Pharmacologic Bases of Antiarrhythmic Therapy
Antiarrhythmic Drugs: Introduction
Na+ Channel Blockers: Class I AADs
K+ Channel Blockers: Class III Antiarrhythmic Effects
Calcium Channel Blockers: Class IV AADS
Gap-Junction Coupling Enhancers
Stretch-Activated Ion Channels
Modulation of Ion Channel Trafficking
Targeting Intracellular Calcium Handling
Pharmacologic Treatment of Inherited Cardiac Arrhythmia Syndromes
Target Cardiac Remodeling: Upstream Therapies
Antiarrhythmic Drugs: Introduction
Cardiac arrhythmias represent a problem in current clinical practice; they are a major cause of morbidity and mortality and are an increasing economic burden for health care systems.1 Treatment of cardiac arrhythmias with antiarrhythmic drugs (AADs) has two main objectives: relieve symptoms and complications (improve quality of life) and reduce mortality directly related to the arrhythmia. A basic principle in pharmacology is that the best treatment is targeted specifically to disease mechanisms. However, in many patients the ultimate underlying mechanisms of the arrhythmia remain incompletely understood. Thus, the choice of a given AAD is empiric and based on the characteristics of the arrhythmia, the pharmacologic properties of the AAD, and, above all, its safety profile. Moreover, triggers and arrhythmogenic substrates can vary among patients with the same arrhythmia depending on the underlying structural heart disease (i.e., coronary artery disease [CAD], heart failure [HF], left ventricular [LV] hypertrophy, hypertension). This variation could explain why AADs produce widely divergent effects on different patients ranging from termination of the arrhythmia to inefficacy, to exacerbation of the treated arrhythmias or generation of entirely new ones (proarrhythmia).
Until recently, arrhythmias were primarily considered a purely electrophysiological problem, and AADs mainly target cardiac Na+, Ca2+, and K+ ion channels (Figure 54-1). They bind to specific receptor sites within the channel drug affinity being strongly modulated by the channel state in a time- and voltage-dependent manner.2 In addition, some AADs modulate the autonomic tone, primarily by antagonizing β1-adrenoceptors (β-blockers) or muscarinic receptors (atropine, disopyramide) or by stimulating adenosine A1 receptors (adenosine).
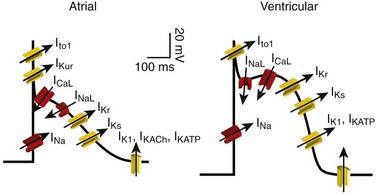
Figure 54-1 Ionic currents involved in shaping of human atrial (A) and ventricular (B) action potentials. The initial upstroke of the action potential is due to the activation of the peak inward Na+ current (INa). Repolarization is determined by the balance between inward-depolarizing L-type Ca2+ (ICaL) and late Na+ (INaL) currents and outward-repolarizing K+ currents, including the rapidly activating and inactivating transient current (Ito1), ultrarapid (IKur), rapid (IKr), and slow (IKs) components of the delayed rectifier, the inward rectifier current (IK1) and the ligand-gated currents activated by a decrease in the intracellular concentration of adenosine triphosphate (IKATP) or activated by acetylcholine (IKAch) or adenosine (IKAdo). Some currents are exclusive to the atria (IKur) or more important (Ito1, IKACh) in the atria than in the ventricles and vice versa (IKr). Arrows indicate the ion movement direction.
Conventional AADs are most commonly grouped according to the Vaughan-Williams classification in four different groups (Table 54-1): Na+ channel-blocking drugs (class I), β-blockers (class II), K+ channel blockers (class III) that prolong action potential (AP) duration (APD) and refractoriness, and L-type Ca2+ channel blockers (class IV). However, most AADs are “dirty drugs” that in a narrow range of concentrations simultaneously block different types of channels and modulate adrenergic and/or muscarinic receptors (e.g., quinidine, disopyramide, propafenone, amiodarone, dronedarone). Moreover, some old (adenosine, digoxin) and new AADs (ivabradine) cannot be listed in any of the original four classes. Table 54-2 shows drug selection based on the mechanisms underlying cardiac arrhythmias, and Figure 54-2 shows the main cellular targets for AADs.
