Physiology of The Newborn
SGA newborns are thought to suffer intrauterine growth retardation (IUGR) as a result of placental, maternal, or fetal abnormalities. Conditions associated with IUGR are shown in Figure 1-1. SGA infants have a body weight below what is appropriate for their age, yet their body length and head circumference are age appropriate. To classify an infant as SGA, the gestational age must be estimated by the physical findings summarized in Table 1-1.
TABLE 1-1
Clinical Criteria for Classification of Low Birth Weight Infants

Adapted from Avery ME, Villee D, Baker S, et al. Neonatology. In: Avery ME, First LR, editors. Pediatric Medicine. Baltimore: William & Wilkins; 1989. p. 148.
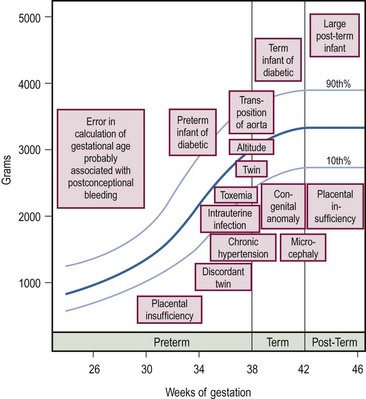
FIGURE 1-1 Graph of conditions associated with deviations in intrauterine growth. The boxes indicate the approximate birth weight and gestational age at which the condition is likely to occur. (Adapted from Avery ME, Villee D, Baker S, et al. Neonatology. In: Avery ME, First LR, editors. Pediatric Medicine. Baltimore: Williams & Wilkins; 1989. p. 148.)
2. Inadequate gastrointestinal absorption
3. Hyaline membrane disease (HMD)
Specific Physiologic Problems of the Newborn
Glucose Metabolism
Hypoglycemia
Clinical signs of hypoglycemia are nonspecific and subtle. Seizure and coma are the most common manifestations of severe hypoglycemia. Neonatal hypoglycemia is generally defined as a glucose level lower than 50 mg/dL.1 Infants who are at high risk for developing hypoglycemia are those who are premature, SGA, and born to mothers with gestational diabetes, severe preeclampsia, and HELLP (hemolysis, elevated liver enzymes, low platelet count). Newborns that require surgical procedures are at particular risk of developing hypoglycemia; therefore, a 10% glucose infusion is typically started on admission to the hospital. Hypoglycemia is treated with an infusion of 1–2 mL/kg (4–8 mg/kg/min) of 10% glucose. If an emergency operation is required, concentrations of up to 25% glucose may be used. Traditionally, central venous access has been a prerequisite for glucose infusions exceeding 12.5%. During the first 36 to 48 hours after a major surgical procedure, it is common to see wide variations in serum glucose levels.
Hyperglycemia
Hyperglycemia is a common problem with the use of parenteral nutrition in very immature infants who are less than 30 weeks’ gestation and less than 1.1 kg birth weight. These infants are usually fewer than 3 days of age and are frequently septic.2 This hyperglycemia appears to be associated with both insulin resistance and relative insulin deficiency, reflecting the prolonged catabolism seen in very low birth weight infants.3 Historically, neonatal hyperglycemia has been linked to intraventricular hemorrhage, dehydration, and electrolyte losses; however, a causal relationship has not been established. Congenital hyperinsulinism refers to an inherited disorder that is the most common cause of recurrent hypoglycemia in the infant. This group of disorders was previously referred to as nesidioblastosis, which is a misnomer. Nesidioblastosis is a term used to describe hyperinsulinemic hypoglycemia attributed to dysfunctional pancreatic beta cells with a characteristically abnormal histological appearance.
Calcium
Calcium is actively transported across the placenta. Of the total amount of calcium transferred across the placenta, 75% occurs after 28 weeks’ gestation.4 This observation partially accounts for the high incidence of hypocalcemia in preterm infants. Neonates are predisposed to hypocalcemia due to limited calcium stores, renal immaturity, and relative hypoparathyroidism secondary to suppression by high fetal calcium levels. Some infants are at further risk for neonatal calcium disturbances due to the presence of genetic defects, pathological intrauterine conditions, or birth trauma.5 Hypocalcemia is defined as an ionized calcium level of less than 1.22 mmol/L (4.9 mg/dL).6 At greatest risk for hypocalcemia are preterm infants, newborn surgical patients, and infants of complicated pregnancies, such as those of diabetic mothers or those receiving bicarbonate infusions. Calcitonin, which inhibits calcium mobilization from the bone, is increased in premature and asphyxiated infants.
Signs of hypocalcemia are similar to those of hypoglycemia and may include jitteriness, seizures, cyanosis, vomiting, and myocardial arrhythmias. Hypocalcemic infants have increased muscle tone, which helps differentiate infants with hypocalcemia from those with hypoglycemia. Symptomatic hypocalcemia is treated with 10% calcium gluconate administered IV at a dosage of 1–2 mL/kg (100–200 mg/kg) over 30 minutes while monitoring the electrocardiogram for bradycardia.1 Asymptomatic hypocalcemia is best treated with calcium gluconate in a dose of 50 mg of elemental calcium/kg/day added to the maintenance fluid: 1 mL of 10% calcium gluconate contains 9 mg of elemental calcium. If possible, parenteral calcium should be given through a central venous line given necrosis that may occur should the peripheral IV infiltrate.
Blood volume
Total RBC volume is at its highest point at delivery. Estimation of blood volume for premature infants, term neonates, and infants are summarized in Table 1-2. By about 3 months of age, total blood volume per kilogram is nearly equal to adult levels as they recover from their postpartum physiologic nadir. The newborn blood volume is affected by shifts of blood between the placenta and the baby prior to clamping the cord. Infants with delayed cord clamping have higher hemoglobin levels.7 A hematocrit greater than 50% suggests placental transfusion has occurred.
TABLE 1-2
| Group | Blood Volume (mL/kg) |
| Premature infants | 85–100 |
| Term newborns | 85 |
| >1 month | 75 |
| 3 months to adult | 70 |
Adapted from Rowe PC, editor. The Harriet Lane Handbook. 11th eds. Chicago: Year Book Medical; 1987. p. 25.
Hemoglobin
At birth, nearly 80% of circulating hemoglobin is fetal (a2Aγ2F). When infant erythropoiesis resumes at about 2 to 3 months of age, most new hemoglobin is adult. When the oxygen level is 27 mmHg, 50% of the bound oxygen is released from adult hemoglobin (P50 = 27 mmHg). Reduction of hemoglobin’s affinity for oxygen allows more oxygen to be released into the tissues at a given oxygen level as shown in Figure 1-2.
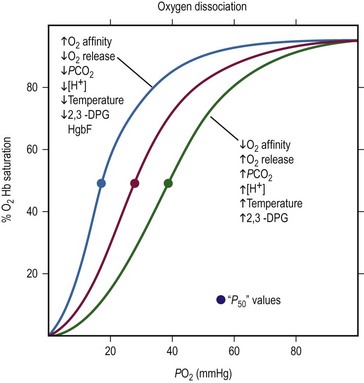
FIGURE 1-2 The oxygen dissociation curve of normal adult blood is shown in red. The P50, the oxygen tension at 50% oxygen saturation, is approximately 27 mmHg. As the curve shifts to the right, the affinity of hemoglobin for oxygen decreases and more oxygen is released. Increases in PCO2, temperature, 2,3-DPG, and hydrogen ion concentration facilitates the unloading of O2 from arterial blood to the tissue. With a shift to the left, unloading of O2 from arterial blood into the tissues is more difficult. Causes of a shift to the left are mirror images of those that cause a shift to the right: decreases in temperature, 2,3-DPG, and hydrogen ion concentration. (Modified from Glancette V, Zipursky A. Neonatal hematology. In: Avery GB, editor, Neonatology. Philadelphia: JB Lippincott; 1986. p. 663.)
