Pharmacology
1. Pharmacology: The study of the interaction of drugs with the organism.
2. Drug: Any chemical compound that may be administered to or used in an individual to aid in the diagnosis, treatment, or prevention of disease, to relieve pain, or to control or improve any physiologic disorder or pathologic condition.
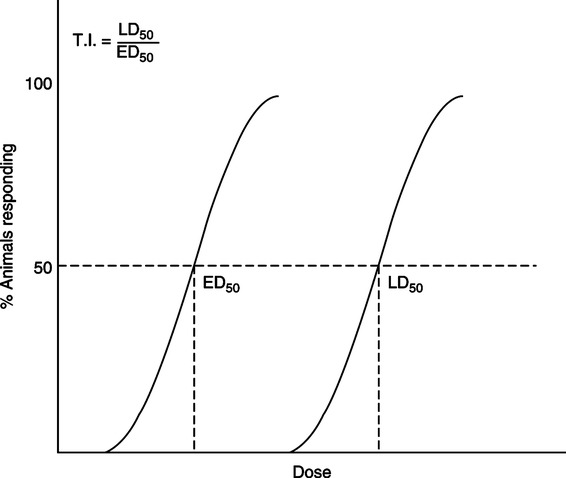
3. LD50: The dosage of a drug that would be lethal to 50% of a test population.
4. ED50: The dosage of a drug that would have therapeutic effects for 50% of a test population.
5. Therapeutic index (TI): The numerical ratio of the LD50 to the ED50 (LD50/ED50). This ratio shows how close the lethal and therapeutic doses of a drug are for a test population. Low indices mean the therapeutic and lethal doses are similar and the drug has a high potential for overdose or toxic side effects (Figure 17-1).
6. Side effect: Any physiologic response other than that for which the drug was administered.
1. Chemical name: Name illustrative of chemical structural formula of the drug.
2. Code name: An investigational designation, usually alphanumeric.
3. Generic name: An assigned name given for clinical investigation of a promising chemical.
4. Official name: Usually the generic name after the drug is accepted for general use.
5. Trade, brand, or proprietary name: The name under which a particular manufacturer markets the drug.
C Principles of drug action: There are three phases of drug action from initial dosing to pharmacologic effect. Each phase includes aspects of the pharmacology of the drug.
1. Pharmaceutical phase: Administering the drug.
a. Dosage forms: Tablet, capsule, liquid, powder, or ointment
b. Route of administration (listed in order of speed in obtaining blood levels)
2. Pharmacokinetic phase: The drug movement phase, including entry into or elimination from the body. This phase generally includes absorption, distribution, metabolism, and elimination of a drug. These factors determine onset of action, peak plasma drug level, and duration of drug action.
a. Absorption: Rate of absorption is determined by the specific physical and chemical characteristics of a drug.
(1) The lung mucosa provides an excellent surface for absorption of inhaled drugs.
(2) A substantial portion of an inhaled drug may be absorbed systemically in the mouth or proximal airways, thereby reducing its ability to affect the distal airways.
(3) Only particles <3 μm in diameter are carried to the distal airways.
b. Distribution: Movement of the drug to an area of desired pharmacologic activity.
(1) The primary mechanism for distribution is the circulatory system.
(2) Topical administration for effect on skin or mucous membrane decreases the likelihood of further undesired distribution.
c. Metabolism: Inactivation of the drug by the body.
(1) The primary organ for detoxification is the liver.
(2) Secondary organs for detoxification are the kidneys and gastrointestinal (GI) tract.
(3) The body’s cells or plasma proteins may inactivate many drugs.
d. Excretion or clearance: Mechanism for elimination of the drug from the body.
3. Pharmacodynamic phase: Drug-receptor interaction
a. Drugs usually create their effect by stimulating or blocking a receptor.
b. Drug-receptor types (Figure 17-2)
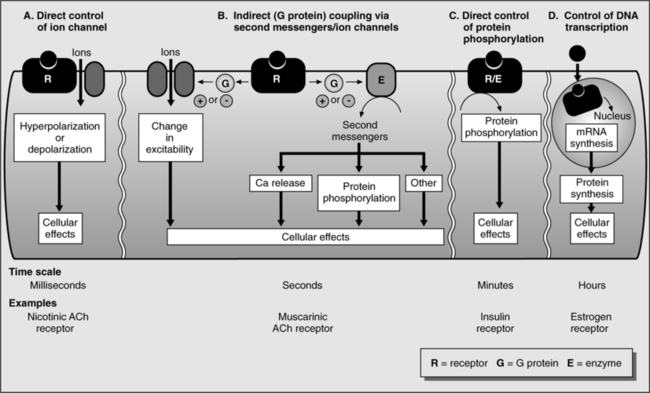
(1) Ligand-gated channel receptors: These transmembrane receptors traverse the cell membrane, acting as a high-speed conduit for transfer of specific chemicals, often ions, into and out of the cell.
(2) Tyrosine kinase linked receptor: Also a transmembrane receptor; binding of a drug to its extracellular receptor site enzymatically activates its intracellular end, acting as a tyrosine kinase. In this example, a phosphate group is added to (phosphorylates) the amino acid tyrosine in proteins that this receptor contacts, changing the protein to produce the clinical effect.
(3) G protein-coupled receptors: Transmembrane receptors that act through an intermediary, the G protein, located within the cell membrane, bound to a molecule of guanosine diphosphate. In this example a drug-receptor binding stimulates the G protein to exchange a guanosine diphosphate molecule for a guanosine triphosphate molecule. Once phosphorylated, the G protein migrates to a separate protein target to create the end drug effect.
(4) Steroid-receptor complex: Lipophilic steroid molecules readily pass through cell membranes and bind to receptors in the cell cytoplasm. Translocation of the steroid-receptor complex into the nucleus allows the complex to affect DNA transcription. The end effect of the drug occurs only after translation of new proteins from the affected DNA, making this a slow process.
(1) Affinity: Tendency of a drug to combine with a matching receptor.
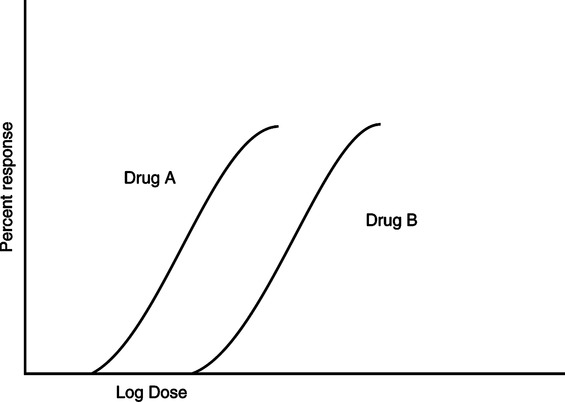
(2) Potency: The activity of a drug per unit weight. A potent drug has a large biologic activity at a small unit dose (Figure 17-3).
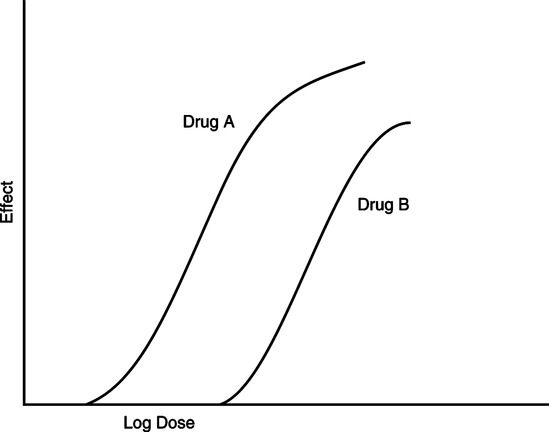
(3) Efficacy: The maximum effect produced by a drug regardless of dose (Figure 17-4).
(4) Cumulation: A gradual increase in the body’s total drug level that occurs when the administration rate of the drug is greater than the body’s rate of removal.
(5) Tolerance: The body’s ability to increase its metabolism of a drug. Increasing amounts of the drug are required to produce the same effect.
1. Additive: Two drugs, when given together, produce an effect equal to the sum of their individual effects.
2. Potentiation: Potentiation occurs when a drug active at a specific receptor site is given with a drug inactive at that receptor site, and the resulting effect is greater than that of the active drug alone (1 + 0 = 3).
3. Synergism: Two drugs active at a receptor site, when given together, cause an effect greater than the sum of their individual effects (1 + 2 = 6).
4. Antagonist: This is a drug with affinity but no efficacy (i.e., it blocks an effect).
a. Competitive: An antagonist whose effects are directly related to dosage. A competitive antagonist decreases potency but does not affect efficacy of the other drug.
b. Noncompetitive: An antagonist whose effects are not dose related. Potency and efficacy of the other drug are decreased.
5. Agonist: This is a drug with affinity and efficacy (i.e., causes an effect).
E The prescription: This is the written order for a drug composed of:
1. The patient’s name, address, and the date.
2. Rx: “Take thou”; take and prepare the medication.
3. Inscription: Name and quantity of the drug.
4. Subscription: Directions (when applicable) to the pharmacist for compounding the drug.
5. Sig: Transcription, “write”; instructions to the patient for taking the drug.
II Administering Aerosolized Drugs (see Chapter 35)
1. Ultrasonic nebulizers (not recommended for bronchodilators)
2. Gas-powered small volume nebulizers
3. Metered dose inhaler (MDI), with or without extension (spacer) device
B Particle size and deposition
C Basic protocol of administration
1. Gas-powered small reservoir nebulizers
b. Nebulizing flow: 6 to 8 L/min (viscous antibiotics, 12 L/min)
a. Assemble, shake canister, and charge with one actuation.
b. Hold 4 cm in front of open mouth or rest on mandibular teeth of open mouth.
d. Begin slow deep breath, and activate MDI.
e. Inspire to total lung capacity, and hold breath at least 3 to 5 seconds.
3. General notes regarding administration
a. Bronchodilators: Wait at least 1 minute, and then take a second puff.
b. Corticosteroids: Take after bronchodilators, hyperextending the neck while inhaling; use a spacer; rinse mouth and throat after dosing.
c. Dry powder inhalers: These require rapid inspiration with high flow rates (60 to 120 L/min) for dispersion.
d. Spacers or extensions with MDIs reduce the need for coordination and oropharyngeal impaction.
III Wetting Agents and Diluents
A Isotonic solution: A solution equivalent to a 0.9% weight/volume (w/v) solution of NaCl. Isotonic solutions are used in respiratory therapy primarily in small-volume nebulizers to increase aerosol volume during drug delivery.
B Hypertonic solution: A solution with a concentration >0.9% w/v solution of NaCl. Hypertonic solutions are used in respiratory therapy primarily for sputum induction.
C Hypotonic solution: A solution with a concentration <0.9% w/v solution of NaCl. Hypotonic solutions are most commonly used in respiratory therapy in large-volume aerosol generators and seem to have less effect on increasing airway resistance than normal saline or water.
D Distilled water: Used in respiratory therapy in all types of humidifiers.
1. Replace and replenish a deficient endogenous surfactant pool in neonatal respiratory distress syndrome (RDS).
2. Endogenous surfactant is normally recycled by reentering the type II alveolar cell as small vesicles. Exogenously administered surfactant can restore a depleted endogenous pool of surfactant, relieving RDS.
B Composition of pulmonary surfactant
1. Surfactant, a complex mixture of lipids and proteins produced by type II alveolar cells, regulates the surface tension forces of the lipid alveolar lining (see Chapter 5).
a. A film of liquid exists at the alveolar-air interface.
b. As the alveolus is compressed during expiration, surfactant decreases surface tension, requiring less pressure and effort to reexpand the alveolus during the next inspiration.
2. Surfactant is composed of lipids (85% to 90%) and proteins (10% to 15%)
(1) 85% to 90% of surfactant by weight.
(2) 90% of lipids are phospholipids.
(3) Phosphatidylcholine comprises 75% to 80% of the phospholipids.
