CHAPTER 4 Physiology of the Heart
CARDIAC ELECTROPHYSIOLOGY
Electrical depolarization initiated by the SA node first travels through the CS of the atria. This part of the electrical cycle is represented on the ECG by the P wave. Figure 4-1 is a diagram depicting the functional anatomy of the conduction system. The normal ECG appearance is shown in Figure 4-2.
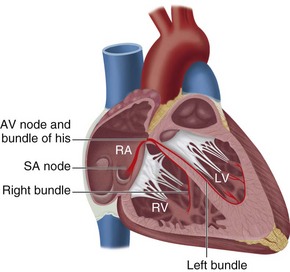
 FIGURE 4-1
FIGURE 4-1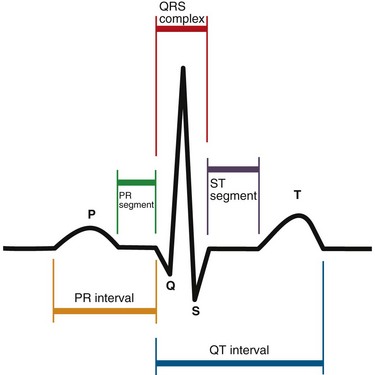
 FIGURE 4-2
FIGURE 4-2MYOCARDIAL AND VALVULAR FUNCTION
Cardiac myocytes are linked with each other by a network of extracellular matrix. The matrix consists primarily of collagen and proteoglycans synthesized by fibroblasts and smooth muscle cells. Individual myocytes are linked to the extracellular matrix by molecules called integrins. When triggered by electrical membrane depolarization, a rapid intracellular increase in calcium causes sequential contraction of the sarcomere through actin/myosin interactions (Fig. 4-3). Coordinated contraction of individual myocytes, connected to each other through the extracellular matrix, results in ventricular systole and propulsion of blood through the cardiac chambers.
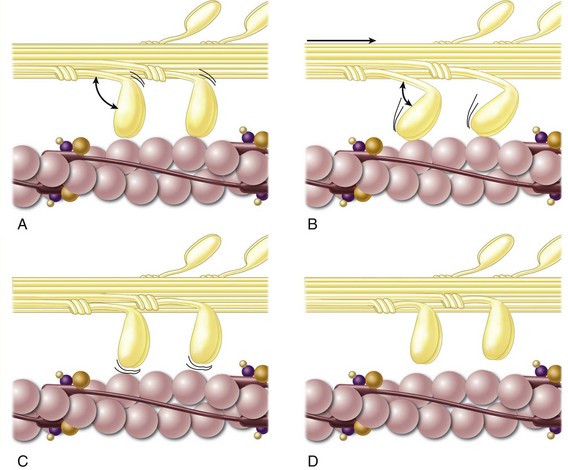
 FIGURE 4-3
FIGURE 4-3Several variables or parameters can be used to quantify myocardial function. Stroke volume is defined as the difference between end-diastolic and end-systolic volumes (the volume ejected from the ventricular cavity between the beginning and end of systole). Dividing the stroke volume by the end-diastolic volume gives the ejection fraction (EF), the percentage of end-diastolic volume ejected. The EF is the most commonly used measure of systolic function.1
Sequential flow of blood through the cardiac chambers is driven by myocardial contraction and opening and closing of the cardiac valves (Fig. 4-4). The cardiac valves are one-way valves that open and close passively in response to pressure differences between the chambers they connect. Under normal circumstances, the cardiac valves permit forward flow only and prevent backward flow of blood.
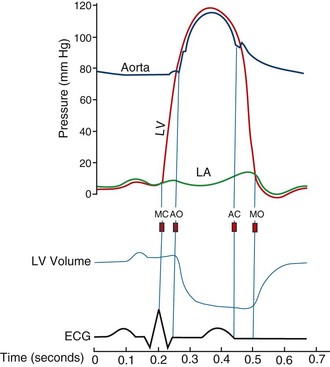
 FIGURE 4-4
FIGURE 4-4During ventricular diastole, pressure in the ventricles decreases to less than the pressure in the atria, causing opening of the AV valves and inflow of blood from the atria into the ventricles. Near the end of diastole, ventricular filling is augmented by atrial contraction. This “atrial kick” increases preload by distending the ventricular cavity at end-diastole, increasing the effectiveness of systolic contraction as a result of the Frank-Starling mechanism.2 At the beginning of ventricular systole, the rapid increase of pressure within the ventricular cavity causes closure of the AV valves. After a brief period of isovolumic contraction, pressure within the ventricular cavity exceeds the aortic and pulmonary arterial pressures, resulting in opening of the aortic and pulmonic valves and forward flow of blood into the aorta and pulmonary artery.
