CHAPTER 11 Pharmacology
Anaesthetic Gases
Oxygen
Discovered by Joseph Priestley in 1777. Manufactured by:
Nitrous oxide
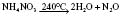
Entonox
Analgesia is due to release of endogenous opioids and direct effect at opioid receptors.
Side-effects
Banks A., Hardman J.G. Nitrous oxide. Contin Educ Anaesth, Crit Care Pain. 2005;5:145-148.
Harris P.D., Barnes R. The uses of helium and xenon in current clinical practice. Anaesthesia. 2008;63:284-293.
Ma D., Wilhelm S., Maze M., et al. Neuroprotective and neurotoxic properties of the ‘inert’ gas, xenon. Br J Anaesth. 2002;89:739-746.
Maze M., Fujinaga M. Recent advances in understanding the actions and toxicity of nitrous oxide. Anaesthesia. 2000;55:311-314.
O’Driscoll B.R., Howard L.S., Davison A.G. British Thoracic Society: British Thoracic Society guideline for emergency oxygen use in adult patients. Thorax. 2008;63(Suppl VI):vi1-vi68. on behalf of the
Sanders R.D., Franks N.P., Maze M. Xenon: no stranger to anaesthesia. Br J Anaesth. 2003;91:709-717.
Sanders R.D., Weimann J., Maze M. Biologic effects of nitrous oxide: a mechanistic and toxicologic review. Anesthesiology. 2009;109:707-722.
Taneja R., Vaughan R.S. Oxygen. BJA CEPD Rev. 2001;1:104-107.
Drug Interactions
Monoamine oxidase inhibitors (MAOIS)
Antiplatelet agents
Tricyclics
Therefore tricyclics result in an increased risk of arrhythmias and hypotension.
Selective serotonin reuptake inhibitors (fluoxetine (Prozac), sertraline, paroxetine)
Anticonvulsants
H2 antagonists
Ranitidine. Causes sinus bradycardia and AV block, especially following i.v. administration.
Cimetidine. Inhibits hepatic cytochrome P450, increasing levels and thus toxicity of lidocaine, nifedipine and propanolol (Table 11.1). Potentiation of action of warfarin and theophyllines. Cimetidine competes with creatinine for renal excretion.
| Hepatic enzyme induction | Hepatic enzyme inhibition |
|---|---|
| Alcohol | Cimetidine |
| Barbiturates | Erythromycin |
| Phenytoin | Ciprofloxacin |
| Carbamazepine | |
| Sodium valproate |
Fee J.P.H., McCaughey W. Preoperative preparation, premedication and concurrent drug therapy. In: Nimmo W.S., Rowbotham D.J., Smith G., editors. Anaesthesia. ed 2. Oxford: Blackwell Scientific; 1994:677-703.
Kam P.C.A., Chang G.W.M. Selective serotonin reuptake inhibitors. Anaesthesia. 1997;52:982-988.
Licker M., Morel D.R. Inhibitors of the renin angiotensin system: implications for the anaesthesiologist. Curr Opin Anaesth. 1998;11:321-326.
Vuyk J. Drug interactions in anaesthesia. Curr Opin Anaesth. 1997;10:267-270.
Ecstasy
Main problems in the management of these patients are:
Intravenous Induction Agents
Propofol (di-isopropylphenol)
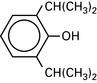
Emulsified in soya bean oil and egg phosphatide (formerly Cremphor EL).
t½ = 4 h. Induction dose 2–3 mg.kg−1. Highly lipid-soluble.
Metabolized by liver and extrahepatic sites (?lung). Hepatic excretion.
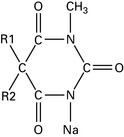
Etomidate
Carboxylated imidazole; pH 3.3. Dissolved in propylene glycol.
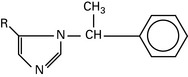
Midazolam
t½ = 2.5 h; 94% protein bound. Hydroxylated by the liver to active metabolites. Renal excretion.
Craven R. Ketamine. Anaesthesia. 2007;6(Suppl 1):48-53.
Jabre P., Combes X., Lapostolle F., et al. Etomidate versus ketamine for rapid sequence intubation in acutely ill patients: a multicentre randomised controlled trial. Lancet. 2009;374:293-300.
