[level-membership-for-obstetrics-gynecology-category]
FIGURE 1.1 Graphic representation of variation in pituitary and ovarian hormonal levels during menstrual cycle (not to scale).
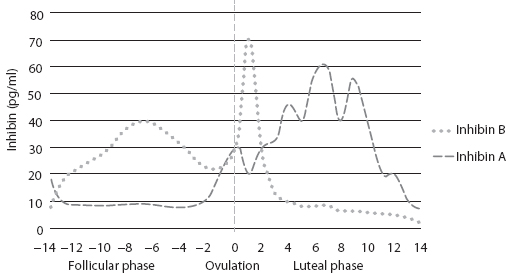
FIGURE 1.2 Graphic representation of inhibin A and B levels during menstrual cycle (not to scale) (2,3).
The LH Surge and the Luteal Phase
Rising levels of E2, produced by the granulosa cells of the preovulatory dominant follicle, cause the release of fast pulse frequencies (>1 pulse per hour) and high amplitude GnRH, favoring increased LH production (2). This causes a switch from a negative to positive feedback effect, resulting in a rapid rise in LH release and the so-called LH surge. The initial onset of the LH surge (not the peak level, which is reached 10–12 hours before ovulation) induces the ovulation about 34–36 hours later. The mean duration of the LH surge is 48 hours. Besides triggering ovulation, the LH surge induces the formation of the corpus luteum, for which an adequate amplitude and duration of LH surge is essential. The wall of the follicle collapses, and capillaries invade the developing corpus luteum probably under the influence of angiogenic and mitogenic factors. The differentiated granulosa cells in the corpus luteum will produce progesterone (P) in increasing amounts, E2, and inhibin A (1,2).
In some species, such as rodents, prolactin-like hormones play a principal role in the luteotropic process, and luteal regression involves a uterine signal such as prostaglandin F2 alpha (1). In contrast to this, in humans, LH is the principle hormone responsible for the following events. First, the mid-cycle surge of gonadotropins (notably, LH) stimulates the resumption of oocyte meiotic maturation; rupture of the dominant follicle, allowing the release of oocyte (ovulation), and corpus luteum formation (i.e., luteinization). Second, the pulsatile secretion of pituitary LH during the luteal phase of the menstrual cycle promotes the continued development and normal functional lifespan of the corpus luteum (1,2).
And finally, the exponential rise in circulating levels of the LH-like hormone, chorionic gonadotropin (CG), secreted by the implanting blastocyst and syncytiotrophoblast of the developing placenta, extends the functional lifespan of the corpus luteum in early pregnancy until luteal activities are assumed by the placenta, that is, at the luteal–placental shift (1,4).
Studies during the 1970s and 1980s and more recent experiments using GnRH antagonists and pure recombinant human LH or human CG (hCG) have strengthened the critical role of LH/CG in regulating primate luteal structure–function and is increasingly being applied in ovarian stimulation for IVF (1).
Although the maturing dominant follicle may be less sensitive to acute LH withdrawal at mid-cycle, a GnRH-induced LH surge of substantial length is required for ovulation and development of normal luteal function. What remains unclear is how duration and/or amplitude of the mid-cycle LH surge influences peri-ovulatory events. Initial monkey and human studies on GnRH-induced LH surges or administering exogenous LH/CG suggest that surges of lesser duration (<24 hours) and amplitude are sufficient to reinitiate oocyte meiosis and granulosa cell luteinization, but surges of greater duration (>24 hours) and amplitude improve oocyte recovery, fertilization, and corpus luteum development (1,2). Although the LH surge is believed to be the physiological signal for peri-ovulatory events, studies in rodents showed that a mid-cycle bolus of FSH can replace LH and elicit oocyte maturation, ovulation, and successful pregnancy (1). The clinical relevance of these physiological observations has recently been brought into sharper focus by the use of GnRHa agonists to induce a mid-cycle LH surge in IVF cycles. Although the short period of stimulation reduces the risk of developing ovarian hyperstimulation syndrome, it is clear that it may impact detrimentally on the luteal phase, compared with the use of hCG as a trigger, which remains bioactive for a longer period. These clinical issues are addressed elsewhere.