ATHEROSCLEROTIC CORONARY DISEASE
Coronary artery disease (CAD) is the largest contributor to cardiac mortality in developed countries, and was the cause of one of every four deaths in the United States in 2005.3 The morbidity and mortality of CAD can manifest in two forms: (1) chronically with reduction of coronary flow or flow reserve resulting from stenoses of the coronary arteries, and (2) acutely with atherothrombotic occlusion of a coronary artery. The stages of development of atherosclerosis and the mechanisms of disease manifestation are briefly reviewed.
Vascular Biology of Atherosclerosis
The normal blood vessel wall consists of three distinct layers—intima, tunica media, and tunica adventitia. The innermost layer, the intima, is the site where atherosclerosis first manifests (Fig. 4-5). The intima consists mainly of endothelial cells, whereas the media contains smooth muscle cells, macrophages, mast cells, and extracellular matrix (primarily collagen and proteoglycans). The adventitia is composed primarily of extracellular connective tissue, but it also contains the vasa vasorum (vascular supply to the blood vessel wall itself) and nerve fibers involved in vasoregulation.
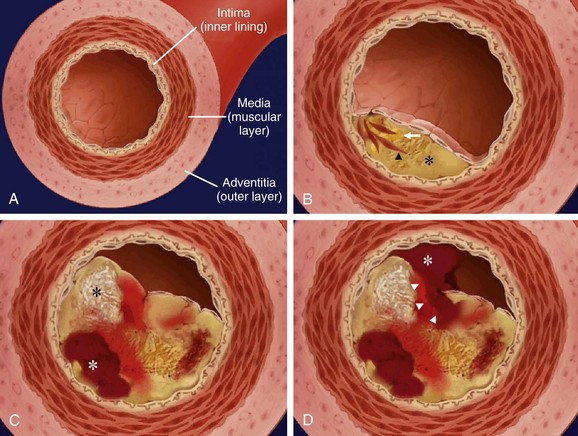
 FIGURE 4-5
FIGURE 4-5In areas of low shear stress (e.g., at vessel bifurcations or near the ostia of side branches), the endothelial layer may thicken (often eccentrically). These areas of thickened endothelium often show early findings of atherosclerosis.4 The earliest atherosclerotic manifestation is the “fatty streak,” characterized by intimal deposition of lipid-laden macrophages and T lymphocytes.5 Fatty streaks occur in areas of intimal thickening because of multifactorial endothelial dysfunction and denudation, allowing intraintimal deposition of serum lipoproteins. Toxic damage from tobacco smoking and oxidative damage from very-low-density lipoproteins can be contributory factors. This initial endothelial injury incites an inflammatory response mediated by vascular cell adhesion molecules, selectins, and cytokines. The inflammatory response leads to ingestion of the lipoproteins by monocytes and macrophages, producing characteristic lipid-laden macrophages (“foam cells”) on histology.6
Over time, demise and disintegration of lipid-laden macrophages and further deposition of serum lipoproteins lead to accumulation of extracellular lipid in the growing “plaque.” Connective tissue, with varying portions of smooth muscle, collagen, and other extracellular proteins, also proliferates. As the plaque grows in size, the lumen of the vessel can become narrowed, although adaptive, positive remodeling of the vessel lumen by outward expansion of the vessel wall with an increase of vessel cross-section area (see Fig. 4-5) may initially prevent stenosis.7 Paradoxically, positive remodeling may occur in an effort to resist endothelial shear stress, but the lower shear stress created by positive remodeling may lead to further lipid accumulation.4 Vascular remodeling can be examined well with cross-sectional imaging modalities that show lumen and vessel wall (Fig. 4-6). On invasive, selective coronary angiography, the vessel wall is not imaged, and plaque “hidden” by positive remodeling may not be recognized.
