Physiological Basis for Oxygenation and Lung Protection Strategies
After reading this chapter, you will be able to:
• Explain why monitoring arterial carbon dioxide pressure (PaCO2) is an important consideration when monitoring oxygen therapy in patients with chronic obstructive pulmonary disease (COPD) who are chronically hypercapnic
• Explain why a change in body position might improve oxygenation in unilateral lung disease
• Describe how various inflammatory processes injure the alveolar capillary membrane and produce severe shunting and hypoxemia in acute respiratory distress syndrome (ARDS)
• Explain how positive end expiratory pressure (PEEP) and continuous positive airway pressure (CPAP) improve oxygenation in ARDS
• Differentiate between PEEP and CPAP indications and mechanisms of action
• Describe how the interplay between alveolar and intrapleural pressures determines lung volume in both spontaneous and mechanically induced ventilation
• Explain why pressure and volume cannot function as independent variables in producing alveolar stretch injury
• Explain how to monitor alveolar pressure clinically and the rationale for its measurement
• Describe the mechanisms whereby alveolar overdistention, atelectrauma, and biotrauma are involved in ventilator-induced lung injury (VILI), and explain how they can be prevented during mechanical ventilation
• Explain why the accepted standard of care in mechanically ventilating patients with ARDS might require the development of hypercapnia and acidosis
• Describe the mechanisms whereby prone positioning of a mechanically ventilated patient with ARDS improves oxygenation and reduces VILI
• Differentiate both mechanically and functionally between volume-targeted and pressure-targeted mechanical ventilation
• Discuss which protective lung ventilation strategies have been conclusively shown to reduce mortality in ARDS
Treatment of Hypoxemia and Severe Shunting
Severe shunting impairs the lung’s ability to transfer oxygen to the blood, resulting in hypoxemia that responds poorly to oxygen therapy, which is known as refractory hypoxemia (see Chapter 12). The primary defect in shunt-induced lung disease is the absence of ventilation in a large number of alveoli because they are either collapsed or filled with fluid. The first priority in treating this type of hypoxemia is to reestablish ventilation in these alveoli without injuring the lung or decreasing oxygen delivery to the tissues. The clinician must find a way to increase the arterial oxygen content while maintaining an adequate cardiac output. An important but less urgent priority is reversal of the underlying defect that caused hypoxemia.
Oxygen Therapy
Generally, oxygen therapy is used to maintain a PaO2 of at least 60 mm Hg (which corresponds to an oxygen saturation in arterial blood [SaO2] of approximately 90%).1 However, in anemia, cardiac insufficiency, and coronary artery disease (hypoxic heart muscle), it is overly conservative to limit arterial oxygenation to these levels, especially if the FIO2 required to achieve a PaO2 of 100 mm Hg (SaO2 = 98%) is 0.40 or less. The nearly 10% increase in arterial oxygen content accompanying a PaO2 increase from 60 mm Hg to 100 mm Hg is clinically significant in such instances.
Oxygen therapy is effective in < ?xml:namespace prefix = "mml" />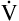 A/
A/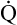 C mismatch but not if a large amount of absolute shunting is present (see Chapter 12). Oxygen therapy is also effective in diffusion defects and hypoventilation. However, oxygen therapy is not the appropriate way to treat hypoventilation because inadequate alveolar ventilation is the primary problem, not inability to oxygenate; that is, oxygen transfer across the alveolar capillary membrane is not impaired in pure uncomplicated hypoventilation.
C mismatch but not if a large amount of absolute shunting is present (see Chapter 12). Oxygen therapy is also effective in diffusion defects and hypoventilation. However, oxygen therapy is not the appropriate way to treat hypoventilation because inadequate alveolar ventilation is the primary problem, not inability to oxygenate; that is, oxygen transfer across the alveolar capillary membrane is not impaired in pure uncomplicated hypoventilation.
Oxygen therapy must be closely monitored when treating patients who are chronically hypoxemic and hypercapnic (e.g., patients with severe chronic obstructive pulmonary disease [COPD]) because it can cause acute carbon dioxide retention and acidosis through mechanisms described in Chapter 11. However, oxygen should never be withheld from severely hypoxemic individuals, regardless of the cause. Hypercapnia and respiratory acidosis are of secondary importance if cerebral oxygenation is threatened. Oxygen should be administered to hypercapnic patients with COPD via devices that can produce relatively low, fixed, inspired oxygen concentrations not influenced by the patient’s ventilatory pattern. In patients with severe hypoxemia, low inspired oxygen concentrations are effective because arterial hemoglobin saturation is located on the steep part of the oxygen-hemoglobin equilibrium curve. For example, a relatively large amount of oxygen is added to the blood when PaO2 is increased from 30 to 60 mm Hg (see Chapter 8). A periodic PaCO2 analysis is important in monitoring oxygen-induced hypercapnia.
CONCEPT QUESTION 14-1
Why is oxygen therapy less effective in absolute shunt than in 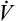 A/
A/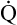 C mismatch?
C mismatch?
Excessively High FIO2
FIO2 values greater than 0.50 for prolonged periods may produce toxic damage to alveolar cells, known as pulmonary oxygen toxicity. High oxygen concentrations lead to the formation of free oxygen radicals, such as superoxide, hydrogen peroxide, and hydroxyl ions; the strong oxidizing effects of these ions damage the ultrastructure of the lung, causing effects similar to the effects seen in acute lung injury (ALI). Oxygen toxicity is marked by an inflammatory response of the lung that leads to alveolar capillary membrane injury and pulmonary edema, greatly impairing oxygen-transfer efficiency and lung compliance. Individual susceptibility to oxygen toxicity varies considerably; generally, FIO2 of 0.50 or less for 2 to 7 days does not produce significant lung impairment.1 For this reason, the goal of oxygen therapy is to achieve a hemoglobin saturation of at least 90% (PaO2 of at least 60 mm Hg) at FIO2 of 0.50 or less. This goal may not always be clinically feasible, and higher FIO2 values may be required. Such instances mark the presence of severe shunting and must be addressed by other therapeutic measures (discussed later in this chapter). Concerns about oxygen toxicity should never supersede concerns about tissue hypoxia.
