SM.
Diagnosis of SM is based on bone marrow examination that shows clusters of morphologically abnormal, spindle-shaped mast cells that are best evaluated by the use of immunohistochemical stains that are specific to mast cells (tryptase, CD117). In addition, mast cell immunophenotyping reveals aberrant CD25 expression by neoplastic mast cells. Other laboratory findings in SM include increased levels of serum tryptase, histamine and urine histamine metabolites, and prostaglandins. SM is associated with KIT mutations, usually KIT D816V, in the majority of patients. Accordingly, mutation screening for KIT D816V is diagnostically useful. However, the ability to detect KIT D816V depends on assay sensitivity and mast cell content of the test sample.
Both indolent and aggressive SM patients might experience mast cell mediator release symptoms, which are usually managed by both H1 and H2 histamine receptor blockers as well as cromolyn sodium. In addition, patients with propensity to vasodilatory shock should wear a medical alert bracelet and carry an Epi-Pen self-injector for self-administration of subcutaneous epinephrine. Urticaria pigmentosa shows variable response to both topical and systemic glucocorticoid therapy. Cytoreductive therapy is not recommended for indolent SM. In aggressive SM, either interferon α or cladribine is considered first-line therapy and benefits the majority of patients. In contrast, imatinib is ineffective in the treatment of PDGFR-unmutated SM.
DENDRITIC AND HISTIOCYTIC NEOPLASMS
Dendritic cell (DC) and histiocyte/macrophage neoplasms are extremely rare. DCs are antigen-presenting cells, whereas histiocyte/macrophages are antigen-processing cells. Bone marrow myeloid stem cells (CD34+) give rise to monocyte (CD14+, CD68+, CD11c+, CD1a–) and DC (CD14–, CD11c+/–, CD1a+/c) precursors. Monocyte precursors, in turn, give rise to macrophages (CD14+, CD68+, CD11c+, CD163+, lysozyme+) and interstitial DCs (CD68+, CD1a–). DC precursors give rise to Langerhans cell DCs (Birbeck granules, CD1a+, S100+, langerin+) and plasmacytoid DCs (CD68+, CD123+). Follicular DCs (CD21+, CD23+, CD35+) originate from mesenchymal stem cells. Dendritic and histiocytic neoplasms are operationally classified into macrophage/histiocyte-related and DC-related neoplasms. The former includes histiocytic sarcoma/malignant histiocytosis and the latter Langerhans cell histiocytosis, Langerhans cell sarcoma, interdigitating DC sarcoma, and follicular DC sarcoma.
Histiocytic Sarcoma/Malignant Histiocytosis Histiocytic sarcoma represents malignant proliferation of mature tissue histiocytes and is often localized. Median age at diagnosis is estimated at 46 years with slight male predilection. Some patients might have history of lymphoma, MDS, or germ cell tumors at time of disease presentation. The three typical disease sites are lymph nodes, skin, and the gastrointestinal system. Patients may or may not have systemic symptoms including fever and weight loss, and other symptoms include hepatosplenomegaly, lytic bone lesions, and pancytopenia. Immunophenotype includes presence of histiocytic markers (CD68, lysozyme, CD11c, CD14) and absence of myeloid or lymphoid markers. Prognosis is poor, and treatment is often ineffective. The term malignant histiocytosis refers to a disseminated disease and systemic symptoms. Lymphoma-like treatment induces complete remissions in some patients, and median survival is estimated at 2 years.
Langerhans Cell Histiocytosis Langerhans cells (LCs) are specialized DCs that reside in mucocutaneous tissue and upon activation become specialized for antigen presentation to T cells. LC histiocytosis (LCH; also known as histiocytosis X) represents neoplastic proliferation of LCs (S-100+, CD1a+, and Birbeck granules on electron microscopy). LCH incidence is estimated at 5 per million, and the disease typically affects children with a male predilection. Presentation can be either unifocal (eosinophilic granuloma) or multifocal. The former usually affects bones and less frequently lymph nodes, skin, and lung, whereas the latter is more disseminated. Unifocal disease often affects older children and adults, whereas multisystem disease affects infants. LCH of the lung in adults is characterized by bilateral nodules. Prognosis depends on organs involved. Only 10% of patients progress from unifocal to multiorgan disease. LCH of the lung might improve upon cessation of smoking.
Langerhans Cell Sarcoma Langerhans cell sarcoma (LCS) also represents neoplastic proliferation of LCs with overtly malignant morphology. The disease can present de novo or progress from antecedent LCH. There is a female predilection, and median age at diagnosis is estimated at 41 years. Immunophenotype is similar to that seen in LCH, and liver, spleen, lung, and bone are the usual sites of disease. Prognosis is poor, and treatment is generally ineffective.
Interdigitating Dendritic Cell Sarcoma Interdigitating DC sarcoma (IDCS), also known as reticulum cell sarcoma, represents neoplastic proliferation of interdigitating DCs. The disease is extremely rare and affects elderly adults with no sex predilection. Typical presentation is asymptomatic solitary lymphadenopathy. Immunophenotype includes S-100+ and negative for vimentin and CD1a. Prognosis ranges from benign local disease to widespread lethal disease.
Follicular Dendritic Cell Neoplasm Follicular DCs (FDCs) reside in B-cell follicles and present antigen to B cells. FDC neoplasms (FDCNs) are usually localized and often affect adults. FDCN might be associated with Castleman’s disease in 10–20% of cases, and increased incidence in schizophrenia has been reported. Cervical lymph nodes are the most frequent site of involvement in FDCN, and other sites include maxillary, mediastinal, and retroperitoneal lymph nodes; oral cavity; gastrointestinal system; skin; and breast. Sites of metastasis include lung and liver. Immunophenotype includes CD21, CD35, and CD23. Clinical course is typically indolent, and treatment includes surgical excision followed by regional radiotherapy and sometimes systemic chemotherapy.
Hemophagocytic Syndromes Hemophagocytic syndrome (HPS) represents nonneoplastic proliferation and activation of macrophages that induce cytokine-mediated bone marrow suppression and features of intense phagocytosis in bone marrow and liver. HPS may result from genetic or acquired disorders of macrophages. The former entail genetically determined inability to regulate macrophage proliferation and activation. Acquired HPS is often precipitated by viral infections, most notably Epstein-Barr virus. HPS might also accompany certain malignancies such as T-cell lymphoma. Clinical course is often fulminant and fatal.
136 |
Plasma Cell Disorders |
The plasma cell disorders are monoclonal neoplasms related to each other by virtue of their development from common progenitors in the B-lymphocyte lineage. Multiple myeloma, Waldenström’s macroglobulinemia, primary amyloidosis (Chap. 137), and the heavy chain diseases comprise this group and may be designated by a variety of synonyms such as monoclonal gammopathies, paraproteinemias, plasma cell dyscrasias, and dysproteinemias. Mature B lymphocytes destined to produce IgG bear surface immunoglobulin molecules of both M and G heavy chain isotypes with both isotypes having identical idiotypes (variable regions). Under normal circumstances, maturation to antibody-secreting plasma cells and their proliferation is stimulated by exposure to the antigen for which the surface immunoglobulin is specific; however, in the plasma cell disorders, the control over this process is lost. The clinical manifestations of all the plasma cell disorders relate to the expansion of the neoplastic cells, to the secretion of cell products (immunoglobulin molecules or subunits, lymphokines), and to some extent to the host’s response to the tumor. Normal development of B lymphocytes is discussed in Chap. 372e and depicted in Fig. 134-2.
There are three categories of structural variation among immunoglobulin molecules that form antigenic determinants, and these are used to classify immunoglobulins. Isotypes are those determinants that distinguish among the main classes of antibodies of a given species and are the same in all normal individuals of that species. Therefore, isotypic determinants are, by definition, recognized by antibodies from a distinct species (heterologous sera) but not by antibodies from the same species (homologous sera). There are five heavy chain isotypes (M, G, A, D, E) and two light chain isotypes (κ, λ). Allotypes are distinct determinants that reflect regular small differences between individuals of the same species in the amino acid sequences of otherwise similar immunoglobulins. These differences are determined by allelic genes; by definition, they are detected by antibodies made in the same species. Idiotypes are the third category of antigenic determinants. They are unique to the molecules produced by a given clone of antibody-producing cells. Idiotypes are formed by the unique structure of the antigen-binding portion of the molecule.
Antibody molecules (Fig. 136-1) are composed of two heavy chains (~50,000 mol wt) and two light chains (~25,000 mol wt). Each chain has a constant portion (limited amino acid sequence variability) and a variable region (extensive sequence variability). The light and heavy chains are linked by disulfide bonds and are aligned so that their variable regions are adjacent to one another. This variable region forms the antigen recognition site of the antibody molecule; its unique structural features form idiotypes that are reliable markers for a particular clone of cells because each antibody is formed and secreted by a single clone. Because of the mechanics of the gene rearrangements necessary to specify the immunoglobulin variable regions (VDJ joining for the heavy chain, VJ joining for the light chain), a particular clone rearranges only one of the two chromosomes to produce an immunoglobulin molecule of only one light chain isotype and only one allotype (allelic exclusion) (Fig. 136-1). After exposure to antigen, the variable region may become associated with a new heavy chain isotype (class switch). Each clone of cells performs these sequential gene arrangements in a unique way. This results in each clone producing a unique immunoglobulin molecule. In most plasma cells, light chains are synthesized in slight excess, secreted as free light chains, and cleared by the kidney, but <10 mg of such light chains is excreted per day.
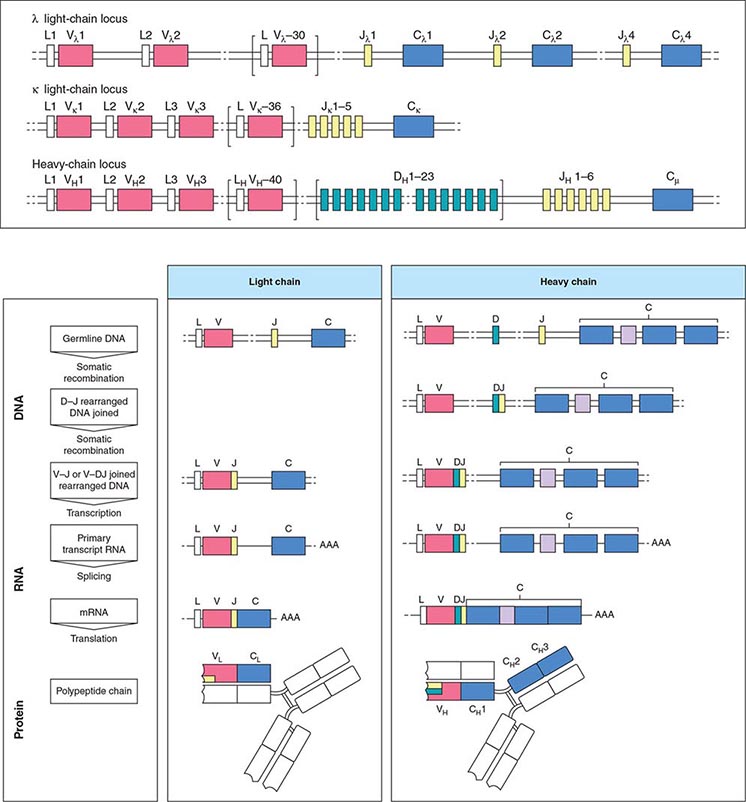
FIGURE 136-1 Immunoglobulin genetics and the relationship of gene segments to the antibody protein. The top portion of the figure is a schematic of the organization of the immunoglobulin genes, λ on chromosome 22, κ on chromosome 2, and the heavy chain locus on chromosome 14. The heavy chain locus is longer than 2 megabases, and some of the D region gene segments are only a few bases long, so the figure depicts the schematic relationship among the segments, not their actual size. The bottom portion of the figure outlines the steps in going from the noncontiguous germline gene segments to an intact antibody molecule. Two recombination events juxtapose the V-D-J (or V-J for light chains) segments. The rearranged gene is transcribed, and RNA splicing cuts out intervening sequences to produce an mRNA, which is then translated into an antibody light or heavy chain. The sites on the antibody that bind to antigen (the so called CDR3 regions) are encoded by D and J segments for heavy chains and the J segments for light chains. (From K Murphy: Janeway’s Immunobiology, 8th ed. Garland Science, 2011.)
Electrophoretic analysis permits separation of components of the serum proteins (Fig. 136-2). The immunoglobulins move heterogeneously in an electric field and form a broad peak in the gamma region, which is usually increased in the sera of patients with plasma cell tumors. There is a sharp spike in this region called an M component (M for monoclonal). Less commonly, the M component may appear in the β2 or α2 globulin region. The monoclonal antibody must be present at a concentration of at least 5 g/L (0.5 g/dL) to be accurately quantitated by this method. This corresponds to ~109 cells producing the antibody. Confirmation of the type of immunoglobulin and that it is truly monoclonal is determined by immunoelectrophoresis that reveals a single heavy and/or light chain type. Hence immunoelectrophoresis and electrophoresis provide qualitative and quantitative assessment of the M component, respectively. Once the presence of an M component has been confirmed, the amount of M component in the serum is a reliable measure of the tumor burden, making M component an excellent tumor marker to manage therapy, yet it is not specific enough to be used to screen asymptomatic patients. In addition to the plasma cell disorders, M components may be detected in other lymphoid neoplasms such as chronic lymphocytic leukemia and lymphomas of B- or T-cell origin; nonlymphoid neoplasms such as chronic myeloid leukemia, breast cancer, and colon cancer; a variety of nonneoplastic conditions such as cirrhosis, sarcoidosis, parasitic diseases, Gaucher’s disease, and pyoderma gangrenosum; and a number of autoimmune conditions, including rheumatoid arthritis, myasthenia gravis, and cold agglutinin disease. Monoclonal proteins are also observed in immunosuppressed patients after organ transplant and, rarely, allogeneic transplant. At least two very rare skin diseases—lichen myxedematosus (also known as papular mucinosis) and necrobiotic xanthogranuloma—are associated with a monoclonal gammopathy. In papular mucinosis, highly cationic IgG is deposited in the dermis of patients. This organ specificity may reflect the specificity of the antibody for some antigenic component of the dermis. Necrobiotic xanthogranuloma is a histiocytic infiltration of the skin, usually of the face, that produces red or yellow nodules that can enlarge to plaques. Approximately 10% progress to myeloma. Five percent of patients with sensory motor neuropathy also have a monoclonal paraprotein.
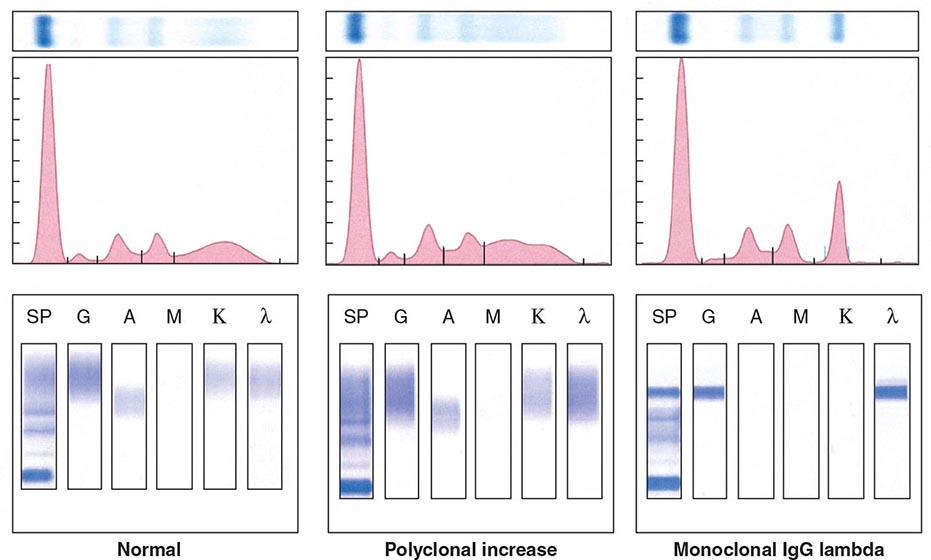
FIGURE 136-2 Representative patterns of serum electrophoresis and immunofixation. The upper panels represent agarose gel, middle panels are the densitometric tracing of the gel, and lower panels are immunofixation patterns. Panel on the left illustrates the normal pattern of serum protein on electrophoresis. Because there are many different immunoglobulins in the serum, their differing mobilities in an electric field produce a broad peak. In conditions associated with increases in polyclonal immunoglobulin, the broad peak is more prominent (middle panel). In monoclonal gammopathies, the predominance of a product of a single cell produces a “church spire” sharp peak, usually in the γ globulin region (right panel). The immunofixation (lower panel) identifies the type of immunoglobulin. For example, normal and polyclonal increase in immunoglobulins produce no distinct bands; however, the right panel shows distinct bands in IgG and lambda protein lanes, confirming the presence of IgG lambda monoclonal protein. (Courtesy of Dr. Neal I. Lindeman; with permission.)
The nature of the M component is variable in plasma cell disorders. It may be an intact antibody molecule of any heavy chain subclass, or it may be an altered antibody or fragment. Isolated light or heavy chains may be produced. In some plasma cell tumors such as extramedullary or solitary bone plasmacytomas, less than one-third of patients will have an M component. In ~20% of myelomas, only light chains are produced and, in most cases, are secreted in the urine as Bence Jones proteins. The frequency of myelomas of a particular heavy chain class is roughly proportional to the serum concentration, and therefore, IgG myelomas are more common than IgA and IgD myelomas. In approximately 1% of patients with myeloma, biclonal or triclonal gammopathy is observed.
MULTIPLE MYELOMA
DEFINITION
Multiple myeloma represents a malignant proliferation of plasma cells derived from a single clone. The tumor, its products, and the host response to it result in a number of organ dysfunctions and symptoms, including bone pain or fracture, renal failure, susceptibility to infection, anemia, hypercalcemia, and occasionally clotting abnormalities, neurologic symptoms, and manifestations of hyperviscosity.
ETIOLOGY
The cause of myeloma is not known. Myeloma occurred with increased frequency in those exposed to the radiation of nuclear warheads in World War II after a 20-year latency. Myeloma has been seen more commonly than expected among farmers, wood workers, leather workers, and those exposed to petroleum products. A variety of chromosomal alterations have been found in patients with myeloma: hyperdiploidy, 13q14 deletions, translocations t(11;14)(q13;q32), t(4;14)(p16;q32), and t(14;16), and 17p13 deletions. Evidence is strong that errors in switch recombination—the genetic mechanism to change antibody heavy chain isotype—participate in the transformation process. However, no common molecular pathogenetic pathway has yet emerged. Genome sequencing studies have failed to identify any recurrent mutation with frequency >20%; N-ras, K-ras, and B-raf mutations are most common and combined occur in over 40% of patients. There is also evidence of complex clusters of subclonal variants at diagnosis that acquire additional mutations over time, indicative of genomic evolution that may drive disease progression. The neoplastic event in myeloma may involve cells earlier in B-cell differentiation than the plasma cell. Interleukin (IL) 6 may play a role in driving myeloma cell proliferation. It remains difficult to distinguish benign from malignant plasma cells based on morphologic criteria in all but a few cases (Fig. 136-3).