Fetal hemoglobin has a P50 value 6–8 mmHg lower than that of adult hemoglobin. This lower P50 value allows more efficient oxygen delivery from the placenta to the fetal tissues. The fetal hemoglobin equilibrium curve is shifted to the left of the normal adult hemoglobin equilibrium curve. Fetal hemoglobin binds less avidly to 2,3-diphosphoglycerate (2,3-DPG) compared to adult hemoglobin causing a decrease in P50.8 This is somewhat of a disadvantage to the newborn because lower peripheral oxygen levels are needed before oxygen is released from fetal hemoglobin. By 4 to 6 months of age in a term infant, the hemoglobin equilibrium curve gradually shifts to the right and the P50 value approximates that of a normal adult.
Anemia
Anemia present at birth is due to hemolysis, blood loss, or decreased erythrocyte production.
Hemolytic Anemia
Hemolytic anemia is most often a result of placental transfer of maternal antibodies that are destroying the infant’s erythrocytes. This can be determined by the direct Coombs test. The most common severe anemia is Rh incompatibility. Hemolytic disease in the newborn produces jaundice, pallor, and hepatosplenomegaly. The most severely affected infants manifest hydrops. This massive edema is not strictly related to the hemoglobin level of this infant. ABO incompatibility frequently results in hyperbilirubinemia but rarely causes anemia.
Anemia of Prematurity
Decreased RBC production frequently contributes to anemia of prematurity. Erythropoietin is not released until a gestational age of 30 to 34 weeks has been reached. These preterm infants have large numbers of erythropoietin-sensitive RBC progenitors. Research has focused on the role of recombinant erythropoietin (epoetin alpha) in treating anemia in preterm infants.9–11 Successful increases in hematocrit levels using epoetin may obviate the need for blood transfusions and reduce the risk of blood borne infections and reactions. Studies suggest that routine use of epoetin is probably helpful for the very low birth weight infant (<750 g), but its regular use for other preterm infants is not likely to significantly reduce the transfusion rate.9–11
Jaundice
The newborn’s liver has a metabolic excretory capacity for bilirubin that is not equal to its task. Even healthy full-term infants usually have an elevated unconjugated bilirubin level. This peaks about the third day of life at approximately 6.5–7.0 mg/dL and does not return to normal until the tenth day of life. A total bilirubin level greater than 7 mg/dL in the first 24 hours or greater than 13 mg/dL at any time in full-term newborns often prompts an investigation for the cause. Breast-fed infants usually have serum bilirubin levels 1–2 mg/dL greater than formula-fed babies. The common causes of prolonged indirect hyperbilirubinemia are listed in Table 1-3.
TABLE 1-3
Causes of Prolonged Indirect Hyperbilirubinemia
| Breast milk jaundice | Pyloric stenosis |
| Hemolytic disease | Crigler–Najjar syndrome |
| Hypothyroidism | Extravascular blood |
Data from Maisels MJ. Neonatal jaundice. In: Avery GB, editor. Neonatology. Pathophysiology and Management of the Newborn. Philadelphia: JB Lippincott; 1987. p. 566.
Pathologic jaundice within the first 36 hours of life is usually due to excessive production of bilirubin. Hyperbilirubinemia is managed based on the infant’s weight. While specific cutoffs defining the need for therapy have not been universally accepted, the following recommendations are consistent with most practice patterns.12 Phototherapy is initiated for newborns: (1) less than 1500 g, when the serum bilirubin level reaches 5 mg/dL; (2) 1500–2000 g, when the serum bilirubin level reaches 8 mg/dL; or (3) 2000–2500 g, when the serum bilirubin level reaches 10 mg/dL. Formula-fed term infants without hemolytic disease are treated by phototherapy when levels reach 13 mg/dL. For hemolytic-related hyperbilirubinemia, phototherapy is recommended when the serum bilirubin level exceeds 10 mg/dL by 12 hours of life, 12 mg/dL by 18 hours, 14 mg/dL by 24 hours, or 15 mg/dL by 36 hours.13 An absolute bilirubin level that triggers exchange transfusion is still not established, but most exchange transfusion decisions are based on the serum bilirubin level and its rate of rise.
Retinopathy of Prematurity
Retinopathy of prematurity (ROP) develops during the active phases of retinal vascular development from the 16th week of gestation. In full-term infants the retina is fully developed and ROP cannot occur. The exact causes are unknown, but oxygen exposure (greater than 93–95%) and extreme prematurity are two risk factors that have been demonstrated.14 The risk and extent of ROP is probably related to the degree of vascular immaturity and abnormal retinal angiogenesis in response to hypoxia. ROP is found in 1.9% of premature infants in large neonatal units.15 Retrolental fibroplasia (RLF) is the pathologic change observed in the retina and overlying vitreous after the acute phases of ROP subsides. Treatment of ROP with laser photocoagulation has been shown to have the added benefit of superior visual acuity and less myopia when compared to cryotherapy in long-term follow-up studies.16–19 The American Academy of Pediatrics’ guidelines recommends a screening examination for all infants who received oxygen therapy who weigh less than 1500 g and are fewer than 32 weeks’ gestation, and selected infants with a birth weight between 1500 and 2000 g or gestational age of more than 32 weeks with an unstable clinical course, including those requiring cardiorespiratory support.20
Thermoregulation
Newborns have difficulty maintaining body temperature due to their relatively large surface area, poor thermal regulation, and small mass to act as a heat sink. Heat loss may occur owing to: (1) evaporation (wet newborn); (2) conduction (skin contact with cool surface); (3) convection (air currents blowing over newborn); and (4) radiation (non-contact loss of heat to cooler surface, which is the most difficult factor to control). Thermoneutrality is the range of ambient temperatures that the newborn can maintain a normal body temperature with a minimal metabolic rate by vasomotor control. The critical temperature is the temperature that requires adaptive metabolic responses to the cold in an effort to replace lost heat. Infants produce heat by increasing metabolic activity by shivering like an adult, nonshivering thermogenesis, and futile cycling of ions in skeletal muscle.21 Brown adipose tissue (BAT) may be involved in thermoregulatory feeding and sleep cycles in the infant with an increase in body temperature signaling an increase in metabolic demand.22 The uncoupling of mitochondrial respiration that occurs in BAT where energy is not conserved in ATP but rather is released as heat may be rendered inactive by vasopressors, anesthetic agents, and nutritional depletion.23–25 Failure to maintain thermoneutrality leads to serious metabolic and physiologic consequences. Double-walled incubators offer the best thermoneutral environment, whereas radiant warmers cannot prevent convection heat loss and lead to higher insensible water loss. In the operating room, special care must be exercised to maintain the neonate’s body temperature in the normal range.
Fluids and Electrolytes
At 12 weeks of gestation, the fetus has a total body water content that is 94% of body weight. This amount decreases to 80% by 32 weeks’ gestation and 78% by term (Fig. 1-3). A further 3–5% reduction in total body water content occurs in the first 3 to 5 days of life. Body water continues to decline and reaches adult levels (approximately 60% of body weight) by 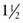 years of age. Extracellular water also declines by 1 to 3 years of age. Premature delivery requires the newborn to complete both fetal and term water unloading tasks. Surprisingly, the premature infant can complete fetal water unloading by one week following birth. Postnatal reduction in extracellular fluid volume has such a high physiologic priority that it occurs even in the presence of relatively large variations of fluid intake.26
years of age. Extracellular water also declines by 1 to 3 years of age. Premature delivery requires the newborn to complete both fetal and term water unloading tasks. Surprisingly, the premature infant can complete fetal water unloading by one week following birth. Postnatal reduction in extracellular fluid volume has such a high physiologic priority that it occurs even in the presence of relatively large variations of fluid intake.26
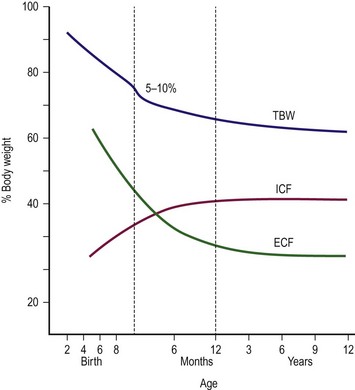
FIGURE 1-3 Friss–Hansen’s classic chart relating total body weight (TBW) and extracellular (ECF) and intracellular (ICF) fluid to percentage of body weight, from early gestation to adolescence. (Adapted from Welch KJ, Randolph JG, Ravitch MM, et al, editors. Pediatric Surgery. 4th ed. Chicago: Year Book Medical; 1986. p. 24.)