The LH-stimulated luteinization of granulosa cells around ovulation includes enhanced vascular endothelial growth factor (VEGF) production, which is likely essential for the angiogenic process within the corpus luteum. With regards to hCG, a mid-cycle bolus in ovarian stimulation cycles increased expression of the endogenous angiopoietin agonist, Ang-1, without altering that of the endogenous antagonist, Ang-2, in macaque granulosa cells. These factors control not only the development or maintenance of the vasculature in developing tissue beds, but also vascular integrity, maturity, and permeability (1). It has been proposed that overexpression, increased bioavailability, or a change in the ratio of angiogenic factors, notably VEGF-A, is a cause of ovarian hyperstimulation syndrome (OHSS), a serious side effect of ovarian stimulation characterized by intravascular volume loss and extravascular fluid accumulation. The early or late occurrence of OHSS in ovarian stimulation cycles has been linked to the ovulatory hCG bolus and endogenous CG production at pregnancy recognition, respectively (1,2).
The Endometrium
The basalis layer that remains after shedding of the more superficial endometrium at menstruation needs to regrow and differentiate (proliferative phase) under the E2 influence secreted by the growing follicles in the follicular phase of the ovarian cycle. The highest E2 response is in the glands via increased E2 receptor (ER) expression (4). First, there is an increase in the mitotic activity, and second, there is formation of a loose capillary network in the spiral vessels. After ovulation, the progesterone (P) produced by the corpus luteum causes the cessation of endometrial proliferation and initiates glandular secretion (secretory phase). The endometrial glands become tortuous and spiral vessels coiled. During the secretory phase, a short specific period of uterine receptivity toward embryonic implantation is designated as the “implantation window” (2,4). Available evidence based on experimental studies supports the discrete time in the cycle between 6 and 10 days after the LH surge that defines this window of implantation (1). Coinciding with this window, the expression of cell adhesion molecules such as integrins, under endocrine and paracrine control, may assist in the initial stages of implantation of the embryo. If conception and implantation occur, the developing blastocyst secretes human chorionic gonadotrophin (hCG), which, in turn, supports and maintains the corpus luteum until the developing placenta takes over steroidogenesis. Continued progesterone exposure also promotes local vasodilatation and uterine musculature quiescence by inducing nitric oxide synthesis in the decidua (1,4).
In the absence of implantation, luteolysis is initiated after 12 days, and P and inhibin A levels decrease, resulting in menstruation. As a result of a reduced negative feedback effect, GnRH pulse frequency will increase, and subsequently, pituitary FSH production will increase and, again, the inter-cycle will rise (2).
The major roles of E2 are for endometrial growth and for enabling P to act on the endometrial tissue—both stroma and glands. To accomplish these goals, E2 induces progesterone receptor (PR) expression and promotes cellular proliferation in the tissue directly through its cognate receptors and indirectly by induction of growth factors that act as autocrine and/or paracrine modulators (1).
With regard to PR, peak expression in human endometrium induced by E2 is at the time of ovulation. PR is most prominent in the glandular epithelium in the proliferative phase. In contrast, stromal cells have high levels of PR in the follicular phase and throughout the luteal phase. Timely downregulation of epithelial PR coincides with the opening of the window of implantation and uterine receptivity for embryonic implantation, and histological delay of the endometrium (a clinically abnormal state) is associated with a failure of such PR downregulation (1).
Recent Advances
The pulsatile release of GnRH is key to starting the menstrual cycle at puberty. Any disruption in the pulse frequency will give rise to reproductive dysfunction. Kisspeptin and neurokinin B (NKB), neuro-peptides secreted by the same neuronal population in the ventral hypothalamus, have recently emerged as crucial central regulators of GnRH and hence gonadotropin secretion. Patients with mutations resulting in loss of signaling by either of these neuroendocrine peptides fail to attain puberty, but the mechanisms mediating this remain unclear (5).
Recent animal and human studies have shown that continuous kisspeptin infusion restores gonadotropin pulsatility in patients with loss of function mutations in NKB or its receptor, indicating that kisspeptin on its own is sufficient to stimulate pulsatile GnRH secretion (Figures 1.3 and 1.4) (5,6). The studies also suggest that NKB action is proximal to kisspeptin in the reproductive neuroendocrine cascade regulating GnRH secretion and may act as an autocrine modulator of kisspeptin secretion. The ability of continuous kisspeptin infusion to induce pulsatile gonadotropin secretion further indicates that GnRH neurons are able to set up pulsatile secretion in the absence of pulsatile exogenous kisspeptin (5,6).
These findings suggest a potential future role of kisspeptin and NKB in treating dysfunctions of the reproductive system and hormone-dependent diseases (5,6).