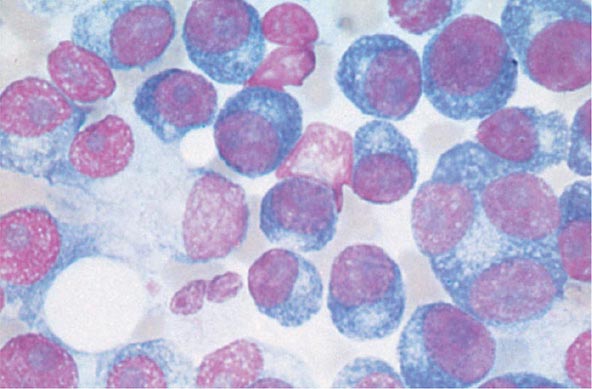
FIGURE 136-3 Multiple myeloma (marrow). The cells bear characteristic morphologic features of plasma cells, round or oval cells with an eccentric nucleus composed of coarsely clumped chromatin, a densely basophilic cytoplasm, and a perinuclear clear zone containing the Golgi apparatus. Binucleate and multinucleate malignant plasma cells can be seen.
INCIDENCE AND PREVALENCE
An estimated 24,050 new cases of myeloma were diagnosed in 2014, and 11,090 people died from the disease in the United States. Myeloma increases in incidence with age. The median age at diagnosis is 70 years; it is uncommon under age 40. Males are more commonly affected than females, and blacks have nearly twice the incidence of whites. Myeloma accounts for 1.3% of all malignancies in whites and 2% in blacks, and 13% of all hematologic cancers in whites and 33% in blacks.
GLOBAL CONSIDERATIONS
![]() The incidence of myeloma is highest in African Americans and Pacific Islanders; intermediate in Europeans and North American whites; and lowest in people from developing countries including Asia. The higher incidence in more developed countries may result from the combination of a longer life expectancy and more frequent medical surveillance. Incidence of multiple myeloma in other ethnic groups including native Hawaiians, female Hispanics, American Indians from New Mexico, and Alaskan natives is higher relative to U.S. whites in the same geographic area. Chinese and Japanese populations have a lower incidence than whites. Immunoproliferative small-intestinal disease with alpha heavy chain disease is most prevalent in the Mediterranean area. Despite these differences in prevalence, the characteristics, response to therapy, and prognosis of myeloma are similar worldwide.
The incidence of myeloma is highest in African Americans and Pacific Islanders; intermediate in Europeans and North American whites; and lowest in people from developing countries including Asia. The higher incidence in more developed countries may result from the combination of a longer life expectancy and more frequent medical surveillance. Incidence of multiple myeloma in other ethnic groups including native Hawaiians, female Hispanics, American Indians from New Mexico, and Alaskan natives is higher relative to U.S. whites in the same geographic area. Chinese and Japanese populations have a lower incidence than whites. Immunoproliferative small-intestinal disease with alpha heavy chain disease is most prevalent in the Mediterranean area. Despite these differences in prevalence, the characteristics, response to therapy, and prognosis of myeloma are similar worldwide.
PATHOGENESIS AND CLINICAL MANIFESTATIONS
Multiple myeloma (MM) cells bind via cell-surface adhesion molecules to bone marrow stromal cells (BMSCs) and extracellular matrix (ECM), which triggers MM cell growth, survival, drug resistance, and migration in the bone marrow milieu (Fig. 136-4). These effects are due both to direct MM cell–BMSC binding and to induction of various cytokines, including IL-6, insulin-like growth factor type I (IGF-I), vascular endothelial growth factor (VEGF), and stromal cell–derived growth factor (SDF)-1α. Growth, drug resistance, and migration are mediated via Ras/Raf/mitogen-activated protein kinase, PI3K/Akt, and protein kinase C signaling cascades, respectively.
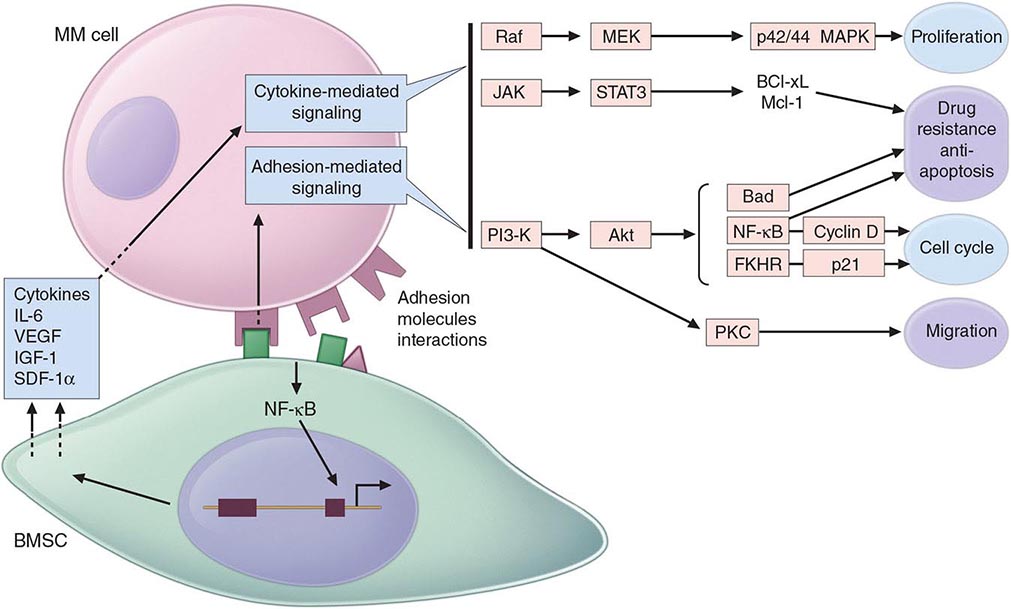
FIGURE 136-4 Pathogenesis of multiple myeloma. Multiple myeloma (MM) cells interact with bone marrow stromal cells (BMSCs) and extracellular matrix proteins via adhesion molecules, triggering adhesion-mediated signaling as well as cytokine production. This triggers cytokine-mediated signaling that provides growth, survival, and antiapoptotic effects as well as development of drug resistance.
Bone pain is the most common symptom in myeloma, affecting nearly 70% of patients. Unlike the pain of metastatic carcinoma, which often is worse at night, the pain of myeloma is precipitated by movement. Persistent localized pain in a patient with myeloma usually signifies a pathologic fracture. The bone lesions of myeloma are caused by the proliferation of tumor cells, activation of osteoclasts that destroy bone, and suppression of osteoblasts that form new bone. The increased osteoclast activity is mediated by osteoclast activating factors (OAFs) made by the myeloma cells (OAF activity can be mediated by several cytokines, including IL-1, lymphotoxin, VEGF, receptor activator of NF-κB [RANK] ligand, macrophage inhibitory factor [MIP]-1α, and tumor necrosis factor [TNF]). The bone lesions are lytic in nature and are rarely associated with osteoblastic new bone formation due to their suppression by dickhoff-1 (DKK-1) produced by myeloma cells. Therefore, radioisotopic bone scanning is less useful in diagnosis than is plain radiography. The bony lysis results in substantial mobilization of calcium from bone, and serious acute and chronic complications of hypercalcemia may dominate the clinical picture (see below). Localized bone lesions may expand to the point that mass lesions may be palpated, especially on the skull (Fig. 136-5), clavicles, and sternum; and the collapse of vertebrae may lead to spinal cord compression. The next most common clinical problem in patients with myeloma is susceptibility to bacterial infections. The most common infections are pneumonias and pyelonephritis, and the most frequent pathogens are Streptococcus pneumoniae, Staphylococcus aureus, and Klebsiella pneumoniae in the lungs and Escherichia coli and other gram-negative organisms in the urinary tract. In ~25% of patients, recurrent infections are the presenting features, and >75% of patients will have a serious infection at some time in their course. The susceptibility to infection has several contributing causes. First, patients with myeloma have diffuse hypogammaglobulinemia if the M component is excluded. The hypogammaglobulinemia is related to both decreased production and increased destruction of normal antibodies. Moreover, some patients generate a population of circulating regulatory cells in response to their myeloma that can suppress normal antibody synthesis. In the case of IgG myeloma, normal IgG antibodies are broken down more rapidly than normal because the catabolic rate for IgG antibodies varies directly with the serum concentration. The large M component results in fractional catabolic rates of 8–16% instead of the normal 2%. These patients have very poor antibody responses, especially to polysaccharide antigens such as those on bacterial cell walls. Most measures of T-cell function in myeloma are normal, but a subset of CD4+ cells may be decreased. Granulocyte lysozyme content is low, and granulocyte migration is not as rapid as normal in patients with myeloma, probably the result of a tumor product. There are also a variety of abnormalities in complement functions in myeloma patients. All these factors contribute to the immune deficiency of these patients. Some commonly used therapeutic agents, e.g., dexamethasone, suppress immune responses and increase susceptibility to bacterial and fungal infection, and bortezomib predisposes to herpesvirus reactivation.
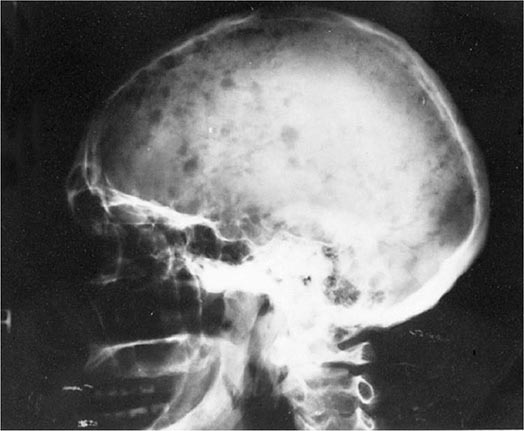
FIGURE 136-5 Bony lesions in multiple myeloma. The skull demonstrates the typical “punched out” lesions characteristic of multiple myeloma. The lesion represents a purely osteolytic lesion with little or no osteoblastic activity. (Courtesy of Dr. Geraldine Schechter; with permission.)
Renal failure occurs in nearly 25% of myeloma patients, and some renal pathology is noted in more than 50%. Many factors contribute to this. Hypercalcemia is the most common cause of renal failure. Glomerular deposits of amyloid, hyperuricemia, recurrent infections, frequent use of nonsteroidal anti-inflammatory agents for pain control, use of iodinated contrast dye for imaging, bisphosphonate use, and occasional infiltration of the kidney by myeloma cells all may contribute to renal dysfunction. However, tubular damage associated with the excretion of light chains is almost always present. Normally, light chains are filtered, reabsorbed in the tubules, and catabolized. With the increase in the amount of light chains presented to the tubule, the tubular cells become overloaded with these proteins, and tubular damage results either directly from light chain toxic effects or indirectly from the release of intracellular lysosomal enzymes. The earliest manifestation of this tubular damage is the adult Fanconi’s syndrome (a type 2 proximal renal tubular acidosis), with loss of glucose and amino acids, as well as defects in the ability of the kidney to acidify and concentrate the urine. The proteinuria is not accompanied by hypertension, and the protein is nearly all light chains. Generally, very little albumin is in the urine because glomerular function is usually normal. When the glomeruli are involved, nonselective proteinuria is also observed. Patients with myeloma also have a decreased anion gap [i.e., Na+ – (Cl– + HCO3–)] because the M component is cationic, resulting in retention of chloride. This is often accompanied by hyponatremia that is felt to be artificial (pseudohyponatremia) because each volume of serum has less water as a result of the increased protein. Renal dysfunction due to light chain deposition disease, light chain cast nephropathy, and amyloidosis is partially reversible with effective therapy. Myeloma patients are susceptible to developing acute renal failure if they become dehydrated.
Normocytic and normochromic anemia occurs in ~80% of myeloma patients. It is usually related to the replacement of normal marrow by expanding tumor cells, to the inhibition of hematopoiesis by factors made by the tumor, to reduced production of erythropoietin by the kidney, and to the effects of long-term therapy. In addition, mild hemolysis may contribute to the anemia. A larger than expected fraction of patients may have megaloblastic anemia due to either folate or vitamin B12 deficiency. Granulocytopenia and thrombocytopenia are rare except when therapy-induced. Clotting abnormalities may be seen due to the failure of antibody-coated platelets to function properly; the interaction of the M component with clotting factors I, II, V, VII, or VIII; antibody to clotting factors; or amyloid damage of endothelium. Deep venous thrombosis is also observed with use of thalidomide, lenalidomide, or pomalidomide in combination with dexamethasone. Raynaud’s phenomenon and impaired circulation may result if the M component forms cryoglobulins, and hyperviscosity syndromes may develop depending on the physical properties of the M component (most common with IgM, IgG3, and IgA paraproteins). Hyperviscosity is defined based on the relative viscosity of serum as compared with water. Normal relative serum viscosity is 1.8 (i.e., serum is normally almost twice as viscous as water). Symptoms of hyperviscosity occur at a level greater than 4 centipoise (cP), which is usually reached at paraprotein concentrations of ~40 g/L (4 g/dL) for IgM, 50 g/L (5 g/dL) for IgG3, and 70 g/L (7 g/dL) for IgA; however, depending on chemical and physical properties of the paraprotein molecule, it can occasionally be observed at lower levels.
Although neurologic symptoms occur in a minority of patients, they may have many causes. Hypercalcemia may produce lethargy, weakness, depression, and confusion. Hyperviscosity may lead to headache, fatigue, shortness of breath, exacerbation or precipitation of heart failure, visual disturbances, ataxia, vertigo, retinopathy, somnolence, and coma. Bony damage and collapse may lead to cord compression, radicular pain, and loss of bowel and bladder control. Infiltration of peripheral nerves by amyloid can be a cause of carpal tunnel syndrome and other sensorimotor mono- and polyneuropathies. Neuropathy associated with monoclonal gammopathy of undetermined significance (MGUS) and myeloma is more frequently sensory than motor neuropathy and is associated with IgM more than other isotypes. In >50% of patients with neuropathy, the IgM monoclonal protein is directed against myelin-associated globulin (MAG). Sensory neuropathy is also a side effect of thalidomide and bortezomib therapy.
Many of the clinical features of myeloma, e.g., cord compression, pathologic fractures, hyperviscosity, sepsis, and hypercalcemia, can present as medical emergencies. Despite the widespread distribution of plasma cells in the body, tumor expansion is dominantly within bone and bone marrow and, for reasons unknown, rarely causes enlargement of spleen, lymph nodes, or gut-associated lymphatic tissue.
DIAGNOSIS AND STAGING
The diagnosis of myeloma requires marrow plasmacytosis (>10%), a serum and/or urine M component, and end organ damage detailed in Table 136-1. Bone marrow plasma cells are CD138 and either monoclonal kappa or lambda light chain positive. The most important differential diagnosis in patients with myeloma involves their separation from individuals with MGUS or smoldering multiple myeloma (SMM). MGUS is vastly more common than myeloma, occurring in 1% of the population older than age 50 years and in up to 10% of individuals older than age 75 years. The diagnostic criteria for MGUS, SMM, and myeloma are described in Table 136-1. Although ~1% of patients per year with MGUS go on to develop myeloma, all myeloma is preceded by MGUS. Non-IgG subtype, abnormal kappa/lambda free light chain ratio, and serum M protein >15 g/L (1.5 g/dL) are associated with higher incidence of progression of MGUS to myeloma. Absence of all three features predicts a 5% chance of progression, whereas higher risk MGUS with the presence of all three features predicts a 60% chance of progression over 20 years. The features responsible for higher risk of progression from SMM to MM are bone marrow plasmacytosis >10%, abnormal kappa/lambda free light chain ratio, and serum M protein >30 g/L (3 g/dL). Patients with only one of these three features have a 25% chance of progression to MM in 5 years, whereas patients with high-risk SMM with all three features have a 76% chance of progression. There are two important variants of myeloma—solitary bone plasmacytoma and solitary extramedullary plasmacytoma. These lesions are associated with an M component in <30% of the cases, they may affect younger individuals, and both are associated with median survivals of ≥10 years. Solitary bone plasmacytoma is a single lytic bone lesion without marrow plasmacytosis. Extramedullary plasmacytomas usually involve the submucosal lymphoid tissue of the nasopharynx or paranasal sinuses without marrow plasmacytosis. Both tumors are highly responsive to local radiation therapy. If an M component is present, it should disappear after treatment. Solitary bone plasmacytomas may recur in other bony sites or evolve into myeloma. Extramedullary plasmacytomas rarely recur or progress.
|
DIAGNOSTIC CRITERIA FOR MULTIPLE MYELOMA, MYELOMA VARIANTS, AND MONOCLONAL GAMMOPATHY OF UNDETERMINED SIGNIFICANCE |
aMyeloma-related organ or tissue impairment (end organ damage): calcium levels increased: serum calcium >0.25 mmol/L above the upper limit of normal or >2.75 mmol/L; renal insufficiency: creatinine >173 mmol/L; anemia: hemoglobin 2 g/dL below the lower limit of normal or hemoglobin <10 g/dL; bone lesions: lytic lesions or osteoporosis with compression fractures (magnetic resonance imaging or computed tomography may clarify); other: symptomatic hyperviscosity, amyloidosis, recurrent bacterial infections (>2 episodes in 12 months). bIf flow cytometry is performed, most plasma cells (>90%) will show a “neoplastic” phenotype. cA small M component may sometimes be present. dThese features should have no attributable other causes and have temporal relation with each other.
Abbreviation: POEMS, polyneuropathy, organomegaly, endocrinopathy, M-protein, and skin changes.
The clinical evaluation of patients with myeloma includes a careful physical examination searching for tender bones and masses. Chest and bone radiographs may reveal lytic lesions or diffuse osteopenia. Magnetic resonance imaging (MRI) offers a sensitive means to document extent of bone marrow infiltration and cord or root compression in patients with pain syndromes. A complete blood count with differential may reveal anemia. Erythrocyte sedimentation rate is elevated. Rare patients (~1%) may have plasma cell leukemia with >2000 plasma cells/μL. This may be seen in disproportionate frequency in IgD (12%) and IgE (25%) myelomas. Serum calcium, urea nitrogen, creatinine, and uric acid levels may be elevated. Protein electrophoresis and measurement of serum immunoglobulins and free light chains are useful for detecting and characterizing M spikes, supplemented by immunoelectrophoresis, which is especially sensitive for identifying low concentrations of M components not detectable by protein electrophoresis. A 24-h urine specimen is necessary to quantitate Bence Jones protein excretion. Serum alkaline phosphatase is usually normal even with extensive bone involvement because of the absence of osteoblastic activity. It is also important to quantitate serum β2-microglobulin and albumin (see below).
The serum M component will be IgG in 53% of patients, IgA in 25%, and IgD in 1%; 20% of patients will have only light chains in serum and urine. Dipsticks for detecting proteinuria are not reliable at identifying light chains, and the heat test for detecting Bence Jones protein is falsely negative in ~50% of patients with light chain myeloma. Fewer than 1% of patients have no identifiable M component; these patients usually have light chain myeloma in which renal catabolism has made the light chains undetectable in the urine. In most of these patients, light chains can now be detected by serum free light chain assay. IgD myeloma may also present with light chain disease. About two-thirds of patients with serum M components also have urinary light chains. The light chain isotype may have an impact on survival. Patients secreting lambda light chains have a significantly shorter overall survival than those secreting kappa light chains. Whether this is due to some genetically important determinant of cell proliferation or because lambda light chains are more likely to cause renal damage and form amyloid than are kappa light chains is unclear. The heavy chain isotype may have an impact on patient management as well. About half of patients with IgM paraproteins develop hyperviscosity compared with only 2–4% of patients with IgA and IgG M components. Among IgG myelomas, it is the IgG3 subclass that has the highest tendency to form both concentration- and temperature-dependent aggregates, leading to hyperviscosity and cold agglutination at lower serum concentrations.