Table 54-1
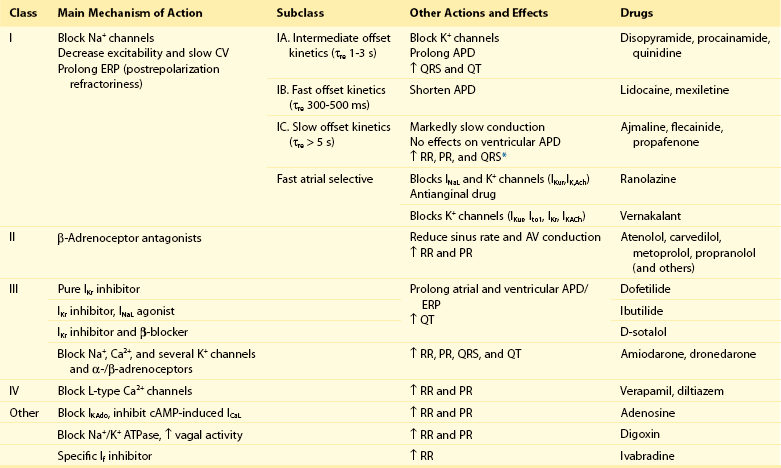
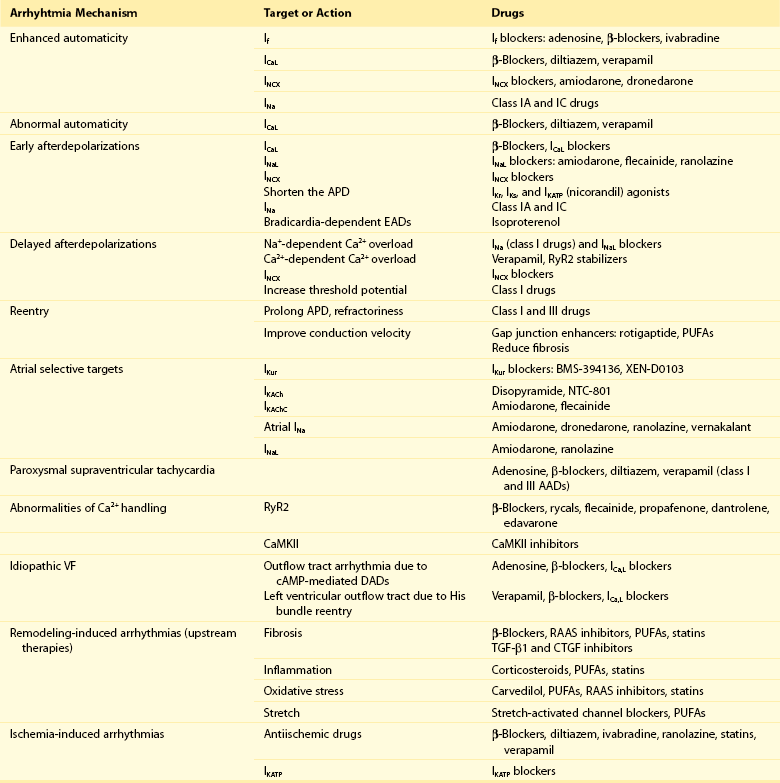
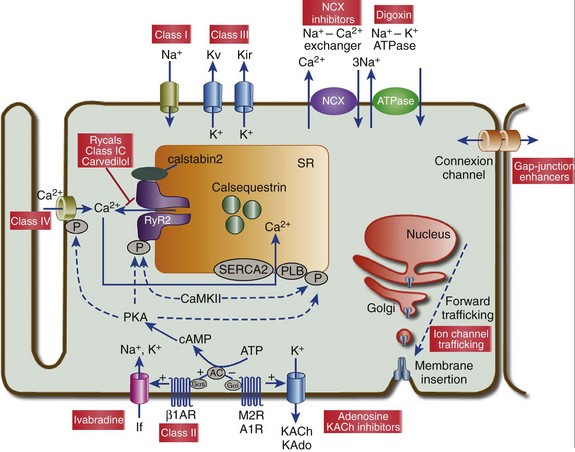
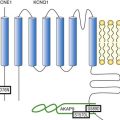
Figure 54-2 Cellular targets of antiarrhythmic drugs (AADs). Most available AADs modify the conductance of Na+ (class I), Ca2+ (class IV), and K+ ion channels (class III) located in the sarcolemma, leading to a decrease or increase in a given ionic current, or they block β-adrenergic (class II), adenosine-A1, and muscarinic-M2 receptors. Some AADs inhibit abnormal persistent Na+ entry and diastolic Ca2+ release via the ryanodine receptor RyR2, with minimal effects on normal channel function. Furthermore, they can inhibit ion exchangers (Na+-Ca2+ [NCX]) or pumps (Na+-K+ ATPase), increase gap junction conductance, and correct ion channel expression within the membrane by modulating trafficking pathways. A1R, adenosine A1 receptor; AC, adenylyl cyclase; ATPase, Na+-K+ ATPase; β1AR, β1-adrenergic receptor; CaMKII, Ca2+/calmodulin kinase II; CSQ, calsequestrin; Gs, stimulatory G protein; M2R, muscarinic type-2 receptor; PBL, phospholamban; PKA, protein kinase A; RyR2, ryanodine receptor; SERCA2a, SR Ca2+-ATPase; SR, sarcoplasmic reticulum.