(4) Depalmitoylphosphatidylcholine (DPPC or lecithin) is the component predominantly responsible for decreasing alveolar surface tension.
(1) 10% of surfactant by weight.
(2) Only 20% of the proteins are surfactant-specific proteins; 80% are serum proteins.
(3) The four surfactant-specific proteins are SP-A, SP-B, SP-C, and SP-D.
c. The alveolar type II cell synthesizes all surfactant components.
C Exogenous surfactant preparations
1. Colfosceril palmitate (Exosurf Neonatal)
a. A protein-free, synthetic lyophilized powder reconstituted with 8 ml of preservative-free sterile water to form a milky white suspension.
b. Colfosceril palmitate is depalmitoylphosphatidylcholine.
(1) Prophylactic therapy for infants with birth weight <1350 g.
(2) Prophylactic therapy for infants with birth weight >1350 g and evidence of pulmonary immaturity and risk for RDS.
(1) The first dose is 5 ml/kg, given as two divided doses.
(2) A single vial of reconstituted Exosurf has a volume of 8 ml and can treat infants with birth weight up to 1600 g.
(3) A second dose of 5 ml/kg is given 12 hours after the first.
(4) A third dose, if necessary, is given 12 hours after the second dose.
(1) Instilled directly into the endotracheal tube (ETT) through a side-port adaptor that is Luer-Lok fitted to the ETT.
(2) With the infant in the midline position, the first half dose is sequentially instilled in synchrony with the inspiratory phase of the ventilator.
(3) The infant is then rotated to one side and ventilated for 30 seconds.
(4) Repositioned to midline, the infant is given the second half dose, then rotated to the opposite side, and ventilated for 30 seconds.
a. A natural bovine lung extract mixed with colfosceril palmitate (DPPC), palmitic acid, and tripalmitin.
b. Contains surfactant proteins SP-B and SP-C but not SP-A.
(1) Used prophylactically for premature infants with birth weight <1250 g.
(2) Used for infants with evidence of surfactant deficiency and risk of RDS.
d. As Survanta, this surfactant preparation comes in an 8-ml solution suspended in 0.9% sodium chloride, having a concentration of 25 mg/ml. Thus one vial has 200 mg.
(1) To gently warm the refrigerated suspension, it should be set out at room temperature for 20 minutes.
(2) Settling of the suspension should be countered by gentle agitation of the vial. The container should not be vigorously shaken.
(3) The dose is given in four aliquots, instilled directly into the trachea through a 5-French catheter.
(4) After each of the aliquots, the catheter is withdrawn, and the infant is ventilated and stabilized for at least 30 seconds.
a. An organic solvent extract of calf lung obtained by bronchoalveolar lavage.
b. This surfactant preparation is a suspension containing SP-B and SP-C.
(1) Used prophylactically in infants <29 weeks gestational age.
(2) Used for rescue of intubated premature infants aged <3 days who develop RDS.
(2) Repeat doses can be given 12 hours apart or as early as 6 hours for infants in continued distress.
(3) Each 6-ml vial contains 210 mg phospholipids, enough to treat a 2-kg infant.
a. A natural surfactant extracted from porcine lung, consisting of 99% phospholipids and 1% surfactant proteins SP-B and SP-C.
(1) Rescue or treatment of premature infants with RDS.
(2) Used off-label for prophylaxis in RDS, acute respiratory distress syndrome (ARDS) from viral pneumonia, HIV-infected infants with Pneumocystis carinii pneumonia, and management of ARDS after near drowning. Efficacy in all of these settings is controversial.
(2) Two subsequent doses of 1.25 ml/kg birth weight can be given in 12-hour intervals for continued distress.
(1) Two equal aliquots are given through a 5-French catheter whose tip is positioned at the tip of the ETT.
(2) Each aliquot is given to an alternate side, positioned dependently, stabilizing the infant by mechanically ventilating with 100% oxygen for at least 1 minute between doses.
(3) Airway suctioning should be avoided for 1 hour after treatment, provided significant airway obstruction does not occur.
2. Generic name: Acetylcysteine (N-acetyl-L-cysteine)
3. Mechanism of action: Lyses disulfide bonds holding mucoproteins together, thus increasing fluidity of mucoid sputum.
4. Concentration: 10% or 20% w/v solution
a. Standard dosage: 4 ml of 10% w/v or 2 ml 20% w/v solution with 2 ml water or normal saline solution
6. Indications: Thick, retained mucoid or mucopurulent secretions
7. Contraindications: Hypersensitivity
a. Questionable efficacy when aerosolized.
(1) Mucolytic activity well documented in vitro.
(2) No data clearly demonstrate the clinical efficacy of aerosolized Mucomyst.
(3) May be useful for direct bronchial instillation during bronchoscopy.
b. Highly recommended that drug is administered in conjunction with bronchodilator.
c. Should be refrigerated after opening.
e. Should be administered in glass, plastic, or nontarnishable metal containers because it reacts with rubber, some plastics, and iron.
f. Ineffective on predominantly purulent secretions.
g. Supplied in 4-, 10-, and 30-ml vials.
h. Should be administered after pretreatment with a bronchodilator.
2. Generic name: Dornase alfa—originally rhDNase (recombinant human DNase)
3. Mechanism of action: A genetically engineered clone of human pancreatic DNase enzyme, it is a peptide proteolytic enzyme that can break down extracellular DNA and F-actin polymers from neutrophils found in purulent secretions.
4. Indication: Cystic fibrosis management to manage purulent mucoid secretions; more effective than Mucomyst in reducing the viscosity of sputum in cystic fibrosis.
a. 2.5 mg/ampule, 1 ampule daily via recommended nebulizer system (e.g., Hudson T Updraft II, Marquest II, or PARI LC Jet Plus).
b. 3 to 5 ml of 5% to 10% w/v solution in 0.45% or 0.9% saline solution.
6. Contraindication: Hypersensitivity to dornase or other components of the drug preparation.
a. Unlike the earlier animal pancreatic dornase (Dornavac), dornase alfa does not cause antibody-mediated allergic reactions, such as bronchospasm.
b. Pharyngitis and vocal alterations
g. Less commonly a variety of respiratory symptoms (e.g., cough, dyspnea, rhinitis, and sinusitis), GI obstruction, hypoxia, and weight loss.
VI Mast Cell Stabilizers (Table 17-1)
TABLE 17-1
| Drug | Brand Name | Formulation and Dosage |
| Cromolyn sodium | Intal | MDI: 800 μg/actuation |
| Adults and children ≥5 yr: 2 inhalations QID | ||
| SVN: 20 mg/ampule or 20 mg/vial | ||
| Nasalcrom | Spray: 40 mg/ml (4%) | |
| Adults and children ≥6 yr: 1 spray each nostril 3-6 times daily every 4-6 hr | ||
| Nedocromil sodium | Tilade | MDI: 1.75 mg/actuation |
| Adults and children ≥12 yr: 2 inhalations QID |
MDI, Metered dose inhaler; QID, four times daily; SVN, small-volume nebulizer.
From Wilkins RL, et al: Egan’s Fundamentals of Respiratory Care, ed 8. St. Louis, Mosby, 2003.
1. Generic name: Cromolyn sodium
a. Inhibits release of histamine and leukotrienes during allergic, immunoglobulin (Ig) E-mediated responses of pulmonary mast cells.
b. Suppresses response of mast cell to antigen-antibody reaction.
3. Concentration: 20-mg capsules (powder) or 20 mg/2 ml H2O ampules (liquid).
4. Standard dosage: 20 mg three or four times daily.
6. Primary indications: Prophylactic maintenance for patients with severe bronchial asthma exercise-induced bronchospasm.
7. Secondary indications: Result of the effect of cromolyn sodium on all mast cells.
8. Contraindications: Hypersensitivity
1. Generic name: Nedocromil sodium
a. Inhibits most cell cytokine release
b. Modulates the synthesis and release of proinflammatory cytokines
3. Concentration: 1.75 mg per MDI actuation (only preparation)
4. Standard dosage: Two inhalations four times per day
6. Primary indication: Prophylactic therapy for the management of mild to moderate asthma.
VII Leukotriene Inhibitors (Antileukotrienes) (Table 17-2)
TABLE 17-2
| Drug | Brand Name | Formulation and Dosage |
| Zafirlukast | Accolate | Tablets: 10 mg, 20 mg |
| Adults and children >12 yr: 20 mg (1 tablet) BID, without food | ||
| Children 5-11 yr: 10 mg BID | ||
| Montelukast | Singulair | Tablets: 10 mg, 5 mg, and 4 mg (cherry-flavored chewable) |
| Adults and children >15 yr: 1 10-mg tablet each evening | ||
| Children 6-14 yr: 1 5-mg chewable tablet each evening | ||
| Children 2-5 yr: 1 4-mg chewable tablet each evening | ||
| Children 12-23 mo: 4 mg oral granules each evening | ||
| Zileuton | Zyflo | Tablets: 600 mg |
| Adults and children ≥12 yr: 1 600-mg tablet QID |
BID, Twice daily; QID, four times daily.
From Wilkins RL, et al: Egan’s Fundamentals of Respiratory Care, ed 8. St. Louis, Mosby, 2003.
1. Originally isolated from leukocytes, leukotrienes have a carbon chain skeleton with three double bonds in series, a triene.
2. Chemically they are lipid mediators of inflammation synthesized from arachidonic acid (AA).
3. Potent bronchoconstrictors, they stimulate airway edema, mucus secretion, and recruitment of other inflammatory cells.
4. They are particularly effective in controlling exercise-induced, aspirin-induced, and, less effectively, allergen-induced asthma.
B Leukotriene production (Figure 17-5)
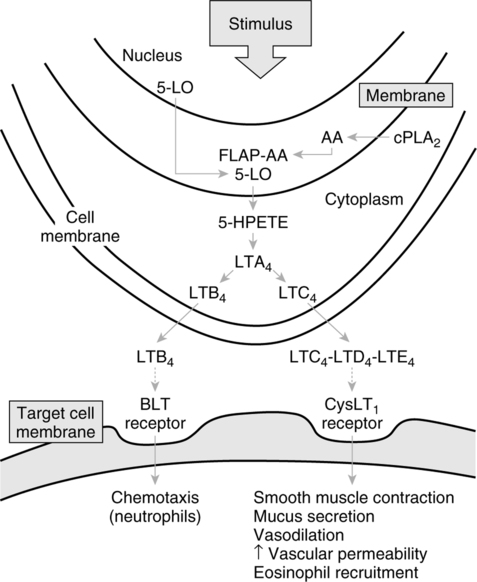
1. In cell cytoplasm, phospholipase A2 (PLA2) moves to the cell nuclear membrane, where it hydrolyzes phospholipids to form free AA. AA binds to 5-lipoxygenase (5-LO)-activating protein (FLAP; AA-FLAP). 5-LO moves to the nuclear membrane and oxygenates AA of the AA-FLAP complex, resulting in 5-hydroperoxyeicosatetraenoic acid (HPETE). HPETE is converted to leukotriene A4 (LTA4), an unstable intermediate that is the source of all leukotrienes. LTA4 is converted into leukotriene B4 (LTB4) or leukotriene C4 (LTC4), both of which move to the extracellular space. LTC4, which structurally contains the amino acid cysteine, can be converted to leukotrienes D4 and E4 (LTD4 and LTE4). These three are known as the cysteinyl leukotrienes (CysLTs). The three CysLTs (LTC4, LTD4, and LTE4) are the components previously known as slow reacting substance of anaphylaxis (SRS-A).
1. Leukotrienes bind to leukotriene receptors to exert inflammatory effects.
a. Increased bronchial hyperresponsiveness to other irritants such as histamine.
c. Increased vascular permeability causing airway wall edema.
d. Plasma exudation into airway lumen.
e. Cumulative effect is increased secretion viscosity and possible airway occlusion often seen in asthma.