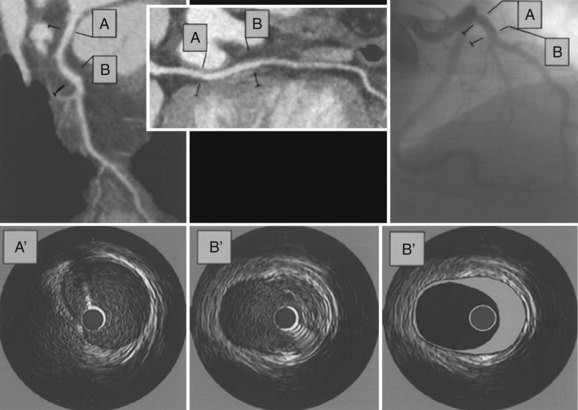
 FIGURE 4-6
FIGURE 4-6Calcification frequently occurs in atherosclerotic plaque, and can occur in lipid-rich and predominantly fibrotic plaques. The quantity of coronary calcification is nonlinearly proportional to overall atherosclerotic burden. Coronary calcification represents approximately 20% of plaque volume.8 The location of calcification does not predict the location of high-grade stenosis or plaque prone to rupture. The relationship between coronary calcification and overall atherosclerotic burden provides the rationale for the practice of coronary artery calcium screening for risk stratification.9
The plaque core eventually becomes hypoxic as it outstrips the nutrient supply by the vascular network within the blood vessel wall, the vasa vasorum. Subsequent hypoxia-induced cellular death within the plaque core and recruitment of fragile, immature neovasculature predisposes to intraplaque hemorrhage. Necrosis of the plaque core and intraplaque hemorrhage are the key contributors to plaque growth at later stages and eventually to rupture of the “unstable” plaque. When the plaque ruptures, the exposed lipid-rich core triggers platelet aggregation and intravascular thrombosis.10
Plaque Rupture and Acute Coronary Syndromes
Counterintuitively, plaque rupture leading to acute cardiac events is more likely to occur in coronary segments with low-grade stenoses than in segments with high-grade coronary stenoses. An analysis of factors associated with progression of CAD in 2938 coronary segments in 298 patients who had not undergone coronary bypass from the Coronary Artery Surgery Study (CASS) showed that although individual segments with high-grade stenoses were more likely to become occluded than individual segments with low-grade stenoses, occlusion was overall much more likely to occur in segments with low-grade stenoses because low-grade stenoses are much more common.11
The acute interruption of coronary blood flow, typically by plaque rupture, without adequate compensatory mechanisms to maintain oxygen supply to the myocardium results in myocardial infarction, defined as myocyte necrosis. The diagnostic criteria for myocardial infarction include prolonged severe chest discomfort of acute onset; elevation of the ST segment on the ECG; and elevated blood concentrations of enzymes specific for myocyte demise, such as the MB-fraction of creatine kinase or the troponins.12 Infarction occurs within minutes to hours from the inciting event. Because myocardial oxygen is supplied from the epicardial vessels toward the subendocardial perforators, the subendocardial myocardium is the layer affected first as the end-vessel territory. With prolonged severe ischemia, myocyte necrosis spreads transmurally from the endocardium toward the mesocardial and epicardial layers. The treatment of choice is revascularization by percutaneous coronary intervention within 4 to 6 hours of symptom onset if available. Intravenous administration of thrombolytic agents, such as recombinant tissue plasminogen activator (rTPA), is an acceptable alternative.13
Chronic Coronary Artery Disease
In the setting of chronic CAD with progressive narrowing of the coronary lumen but without sudden complete occlusion, the typical clinical feature is chronic stable angina. “Typical” angina is defined as retrosternal discomfort that is provoked by exertion and relieved by rest or administration of nitroglycerin. Episodes of typical angina last less than 30 minutes. It can be difficult to determine with anatomic imaging modalities such as coronary CT or MRI whether a stenosis caused by plaque is “significant”—severe enough to limit oxygen delivery and be the cause of angina or myocardial dysfunction (see later section on myocardial viability) or both. At rest, the myocardial oxygen extraction rate is approximately 60%.14 An increase in oxygen consumption, such as occurs with physical activity, requires an increase in oxygen delivery, which is accomplished by vasodilation under normal circumstances. The ability of the coronary vasculature to increase coronary blood flow, expressed as the ratio of maximal coronary flow to resting flow, is referred to as coronary flow reserve.15 In the presence of coronary stenosis, vasodilation to improve blood flow distal to the narrowed segment occurs at rest. This vasodilation decreases coronary flow reserve because the compensatory mechanism is already employed at baseline. The ability to elicit and document abnormal coronary flow reserve is the basic principle underlying stress testing as a functional means for assessing CAD.16