Glomerular Filtration Rate and Early Renal Function
The glomerular filtration rate (GFR) of newborns is slower than that of adults.27 From 21 mL/min/1.73 m2 at birth in the term infant, GFR quickly increases to 60 mL/min/1.73 m2 by 2 weeks of age. GFR reaches adult levels by 18 months to 2 years of age. A preterm infant has a GFR that is only slightly slower than that of a full-term infant. In addition to this difference in GFR, the concentrating capacity of the preterm and the full-term infant is well below that of the adult. An infant responding to water deprivation increases urine osmolarity to a maximum of 600 mOsm/kg. This is in contrast to the adult, whose urine concentration can reach 1200 mOsm/kg. It appears that the difference in concentrating capacity is due to the insensitivity of the collecting tubules of the newborn to antidiuretic hormone. Although the newborn cannot concentrate urine as efficiently as the adult, the newborn can excrete very dilute urine at 30–50 mOsm/kg. Newborns are unable to excrete excess sodium, an inability thought to be due to a tubular defect. Term babies are able to conserve sodium, but premature infants are considered ‘salt wasters’ because they have an inappropriate urinary sodium excretion, even with restricted sodium intake.
Neonatal Fluid Requirements
To estimate fluid requirements in the newborn requires an understanding of: (1) any preexisting fluid deficits or excesses; (2) metabolic demands; and (3) losses. Because these factors change quickly in the critically ill newborn, frequent adjustments in fluid management are necessary (Table 1-4). Hourly monitoring of intake and output allows early recognition of fluid balance that will affect treatment decisions. This dynamic approach requires two components: (1) an initial hourly fluid intake that is safe and (2) a monitoring system to detect the patient’s response to the treatment program selected. No ‘normal’ urine output exists for a given neonate, yet one may generally target 1–2 mL/kg/h.
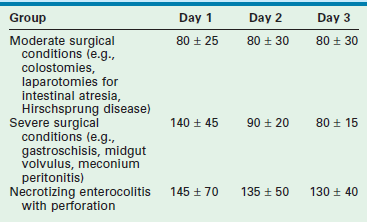
Illustrative Examples
Renal Failure.
Fractional Na excretion (FE Na):
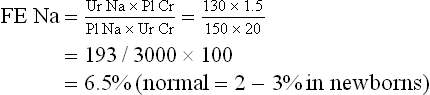
FE Na less than 1% usually indicates a prerenal cause of oliguria, whereas greater than 3% usually implies a renal cause (e.g., acute tubular necrosis). This patient is in acute renal failure. The plan is to restrict fluids to insensible losses plus measured losses for the next 4 hours and to then reassess the plan using both urine and serum studies. Of note, while the FE urea may be a better predictor of prerenal failure in this population, both FE urea and the FE Na have limited utility in neonates, reflecting the relative immaturity of neonatal renal function.28
Pulmonary System of the Newborn
Maturation of the lungs is generally divided into five periods:
• Embryonic phase (begins approximately week 3)
• Pseudoglandular phase (5–17 weeks)
• Canalicular phase (16–25 weeks)
Pulmonary development begins in the third week (embryonic phase) when a ventral diverticulum develops off the foregut (laryngotracheal groove), initiating tracheal development. During the pseudoglandular phase, all of the major elements of the lung form except those involved in gas exchange. The dichotomous branching of the bronchial tree that develops during the fourth week from the primitive trachea is usually completed by 17 weeks’ gestation. Fetuses born during this phase are unable to survive because respiration is not possible. In the canalicular phase, respiration is made possible because thin-walled terminal sacs (primordial alveoli) have developed at the ends of the respiratory bronchioles and the lung tissue is well vascularized. No actual alveoli are seen until 24 to 26 weeks’ gestation, during the terminal saccular phase. The air–blood surface area for gas diffusion is limited should the fetus be delivered at this age. The terminal saccular phase is defined by the establishment of the blood–air barrier that allows gas exchange for the survival of the fetus should it be born prematurely. Between 24 and 28 weeks, the cuboidal and columnar cells flatten and differentiate into type I (lining cells) and/or type II (granular) pneumocytes. Between 26 and 32 weeks of gestation, terminal air sacs begin to give way to air spaces. At the same time, the phospholipids that constitute pulmonary surfactant begin to line the terminal lung air spaces. Surfactant is produced by type II pneumocytes and is extremely important in maintaining alveolar stability. During the alveolar phase, further budding of these air spaces occurs and alveoli become numerous, a process that continues postnatally until the age of 3 to 8 years.29
The change in the ratio of the amniotic phospholipids (lecithin: sphingomyelin) is used to assess fetal lung maturity. A ratio greater than 2 is considered compatible with mature lung function. Absence of adequate surfactant leads to HMD or respiratory distress syndrome (RDS). HMD is present in 10% of premature infants. Other conditions associated with pulmonary distress in the newborn include delayed fetal lung absorption, meconium aspiration syndrome, intrapartum pneumonia, and developmental structural anomalies (e.g., congenital diaphragmatic hernia (CDH) and congenital lobar emphysema). In all of these conditions, endotracheal intubation and mechanical ventilation may be required for hypoxia, CO2 retention, or apnea. Ventilator options and management depend on the clinical context and are further discussed in Chapter 7.
Surfactant
The most efficacious administration method is currently under investigation. The standard approach is to instill aliquots into an endotracheal tube. The indications for the use of surfactant include: (1) intubated infants with RDS; (2) intubated infants with meconium aspiration syndrome (MAS) requiring more than 50% oxygen; (3) intubated infants with pneumonia and an oxygen index great than 15; and (4) intubated infants with pulmonary hemorrhage that have clinically deteriorated. Its efficacy is uncertain in neonates with pulmonary hemorrhage and pneumonia. Worse outcomes are associated with surfactant use in CDH.30,31
The acute pulmonary effects of surfactant therapy are improved lung function and alveolar expansion leading to improved oxygenation, which results in a reduction in the need for mechanical ventilation and extracorporeal oxygenation.32–34
Two meta-analyses support the use of surfactant therapy in infants with RDS to reduce air leak syndromes, pneumothorax, bronchopulmonary dysplasia (BPD), pulmonary interstitial emphysema, and mortality.35,36 The INSURE (INtubate, SURfactant, Extubate) technique consists of administration of surfactant followed by extubation within one hour to nasal continuous positive airway pressure (nCPAP). Another randomized trial demonstrated the reduced mortality and air leaks for infants assigned to early surfactant treatment versus nCPAP alone.37 In large trials that reflect the current practice of treating infants at risk for the development of RDS (administration of maternal steroids and the routine stabilization on CPAP), the selective use of surfactant in infants with established RDS demonstrates a decreased risk of chronic lung disease or death when compared to infants who are more aggressively treated with prophylactic administration of surfactant.38
Several adverse outcomes have been associated with the use of surfactant (Table 1-5). Intraventricular hemorrhage is one of the most worrisome potential side effects. However, meta-analyses of multiple trials have not shown a statistically significant increase in this risk.35,36

Monitoring
Arterial Blood Gases and Derived Indices
Capillary blood samples are ‘arterialized’ by topical vasodilators or heat to increase blood flow to a peripheral site. Blood flowing sluggishly and exposed to atmospheric oxygen falsely raises the PaO2 from a capillary sample, especially in the 40–60 mmHg range.39 Capillary blood pH and carbon dioxide tension (PCO2) correlate well with arterial samples, unless perfusion is poor. PaO2 is least reliable when determined by capillary blood gas. In patients receiving oxygen therapy in which arterial PaO2 exceeds 60 mmHg, the capillary PaO2 correlates poorly with the arterial measurement.40,41
Pulse Oximetry
The noninvasive determination of oxygen saturation (SaO2) gives moment-to-moment information regarding the availability of O2 to the tissues. If the PaO2 is plotted against the oxygen saturation of hemoglobin, the S-shaped hemoglobin dissociation curve is obtained (see Fig. 1-2). From this curve, it is evident that hemoglobin is 50% saturated at 27 mmHg PaO2 and 90% saturated at 50 mmHg. Pulse oximetry has a rapid (5-7 seconds) response time, requires no calibration, and may be left in place continuously.