2. The CysLT1 receptor, located on smooth muscle cells in the airway, mediates the proasthmatic action of the CysLTs.
a. Stimulation of the CysLT1 receptor causes bronchoconstriction.
b. The CysLTs are more potent airway constrictors than histamine.
a. The CysLTs are produced by eosinophils, mast cells, and macrophages, all of which are seen in the airways of persons with asthma.
b. The CysLT2 receptor subtype mediates constriction of vascular smooth muscle.
c. Drugs that block the binding of leukotrienes to CysLT1 have the generic suffix –leukast.
a. Approved for use in early 1997.
b. Orally active inhibitor of 5-LO, preventing catalysis of leukotrienes from AA.
c. Indicated for prophylactic and long-term management of asthma in adults and children aged >12 years.
a. Available as a single tablet of 600 mg.
b. One 600-mg tablet four times daily for total daily dose of 2400 mg.
4. Precautions: Patients should be monitored for liver injury.
a. Hepatic transaminase enzymes should be evaluated before initiation of treatment, monthly for the first 3 months, and every 2 to 3 months thereafter for the first year of treatment.
b. With clinical signs of liver injury (e.g., right upper quadrant pain, nausea, fatigue, lethargy, pruritus, jaundice, or flulike symptoms), discontinue use.
a. Headache, abdominal pain, loss of strength, and dyspepsia.
b. Elevated liver function test values.
(1) Hepatic transaminase enzymes should be monitored before and during treatment.
(2) Liver function test values may return to normal either during or after treatment.
c. Contraindicated in patients with acute liver disease or transaminase levels greater than three times normal.
a. Approved for use in the United States in 1996 as Accolate.
b. Indicated for prophylaxis and long-term management of asthma in adults or children aged ≥12 years.
a. Leukotriene receptor antagonist that blocks inflammatory effects of leukotrienes.
b. Binds to the CysLT1 receptors.
c. Competitively inhibits leukotrienes LTC4, LTD4, and LTE4, blocking inflammation.
a. Rapidly absorbed orally, reaching peak plasma levels in 3 hours.
b. Half-life approximately 10 hours.
d. 10% excreted in urine, remainder in feces.
e. Administration of zafirlukast with food reduces mean bioavailability by 40%.
a. Headache, infection (primarily respiratory), nausea, diarrhea, and generalized and abdominal pain.
b. Liver disease will increase drug plasma levels.
c. Doses >40 mg daily can cause elevations in serum liver enzymes.
d. Instances of hepatitis and hyperbilirubinemia reported in patients receiving 40 mg/day for 100 days.
a. Approved for use in February 1998.
b. Orally active leukotriene receptor antagonist.
c. Indicated for prophylaxis and long-term management of asthma.
(1) Has no bronchodilatory effect in short-term asthma treatment.
(2) Approved for use in pediatric patients (children aged ≥2 years).
(3) Efficacious in management of mild to moderate asthma and exercise-induced bronchial constriction.
(4) Exhibits improved asthma control in children aged 6 to 14 years and in adults and adolescents aged >15 years.
a. Supplied in 10-mg oral tablet or 4-mg and 5-mg chewable cherry-flavored tablets.
b. Adults and adolescents aged >15 years: one 10-mg tablet daily, taken in the evening.
c. Pediatric patients aged 6 to 14 years: one 5-mg chewable tablet daily, taken in the evening.
d. Pediatric patients aged 2 to 5 years: one 4-mg chewable tablet daily, taken in the evening.
a. Competitive antagonist for the CysLTs LTC4, LTD4, and LTE4.
b. Binds to the CysLT1 receptor subtype, preventing leukotriene receptor stimulation in airway smooth muscle and secretory gland cells.
c. Shown to inhibit early- and late-phase asthmatic bronchoconstriction.
(1) A 10-mg dose reaches peak plasma concentration in 3 to 4 hours.
(2) A 10-mg dose has mean bioavailability of 64%, not influenced by intake of food.
b. A 5-mg fasting dose had slightly higher concentrations than when taken with food.
c. A 4-mg tablet reached peak plasma concentrations in 2 hours.
d. Mean plasma life in adults: 2.7 to 5.5 hours.
e. Metabolized in liver and excreted via bile.
f. Mild to moderate liver disease will increase plasma levels.
VIII Aerosolized Antimicrobial Agents
1. Aerosolized delivery achieves high drug concentration in lung tissue.
2. Antibiotics effective against particular organisms, such as Pseudomonas; have poor lung bioavailability when taken orally.
1. Nebulization of antibiotics should occur in contained rooms to prevent escape of the drug and possible development of resistant organisms within the local environment.
2. Caregivers should be protected to prevent development of sensitivity reactions or resistance to antibiotics.
3. Antibiotic solutions vary in viscosity from the aqueous solutions for which disposable nebulizers were designed.
a. An aminoglycoside used for management of Pseudomonas aeruginosa, a pathogen often associated with chronic infection of cystic fibrosis patients.
b. Tobramycin solution for inhalation (TOBI) is a preservative-free preparation that may reduce the risk of adverse effects.
c. TOBI should be administered in the PARI LC Plus nebulizer to ensure adequate drug output and particle size.
3. Contraindicated in patients with hypersensitivity to aminoglycosides.
4. Dose: 300 mg twice daily in repeated cycles of 28 days on drug, followed by 28 days off drug.
D Colistimethate (Coly-Mycin, Colistin)
a. A polypeptide antibiotic used in aerosol form for management of Pseudomonas pneumonia.
b. Most often used for immunocompromised patients or patients with cystic fibrosis.
2. Contraindicated in patients with known hypersensitivity to the drug.
1. Characteristics: An antifungal agent given prophylactically to prevent fungal pneumonia in immunocompromised patients.
2. Contraindicated in patients with known hypersensitivity to the drug.
a. Usual dose: 10 mg twice daily.
b. A vial with 50 mg powder is reconstituted in 10 ml sterile water for inhalation, creating a concentration of 5 mg/ml.
a. Extubated patients: Via Pari nebulizer.
(1) Using a small-volume nebulizer, dilute the 2-ml dose (5 mg/ml) with 2 ml water for a final solution of 4 ml.
(2) Use additional ventilator expiratory limb filter to prevent drug interference with exhalation valve or flow sensor.
c. Amphotericin B, incompatible with most drugs, should not be mixed with other drugs in the nebulizer.
a. An antiprotozoal agent used for prophylaxis of P. carinii pneumonia, often seen in immunocompromised patients.
b. Has been replaced by trimethoprim-sulfamethoxazole (TMP/SMX) for treatment (IV) and prophylaxis (nebulized) in patients allergic to sulfa drugs.
I Table 17-3 lists commonly used antibacterial agents with their mechanisms of action, spectra of activity, and toxicities.
TABLE 17-3
Common Antibacterial Antibiotics, Mechanisms, Spectra of Activity, and Toxicities
| Class | Examples | Mechanism of Action | Spectrum of Activity | Toxicity |
| Penicillins | Penicillin, amoxicillin, amoxicillin-clavulanate (Augmentin), piperacillin, ticarcillin | Cleave bonds contained in peptidoglycan cell wall, disrupting bacterial structural integrity | G+/G−, and anaerobic activities increasing with successive generations | Hypersensitivity, GI intolerance, hepatitis with prolonged use |
| Cephalosporins | Cefazolin (Ancef), cephalexin (Keflex), ceftriaxone (Rocephin), ceftazidime | Same as penicillins | Same as penicillins | Rare cytopenias |
| Carbapenems | Imipenem | Same as penicillins | Broad G+, G−, anaerobic coverage | Seizure, GI intolerance, occasional cytopenias |
| Monobactams | Aztreonam | Same as penicillins | Broad G+, G−, anaerobic coverage | Similar to penicillins |
| Aminoglycosides | Gentamicin, tobramycin (TOBI) | Antiribosomal: interfere with translation of proteins from mRNA by binding the bacterial ribosome 30S subunit, bacteriocidal | G− | Nephrotoxicity, ototoxicity, rare paralysis; levels requiring monitoring |
| Tetracyclines | Tetracycline, doxycycline, minocycline | Antiribosomal; bind the 30S subunit; bacteriocidal at high concentrations | G+, atypicals | Tooth and bone defects, phototoxicity, GI intolerance, benign intracranial hypertension, mild hepatitis |
| Sulfonamides | Trimethoprim/sulfamethoxazole (TMP/SMX, Bactrim, Septra) | Block metabolic pathway for folate production | G+, G−, Pneumocystis carinii | Hypersensitivity, possible trigger of hemolysis in G6PD deficiency |
| Fluoroquinolones | Ciprofloxacin, levofloxacin, gatifloxacin | Inhibit DNA gyrase, interfere with DNA synthesis | G−, G+, atypicals | Cartilage erosion in animals; not administered to children, except those with cystic fibrosis |
| Macrolides | Erythromycin, clarithromycin (Biaxin), azithromycin (Zithromax) | Antiribosomal: bind 50S subunit; bacteriostatic | G+, some G−; good atypical coverage, including Legionella species | GI intolerance |
| Others | Vancomycin | Inhibits cell wall synthesis | G+, including MRSA; Enterococcus species | Hypotension and rash with infusion, ototoxicity |
| Metronidazole | Anaerobes | Ethanol intolerance, seizures, peripheral neuropathy | ||
| Chloramphenicol | Antiribosomal: binds 50S subunit | G+, many G−, anaerobes, rickettsiae | Aplastic anemia; used only when strongly indicated | |
| Clindamycin | Antiribosomal: bacteriocidal at high concentrations | G+ and anaerobes | Clostridium difficile colitis |
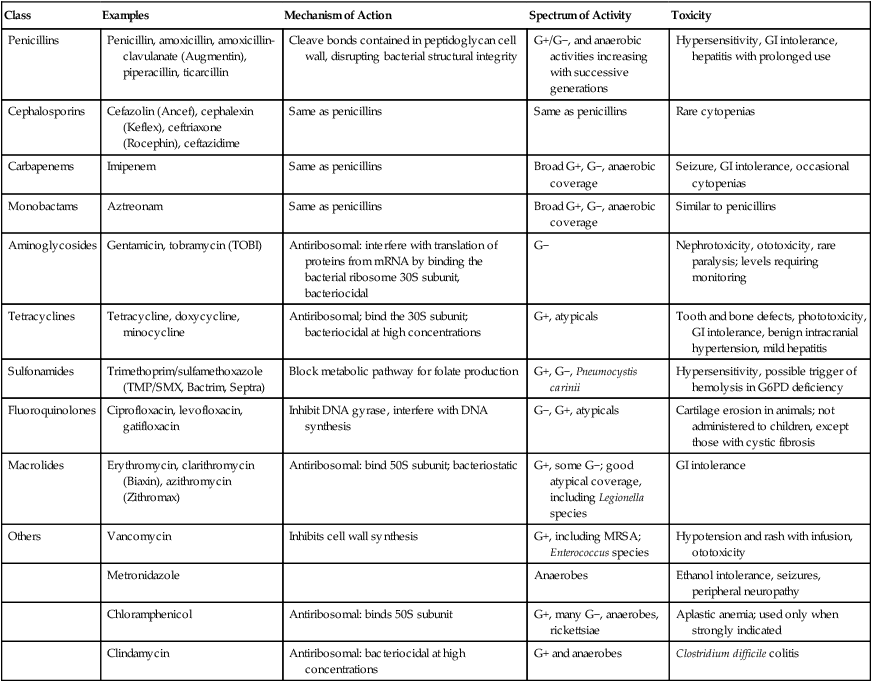
From Hess DR, et al: Respiratory Care: Principles and Practices. Philadelphia, WB Saunders, 2002.