Although numerous studies have shown a correlation between percent stenosis and decrease in coronary flow reserve, the correlation is nonlinear. Functional severity of a given percent stenosis may vary among patients, depending on many factors, including coronary perfusion pressure, narrowest lumen diameter, and number and length of stenoses. Although it is generally accepted that stenoses greater than 70% reduce coronary flow reserve to a clinically relevant degree, the clinical “significance” of stenoses of intermediate severity (50% to 70%) can be difficult to ascertain. Qualitative or quantitative assessment of myocardial perfusion during stress and at rest using nuclear medicine techniques or MRI can provide information that is complementary to arterial illustration using conventional x-ray or CT angiography, and has important implications for patient management.17,18
Myocardial infarction in the setting of chronic CAD with preexisting high-grade coronary stenoses can occur (1) as a consequence of plaque rupture, or (2) in situations where myocardial oxygen demand exceeds the oxygen supply that is limited by coronary stenosis, such as during unusual physical exertion or other physiologic stress, including high-risk surgery or severe systemic illness. The latter scenario often results in so-called non–ST segment elevation myocardial infarction.19,20
MYOCARDIAL DISEASE
Ischemic Cardiomyopathy
Information on the degree of left ventricular dysfunction is crucial for patient management. Poor left ventricular function predicts poor outcome. At an EF of less than 20%, the average 1-year survival is less than 50%. In these patients, ventricular dysrhythmia is at least as important a cause of death as is heart failure. Placement of implantable cardioverter defibrillators can improve survival, and is recommended for prevention of sudden cardiac death in patients with EF less than 30% to 40% and New York Heart Association (NYHA) class II-III heart failure or EF less than 30% to 35% and NYHA class I heart failure.21
Myocardial Viability, Stunning, and Hibernation
In chronic CAD, compensatory changes occur at the myocardial level, such as decreased oxygen demand and increased myocardial oxygen extraction, cell loss, and increased glycogen storage. With more severe chronic ischemia, hemodynamic and functional adaptations, including elevation of end-diastolic pressure, decreased stroke volume, and delayed myocardial contraction and relaxation, help cope further with reduced oxygen delivery. The net effect of these mechanisms representing “myocardial hibernation” decreased systolic function, which may be reversible if adequate oxygen supply is restored by revascularization.22 Reversible myocardial dysfunction may also result from “stunning” in the setting of acute but transient ischemic events. Stunning may be the only consequence of an acute coronary syndrome or may coexist with irreversible myocyte damage.
Pressure and Volume Overload
The response of the heart to increased work demand is hypertrophy—an increase in myocardial mass accomplished by enlargement of individual myocytes. Hypertrophy can represent a physiologic response to repetitive exercise, and probably plays a role in the normal enlargement of the heart through childhood and adolescence as well as in physiologic states such as pregnancy. The hypertrophic response can result in decompensated hypertrophy, however, in the setting of pressure or volume overload, or when it occurs as a compensatory mechanism after infarction of another coronary perfusion territory. The relationship between pressure and volume can be quite complex, and although the classification into pressure and volume overload is a useful conceptual construct, many patients have elements of both processes and may not fit neatly into one category or the other.23
PHYSIOLOGY OF DIASTOLE
Diastolic dysfunction resulting from myocardial disease occurs in the setting of increased myocardial stiffness. Increased myocardial stiffness may be due to myocardial fibrosis at preserved left ventricular wall-to-cavity ratio or due to left ventricular hypertrophy. Left ventricular hypertrophy (see earlier) most often results from long-standing hypertension, but may also be due to infiltrative diseases such as amyloidosis or primary genetic disorders. Diastolic dysfunction is probably under-recognized in many settings. Although there is controversy about the clinical importance of diastolic dysfunction, its presence seems to increase mortality.24
PERICARDIAL DISEASE
The pericardium is a sac of fibrous tissue composed of visceral and parietal layers. It surrounds the heart and the proximal great vessels, and typically contains a physiologic small amount of fluid (Fig. 4-7). In normal patients, the pericardium does not play a significant role in cardiac physiology; patients with complete or partial congenital absence of the pericardium rarely have clinical signs or symptoms attributable to abnormal diastolic function.25 The pericardium or its contents can impede normal diastolic and systolic function, however. The two principal manifestations of pericardial disease are tamponade and constriction.