PROGNOSIS
Serum β2-microglobulin is the single most powerful predictor of survival and can substitute for staging. β2-Microglobulin is a protein of 11,000 mol wt with homologies to the constant region of immunoglobulins that is the light chain of the class I major histocompatibility antigens (HLA-A, -B, -C) on the surface of every cell. Patients with β2-microglobulin levels <0.004 g/L have a median survival of 43 months, and those with levels >0.004 g/L have a survival of only 12 months. Combination of serum β2-microglobulin and albumin levels forms the basis for a three-stage International Staging System (ISS) (Table 136-2) that predicts survival. With the use of high-dose therapy and the newer agents, the Durie-Salmon staging system is unable to predict outcome and thus is no longer used. High labeling index, circulating plasma cells, performance status, and high levels of lactate dehydrogenase are also associated with poor prognosis.
|
RISK STRATIFICATION IN MYELOMA |
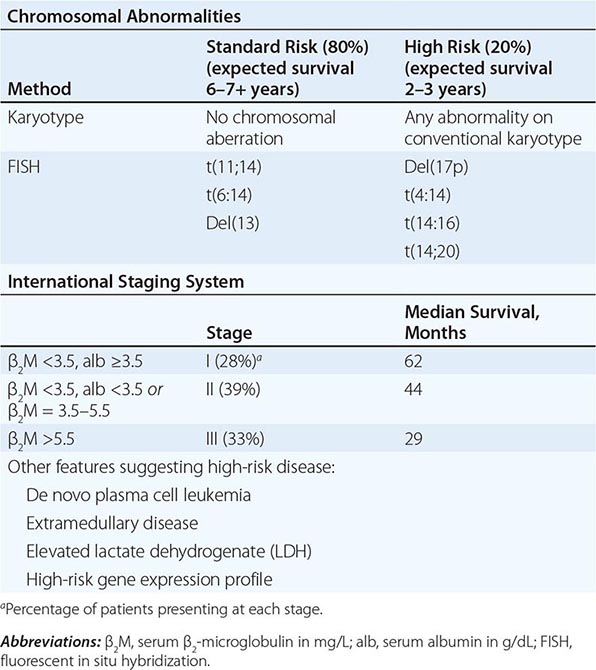
Other factors that may influence prognosis are the presence of cytogenetic abnormalities and hypodiploidy by karyotype, fluorescent in situ hybridization (FISH)–identified chromosome 17p deletion, and translocations t(4;14), (14;16), and t(14;20). Chromosome 13q deletion, previously thought to predict poor outcome, is not a predictor following the use of newer agents. Microarray profiling and comparative genomic hybridization have formed the basis for RNA- and DNA-based prognostic staging systems, respectively. The ISS system, along with cytogenetic changes, is the most widely used method for assessing prognosis (Table 136-2).
WALDENSTRÖM’S MACROGLOBULINEMIA
In 1948, Waldenström described a malignancy of lymphoplasmacytoid cells that secreted IgM. In contrast to myeloma, the disease was associated with lymphadenopathy and hepatosplenomegaly, but the major clinical manifestation was hyperviscosity syndrome. The disease resembles the related diseases chronic lymphocytic leukemia, myeloma, and lymphocytic lymphoma. It originates from a post–germinal center B cell that has undergone somatic mutations and antigenic selection in the lymphoid follicle and has the characteristics of an IgM-bearing memory B cell. Waldenström’s macroglobulinemia (WM) and IgM myeloma follow a similar clinical course, but therapeutic options are different. The diagnosis of IgM myeloma is usually reserved for patients with lytic bone lesions and predominant infiltration with CD138+ plasma cells in the bone marrow. Such patients are at greater risk of pathologic fractures than patients with WM.
A familial occurrence is common in WM, but its molecular bases are yet unclear. A distinct MYD88 L265P somatic mutation has been reported in over 90% of patients with WM and the majority of IgM MGUS. Presence of this mutation is now used as a diagnostic test to discriminate WM from marginal zone lymphomas (MZLs), IgM-secreting myeloma, and chronic lymphocytic leukemia (CLL) with plasmacytic differentiation. This mutation also explains the molecular pathogenesis of the disease, with involvement of Toll-like receptor (TLR) and interleukin 1 receptor (IL-1R) signaling leading to activation of IL-1R–associated kinase (IRAK) 4 and IRAK1 followed by nuclear factor-κB (NF-κB) activation. The disease is similar to myeloma in being slightly more common in men and occurring with increased incidence with increasing age (median 64 years). There have been reports that the IgM in some patients with macroglobulinemia may have specificity for myelin-associated glycoprotein (MAG), a protein that has been associated with demyelinating disease of the peripheral nervous system and may be lost earlier and to a greater extent than the better known myelin basic protein in patients with multiple sclerosis. Sometimes patients with macroglobulinemia develop a peripheral neuropathy, and half of these patients are positive for anti-MAG antibody. The neuropathy may precede the appearance of the neoplasm. There is speculation that the whole process begins with a viral infection that may elicit an antibody response that cross-reacts with a normal tissue component.
Like myeloma, the disease involves the bone marrow, but unlike myeloma, it does not cause bone lesions or hypercalcemia. Bone marrow shows >10% infiltration with lymphoplasmacytic cells (surface IgM+, CD19+, CD20+, and CD22+, rarely CD5+, but CD10– and CD23–) with an increase in number of mast cells. Like myeloma, an M component is present in the serum in excess of 30 g/L (3 g/dL), but unlike myeloma, the size of the IgM paraprotein results in little renal excretion, and only ~20% of patients excrete light chains. Therefore, renal disease is not common. The light chain isotype is kappa in 80% of the cases. Patients present with weakness, fatigue, and recurrent infections similar to myeloma patients, but epistaxis, visual disturbances, and neurologic symptoms such as peripheral neuropathy, dizziness, headache, and transient paresis are much more common in macroglobulinemia. Physical examination reveals adenopathy and hepatosplenomegaly, and ophthalmoscopic examination may reveal vascular segmentation and dilation of the retinal veins characteristic of hyperviscosity states. Patients may have a normocytic, normochromic anemia, but rouleaux formation and a positive Coombs’ test are much more common than in myeloma. Malignant lymphocytes are usually present in the peripheral blood. About 10% of macroglobulins are cryoglobulins. These are pure M components and are not the mixed cryoglobulins seen in rheumatoid arthritis and other autoimmune diseases. Mixed cryoglobulins are composed of IgM or IgA complexed with IgG, for which they are specific. In both cases, Raynaud’s phenomenon and serious vascular symptoms precipitated by the cold may occur, but mixed cryoglobulins are not commonly associated with malignancy. Patients suspected of having a cryoglobulin based on history and physical examination should have their blood drawn into a warm syringe and delivered to the laboratory in a container of warm water to avoid errors in quantitating the cryoglobulin.
POEMS SYNDROME
The features of this syndrome are polyneuropathy, organomegaly, endocrinopathy, M-protein, and skin changes (POEMS). Diagnostic criteria are described in Table 136-1. Patients usually have a severe, progressive sensorimotor polyneuropathy associated with sclerotic bone lesions from myeloma. Polyneuropathy occurs in ~1.4% of myelomas, but the POEMS syndrome is only a rare subset of that group. Unlike typical myeloma, hepatomegaly and lymphadenopathy occur in about two-thirds of patients, and splenomegaly is seen in one-third. The lymphadenopathy frequently resembles Castleman’s disease histologically, a condition that has been linked to IL-6 overproduction. The endocrine manifestations include amenorrhea in women and impotence and gynecomastia in men. Hyperprolactinemia due to loss of normal inhibitory control by the hypothalamus may be associated with other central nervous system manifestations such as papilledema and elevated cerebrospinal fluid pressure and protein. Type 2 diabetes mellitus occurs in about one-third of patients. Hypothyroidism and adrenal insufficiency are occasionally noted. Skin changes are diverse: hyperpigmentation, hypertrichosis, skin thickening, and digital clubbing. Other manifestations include peripheral edema, ascites, pleural effusions, fever, and thrombocytosis. Not all the components of POEMS syndrome may be present initially.
The pathogenesis of the disease is unclear, but high circulating levels of the proinflammatory cytokines IL-1, IL-6, VEGF, and TNF have been documented, and levels of the inhibitory cytokine transforming growth factor β are lower than expected. Treatment of the myeloma may result in an improvement in the other disease manifestations.
Patients are often treated similarly to those with myeloma. Plasmapheresis does not appear to be of benefit in POEMS syndrome. Patients presenting with isolated sclerotic lesions may have resolution of neuropathic symptoms after local therapy for plasmacytoma with radiotherapy. Similar to multiple myeloma, novel agents and high-dose therapy with autologous stem cell transplantation have been pursued in selected patients and have been associated with prolonged progression-free survival.
HEAVY CHAIN DISEASES
The heavy chain diseases are rare lymphoplasmacytic malignancies. Their clinical manifestations vary with the heavy chain isotype. Patients have absence of light chain and secrete a defective heavy chain that usually has an intact Fc fragment and a deletion in the Fd region. Gamma, alpha, and mu heavy chain diseases have been described, but no reports of delta or epsilon heavy chain diseases have appeared. Molecular biologic analysis of these tumors has revealed structural genetic defects that may account for the aberrant chain secreted.
GAMMA HEAVY CHAIN DISEASE (FRANKLIN’S DISEASE)
This disease affects individuals of widely different age groups and countries of origin. It is characterized by lymphadenopathy, fever, anemia, malaise, hepatosplenomegaly, and weakness. It is frequently associated with autoimmune diseases, especially rheumatoid arthritis. Its most distinctive symptom is palatal edema, resulting from involvement of nodes in Waldeyer’s ring, and this may progress to produce respiratory compromise. The diagnosis depends on the demonstration of an anomalous serum M component (often <20 g/L [<2 g/dL]) that reacts with anti-IgG but not anti–light chain reagents. The M component is typically present in both serum and urine. Most of the paraproteins have been of the γ1 subclass, but other subclasses have been seen. The patients may have thrombocytopenia, eosinophilia, and nondiagnostic bone marrow that may show increased numbers of lymphocytes or plasma cells that do not stain for light chain. Patients usually have a rapid downhill course and die of infection; however, some patients have survived 5 years with chemotherapy. Therapy is indicated when symptomatic and involves chemotherapeutic combinations used in low-grade lymphoma. Rituximab has also been reported to show efficacy.
ALPHA HEAVY CHAIN DISEASE (SELIGMANN’S DISEASE)
This is the most common of the heavy chain diseases. It is closely related to a malignancy known as Mediterranean lymphoma, a disease that affects young persons in parts of the world where intestinal parasites are common, such as the Mediterranean, Asia, and South America. The disease is characterized by an infiltration of the lamina propria of the small intestine with lymphoplasmacytoid cells that secrete truncated alpha chains. Demonstrating alpha heavy chains is difficult because the alpha chains tend to polymerize and appear as a smear instead of a sharp peak on electrophoretic profiles. Despite the polymerization, hyperviscosity is not a common problem in alpha heavy chain disease. Without J chain–facilitated dimerization, viscosity does not increase dramatically. Light chains are absent from serum and urine. The patients present with chronic diarrhea, weight loss, and malabsorption and have extensive mesenteric and paraaortic adenopathy. Respiratory tract involvement occurs rarely. Patients may vary widely in their clinical course. Some may develop diffuse aggressive histologies of malignant lymphoma. Chemotherapy may produce long-term remissions. Rare patients appear to have responded to antibiotic therapy, raising the question of the etiologic role of antigenic stimulation, perhaps by some chronic intestinal infection. Chemotherapy plus antibiotics may be more effective than chemotherapy alone. Immunoproliferative small-intestinal disease (IPSID) is recognized as an infectious pathogen–associated human lymphoma that has association with Campylobacter jejuni. It involves mainly the proximal small intestine resulting in malabsorption, diarrhea, and abdominal pain. IPSID is associated with excessive plasma cell differentiation and produces truncated alpha heavy chain proteins lacking the light chains as well as the first constant domain. Early-stage IPSID responds to antibiotics (30–70% complete remission). Most untreated IPSID patients progress to lymphoplasmacytic and immunoblastic lymphoma. Patients not responding to antibiotic therapy are considered for treatment with combination chemotherapy used to treat low-grade lymphoma.
MU HEAVY CHAIN DISEASE
The secretion of isolated mu heavy chains into the serum appears to occur in a very rare subset of patients with CLL. The only features that may distinguish patients with mu heavy chain disease are the presence of vacuoles in the malignant lymphocytes and the excretion of kappa light chains in the urine. The diagnosis requires ultracentrifugation or gel filtration to confirm the nonreactivity of the paraprotein with the light chain reagents, because some intact macroglobulins fail to interact with these serums. The tumor cells seem to have a defect in the assembly of light and heavy chains, because they appear to contain both in their cytoplasm. There is no evidence that such patients should be treated differently from other patients with CLL (Chap. 134).
137 |
Amyloidosis |
GENERAL PRINCIPLES
Amyloidosis is the term for a group of protein folding disorders characterized by the extracellular deposition of insoluble polymeric protein fibrils in tissues and organs. A robust cellular machinery exists to chaperone proteins during the process of synthesis and secretion, to ensure that they achieve correct tertiary conformation and function, and to eliminate proteins that misfold. However, genetic mutation, incorrect processing, and other factors may favor misfolding, with consequent loss of normal protein function and intracellular or extracellular aggregation. Many diseases, ranging from cystic fibrosis to Alzheimer’s disease, are now known to involve protein misfolding. In the amyloidoses, the aggregates are typically extracellular, and the misfolded protein subunits assume a common antiparallel, β-pleated sheet–rich structural conformation that leads to the formation of higher-order oligomers and then of fibrils with unique staining properties. The term amyloid was coined around 1854 by the pathologist Rudolf Virchow, who thought that these deposits resembled starch (Latin amylum) under the microscope.
Amyloid diseases, defined by the biochemical nature of the protein composing the fibril deposits, are classified according to whether they are systemic or localized, whether they are acquired or inherited, and their clinical patterns (Table 137-1). The standard nomenclature is AX, where A indicates amyloidosis and X represents the protein present in the fibril. This chapter focuses primarily on the systemic forms. AL refers to amyloid composed of immunoglobulin light chains (LCs); this disorder, formerly termed primary systemic amyloidosis, arises from a clonal B cell or plasma cell disorder and can be associated with myeloma or lymphoma. AF refers to the familial amyloidoses, which are most commonly due to mutations in transthyretin (TTR), the transport protein for thyroid hormone and retinol-binding protein. AA amyloid is composed of the acute-phase reactant protein serum amyloid A (SAA) and occurs in the setting of chronic inflammatory or infectious diseases; for this reason, this type was formerly known as secondary amyloidosis. Aβ2M amyloid results from misfolded β2-microglobulin, occurring in individuals with long-standing renal disease who have undergone dialysis, typically for years. Aβ, the most common form of localized amyloidosis, is found in the brain of patients with Alzheimer’s disease after abnormal proteolytic processing and aggregation of polypeptides derived from the amyloid precursor protein.
|
AMYLOID PRECURSOR PROTEINS AND THEIR CLINICAL SYNDROMES |
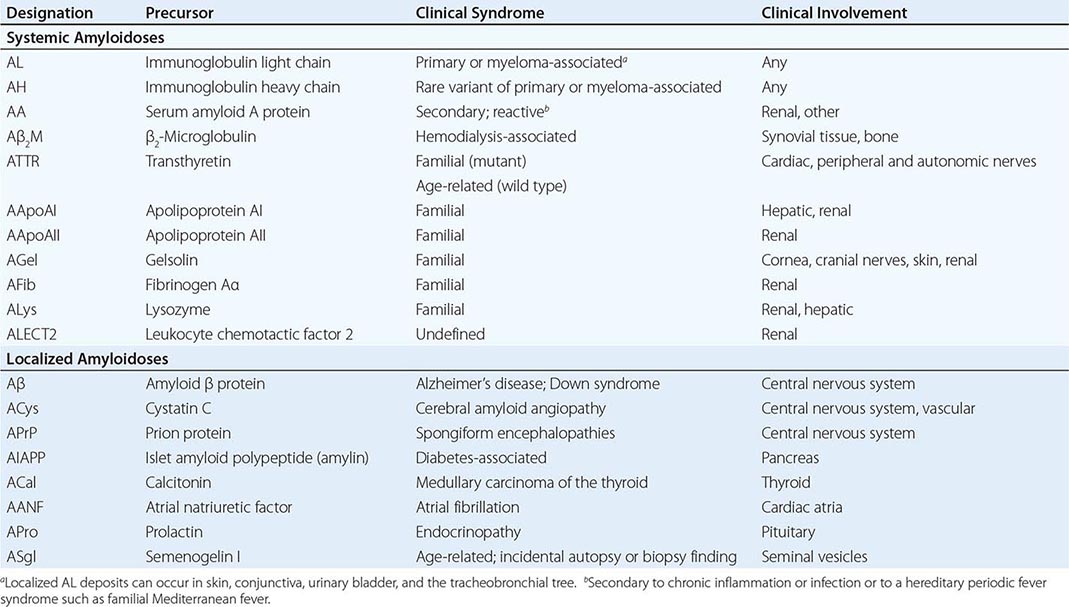
Diagnosis and treatment of the amyloidoses rest upon the histopathologic identification of amyloid deposits and immunohistochemical, biochemical, or genetic determination of amyloid type (Fig. 137-1). In the systemic amyloidoses, the clinically involved organs can be biopsied, but amyloid deposits may be found in any tissue of the body. Historically, blood vessels of the gingiva or rectal mucosa were often examined, but the most easily accessible tissue—positive in more than 80% of patients with systemic amyloidosis—is fat. After local anesthesia, fat is aspirated from the abdominal pannus with a 16-gauge needle. Fat globules expelled onto a glass slide can be stained, thus avoiding a surgical procedure. If this material is negative, more invasive biopsies of the kidney, heart, liver, or gastrointestinal tract can be considered in patients in whom amyloidosis is suspected. The regular β-sheet structure of amyloid deposits exhibits a unique “apple green” birefringence by polarized light microscopy when stained with Congo red dye; other regular protein structures (e.g., collagen) appear white under these conditions. The 10-nm-diameter fibrils can also be visualized by electron microscopy of paraformaldehyde-fixed tissue. Once amyloid is found, the protein type must be determined by immunohistochemistry, immunoelectron microscopy, or extraction and biochemical analysis employing mass spectrometry; gene sequencing is used to identify mutants causing AF amyloid. The patient’s history, physical findings, and clinical presentation, including age and ethnic origin, organ system involvement, underlying diseases, and family history, may provide helpful clues as to the type of amyloid. However, there can be considerable overlap in clinical presentations, and accurate typing is essential to guide appropriate therapy.