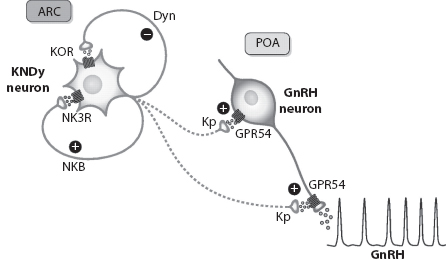
FIGURE 1.3 Suggested model for the role of kisspeptin neurons (KNDy) expressing both neurokinin B (NKB) and dynorphin A (Dyn) neurons in the control of pulsatile GnRH secretion. A schematic model for the likely roles of NKB and Dyn as co-transmitters and possible regulators of the secretory activity of Kiss1 neurons, located at the arcuate nuclei (ARC) as a major driving signal for GnRH pulsatile release by GnRH neurons sited in the preoptic area (POA). (From http://physrev.physiology.org/content/92/3/1235, with permission.)
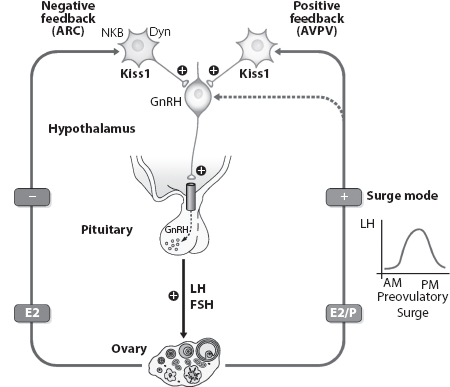
FIGURE 1.4 Differential regulation and actions of ARC versus anteroventral peri-ventricular nucleus (AVPV) Kiss1 neurons in the control of GnRH in rodents. A schematic illustration of the roles of Kiss1 neurons in the ARC and AVPV in mediating the negative and positive feedback effects of ovarian sex steroids on GnRH and gonadotropin secretion as suggested on the basis of female rodent data. (From http://physrev.physiology.org/content/92/3/1235, with permission.)
Conclusion
This chapter outlines the endocrine and paracrine control of the ovarian and concurrent menstrual cycles in the light of available evidence. This acknowledges the complex molecular interactions, and how this could be implied in the context of ART as well as looking into the scope of further research, where the mechanisms are still less clearly understood.
Level of Evidence of Statements
|
Statement |
Level of Evidence |
|
The initial growth of primordial follicles (also referred to as primary recruitment) is random, being independent of FSH. The cohort size of healthy early antral follicles recruited during the luteofollicular transition is around 10 per ovary. |
3 |
|
Inhibin A, secreted by maturing follicle and corpus luteum, has a direct endocrine role in the negative feedback on pituitary FSH production. |
2b |
|
Although the LH surge is believed to be the physiological signal for peri-ovulatory events, a mid-cycle bolus of FSH can replace LH and elicit oocyte maturation, ovulation, early luteinization of granulosa cells, and successful pregnancy. |
2b |
|
The major roles of E2 on uterine endometrium are for endometrial growth and for enabling P to act on the tissue. |
2b |
|
Kisspeptin and neurokinin B (NKB), neuropeptides secreted by the same neuronal population in the ventral hypothalamus, have emerged recently as critical central regulators of GnRH and thus gonadotropin secretion (5,6). |
2b |
REFERENCES
1. Macklon NS, Stouffer RL, Giudice LC, Fauser BCJM. The science behind 25 years of ovarian stimulation for in vitro fertilization Endocrine Reviews. 2006; 27(2):170–207. doi: 10.1210/er.2005-0015.
2. Beckers NGM, Macklon NS, Fauser BCJM. Follicular and luteal phase aspects of ovarian stimulation for in vitro fertilisation. 2006; 11–3.
3. Ledger LW. Ovarian and Menstrual Cycles, Chapter 49, pp. 509–513.
4. Macklon NS, Greer IA, Steegers EAP. Textbook of Periconceptional Medicine. Chapter 21, The Luteal Phase, pp. 298–9.
5. Young J, George JT, Tello JA, Francou B, Bouligand J, Guiochon-Mantel A et al. Kisspeptin restores pulsatile LH secretion in patients with neurokinin B signaling deficiencies: Physiological, pathophysiological and therapeutic implications. Neuroendocrinology. 2013 Mar; 97(2):193–202. Published online Feb 24, 2012. doi: 10.1159/000336376 PMCID: PMC3902960.