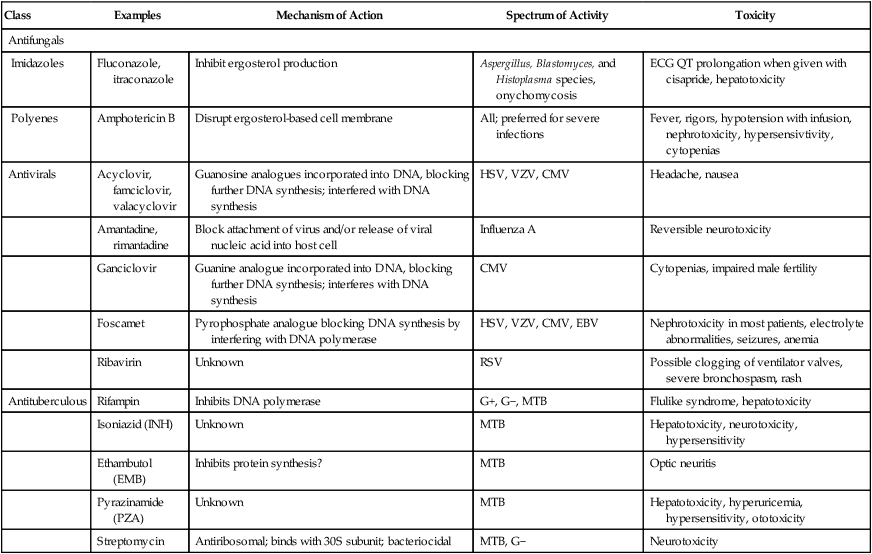
J Table 17-4 lists commonly used antifungal agents and antituberculous agents with their mechanisms of action, spectra of activity, and toxicities.
TABLE 17-4
Common Antibiotics, Mechanisms, Spectra of Activity, and Toxicities
| Class | Examples | Mechanism of Action | Spectrum of Activity | Toxicity |
| Antifungals | ||||
| Imidazoles | Fluconazole, itraconazole | Inhibit ergosterol production | Aspergillus, Blastomyces, and Histoplasma species, onychomycosis | ECG QT prolongation when given with cisapride, hepatotoxicity |
| Polyenes | Amphotericin B | Disrupt ergosterol-based cell membrane | All; preferred for severe infections | Fever, rigors, hypotension with infusion, nephrotoxicity, hypersensivtivity, cytopenias |
| Antivirals | Acyclovir, famciclovir, valacyclovir | Guanosine analogues incorporated into DNA, blocking further DNA synthesis; interfered with DNA synthesis | HSV, VZV, CMV | Headache, nausea |
| Amantadine, rimantadine | Block attachment of virus and/or release of viral nucleic acid into host cell | Influenza A | Reversible neurotoxicity | |
| Ganciclovir | Guanine analogue incorporated into DNA, blocking further DNA synthesis; interferes with DNA synthesis | CMV | Cytopenias, impaired male fertility | |
| Foscamet | Pyrophosphate analogue blocking DNA synthesis by interfering with DNA polymerase | HSV, VZV, CMV, EBV | Nephrotoxicity in most patients, electrolyte abnormalities, seizures, anemia | |
| Ribavirin | Unknown | RSV | Possible clogging of ventilator valves, severe bronchospasm, rash | |
| Antituberculous | Rifampin | Inhibits DNA polymerase | G+, G−, MTB | Flulike syndrome, hepatotoxicity |
| Isoniazid (INH) | Unknown | MTB | Hepatotoxicity, neurotoxicity, hypersensitivity | |
| Ethambutol (EMB) | Inhibits protein synthesis? | MTB | Optic neuritis | |
| Pyrazinamide (PZA) | Unknown | MTB | Hepatotoxicity, hyperuricemia, hypersensitivity, ototoxicity | |
| Streptomycin | Antiribosomal; binds with 30S subunit; bacteriocidal | MTB, G− | Neurotoxicity | |
IX Adrenergic Bronchodilators (β-Adrenergic Agonists): General Considerations (Table 17-5)
TABLE 17-5
Inhaled Adrenergic Bronchodilator Agents Currently Available in the United States
| Drug | Brand Name | Receptor Preference | Adult Dosage | Time Course (Onset, Peak, Duration) |
| Epinephrine | Adrenalin Cl | (α, β) | SVN: 1% solution (1:100), 0.25-0.5 ml (2.5-5.0 mg) QID; MDI: 0.2 mg/puff; 2 puffs as ordered or needed | 3-5 min, 5-20 min, 1-3 hr |
| Racemic epinephrine | MicroNefrin, AsthmaNefrin, various | (α, β) | SVN: 2.25% solution, 0.25-0.5 ml (5.63-11.25 mg) QID | 3-5 min, 5-20 min, 0.5-2 hr |
| Isoproterenol | Isuprel, Isuprel Mistometer | (β) | SVN: 0.5% solution (1:200), 0.25-0.5 ml (1.25-2.5 mg) QID, MDI; 103 μg/puff QID | 2-5 min, 5-30 min, 0.5-2 hr |
| Isoetharine | Isoetharine HCl | (β2) | SVN: 1% solution, 0.25-0.5 ml (2.5-5.0 mg) QID | 1-6 min, 15-60 min |
| Terbutaline | Brethaire | (β2) | MDI; 200 μg/puff, 2 puffs every 4-6 hr; TABS: 2.5 or 5 mg, 5 mg every 6 hr; INJ: 1 mg/mL, 0.25 mg SC | 5-30 min, 30-60 min, 3-6 hr |
| Metaproterenol | Alupent | (β2) | SVN; 5% solution, 0.3 ml (15 mg), TID, QID; MDI: 650 μg/puff, 2-3 puffs TID, QID; TABS: 10 or 20 mg, 20 mg TID, QID; SYRUP: 10 mg/5 ml, 2 tsp TID, QID | 1-5 min, 60 min, 2-6 hr |
| Albuterol | Proventil, Proventil HFA, Ventolin | (β2) | SVN: 0.5% solution, 0.5 ml (2.5 mg) TID, QID; MDI: 90 μg/puff, 2 puffs TID, QID; TABS: 2 mg, 4 mg TID, QID; SYRUP: 2 mg/5 ml, 1-2 tsp TID, QID | 15 min, 30-60 min |
| Bitolterol | Tornalate | (β2) | SVN: 0.31 mg and 0.2% solution, 1.25 ml (2.5 mg) BID-QID; MDI: 370 μg/puff, 2 puffs every 8 hr | 3-4 min, 30-60 min, 5-8 hr |
| Pirbuterol | Maxair | (β2) | MDI: 200 μg/puff, 2 puffs every 4-6 hr | 5 min, 30 min, 5 hr |
| Levalbuterol | Xopenex | (β2) | SVN: 0.31 mg and 0.63 mg/3 ml TID or 1.25 mg/3 ml TID | 15 min, 30-60 min, 5-8 hr |
| Salmeterol | Serevent | (β2) | MDI: 25 μg/puff, 2 puffs BID; DPI: 50 μg/blister BID | 20 min, 3-5 min, 12 hr |
| Formoterol | Foradil | (β2) | DPI: 12 μg/inhalation BID | 5 min, 30-60 min, 12 hr |
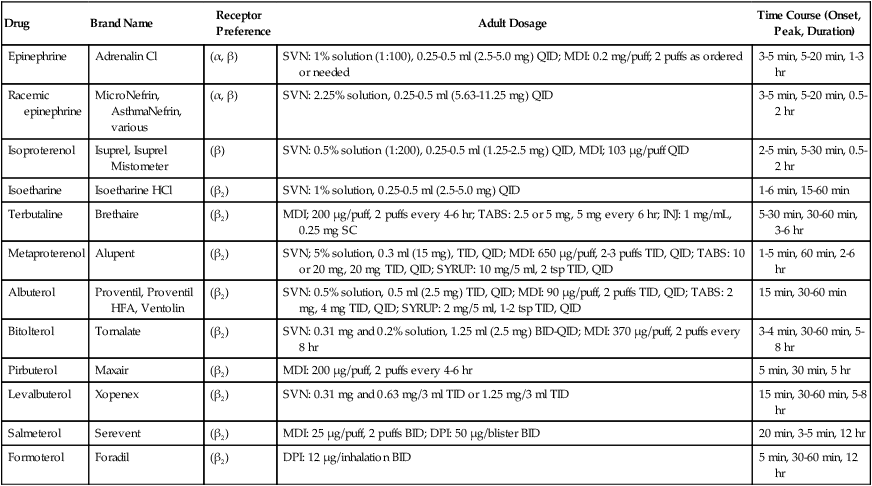
From Wilkins RL, et al: Egan’s Fundamentals of Respiratory Care, ed 8. St. Louis, Mosby, 2003.
1. The most widely prescribed class of bronchodilator.
2. Indicated to relieve airflow obstruction in diseases such as asthma, acute and chronic bronchitis, emphysema, bronchiectasis, and cystic fibrosis.
3. Divided into two main categories by the 1997 National Asthma Education and Prevention Program Expert Panel II guidelines.
1. α-Receptor stimulation: Causes vasoconstriction and increased blood pressure (vasopressor effect).
2. β1-Receptor stimulation: Causes increased heart rate and myocontractility (increased chronotropic and inotropic effects).
3. β2-Receptor stimulation: Relaxes bronchial smooth muscle, stimulates mucociliary activity, and has some inhibitory action on inflammatory mediator release.
C Mechanism of action for β2-receptor-mediated relaxation of airway smooth muscle (Figure 17-6)
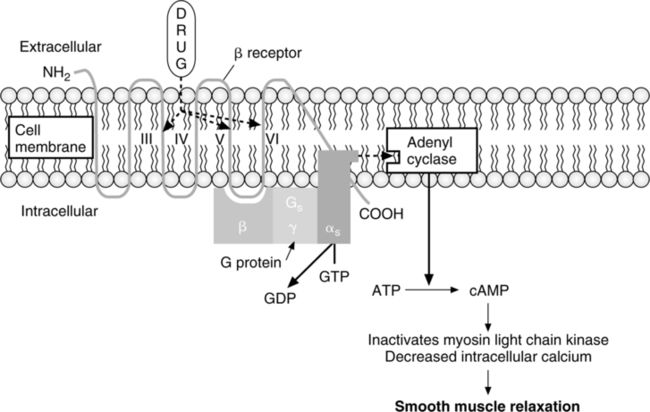
D Specific adrenergic bronchodilators (see Table 17-5)
E Ultra-short-acting catecholamines
a. All are catecholamines that lack β2 specificity, commonly resulting in tachycardia and increased blood pressure.
b. All have a short duration of action because they are metabolized by the enzyme catechol-O-methyl-transferase.
a. Used as a bronchodilator since the start of the 20th century.
b. Stimulates α-adrenergic and β-adrenergic receptors.
c. Racemic epinephrine is often used for its vasoconstrictive action in management of stridor associated with laryngeal edema and croup.
a. Widely used for management of asthma until the 1970s.
b. Possesses long-acting stimulant effects and was sold over-the-counter as a central nervous stimulant in the 1970s.
c. Removed from over-the-counter sales in the 1980s because of its abuse as a stimulant and because it was used as a precursor in the synthesis of amphetamine.
b. First bronchodilator with only β agonist effects.
c. Causes bronchodilation by relaxing bronchial smooth muscle but also relaxed vascular smooth muscle, causing decreased peripheral vascular resistance and lower blood pressure.
d. Stimulation of β1 receptors could cause increased heart rate and contractility.
F Short-acting noncatecholamines
a. Longer-acting β2-specific drugs, with duration of action of 4 to 6 hours.
b. Better suited to maintenance therapy than catecholamines.
c. Modest duration of action results in loss of bronchodilatation overnight.
a. A rapid-acting β agonist primarily used for symptomatic relief of bronchospasm.
b. The most commonly prescribed pulmonary medication in the United States, with few side effects.
a. The R-enantiomer of albuterol, a single isomer β2-selective agonist.
b. The R-enantiomer form causes most of the bronchodilatory effect on airways.
c. Levalbuterol binds with greater affinity to receptor sites, providing effective bronchodilation at lower doses than albuterol.
d. Clinically often used at half the dosage of albuterol.
e. Laboratory evidence indicates that a 0.63-mg dose produces a forced expiratory volume in one sec change equivalent to a 2.5-mg dose of albuterol.