A study comparing pulse oximetry with PaO2 from indwelling arterial catheters has shown that SaO2 greater than or equal to 85% corresponds to a PaO2 greater than 55 mmHg, and saturations less than or equal to 90% correspond with a PaO2 less than 80 mmHg.42 Guidelines for monitoring infants using pulse oximetry have been suggested for the following three conditions:
1. In the infant with acute respiratory distress without direct arterial access, saturation limits of 85% (lower) and 92% (upper) should be set
2. In the older infant with chronic respiratory distress who is at low risk for ROP, the upper saturation limit may be set at 95%; the lower limit should be set at 87% to avoid pulmonary vasoconstriction and pulmonary hypertension
3. As the concentration of fetal hemoglobin in newborns affects the accuracy of pulse oximetry, infants with arterial access should have both PaO2 and SaO2 monitored closely. A graph should be kept at the bedside documenting the SaO2 each time the PaO2 is measured. Limits for the SaO2 alarm can be changed because the characteristics of this relationship change.
End-Tidal Carbon Dioxide
Measuring expired CO2 by capnography provides a noninvasive means of continuously monitoring alveolar PCO2. Capnometry measures CO2 by an infrared sensor either placed in-line between the ventilator circuit and the endotracheal tube or off to the side of the air flow, both of which are applicable only to the intubated patient. A comparative study of end-tidal carbon dioxide in critically ill neonates demonstrated that both sidestream and mainstream end-tidal carbon dioxide measurements approximated PaCO2.43 When the mainstream sensor was inserted into the breathing circuit, the PaCO2 increased an average of 2 mmHg.
Central Venous Catheter
Indications for central venous catheter placement include: (1) hemodynamic monitoring; (2) inability to establish other venous access; (3) TPN; and (4) infusion of inotropic drugs or other medications that cannot be given peripherally. Measuring central venous pressure (CVP) to monitor volume status is frequently used in the resuscitation of a critically ill patient. A catheter placed in the superior vena cava or right atrium measures the filling pressure of the right side of the heart, which usually reflects left atrial and filling pressure of the left ventricle. Often, a wide discrepancy exists between left and right atrial pressure if pulmonary disease, overwhelming sepsis, or cardiac anomalies are present. Positive-pressure ventilation, pneumothorax, abdominal distention, or pericardial tamponade all elevate CVP.
Pulmonary Artery
Another study concluded that using right heart catheters in treating critically ill adult patients resulted in an increased mortality.44 However, a consensus committee report documents the continued safety and efficacy of right heart catheters in the care of critically ill children.45 A newer technique of deriving some of these data employs femoral arterial access and is gaining popularity in the pediatric intensive care unit: transcardiopulmonary thermodilution monitoring device (pulse contour cardiac output [PCCO]).
A proprietary PiCCO® device has been developed, and employs a standard central venous catheter and a proprietary thermistor-tipped arterial catheter to assess hemodynamic parameters via transpulmonary thermodilution. Manual calibration is required and must be performed frequently (every hour) for reasonably accurate data.46 It is recommended to recalibrate the curve after interventions are performed.47 This device may give incorrect thermodilution measurements if blood is either extracted from or infused back into the cardiopulmonary circulation as seen with an intracardiac shunt, aortic stenosis, lung embolism, and extracorporeal membrane oxygenation (ECMO).48
Venous Oximetry
Reflectance spectrophotometry is currently used for continuous venous oximetry. Multiple wavelengths of light are transmitted at a known intensity by means of fiber optic bundles in a special pulmonary artery or right atrial catheter. The light is reflected by RBCs flowing past the tip of the catheter. The wavelengths of light are chosen so that both oxyhemoglobin and deoxyhemoglobin are measured to determine the fraction of hemoglobin saturated with oxygen. The system requires either in vitro calibration by reflecting light from a standardized target that represents a known oxygen saturation or in vivo calibration by withdrawing blood from the pulmonary artery catheter and measuring the saturation by laboratory co-oximetry.
Mixed venous oxygen saturation values within the normal range (68–77%) indicate a normal balance between oxygen supply and demand, provided that vasoregulation is intact and distribution of peripheral blood flow is normal. Values greater than 77% are most commonly associated with syndromes of vasoderegulation, such as sepsis. Uncompensated changes in O2 saturation, hemoglobin level, or cardiac output lead to a decrease in SvO2. A sustained decrease in SvO2 greater than 10% should lead to measuring SaO2, hemoglobin level, and cardiac output to determine the cause of the decline.49 The most common sources of error in measuring SvO2 are calibration and catheter malposition. The most important concept in SvO2 monitoring is the advantage of continuous monitoring, which allows early warning of a developing problem.50
Although most clinical experience has been with pulmonary artery catheters, right atrial catheters are more easily inserted and may thus provide better information to detect hemodynamic deterioration earlier and permit more rapid treatment of physiologic derangements.51 A study has shown that, when oxygen consumption was monitored and maintained at a consistent level, the right atrial venous saturation was found to be an excellent monitor.52
Shock
Shock is a state in which the cardiac output is insufficient to deliver adequate oxygen to meet metabolic demands of the tissues. Cardiovascular function is determined by preload, cardiac contractility, heart rate, and afterload. Shock may be classified broadly as hypovolemic, cardiogenic, or distributive (systemic inflammatory response syndrome [SIRS]—septic or neurogenic).
Cardiogenic Shock
Adrenergic receptors are important in regulating calcium flux, which, in turn, is important in controlling myocardial contractility. The α and β receptors are proteins present in the sarcolemma of myocardial and vascular smooth muscle cells. The β1 receptors are predominantly in the heart and, when stimulated, result in increased contractility of myocardium. The β2 receptors are predominately in respiratory and vascular smooth muscle. When stimulated, these receptors result in bronchodilation and vasodilation. The α1-adrenergic receptors are located on vascular smooth muscle and result in vascular constriction when stimulated. The α2 receptors are found mainly on prejunctional sympathetic nerve terminals. The concept of dopaminergic receptors has also been used to account for the cardiovascular effects of dopamine not mediated through α or β receptors. Activation of dopaminergic receptors results in decreased renal and mesenteric vascular resistance and, usually, increased blood flow. The most commonly used inotropic and vasoactive drugs are listed in Table 1-6.
TABLE 1-6
Vasoactive Medications Commonly Used in the Newborn
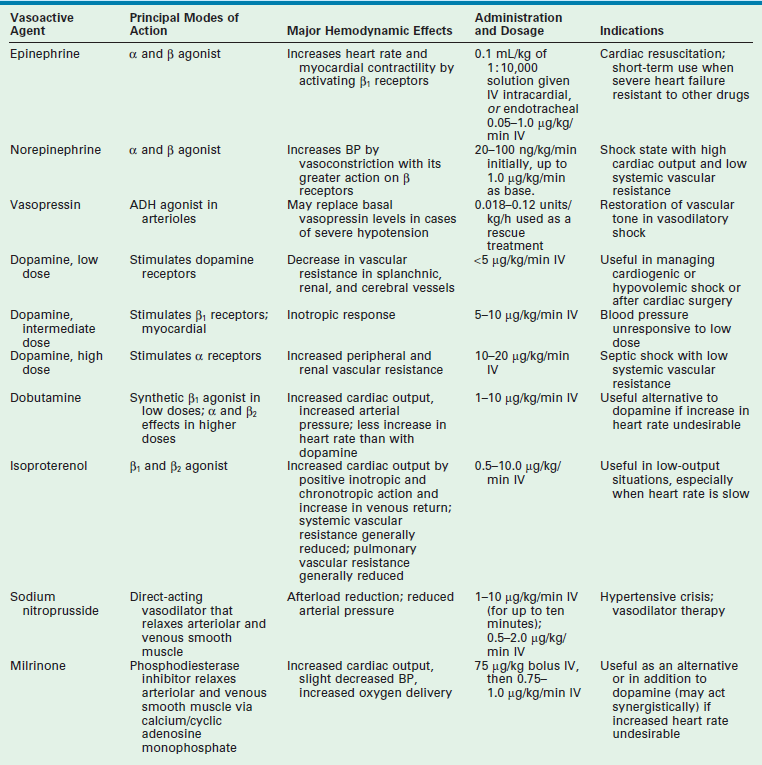
ADH, antidiuretic hormone; BP, blood pressure; IV, intravenous.