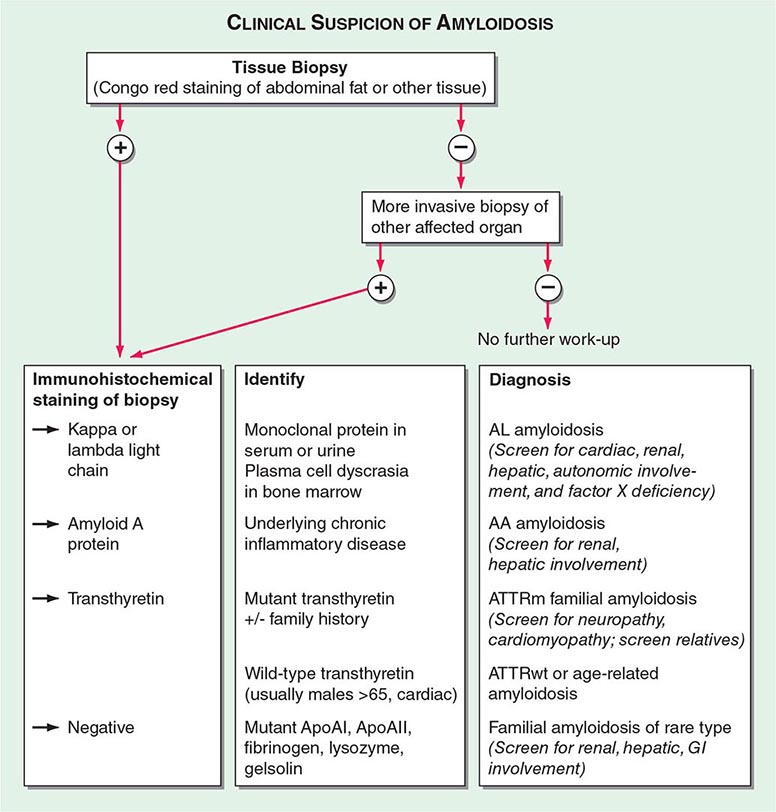
FIGURE 137-1 Algorithm for the diagnosis of amyloidosis and determination of type. Clinical suspicion: unexplained nephropathy, cardiomyopathy, neuropathy, enteropathy, arthropathy, and macroglossia. ApoAI, apolipoprotein AI; ApoAII, apolipoprotein AII; GI, gastrointestinal.
The mechanisms of fibril formation and tissue toxicity remain controversial. The “amyloid hypothesis,” as it is currently understood, proposes that precursor proteins undergo a process of reversible unfolding or misfolding; misfolded proteins form oligomeric aggregates, higher-order polymers, and then fibrils that deposit in tissues. Accumulating evidence suggests that the oligomeric intermediates may constitute the most toxic species. Oligomers are more capable than large fibrils of interacting with cells and inducing formation of reactive oxygen species and stress signaling. Ultimately, the fibrillar tissue deposits are likely to interfere with normal organ function. A more sophisticated understanding of the mechanisms leading to amyloid formation and cell and tissue dysfunction will continue to provide new targets for therapies.
The clinical syndromes of the amyloidoses are associated with relatively nonspecific alterations in routine laboratory tests. Blood counts are usually normal, although the erythrocyte sedimentation rate is frequently elevated. Patients with glomerular kidney involvement generally have proteinuria, often in the nephrotic range, leading to hypoalbuminemia that may be severe; patients with serum albumin levels below 2 g/dL generally have pedal edema or anasarca. Amyloid cardiomyopathy is characterized by concentric ventricular hypertrophy and diastolic dysfunction associated with elevation of brain natriuretic peptide or N-terminal pro–brain natriuretic peptide as well as troponin. These cardiac biomarkers can be used for disease staging, prognostication, and disease activity monitoring in patients with AL amyloidosis. Notably, renal insufficiency can falsely elevate levels of these biomarkers. Recently, biomarkers of cardiac remodeling—i.e., matrix metalloproteinases and tissue inhibitors of metalloproteinases—have been found to be altered in the serum of patients with amyloid cardiomyopathy. Electrocardiographic and echocardiographic features of amyloid cardiomyopathy are described below. Patients with liver involvement, even when advanced, usually develop cholestasis with an elevated alkaline phosphatase concentration but minimal alteration of the aminotransferases and preservation of synthetic function. In AL amyloidosis, endocrine organs may be infiltrated with fibrils, and hypothyroidism, hypoadrenalism, or even hypopituitarism can occur. Although none of these findings is specific for amyloidosis, the presence of abnormalities in multiple organ systems should raise suspicion regarding this diagnosis.
AL AMYLOIDOSIS
Etiology and Incidence AL amyloidosis is most frequently caused by a clonal expansion of bone-marrow plasma cells that secrete a monoclonal immunoglobulin LC depositing as amyloid fibrils in tissues. Whether the clonal plasma cells produce an LC that misfolds and leads to AL amyloidosis or an LC that folds properly, allowing the cells to inexorably expand over time and develop into multiple myeloma (Chap. 136), may depend upon primary sequence or other genetic or epigenetic factors. AL amyloidosis can occur with multiple myeloma or other B lymphoproliferative diseases, including non-Hodgkin’s lymphoma (Chap. 134) and Waldenström’s macroglobulinemia (Chap. 136). AL amyloidosis is the most common type of systemic amyloidosis diagnosed in North America. Its incidence has been estimated at 4.5 cases/100,000 population; however, ascertainment continues to be inadequate, and the true incidence may be much higher. AL amyloidosis, like other plasma cell diseases, usually occurs after age 40 and is often rapidly progressive and fatal if untreated.
Pathology and Clinical Features Amyloid deposits are usually widespread in AL amyloidosis and can be present in the interstitium of any organ outside the central nervous system. The amyloid fibril deposits are composed of full-length 23-kDa monoclonal immunoglobulin LCs as well as fragments. Accessory molecules co-deposited with LC fibrils (as well as with other amyloid fibrils) include serum amyloid P component, other proteins, glycosaminoglycans, and metal ions. Although all kappa and lambda LC subtypes have been identified in AL amyloid fibrils, lambda subtypes predominate. The lambda 6 subtype appears to have unique structural properties that predispose it to fibril formation, often in the kidney.
AL amyloidosis is often a rapidly progressive disease that presents as a pleiotropic set of clinical syndromes, recognition of which is key for initiation of the appropriate workup. Nonspecific symptoms of fatigue and weight loss are common; however, the diagnosis is rarely considered until symptoms referable to a specific organ develop. The kidneys are the most frequently involved organ and are affected in 70–80% of patients. Renal amyloidosis usually manifests as proteinuria, often in the nephrotic range and associated with hypoalbuminemia, secondary hypercholesterolemia and hypertriglyceridemia, and edema or anasarca. In some patients, interstitial rather than glomerular amyloid deposition can produce azotemia without proteinuria. The heart is the second most commonly affected organ (50–60% of patients), and cardiac involvement is the leading cause of death from AL amyloidosis. Early on, the electrocardiogram may show low voltage in the limb leads with a pseudo-infarct pattern. Echocardiographic features of disease include concentrically thickened ventricles and diastolic dysfunction with an abnormal strain pattern a “sparkly” appearance has been described but is often not seen with modern high-resolution echocardiographic techniques. Poor atrial contractility occurs even in sinus rhythm, and patients with cardiac amyloidosis are at risk for development of atrial thrombi and stroke. Cardiac MRI can show increased wall thickness, and characteristic enhancement of the subendocardium has been described following injection of gadolinium contrast. Nervous system symptoms include peripheral sensorimotor neuropathy and/or autonomic dysfunction manifesting as gastrointestinal motility disturbances (early satiety, diarrhea, constipation), impotence, orthostatic hypotension, and/or neurogenic bladder. Macroglossia (Fig. 137-2A), a pathognomonic sign of AL amyloidosis, is seen in only ~10% of patients. Liver involvement causes cholestasis and hepatomegaly. The spleen is frequently involved, and there may be functional hyposplenism in the absence of significant splenomegaly. Many patients experience “easy bruising” due to amyloid deposits in capillaries or deficiency of clotting factor X, which can bind to amyloid fibrils; cutaneous ecchymoses appear, particularly around the eyes, producing the “raccoon-eye” sign Fig. 137-2B), another uncommon but pathognomonic finding. Other findings include nail dystrophy (Fig. 137-2C), alopecia, and amyloid arthropathy with thickening of synovial membranes in the wrists and shoulders. The presence of a multisystemic illness or general fatigue along with any of these clinical syndromes should prompt a workup for amyloidosis.
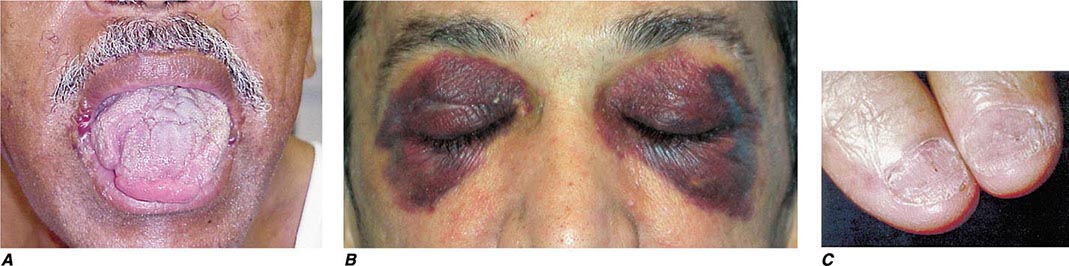
FIGURE 137-2 Clinical signs of AL amyloidosis. A. Macroglossia. B. Periorbital ecchymoses. C. Fingernail dystrophy.
Diagnosis Identification of an underlying clonal plasma cell or B lymphoproliferative process and a clonal LC are key to the diagnosis of AL amyloidosis. Serum protein electrophoresis and urine protein electrophoresis, although of value in multiple myeloma, are not useful screening tests if AL amyloidosis is suspected because the clonal LC or whole immunoglobulin often is not present in sufficient amounts to produce a monoclonal “M-spike” in the serum or LC (Bence Jones) protein in the urine. However, more than 90% of patients with AL amyloidosis have serum or urine monoclonal LC or whole immunoglobulin detectable by immunofixation electrophoresis of serum (SIFE) or urine (UIFE) (Fig. 137-3A) or by nephelometric measurement of “free” LCs (i.e., LCs circulating in monomeric form rather than in an immunoglobulin tetramer with heavy chain). Examining the ratio as well as the absolute amount of free LCs is essential, as renal insufficiency reduces LC clearance, elevating both isotypes. In addition, an increased percentage of plasma cells in the bone marrow—typically 5–30% of nucleated cells—is found in ~90% of patients. Kappa or lambda clonality should be demonstrated by flow cytometry, immunohistochemistry, or in situ hybridization for LC mRNA (Fig. 137-3B).
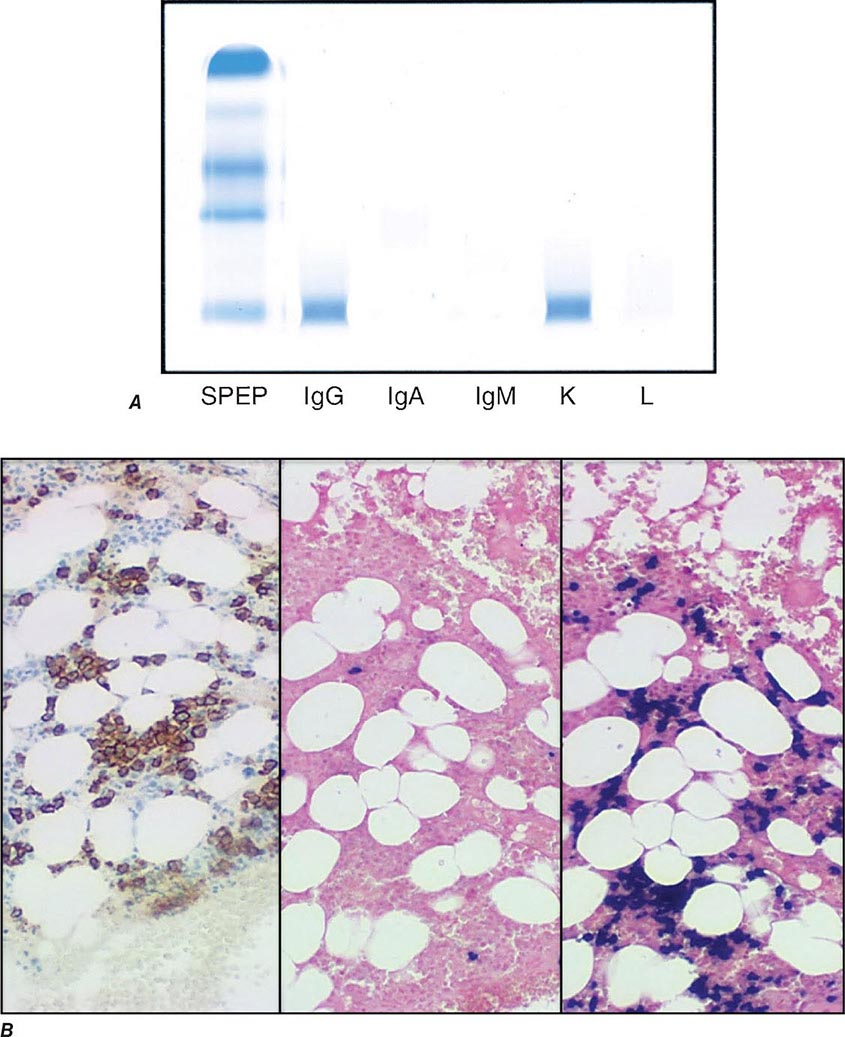
FIGURE 137-3 Laboratory features of AL amyloidosis. A. Serum immunofixation electrophoresis reveals an IgGκ monoclonal protein in this example; serum protein electrophoresis is often normal. B. Bone-marrow biopsy sections stained by immunohistochemistry with antibody to CD138 (syndecan, highly expressed on plasma cells) (left) or by in situ hybridization with fluorescein-tagged probes (Ventana Medical Systems) binding to κ mRNA (center) and λ mRNA (right) in plasma cells. (Photomicrograph courtesy of C. O’Hara; with permission.)
A monoclonal serum protein by itself is not diagnostic of amyloidosis, since monoclonal gammopathy of uncertain significance is common in older patients (Chap. 136). However, when monoclonal gammopathy of uncertain significance is found in patients with biopsy-proven amyloidosis, the AL type should be strongly suspected. Similarly, patients thought to have “smoldering myeloma” because of a modest elevation of bone-marrow plasma cells should be screened for AL amyloidosis if they have signs or symptoms of renal, cardiac, or neurologic disease. Accurate tissue amyloid typing is essential for appropriate treatment. Immunohistochemical staining of the amyloid deposits is useful if they bind one LC antibody in preference to the other; some AL deposits bind antibodies nonspecifically. Immunoelectron microscopy is more reliable, and mass spectrometry–based microsequencing of small amounts of protein extracted from fibril deposits can also be undertaken. In ambiguous cases, other forms of amyloidosis should be thoroughly excluded with appropriate genetic and other testing.
AA AMYLOIDOSIS
Etiology and Incidence AA amyloidosis can occur in association with almost any chronic inflammatory state (e.g., rheumatoid arthritis, inflammatory bowel disease, familial Mediterranean fever [Chap. 392], or other periodic fever syndromes) or chronic infections such as tuberculosis or subacute bacterial endocarditis. In the United States and Europe, AA amyloidosis has become less common, occurring in fewer than 2% of patients with these diseases, presumably because of advances in anti-inflammatory and antimicrobial therapies. It has also been described in association with Castleman’s disease, and patients with AA amyloidosis should undergo CT scanning to look for such tumors as well as serologic and microbiologic studies. AA amyloidosis can also be seen without any identifiable underlying disease. AA is the only type of systemic amyloidosis that occurs in children.
Pathology and Clinical Features Organ involvement in AA amyloidosis usually begins in the kidneys. Hepatomegaly, splenomegaly, and autonomic neuropathy can also occur as the disease progresses; cardiomyopathy occurs, albeit rarely. The symptoms and signs of AA disease cannot be reliably distinguished from those of AL amyloidosis. AA amyloid fibrils are usually composed of an 8-kDa, 76-amino-acid N-terminal portion of the 12-kDa precursor protein SAA. This acute-phase apoprotein is synthesized in the liver and transported by high-density lipoprotein (HDL3) in the plasma. Several years of an underlying inflammatory disease causing chronic elevation of SAA levels usually precede fibril formation, although infections can lead to AA deposition more rapidly.
ATTR AND AF AMYLOIDOSIS
![]() The familial amyloidoses are autosomal dominant diseases in which, beginning in midlife, a variant (FINE) plasma protein forms amyloid deposits. These diseases are rare, with an estimated incidence of <1 case/100,000 population in the United States, although founder effects in isolated areas of Portugal, Sweden, and Japan have led to a much higher incidence. The most common form of AF amyloidosis is ATTRm in the updated nomenclature, caused by mutation of the abundant plasma protein transthyretin (TTR, also known as prealbumin). More than 100 TTR mutations are known, and most are associated with ATTR amyloidosis. One variant, V122I, has a carrier frequency that may be as high as 4% in the African-American population and is associated with late-onset cardiac amyloidosis. The actual incidence and penetrance of disease in the African-American population is the subject of ongoing research, but ATTR amyloidosis warrants consideration in the differential diagnosis of African-American patients who present with concentric cardiac hypertrophy and evidence of diastolic dysfunction, particularly in the absence of a history of hypertension. Other familial amyloidoses, caused by variant apolipoproteins AI or AII, gelsolin, fibrinogen Aα, or lysozyme, are reported in only a few families worldwide. New amyloidogenic serum proteins continue to be identified periodically, including recently the leukocyte chemotactic factor LECT2, a cause of renal amyloidosis in Hispanic and Pakistani populations. To date, no mutation in the coding sequence for the LECT2 gene has been identified, so the heritability of ALECT2 is uncertain.
The familial amyloidoses are autosomal dominant diseases in which, beginning in midlife, a variant (FINE) plasma protein forms amyloid deposits. These diseases are rare, with an estimated incidence of <1 case/100,000 population in the United States, although founder effects in isolated areas of Portugal, Sweden, and Japan have led to a much higher incidence. The most common form of AF amyloidosis is ATTRm in the updated nomenclature, caused by mutation of the abundant plasma protein transthyretin (TTR, also known as prealbumin). More than 100 TTR mutations are known, and most are associated with ATTR amyloidosis. One variant, V122I, has a carrier frequency that may be as high as 4% in the African-American population and is associated with late-onset cardiac amyloidosis. The actual incidence and penetrance of disease in the African-American population is the subject of ongoing research, but ATTR amyloidosis warrants consideration in the differential diagnosis of African-American patients who present with concentric cardiac hypertrophy and evidence of diastolic dysfunction, particularly in the absence of a history of hypertension. Other familial amyloidoses, caused by variant apolipoproteins AI or AII, gelsolin, fibrinogen Aα, or lysozyme, are reported in only a few families worldwide. New amyloidogenic serum proteins continue to be identified periodically, including recently the leukocyte chemotactic factor LECT2, a cause of renal amyloidosis in Hispanic and Pakistani populations. To date, no mutation in the coding sequence for the LECT2 gene has been identified, so the heritability of ALECT2 is uncertain.