Kam P.C.A., Cardon D. Propofol infusion syndrome. Anaesthesia. 2007;62:690-701.
Mather L.E., Edwards S.R. Chirality in anaesthesia – ropivacaine, ketamine and thiopentone. Curr Opin Anaesth. 1998;11:383-390.
Morris C., McAllister C. Etomidate for emergency anaesthesia; mad, bad and dangerous to know. Anaesthesia. 2005;60:737-740.
Pai A., Heining M. Ketamine. Contin Educ Anaesth, Crit Care Pain. 2007;7:59-63.
Sneyd J.R. Recent advances in intravenous anaesthesia. Br J Anaesth. 2004;93:725-736.
Local Anaesthetics
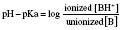
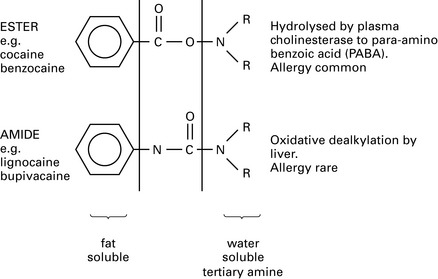
Specific drugs
Ropivacaine
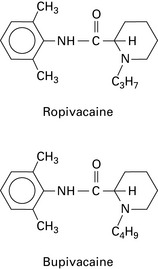
An amide anaesthetic structurally similar to bupivacaine, with similar potency and duration as bupivacaine but less cardiotoxicity (Fig. 11.2). This may be because it is manufactured in the S (–) form, whereas bupivacaine exists in the racemic (RS) form. The sensory block is similar to that provided by bupivacaine but the motor block is slower in onset, less intense and shorter in duration. It is an effective vasoconstrictor (bupivacaine vasodilates) and has no detrimental effect on placental blood flow. Cardiotoxicity and CNS symptoms occur at higher doses compared with bupivacaine.
Maximum recommended doses
Table 11.2 Maximum recommended doses of some common local anaesthetics
| Plain | With adrenaline | |
|---|---|---|
| Lidocaine | 3 mg/kg | 7 mg/kg |
| Bupivacaine | 2 mg/kg | 2 mg/kg |
| Prilocaine | 5 mg/kg | 8 mg/kg |
| Cocaine | 2 mg/kg | |
| Tetracaine | 1.5 mg/kg |
Guidelines for the Management of Severe Local Anaesthetic Toxicity
Association of Anaesthetists of Great Britain and Ireland 2007
Signs of severe toxicity:
Immediate management:
Management of cardiac arrest associated with LA injection
Treatment of cardiac arrest with lipid emulsion: (approximate doses for a 70-kg patient)
Follow-up action:
Association of Anaesthetists of Great Britain and Ireland. Guidelines for the management of severe local anaesthetic toxicity. Reproduced with the kind permission of the Association of anaesthetists of Great Britain and Ireland, 2007.
Heavener J.E. Local anesthetics. Curr Opin Anaesth. 2007;20:336-342.
Leskiw U., Weinberg G.L. Lipid resuscitation for local anesthetic toxicity: is it really lifesaving? Curr Opin Anesth. 2009;22:667-671.
McLeod G.A., Burke D. Levobupivacaine. Anaesthesia. 2001;56:331-341.
Picard J., Harrop-Griffith W. Lipid emulsion to treat drug overdose: past, present and future. Anaesthesia. 2009;64:119-121.
Whiteside J.B., Wildsmith J.A.W. Developments in local anaesthetic drugs. Br J Anaesth. 2001;87:27-35.