G Long-acting adrenergic bronchodilators
a. Long-acting β2 agonists with slower onset of action.
b. Indicated for maintenance bronchodilation and control of nocturnal symptoms in asthma or other obstructive diseases.
c. Often combined with antiinflammatory agents for control of airway inflammation and bronchospasm.
H General therapeutic uses of sympathomimetics
1. Management of generalized bronchoconstriction
2. Management of mucosal congestion
X Anticholinergic Bronchodilators (Table 17-6)
TABLE 17-6
Inhaled Anticholinergic Bronchodilator Agents*
| Drug | Brand Name | Adult Dosage | Time Course (Onset, Peak, Duration) |
| Ipratropium bromide | Atrovent | MDI: 18 μg/puff, 2 puffs QID; SVN: 0.02% solution (0.2 mg/ml), 500 μg TID, QID; Nasal spray: 0.03%, 0.06%; 2 sprays per nostril 2 to 4 times daily (dosage varies) | Onset: 15 min, peak: 1-2 hr, duration: 4-6 hr |
| Ipratropium bromide and albuterol | Combivent | MDI: ipratropium 18 μg/puff and albuterol 90 μg/puff, 2 puffs QID | Onset: 15 min, peak: 1-2 hr, duration: 4-6 hr |
| DuoNeb | SVN: ipratropium 0.5 mg and albuterol 3.0 mg (equal to 2.5 mg albuterol base) | ||
| Oxitropium bromide† | MDI: 100 μg/puff, 2 puffs BID, TID | Onset: 15 min, peak: 1-2 hr, duration: 8 hr | |
| Tiotropium bromide‡ | Spiriva | DPI: 18 μg/inhalation, 1 inhalation daily | Onset: 30 min, peak: 3 hr, duration: 24 hr |
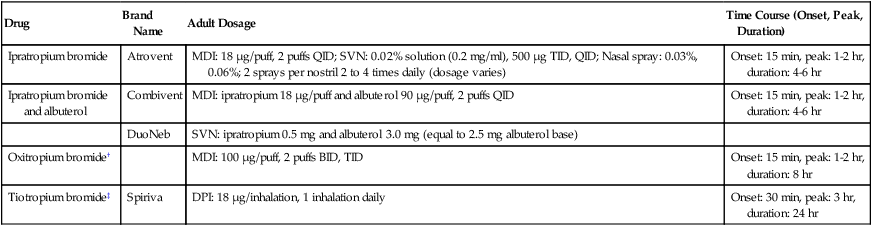
*Ipratropium bromide is the only agent currently approved for use in the United States as an inhaled bronchodilator. A holding chamber is recommended with MDI administration to prevent accidental eye exposure.
†Available outside the United States.
From Wilkins RL et al: Egan’s Fundamentals of Respiratory Care, ed 8. St. Louis, Mosby, 2003.
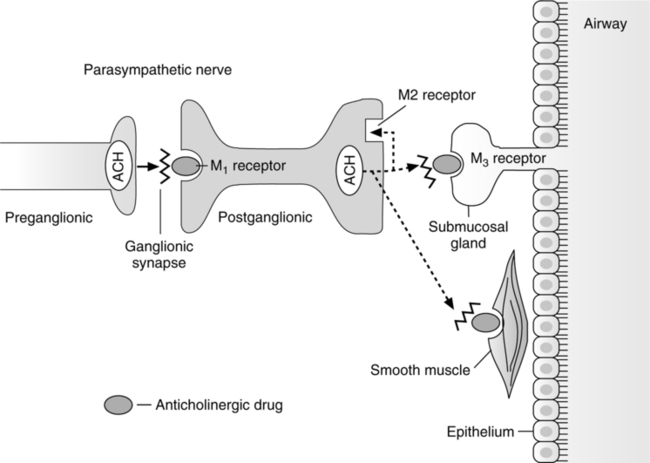
1. Blocks cholinergic-induced bronchoconstriction.
a. Indicated for maintenance management of chronic obstructive pulmonary disease (COPD).
b. As effective as the β agonists for COPD patients but less effective for those with asthma.
2. Mode of action: Competitive antagonist for acetylcholine at muscarinic receptors on airway smooth muscle (antimuscarinic agents) (Figure 17-7).
1. A tertiary ammonium compound long known to have bronchodilatory action.
2. Multiple undesirable side effects rendered it obsolete as a bronchodilator once ipratropium was developed in the 1980s.
1. The primary anticholinergic agent used for management of airway obstructive conditions.
2. Ipratropium’s quaternary ammonium structure allows it to carry a positive charge, thus inhibiting its diffusion across cell membranes.
a. This feature restricts ipratropium’s distribution, limiting side effects.
b. By not being able to cross the blood-brain barrier, absorption to the CNS is also limited.
3. Half-life of 3 hours requires dosing every 4 to 6 hours.
4. Also available in a metered dose inhaler formulation combined with albuterol (Combivent).
5. Given only by inhalation because it is poorly absorbed through the oral mucosa.
6. Does not have the secretion drying effects of atropine or glycopyrrolate.
D Tiotropium bromide (Spiriva): Structurally similar to ipratropium but selective for muscarinic receptors M1 and M3.
1. Tiotropium can confer bronchodilation for up to 24 hours in COPD patients.
2. Has a slower onset than ipratropium but after onset maintains a higher level of baseline bronchodilation.
E Oxitropium bromide: A derivative of scopolamine used outside the United States as an aerosolized anticholinergic bronchodilator for management of COPD.
XI Inhaled Corticosteroids (Table 17-7)
TABLE 17-7
Corticosteroids Available by Aerosol for Oral Inhalation
| Drug | Brand Name | Formulation and Dosage |
| Beclomethasone dipropionate | OVAR | MDI: 40 μg/puff and 80 μg/puff |
| Adults ≥12 yr: 40-80 μg BID* or 40-160 μg BID† | ||
| Children 5-11 yr: 40 μg BID*† | ||
| Triamcinolone acetonide | Azmacort | MDI: 100 μg/puff |
| Adults: 2 puffs TID or QID | ||
| Children: 1-2 puffs TID or QID | ||
| Flunisolide | AeroBid, AeroBid-M | MDI: 250 mg/puff |
| Adults: 2 puffs BID | ||
| Children: 2 puffs BID | ||
| Fluticasone propionate | Flovent | MDI: 44 μg/puff, 110 μg/puff, 220 μg/puff |
| Adults ≥12 yr: 88 μg BID,* 88-220 μg BID,† 880 μg BID‡ | ||
| Flovent Rotadisk | DPI: 50 μg, 100 μg, 250 μg | |
| Adults: 100 μg BID,* 100-250 μg BID,† 1000 μg BID‡ | ||
| Children 4-11 yr: 50 μg BID | ||
| Budesonide | Pulmicort Tubuhaler | DPI: 200 μg/actuation |
| Adults: 200-400 μg BID,* 200-400 μg BID,† | ||
| 400-800 μg BID‡ | ||
| Children ≥ 6 yr: 200 μg BID | ||
| Pulmicort Respules | SVN: 0.25 mg/2 ml, 0.5 mg/2 ml | |
| Children 1-8 yr: 0.5 mg total dose given daily or BID in divided doses*†; 1 mg given as 0.5 mg BID or daily‡ | ||
| Fluticasone propionate/salmeterol | Advair Diskus | DPI: 100 μg fluticasone/50 μg salmeterol, 250 μg fluticasone/50 μg salmeterol, or 500 μg fluticasone/50 μg salmeterol |
| Adults and children >12 yr: 100 μg fluticasone/50 μg salmeterol 1 inhalation BID about 12 hr apart (starting dose if not currently on inhaled corticosteroids); maximum recommended dose is 500 μg fluticasone/50 μg salmeterol BID |
*Recommended starting dose if on bronchodilators alone.
†Recommended starting dose if on inhaled corticosteroids previously.
‡Recommended starting dose if on oral corticosteroids previously.
From Wilkins RL, et al: Egan’s Fundamentals of Respiratory Care, ed 8. St. Louis, Mosby, 2003.
1. Development of inhaled corticosteroids allowed control of airway inflammation without the side effects associated with systemic steroid use. Direct delivery to the lungs reduced systemic absorption and allowed long-term steroid use with minimal side effects.
2. Continuous treatment by inhaled corticosteroids has become standard maintenance therapy for those with moderate and severe asthma and is indicated for short-term use in exacerbations of asthma.
a. In exacerbations of asthma, the underlying poorly controlled inflammation should be managed by an increase in corticosteroids.
(1) Exacerbations of asthma are usually preceded by a period of increasing bronchial inflammation.
(2) The inflammatory process is often treated with a 5- to 20-day course of oral corticosteroids, initially dosed at 20 to 40 mg prednisone daily.
b. The antiinflammatory effects of steroids may take hours to develop. Therefore, serious asthmatic attacks should be managed with a short-acting bronchodilator to prevent acute respiratory failure.
3. All corticosteroids used for management of asthma are glucocorticoids.
1. Glucocorticoids are lipid-soluble drugs that exert effects on intracellular receptors (Figure 17-8).
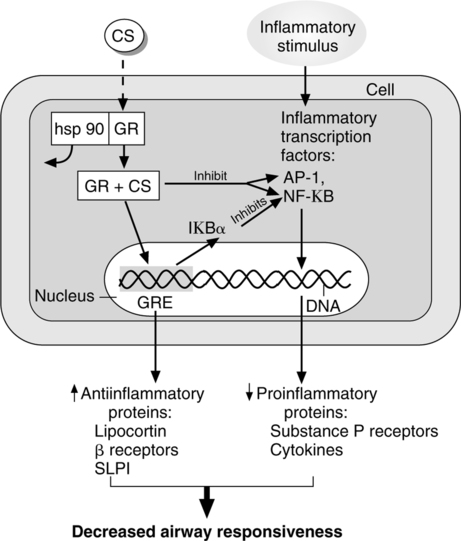
2. Full antiinflammatory effects do not occur for hours to days after beginning treatment, making them unsuitable for use as a rescue drug.
C Specific inhaled corticosteroids: New synthetic hydrocortisone analogues have high antiinflammatory action with few systemic side effects.
a. The first corticosteroid developed for inhalation.
b. Its high level of systemic absorption rendered it obsolete once other inhaled corticosteroids were developed.
2. Beclomethasone dipropionate (Vanceril, Beclovent, QVAR)
a. The second inhaled corticosteroid available in the United States, after the Decadron (dexamethasone) Respihaler, which is no longer available.
b. A medium potency inhaled corticosteroid that is rapidly metabolized in the GI tract, preventing its use in an oral form.
c. Available in MDI formulation as Vanceril and Beclovent.
d. Reformulated with a hydrofluoroalkane propellant MDI in 40-μg and 80-μg strength as QVAR.
e. Available for nasal inhalation as Vancenase and Beconase to manage inflammation of the nasopharynx and sinuses.
3. Triamcinolone acetonide (Azmacort)
a. Low systemic absorption because of its nonpolar and water-insoluble nature.
b. Less topically active than beclomethasone.
c. Its low potency and short duration of action require high initial doses (12 to 16 inhalations/day) for those with severe asthma.
a. Also a low potency agent, similar in potency to triamcinolone, but with a longer duration of action.
b. Reaches peak plasma level between 2 and 60 minutes after inhalation.
c. The plasma half-life is approximately 1.8 hours.
d. Poor taste complicates its regular use in pediatric patients.
a. A high potency agent with a potency greater than beclomethasone, triamcinolone, or flunisolide but less potent than fluticasone.
b. Delivered by Pulmicort Turbuhaler, a breath-activated MDI.
c. Peak plasma concentrations occur between 15 and 45 minutes after inhalation.
6. Fluticasone propionate (Flovent, Flonase)
a. The highest potency inhaled glucocorticoid agent, available in MDI form.
7. Fluticasone propionate/salmeterol (Advair)
a. A combination of fluticasone and salmeterol, a long-acting β2 agonist.
b. Glucocorticoids and β-adrenergic agonists have beneficial complementary interactions.