Body Position
A simple change in body position may improve oxygenation by improving the match between ventilation and blood flow. Gravity affects the distribution of ventilation and perfusion, as discussed in Chapters 3 and 6. For example, cardiac output is greater in the supine than in the sitting position because venous blood does not have to be pumped “uphill,” from the trunk and lower extremities up to the heart. The supine position improves venous return, pulmonary blood flow, and PaO2 in patients with hypovolemic shock. The same position decreases PaO2 in patients with congestive heart failure because increased venous return overwhelms the heart’s pumping capacity, causing pulmonary edema (see Chapter 6).
Treatment of Shunting in Acute Lung Injury
Acute lung injury (ALI) is characterized by alveolar inflammation, alveolar-capillary membrane injury, high-permeability pulmonary edema, widespread atelectasis, and low lung compliance; the end result is severe shunting and refractory hypoxemia. Acute respiratory distress syndrome (ARDS) is the most severe form of ALI. According to the American-European Consensus Conference on ARDS, the PaO2/FIO2 in ALI, regardless of applied positive end expiratory pressure (PEEP), is 300 mm Hg or less, whereas it is 200 mm Hg or less in ARDS.2 ARDS is not a single disease entity; rather, it is a syndrome set in motion by any one of a wide variety of physiological insults to the lung (Table 14-1). The pathological feature common to each of these insults is injury to and increased permeability of the alveolar capillary membrane; the injury can be either direct (e.g., aspiration of gastric contents) or indirect (e.g., circulating bacterial endotoxins in sepsis).
TABLE 14-1
Conditions Associated with Acute Respiratory Distress Syndrome and Acute Lung Injury
| Direct Injury | Indirect Injury |
| Pneumonia | Sepsis |
| Gastric content aspiration | Major nonpulmonary trauma |
| Pulmonary contusion | Multiple blood transfusions |
| Toxic gas/smoke inhalation Near drowning |
Pancreatitis Heart-lung bypass machine Adverse drug effects Liver failure |
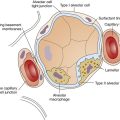
Any of the risk factors listed in Table 14-1 can initiate the release of inflammatory mediators that attract and activate neutrophils (neutrophils are circulating white blood cells essential for phagocytosis and destruction of bacteria and other pathological substances). Prostaglandins, endotoxins, and cytokines are examples of inflammatory mediators. Prostaglandins are substances that can change capillary permeability, cause platelet aggregation (which increases the risk for intravascular clotting), and change vascular smooth muscle tone. Endotoxins are contained in the cell walls of gram-negative bacteria and when released can injure the vascular endothelium and induce inappropriate synthesis of nitric oxide; the result is massive vasodilation (septic shock). Cytokines are any of several regulatory proteins released by immune system cells (e.g., T cells) in generating an inflammatory response. Interleukins are a subgroup of cytokines produced mainly by T cells (T cells are thymus-derived lymphocytes of the body’s immune system that destroy or neutralize foreign substances); interleukins are signaling molecules that facilitate communication among neutrophils, causing them to migrate to the site of injury. Interleukins are thus important in mounting the inflammatory response.
Circulating neutrophils contain an array of toxic substances essential for destroying bacteria and other microorganisms. A hallmark of ARDS is the adherence of neutrophils to the pulmonary capillary endothelium (neutrophils become “sticky”).2 Toxic compounds released by neutrophils damage and increase the permeability of the capillary endothelium and alveolar epithelium as neutrophils transmigrate from the bloodstream into the alveoli. Among these compounds are reactive oxygen ions and proteolytic enzymes that degrade and destroy tissues (Figure 14-1). The end result is increased alveolar capillary membrane permeability and flooding of the alveolar airspaces with protein-rich fluids. These fluids interfere with surfactant function, increasing surface tension and inducing widespread atelectasis. Shunting is the result of both alveolar filling and alveolar collapsing processes. The loss of alveolar airspace greatly reduces FRC and lung compliance in ARDS.
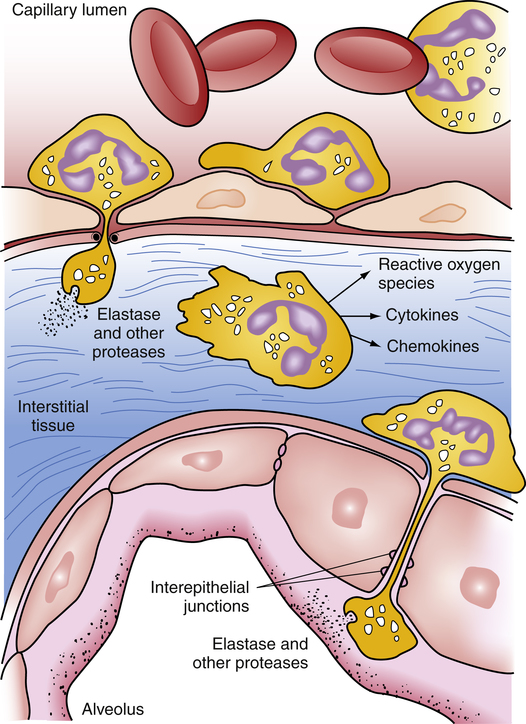
The hypoxemic effect of shunt in ARDS is especially severe because the normal hypoxic pulmonary vasoconstriction (HPV) mechanism is severely blunted.3,4 As noted in Chapter 6, the physiological function of HPV is to match blood flow and ventilation better. HPV normally reduces blood flow to nonventilated alveoli, lessening the hypoxemic effect they can exert on arterial blood. In ARDS, blood flows more freely through the capillaries of nonventilated alveoli, contributing to the profound refractory hypoxemia characteristic of ARDS.