TTR deposits composed of unmutated fibrils occur with aging, and ATTRwt is being diagnosed with increasing frequency in Caucasian men >65 years of age with amyloid cardiomyopathy. Formerly termed senile systemic amyloidosis, ATTRwt has been found at autopsy in 25% of hearts from patients older than age 80 years. Why a wild type protein becomes amyloidogenic, and why patients bearing mutant TTR genes do not express disease until adulthood, remains a mystery.
Clinical Features and Diagnosis AF amyloidosis has a presentation that is variable but is usually consistent within kindreds affected by the same mutant protein. A family history makes AF disease more likely, but many patients present sporadically with new mutations. ATTR amyloidosis typically presents as a syndrome of familial amyloidotic polyneuropathy or familial amyloidotic cardiomyopathy. Peripheral neuropathy begins as a small-fiber lower-extremity sensory and motor neuropathy and progresses to the upper extremities. Autonomic neuropathy manifests as diarrhea with weight loss and orthostatic hypotension. Patients with TTR V30M, the most common mutation, have normal electrocardiograms but may develop conduction defects late in the disease. Patients with TTR T60A and several other mutations have myocardial thickening similar to that caused by AL amyloidosis, although heart failure is less common and long-term survival rates are usually better. Vitreous opacities caused by amyloid deposits are pathognomonic for ATTR amyloidosis.
Typical syndromes associated with other forms of AF disease include renal amyloidosis with mutant fibrinogen, lysozyme, or apolipoproteins; hepatic amyloidosis with apolipoprotein AI; and amyloidosis of cranial nerves and cornea with gelsolin. Patients with AF amyloidosis can present with clinical syndromes that mimic those of patients with AL disease. Rarely, AF carriers can develop AL disease or AF patients may have monoclonal gammopathy without AL. Thus, it is important to screen both for plasma cell disorders and for mutations in patients with amyloidosis. Variant TTRs can usually be detected by isoelectric focusing, but DNA sequencing is now standard for diagnosis of ATTR and other AF mutations.
Aβ2M AMYLOIDOSIS
Aβ2M amyloid is composed of β2-microglobulin, the invariant chain of class I human leukocyte antigens, and produces rheumatologic manifestations in patients undergoing long-term hemodialysis. β2-Microglobulin is excreted by the kidney, and levels become elevated in end-stage renal disease. The molecular mass of β2M is 11.8 kDa—above the cutoff of some dialysis membranes. The incidence of this disease appears to be declining with the use of newer membranes in high-flow dialysis techniques. Aβ2M amyloidosis usually presents as carpal tunnel syndrome, persistent joint effusions, spondyloarthropathy, or cystic bone lesions. Carpal tunnel syndrome is often the first symptom. In the past, persistent joint effusions accompanied by mild discomfort were found in up to 50% of patients who had undergone dialysis for >12 years. Involvement is bilateral, and large joints (shoulders, knees, wrists, and hips) are most frequently affected. The synovial fluid is noninflammatory, and β2M amyloid can be found if the sediment is stained with Congo red. Although less common, visceral β2M amyloid deposits do occasionally occur in the gastrointestinal tract, heart, tendons, and subcutaneous tissues of the buttocks. There is no specific therapy for Aβ2M amyloidosis, but cessation of dialysis after renal allografting may lead to symptomatic improvement.
SUMMARY
A diagnosis of amyloidosis should be considered in patients with unexplained nephropathy, cardiomyopathy (particularly with diastolic dysfunction), neuropathy (either peripheral or autonomic), enteropathy, or the pathognomonic soft tissue findings of macroglossia or periorbital ecchymoses. Pathologic identification of amyloid fibrils can be made with Congo red staining of aspirated abdominal fat or of an involved-organ biopsy specimen. Accurate typing by a combination of immunologic, biochemical, and genetic testing is essential in selecting appropriate therapy (Fig. 137-1). Systemic amyloidosis should not be considered an untreatable condition, as anti–plasma cell chemotherapy is highly effective in AL disease and targeted therapies are being developed for AA and ATTR disease. Tertiary referral centers can provide specialized diagnostic techniques and access to clinical trials for patients with these rare diseases.
ACKNOWLEDGMENT
This chapter represents a revised version of a chapter that was co-authored by Dr. Martha Skinner and Dr. David Seldin in previous editions of Harrison’s Principles of Internal Medicine.
138e |
Transfusion Biology and Therapy |
BLOOD GROUP ANTIGENS AND ANTIBODIES
The study of red blood cell (RBC) antigens and antibodies forms the foundation of transfusion medicine. Serologic studies initially characterized these antigens, but now the molecular composition and structure of many are known. Antigens, either carbohydrate or protein, are assigned to a blood group system based on the structure and similarity of the determinant epitopes. Other cellular blood elements and plasma proteins are also antigenic and can result in alloimmunization, the production of antibodies directed against the blood group antigens of another individual. These antibodies are called alloantibodies.
Antibodies directed against RBC antigens may result from “natural” exposure, particularly to carbohydrates that mimic some blood group antigens. Those antibodies that occur via natural stimuli are usually produced by a T cell–independent response (thus, generating no memory) and are IgM isotype. Autoantibodies (antibodies against autologous blood group antigens) arise spontaneously or as the result of infectious sequelae (e.g., from Mycoplasma pneumoniae) and are also often IgM. These antibodies are often clinically insignificant due to their low affinity for antigen at body temperature. However, IgM antibodies can activate the complement cascade and result in hemolysis. Antibodies that result from allogeneic exposure, such as transfusion or pregnancy, are usually IgG. IgG antibodies commonly bind to antigen at warmer temperatures and may hemolyze RBCs. Unlike IgM antibodies, IgG antibodies can cross the placenta and bind fetal erythrocytes bearing the corresponding antigen, resulting in hemolytic disease of the newborn, or hydrops fetalis.
Alloimmunization to leukocytes, platelets, and plasma proteins may also result in transfusion complications such as fevers and urticaria but generally does not cause hemolysis. Assay for these other alloantibodies is not routinely performed; however, they may be detected using special assays.
ABO ANTIGENS AND ANTIBODIES
The first blood group antigen system, recognized in 1900, was ABO, the most important in transfusion medicine. The major blood groups of this system are A, B, AB, and O. O type RBCs lack A or B antigens. These antigens are carbohydrates attached to a precursor backbone, may be found on the cellular membrane either as glycosphingolipids or glycoproteins, and are secreted into plasma and body fluids as glycoproteins. H substance is the immediate precursor on which the A and B antigens are added. This H substance is formed by the addition of fucose to the glycolipid or glycoprotein backbone. The subsequent addition of N-acetylgalactosamine creates the A antigen, whereas the addition of galactose produces the B antigen.
The genes that determine the A and B phenotypes are found on chromosome 9p and are expressed in a Mendelian codominant manner. The gene products are glycosyl transferases, which confer the enzymatic capability of attaching the specific antigenic carbohydrate. Individuals who lack the “A” and “B” transferases are phenotypically type “O,” whereas those who inherit both transferases are type “AB.” Rare individuals lack the H gene, which codes for fucose transferase, and cannot form H substance. These individuals are homozygous for the silent h allele (hh) and have Bombay phenotype (Oh).
The ABO blood group system is important because essentially all individuals produce antibodies to the ABH carbohydrate antigen that they lack. The naturally occurring anti-A and anti-B antibodies are termed isoagglutinins. Thus, type A individuals produce anti-B, whereas type B individuals make anti-A. Neither isoagglutinin is found in type AB individuals, whereas type O individuals produce both anti-A and anti-B. Thus, persons with type AB are “universal recipients” because they do not have antibodies against any ABO phenotype, whereas persons with type O blood can donate to essentially all recipients because their cells are not recognized by any ABO isoagglutinins. The rare individuals with Bombay phenotype produce antibodies to H substance (which is present on all red cells except those of hh phenotype) as well as to both A and B antigens and are therefore compatible only with other hh donors.
In most people, A and B antigens are secreted by the cells and are present in the circulation. Nonsecretors are susceptible to a variety of infections (e.g., Candida albicans, Neisseria meningitidis, Streptococcus pneumoniae, Haemophilus influenzae) because many organisms may bind to polysaccharides on cells. Soluble blood group antigens may block this binding.
RH SYSTEM
The Rh system is the second most important blood group system in pretransfusion testing. The Rh antigens are found on a 30- to 32-kDa RBC membrane protein that has no defined function. Although >40 different antigens in the Rh system have been described, five determinants account for the vast majority of phenotypes. The presence of the D antigen confers Rh “positivity,” whereas persons who lack the D antigen are Rh negative. Two allelic antigen pairs, E/e and C/c, are also found on the Rh protein. The three Rh genes, E/e, D, and C/c, are arranged in tandem on chromosome 1 and inherited as a haplotype, i.e., cDE or Cde. Two haplotypes can result in the phenotypic expression of two to five Rh antigens.
The D antigen is a potent alloantigen. About 15% of individuals lack this antigen. Exposure of these Rh-negative people to even small amounts of Rh-positive cells, by either transfusion or pregnancy, can result in the production of anti-D alloantibody.
OTHER BLOOD GROUP SYSTEMS AND ALLOANTIBODIES
More than 100 blood group systems are recognized, composed of more than 500 antigens. The presence or absence of certain antigens has been associated with various diseases and anomalies; antigens also act as receptors for infectious agents. Alloantibodies of importance in routine clinical practice are listed in Table 138e-1.
|
RBC BLOOD GROUP SYSTEMS AND ALLOANTIGENS |
Antibodies to Lewis system carbohydrate antigens are the most common cause of incompatibility during pretransfusion screening. The Lewis gene product is a fucosyl transferase and maps to chromosome 19. The antigen is not an integral membrane structure but is adsorbed to the RBC membrane from the plasma. Antibodies to Lewis antigens are usually IgM and cannot cross the placenta. Lewis antigens may be adsorbed onto tumor cells and may be targets of therapy.
I system antigens are also oligosaccharides related to H, A, B, and Le. I and i are not allelic pairs but are carbohydrate antigens that differ only in the extent of branching. The i antigen is an unbranched chain that is converted by the I gene product, a glycosyltransferase, into a branched chain. The branching process affects all the ABH antigens, which become progressively more branched in the first 2 years of life. Some patients with cold agglutinin disease or lymphomas can produce anti-I autoantibodies that cause RBC destruction. Occasional patients with mononucleosis or Mycoplasma pneumonia may develop cold agglutinins of either anti-I or anti-i specificity. Most adults lack i expression; thus, finding a donor for patients with anti-i is not difficult. Even though most adults express I antigen, binding is generally low at body temperature. Thus, administration of warm blood prevents isoagglutination.
The P system is another group of carbohydrate antigens controlled by specific glycosyltransferases. Its clinical significance is in rare cases of syphilis and viral infection that lead to paroxysmal cold hemoglobinuria. In these cases, an unusual autoantibody to P is produced that binds to RBCs in the cold and fixes complement upon warming. Antibodies with these biphasic properties are called Donath-Landsteiner antibodies. The P antigen is the cellular receptor of parvovirus B19 and also may be a receptor for Escherichia coli binding to urothelial cells.
The MNSsU system is regulated by genes on chromosome 4. M and N are determinants on glycophorin A, an RBC membrane protein, and S and s are determinants on glycophorin B. Anti-S and anti-s IgG antibodies may develop after pregnancy or transfusion and lead to hemolysis. Anti-U antibodies are rare but problematic; virtually every donor is incompatible because nearly all persons express U.
The Kell protein is very large (720 amino acids), and its secondary structure contains many different antigenic epitopes. The immunogenicity of Kell is third behind the ABO and Rh systems. The absence of the Kell precursor protein (controlled by a gene on X) is associated with acanthocytosis, shortened RBC survival, and a progressive form of muscular dystrophy that includes cardiac defects. This rare condition is called the McLeod phenotype. The Kx gene is linked to the 91-kDa component of the NADPH-oxidase on the × chromosome, deletion or mutation of which accounts for about 60% of cases of chronic granulomatous disease.
The Duffy antigens are codominant alleles, Fya and Fyb, that also serve as receptors for Plasmodium vivax. More than 70% of persons in malaria-endemic areas lack these antigens, probably from selective influences of the infection on the population. For unknown reasons, the lack of the Duffy antigen receptor for cytokines (DARC) is associated with mild neutropenia.
The Kidd antigens, Jka and Jkb, may elicit antibodies transiently. A delayed hemolytic transfusion reaction that occurs with blood tested as compatible is often related to delayed appearance of anti-Jka.
PRETRANSFUSION TESTING
Pretransfusion testing of a potential recipient consists of the “type and screen.” The “forward type” determines the ABO and Rh phenotype of the recipient’s RBC by using antisera directed against the A, B, and D antigens. The “reverse type” detects isoagglutinins in the patient’s serum and should correlate with the ABO phenotype, or forward type.
The alloantibody screen identifies antibodies directed against other RBC antigens. The alloantibody screen is performed by mixing patient serum with type O RBCs that contain the major antigens of most blood group systems and whose extended phenotype is known. The specificity of the alloantibody is identified by correlating the presence or absence of antigen with the results of the agglutination.
Cross-matching is ordered when there is a high probability that the patient will require a packed RBC (PRBC) transfusion. Blood selected for cross-matching must be ABO compatible and lack antigens for which the patient has alloantibodies. Nonreactive cross-matching confirms the absence of any major incompatibility and reserves that unit for the patient.
In the case of Rh-negative patients, every attempt must be made to provide Rh-negative blood components to prevent alloimmunization to the D antigen. In an emergency, Rh-positive blood can be safely transfused to an Rh-negative patient who lacks anti-D; however, the recipient is likely to become alloimmunized and produce anti-D. Rh-negative women of childbearing age who are transfused with products containing Rh-positive RBCs should receive passive immunization with anti-D (RhoGam or WinRho) to reduce or prevent sensitization.
BLOOD COMPONENTS
Blood products intended for transfusion are routinely collected as whole blood (450 mL) in various anticoagulants. Most donated blood is processed into components: PRBCs, platelets, and fresh-frozen plasma (FFP) or cryoprecipitate (Table 138e-2). Whole blood is first separated into PRBCs and platelet-rich plasma by slow centrifugation. The platelet-rich plasma is then centrifuged at high speed to yield one unit of random donor (RD) platelets and one unit of FFP. Cryoprecipitate is produced by thawing FFP to precipitate the plasma proteins and then separated by centrifugation.
|
CHARACTERISTICS OF SELECTED BLOOD COMPONENTS |
Apheresis technology is used for the collection of multiple units of platelets from a single donor. These single-donor apheresis platelets (SDAP) contain the equivalent of at least six units of RD platelets and have fewer contaminating leukocytes than pooled RD platelets.
Plasma may also be collected by apheresis. Plasma derivatives such as albumin, intravenous immunoglobulin, antithrombin, and coagulation factor concentrates are prepared from pooled plasma from many donors and are treated to eliminate infectious agents.
WHOLE BLOOD
Whole blood provides both oxygen-carrying capacity and volume expansion. It is the ideal component for patients who have sustained acute hemorrhage of ≥25% total blood volume loss. Whole blood is stored at 4°C to maintain erythrocyte viability, but platelet dysfunction and degradation of some coagulation factors occurs. In addition, 2,3-bisphosphoglycerate levels fall over time, leading to an increase in the oxygen affinity of the hemoglobin and a decreased capacity to deliver oxygen to the tissues, a problem with all red cell storage. Fresh whole blood avoids these problems, but it is typically used only in emergency settings (i.e., military). Whole blood is not readily available, because it is routinely processed into components.
PACKED RED BLOOD CELLS
This product increases oxygen-carrying capacity in the anemic patient. Adequate oxygenation can be maintained with a hemoglobin content of 70 g/L in the normovolemic patient without cardiac disease; however, comorbid factors may necessitate transfusion at a higher threshold. The decision to transfuse should be guided by the clinical situation and not by an arbitrary laboratory value. In the critical care setting, liberal use of transfusions to maintain near-normal levels of hemoglobin has not proven advantageous. In most patients requiring transfusion, levels of hemoglobin of 100 g/L are sufficient to keep oxygen supply from being critically low.
PRBCs may be modified to prevent certain adverse reactions. The majority of cellular blood products are now leukocyte reduced, and universal prestorage leukocyte reduction has been recommended. Prestorage filtration appears superior to bedside filtration as smaller amounts of cytokines are generated in the stored product. These PRBC units contain <5 × 106 donor white blood cells (WBCs), and their use lowers the incidence of posttransfusion fever, cytomegalovirus (CMV) infections, and alloimmunization. Other theoretical benefits include less immunosuppression in the recipient and lower risk of infections. Plasma, which may cause allergic reactions, can be removed from cellular blood components by washing.
PLATELETS
Thrombocytopenia is a risk factor for hemorrhage, and platelet transfusion reduces the incidence of bleeding. The threshold for prophylactic platelet transfusion is 10,000/μL. In patients without fever or infections, a threshold of 5000/μL may be sufficient to prevent spontaneous hemorrhage. For invasive procedures, the usual target level is 50,000/μL platelets.
Platelets are given either as pools prepared from RDs or as SDAPs from a single donor. In an unsensitized patient without increased platelet consumption (splenomegaly, fever, disseminated intravascular coagulation [DIC]), two units of transfused RD per square-meter body surface area (BSA) is anticipated to increase the platelet count by approximately 10,000/μL. Patients who have received multiple transfusions may be alloimmunized to many HLA- and platelet-specific antigens and have little or no increase in their posttransfusion platelet counts. Patients who may require multiple transfusions are best served by receiving SDAP and leukocyte-reduced components to lower the risk of alloimmunization.
Refractoriness to platelet transfusion may be evaluated using the corrected count increment (CCI):
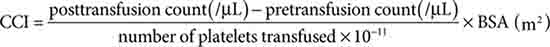
where BSA is body surface area measured in square meters. The platelet count performed 1 h after the transfusion is acceptable if the CCI is 10 × 109/mL, and after 18–24 h an increment of 7.5 × 109/mL is expected. Patients who have suboptimal responses are likely to have received multiple transfusions and have antibodies directed against class I HLA antigens. Refractoriness can be investigated by detecting anti-HLA antibodies in the recipient’s serum. Patients who are sensitized will often react with 100% of the lymphocytes used for the HLA-antibody screen, and HLA-matched SDAPs should be considered for patients who require transfusion. Although ABO-identical HLA-matched SDAPs provide the best chance for increasing the platelet count, locating these products is difficult. Platelet cross-matching is available in some centers. Additional clinical causes for a low platelet CCI include fever, bleeding, splenomegaly, DIC, or medications in the recipient.
FRESH-FROZEN PLASMA
FFP contains stable coagulation factors and plasma proteins: fibrinogen, antithrombin, albumin, and proteins C and S. Indications for FFP include correction of coagulopathies, including the rapid reversal of warfarin; supplying deficient plasma proteins; and treatment of thrombotic thrombocytopenic purpura. FFP should not be routinely used to expand blood volume. FFP is an acellular component and does not transmit intracellular infections, e.g., CMV. Patients who are IgA-deficient and require plasma support should receive FFP from IgA-deficient donors to prevent anaphylaxis (see below).