Na+ Channel Blockers: Class I AADs
Na+ channel blockers bind and unbind in a strongly time- and voltage-dependent manner to a receptor site within the pore-forming subunit (Nav1.5) of the channel when channels are in the activated or inactivated state.2 Na+ channel blockers also slow Na+ channel reactivation (transition from the inactivated to the resting state) upon repolarization, which prolongs refractoriness independently from changes in APD—that is, they increase the effective ratio of refractory period (ERP) to APD (postrepolarization refractoriness). As a consequence, the effects of class I AAD would be expected to increase: (1) at faster rates of stimulation (use-dependent block), because Na+ channels spend more time in activated and inactivated states, and the diastolic time for recovery from drug-induced block is shortened; and (2) in depolarized cardiac tissues (voltage-dependent block) because membrane depolarization inactivates Na+ channels and slows recovery from block.
Clinical and experimental data suggest that block of atrial Na+ channels can terminate AF. Indeed, propafenone and flecainide are first-choice drugs for AF cardioversion, but only in patients without structural heart disease. Conversely, ventricular Na+ channel block is associated with proarrhythmic effects. Class I AADs decrease excitability and slow ventricular conduction and can increase all-cause mortality and, therefore, they have no role (or are contraindicated) in the primary prevention of sudden cardiac death (SCD) because of life-threatening ventricular tachyarrhythmias (VT or ventricular fibrillation [VF]) in high-risk patients with post–myocardial infarction.3 In secondary prevention, the use of class I AADs is limited because of their low efficacy and serious adverse effects that may offset the therapeutic benefit, including proarrhythmia, multiorgan toxicity, reduced cardiac function, or worsening coexisting diseases.4–6
Class I drugs are subdivided into drugs with intermediate (IA), fast (IB), and slow (IC) onset/offset kinetics of Na+ channel block (see Table 54-1). Recently, it has been demonstrated experimentally that drugs with fast onset/offset kinetics of Na+ channel block, such as vernakalant and ranolazine, could selectively target atrial versus ventricular Na+ channels.7,8 This targeting would allow them to exhibit a disease-specific component of atrial-selectivity because of the high atrial rate in AF, which enhances block of atrial over ventricular Na+ channels. Indeed, it has been proposed that the faster the AADs dissociation kinetics the more selective the atrial effect during AF and the fewer the ventricular proarrhythmic effects, because the fast dissociation kinetics allows recovery from block at slower frequencies (e.g., those of the ventricles either at sinus rhythm [SR] or during AF). Moreover, atrial cells exhibit a slightly more depolarized resting membrane potential (RMP) and a more gradual phase 3 of the AP, which at rapid atrial rates results in a less negative take-off potential.7,8 In addition, atrial cells exhibit a more negative potential for half-maximum inactivation of Na+ channels than ventricular myocytes. Because reactivation depends on membrane potential, fewer Na+ channels recover from the inactivated state during diastole in atria than in ventricles. Therefore, Na+ channel blockers that preferentially bind to the inactivated state of the channel and have a fast dissociation rate will exhibit atrial selectivity, because steady-state drug binding and consequently channel block will be larger in atria than in ventricles.8 However, block of K+ channels, particularly those preferentially or exclusively present in the atria, seems to be also required to meet the “class I atrial selective” profile. Otherwise lidocaine, which is highly selective for the inactivated state and exhibits a fast onset and offset kinetics, would be also atrial selective.