ARDS does not affect the lung uniformly; computed tomography scans reveal that both normal-appearing lung and heavily consolidated lung coexist.2 ARDS is often characterized as a heterogeneous disease (i.e., interspersed normal and abnormal regions) as opposed to a homogeneous disease. In effect, reduced open airspace creates a “small” lung in ARDS; in other words, one can think of the ARDS lung as a relatively small “normal” lung sharing the same major airways with an abnormal consolidated or atelectatic lung. This means the ARDS lung has marked regional differences in lung compliance, which presents an especially difficult challenge in mechanical ventilation of these patients. A mechanical positive pressure breath preferentially inflates the most compliant lung areas, potentially overdistending and damaging these “normal” regions. Thus, much attention has been given to the phenomenon of ventilator-induced lung injury (VILI) and to protective ventilation strategies. The remaining sections of this chapter address the physiological rationale for various therapeutic strategies designed to improve oxygenation and to protect the mechanically ventilated ARDS lung from further injury.
Positive End Expiratory Pressure
Mechanism of Action
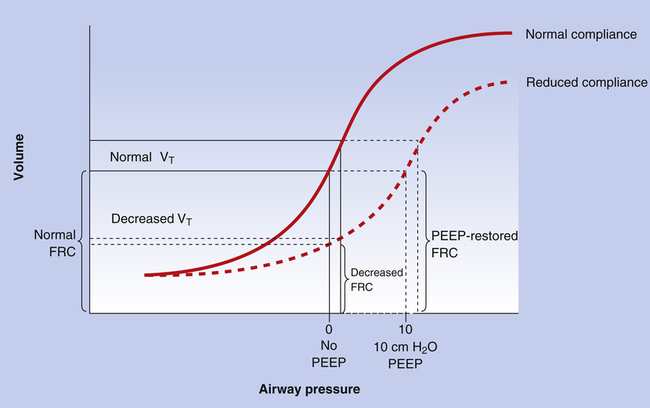
In the atelectatic lung, PEEP increases FRC and lung compliance by increasing transpulmonary pressure (PA − Ppl) at the end of expiration. This effect is beneficial only in people who already have abnormally low FRC and low lung compliance. In such individuals, PEEP may prevent alveolar collapse at the end of expiration and at sufficiently high levels may help to reopen collapsed alveoli.5 The reopening of previously collapsed alveoli reduces intrapulmonary shunt and improves PaO2. In addition, PEEP improves lung compliance (i.e., less pressure is required to inflate the lung to a given volume) by restoring the FRC of the diseased lung; that is, PEEP places the tidal volume on a steeper portion of the lung compliance curve (Figure 14-2), requiring less pressure to achieve a given tidal volume. As collapsed alveoli are reopened, the lung’s compliance curve shifts back toward its normal position.
Continuous Positive Airway Pressure
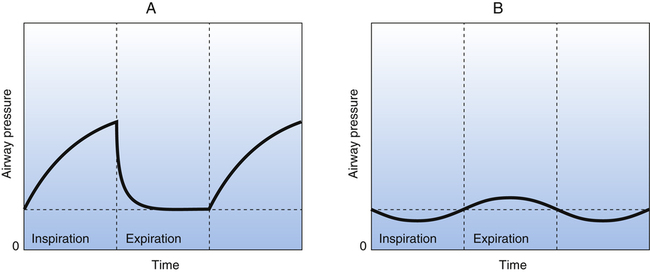
A variation of PEEP that produces the same effect is continuous positive airway pressure (CPAP), which is applied to spontaneously breathing individuals. In CPAP, a relatively constant positive airway pressure is maintained throughout inspiration and expiration. CPAP can be applied through a facemask or an endotracheal tube. The beneficial effects of CPAP are similar to the effects of PEEP; alveolar collapse is prevented, and collapsed alveoli may be recruited. The improved lung compliance illustrated in Figure 14-2 decreases the patient’s work of breathing. The major difference between CPAP and PEEP is that airway pressure does not increase during inspiration with CPAP as it does with PEEP. Therefore CPAP is associated with lower mean airway pressure than PEEP. Figure 14-3 illustrates PEEP and CPAP pressure waveforms.
How PEEP and CPAP Devices Work
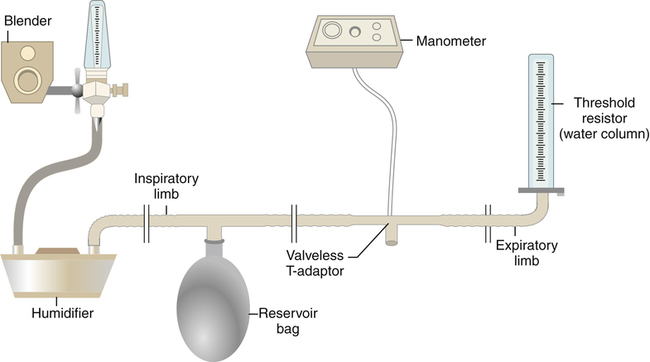
A CPAP device may consist of a continuous flow apparatus or a demand valve system. In a continuous flow device, a source gas flows through a tube past the patient’s airway (either through a mask or, as shown in Figure 14-4, through an adapter attached to an endotracheal tube) and out of a CPAP valve that can be adjusted to release gas at a given pressure threshold. A CPAP valve functions in a manner similar to a PEEP valve. For example, if the CPAP valve allows gas to escape only when the tubing pressure exceeds 10 cm H2O, this pressure is maintained in the breathing circuit as long as the source gas flow rate exceeds the patient’s peak inspiratory flow demand.