CRYOPRECIPITATE
Cryoprecipitate is a source of fibrinogen, factor VIII, and von Willebrand factor (VWF). It is ideal for supplying fibrinogen to the volume-sensitive patient. When factor VIII concentrates are not available, cryoprecipitate may be used because each unit contains approximately 80 units of factor VIII. Cryoprecipitate may also supply VWF to patients with dysfunctional (type II) or absent (type III) von Willebrand’s disease.
PLASMA DERIVATIVES
Plasma from thousands of donors may be pooled to derive specific protein concentrates, including albumin, intravenous immunoglobulin, antithrombin, and coagulation factors. In addition, donors who have high-titer antibodies to specific agents or antigens provide hyperimmune globulins, such as anti-D (RhoGam, WinRho), and antisera to hepatitis B virus (HBV), varicella-zoster virus, CMV, and other infectious agents.
ADVERSE REACTIONS TO BLOOD TRANSFUSION
Adverse reactions to transfused blood components occur despite multiple tests, inspections, and checks. Fortunately, the most common reactions are not life threatening, although serious reactions can present with mild symptoms and signs. Some reactions can be reduced or prevented by modified (filtered, washed, or irradiated) blood components. When an adverse reaction is suspected, the transfusion should be stopped and reported to the blood bank for investigation.
Transfusion reactions may result from immune and nonimmune mechanisms. Immune-mediated reactions are often due to preformed donor or recipient antibody; however, cellular elements may also cause adverse effects. Nonimmune causes of reactions are due to the chemical and physical properties of the stored blood component and its additives.
Transfusion-transmitted viral infections are increasingly rare due to improved screening and testing. As the risk of viral infection is reduced, the relative risk of other reactions increases, such as hemolytic transfusion reactions and sepsis from bacterially contaminated components. Pretransfusion quality assurance improvements further increase the safety of transfusion therapy. Infections, like any adverse transfusion reaction, must be brought to the attention of the blood bank for appropriate studies (Table 138e-3).
|
RISKS OF TRANSFUSION COMPLICATIONS |
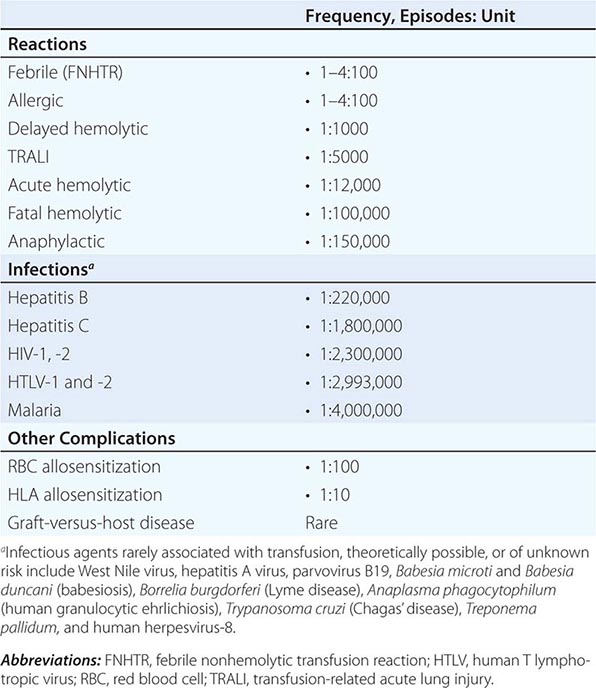
IMMUNE-MEDIATED REACTIONS
Acute Hemolytic Transfusion Reactions Immune-mediated hemolysis occurs when the recipient has preformed antibodies that lyse donor erythrocytes. The ABO isoagglutinins are responsible for the majority of these reactions. However, alloantibodies directed against other RBC antigens, i.e., Rh, Kell, and Duffy, are responsible for more fatal hemolytic transfusion reactions.
Acute hemolytic reactions may present with hypotension, tachypnea, tachycardia, fever, chills, hemoglobinemia, hemoglobinuria, chest and/or flank pain, and discomfort at the infusion site. Monitoring the patient’s vital signs before and during the transfusion is important to identify reactions promptly. When acute hemolysis is suspected, the transfusion must be stopped immediately, intravenous access maintained, and the reaction reported to the blood bank. A correctly labeled posttransfusion blood sample and any untransfused blood should be sent to the blood bank for analysis. The laboratory evaluation for hemolysis includes the measurement of serum haptoglobin, lactate dehydrogenase (LDH), and indirect bilirubin levels.
The immune complexes that result in RBC lysis can cause renal dysfunction and failure. Diuresis should be induced with intravenous fluids and furosemide or mannitol. Tissue factor released from the lysed erythrocytes may initiate DIC. Coagulation studies including prothrombin time (PT), activated partial thromboplastin time (aPTT), fibrinogen, and platelet count should be monitored in patients with hemolytic reactions.
Errors at the patient’s bedside, such as mislabeling the sample or transfusing the wrong patient, are responsible for the majority of these reactions. The blood bank investigation of these reactions includes examination of the pre- and posttransfusion samples for hemolysis and repeat typing of the patient samples; direct antiglobulin test (DAT), sometimes called the direct Coombs test, of the posttransfusion sample; repeating the cross-matching of the blood component; and checking all clerical records for errors. DAT detects the presence of antibody or complement bound to RBCs in vivo (Fig. 138e-1).
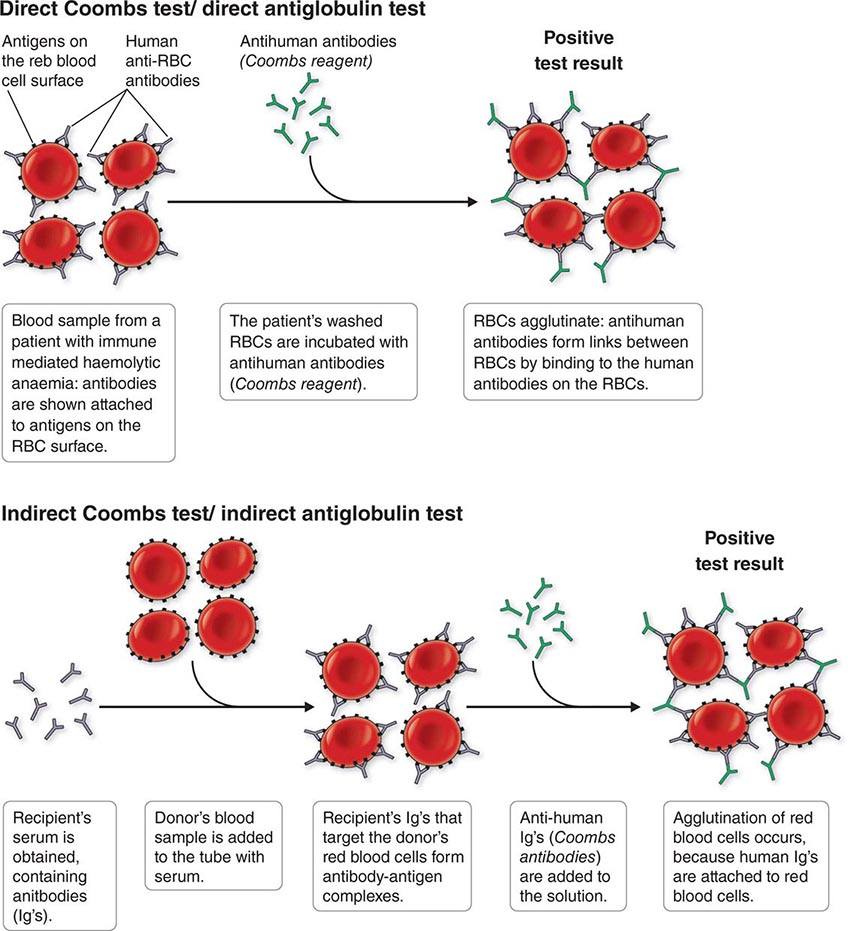
FIGURE 138e-1 Direct and indirect Coombs test. The direct Coombs (antiglobulin) test detects the presence of antibodies (or complement) on the surface of erythrocytes. The indirect Coombs (antiglobulin) test detects antibodies in the serum that may bind to donor erythrocytes. RBC, red blood cell. (Adapted from http://upload.wikimedia.org/wikipedia/commons/1/1c/coombs_test_schematic.png.)
Delayed Hemolytic and Serologic Transfusion Reactions Delayed hemolytic transfusion reactions (DHTRs) are not completely preventable. These reactions occur in patients previously sensitized to RBC alloantigens who have a negative alloantibody screen due to low antibody levels. When the patient is transfused with antigen-positive blood, an anamnestic response results in the early production of alloantibody that binds donor RBCs. The alloantibody is detectable 1–2 weeks following the transfusion, and the posttransfusion DAT may become positive due to circulating donor RBCs coated with antibody or complement. The transfused, alloantibody-coated erythrocytes are cleared by the reticuloendothelial system. These reactions are detected most commonly in the blood bank when a subsequent patient sample reveals a positive alloantibody screen or a new alloantibody in a recently transfused recipient.
No specific therapy is usually required, although additional RBC transfusions may be necessary. Delayed serologic transfusion reactions are similar to DHTR, because the DAT is positive and alloantibody is detected; however, RBC clearance is not increased.
Febrile Nonhemolytic Transfusion Reaction The most frequent reaction associated with the transfusion of cellular blood components is a febrile nonhemolytic transfusion reaction (FNHTR). These reactions are characterized by chills and rigors and a ≥1°C rise in temperature. FNHTR is diagnosed when other causes of fever in the transfused patient are ruled out. Antibodies directed against donor leukocyte and HLA antigens may mediate these reactions; thus, multiply transfused patients and multiparous women are felt to be at increased risk. Although anti-HLA antibodies may be demonstrated in the recipient’s serum, investigation is not routinely done because of the mild nature of most FNHTR. The use of leukocyte-reduced blood products may prevent or delay sensitization to leukocyte antigens and thereby reduce the incidence of these febrile episodes. Cytokines released from cells within stored blood components may mediate FNHTR; thus, leukoreduction before storage may prevent these reactions.
Allergic Reactions Urticarial reactions are related to plasma proteins found in transfused components. Mild reactions may be treated symptomatically by temporarily stopping the transfusion and administering antihistamines (diphenhydramine, 50 mg orally or intramuscularly). The transfusion may be completed after the signs and/or symptoms resolve. Patients with a history of allergic transfusion reaction should be premedicated with an antihistamine. Cellular components can be washed to remove residual plasma for the extremely sensitized patient.
Anaphylactic Reaction This severe reaction presents after transfusion of only a few milliliters of the blood component. Symptoms and signs include difficulty breathing, coughing, nausea and vomiting, hypotension, bronchospasm, loss of consciousness, respiratory arrest, and shock. Treatment includes stopping the transfusion, maintaining vascular access, and administering epinephrine (0.5–1 mL of 1:1000 dilution subcutaneously). Glucocorticoids may be required in severe cases.
Patients who are IgA-deficient, <1% of the population, may be sensitized to this Ig class and are at risk for anaphylactic reactions associated with plasma transfusion. Individuals with severe IgA deficiency should therefore receive only IgA-deficient plasma and washed cellular blood components. Patients who have anaphylactic or repeated allergic reactions to blood components should be tested for IgA deficiency.
Graft-Versus-Host Disease Graft-versus-host disease (GVHD) is a frequent complication of allogeneic stem cell transplantation, in which lymphocytes from the donor attack and cannot be eliminated by an immunodeficient host. Transfusion-related GVHD is mediated by donor T lymphocytes that recognize host HLA antigens as foreign and mount an immune response, which is manifested clinically by the development of fever, a characteristic cutaneous eruption, diarrhea, and liver function abnormalities. GVHD can also occur when blood components that contain viable T lymphocytes are transfused to immunodeficient recipients or to immunocompetent recipients who share HLA antigens with the donor (e.g., a family donor). In addition to the aforementioned clinical features of GVHD, transfusion-associated GVHD (TA-GVHD) is characterized by marrow aplasia and pancytopenia. TA-GVHD is highly resistant to treatment with immunosuppressive therapies, including glucocorticoids, cyclosporine, antithymocyte globulin, and ablative therapy followed by allogeneic bone marrow transplantation. Clinical manifestations appear at 8–10 days, and death occurs at 3–4 weeks after transfusion.
TA-GVHD can be prevented by irradiation of cellular components (minimum of 2500 cGy) before transfusion to patients at risk. Patients at risk for TA-GVHD include fetuses receiving intrauterine transfusions, selected immunocompetent (e.g., lymphoma patients) or immunocompromised recipients, recipients of donor units known to be from a blood relative, and recipients who have undergone marrow transplantation. Directed donations by family members should be discouraged (they are not less likely to transmit infection); lacking other options, the blood products from family members should always be irradiated.
Transfusion-Related Acute Lung Injury Transfusion-related acute lung injury (TRALI) is the most common cause of transfusion related fatalities. The recipient develops symptoms of hypoxia (Pao2/FIo2 <300 mmHg) and signs of noncardiogenic pulmonary edema, including bilateral interstitial infiltrates on chest x-ray, either during or within 6 h of transfusion. Treatment is supportive, and patients usually recover without sequelae. TRALI usually results from the transfusion of donor plasma that contains high-titer anti-HLA class II antibodies that bind recipient leukocytes. The leukocytes aggregate in the pulmonary vasculature and release mediators that increase capillary permeability. Testing the donor’s plasma for anti-HLA antibodies can support this diagnosis. The implicated donors are frequently multiparous women. The transfusion of plasma from male and nulliparous women donors reduces the risk of TRALI. Recipient factors that are associated with increased risk of TRALI include smoking, chronic alcohol use, shock, liver surgery (transplantation), mechanical ventilation with >30 cmH2O pressure support and positive fluid balance.
Posttransfusion Purpura This reaction presents as thrombocytopenia 7–10 days after platelet transfusion and occurs predominantly in women. Platelet-specific antibodies are found in the recipient’s serum, and the most frequently recognized antigen is HPA-1a found on the platelet glycoprotein IIIa receptor. The delayed thrombocytopenia is due to the production of antibodies that react to both donor and recipient platelets. Additional platelet transfusions can worsen the thrombocytopenia and should be avoided. Treatment with intravenous immunoglobulin may neutralize the effector antibodies, or plasmapheresis can be used to remove the antibodies.
Alloimmunization A recipient may become alloimmunized to a number of antigens on cellular blood elements and plasma proteins. Alloantibodies to RBC antigens are detected during pretransfusion testing, and their presence may delay finding antigen-negative cross-match-compatible products for transfusion. Women of childbearing age who are sensitized to certain RBC antigens (i.e., D, c, E, Kell, or Duffy) are at risk for bearing a fetus with hemolytic disease of the newborn. Matching for D antigen is the only pretransfusion selection test to prevent RBC alloimmunization.
Alloimmunization to antigens on leukocytes and platelets can result in refractoriness to platelet transfusions. Once alloimmunization has developed, HLA-compatible platelets from donors who share similar antigens with the recipient may be difficult to find. Hence, prudent transfusion practice is directed at preventing sensitization through the use of leukocyte-reduced cellular components, as well as limiting antigenic exposure by the judicious use of transfusions and use of SDAPs.
NONIMMUNOLOGIC REACTIONS
Fluid Overload Blood components are excellent volume expanders, and transfusion may quickly lead to transfusion-associated circulatory overload (TACO). Dyspnea with PaO2 <90% on room air, bilateral infiltrates on chest x-ray, and systolic hypertension are found with TACO. Brain natriuretic peptide (BNP) is often elevated (>1.5) compared with pretransfusion levels. Monitoring the rate and volume of the transfusion and using a diuretic can minimize this problem.
Hypothermia Refrigerated (4°C) or frozen (–18°C or below) blood components can result in hypothermia when rapidly infused. Cardiac dysrhythmias can result from exposing the sinoatrial node to cold fluid. Use of an in-line warmer will prevent this complication.
Electrolyte Toxicity RBC leakage during storage increases the concentration of potassium in the unit. Neonates and patients in renal failure are at risk for hyperkalemia. Preventive measures, such as using fresh or washed RBCs, are warranted for neonatal transfusions because this complication can be fatal.
Citrate, commonly used to anticoagulate blood components, chelates calcium and thereby inhibits the coagulation cascade. Hypocalcemia, manifested by circumoral numbness and/or tingling sensation of the fingers and toes, may result from multiple rapid transfusions. Because citrate is quickly metabolized to bicarbonate, calcium infusion is seldom required in this setting. If calcium or any other intravenous infusion is necessary, it must be given through a separate line.
Iron Overload Each unit of RBCs contains 200–250 mg of iron. Symptoms and signs of iron overload affecting endocrine, hepatic, and cardiac function are common after 100 units of RBCs have been transfused (total-body iron load of 20 g). Preventing this complication by using alternative therapies (e.g., erythropoietin) and judicious transfusion is preferable and cost effective. Chelating agents, such as deferoxamine and deferasirox, are available, but the response is often suboptimal.
Hypotensive Reactions Transient hypotension may be noted among transfused patients who take angiotensin-converting enzyme (ACE) inhibitors. Because blood products contain bradykinin that is normally degraded by ACE, patients on ACE inhibitors may have increased bradykinin levels that cause hypotension in the recipient. The blood pressure typically returns to normal without intervention.
Immunomodulation Transfusion of allogeneic blood is immunosuppressive. Multiply transfused renal transplant recipients are less likely to reject the graft, and transfusion may result in poorer outcomes in cancer patients and increase the risk of infections. Transfusion-related immunomodulation is thought to be mediated by transfused leukocytes. Leukocyte-depleted cellular products may cause less immunosuppression, although controlled data are unlikely to be obtained as the blood supply becomes universally leukocyte depleted.
INFECTIOUS COMPLICATIONS
The blood supply is initially screened by selecting healthy donors without high-risk lifestyles, medical conditions, or exposure to transmissible pathogens, such as intravenous drug use or visiting malaria endemic areas. Multiple tests performed on donated blood to detect the presence of infectious agents using nucleic acid amplification testing (NAAT) or evidence of prior infections by testing for antibodies to pathogens and sterility of platelet products further reduce the risk of transfusion-acquired infections.
Viral Infections • HEPATITIS C VIRUS (HCV) Blood donations are tested for antibodies to HCV and HCV RNA. The risk of acquiring HCV through transfusion is now calculated to be approximately 1 in 2,000,000 units. Infection with HCV may be asymptomatic or lead to chronic active hepatitis, cirrhosis, and liver failure.
HUMAN IMMUNODEFICIENCY VIRUS TYPE 1 (HIV-1) Donated blood is tested for antibodies to HIV-1, HIV-1 p24 antigen, and HIV RNA using NAAT. Approximately a dozen seronegative donors have been shown to harbor HIV RNA. The risk of HIV-1 infection per transfusion episode is 1 in 2,000,000. Antibodies to HIV-2 are also measured in donated blood. No cases of HIV-2 infection have been reported in the United States since 1992.