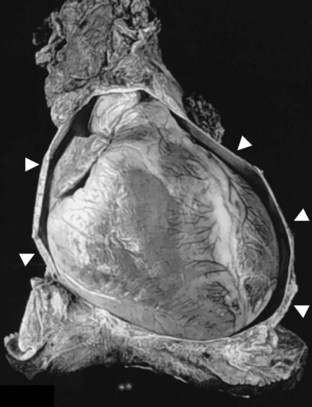
 FIGURE 4-7
FIGURE 4-7Tamponade
Acute cardiac tamponade is a medical emergency. The treatment of choice is emergent pericardiocentesis, preferably under echocardiographic guidance if available. Chronic tamponade can have numerous causes (Table 4-1). Because the pericardium can gradually expand to accommodate slowly growing effusions, however, the volume of pericardial effusion can be large (2 L), and is generally less important than the rate of increase for determining the physiologic consequences (Fig. 4-8). In this situation, removal of even small amounts of fluid can result in impressive improvement of diastolic filling and cardiac output.
TABLE 4-1 Causes of Cardiac Tamponade and Constrictive Pericarditis
| Causes of Cardiac Tamponade | Causes of Constrictive Pericarditis | |
|---|---|---|
| Acute | Chronic | |
| Trauma | Heart failure | Postoperative |
| Iatrogenic | Uremia | Trauma |
| Infection | Metastasis | Radiation |
| Myocardial infarction with rupture | Infection Radiation |
Infection (particularly tuberculosis) |
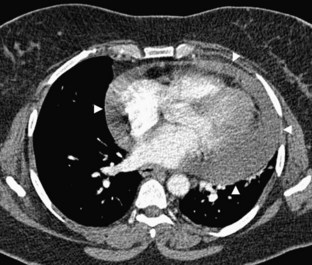
 FIGURE 4-8
FIGURE 4-8Constrictive Pericarditis
Constrictive pericarditis results from pericardial scarring, which often has a nodular and calcified appearance on imaging (Fig. 4-9). Constrictive pericarditis can occur secondary to many causes, all of which functionally eliminate the pericardial space and “constrict” the volume of the heart (Table 4-2). In contrast to tamponade, early diastolic filling not only is unimpeded by pressure equalization, but also is even more rapid than usual. Mid-diastolic and late diastolic filling is decreased, however, when the volume of the heart approaches the fixed volume of the constricting pericardium.
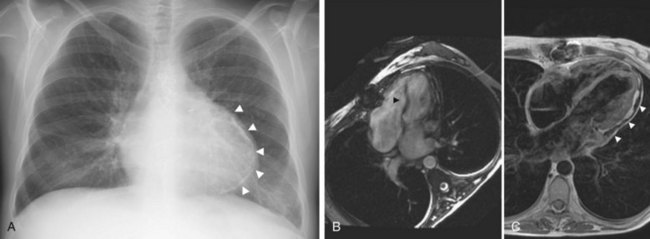
 FIGURE 4-9
FIGURE 4-9| Normal Ventricular Pressures in Recumbent Adults: | ||
|---|---|---|
| Right Ventricle | Left Ventricle | |
| Diastole | 0-8 mm Hg | 5-12 mm Hg |
| Systole | 15-28 mm Hg | 90-120 mm Hg |
| Normal Values | Male | Female |
|---|---|---|
| Ejection fraction (%) | 56-78 | 56-78 |
| Stroke volume (mL) | 51-133 | 33-97 |
As a feature distinguishing the physiologies of tamponade and constrictive pericarditis, systemic venous return does not increase during inspiration in the latter. Clinically, patients often present with findings of venous congestion and severe right heart failure, which can mimic hepatic failure. As might be expected, the clinical presentation is similar to restrictive cardiomyopathy. In contrast to restrictive cardiomyopathy, constrictive pericarditis can generally be treated successfully with a pericardial resection. It is important to distinguish constrictive from merely restrictive physiology when diastolic dysfunction is present. Imaging can be useful to confirm the presence of pericardial thickening or calcifications to support a diagnosis of pericardial constriction. The most compelling data for the clinician are determined by cardiac catheterization; specific patterns of the left ventricular pressure curve, equalization of diastolic pressure in all chambers, and greatly increased “ventricular interdependence” during respiration are features diagnostic of constrictive pericarditis.26
VALVULAR DYSFUNCTION
Dysfunction of the cardiac valves primarily manifests as insufficiency or stenosis. Insufficiency, also referred to as regurgitation, is characterized by insufficient valve closure, allowing inappropriate retrograde flow of blood through the AV valves during systole or the semilunar valves during diastole. Stenosis is characterized by inadequate valve opening, creating an obstacle to antegrade flow across the valve and increasing flow velocity through the valve orifice. Left-sided (mitral or aortic) valvular dysfunction is more common and more relevant to clinical practice than right-sided valvular dysfunction. The left-sided valvular abnormalities encountered most frequently are mitral insufficiency and aortic stenosis.27