Adapted from Lees MH, King DH. Cardiogenic shock in the neonate. Pediatr Rev 1988;9:263; Yager P, Noviski N. Shock. Pediatrics in Review 2010;21:311–18; and Piastra M, Luca E, Mensi S, et al. Inotropic and Vasoactive Drugs in Pediatric ICU. Current Drug Targets 2012;13:900–5.
Dopamine
Dopamine is an endogenous catecholamine with β-adrenergic, α-adrenergic, and dopaminergic effects. It is both a direct and an indirect β-adrenergic agonist. Dopamine elicits positive inotropic and chronotropic responses by direct interaction with the β receptor (direct effect) and by stimulating the release of norepinephrine from the sympathetic nerve endings, which interacts with the β receptor (indirect effect). At low dosages (<5 µg/kg/min), the dopaminergic effect of the drug predominates, resulting in reduced renal and mesenteric vascular resistance and further blood flow to these organs. The β-adrenergic effects become more prominent at intermediate dosages (5–10 µg/kg/min), producing a higher cardiac output. At relatively high dosages (10–20 µg/kg/min), the α-adrenergic effects become prominent with peripheral vasoconstriction.
Experience with the use of dopamine in pediatric patients suggests that it is effective in increasing blood pressure in neonates, infants, and children. The precise dosages at which the desired hemodynamic effects are maximized are not known. The effects of low dosages of dopamine on blood pressure, heart rate, and renal function were studied in 18 hypotensive, preterm infants.53 The blood pressure and diuretic effects were observed at 2, 4, and 8 µg/kg/min. Elevations in heart rate were seen only at 8 µg/kg/min. Further work is needed to better characterize the pharmacokinetics and pharmacodynamics of dopamine in children, especially in newborns.
Dobutamine
Dobutamine, a synthetic catecholamine, has predominantly β-adrenergic effects with minimal α-adrenergic effects. The hemodynamic effect of dobutamine in infants and children with shock has been studied.54 Dobutamine infusion significantly increased cardiac index, stroke index, and pulmonary capillary wedge pressure, and it decreased SVR. The drug appears more efficacious in treating cardiogenic shock than septic shock. The advantage of dobutamine over isoproterenol is its lesser chronotropic effect and its tendency to maintain systemic pressure. The advantage over dopamine is dobutamine’s lesser peripheral vasoconstrictor effect. The usual range of dosages for dobutamine is 1–10 µg/kg/min. The combination of dopamine and dobutamine has been increasingly used; however, little information regarding their combined advantages or effectiveness in the neonate and infant has been published.
Milrinone
Milrinone, a phosphodiesterase inhibitor, is a potent positive inotrope and vasodilator (hence, also known as an ino-dilator) that has been shown to improve cardiac function in infants and children.55–57 The proposed action is due, in part, to an increase in intracellular cyclic adenosine monophosphate and calcium transport secondary to inhibition of cardiac phosphodiesterase. This effect is independent of β-agonist stimulation and, in fact, may act synergistically with the β agonist to improve cardiac performance. Milrinone increases cardiac index and oxygen delivery without affecting heart rate, blood pressure, or pulmonary wedge pressure. Milrinone is administered as a 75 µg/kg bolus followed by infusion of 0.75–1.0 g/kg/min.
Septic Shock
Septic shock is a distributive form of shock that differs from other forms of shock. Cardiogenic and hypovolemic shock lead to increased SVR and decreased cardiac output. Septic shock results from a severe decrease in SVR and a generalized maldistribution of blood and leads to a hyperdynamic state.58 The pathophysiology of septic shock begins with a nidus of infection. Organisms may invade the blood stream, or they may proliferate at the infected site and release various mediators into the blood stream. Substances produced by microorganisms, such as lipopolysaccharide, endotoxin, exotoxin, lipid moieties, and other products can induce septic shock by stimulating host cells to release numerous cytokines, chemokines, leukotrienes, and endorphins.
Endotoxin is a lipopolysaccharide found in the outer membrane of Gram-negative bacteria. Functionally, the molecule is divided into three parts: (1) the highly variable O-specific polysaccharide side chain (conveys serotypic specificity to bacteria and can activate the alternate pathway of complement); (2) the R-core region (less variable among different gram-negative bacteria; antibodies to this region could be cross protective); and (3) lipid-A (responsible for most of the toxicity of endotoxin). Endotoxin stimulates tumor necrosis factor (TNF) and can directly activate the classic complement pathway in the absence of antibody. Endotoxin has been implicated as an important factor in the pathogenesis of human septic shock and Gram-negative sepsis.59 Therapy has focused on developing antibodies to endotoxin to treat septic shock. Antibodies to endotoxin have been used in clinical trials of sepsis with variable results.60–62
Cytokines, especially TNF, play a dominant role in the host’s response. Endotoxin and exotoxin both induce TNF release in vivo and produce many other toxic effects via this endogenous mediator.63–65 TNF is released primarily from monocytes and macrophages. It is also released from natural killer cells, mast cells, and some activated T-lymphocytes. Antibodies against TNF protect animals from exotoxin and bacterial challenge.66,67 Other stimuli for its release include viruses, fungi, parasites, and interleukin-1 (IL-1). In sepsis, the effects of TNF release may include cardiac dysfunction, disseminated intravascular coagulation, and cardiovascular collapse. TNF release also causes the release of granulocyte–macrophage colony-stimulating factor (GM-CSF), interferon-α, and IL-1.
IL-2, also known as T-cell growth factor, is produced by activated T-lymphocytes and strengthens the immune response by stimulating cell proliferation. Its clinically apparent side effects include capillary leak syndrome, tachycardia, hypotension, increased cardiac index, decreased SVR, and decreased left ventricular ejection fraction.68,69
Studies in dogs have suggested that in immature animals, septic shock is more lethal and has different mechanisms of tissue injury.70 These include more dramatic aberrations in blood pressure (more constant decline), heart rate (progressive, persistent tachycardia), blood sugar level (severe, progressive hypoglycemia), acid–base status (severe acidosis), and oxygenation (severe hypoxemia). These changes are significantly different from those seen in the adult animals that also experience an improved survival of almost 600% (18.5 vs 3.1 hours) compared with the immature animal.