(1) Steroids increase upregulation of β receptors.
(2) Development of tolerance to β-adrenergic agonists is suppressed by steroid use.
(3) Salmeterol initiates an antiinflammatory effect in vascular cells, even in the absence of glucocorticoids, by promoting binding of the glucocorticoid receptor to the response element of the cell’s DNA.
XII Sympathomimetics Used for Their Effects on the Cardiovascular System
1. Trade name: Levophed, Noradrenalin
2. Generic name: Norepinephrine
1. Trade name: Intropin, Dopastat
a. A precursor of norepinephrine; stimulates dopaminergic, β1-adrenergic, and α-adrenergic receptors in a dose-dependent fashion.
a. Low dosages (1 to 2 μg/kg/min)
(1) Stimulates dopaminergic receptors, increasing cerebral, renal, and mesenteric vasodilation.
(2) Increases venous tone because of β-adrenergic stimulation.
(3) Urine output may increase, but heart rate and blood pressure are usually unchanged.
(4) May cause a mild increase in cardiac output because of vasodilation.
b. Moderate dosages (2 to 10 μg/kg/min)
(1) Stimulates β1-and α-adrenergic receptors.
(2) β1-adrenergic stimulation increases cardiac output, partially antagonizing α-adrenergic-mediated vasoconstriction.
(3) Substantial increases in venous tone and central venous pressures occur at doses >2.5 μg/kg/min.
(4) Resultant effect is enhanced cardiac output with a modest increase in systemic vascular resistance.
c. High dosages: Systemic vasoconstriction
(1) α-Adrenergic effects predominate at doses >10 μg/kg/min, resulting in renal, mesenteric, and peripheral arterial and venous vasoconstriction.
(2) Hemodynamic effects similar to norepinephrine occur at doses >20 μg/kg/min.
d. Dopamine increases myocardial work without compensatory increases in coronary blood flow, leading to an imbalance between oxygen supply and demand that may create myocardial ischemia.
6. Administration: IV only in appropriate dilution of nonalkaline solutions.
C Dobutamine: A synthetic sympathomimetic catecholamine.
3. Action: Potent inotrope; stimulates β1– and α-adrenergic myocardial receptors.
a. Mild vasodilator because of dual effects of stimulating α-adrenergic peripheral receptors but antagonistically stimulating more potent β2-adrenergic receptors.
b. Increases cardiac output and decreases peripheral vascular resistance.
(1) Increases renal and mesenteric blood flow by increasing cardiac output.
(2) At conventional doses (2 to 20 μg/kg/min), less apt to induce tachycardia than Isuprel or dopamine.
(3) Net hemodynamic effects are similar to dopamine combined with a vasodilator such as nitroprusside.
c. Does not increase myocardial oxygen demand by inducing norepinephrine release, as occurs with dopamine.
XIII Noncatecholamines Affecting the Sympathetic Nervous System
3. Mechanism of action: Causes release of epinephrine and norepinephrine stored throughout the body.
B Methylxanthines (primarily theophylline)
3. Role: Traditionally used for treatment of patients with asthma and COPD; its current clinical role has been reduced to second- or third-line agents.
(1) Methylxanthines are not indicated for acute exacerbations of asthma. Theophylline has a relatively weak bronchodilating effect when compared with β agonists.
(2) Sustained-release theophylline has been used as a long-term controller drug for management of asthma.
(3) Low-dose inhaled corticosteroids or cromolyn-like agents are preferred to theophylline for management of stable asthma.
(4) Antileukotrienes are more therapeutic and have fewer side effects than theophylline.
4. Mechanism of action unknown
a. Formerly thought that theophylline inhibited the enzyme phosphodiesterase, which led to increased intracellular cyclic adenosine monophosphate (cAMP), that causes relaxation of bronchial smooth muscle.
b. At clinical dosage levels in humans, theophylline is a poor inhibitor of cAMP-specific phosphodiesterase, invalidating the aforementioned explanation.
c. Theophylline is a weak and nonselective inhibitor of cAMP-specific phosphodiesterase.
d. Currently there is no definitive explanation for the bronchodilatory effects of theophylline.
a. It is difficult to determine therapeutic doses of theophylline because people metabolize theophylline at different rates.
b. The recommended therapeutic range for management of asthma is 5 to 15 μg/ml.
c. For COPD, the American Thoracic Society (2001) recommends a therapeutic range of 10 to 12 μg/ml.
8. Side effects (normally noted if therapeutic level is exceeded)
XIV Parasympathomimetics (Cholinergic Agents)
A Action: Enhance effects of parasympathetic nervous system (Figure 17-9).
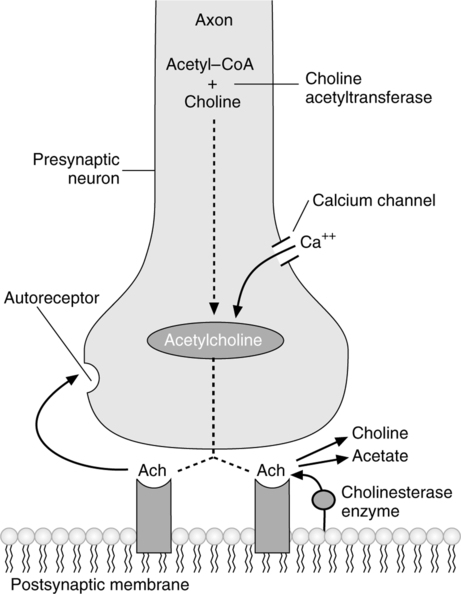
1. In the parasympathetic nervous system, the neurotransmitter acetylcholine conducts nerve impulses across synapses.
a. A nerve impulse (action potential) arriving at the presynaptic neuron site causes the release of calcium.
b. Calcium affects the release of acetylcholine at the end of the nerve fiber.
c. Acetylcholine attaches to receptors on the postsynaptic membrane, creating an effect at the tissue or organ site.
2. Acetylcholinesterase in the synapse enzymatically cleaves acetylcholine, terminating the effect at the receptor site.
1. Muscarinic: Affects parasympathetic postganglionic fibers, thus stimulating only the parasympathetic nervous system.
2. Nicotinic: Affects acetylcholine receptors at autonomic ganglia and skeletal neuromuscular junctions where acetylcholine is the transmitter substance, thus stimulating sites outside the parasympathetic nervous system.
1. Direct acting: Mimic acetylcholine, thereby binding to muscarinic or nicotinic receptors directly.
1. Physostigmine: Stimulates muscarinic receptors in the iris and ciliary muscle of the eye, causing constriction of the pupil and thickening of the lens.
3. Neostigmine increases acetylcholine at the neuromuscular junction, reversing neuromuscular blockade caused by paralytics such as pancuronium.
a. Short acting (5 to 15 minutes), thereby useful as a diagnostic agent.
(1) Inhibition of cholinesterase creates an excess of acetylcholine, causing receptor fatigue, leading to muscle weakness.
(2) If Tensilon is given and muscle strength is improved, insufficient cholinergic agents are present.
(3) If Tensilon is given and muscle strength is not improved, excess anticholinergic agents may be present.
E Side effects associated with excessive stimulation of parasympathetic nervous system
XV Parasympatholytics (Anticholinergic Agents)
A Action: Inhibition of parasympathetic nervous system.
1. Blocks acetylcholine receptors (cholinergic antagonists) by competitive inhibition of acetylcholine at muscarinic sites only.
2. Parasympatholytic agents are solely antimuscarinic because of being limited to parasympathetic terminal fiber sites.
3. Ophthalmologic (pupil dilation)
6. Genitourinary tract disorders
8. Over-the-counter sleeping pills
9. Motion sickness, nausea, and vomiting
11. Used with parasympathomimetics for reversal of neuromuscular-blocking agents
E Representative parasympatholytic drugs
1. Atropine, Atropa belladonna
a. A competitive antagonist of acetylcholine at muscarinic receptor sites.
b. Nonspecific for muscarinic receptor subtypes (blocks M1, M2, and M3).
c. Structurally a tertiary ammonium compound that carries no electrical charge, allowing it to be easily absorbed, creating the possibility of many side effects.
d. Blocks parasympathetic component of smooth muscle tone, causing relaxation of bronchial smooth muscle. Since the development of ipratropium, no longer used as a bronchodilator.
e. Blocks vagal component of cardiac innervation, increasing heart rate.
f. Decreases mucous gland secretions, creating “dry mouth” because salivary gland secretions are reduced.
g. Decreases smooth muscle tone, mobility, and acid secretion within the GI tract.
h. Reduces smooth muscle tone of the bladder, causing urinary retention.
i. Reduces perspiration by blocking acetylcholine receptors of sweat glands.
2. Ipratropium bromide (Atrovent, see Section X, Anticholinergic bronchodilators)
a. A derivative of atropine that is also not well absorbed across cell membranes, leading to fewer side effects than atropine.
b. Used during reversal of neuromuscular blockade as an antimuscarinic agent with few CNS side effects.
c. Not approved for inhalation but has been used IV as a less expensive alternative to Atrovent.
d. Onset of action: 15 to 30 minutes
4. Scopolamine hydrobromide (Hyoscine)
a. A tertiary ammonium compound similar in structure to atropine.
b. Can produce drowsiness or amnesia.
c. Can reduce motion sickness through its antimuscarinic effects on the vestibular system of the ear.
XVI Sympatholytics (Adrenergic-Blocking Agents)
A Action: Inhibition of the sympathetic nervous system.
B α-Receptor subtypes: α receptors are divided into α1 and α2 subtypes based on location and variation in pharmacologic response.
1. α-Receptor antagonists may produce opposite effects based on whether they affect α1– or α2-receptor subtypes.
a. Prazosin prevents vasoconstriction (decreases blood pressure) via blockade of α1-excitatory receptors.
b. Yohimbine prevents vasodilation (increases blood pressure) via blockade of α2-inhibitory receptors.
D β-Adrenergic blocking agents
1. Mechanism of action: Competitive inhibition at β1– and β2-adrenergic receptor sites.
(1) Blockade of myocardial β receptors decreases rate and contractility, reducing cardiac output.
(2) Blocks β receptors on renal juxtaglomerular cells, inhibiting renin production, resulting in decreased angiotensin II concentration.
(3) Blockade of CNS β receptors decreases sympathetic tone.
(4) Decreases norepinephrine production via blockade of peripheral β receptors.
b. Cardiac arrhythmias: Slows A-V nodal conduction, helping prevent atrial fibrillation, atrial flutter, and paroxysmal supraventricular tachycardia (PSVT).
a. CHF and associated pulmonary edema
(1) Exacerbation of stable asthma or COPD may occur with use of β blockade.
(2) β1-selective drugs (e.g., atenolol and metoprolol) have fewer pulmonary side effects at low doses but are less β1 specific at higher doses.
d. Contraindicated in patients with bradycardia or significant A-V block.
a. Propranolol (Inderal): Nonselective blockade of β1 and β2 receptors, thus potentially affecting cardiac and pulmonary systems.
b. Metoprolol (Lopressor): β1 selective at low doses.
c. Atenolol (Tenormin): β1 selective at low doses.
d. Timolol (Bleocardin, Timolide)
f. Pindolol (Visken): Possesses intrinsic sympathomimetic activity, decreasing the intensity of its β blockade.
E α- and β-adrenergic blockers: labetalol (Normodyne, Trandate) and carvedilol (Coreg)
XVII Advanced Cardiac Life Support Medications
1. Amiodarone: A complex drug with effects on sodium, potassium, and calcium channels and has α- and β-adrenergic blocking effects.
a. Has broad atrial and ventricular antiarrhythmic effects and lesser negative inotropic effects.
b. Alters accessory conductive pathways, thus effective in controlling rapid ventricular rate in supraventricular tachycardia (SVT).
(1) 150 mg over 10 minutes, followed by infusion of 1 mg/min for 6 hours, then 0.5 mg/min continuous infusion.