Indications for PEEP and CPAP
The general indication for this type of airway pressure therapy is the presence of a significant amount of absolute shunting. Generally, if PaO2 of at least 60 mm Hg with FIO2 of 0.5 or less cannot be achieved, PEEP or CPAP should be considered. Refractory hypoxemia combined with bilateral, uniform lung disease and uniformly low lung compliance are good indications for the use of PEEP or CPAP.6 A classic indication for PEEP is diagnosed ARDS. As previously described, ARDS is associated with alveolar instability and collapse and extensive intrapulmonary shunting and refractory hypoxemia.
Poor candidates for PEEP and CPAP are patients with unilateral or localized lung disease (involving only one lung or one lobe) and patients with normally or highly compliant lungs.6 In such individuals, PEEP preferentially affects the most compliant regions and may result in overdistention of normal alveoli. This overdistention leads to a high risk of pulmonary air leaks, lung rupture, and pneumothorax.
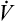
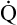
Concepts and Mechanisms of Ventilator-Induced Lung Injury
Basic Concepts of Hyperinflation Injury
Few subjects have been made more complicated than the cause of hyperinflation lung injury. In thinking about what causes overdistention of the lung, during mechanical ventilation, one must keep in mind that changes in lung volume cannot be caused by any factor other than changes in alveolar distending pressure i.e., transpulmonary pressure (see Chapter 3). That is, if transpulmonary pressure does not change, volume cannot change. One must be clear about cause and effect: Pressure changes are the cause of lung volume changes; the lung cannot somehow gain volume independent of a pressure change. The separation of pressure changes from volume changes during ventilation, whether spontaneous or mechanical, is conceptually incorrect. Lung inflation, and thus stretch of the alveolar epithelium, can occur only when the transpulmonary pressure gradient (PA − Ppl) increases. Several references in the medical literature erroneously imply that high volume produces alveolar trauma, not high pressure—as though pressure and volume changes are independent of one another.
A study sometimes cited in support of the idea that pressure and volume have separate effects on the lung is one in which rats (presumably with similar lung compliance) were ventilated with (1) high airway pressures and high tidal volumes; (2) low airway pressures and similarly high tidal volumes—accomplished with a negative pressure ventilator that mimicked spontaneous breathing mechanics; and (3) high airway pressures and low tidal volumes—accomplished by strapping the rats’ chest walls with rubber bands.7 The results showed that only the lungs exposed to high volumes sustained injury, as was evidenced by the presence of high-permeability pulmonary edema.8 That is, the rats in group 3, exposed to high airway pressures but low tidal volumes (because their chest walls could not expand), did not develop edema and were presumed to be uninjured.
These findings have been interpreted to mean that lung volume, not pressure, is the causative agent that produces lung injury. Although it is true that lung volume must increase to produce alveolar overdistention, it cannot do so without an increase in distending pressure, which should not be confused with the absolute alveolar pressure. The conclusion that volume is more important than pressure in producing lung injury is simplistic; it overlooks the inseparable relationship between transpulmonary pressures and lung volumes. The results of the rat study are not surprising when one realizes that transpulmonary pressure determined the alveolar volume and stretch in each ventilation method. As shown in Figure 14-5, ventilation with positive pressure (method 1) and with negative pressure (method 2) are the same with respect to their effects on alveolar epithelial stretch because their transpulmonary pressures were the same and thus produced the same tidal volumes. In ventilation method 3, the strapped chest walls caused alveolar and intrapleural pressures to increase together, keeping transpulmonary pressures (and thus tidal volumes and alveolar stretch) relatively small; therefore, this lung sustained no injury.
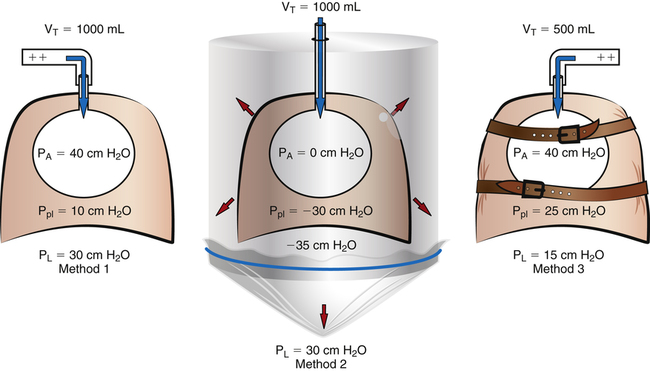
Figure 14-5 oversimplifies the lung’s extremely complex architecture; that is, alveoli do not behave like individual balloons; they have shared walls. An intricate framework of connective tissue binds adjacent alveoli together such that they are interdependent. The mechanism of lung inflation does not involve balloon-like expansion and contraction of alveoli in the normal lung (see Chapter 3). However, regardless of inflation mechanism, lung volume changes can occur only as a result of transpulmonary pressure changes.
To summarize, the rat study8 did not distinguish between the effects of pressure and volume on alveolar injury. Instead, it demonstrated that an increase in transpulmonary pressure, no matter how it is produced, generates an increase in lung volume, which, if great enough, injures the alveoli. It should be apparent that lung volume is a function of distending pressure and that volume and pressure cannot be separated in the roles they play in VILI. The idea that pressure and volume are independent of one another led to the suggestion that a mechanical ventilator can be engineered to guarantee delivery of a desired tidal volume without exceeding a preselected maximal pressure. In actuality, the microprocessor in such ventilators computes lung compliance and then determines whether or not the
selected volume can be delivered without exceeding the targeted pressure; if it cannot, the microprocessor simply adjusts the pressure target upward (within a factory preset limit) to deliver the desired volume. If still more inflation pressure is required to deliver the desired volume, the ventilator delivers only the volume that the lung can accept within the preset pressure limit (i.e., not all of the desired volume is delivered).