Indeed, class IA and IC, like class III AADs, also block several K+ channels and prolong ventricular (quinidine) or atrial (propafenone and flecainide) APD, depending on the type of K+ channel they preferentially block (discussed later). Furthermore, amiodarone, dronedarone, ranolazine, and vernakalant, which produce an atrial-selective INa block and in animal models suppress AF with minor ventricular effects, also inhibit the rapid component of the delayed rectifier current (IKr) and other K+ currents prolonging atrial APD. This effect that enhances the use-dependent INa block by further reducing the diastolic interval at fast rates.8,9
Late Na+ Current (INaL) Inhibition
Na+ channels open for a few milliseconds after membrane depolarization, generating peak INa, and then most of them undergo fast inactivation. However, some Na+ channels do not inactivate or inactivate but reopen during depolarization generating the late Na+ current (INaL), which presents slow inactivation and recovery kinetics.10 INaL increases under pathologic conditions, including myocardial ischemia, LV hypertrophy, HF, AF, and some variants of the long QT syndrome (LQTS).10,11 In the ventricular wall, M cells present a larger INaL than do epicardial and endocardial cells, and an increase in INaL increases transmural dispersion of repolarization (TDR) facilitating reentry.11 During myocardial ischemia, enhanced INaL increases Na+ influx and, via the Na+/Ca2+ exchanger (NCX), produces a Na+-mediated Ca2+ overload that prolongs ventricular APD. APD prolongation enhances triggered activity that can occur during phases 2 or 3 of the AP (early afterdepolarizations [EADs]), whereas intracellular Ca2+ (Cai2+) increase enhances triggered activity after AP repolarization (delayed afterdepolarizations [DADs]).10 Ranolazine is an antianginal drug that, besides its effects on peak INa at fast rates, blocks INaL and suppresses atrial and ventricular arrhythmias in a variety of conditions (e.g., ischemia, reperfusion, HF, AF, type 3 LQTS).10,11 During myocardial ischemia, ranolazine reduces intracellular Na+ and Ca2+ overload and improves mechanical dysfunction, but has no effect on INa, excitability or conduction velocity.11 In non−ST-elevation acute coronary syndromes12 ranolazine reduces non-sustained VT and supraventricular tachycardias, and preliminary evidence suggests that it safely converts paroxysmal AF and prevents AF recurrences.11 Moreover, ranolazine prevents ventricular APD (QT) prolongation, reduces TDR, and suppresses EADs produced by IKr inhibitors and in patients with type 3 LQTS.
Antiarrhythmic Effects of Class I AADs
Describing the ultimate mechanisms responsible for the antiarrhythmic effects of class I AADs in reentry is a big challenge. Indeed, the underlying mechanism responsible for reentry itself is a matter of debate. Class I AADs markedly slow conduction and prolong refractoriness. The leading-circle theory of reentry predicts that because Na+ channel blockers slow conduction they should, if anything, favor reentry by decreasing the wavelength (product of refractory period and conduction velocity), resulting in proarrhythmic effects. The proarrhythmia risk increases with drugs that produce marked conduction slowing in depolarized-ischemic cardiac tissues. Experimental and clinical evidence, however, shows that class I AADs can terminate reentrant arrhythmias, such as AF, without increasing the wavelength. The most characteristic electrophysiological change that they produce before AF termination is an increase in the temporal excitable gap.13,14 According to the multiple wavelets theory of the origin of AF, this widening will lower the chance that reentrant waves encounter areas of partially refractory tissue, so that slowing of conduction and fractionation of wavelets will occur less frequently. This widening decreases the number of fibrillation waves by promoting their fusion, which increases the chance to terminate the arrhythmia.