Neuromuscular Blockade
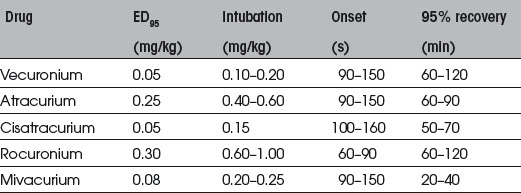
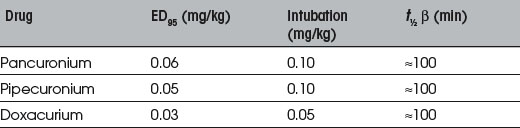
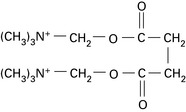
Suxamethonium
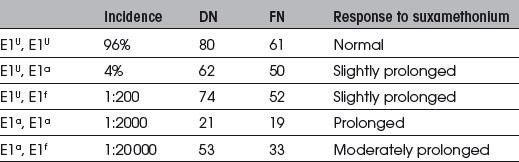
Congenital. Several genes control the structure of plasma cholinesterase (Table 11.5). Normal homozygote genetic structure is E1U, E1U. Common abnormal variants are:
Monitoring
Patterns of nerve stimulation
| Depolarizing block | Non-depolarizing block |
|---|---|
| Reduced twitch height | Reduced twitch height |
| No fade of TO4/tetanus | Fade of TO4/tetanus |
| No post-tetanic potentiation | Post-tetanic potentiation |
Donati F., Bevan D.R. Not all muscles are the same. Editorial,. Br J Anaesth. 1992;68:235-236.
Fuchs-Buder T., Schreiber J.U., Meistelman C. Monitoring neuromuscular block: an update. Anaesthesia. 2009;64(Suppl 1):82-89.
Kopman A.F., Eikermann M. Antagonism of non-depolarising neuromuscular block: current practice. Anaesthesia. 2009;64(Suppl 1):22-30.
Kopman A.F. Sugammadex: A revolutionary approach to neuromuscular antagonism. Anesthesiology. 2006;104:631-633.
Martyn J.A.J., Fagerlund M.J., Eriksson L.I. Basic principles of neuromuscular transmission. Anaesthesia. 2009;64(Suppl 1):1-9.
Martyn J., Durieux M.E. Succinylcholine: new insights into mechanisms of action of an old drug. Anesthesiology. 2006;104:633-634.
McGrath C.D., Hunter J.M. Monitoring of neuromuscular block. Contin Educ Anaesthesia, Crit Care Pain. 2006;6:7-12.
Mirakhur R.K. Sugammadex in clinical practice. Anaesthesia. 2009;64(Suppl 1):45-54.
Mirakhur R.K., Harrop-Griffiths W. Management of neuromuscular block: time for a change? Anaesthesia. 2009;64(Suppl 1):iv-v.
Opioids and Other Analgesics
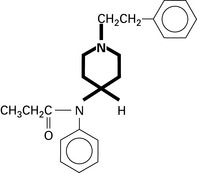
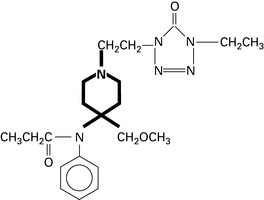
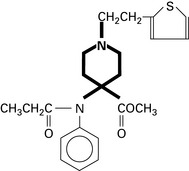
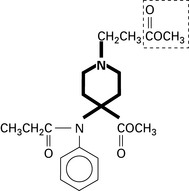
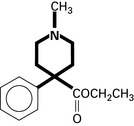
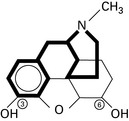
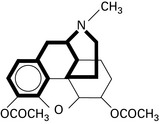
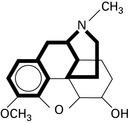
Tramadol
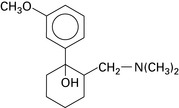
Budd K., Langford R. Tramadol revisited. Br J Anaesth. 1999;82:493-496.
Komatsu R., Turan A.M., Orhan-Sungur M., et al. Remifentanil for general anaesthesia: a systematic review. Anaesthesia. 2007;62:1266-1280.
Sear J.W. Recent advances and developments in the clinical use of i.v. opioids during the preoperative period. Br J Anaesth. 1998;81:38-50.