On the other hand, the rat study8 points out the important fact that high alveolar pressure is not always associated with high lung volume. That is, high alveolar pressure does not always mean distention (transpulmonary) pressure is high, as demonstrated by the ventilation of rats with rubber-banded chest walls. Only when extrathoracic forces hinder chest wall expansion or hinder diaphragmatic displacement (e.g., rib cage deformities, severe abdominal distention, morbid obesity) is it possible for high alveolar pressures to be associated with a relatively low distention pressure. (Two other ordinary examples are the Valsalva maneuver, in which a person strains to exhale against a closed glottis, and the playing of a wind instrument such as an oboe, which presents high expiratory resistance; both situations increase the alveolar pressure without increasing transpulmonary pressure, as is evidenced by the lack of lung expansion.)
In mechanically ventilated patients, the correlate of alveolar pressure is plateau pressure, measured at the endotracheal tube at end-inspiration under zero airflow conditions (see Chapter 3). In all patients except patients with restricted chest wall and diaphragmatic movement, plateau pressure measurement is a reliable surrogate for assessing overdistention of the lung. It is generally accepted that the incidence of stretch-related lung injury is minimized if inspiratory plateau pressure is limited to less than 30 cm H2O.9,10
Mechanisms of Lung Injury
Mechanisms of VILI fall into three general categories: (1) mechanical stretch injury from hyperinflation (barotrauma/volutrauma); (2) repetitive collapse and reinflation of alveoli (atelectrauma); and (3) toxic effects of inflammatory mediators on pulmonary tissues (biotrauma). As already indicated, mechanical overdistention of the alveoli disrupts the structural integrity of the lung, allowing air to dissect through lung tissues into the pleural space and mediastinum, producing pneumothorax and possibly dissection of air into subcutaneous tissues around the neck and upper chest (subcutaneous emphysema). In addition, overstretch of the alveoli disrupts the alveolar capillary membrane and induces the release of inflammatory mediators, both of which increase membrane permeability.9–11 The heterogeneous distribution of diseased lung regions in ARDS makes the lung especially susceptible to this type of injury because of regional compliance differences; that is, a mechanically delivered tidal volume is preferentially distributed to the most compliant alveoli, placing them at greater risk of overdistention.
The repetitive collapse and reopening of alveoli (recruitment and derecruitment) during mechanical ventilation is another mechanism of VILI.2,10,12 This phenomenon has led to the term atelectrauma. Such opening and closing of alveoli subjects the lung fibroskeleton, microvasculature, and delicate juxtaalveolar tissues to mechanical sheer stress forces, especially at the junctions of regions with differing compliances and anchoring attachments.10
The term biotrauma was coined to describe the damage brought about by the release of inflammatory mediators in the lung, such as cytokines, in response to atelectrauma and hyperinflation injury.12 It has been shown in animal models that physical stretch of the lung during mechanical ventilation greatly increases lung epithelial release of tumor necrosis factor-alpha (TNF alpha), a key cytokine that mediates the effects of sepsis.12
Other cytokines, such as interleukins, may also be involved. Inflammatory mediators not only can affect the lung but also can affect other body organs; if alveolar disruption allows inflammatory mediators to translocate from the lung into the systemic circulation, it is possible that they can damage other body organs. It has been shown in adult patients with ARDS that a minimal stress mechanical ventilation strategy (low tidal volumes and the use of PEEP to prevent alveolar collapse) resulted in lower levels of cytokines in the systemic circulation than an injurious ventilation strategy (high tidal volumes with no PEEP).12 Inflammatory mediators in the systemic circulation attract neutrophils and other inflammatory cells, potentially leading to injury of other organs, such as the kidneys and liver. This phenomenon may explain the increased incidence of multisystem organ dysfunction syndrome (MODS) seen in mechanically ventilated patients with severe ARDS.12 A more recent study confirmed that a low tidal volume ventilation strategy coupled with adequate PEEP to prevent alveolar collapse during expiration reduces circulating cytokines.13
Physiological Rationale for Protective Ventilation Strategies
It stands to reason that if one can prevent lung overdistention, repetitive collapse and reopening of alveoli, and uneven distribution of the tidal volume, one can greatly reduce the incidence of VILI and its detrimental systemic effects. Although there is strong evidence that alveoli of normal lungs are very stable and do not change size during ventilation, alveoli do undergo large size changes in ALI, and alveolar recruitment and derecruitment is widespread.14 It is reasonable to believe that if one can find a way to ventilate the lungs while keeping alveolar size stable, the risk of alveolar injury should be reduced.