(2) Recurrent arrhythmias treated with a repeated 150-mg infusion up to a maximum daily dose of 2 g.
(3) In pulseless ventricular tachycardia or ventricular fibrillation, give a 300-mg rapid infusion diluted with 20 to 30 ml saline.
a. A primary agent for management of wide complex tachycardia. Suppresses ventricular arrhythmias associated with acute myocardial ischemia and infarction (AMI).
(1) Its antiventricular arrhythmic use in AMI reduces the incidence of ventricular fibrillation but does not decrease mortality rate.
(2) Routine prophylactic use in patients with AMI is not recommended.
b. Appropriate as a second-line agent for controlling hemodynamically stable ventricular tachycardia.
c. Useful for controlling hemodynamically compromising premature ventricular contractions.
d. Not indicated for management of supraventricular tachycardia.
a. Effective for management of supraventricular arrhythmias and ventricular tachycardia.
b. Alters conduction across an accessory pathway, thus effective in breaking SVT.
c. Has vasodilatory and negative inotropic effects that may lead to hypotension.
4. Adenosine: An endogenous purine nucleoside that depresses sinus and A-V node conduction.
a. Used to manage narrow complex tachycardia, narrow complex SVT, and wide complex tachycardia that is supraventricular in origin.
b. Produces a short-lived pharmacologic conductive block at the A-V node.
c. Initial dose: 6 mg bolus over 1 to 3 seconds followed by 20-ml saline flush.
d. A repeat dose of 12 mg may be given twice if no response is obtained.
5. Atropine (also see section XVI, E, Anticholinergic Agents)
a. Reverses cholinergic-mediated bradycardia, decreased systemic vascular resistance, and decreased blood pressure.
(1) For asystole and pulseless electrical activity: 1.0 mg IV, repeated in 3 to 5 minutes.
(2) For bradycardia: 0.5 to 1.0 g IV every 3 to 5 minutes to a maximum of 0.04 mg/kg.
c. Well absorbed through the tracheal route of administration.
d. Should be used carefully for AMI; excessive increases in heart rate may worsen ischemia or increase a zone of infarction.
e. Contraindicated in type II A-V block and third-degree block with new wide QRS complexes.
6. β-Adrenergic blockers (also see Section XVII, D, Sympatholytics)
a. Indicated for all patients with AMI and high-risk unstable angina.
b. Atenolol, metoprolol, and propranolol significantly reduce ventricular fibrillation in post-MI patients who did not receive fibrinolytic agents.
c. Esmolol: A short-acting (half-life, 2 to 9 minutes) β1-selective β blocker used for management of SVTs.
d. Side effects of β blockade: Bradycardia, A-V conduction delay, and hypotension.
e. Contraindicated in second- or third-degree heart block, severe CHF, and lung disease associated with severe bronchospasm.
7. Verapamil and diltiazem: Calcium channel blockers
a. Action: Slows conduction and increases refractoriness within the A-V node by blocking calcium channels.
(1) Can terminate A-V nodal reentry arrhythmias.
(2) Can control ventricular response rate in patients with atrial fibrillation, atrial flutter, or multifocal atrial tachycardia (MAT).
(3) May decrease myocardial contractility, thus may worsen CHF in patients with severe left ventricular dysfunction.
b. Verapamil initial dose: 2.5 to 5 mg IV given over 2 minutes. Repeated doses of 5 to 10 mg may be given every 15 to 30 minutes. Maximum cumulative dose: 20 mg.
(1) Indicated only for narrow complex PSVT or supraventricular arrhythmias.
(2) Contraindicated in patients with impaired left ventricular function or CHF.
c. Diltiazem initial dose: 0.25 mg/kg, followed by a second dose of 0.35 mg/kg.
8. Dopamine (also see Section XII, B, Sympathomimetics utilized for their effects on the cardiovascular system.)
a. An endogenous catecholamine with dopaminergic, α- and β-adrenergic agonist activity.
b. Currently the preferred catecholamine for bradycardia in which atropine is contraindicated or ineffective.
a. A deficiency of magnesium can be associated with ventricular arrhythmias.
b. Considered the hallmark therapy for ventricular dysrhythmia, even in the absence of magnesium deficiency.
c. Dose: 1 to 2 g magnesium sulfate, diluted in 100 ml D5W, given over 1 to 2 minutes.
a. A nonselective β blocker approved for ventricular and supraventricular arrhythmias.
b. Prolongs action potential and increases refractoriness of cardiac tissue.
c. Dose: Given IV, 1 to 1.5 mg/kg at a rate of 10 mg/min.
d. Its slow infusion rate renders it impractical during cardiopulmonary resuscitation (CPR) or for emergent hypotensive crises.
B Agents for optimizing cardiac output and blood pressure
1. Vasopressin: The endogenous antidiuretic hormone, acts as a nonadrenergic peripheral vasoconstrictor when given in unnaturally high doses.
a. Directly stimulates smooth muscle vasopressin V1 receptors.
b. Does not vasodilate skeletal muscle vessels or increase myocardial oxygen consumption during CPR because it has no β-adrenergic activity.
(1) During CPR, vasopressin causes peripheral vasoconstriction in skeletal muscle, skin, intestine, and fat, with less constriction of coronary and renal vasculature, but vasodilates the cerebral vasculature.
(2) Repeated doses of vasopressin are more effective than epinephrine to maintain coronary perfusion pressure.
c. Half-life in animal models is 10 to 20 minutes (much longer than epinephrine).
d. Used as a continuous infusion to increase vascular tone and support hemodynamics in septic shock and sepsis syndrome (systemic inflammatory response syndrome).
2. Epinephrine: An endogenous catecholamine synthesized in the adrenal medulla.
a. Has potent α and β effects.
b. Increases myocardial oxygen demand but may not increase perfusion enough to prevent myocardial hypoxia.
c. Traditionally has been the first-line pressor and cardiac stimulant in CPR but is currently being supplanted by vasopressin.
3. Norepinephrine (see Section XII, A, Sympathomimetics utilized for their effects on the cardiovascular system)
4. Dopamine (see Section XII, B, Sympathomimetics utilized for their effects on the cardiovascular system)
5. Dobutamine (see Section XII, C, Sympathomimetics utilized for their effects on the cardiovascular system)
6. Amrinone and milrinone (phosphodiesterase inhibitors)
a. Positive inotropic agents that selectively inhibit cAMP phosphodiesterase, the enzyme that hydrolyzes cAMP.
(1) Elevated cAMP increases intracellular calcium, enhancing myocardial contractility.
(2) Elevated cAMP causes vasodilation of arterial smooth muscle, which may result in hypotension.
(3) Phosphodiesterase inhibitors are an alternative to dobutamine-induced tachycardia.
b. Have a more significant effect on preload than catecholamines.
c. Hemodynamic effects are similar to dobutamine.
d. Indicated in severe heart failure or cardiogenic shock that is unresponsive to standard treatment.
e. Amrinone may worsen myocardial ischemia or increase ventricular ectopy.
a. Important in myocardial contractility and impulse generation.
b. In cardiac arrest administration of calcium has demonstrated no benefit.
c. If hyperkalemia, hypocalcemia, or calcium channel blocker toxicity exists, administration of calcium may be helpful.
a. Slows atrioventricular node conduction, thereby decreases the ventricular rate in atrial fibrillation or flutter.
b. Effectively controls ventricular response rate in patients with chronic atrial fibrillation.
(1) Less effective in patients with paroxysmal atrial fibrillation.
(2) Not effective for rate control in CHF, hyperthyroidism, or during exercise (high adrenergic states).
c. Calcium channel blockers or β-adrenergic blockers should be used for initial ventricular rate control in atrial fibrillation.
d. Digitalis toxic-to-therapeutic ratio is narrow, particularly during hypokalemia.
e. Digitalis toxicity can cause cardiac arrest or ventricular arrhythmias.
a. A potent venodilator, vasodilates coronary vessels and collaterals, decreasing end-diastolic pressure and myocardial oxygen demand.
b. Indicated in AMI, angina, hypertension, and acute heart failure.
c. Nitroglycerin effects depend on intravascular volume status.
(1) In hypovolemic states, vasodilation increases the risk of hypotension.
(2) Further hypotension may reduce coronary blood flow and increase myocardial ischemia.
d. Delivered by sublingual tablets for relief of acute angina.
(1) Each nitroglycerin tablet contains 0.3 to 0.4 mg.
(2) Provides immediate onset of action with duration of action approximately 30 minutes.
(3) Tablets should be repeated every 5 minutes until angina is relieved.
(4) If symptoms are not relieved after a maximum of three doses within 15 minutes, emergency care should be summoned.
e. Adverse reactions may include palpitations, hypotension, dizziness, tachycardia, and headache.
a. A potent peripheral vasodilator for management of severe heart failure and acute hypertensive crises.
b. Venodilation reduces preload, potentially relieving pulmonary congestion and reducing left ventricular pressure and volume.
c. Higher doses cause arterial dilation, decreasing arterial resistance to flow (afterload).
(1) Decreased afterload increases systolic ejection, reducing left ventricular distention.
(2) Myocardial oxygen consumption is reduced by more efficient myocardial contractility.
d. Nitroprusside affects the pulmonary system by dilating pulmonary arteries, reversing hypoxic pulmonary vasoconstriction, thereby increasing intrapulmonary shunt, resulting in decreased Pao2.
A Adrenocorticotropic hormone (ACTH) and adrenocorticosteroids
1. ACTH (corticotropin) is produced and released from the adenohypophysis (anterior pituitary).
2. Physiology of steroid regulation
a. Release of ACTH into the blood causes the adrenal cortex to release steroids into the bloodstream.
b. Release of ACTH is affected directly by corticotropin-releasing factor (CRF), which is secreted by the hypothalamus.
c. Blood level of CRF is indirectly affected by blood steroid levels.
d. Thus there is a cyclic relation between ACTH, CRF, and steroid levels.
e. An increase in steroid level causes a decrease in CRF levels, which causes a decrease in ACTH levels. This results in a decrease in steroid levels, which causes an increase in CRF levels, and so forth, thus maintaining a normal equilibrium.
a. Stimulate glucose formation (i.e., increase blood glucose levels).
b. Decrease glucose utilization.
c. Promote storage of glucose in liver.
d. Regulate rate of synthesis of proteins.
e. Control distribution of body fat.
g. Regulate reabsorption of sodium ions from kidney tubules.
h. Increase urinary excretion of potassium and hydrogen.
i. Maintain normal function of skeletal muscles.
j. Increase hemoglobin and red blood cell content of blood.
k. Maintain normal lymphoid tissue.
l. Prevent or suppress inflammatory responses caused by hypersensitivity.
a. Modification of cell function protein production and direct stimulation of tissue receptor sites to inhibit the complex mechanisms of inflammation.
b. Allergic disorders: Has a catabolic effect on lymphoid tissue.
c. Edema: Decreases capillary permeability by an unknown mechanism.
b. Acute and chronic adrenal insufficiency
c. Suppression of immune response in organ transplant patients
d. Congenital adrenal hyperplasia
e. Adrenal insufficiency secondary to anterior pituitary insufficiency
7. Representative aerosolized drugs (see Section XI, Inhaled Corticosteroids)
XIX Neuromuscular-Blocking Agents
A Major action: Interruption of transmission of nerve impulse at skeletal neuromuscular junction, resulting in paralysis.
a. Mechanism of action: Competitive inhibition of acetylcholine at muscle postsynaptic receptor sites. The muscle tissue itself remains sensitive to external stimulation.
(1) Nondepolarizing agents act by competitive inhibition, thus their effects are dose related.
(2) Maximal effects are attained in 2 to 10 minutes and persist for 30 to 60 minutes.
(3) Hypotension as a result of histamine release is seen occasionally with d-tubocurarine.
(4) Bradycardia occasionally is seen with pancuronium.