Volatile Agents
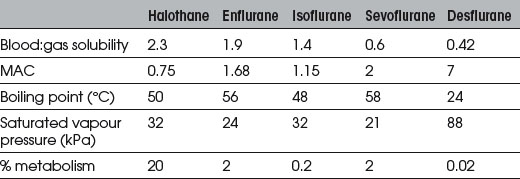
Halothane
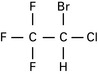
Halothane hepatitis
There are two patterns of hepatitis:
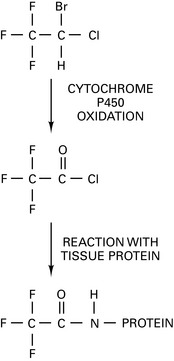
Some 75% of patients with halothane hepatitis have antibodies reacting to halothane-altered antigens. Route of metabolism of halothane depends upon O2 tension in liver. At high O2 tension, an oxidative route generates trifluoroacetyl halide (TFAH) (Fig. 11.3), which covalently binds to liver proteins, forming haptens. Halothane-directed antibodies detectable by ELISA test have been identified that are directed against TFAH antigens. Present in 70% of cases of halothane-induced FHF. The significance of the more minor reductive route at low O2 tension is debated. It may be associated with direct liver damage with release of fluoride. National database of FHF patients set up at St Mary’s Hospital, London (Prof. R.M. Jones), to whom these patients should be reported.
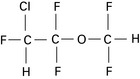
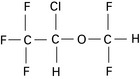
Desflurane
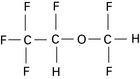
May generate significant amounts of carbon monoxide in the presence of dry baralyme or soda lime (see Ch. 12).
Sevoflurane
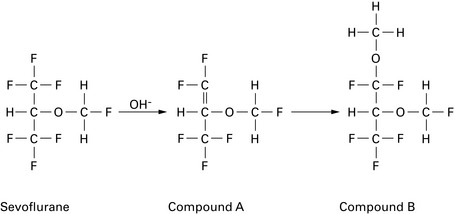
Has similar CVS effects to isoflurane but less tachycardia and coronary vasodilation (no evidence for coronary steal). Less myocardial depression than halothane. Not arrhythmogenic, even with adrenaline. No more irritant than halothane to upper respiratory tract. Greater respiratory depression than halothane but faster elimination results in less postoperative respiratory depression. EEG effects similar to isoflurane, with dose-related depression. Decomposed by soda lime to compounds A and B (Fig. 11.4; and see Ch. 12); 2% metabolized. Levels of fluoride ions >50 μmol.L−1 (thought to be the threshold for nephrotoxicity) and post-anaesthetic albuminuria have been reported, but there is no evidence of significant post-anaesthetic renal impairment. However, consider avoiding in patients with renal failure (fresh gas flow rates <1 L.min−1 are not recommended). Also reported to cause small post-anaesthetic increase in serum ALT, suggesting mild transient hepatic injury.
Use of sevoflurane is associated with more rapid recovery than either propofol or isoflurane.
Bedford R.F., Ives H.E. The renal safety of sevoflurane. Anesth Analg. 2000;90:505-508.
Committee on Safety of Medicines. Guidelines for halothane exposure. Curr Prob. 1997;23:7.
Coppens M.J., Versichelen L.F., Rolly G., et al. The mechanisms of carbon monoxide production by inhalational agents. Anaesthesia. 2006;61:462-468.
Elliot R.H., Strunin L. Hepatotoxicity of volatile anaesthetics. Review. Br J Anaesth. 1993;70:339-348.
Jones R. The new inhalational agents: desflurane and sevoflurane. What is the clinical role of 3rd generation fluorinated anaesthetics and how do they compare with 2nd generation agents. Acta Anaesth Scand. 1995;39(Suppl):130-131.
Kharasch E.D. Adverse drug reactions with halogenated anesthetics. Clin Pharmacol Ther. 2008;84:158-162.
Langbein T., Sonntag H., Trapp D., et al. Volatile anaesthetics and the atmosphere: atmospheric lifetimes and atmospheric effects of halothane, enflurane, isoflurane, desflurane and sevoflurane. Br J Anaesth. 1999;82:66-73.
Neuberger J., Williams R. Halothane hepatitis. Dig Dis. 1988;6:52-54.
Smith I., Nathanson M., White P.F. Sevoflurane – a long-awaited volatile anaesthetic. Br J Anaesth. 1996;76:435-445.
Terrell R.C. The invention and development of enflurane, isoflurane, sevoflurane, and desflurane. Anesthesiology. 2008;108:531-533.
Young C.J., Apfelbaum J.L. A comparative review of the newer inhalational anaesthetics. CNS Drugs. 1998;10:287-310.