Preventing Overdistention
A landmark study published by the ARDS Network in 2000 demonstrated convincingly that in ALI, mechanical ventilation with lower tidal volumes and inspiratory pressures than traditionally used significantly decreases mortality and the number of days required on the ventilator.11 This study confirmed the results of earlier studies in which low volumes were used despite the resulting hypercapnia and acidosis.15 Ventilation with traditional volumes (12 mL/kg predicted body weight) was compared with lower ventilation volumes (6 mL/kg). In addition, the inspiratory plateau (alveolar) pressure in the traditional group was limited to 50 cm H2O or less, whereas it was limited to 30 cm H2O or less in the low-volume group. These pressure ceilings meant that the full calculated tidal volume could not be delivered if lung compliance was too low to permit its delivery; the low-volume ventilation group was most affected by this limitation as was evidenced by significantly higher PaCO2 and lower arterial pH. Nevertheless, mortality was reduced by 22% in the low-volume group.11 More recently, ARDS Network guidelines that limit plateau pressures to 30 cm H2O or less were validated with regard to significant reduction in hospital mortality.16 Ventilation with low volumes and the limitation of plateau pressures to 30 cm H2O or less is considered the standard of care in the mechanical ventilation of patients with ARDS.17
If the low tidal volume ventilation strategy causes hypercapnia and acidosis, it is often referred to as permissive hypercapnia.15 In other words, the adverse effects of hypercapnic acidosis that may result from low ventilation volumes are considered to be less detrimental to the patient than the alternative of overdistention lung injury. No safe upper limit for hypercapnia has been established, although some authorities believe the accompanying acidosis should not be allowed to fall below a pH of 7.20; infusion of sodium bicarbonate has been used to prevent arterial pH from falling below this level.11,17 However, more recent evidence shows that hypercapnic acidosis may actually have a protective effect independent of lung volume.18
Preventing Repetitive Collapse and Reopening of Alveoli (Atelectrauma)
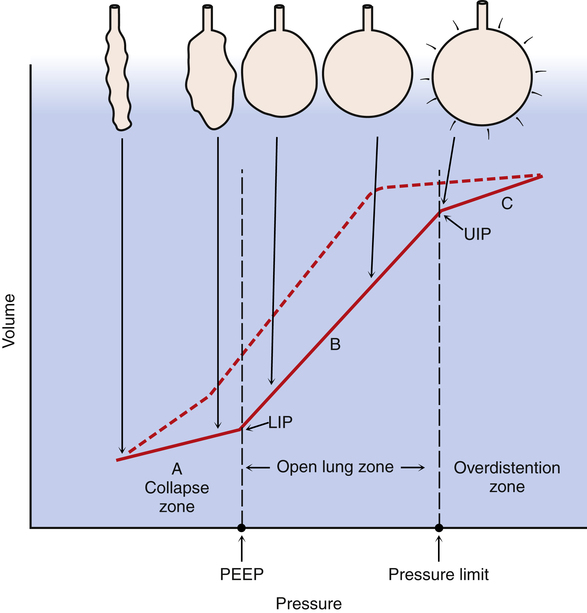
It stands to reason that a properly set PEEP level would prevent alveoli from collapsing at the end of expiration. Coupled with the low tidal volume strategy just outlined, the lung in ARDS would be protected from both overdistention and atelectrauma. That is, the lung would always be “open.” This open-lung approach was first advocated in 199219; an often cited study published in 1998 confirmed the protective effect of this ventilatory approach.20 In this study, the investigator set PEEP just above the lower inflection point on the pressure-volume curve (see Chapter 3) to prevent repetitive alveolar collapse and reopening. The 28-day mortality rate was significantly lower in patients with ARDS ventilated with an open-lung approach (38%) compared with a control group ventilated in the conventional manner (71%). The open-lung concept, or the use of enough PEEP to prevent alveolar collapse, and the limitation of plateau pressure to prevent overdistention—permitting hypercapnia if necessary—is now considered to be the standard of care against which all other strategies are measured in ventilating patients with ARDS (Figure 14-6).21 As discussed in Chapter 3, it is now questioned whether the lower inflection point on the inspiratory pressure-volume curve is an appropriate criterion for setting PEEP at a level that prevents alveolar collapse; the hysteresis of the pressure-volume curve shows that lung volume is considerably greater at a given airway pressure on the expiratory limb of the curve than it is on the inspiratory limb. Nevertheless, the lower inflection point continues to be used by some practitioners as a criterion for setting PEEP in ALI,22 although its use in this regard is controversial.
Prone Position: Improved V . A / Q . C 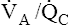 Match and Reduced Ventilator-Induced Lung Injury
Match and Reduced Ventilator-Induced Lung Injury
Conventional mechanical ventilation is accomplished with the patient in the semisitting supine position. However, the prone position has been shown to improve oxygenation significantly and to delay VILI.2,23–27
Generally, the anterior (ventral) chest wall is more compliant and free to move than the posterior (dorsal) chest wall. The vertebral column limits dorsal rib cage movements much more than the sternum limits ventral rib cage movements. This condition is amplified in the supine position in which movements of the rigid dorsal rib cage are further restricted by the body’s weight against the bed’s surface. For these reasons, ventral alveoli receive more ventilation than their dorsal counterparts (Figure 14-7, A). Moreover, in the supine position, the heart and the edematous ARDS lung weigh heavily on dorsal lung regions, possibly creating positive intrapleural pressure in this region. Consequently, dorsal alveoli tend to collapse. In mechanical ventilation, most of the tidal volume enters the more compliant ventral areas—both the lung and the rib cage are more compliant. Dorsal alveoli may open late in the inspiratory phase but then close again during expiration. Cyclical opening and closing of dorsal alveoli leads to lung injury, as previously described. For these reasons, VILI is most severe in dorsal lung regions of supine patients.2
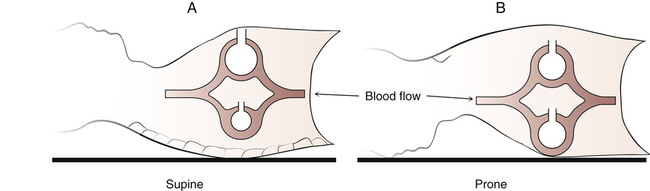
If the patient is turned to the prone position, the stiff dorsal component of the rib cage is freer to move than before, and the heart no longer compresses dorsal tissue (see Figure 14-7, B). The ventral rib cage is held against the bed surface,