Mitral Valve Disease
The principal causes of mitral insufficiency are abnormalities of the mitral valve (e.g., mitral valve prolapse) or the mitral valve annulus, defective tensor apparatus (papillary muscles and chordae), and altered left atrial or ventricular size or geometry (dilative or hypertrophic cardiomyopathies). In addition, chronic elevation of end-systolic pressure from hypertension and obesity may contribute to mild, asymptomatic valvular regurgitation in many patients. Physical examination shows a systolic murmur, often holosystolic or preceded by a “mid-systolic click,” depending on the mechanism of mitral insufficiency. Because of the chronic volume overload resulting from continuous back-and-forth flow of blood across the insufficient valve, the left atrium and left ventricle enlarge, and stroke volume and EF are supranormal. The treatment of choice is valve repair (where possible) or valve replacement. Optimal timing of surgery is when mitral regurgitation is “severe” (stage 4) while the patient is still asymptomatic.28 Acute mitral regurgitation can occur as a complication of myocardial infarction, endocarditis, or trauma, and often causes severe hemodynamic instability and congestive heart failure. This group of patients requires aggressive management and early surgery.
Aortic Valve Disease
Aortic stenosis can be subvalvular (which is different from the dynamic outflow tract obstruction in hypertrophic cardiomyopathy), valvular, or supravalvular. The valvular form is most common, and the most common cause is senile degeneration. Rarer causes of aortic stenosis include congenitally bicuspid valve and rheumatic fever. Physical examination is remarkable for a systolic murmur, the characteristics of which vary and can provide important clues to distinguishing aortic stenosis from hypertrophic cardiomyopathy. The increased afterload leads to compensatory left ventricular hypertrophy (which serves to normalize wall stress.) Chamber size is typically normal until compensatory mechanisms are exhausted, and left ventricular dilation and dysfunction occur. The proximal aorta may show typical “poststenotic” dilation. The treatment of choice is valve replacement. Valve replacement is indicated for all patients with “critical” aortic stenosis (pressure gradient between left ventricular outflow tract and thoracic aorta >80 mm Hg or aortic valve area <0.7 cm2). Traditionally, severe aortic stenosis (pressure gradient >40 mm Hg or aortic valve area <1 cm2) was an indication only for patients symptomatic with dyspnea, chest pain, or syncope. More recent studies of the natural history of severe aortic stenosis suggest, however, that asymptomatic patients with severe aortic stenosis should also undergo valve replacement.29
Imaging Assessment of Valve Disease
Clinically, MRI is increasingly used to evaluate valvular function in various clinical scenarios.30 These examinations classically are performed using phase-contrast sequences that allow quantification of flow (Fig. 4-10). Even in the absence of such dedicated sequences, many “bright blood” gradient-echo sequences show intravoxel dephasing from turbulent flow. These findings can provide important clues to valvular abnormalities on conventional MRI examinations, on which the valve leaflets themselves are often seen only partially or not at all (Fig. 4-11). Assessment of valvular disease by CT is currently not part of routine clinical practice.