HEPATITIS B VIRUS (HBV) Donated blood is screened for HBV using assays for hepatitis B surface antigen (HbsAg). NAAT testing is not practical because of slow viral replication and lower levels of viremia. The risk of transfusion-associated HBV infection is several times greater than for HCV. Vaccination of individuals who require long-term transfusion therapy can prevent this complication.
OTHER HEPATITIS VIRUSES Hepatitis A virus is rarely transmitted by transfusion; infection is typically asymptomatic and does not lead to chronic disease. Other transfusion-transmitted viruses—TT virus, SEN virus, and GB virus C—do not cause chronic hepatitis or other disease states. Routine testing does not appear to be warranted.
WEST NILE VIRUS (WNV) Transfusion-transmitted WNV infections were documented in 2002. This RNA virus can be detected using NAAT; routine screening began in 2003. WNV infections range in severity from asymptomatic to fatal, with the older population at greater risk.
CYTOMEGALOVIRUS This ubiquitous virus infects ≥50% of the general population and is transmitted by the infected “passenger” WBCs found in transfused PRBCs or platelet components. Cellular components that are leukocyte-reduced have a decreased risk of transmitting CMV, regardless of the serologic status of the donor. Groups at risk for CMV infections include immunosuppressed patients, CMV-seronegative transplant recipients, and neonates; these patients should receive leukocyte-depleted components or CMV seronegative products.
HUMAN T LYMPHOTROPIC VIRUS (HTLV) TYPE 1 Assays to detect HTLV-1 and -2 are used to screen all donated blood. HTLV-1 is associated with adult T cell leukemia/lymphoma and tropical spastic paraparesis in a small percentage of infected persons (Chap. 225e). The risk of HTLV-1 infection via transfusion is 1 in 641,000 transfusion episodes. HTLV-2 is not clearly associated with any disease.
PARVOVIRUS B19 Blood components and pooled plasma products can transmit this virus, the etiologic agent of erythema infectiosum, or fifth disease, in children. Parvovirus B19 shows tropism for erythroid precursors and inhibits both erythrocyte production and maturation. Pure red cell aplasia, presenting either as acute aplastic crisis or chronic anemia with shortened RBC survival, may occur in individuals with an underlying hematologic disease, such as sickle cell disease or thalassemia (Chap. 130). The fetus of a seronegative woman is at risk for developing hydrops from this virus.
Bacterial Contamination The relative risk of transfusion-transmitted bacterial infection has increased as the absolute risk of viral infections has dramatically decreased.
Most bacteria do not grow well at cold temperatures; thus, PRBCs and FFP are not common sources of bacterial contamination. However, some gram-negative bacteria can grow at 1–6°C. Yersinia, Pseudomonas, Serratia, Acinetobacter, and Escherichia species have all been implicated in infections related to PRBC transfusion. Platelet concentrates, which are stored at room temperature, are more likely to contain skin contaminants such as gram-positive organisms, including coagulase-negative staphylococci. It is estimated that 1 in 1000–2000 platelet components is contaminated with bacteria. The risk of death due to transfusion-associated sepsis has been calculated at 1 in 17,000 for single-unit platelets derived from whole blood donation and 1 in 61,000 for apheresis product. Since 2004, blood banks have instituted methods to detect contaminated platelet components.
Recipients of transfusion contaminated with bacteria may develop fever and chills, which can progress to septic shock and DIC. These reactions may occur abruptly, within minutes of initiating the transfusion, or after several hours. The onset of symptoms and signs is often sudden and fulminant, which distinguishes bacterial contamination from an FNHTR. The reactions, particularly those related to gram-negative contaminants, are the result of infused endotoxins formed within the contaminated stored component.
When these reactions are suspected, the transfusion must be stopped immediately. Therapy is directed at reversing any signs of shock, and broad-spectrum antibiotics should be given. The blood bank should be notified to identify any clerical or serologic error. The blood component bag should be sent for culture and Gram stain.
Other Infectious Agents Various parasites, including those causing malaria, babesiosis, and Chagas’ disease, can be transmitted by blood transfusion. Geographic migration and travel of donors shift the incidence of these rare infections. Other agents implicated in transfusion transmission include dengue, chikungunya virus, variant Creutzfeldt-Jakob disease, A. phagocytophilum, and yellow fever vaccine virus, and the list will grow. Tests for some pathogens are available, such as T. cruzi, but not universally required, whereas others are being developed (B. microti). These infections should be considered in the transfused patient in the appropriate clinical setting.
ALTERNATIVES TO TRANSFUSION
Alternatives to allogeneic blood transfusions that avoid homologous donor exposures with attendant immunologic and infectious risks remain attractive. Autologous blood is the best option when transfusion is anticipated. However, the cost-benefit ratio of autologous transfusion remains high. No transfusion is a zero-risk event; clerical errors and bacterial contamination remain potential complications even with autologous transfusions. Additional methods of autologous transfusion in the surgical patient include preoperative hemodilution, recovery of shed blood from sterile surgical sites, and postoperative drainage collection. Directed or designated donation from friends and family of the potential recipient has not been safer than volunteer donor component transfusions. Such directed donations may in fact place the recipient at higher risk for complications such as GVHD and alloimmunization.
Granulocyte and granulocyte-macrophage colony-stimulating factors are clinically useful to hasten leukocyte recovery in patients with leukopenia related to high-dose chemotherapy. Erythropoietin stimulates erythrocyte production in patients with anemia of chronic renal failure and other conditions, thus avoiding or reducing the need for transfusion. This hormone can also stimulate erythropoiesis in the autologous donor to enable additional donation.
139e |
Hematopoietic Cell Transplantation |
Bone marrow transplantation was the original term used to describe the collection and transplantation of hematopoietic stem cells, but with the demonstration that peripheral blood and umbilical cord blood are also useful sources of stem cells, hematopoietic cell transplantation has become the preferred generic term for this process. The procedure is usually carried out for one of two purposes: (1) to replace an abnormal but nonmalignant lymphohematopoietic system with one from a normal donor or (2) to treat malignancy by allowing the administration of higher doses of myelosuppressive therapy than would otherwise be possible. The use of hematopoietic cell transplantation has been increasing, both because of its efficacy in selected diseases and because of increasing availability of donors. The Center for International Blood and Marrow Transplant Research (http://www.cibmtr.org) estimates that about 65,000 transplants are performed each year.
THE HEMATOPOIETIC STEM CELL
Several features of the hematopoietic stem cell make transplantation clinically feasible, including its remarkable regenerative capacity, its ability to home to the marrow space following intravenous injection, and the ability of the stem cell to be cryopreserved (Chap. 89e). Transplantation of a single stem cell can replace the entire lymphohematopoietic system of an adult mouse. In humans, transplantation of a small percentage of a donor’s bone marrow volume regularly results in complete and sustained replacement of the recipient’s entire lymphohematopoietic system, including all red cells, granulocytes, B and T lymphocytes, and platelets, as well as cells comprising the fixed macrophage population, including Kupffer cells of the liver, pulmonary alveolar macrophages, osteoclasts, Langerhans cells of the skin, and brain microglial cells. The ability of the hematopoietic stem cell to home to the marrow following intravenous injection is mediated, in part, by an interaction between CXCL12, also known as stromal cell–derived factor 1, produced by marrow stromal cells and the alpha-chemokine receptor CXCR4 found on stem cells. Homing is also influenced by the interaction of cell-surface molecules, termed selectins, including E- and L-selectin, on bone marrow endothelial cells with ligands, termed integrins, such as VLA-4, on early hematopoietic cells. Human hematopoietic stem cells can survive freezing and thawing with little, if any, damage, making it possible to remove and store a portion of the patient’s own bone marrow for later reinfusion following treatment of the patient with high-dose myelotoxic therapy.
CATEGORIES OF HEMATOPOIETIC CELL TRANSPLANTATION
Hematopoietic cell transplantation can be described according to the relationship between the patient and the donor and by the anatomic source of stem cells. In ~1% of cases, patients have identical twins who can serve as donors. With the use of syngeneic donors, there is no risk of graft-versus-host disease (GVHD), which often complicates allogeneic transplantation, and unlike the use of autologous marrow, there is no risk that the stem cells are contaminated with tumor cells.
Allogeneic transplantation involves a donor and a recipient who are not genetically identical. Following allogeneic transplantation, immune cells transplanted with the stem cells or developing from them can react against the patient, causing GVHD. Alternatively, if the immunosuppressive preparative regimen used to treat the patient before transplant is inadequate, immunocompetent cells of the patient can cause graft rejection. The risks of these complications are greatly influenced by the degree of matching between donor and recipient for antigens encoded by genes of the major histocompatibility complex.
The human leukocyte antigen (HLA) molecules are responsible for binding antigenic proteins and presenting them to T cells. The antigens presented by HLA molecules may derive from exogenous sources (e.g., during active infections) or may be endogenous proteins. If individuals are not HLA-matched, T cells from one individual will react strongly to the mismatched HLA, or “major antigens,” of the second. Even if the individuals are HLA-matched, the T cells of the donor may react to differing endogenous or “minor antigens” presented by the HLA of the recipient. Reactions to minor antigens tend to be less vigorous. The genes of major relevance to transplantation include HLA-A, -B, -C, and -D; they are closely linked and therefore tend to be inherited as haplotypes, with only rare crossovers between them. Thus, the odds that any one full sibling will match a patient are one in four, and the probability that the patient has an HLA-identical sibling is 1 – (0.75)n, where n equals the number of siblings.
With current techniques, the risk of graft rejection is 1–3%, and the risk of severe, life-threatening acute GVHD is ~15% following transplantation between HLA-identical siblings. The incidence of graft rejection and GVHD increases progressively with the use of family member donors mismatched for one, two, or three antigens. Although survival following a one-antigen mismatched transplant is not markedly altered, survival following two- or three-antigen mismatched transplants is significantly reduced. Since the formation of the National Marrow Donor Program and other registries, it has become possible to identify HLA-matched unrelated donors for many patients. The genes encoding HLA antigens are highly polymorphic, and thus the odds of any two unrelated individuals being HLA identical are extremely low, somewhat less than 1 in 10,000. However, by identifying and typing >20 million volunteer donors, HLA-matched donors can now be found for ~60% of patients for whom a search is initiated, with higher rates among whites and lower rates among minorities and patients of mixed race. It takes, on average, 3–4 months to complete a search and schedule and initiate an unrelated donor transplant. With improvements in HLA typing and supportive care measures, survival following matched unrelated donor transplantation is essentially the same as that seen with HLA-matched siblings.
Autologous transplantation involves the removal and storage of the patient’s own stem cells with subsequent reinfusion after the patient receives high-dose myeloablative therapy. Unlike allogeneic transplantation, there is no risk of GVHD or graft rejection with autologous transplantation. On the other hand, autologous transplantation lacks a graft-versus-tumor (GVT) effect, and the autologous stem cell product can be contaminated with tumor cells, which could lead to relapse. A variety of techniques have been developed to “purge” autologous products of tumor cells. Some use antibodies directed at tumor-associated antigens plus complement, antibodies linked to toxins, or antibodies conjugated to immunomagnetic beads. Another technique is positive selection of stem cells using antibodies to CD34, with subsequent column adherence or flow techniques to select normal stem cells while leaving tumor cells behind. All of these approaches can reduce the number of tumor cells from 1000- to 10,000-fold and are clinically feasible; however, no prospective randomized trials have yet shown that any of these approaches results in a decrease in relapse rates or improvements in disease-free or overall survival.
Bone marrow aspirated from the posterior and anterior iliac crests initially was the source of hematopoietic stem cells for transplantation. Typically, anywhere from 1.5 to 5 × 108 nucleated marrow cells per kilogram are collected for allogeneic transplantation. Several studies have found improved survival in the settings of both matched sibling and unrelated transplantation by transplanting higher numbers of bone marrow cells.
Hematopoietic stem cells circulate in the peripheral blood but in very low concentrations. Following the administration of certain hematopoietic growth factors, including granulocyte colony-stimulating factor (G-CSF) or granulocyte-macrophage colony-stimulating factor (GM-CSF), and during recovery from intensive chemotherapy, the concentration of hematopoietic progenitor cells in blood, as measured either by colony-forming units or expression of the CD34 antigen, increases markedly. This has made it possible to harvest adequate numbers of stem cells from the peripheral blood for transplantation. Donors are typically treated with 4 or 5 days of hematopoietic growth factor, following which stem cells are collected in one or two 4-h pheresis sessions. In the autologous setting, transplantation of >2.5 × 106 CD34 cells per kilogram, a number that can be collected in most circumstances, leads to rapid and sustained engraftment in virtually all cases. In the 10–20% of patients who fail to mobilize sufficient CD34+ cells with growth factor alone, the addition of plerixafor, an antagonist of CXCR4, may be useful. Compared to the use of autologous marrow, use of peripheral blood stem cells results in more rapid hematopoietic recovery, with granulocytes recovering to 500/μL by day 12 and platelets recovering to 20,000/μL by day 14. Although this more rapid recovery diminishes the morbidity rate of transplantation, no studies show improved survival.
Hesitation in studying the use of peripheral blood stem cells for allogeneic transplantation occurred because peripheral blood stem cell products contain as much as 1 log more T cells than are contained in the typical marrow harvest; in animal models, the incidence of GVHD is related to the number of T cells transplanted. Nonetheless, clinical trials have shown that the use of growth factor–mobilized peripheral blood stem cells from HLA-matched family members leads to faster engraftment without an increase in acute GVHD. Chronic GVHD may be increased with peripheral blood stem cells, but in trials conducted so far, this has been more than balanced by reductions in relapse rates and nonrelapse mortality rates, with the use of peripheral blood stem cells resulting in improved overall survival. However, in the setting of matched unrelated donor transplantation, use of peripheral blood results in more chronic GVHD without a compensatory survival advantage, favoring the use of bone marrow in this setting.
Umbilical cord blood contains a high concentration of hematopoietic progenitor cells, allowing for its use as a source of stem cells for transplantation. Cord blood transplantation from family members has been explored in the setting where the immediate need for transplantation precludes waiting the 9 or so months generally required for the baby to mature to the point of donating marrow. Use of cord blood results in slower engraftment and peripheral count recovery than seen with marrow but a lower incidence of GVHD, perhaps reflecting the low number of T cells in cord blood. Multiple cord blood banks have been developed to harvest and store cord blood for possible transplantation to unrelated patients from material that would otherwise be discarded. Currently more than 500,000 units are cryopreserved and available for use. The advantages of unrelated cord blood are rapid availability and decreased immune reactivity allowing for the use of partially matched units, which is of particular importance for those without matched unrelated donors. The risks of graft failure and transplant-related mortality are related to the dose of cord blood cells per kilogram, which previously limited the application of single cord blood transplantation to pediatric and smaller adult patients. Subsequent trials have found that the use of double cord transplants diminishes the risk of graft failure and early mortality even though only one of the donors ultimately engrafts. Survival rates are now similar with unrelated donor and cord blood transplantation.
THE TRANSPLANT PREPARATIVE REGIMEN
The treatment regimen administered to patients immediately preceding transplantation is designed to eradicate the patient’s underlying disease and, in the setting of allogeneic transplantation, immunosuppress the patient adequately to prevent rejection of the transplanted marrow. The appropriate regimen therefore depends on the disease setting and source of marrow. For example, when transplantation is performed to treat severe combined immunodeficiency and the donor is a histocompatible sibling, no treatment is needed because no host cells require eradication and the patient is already too immunoincompetent to reject the transplanted marrow. For aplastic anemia, there is no large population of cells to eradicate, and high-dose cyclophosphamide plus antithymocyte globulin are sufficient to immunosuppress the patient adequately to accept the marrow graft. In the setting of thalassemia and sickle cell anemia, high-dose busulfan is frequently added to cyclophosphamide in order to eradicate hyperplastic host hematopoiesis. A variety of different regimens have been developed to treat malignant diseases. Most of these regimens include agents that have high activity against the tumor in question at conventional doses and have myelosuppression as their predominant dose-limiting toxicity. Therefore, these regimens commonly include busulfan, cyclophosphamide, melphalan, thiotepa, carmustine, etoposide, and total-body irradiation in various combinations.
Although high-dose treatment regimens have typically been used in transplantation, the understanding that much of the antitumor effect of transplantation derives from an immunologically mediated GVT response has led investigators to ask if reduced-intensity conditioning regimens might be effective and more tolerable. Evidence for a GVT effect comes from studies showing that posttransplant relapse rates are lowest in patients who develop acute and chronic GVHD, higher in those without GVHD, and higher still in recipients of T cell–depleted allogeneic or syngeneic marrow. The demonstration that complete remissions can be obtained in many patients who have relapsed after transplant by simply administering viable lymphocytes from the original donor further strengthens the argument for a potent GVT effect. Accordingly, a variety of reduced-intensity regimens have been studied, ranging from the very minimum required to achieve engraftment (e.g., fludarabine plus 200 cGy total-body irradiation) to regimens of more immediate intensity (e.g., fludarabine plus melphalan). Studies to date document that engraftment can be readily achieved with less toxicity than seen with conventional transplantation. Furthermore, the severity of acute GVHD appears to be somewhat decreased. Complete sustained responses have been documented in many patients, particularly those with more indolent hematologic malignancies. In general, relapse rates are higher following reduced-intensity conditioning, but transplant-related mortality is lower, favoring the use of reduced-intensity conditioning in older patients and those with significant comorbidities. High-dose regimens are favored in younger, fitter patients.
THE TRANSPLANT PROCEDURE
Marrow is usually collected from the donor’s posterior and sometimes anterior iliac crests, with the donor under general or spinal anesthesia. Typically, 10–15 mL/kg of marrow is aspirated, placed in heparinized media, and filtered through 0.3- and 0.2-mm screens to remove fat and bony spicules. The collected marrow may undergo further processing depending on the clinical situation, such as the removal of red cells to prevent hemolysis in ABO-incompatible transplants, the removal of donor T cells to prevent GVHD, or attempts to remove possible contaminating tumor cells in autologous transplantation. Marrow donation is safe, with only very rare complications reported.
Peripheral blood stem cells are collected by leukapheresis after the donor has been treated with hematopoietic growth factors or, in the setting of autologous transplantation, sometimes after treatment with a combination of chemotherapy and growth factors. Stem cells for transplantation are infused through a large-bore central venous catheter. Such infusions are usually well tolerated, although occasionally patients develop fever, cough, or shortness of breath. These symptoms typically resolve with slowing of the infusion. When the stem cell product has been cryopreserved using dimethyl sulfoxide, patients more often experience short-lived nausea or vomiting due to the odor and taste of the cryoprotectant.
ENGRAFTMENT
Peripheral blood counts usually reach their nadir several days to a week after transplant as a consequence of the preparative regimen; then cells produced by the transplanted stem cells begin to appear in the peripheral blood. The rate of recovery depends on the source of stem cells, the use of posttransplant growth factors, and the form of GVHD prophylaxis used. If marrow is the source of stem cells, recovery to 100 granulocytes/μL occurs on average by day 16 and to 500/μL by day 22. Use of G-CSF–mobilized peripheral blood stem cells speeds the rate of recovery by ~1 week when compared to marrow, whereas engraftment following cord blood transplantation is typically delayed by ~1 week compared to marrow. Use of a myeloid growth factor (G-CSF or GM-CSF) after transplant can accelerate recovery by 3–5 days, whereas use of methotrexate to prevent GVHD delays engraftment by a similar period. Following allogeneic transplantation, engraftment can be documented using fluorescence in situ hybridization of sex chromosomes if donor and recipient are sex-mismatched or by analysis of a variable number of tandem repeats or short tandem repeat polymorphisms after DNA amplification.