decreasing its compliance and freedom of movement. More of the mechanically delivered volume is directed toward dorsal regions than before. However, the natural stiffness of the dorsal rib cage limits how much ventilation can shift in its direction, forcing some of the volume into ventral units as well. The result is a fairly even tidal volume distribution in the lung and a better match between ventilation and blood flow; shunting is reduced, and oxygenation improves. To the extent that dorsal alveoli remain open, lung compliance improves, lowering the required inflation pressures. Because of less closing and reopening of alveoli in dorsal regions, the prone position is associated with less VILI.25
Prone positioning is generally combined with the other protective strategies already outlined, including low tidal volumes to prevent overdistention and PEEP adjusted to prevent alveolar recollapse. It is reasonable to believe that if prone ventilation is effective in improving oxygenation, it should also result in better carbon dioxide elimination, presumably through better ventilation of dorsal regions. It was found that a reduced PaCO2 in response to prone ventilation was predictive of an improved survival rate; that is, patients in whom prone ventilation was associated with decreased PaCO2 had a lower mortality rate than patients in whom PaCO2 remained the same.28 Another predictor of response to prone positioning may be the distribution of disease in the ARDS lung.29 ARDS is categorized as having one of three distribution patterns: (1) a lobar or segmental distribution; (2) a “patchy” distribution, in which lobar and segmental distribution coexist with areas not limited by anatomical boundaries; and (3) a “diffuse” distribution, in which disease is widespread and uniform throughout the lungs. Patients with lobar and patchy distributions are more likely to respond positively to prone positioning; that is, redistribution of densities is not as likely to occur in patients with diffuse, widespread patterns.29
A major meta-analysis of prone ventilation in ARDS did not show improved long-term patient survival, although oxygenation was significantly improved; however, there was a trend for lower mortality in the most severely ill patients when prone ventilation was used.27 Longer duration in the prone position was associated with better outcomes; some authorities recommend maintaining the prone position for 12 to 20 hours per day.10,27 Animal studies show reduced incidence of VILI with prone ventilation,25 and it is thought that the prone position should be used sooner rather than later in the course of ARDS.26,27 Generally, prone positioning is recommended for ARDS patients who require potentially harmful levels of FIO2 and plateau pressure, as long as positional changes do not place them at high risk, as might be the case in patients with spinal cord injuries.10,17 Reversion to supine positioning at least once per day is recommended for patient washing and dressing changes; many patients with ARDS require almost continuous prone positioning for the first few days.10 Of all lung protective ventilation strategies, volume-limiting and pressure-limiting strategies are the most universally accepted and most demonstrably effective in reducing mortality in ARDS.30
Pressure-Targeted versus Volume-Targeted Ventilation in Acute Respiratory Distress Syndrome
Patients with ARDS can be effectively and safely ventilated by either type of ventilator. Low volumes can be used in volume-targeted machines, and high-pressure alarms can alert the clinician to dangerously high pressures. Likewise, desired pressure limits can be set in pressure-targeted ventilators, and low-volume alarms can alert the clinician of changing lung mechanics. However, one major difference between the two types of ventilators is the ventilator’s flow rate response to the patient’s spontaneous effort to breathe. In volume-targeted ventilators, the flow pattern is preset and does not change, whereas flow increases or decreases in response to patient demand in the pressure-targeted ventilator. By design, the pressure-targeted machine must maintain a constant pressure at the endotracheal tube for a specified time, which requires the ventilator to increase its flow output if the patient’s inspiratory effort makes the circuit pressure drop. In the final analysis, the way in which inspiratory pressure is reduced (either by limiting the tidal volume in volume-targeted ventilation or by directly limiting inspiratory pressure in pressure-targeted ventilation) has no effect on mortality.31 The important element influencing mortality is simply the use of low pressures and volumes.
Other Techniques for Treating Severe Oxygenation Failure
Airway Pressure Release Ventilation and Bilevel Positive Airway Pressure
APRV and BIPAP are mechanical ventilation modes that allow the patient to breathe spontaneously on top of the imposed mechanical ventilation pattern. Both modes supply the patient with alternating high and low levels of CPAP, which correspond to mechanical inspiration and expiration.32 The term APRV traditionally refers to the mode in which the high CPAP level (inspiratory phase) lasts longer than the low CPAP level (expiratory phase), producing an inverse ratio pattern (i.e., inspiration lasts longer than expiration). The term BIPAP is used to describe the mode if high and low CPAP levels are applied for equal time periods or if low CPAP time is longer than high CPAP duration. However, these terms are not universally recognized; the numerous ventilator manufacturers tend to coin their own terms for these modes, each claiming some unique distinction that sets their ventilator’s mode apart. For example, APRV is also known as Bi-Level Ventilation, Bi-Vent, and Duo-PAP.32 Overall, APRV and BIPAP create higher mean airway pressures than the conventional low-volume, high-PEEP strategies mentioned earlier in this chapter.