The proliferative rate of the granulocyte–macrophage precursor has been reported to be at near-maximal capacity in the neonate. However, the neutrophil storage pool is markedly reduced in the newborn compared with the adult. After bacterial challenge, newborns fail to increase stem cell proliferation and deplete their already reduced neutrophil storage pool. Numerous in vitro abnormalities have been demonstrated in neonatal polymorphonuclear neutrophils, especially in times of stress or infection.71 These abnormalities include decreased deformability, chemotaxis, phagocytosis, C3b receptor expression, adherence, bacterial killing, and depressed oxidative metabolism. Chemotaxis is impaired in neonatal neutrophils in response to various bacterial organisms and antigen–antibody complexes.72 Granulocytes are activated by their interaction with endothelial cells followed by entry into secondary lymphoid issues via the endothelial venules. Initial adhesion of granulocytes is dependent on their expression of L-selectin, a cell adhesion molecule expressed on the granulocyte cell surface. Evaluation of cord blood has demonstrated a significantly lower expression of L-selectin on granulocyte surfaces when compared to older newborn (5 days old) and adult samples, indicating a depressed level of interaction with vascular endothelial cells at the initial stage of adhesion.73 Although phagocytosis has additionally been demonstrated to be abnormal in neonatal phagocytes, it appears that this phenomenon is most likely secondary to decreased opsonic activity rather than an intrinsic defect of the neonatal polymorphonuclear neutrophils.74,75 Currently, there is inconclusive evidence to support or refute the routine use of granulocyte transfusions in the prevention or treatment of sepsis in the neonate.76
Preterm and term newborns have poor responses to various antigenic stimuli, reduced gamma globulin levels at birth, and reduced maternal immunoglobulin supply from placental transport. Almost 33% of infants with a birth weight less than 1500 g develop substantial hypogammaglobulinemia.77 IgA and IgM levels are also low due to the inability of these two immunoglobulins to cross the placenta. Thus, neonates are usually more susceptible to pyogenic bacterial infections because most of the antibodies that opsonize pyogenic bacterial capsular antigens are IgG and IgM. In addition, neonates do not produce type-specific antibodies because of defects in the differentiation of B-lymphocytes into immunoglobulin-secreting plasma cells and in T-lymphocyte-mediated facilitation of antibody synthesis. In the term infant, total hemolytic complement activity, which measures the classic complement pathway, constitutes approximately 50% of adult activity.78 The activity of the alternative complement pathway, secondary to lowered levels of factor B, is also decreased in the neonate.79 Fibronectin, a plasma protein that promotes reticuloendothelial clearance of invading microorganisms, is deficient in neonatal cord plasma.80
The use of intravenous immunoglobulins (IVIGs) for the prophylaxis and treatment of sepsis in the newborn, especially the preterm, low birth weight infant, has been studied in numerous trials with varied outcomes. In one study, a group of infants weighing 1500 g was treated with 500 mg/kg of IVIG each week for four weeks and compared with infants who were not treated with immunoglobulin.81 The death rate was 16% in the IVIG-treated group compared with 32% in the untreated control group. Another recent analysis examined the role of IVIG to prevent and treat neonatal sepsis.82 A significant (but only marginal) benefit was noted from prophylactic use of IVIG to prevent sepsis in low birth weight premature infants. However, using IVIG to treat neonatal sepsis produced a greater than 6% decrease in the mortality rate. A review of nineteen randomized control trials found a 3% decrease in the incidence of neonatal sepsis in preterm infants without a significant difference in all-cause and infection-related mortality when prophylactic IVIG was administered.83 Based on the marginal reduction of neonatal sepsis without a reduction in mortality, routine use of prophylactic IVIG cannot be recommended.
Colony-stimulating factors (CSFs) are a family of glycoproteins that stimulate proliferation and differentiation of hematopoietic cells of various lineages. GM-CSF and granulocyte CSF (G-CSF) have similar physiologic actions. Both stimulate the proliferation of bone marrow myeloid progenitor cells, induce the release of bone marrow neutrophil storage pools, and enhance mature neutrophil effect or function.82–84 Preliminary studies of GM-CSF in neonatal animals demonstrate enhancement of neutrophil oxidative metabolism as well as priming of neonatal neutrophils for enhanced chemotaxis and bacterial killing. Both GM-CSF and G-CSF induce peripheral neutrophilia within two to six hours of intraperitoneal administration. This enhanced affinity for neutrophils returns to normal baseline level by 24 hours.85 Studies have confirm the efficacy and safety of G-CSF therapy for neonatal sepsis and neutropenia.86 Other investigations have demonstrated no long-term adverse hematologic, immunologic, or developmental effects from G-CSF therapy in the septic neonate. Prolonged prophylactic treatment in the very low birth weight neonate with recombinant GM-CSF has been shown to be well tolerated and have a significant decrease in the rate of nosocomial infections.87,88
Unique to the newborn in septic shock is the persistence of fetal circulation and resultant pulmonary hypertension.89 In fact, the rapid administration of fluid can further exacerbate this problem by causing left to right shunting through a patent ductus arteriosus (PDA) and subsequent congestive heart failure from ventricular overload. Infants in septic shock with a new heart murmur should undergo a cardiac echocardiogram. If present, a PDA may warrant treatment with indomethacin (prostaglandin inhibitor) or surgical ligation to achieve closure, depending on the clinical picture.
The critical care of a neonate/infant in septic shock can be extremely challenging. Septic shock has a distinctive clinical presentation and is characterized by an early compensated stage where one can see a decreased SVR, an increase in cardiac output, tachycardia, warm extremities, and an adequate urine output. Later in the clinical presentation, septic shock is characterized by an uncompensated phase where one will see a decrease in intravascular volume, myocardial depression, high vascular resistance, and a decreasing cardiac output.90 Management of these patients is based on the principles of source control, antibiotics (broad-spectrum, institutionally based when possible and including antifungal agents as warranted), and supportive care.
Patients with severe septic shock often do not respond to conventional forms of volume loading and cardiovascular supportive medications. The administration of arginine vasopressin has been shown to decrease mortality in adult patients with recalcitrant septic shock.91,92 Vasopressin (see Table 1-6), also known as antidiuretic hormone (ADH), is made in the posterior pituitary and plays a primary role in water regulation by the kidneys. In septic shock, vasopressin has profound effects on increasing blood pressure in intravascular depleted states. Sparked initially by a randomized, double-blinded, placebo-controlled study in adults that demonstrated a beneficial effect of vasopressin in recalcitrant septic shock, its utilization in the pediatric population has become common.93,94 While a detailed discussion is beyond the scope of this chapter, current trends suggest that ECMO may serve as rescue therapy in select patients with profound sepsis and cardiopulmonary failure refractory to other measures (reported ELSO database newborn survival 80%, older children 50%).94,95
Given the difficult nature of caring for septic patients, extensive investigation has been launched in attempt to identify patients at risk.96–98 Early serum markers such as C-reactive protein, IL-6, and procalcitonin carry promise but warrant further validation.
References
1. Sarafoglou, K, Hoffmann, G, Roth, K. Pediatric Endocrinology and Inborn Errors of Metabolism. China: The McGraw-Hill Companies, Inc; 2009.
2. Dweck, HS, Cassady, G. Glucose intolerance in infants of very low birth weight, I: Incidence of hyperglycemia in infants of birth weights 1,110 grams or less. Pediatr. 1974; 53:189–195.
3. Beardsall, K, Dunger, D. The physiology and clinical management of glucose metabolism in the newborn. Endocr Dev. 2007; 12:124–137.
4. Ziegler, EE, O’Donnell, AM, Nelson, SE, et al. Body composition of reference fetus. Growth. 1976; 40:329.
5. Hsu, SC, Levine, MA. Perinatal calcium metabolism: Physiology and pathophysiology. Semin Neonatol. 2004; 9:23–26.
6. Thomas, TC, Smith, JM, White, PC, Adhikari, S. Transient neonatal hypocalcemia: Presentation and outcomes. Pediatrics. 2012; 129:e1461–e1467.
7. Colozzi, AE. Clamping of the umbilical cord. Its effect on the placental transfusion. N Engl J Med. 1954; 250:629.
8. Bauer, C, Ludwig, I, Ludwig, M. Different effects of 2,3-diphosphoglycerate and adenosine triphosphate on the oxygen affinity of adult and fetal human hemoglobin. Life Sci. 1968; 7:1339.
9. Asch, J, Wedgwood, JF. Optimizing the approach to anemia in the preterm infant: Is there a role for erythropoietin therapy? J Perinatol. 1997; 17:276–282.
10. Doyle, JJ. The role of erythropoietin in the anemia of prematurity. Semin Perinatol. 1997; 21:20–27.
11. King, PJ, Sullivan, TM, Leftwich, ME, et al. Score for neonatal acute physiology and phlebotomy blood loss predict erythrocyte transfusions in premature infants. Arch Pediatr Adolesc Med. 1997; 151:27–31.
12. Maisels, MJ. What’s in a name? Physiologic and pathologic jaundice: The conundrum of defining normal bilirubin levels in the newborn. Pediatrics. 2006; 118:805–807.
13. Osborn, LM, Lenarsky, C, Oakes, RC, et al. Phototherapy in full-term infants with hemolytic disease secondary to ABO incompatibility. Pediatrics. 1984; 73:520–526.