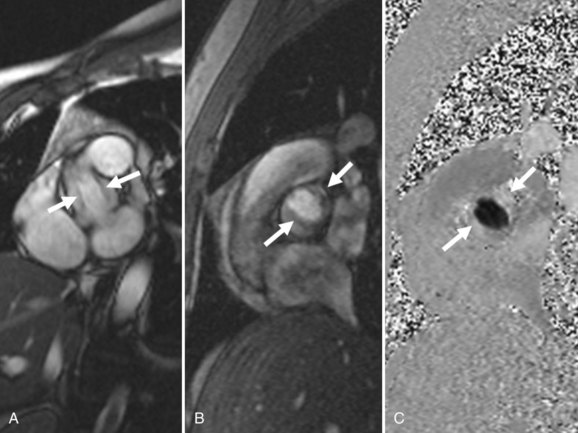
 FIGURE 4-10
FIGURE 4-10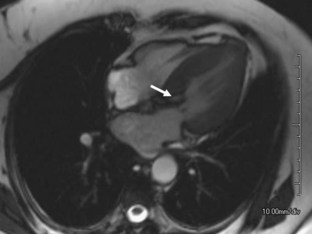
 FIGURE 4-11
FIGURE 4-11CONCLUSION





Chatzizisis YS, Coskun AU, Jonas M, et al. Role of endothelial shear stress in the natural history of coronary atherosclerosis and vascular remodeling. J Am Coll Cardiol. 2007;49:2379-2393.
Fowler SJ, Narula J, Gurudevan SV. Review of noninvasive imaging for hypertrophic cardiac syndromes and restrictive physiology. Heart Fail Clin. 2006;2:215-230.
Gilkeson RC, Markowitz AH, Balgude A, et al. MDCT evaluation of aortic valvular disease. AJR Am J Roentgenol. 2006;186:350-360.
Glockner JF, Johnston DL, McGee KP. Evaluation of cardiac valvular disease with MR imaging: qualitative and quantitative techniques. RadioGraphics. 2003;23:e9.
Raggi P, Berman DS. Computed tomography coronary calcium screening and myocardial perfusion imaging. J Nucl Cardiol. 2005;12:96-103.
Vogel-Claussen J, Pannu H, Spevak PJ, et al. Cardiac valve assessment with MR imaging and 64-section multi-detector row CT. RadioGraphics. 2006;26:1769-1784.
Wang ZJ, Reddy GP, Gotway MB, et al. CT and MR imaging of pericardial disease. RadioGraphics. 2003;23:S167-S180.
1 Rumberger JA, Behrenbeck T, Bell MR, et al. Determination of ventricular ejection fraction: a comparison of available imaging methods. The Cardiovascular Imaging Working Group. Mayo Clin Proc. 1997;72:860-870.
2 Hanft LM, Korte FS, McDonald KS. Cardiac function and modulation of sarcomeric function by length. Cardiovasc Res. 2008;77:627-636.
3 Kung HC, Hoyert DL, Xu J, et al. Deaths: final data for 2005. Natl Vital Stat Rep. 2008;56:1-120.
4 Chatzizisis YS, Coskun AU, Jonas M, et al. Role of endothelial shear stress in the natural history of coronary atherosclerosis and vascular remodeling: molecular, cellular, and vascular behavior. J Am Coll Cardiol. 2007;49:2379-2393.
5 Stary H. Natural history and histological classification of atherosclerotic lesions: an update. Arterioscler Thromb Vasc Biol. 2000;20:1177-1178.
6 Libby P. Inflammation in atherosclerosis. Nature. 2002;6917:868-874.
7 Schoenhagen P, Tuzcu EM, Stillman AE, et al. Non-invasive assessment of plaque morphology and remodeling in mildly stenotic coronary segments: comparison of 16-slice computed tomography and intravascular ultrasound. Coron Artery Dis. 2003;14:459-462.
8 Sangiorgi G, Rumberger JA, Severson A, et al. Arterial calcification and not lumen stenosis is highly correlated with atherosclerotic plaque burden in humans: a histologic study of 723 coronary artery segments using nondecalcifying methodology. J Am Coll Cardiol. 1998;31:126-133.
9 Rumberger JA, Sheedy PF, Breen JF, et al. Electron beam computed tomographic coronary calcium score cutpoints and severity of associated angiographic lumen stenosis. J Am Coll Cardiol. 1997;29:1542-1548.
10 Virmani R, Burke A, Farb A, et al. Pathology of the vulnerable plaque. J Am Coll Cardiol. 2006;47(8 Suppl):C13-C18.
11 Little WC, Constantinescu M, Applegate RJ, et al. Can coronary angiography predict the site of a subsequent myocardial infarction in patients with mild-to-moderate coronary artery disease? Circulation. 1988;78:1157-1166.
12 Thygesen K, Alpert JS, Ryden L, et al. Myocardial infarction redefined—a consensus document of the joint European Society of Cardiology/American College of Cardiology committee for the redefinition of myocardial infarction. J Am Coll Cardiol. 2000;36:959-969.