COMPLICATIONS FOLLOWING HEMATOPOIETIC CELL TRANSPLANT
Early Direct Chemoradiotoxicities The transplant preparative regimen may cause a spectrum of acute toxicities that vary according to intensity of the regimen and the specific agents used, but frequently results in nausea, vomiting, and mild skin erythema (Fig. 139e-1). Regimens that include high-dose cyclophosphamide can result in hemorrhagic cystitis, which can usually be prevented by bladder irrigation or with the sulfhydryl compound mercaptoethanesulfonate (MESNA); rarely, acute hemorrhagic carditis is seen. Most high-dose preparative regimens will result in oral mucositis, which typically develops 5–7 days after transplant and often requires narcotic analgesia. Use of a patient-controlled analgesic pump provides the greatest patient satisfaction and results in a lower cumulative dose of narcotic. Keratinocyte growth factor (palifermin) can shorten the duration of mucositis by several days following autologous transplantation. Patients begin losing their hair 5–6 days after transplant and by 1 week are usually profoundly pancytopenic.
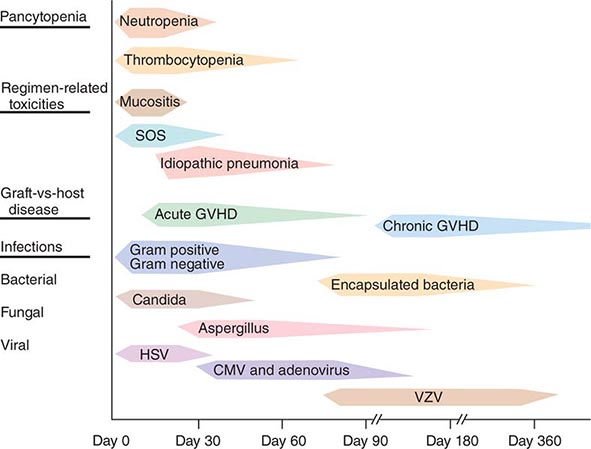
FIGURE 139e-1 Major syndromes complicating marrow transplantation. CMV, cytomegalovirus; GVHD, graft-versus-host disease; HSV, herpes simplex virus; SOS, sinusoidal obstructive syndrome (formerly venoocclusive disease); VZV, varicella-zoster virus. The size of the shaded area roughly reflects the period of risk of the complication.
Depending on the intensity of the conditioning regimen, 3–10% of patients will develop sinusoidal obstruction syndrome (SOS) of the liver (formerly called venoocclusive disease), a syndrome that results from direct cytotoxic injury to hepatic-venular and sinusoidal endothelium, with subsequent deposition of fibrin and the development of a local hypercoagulable state. This chain of events leads to the clinical symptoms of tender hepatomegaly, ascites, jaundice, and fluid retention. These symptoms can develop any time during the first month after transplant, with the peak incidence at day 16. Predisposing factors include prior exposure to intensive chemotherapy, pretransplant hepatitis of any cause, and use of more intense conditioning regimens. The mortality rate of sinusoidal obstruction syndrome is ~30%, with progressive hepatic failure culminating in a terminal hepatorenal syndrome. Both thrombolytic and antithrombotic agents, such as tissue plasminogen activator, heparin, and prostaglandin E, have been studied as therapy, but none has proven of consistent major benefit in controlled trials, and all have significant toxicity. Studies with defibrotide, a polydeoxyribonucleotide, seem encouraging.
Although most pneumonias developing early after transplant are caused by infectious agents, in ~5% of patients a diffuse interstitial pneumonia will develop that is thought to be the result of direct toxicity of high-dose preparative regimens. Bronchoalveolar lavage usually shows alveolar hemorrhage, and biopsies are typically characterized by diffuse alveolar damage, although some cases may have a more clearly interstitial pattern. High-dose glucocorticoids or antitumor necrosis factor therapies are sometimes used as treatment, although randomized trials testing their utility have not been reported.
Late Direct Chemoradiotoxicities Late complications of the preparative regimen include decreased growth velocity in children and delayed development of secondary sex characteristics. These complications can be partly ameliorated with the use of appropriate growth and sex hormone replacement. Most men become azoospermic, and most postpubertal women will develop ovarian failure, which should be treated. However, pregnancy is possible after transplantation, and patients should be counseled accordingly. Thyroid dysfunction, usually well compensated, is sometimes seen. Cataracts develop in 10–20% of patients and are most common in patients treated with total-body irradiation and those who receive glucocorticoid therapy after transplant for treatment of GVHD. Aseptic necrosis of the femoral head is seen in 10% of patients and is particularly frequent in those receiving chronic glucocorticoid therapy. Both acute and late chemoradiotoxicities (except those due to glucocorticoids and other agents used to treat GVHD) are considerably less frequent in recipients of reduced-intensity compared to high-dose preparative regimens.
Graft Failure Although complete and sustained engraftment is usually seen after transplant, occasionally marrow function either does not return or, after a brief period of engraftment, is lost. Graft failure after autologous transplantation can be the result of inadequate numbers of stem cells being transplanted, damage during ex vivo treatment or storage, or exposure of the patient to myelotoxic agents after transplant. Infections with cytomegalovirus (CMV) or human herpesvirus type 6 have also been associated with loss of marrow function. Graft failure after allogeneic transplantation can also be due to immunologic rejection of the graft by immunocompetent host cells. Immunologically based graft rejection is more common following use of less immunosuppressive preparative regimens, in recipients of T cell–depleted stem cell products, and in patients receiving grafts from HLA-mismatched donors or cord blood.
Treatment of graft failure usually involves removing all potentially myelotoxic agents from the patient’s regimen and attempting a short trial of a myeloid growth factor. Persistence of lymphocytes of host origin in allogeneic transplant recipients with graft failure indicates immunologic rejection. Reinfusion of donor stem cells in such patients is usually unsuccessful unless preceded by a second immunosuppressive preparative regimen. Standard high-dose preparative regimens are generally tolerated poorly if administered within 100 days of a first transplant because of cumulative toxicities. However, use of regimens combining, for example, fludarabine plus low-dose total-body irradiation, or cyclophosphamide plus antithymocyte globulin, has been effective in some cases.
Graft-Versus-Host Disease GVHD is the result of allogeneic T cells that are transferred with the donor’s stem cell inoculum reacting with antigenic targets on host cells. Acute GVHD usually occurs within the first 3 months after transplant with a peak onset around 4 weeks and is characterized by an erythematous maculopapular rash; by persistent anorexia or diarrhea, or both; and by liver disease with increased serum levels of bilirubin, alanine and aspartate aminotransferase, and alkaline phosphatase. Because many conditions can mimic acute GVHD, the diagnosis usually requires skin, liver, or endoscopic biopsy for confirmation. In all these organs, endothelial damage and lymphocytic infiltrates are seen. In skin, the epidermis and hair follicles are damaged; in liver, the small bile ducts show segmental disruption; and in intestines, destruction of the crypts and mucosal ulceration may be noted. A commonly used rating system for acute GVHD is shown in Table 139e-1. Grade I acute GVHD is of little clinical significance, does not affect the likelihood of survival, and does not require treatment. In contrast, grades II to IV GVHD are associated with significant symptoms and a poorer probability of survival, and they require aggressive therapy. The incidence of acute GVHD is higher in recipients of stem cells from mismatched or unrelated donors, in older patients, and in patients unable to receive full doses of drugs used to prevent the disease.
|
CLINICAL STAGING AND GRADING OF ACUTE GRAFT-VERSUS-HOST DISEASE |
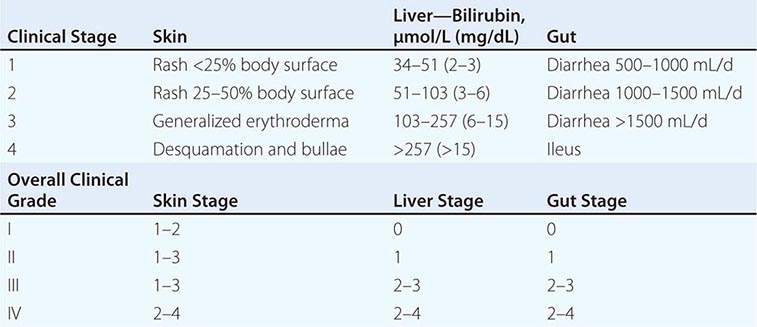
One general approach to the prevention of GVHD is the administration of immunosuppressive drugs early after transplant. Combinations of methotrexate and either cyclosporine or tacrolimus are among the most effective and widely used regimens. Prednisone, anti–T cell antibodies, mycophenolate mofetil, sirolimus, and other immunosuppressive agents have also been or are being studied in various combinations. A second general approach to GVHD prevention is removal of T cells from the stem cell inoculum. While effective in preventing GVHD, T cell depletion has been associated with an increased incidence of graft failure, infectious complications, and tumor recurrence after transplant; as yet, it is unsettled whether T cell depletion improves cure rates in any specific setting.
Despite prophylaxis, significant acute GVHD will develop in ~30% of recipients of stem cells from matched siblings and in as many as 60% of those receiving stem cells from unrelated donors. The disease is usually treated with glucocorticoids, additional immunosuppressants, or monoclonal antibodies targeted against T cells or T cell subsets.
Chronic GVHD occurs most commonly between 3 months and 2 years after allogeneic transplant, developing in 20–50% of recipients. The disease is more common in older patients, in recipients of mismatched or unrelated stem cells, and in those with a preceding episode of acute GVHD. The disease resembles an autoimmune disorder with malar rash, sicca syndrome, arthritis, obliterative bronchiolitis, and bile duct degeneration and cholestasis. Single-agent prednisone or cyclosporine is standard treatment at present, although trials of other agents are under way. Mortality rates from chronic GVHD average around 15%, but range from 5–50% depending on severity. In most patients, chronic GVHD resolves, but it may require 1–3 years of immunosuppressive treatment before these agents can be withdrawn without the disease recurring. Because patients with chronic GVHD are susceptible to significant infection, they should receive prophylactic trimethoprim-sulfamethoxazole, and all suspected infections should be investigated and treated aggressively.
Although onset before or after 3 months after transplant is often used to discriminate between acute and chronic GVHD, occasional patients will develop signs and symptoms of acute GVHD after 3 months (late-onset acute GVHD), whereas others will exhibit signs and symptoms of both acute and chronic GVHD (overlap syndrome). There are as yet no data to suggest that these patients should be treated differently than those with classic acute or chronic GVHD.
From 3–5% of patients will develop an autoimmune disorder following allogeneic HCT, most commonly autoimmune hemolytic anemia or idiopathic thrombocytopenic purpura. Unrelated donor source and chronic GVHD are risk factors, but autoimmune disorders have been reported in patients with no obvious GVHD. Treatment is with prednisone, cyclosporine, or rituximab.
Infection Posttransplant patients, particularly recipients of allogeneic transplantation, require unique approaches to the problem of infection. Early after transplantation, patients are profoundly neutropenic, and because the risk of bacterial infection is so great, most centers initiate antibiotic treatment once the granulocyte count falls to <500/μL. Fluconazole prophylaxis at a dose of 200–400 mg/d reduces the risk of candidal infections. Patients seropositive for herpes simplex should receive acyclovir prophylaxis. One approach to infection prophylaxis is shown in Table 139e-2. Despite these prophylactic measures, most patients will develop fever and signs of infection after transplant. The management of patients who become febrile despite bacterial and fungal prophylaxis is a difficult challenge and is guided by individual aspects of the patient and by the institution’s experience.
|
APPROACH TO INFECTION PROPHYLAXIS IN ALLOGENEIC TRANSPLANT RECIPIENTS |
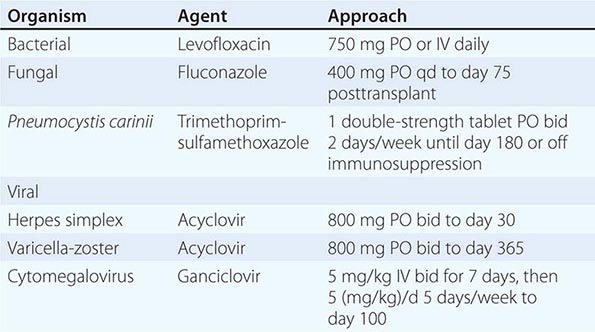
The general problem of infection in the immunocompromised host is discussed in Chap. 169.
Once patients engraft, the incidence of bacterial infection diminishes; however, patients, particularly allogeneic transplant recipients, remain at significant risk of infection. During the period from engraftment until about 3 months after transplant, the most common causes of infection are gram-positive bacteria, fungi (particularly Aspergillus), and viruses including CMV. CMV infection, which in the past was frequently seen and often fatal, can be prevented in seronegative patients transplanted from seronegative donors by the use of either seronegative blood products or products from which the white blood cells have been removed. In seropositive patients or patients transplanted from seropositive donors, the use of ganciclovir, either as prophylaxis beginning at the time of engraftment or initiated when CMV first reactivates as evidenced by development of antigenemia or viremia, can significantly reduce the risk of CMV disease. Foscarnet is effective for some patients who develop CMV antigenemia or infection despite the use of ganciclovir or who cannot tolerate the drug.
Pneumocystis jiroveci pneumonia, once seen in 5–10% of patients, can be prevented by treating patients with oral trimethoprim-sulfamethoxazole for 1 week before transplant and resuming the treatment once patients have engrafted.
The risk of infection diminishes considerably beyond 3 months after transplant unless chronic GVHD develops, requiring continuous immunosuppression. Most transplant centers recommend continuing trimethoprim-sulfamethoxazole prophylaxis while patients are receiving any immunosuppressive drugs and also recommend careful monitoring for late CMV reactivation. In addition, many centers recommend prophylaxis against varicella-zoster, using acyclovir for 1 year after transplant. Patients should be revaccinated against tetanus, diphtheria, Haemophilus influenzae, polio, and pneumococcal pneumonia starting at 12 months after transplant and against measles, mumps, and rubella (MMR), varicella-zoster virus, and possibly pertussis at 24 months.
TREATMENT OF SPECIFIC DISEASES USING HEMATOPOIETIC CELL TRANSPLANTATION
|
TREATMENT |
NONMALIGNANT DISEASES |
IMMUNODEFICIENCY DISORDERS
By replacing abnormal stem cells with cells from a normal donor, hematopoietic cell transplantation can cure patients of a variety of immunodeficiency disorders including severe combined immunodeficiency, Wiskott-Aldrich syndrome, and Chédiak-Higashi syndrome. The widest experience has been with severe combined immunodeficiency disease, where cure rates of 90% can be expected with HLA-identical donors and success rates of 50–70% have been reported using haplotype-mismatched parents as donors (Table 139e-3).
|
ESTIMATED 5-YEAR SURVIVAL RATES FOLLOWING TRANSPLANTATIONa |
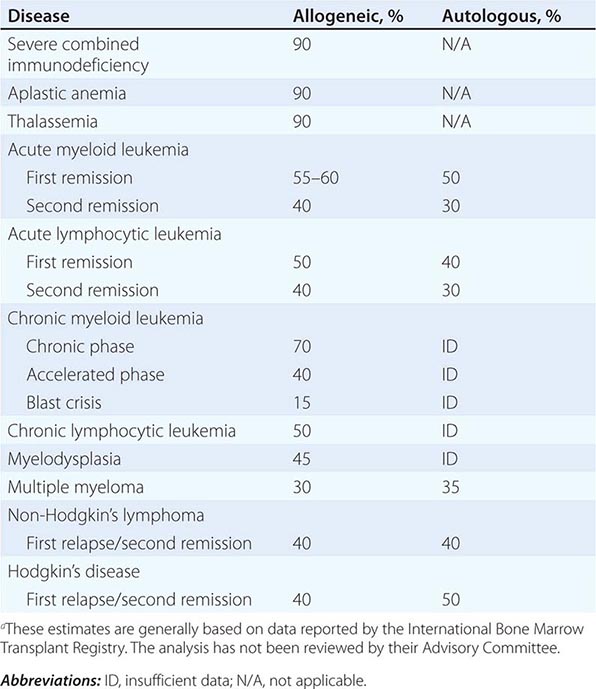
APLASTIC ANEMIA
Transplantation from matched siblings after a preparative regimen of high-dose cyclophosphamide and antithymocyte globulin can cure up to 90% of patients age <40 years with severe aplastic anemia. Results in older patients and in recipients of mismatched family member or unrelated marrow are less favorable; therefore, a trial of immunosuppressive therapy is generally recommended for such patients before considering transplantation. Transplantation is effective in all forms of aplastic anemia including, for example, the syndromes associated with paroxysmal nocturnal hemoglobinuria and Fanconi’s anemia. Patients with Fanconi’s anemia are abnormally sensitive to the toxic effects of alkylating agents, and so less intensive preparative regimens must be used in their treatment (Chap. 130).
HEMOGLOBINOPATHIES
Marrow transplantation from an HLA-identical sibling following a preparative regimen of busulfan and cyclophosphamide can cure 80–90% of patients with thalassemia major. The best outcomes can be expected if patients are transplanted before they develop hepatomegaly or portal fibrosis and if they have been given adequate iron chelation therapy. Among such patients, the probabilities of 5-year survival and disease-free survival are 95 and 90%, respectively. Although prolonged survival can be achieved with aggressive chelation therapy, transplantation is the only curative treatment for thalassemia. Transplantation is being studied as a curative approach to patients with sickle cell anemia. Two-year survival and disease-free survival rates of 90 and 80%, respectively, have been reported following matched sibling or cord blood transplantation. Decisions about patient selection and the timing of transplantation remain difficult, but transplantation represents a reasonable option for younger patients who suffer repeated crises or other significant complications and who have not responded to other interventions (Chap. 127).
OTHER NONMALIGNANT DISEASES
Theoretically, hematopoietic cell transplantation should be able to cure any disease that results from an inborn error of the lymphohematopoietic system. Transplantation has been used successfully to treat congenital disorders of white blood cells such as Kostmann’s syndrome, chronic granulomatous disease, and leukocyte adhesion deficiency. Congenital anemias such as Blackfan-Diamond anemia can also be cured with transplantation. Infantile malignant osteopetrosis is due to an inability of the osteoclast to resorb bone, and because osteoclasts derive from the marrow, transplantation can cure this rare inherited disorder.
Hematopoietic cell transplantation has been used as treatment for a number of storage diseases caused by enzymatic deficiencies, such as Gaucher’s disease, Hurler’s syndrome, Hunter’s syndrome, and infantile metachromatic leukodystrophy. Transplantation for these diseases has not been uniformly successful, but treatment early in the course of these diseases, before irreversible damage to extramedullary organs has occurred, increases the chance for success.
Transplantation is being explored as a treatment for severe acquired autoimmune disorders. These trials are based on studies demonstrating that transplantation can reverse autoimmune disorders in animal models and on the observation that occasional patients with coexisting autoimmune disorders and hematologic malignancies have been cured of both with transplantation.





{kind=link}