14. Saugstad, O. Optimal oxygenation at birth and in the neonatal period. Neonatology. 2007; 91:319–322.
15. Biglan, AW, Cheng, KP, Brown, DR. Update on retinopathy of prematurity. Intern Ophthalmol Clin. 1989; 29:2–4.
16. National Institutes of Health. Cryotherapy for retinopathy of prematurity cooperative group. Multicenter trial of cryotherapy for retinopathy of prematurity. Arch Ophthalmol. 1988; 106:471–479.
17. Ng, E, Connolly, B, McNamara, J, et al. A comparison of laser photocoagulation with cryotherapy for threshold retinopathy of prematurity at 10 years: Part 1. Visual function and structural outcome. Ophthalmol. 2002; 202:928–935.
18. Shalev, B, Farr, A, Repka, M. Randomized comparison of diode laser photocoagulation versus cryotherapy for threshold retinopathy of prematurity: Seven-year outcome. Am J Ophthalmol. 2002; 132:76–80.
19. Connolly, B, McNamara, J, Sharma, S, et al. A comparison of laser phototherapy with trans-scleral cryotherapy for the treatment of threshold retinopathy. Ophthalmol. 1998; 105:1628–1631.
20. American Academy of Pediatrics, Section on Ophthalmology, AAo Ophth, AAo Ophth/Strabismus. Screening examination of premature infants for retinopathy of prematurity. Pediatrics. 2006; 117:572–576.
21. Lowell, BB, Spiegelman, BM. Towards a molecular understanding of adaptive thermogenesis. Nature. 2000; 404:652–660.
22. Chardon, K, Cardot, V, Leke, A, et al. Thermoregulatory controlof feeding and sleep in premature infants. Obesity. 2006; 14:1535–1542.
23. Karlberg, P, Moore, RE, Oliver, TK. The thermogenic response of the newborn infant to noradrenaline. Acta Paediatr Scand. 1962; 51:284.
24. Stein, J, Cheu, H, Lee, M, et al, Effects of muscle relaxants, sedatives, narcotics and anesthetics on neonatal thermogenesis. Surgical Forum. Pannell, M, eds. Surgical Forum; vol. 38. American College of Surgeons, Chicago, 1987:76.
25. Landsberg, L, Young, JB. Fasting, feeding and regulation of the sympathetic nervous system. N Engl J Med. 1978; 198:1295.
26. Lorenz, JM, Kleinman, LI, Kotagal, UR, et al. Water balance in very low birth weight infants: Relationship to water and sodium intake and effect on outcome. J Pediatr. 1982; 101:423–432.
27. Aperia, A, Broberger, O, Herin, P, et al. Postnatal control of water and electrolyte homeostatis in pre-term and full-term infants. Acta Paediatr Scand. 1983; 305:61–65.
28. Fahimi, D, Mohajeri, S, Hajizadeh, N, et al. Comparison between fraction excretions of urea and sodium in children with acute kidney injury. Pediatr Nephrol. 2009; 24:2409–2412.
29. Thurlbeck, WM. Lung growth and development. In Thurlbeck WM, Churg AM, eds.: Pathology of the Lung, 2nd ed, New York: Thieme Medical Publishers, 1995.
30. Meurs, KV, The Congenital Diaphragmatic Hernia Study Group. Is surfactant therapy beneficial in the treatment of the term newborn infant with congenital diaphragmatic hernia? J Pediatr. 2004; 145:312–316.
31. Lally, KP, Lally, PA, Langham, MR, et al. Surfactant does not improve survival rate in preterm infants with congenital diaphragmatic hernia. J Pediatr Surg. 2004; 39:829–833.
32. Tooley, WH, Clements, JA, Muramatsu, K, et al. Lung function in prematurely delivered rabbits treated with a synthetic surfactant. Am Rev Respir Dis. 1987; 136:651–656.
33. Robertson, B, Enhorning, G. The alveolar lining of the premature newborn rabbit after pharyngeal deposition of surfactant. Lab Invest. 1974; 31:54–59.
34. Shahed, AI, Dargaville, PA, Ohlsson, A, et al. Surfactant for meconium aspiration syndrome in full term/near term infants. Cochrane Database Syst Reviews. 2009.
35. Seger, N, Soll, R. Animal derived surfactant extract for treatment of respiratory distress syndrome. Cochrane Database Syst Reviews. (3):2009.
36. Soll, R. Synthetic surfactant for respiratory distress syndrome in preterm infants. Cochrane Database Syst Reviews. (3):2009.
37. Rojas, MA, Lozano, JM, Rojas, MX, et al. Very early surfactant without mandatory ventilation in premature infants treated with early continuous positive airway pressure: A randomized, controlled trial. Pediatrics. 2009; 123:137–142.
38. Rojas-Reyes, MX, Morley, CJ, Soll, R. Prophylactic versus selective use of surfactant in preventing morbidity and mortality in preterm infants. Cochrane Database Syst Reviews. (3):2012.
39. Garg, AK. ‘Arterialized’ capillary blood [letter]. CMAJ. 1972; 107:16.
40. Glasgow, JF, Flynn, DM, Swyer, PR. A comparison of descending aortic and ‘arterialized’ capillary blood in the sick newborn. CMAJ. 1972; 106:660.
41. Siggaard-Andersen, O. Acid-base and blood gas parameters—arterial or capillary blood? Scand J Clin Lab Invest. 1968; 21:289.
42. Reynolds, GJ, Yu, VYH. Guidelines for the use of pulse oximetry in the non-invasive estimation of oxygen saturation in oxygen-dependent newborn infants. Aust Paediatr J. 1988; 24:346–350.
43. McEvedy, BAB, McLeod, ME, Kirpalani, H, et al. End-tidal carbon dioxide measurements in critically ill neonates: A comparison of sidestream capnometers. Can J Anaesth. 1990; 37:322–326.
44. Connors, A. The effectiveness of right heart catheterization in the initial care of critically ill patients. JAMA. 1996; 276:889–897.
45. Thompson, AE. Pulmonary artery catheterization in children. New Horiz. 1997; 5:244–250.
46. Hamzaoui, O, Monnet, X, Richard, C. Effects of changes in vascular tone on the agreement between pulse contour and transpulmonary thermodilution cardiac output measurements within an up to 6-hour calibration-free period. Crit Care Med. 2008; 36:434–440.
47. Bein, B, Meybohm, P, Cavus, E. The reliability of pulse contour-derived cardiac output during hemorrhage and after vasopressors administration. Anesth Analg. 2007; 105:107–113.
48. Gazit, A, Cooper, DS. Emerging technologies. Pediatr Crit Care Med. 2011; 12:S55–S61.
49. Nelson, LD. Application of venous saturation monitoring. In: Civetta JM, Taylor RW, Kirby RR, eds. Critical Care. Philadelphia: JB Lippincott; 1988:327–334.
50. Norfleet, EA, Watson, CB. Continuous mixed venous oxygen saturation measurement: A significant advance in hemodynamic monitoring? J Clin Monit Comput. 1985; 1:245–258.
51. Ko, WJ, Chang, CI, Chiu, IS. Continuous monitoring of venous oxygen saturation in critically-ill infants. J Formos Med Assoc. 1996; 95:258–262.
52. Hirschl, RB, Palmer, P, Heiss, KF, et al. Evaluation of the right atrial venous oxygen saturation as a physiologic monitor in a neonatal model. J Pediatr Surg. 1993; 28:901–905.
53. DiSessa, TG, Leitner, M, Ti, CC, et al. The cardiovascular effects of dopamine in the severely asphyxiated neonate. J Pediatr. 1981; 99:772–776.
54. Perkin, RM, Levin, DL, Webb, R, et al. Dobutamine: A hemodynamic evaluation in children with shock. J Pediatr. 1982; 100:977–983.
55. Osborn, D, Evans, N, Klucklow, M. Randomized trial of dobutamine versus dopamine in preterm infants with low systemic blood flow. J Pediatr. 2002; 140:183–191.
56. Barton, P, Garcia, JK, Kitchen, A, et al. Hemodynamic effects of I.V. milrinone lactate in pediatric patients with septic shock. A prospective double-blinded, randomized, placebo-controlled interventional study. Chest. 1996; 109:1302–1312.