(5) Cholinesterase-inhibiting agents (e.g., neostigmine) may reverse effects of this group of drugs.
| Representative drugs | Duration (minutes) |
| (1) Tubocurarine (d-tubocurarine) | >35 |
| (2) Pancuronium (Pavulon) | >35 |
| (3) Metocurine (Metubine) | >35 |
| (4) Atracurium (Tracrium) | 20-35 |
| (5) Vecuronium (Norcuron) | 20-35 |
| (6) cis-Atracurium | 20-35 |
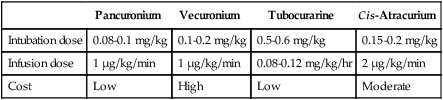
d. Neuromuscular-blocking agents commonly used in the intensive care unit (ICU) (Table 17-8)
TABLE 17-8
Neuromuscular Blocking Agents Commonly Used in the Intensive Care Unit
| Pancuronium | Vecuronium | Tubocurarine | Cis-Atracurium | |
| Intubation dose | 0.08-0.1 mg/kg | 0.1-0.2 mg/kg | 0.5-0.6 mg/kg | 0.15-0.2 mg/kg |
| Infusion dose | 1 μg/kg/min | 1 μg/kg/min | 0.08-0.12 mg/kg/hr | 2 μg/kg/min |
| Cost | Low | High | Low | Moderate |
(1) They cause persistent depolarization of the postjunctional membrane by competitively binding at acetylcholine receptor sites, rendering them incapable of repolarization.
(2) As this wave of depolarization proceeds, a rippling of voluntary muscles occurs (muscle fasciculation), usually from the head downward. Because these agents are chemically similar to acetylcholine, they cause continual stimulation of the motor endplate.
(3) This continual stimulation does not allow time for repolarization; therefore, the muscle develops a flaccid paralysis.
(4) There are no drugs known that reverse depolarizing agents. With time depolarizing agents are metabolized by acetylcholinesterase.
b. The muscle fiber itself is still sensitive to external stimulation.
| Representative drugs | Duration (minutes) |
| (1) Succinylcholine (Anectine) | 10-15 |
| (2) Decamethonium (Syncurine) | 20-35 |
| (a) Available in Europe | |
| (b) Not metabolized by acetylcholinesterase. | |
| (c) Requires hepatic and renal function for elimination. |
A Primary use (narcotics): Analgesia and relief of severe pain.
B Mechanism of action: Unclear, but these drugs affect neurotransmission at specific CNS sites, affect autonomic nervous system transmission, and cause some histamine release.
C General pharmacologic effects
a. Alters the perception of pain.
b. Interferes with the continuance of pain impulses in the spinal cord.
2. Euphoric: Seen at therapeutic dosages.
3. Hypnotic: With increasing dosages, more subjective CNS depression.
4. Metabolic: Transient hyperglycemia.
5. Endocrine: Stimulates the release of antidiuretic hormone.
7. GI tract: Constipation because of decreased overall activity.
8. Nausea and vomiting: Direct stimulation of medullary control center.
a. If patient is well oxygenated and in normal acid-base balance, there are no significant effects.
b. Cardiac arrhythmias may be seen with acid-base abnormalities.
c. Hypotension may result because of direct effect on venous smooth muscle and release of histamine.
a. Direct depression of medullary respiratory center response to CO2 changes.
b. Significant decrease in respiratory rate, tidal volume, and minute volume is seen with large dosages.
11. Cough reflex: Decreased as a result of direct depression of medullary cough center.
F Nonnarcotic analgesics and antiinflammatory agents: All have analgesic properties and are normally used for mild or chronic pain.
5. Nonsteroidal antiinflammatory drugs (NSAIDs)
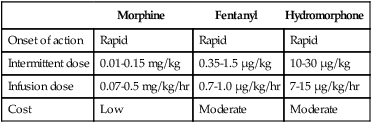
6. Narcotics commonly used in the ICU (Table 17-9)
TABLE 17-9
Narcotics Commonly Used in the Intensive Care Unit
| Morphine | Fentanyl | Hydromorphone | |
| Onset of action | Rapid | Rapid | Rapid |
| Intermittent dose | 0.01-0.15 mg/kg | 0.35-1.5 μg/kg | 10-30 μg/kg |
| Infusion dose | 0.07-0.5 mg/kg/hr | 0.7-1.0 μg/kg/hr | 7-15 μg/kg/hr |
| Cost | Low | Moderate | Moderate |
A Sole use: To reverse effects of narcotics.
B Mechanism of action: Competitive displacement of agonist from receptor site.
1. Cause narcotic-like effects in absence of a narcotic.
2. Cause increased respiratory depression if administered in a nonnarcotic overdose (e.g., barbiturates).
D Pure antagonists: Have only narcotic antagonist properties.
A Solid or liquid substances that cause a longer generalized depression of the CNS than do anesthetic gases.
B Mechanism of action: Selective depression of ascending reticular activating system at either the cellular or synaptic level, resulting in loss of consciousness.
1. Behavioral changes caused by increased dosages
a. Sedation: Generalized decreased responsiveness
b. Disinhibition: Impaired judgment and loss of self-control
2. Electroencephalographic pattern changes consistent with generalized CNS depression
4. Anticonvulsant: Phenobarbital the most effective
5. Withdrawal state with repeated long-term use and abrupt discontinuance
7. Voluntary muscle relaxation from spinal cord depression
8. Depression of respiratory medullary center with larger doses
9. Profound vasomotor depression and shock with larger doses
a. Ultra-short-acting: Anesthetic agents
b. Short-acting: Primarily for sleep induction
c. Intermediate-acting: Relief of anxiety
d. Long-acting: Anticonvulsant
2. Nonbarbiturate sedatives: Hypnotics
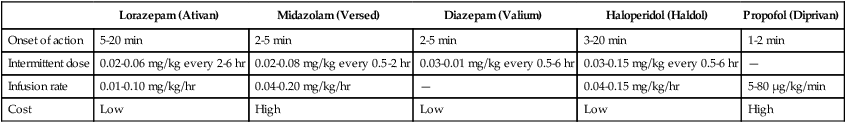
4. Agents commonly used in the ICU (Table 17-10)
TABLE 17-10
Sedatives Commonly Used in the Intensive Care Unit
| Lorazepam (Ativan) | Midazolam (Versed) | Diazepam (Valium) | Haloperidol (Haldol) | Propofol (Diprivan) | |
| Onset of action | 5-20 min | 2-5 min | 2-5 min | 3-20 min | 1-2 min |
| Intermittent dose | 0.02-0.06 mg/kg every 2-6 hr | 0.02-0.08 mg/kg every 0.5-2 hr | 0.03-0.01 mg/kg every 0.5-6 hr | 0.03-0.15 mg/kg every 0.5-6 hr | — |
| Infusion rate | 0.01-0.10 mg/kg/hr | 0.04-0.20 mg/kg/hr | — | 0.04-0.15 mg/kg/hr | 5-80 μg/kg/min |
| Cost | Low | High | Low | Low | High |
XXIII Diuretics and Antihypertensive Agents
A Functional renal unit: The nephron (Figure 17-10)
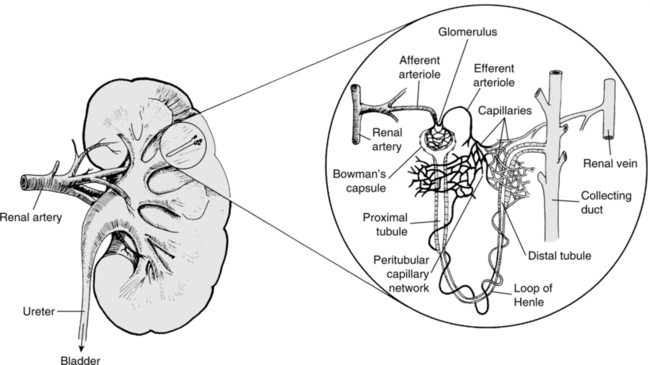
1. Approximately 20% of blood entering the glomerulus is filtered, forming a cell-free ultrafiltrate within the glomerulus.
2. Flowing through the nephrons, >99% of the ultrafiltrate is reabsorbed.
3. Diuretic drugs alter the reabsorption of water within the nephron system.
B Electrolyte balance within the nephron
a. Nearly all ultrafiltrate potassium is reabsorbed in the proximal tubules.
b. Small amounts of potassium are secreted by the distal tubule.
3. Chloride and bicarbonate are reabsorbed passively in proximal and distal tubules.
4. Water reabsorption occurs passively, following osmotic gradients created by electrolytes, primarily sodium. Drugs that decrease sodium reabsorption decrease reabsorption of water.
C Classes of diuretics (Figure 17-11)
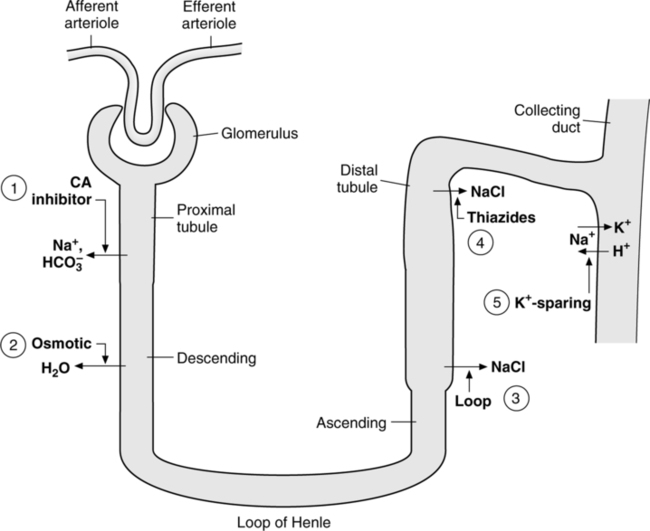
1. Osmotic diuretics: Interfere with water reabsorption in the descending loop of Henle and in the proximal tubule.
a. Once filtered into the nephron, osmotic diuretics stay within the tubule lumen and prevent the reabsorption of sodium.
b. Increased sodium and (because of preservation of electrical neutrality within the tubule) chloride delivered to the distal tubule promote an exchange of sodium for potassium, increasing potassium elimination.
c. Mannitol: The preferred osmotic diuretic because of its low toxicity and short half-life.
2. Carbonic anhydrase inhibitors: Prevent reabsorption of sodium and bicarbonate ions in the proximal tubule.
a. Carbonic anhydrase enzymatically hydrates bicarbonate, which can then be reabsorbed.
b. Sodium is reabsorbed with bicarbonate to preserve electrical neutrality.
c. Carbonic anhydrase inhibitors thus increase the excretion of sodium and bicarbonate, resulting in a metabolic acidemia.
d. Because carbonic anhydrase inhibitors act only on the proximal tubule, sodium can be reabsorbed in the ascending limb of the loop of Henle and in the distal tubule. Thus carbonic anhydrase inhibitors are weak diuretics.
3. Loop diuretics: Inhibit absorption of chloride ions in the ascending loop of Henle (sodium and water passively follow the chloride).
a. Loop diuretics produce an acute vasodilatory effect that may be caused by the renal release of vasodilating prostaglandins.
a. Block sodium and chloride ion reabsorption in the distal tubule.
b. Because only 5% of sodium is reabsorbed in the distal tubule, thiazide diuretics are much less potent than loop diuretics.
5. Potassium-sparing diuretics block sodium reabsorption in the distal tubule and collecting duct.
a. Normally 80% of sodium is absorbed in the proximal tubule. In the distal convoluted tubule, most of the remaining 20% of sodium in the ultrafiltrate is reabsorbed by exchange of Na+ for K+ ions.
b. Diuretics that increase the concentration of sodium at the distal convoluted tubule promote increased exchange of Na+ for K+, which may create hypokalemia.
a. Volume depletion because of diuretic use may result in hypokalemia and hypochloremia. As K+ and Cl− are excreted, HCO3− is retained, creating a metabolic alkalosis.
b. Repletion of K+ or Cl− corrects the alkalosis by allowing excretion of HCO3−.
3. Hyperglycemia has been associated with the use of loop and thiazide diuretics. The mechanism is not clear.