6. Pinilla L, Aguilar E, Dieguez C, Millar RP, Tena-Sempere M. Kisspeptins and reproduction: Physiological roles and regulatory mechanisms. Physiological Reviews. 2012, July; 92(3):1235–316. doi: 10.1152/physrev.00037.2010.
[/level-membership-for-obstetrics-gynecology-category][not-level-membership-for-obstetrics-gynecology-category]
FIGURE 1.1 Graphic representation of variation in pituitary and ovarian hormonal levels during menstrual cycle (not to scale).
FIGURE 1.2 Graphic representation of inhibin A and B levels during menstrual cycle (not to scale) (2,3).
The LH Surge and the Luteal Phase
Rising levels of E2, produced by the granulosa cells of the preovulatory dominant follicle, cause the release of fast pulse frequencies (>1 pulse per hour) and high amplitude GnRH, favoring increased LH production (2). This causes a switch from a negative to positive feedback effect, resulting in a rapid rise in LH release and the so-called LH surge. The initial onset of the LH surge (not the peak level, which is reached 10–12 hours before ovulation) induces the ovulation about 34–36 hours later. The mean duration of the LH surge is 48 hours. Besides triggering ovulation, the LH surge induces the formation of the corpus luteum, for which an adequate amplitude and duration of LH surge is essential. The wall of the follicle collapses, and capillaries invade the developing corpus luteum probably under the influence of angiogenic and mitogenic factors. The differentiated granulosa cells in the corpus luteum will produce progesterone (P) in increasing amounts, E2, and inhibin A (1,2).
In some species, such as rodents, prolactin-like hormones play a principal role in the luteotropic process, and luteal regression involves a uterine signal such as prostaglandin F2 alpha (1). In contrast to this, in humans, LH is the principle hormone responsible for the following events. First, the mid-cycle surge of gonadotropins (notably, LH) stimulates the resumption of oocyte meiotic maturation; rupture of the dominant follicle, allowing the release of oocyte (ovulation), and corpus luteum formation (i.e., luteinization). Second, the pulsatile secretion of pituitary LH during the luteal phase of the menstrual cycle promotes the continued development and normal functional lifespan of the corpus luteum (1,2).
And finally, the exponential rise in circulating levels of the LH-like hormone, chorionic gonadotropin (CG), secreted by the implanting blastocyst and syncytiotrophoblast of the developing placenta, extends the functional lifespan of the corpus luteum in early pregnancy until luteal activities are assumed by the placenta, that is, at the luteal–placental shift (1,4).
Studies during the 1970s and 1980s and more recent experiments using GnRH antagonists and pure recombinant human LH or human CG (hCG) have strengthened the critical role of LH/CG in regulating primate luteal structure–function and is increasingly being applied in ovarian stimulation for IVF (1).
Although the maturing dominant follicle may be less sensitive to acute LH withdrawal at mid-cycle, a GnRH-induced LH surge of substantial length is required for ovulation and development of normal luteal function. What remains unclear is how duration and/or amplitude of the mid-cycle LH surge influences peri-ovulatory events. Initial monkey and human studies on GnRH-induced LH surges or administering exogenous LH/CG suggest that surges of lesser duration (<24 hours) and amplitude are sufficient to reinitiate oocyte meiosis and granulosa cell luteinization, but surges of greater duration (>24 hours) and amplitude improve oocyte recovery, fertilization, and corpus luteum development (1,2). Although the LH surge is believed to be the physiological signal for peri-ovulatory events, studies in rodents showed that a mid-cycle bolus of FSH can replace LH and elicit oocyte maturation, ovulation, and successful pregnancy (1). The clinical relevance of these physiological observations has recently been brought into sharper focus by the use of GnRHa agonists to induce a mid-cycle LH surge in IVF cycles. Although the short period of stimulation reduces the risk of developing ovarian hyperstimulation syndrome, it is clear that it may impact detrimentally on the luteal phase, compared with the use of hCG as a trigger, which remains bioactive for a longer period. These clinical issues are addressed elsewhere.
The LH-stimulated luteinization of granulosa cells around ovulation includes enhanced vascular endothelial growth factor (VEGF) production, which is likely essential for the angiogenic process within the corpus luteum. With regards to hCG, a mid-cycle bolus in ovarian stimulation cycles increased expression of the endogenous angiopoietin agonist, Ang-1, without altering that of the endogenous antagonist, Ang-2, in macaque granulosa cells. These factors control not only the development or maintenance of the vasculature in developing tissue beds, but also vascular integrity, maturity, and permeability (1
[/not-level-membership-for-obstetrics-gynecology-category]