57. Chang, AC, Am, A, Wernovsky, G, et al. Milrinone: Systemic and pulmonary hemodynamic effects in neonates after cardiac surgery. Crit Care Med. 1995; 23:1907–1914.
58. Parrillo, JE. Septic shock in humans. Advances in the understanding of pathogenesis, cardiovascular dysfunction, and therapy. Ann Intern Med. 1990; 113:227–242.
59. Danner, R, Elin, RJ, Hosline, KM, et al. Endotoxin determinations in 100 patients with septic shock. Clin Res. 1988; 36:453A.
60. McCloskey, RV, Straube, KC, Sanders, C, et al. Treatment of septic shock with human monoclonal antibody HA-1A. A randomized, double-blind, placebo-controlled trial. CHESS Trial Study Group. Ann Intern Med. 1994; 121:1–5.
61. Rogy, MA, Moldawer, LL, Oldenburg, HS, et al. Anti-endotoxin therapy in primate bacteremia with HA-1A and BPI. Ann Surg. 1994; 220:77–85.
62. Ziegler, EJ, Fisher, CJ, Jr., Sprung, CL, et al. Treatment of gram-negative bacteremia and septic shock with HA-1A human monoclonal antibody against endotoxin. A randomized, double-blind, placebo-controlled trial. The HA-1A Sepsis Study Group. N Engl J Med. 1991; 324:429–436.
63. Tracey, KJ, Lowry, SF, Cerami, A. Chachectin: A hormone that triggers acute shock and chronic cachexia. J Infect Dis. 1988; 157:413–420.
64. Nedwin, GE, Svedersky, LP, Bringman, TS. Effect of interleukin-2, interferon-gamma and mitogens on the production of tumor necrosis factors alpha and beta. J Immunol. 1985; 135:2492–2497.
65. Jupin, C, Anderson, S, Damais, C, et al. Toxic shock syndrome toxin 1 as an inducer of human tumor necrosis factors and gamma interferon. J Exp Med. 1988; 167:752–761.
66. Tracey, KJ, Fong, Y, Hesse, DG, et al. Anti-cachectin/TNF monoclonal antibodies prevent septic shock during lethal bacteraemia. Nature. 1987; 330:662–664.
67. Beutler, B, Milsaark, IW, Cerami, AC. Passive immunization against cachectin/tumor necrosis factor protects mice from the lethal effects of endotoxin. Science. 1981; 229:869–871.
68. Rosenstein, M, Ettinghausen, SE, Rosenberg, SA. Extravasation of intravascular fluid mediated by the systemic administration of recombinant interleukin-2. Immunology. 1986; 137:1735.
69. Ognibene, FP, Rosenberg, SA, Lotze, M, et al. Interleukin-2 administration causes reversible hemodynamic changes and left ventricular dysfunction similar to those seen in septic shock. Chest. 1988; 94:750.
70. Pryor, RW, Hinshaw, LB. Sepsis/septic shock in adults and children. Pathol Immunopathol Res. 1989; 8:222–230.
71. Hill, HR. Biochemical, structural and functional abnormalities of polymorphonuclear leukocytes in the neonate. Pediatr Res. 1987; 22:375–382.
72. Miller, M. Chemotactic function in the human neonate: Humoral and cellular aspects. Pediatr Res. 1971; 5:487–492.
73. Moriguchi, N, Yamamoto, S, Isokawa, S, et al. Granulocyte function and changes in ability with age in newborns; Report no,1: Flow cytometric analysis of granulocyte functions in whole blood. Pediatr Int. 2006; 48:17–21.
74. Miller, ME. Phagocytosis in the newborn: Humoral and cellular factors. J Pediatr. 1969; 75:255–259.
75. Forman, ML, Stiehm, ER. Impaired opsonic activity but normal phagocytosis in low-birth-weight infants. N Engl J Med. 1969; 281:926–931.
76. Mohan, P, Brocklehurst. Granulocyte transfusions for neonates with confirmed or suspected sepsis. Cochrane Database Syst Reviews. (4):2003.
77. Cates, KL, Rowe, JC, Ballow, M. The premature infant as a compromised host. Curr Probl Pediatr. 1983; 13:1–63.
78. Anderson, DC, Hughes, J, Edwards, MS, et al. Impaired chemotaxigenesis by type III group B streptococci in neonatal sera: Relationship to diminished concentration of specific anticapsular antibody and abnormalities of serum complement. Pediatr Res. 1983; 17:496–502.
79. Stossel, TP, Alper, CH, Rosen, F. Opsonic activity in the newborn: Role of properidin. Pediatr. 1973; 52:134–137.
80. Gerdes, JS, Yoder, MC, Douglas, SD, et al. Decreased plasma fibronectin in neonatal sepsis. Pediatrics. 1983; 72:877–881.
81. Chirico, G, Rondini, G, Plebani, A, et al. Intravenous gamma globulin therapy for prophylaxis of infection in high-risk neonates. J Pediatr. 1987; 110:437–442.
82. Clark, SC, Kamen, R. The human hematopoietic colony-stimulating factors. Science. 1987; 236:1229–1237.
83. Ohlsson, A, Lacy, J. Intravenous immunoglobulin for preventing infection in preterm and or low birth weight infants. Cochrane Database Syst Reviews. (1):2004.
84. Sieff, CA. Hematopoietic growth factors. J Clin Invest. 1987; 79:1549.
85. Barak, Y, Leibovitz, E, Mogilner, B, et al. The in vivo effect of recombinant human granulocyte-colony stimulating factor in neutropenic neonates with sepsis. Eur J Pediatr. 1997; 156:643–646.
86. Wolach, B. Neonatal sepsis: Pathogenesis and supportive therapy [Review]. Semin Perinatol. 1997; 21:28–38.
87. Cairo, M, Seth, T, Fanaroff, A, et al. A double-blinded randomized placebo controlled pilot study of RhGM-CSF in low birth weight neonates (LBWN): Preliminary results demonstrate a significant reduction in nosocomial infections with Rhu-GM-CS. Pediatr Res. 1996; 39:294a.
88. Brancho, F, Goldman, S, Cairo, M. Potential use of granulocyte colony-stimulating factor and granulocyte-macrophage colony-stimulating factor in neonates. Curr Opin Hematology. 1998; 5:315–320.
89. Carcillo, JA, Fields, AI, Task Force Committee Members. Clinical practice parameters for hemodynamic support of pediatric and neonatal patients septic shock. Crit Care Med. 2002; 30:1365–1377.
90. Tobin, JR, Wetzel, RC, Shock and multi-organ system failure In Handbook of Pediatric Intensive Care, Rogers and Helfaer, eds, 1999.
91. Ruokonen, E, Parviainen, I, Usuaro, A. Treatment of impaired perfusion in septic shock. Ann Med. 2002; 34:590–597.
92. Dellinger, RP. Cardiovascular management of septic shock. Crit Care Med. 2003; 31:946–955.
93. Malay, MB, Ashton, RC, Landry, DW, et al. Low-dose vasopressin in the treatment of vasodilatory septic shock. J Trauma. 1999; 47:699–703.
94. Brierley, J, Carcillo, JA, Choong, K, et al. Clinical practice parameters for hemodynamic support of pediatric and neonatal septic shock: 2007 update from the American College of Critical Care Medicine. Crit Care Med. 2009; 37:666–688.
95. Yager, P, Noviski, N. Shock. Pediatr Rev. 2010; 21:311–318.
96. Srinivasan, L, Harris, MC. New technologies for the rapid diagnosis of neonatal sepsis. Curr Opin Pediatr. 2012; 24:165–171.
97. Hofer, N, Zacharias, E, Muller, W, et al. An update on the use of C-reactive protein in early-onset neonatal sepsis: current insights and new tasks. Neonatol. 2012; 102:25–36.
98. Dilli, D, Dilmer, U. The role of interleukin-6 and C-reactive protein in non-thyroidal illness in premature infants followed in neonatal intensive care unit. J Clin Res Pediatr Endocrinol. 2012; 4:66–71.