13 Antman EM, Anbe DT, Armstrong PW, et al. American College of Cardiology; American Heart Association; Canadian Cardiovascular Society. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction—executive summary. A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to revise the 1999 guidelines for the management of patients with acute myocardial infarction). J Am Coll Cardiol. 2004;44:671-719.
14 Porenta G, Cherry S, Czernin J, et al. Noninvasive determination of myocardial blood flow, oxygen consumption and efficiency in normal humans by carbon-11 acetate positron emission tomography imaging. Eur J Nucl Med. 1999;26:1465.
15 Vassalli G, Hess OM. Measurement of coronary flow reserve and its role in patient care. Basic Res Cardiol. 1998;93:339.
16 Jr Fraker TD, SD Fihn. 2002 Chronic Stable Angina Writing Committee; American College of Cardiology; American Heart Association: 2007 chronic angina focused update of the ACC/AHA 2002 guidelines for the management of patients with chronic stable angina: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines Writing Group to develop the focused update of the 2002 guidelines for the management of patients with chronic stable angina. J Am Coll Cardiol. 2007;50:2264-2274.
17 Beanlands RS, Muzik O, Melon P, et al. Noninvasive quantification of regional myocardial flow reserve in patients with coronary atherosclerosis using nitrogen-13 ammonia positron emission tomography: determination of extent of altered vascular reactivity. J Am Coll Cardiol. 1995;26:1465-1475.
18 DiCarli M, Czernin J, Hoh CK, et al. Relation among stenosis severity, myocardial blood flow, and flow reserve in patients with CAD. Circulation. 1995;91:1944-1951.
19 Braunwald E, Antman EM, Beasley JW, et al ACC/AHA guidelines for the management of patients with unstable angina and non-ST-segment elevation myocardial infarction. A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee on the Management of Patients with Unstable Angina) [published correction appears in J Am Coll Cardiol 2001; 38:294-295] J Am Coll Cardiol, 36 2000. 970-1062.
20 Braunwald E, Antman EM, Beasley JW, et al. ACC/AHA guideline update for the management of patients with unstable angina and non-ST-segment elevation myocardial infarction—2002: summary article. A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee on the Management of Patients with Unstable Angina). Circulation. 2002;106:1893-1900.
21 Zipes DP, Camm AJ, Borggrefe M, et al. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death—executive summary. A report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to develop guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death). J Am Coll Cardiol. 2006;48:1064-1108.
22 Camici PG, Prasad SK, Rimoldi OE. Stunning, hibernation, and assessment of myocardial viability. Circulation. 2008;117:103-114.
23 Burkhoff D, Mirsky I, Suga H. Assessment of systolic and diastolic ventricular properties via pressure-volume analysis: a guide for clinical, translational, and basic researchers. Am J Physiol Heart Circ Physiol. 2005;289:501-512.
24 Redfield MM, Jacobsen SJ, Burnett JCJr, et al. Burden of systolic and diastolic ventricular dysfunction in the community appreciating the scope of the heart failure epidemic. JAMA. 2003;289:194-202.
25 Gatzoulis MA, Munk MD, Merchant N, et al. Isolated congenital absence of the pericardium: clinical presentation, diagnosis, and management. Ann Thorac Surg. 2000;69:1209-1215.
26 Talreja DR, Nishimura RA, Oh JK, et al. Constrictive pericarditis in the modern era. J Am Coll Cardiol. 2008;51:315-319.
27 Supino PG, Borer JS, Preibisz J, et al. The epidemiology of valvular heart disease: a growing public health problem. Heart Fail Clin. 2006;2:379-393.
28 Calvinho P, Antunes M. Current surgical management of mitral regurgitation. Expert Rev Cardiovasc Ther. 2008;6:481-490.
29 Brown ML, Pellikka PA, Schaff HV, et al. The benefits of early valve replacement in asymptomatic patients with severe aortic stenosis. J Thorac Cardiovasc Surg. 2008;135:308-315.
30 Stork A, Franzen O, Ruschewski H, et al. Assessment of functional anatomy of the mitral valve in patients with mitral regurgitation with cine magnetic resonance imaging: comparison with transesophageal echocardiography and surgical results. Eur Radiol. 2007;12:3189-3198.