CLINICAL FOCUS 14-3 ARDS Network Mechanical Ventilation Protocol for Acute Lung Injury and Acute Respiratory Distress Syndrome
In 1994, the National Institutes of Health and the National Heart, Lung, and Blood Institute initiated a clinical network (ARDS Network)11 to find the best treatment strategies for ALI and ARDS through multicenter clinical trials. The network conducted a landmark randomized, controlled clinical trial that enrolled 861 subjects to compare lower tidal volume ventilation versus higher tidal volume in the treatment of patients with ALI early in their disease course. The study, published in the New England Journal of Medicine in 2000, conclusively showed for the first time that lower tidal volumes resulted in improved survival. The following mechanical ventilation protocol summary can be accessed on the Internet at www.ardsnet.org:
Ventilator Setup and Adjustment
• Calculate predicted body weight (PBW)
• Set initial VT to 8 mL/kg PBW
• Reduce VT by 1 mL/kg at intervals of 2 hours or less until VT = 6 mL/kg
• Set initial respiratory rate (RR) to approximate baseline minute ventilation not greater than 35 breaths per minute
• Adjust VT and RR to achieve pH and plateau pressure goals
• Set inspiratory flow to exceed patient demand (usually >80 L/min)
Oxygenation Goal: PaO2 55 to 80 mm Hg or SpO2 88% to 95%
| FIO2 | 0.3 | 0.4 | 0.4 | 0.5 | 0.5 | 0.6 | 0.7 | 0.7 |
| PEEP | 5 | 5 | 8 | 8 | 10 | 10 | 10 | 12 |
| FIO2 | 0.7 | 0.8 | 0.9 | 0.9 | 0.9 | 1.0 | 1.0 | 1.0 |
| PEEP | 14 | 14 | 14 | 16 | 18 | 20 | 22 | 24 |
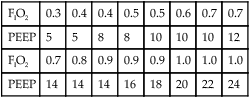
Plateau Pressure (Pplat) Goal: 30 cm H2O or Less
• Check Pplat (0.5 second inspiratory pause), SpO2, RR, VT, and pH at least every 4 hours and after each PEEP or VT change
If Pplat is greater than 30 cm H2O: Decrease VT by 1-mL/kg increments to minimum of 4 mL/kg
If Pplat is less than 25 cm H2O: If VT is less than 6 mL/kg, increase VT by 1-mL/kg increments until Pplat is greater than 25 cm H2O, or VT = 6 mL/kg
If Pplat is less than 30 cm H2O and breath stacking occurs: May increase VT in 1-mL/kg increments to maximum of 8 mL/kg
of these modes. In theory, APRV/BIPAP should make patients feel more comfortable because they can breathe spontaneously, and thus less sedation should be necessary; additionally, the patient’s ability to use ventilatory muscles should improve muscle strength and endurance and shorten time spent on the ventilator.
At the time of this writing, none of the claims for the unique benefits of APRV and BIPAP have been subjected to rigorous scientific scrutiny; there is no strong evidence that these modes reduce the need for sedation, decrease the work of breathing, or reduce the time patients spend on mechanical ventilation.32 Research efforts have been complicated by (1) disagreement among clinicians as to what constitutes APRV or BIPAP, (2) the lack of consistency among researchers in setting inspiratory/expiratory timing ratios, and (3) the overlap of these modes with conventional pressure-targeted mechanical ventilation.32
Inhaled nitric oxide (iNO) may play a limited role in improving ventilation-perfusion relationships and PaO2 in patients with severe ARDS.33 As discussed in Chapter 6, nitric oxide is a potent pulmonary vasodilator and plays an important role in regulating pulmonary vascular resistance. In ARDS, hypoxic vasoconstriction (even though it is blunted in ARDS) reduces pulmonary blood flow in atelectatic regions of the lung. Inhalation of nitric oxide selectively dilates pulmonary vessels in well-ventilated regions where it is distributed; this diverts more blood flow to these regions and should improve their relationships. Studies have shown that iNO does improve  A/
A/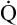 C, as evidenced by increased PaO2; iNO also reduces pulmonary vascular resistance.33 Such improved oxygenation may allow reductions in PEEP and FIO2, reducing the incidence of barotrauma and oxygen toxicity. However, iNO has not been shown to improve the survival rate in patients with ARDS; this is not surprising considering that most patients with ARDS die of multiorgan failure, not hypoxemia.31 At the present time, evidence is insufficient to justify the routine use of iNO in patients with ARDS.33,34
C, as evidenced by increased PaO2; iNO also reduces pulmonary vascular resistance.33 Such improved oxygenation may allow reductions in PEEP and FIO2, reducing the incidence of barotrauma and oxygen toxicity. However, iNO has not been shown to improve the survival rate in patients with ARDS; this is not surprising considering that most patients with ARDS die of multiorgan failure, not hypoxemia.31 At the present time, evidence is insufficient to justify the routine use of iNO in patients with ARDS.33,34
High-Frequency Oscillatory Ventilation
In high-frequency oscillatory ventilation (HFOV), a continuous inspiratory flow (called bias flow) maintains a constant mean pressure in the airway. A rapidly oscillating (3 to 10 cycles per second) piston or diaphragm provides ventilation by creating small peak and trough pressures around the preset mean airway pressure. Gas exchange occurs essentially by diffusion; “tidal volumes” are less than the anatomical dead space volume. The constant mean oscillating airway pressure keeps the lung in a continuous partially inflated condition. Theoretically, HFOV is ideally suited for ARDS because alveoli are maintained in a continuously open state, avoiding atelectrauma and volutrauma because of the very small oscillating volumes. Nevertheless, clinical trials do not provide enough evidence to show improved mortality compared with conventional low-volume, low-pressure ventilation.35
Liquid Ventilation
Liquid ventilation uses an organic fluid that has a very high capacity for dissolving oxygen and carbon dioxide. The most widely used fluid is a perfluorocarbon compound known as perflubron.2 The most common technique used is partial liquid ventilation (PLV), in which the lung is filled with perflubron to the FRC level, and a conventional ventilator is used to deliver gas tidal volumes. Perflubron has a very low surface tension, which theoretically allows it to penetrate dependent lung regions and open collapsed alveoli.2 Once filled with perflubron, alveoli cannot collapse again. The liquid also displaces secretions, making their removal more effective. PLV allows the use of lower pressures and volumes than conventional ventilation.36 In theory, PLV should avoid the factors that produce VILI. However, there is not enough evidence at the present time to support its routine use in ARDS; clinical studies show no improvement in mortality over conventional low-volume, low-pressure ventilation.36