However, there are clinical and experimental data demonstrating that reentry is maintained by the periodic activity of one or a small number of functional reentrant sources with a wavefront rotating around a central core (“rotor”).15,16 The waves emerging from the rotors undergo spatially distributed fragmentation and give rise to fibrillatory conduction. The size of the spiral wave is determined by tissue excitability and refractoriness, so that the rotor will turn faster and in a more stable position, resulting in higher excitability and shorter refractoriness.15,16 In this context, Na+ channel blockers terminate reentry as they (1) enlarge center of rotation, so that the rotor cannot any longer be accommodated by the substrate; (2) decrease anchoring to functional obstacles, increasing meander and extinction at boundaries; and (3) reduce the number of daughter waves that could provide new primary rotors.15,16
β-BLOCKERS: CLASS II AADs
β-Blockers include a heterogeneous group of drugs that inhibit sympathetic effects. Antiarrhythmic effects of β-blockers are the result of their electrophysiological effects (β-adrenoceptor stimulation modulates several ion currents, including hyperpolarization-activated inward current [If], INa, ICaL, IK1, Ito1, IKr, and IKs), inhibition of neurohumoral activation (and perhaps of sympathetic hyperinnervation and sprouting), and their antiischemic (reduce myocardial oxygen demands, increase subendocardial perfusion, and improve cardiac metabolism), antihypertensive, and antiproliferative effects.17,18 Indeed, β-blockers exert a beneficial effect on LV remodeling in patients with myocardial infarction (MI) or HF. Furthermore, in the ischemic myocardium, β-blockers decrease dispersion of ventricular repolarization and increase the VF threshold.
Sympathetic hyperactivity is the predominant change in the autonomic tone preceding malignant ventricular arrhythmias and SCD, whereas reduced vagal activity is associated with increased mortality in HF. β-Blockers remain among the few AADs that are effective for both primary and secondary prevention of SCD in different clinical conditions, including acute and chronic myocardial ischemia, congestive HF or LV dysfunction, and hypertrophic cardiomyopathy.3,17,18 Thus, most patients who have a propensity to develop VT/VF should receive β-blockers because they are a rational, mechanism-based therapy for the treatment of these arrhythmias. Moreover, β-blockers decrease the incidence of postoperative AF, which is explained by the role of an increased adrenergic tone in the genesis of this arrhythmia.
By inhibiting If and ICaL, β-blockers decrease spontaneous activity of the sinoatrial node (SAN), inhibit the ectopic activity of the His-Purkinje system (Purkinje fibers are particularly prone to develop abnormal automaticity during increased sympathetic activation), and suppress abnormal automaticity generated in cardiac cells depolarized to potentials between −60 and −40 mV owing to the activation of ICaL.18 In the atrioventricular node (AVN), they decrease conduction velocity and prolong refractoriness. β-Blockers are widely used to treat inappropriate sinus tachycardia and exercise-induced and supraventricular and ventricular arrhythmias where an increase in sympathetic tone plays a role17; they are the most effective drugs for ventricular rate control in AF and are first-choice drugs in the treatment of supraventricular reentrant tachycardias where the AVN forms part of the circuit.1,17 Finally, β-blockers are effective in controlling the proarrhythmia induced by class I AADs, probably because of their bradycardic effect, which decreases the rate-dependent conduction slowing induced by Na+ channel blockers.
β-Blockers decrease all the proarrhythmic actions secondary to the increase of extracellular Ca2+ influx through L-type Ca2+ channels, sarcoplasmic reticulum Ca2+ release, and intracellular Ca2+ overload.17,18 Thus, they suppress EADs and DADs and are first-choice drugs in drug-induced torsades de pointes (TdP) and in patients with LQTS types 1 and 2 or catecholaminergic polymorphic VT (CPVT).19,20 Surprisingly, and even when there are marked pharmacologic differences among β-blockers, there are few direct comparator data regarding their efficacy.
K+ Channel Blockers: Class III Antiarrhythmic Effects
These drugs prolong cardiac APD and refractoriness effects that, under theoretical bases, can suppress reentry. Some drugs (e.g., dofetilide) are considered pure class III AADs because they selectively block the channels that generate the IKr (named Kv11.1 or HERG), thus prolonging both atrial and ventricular APD and refractoriness in the absence of effects in conduction velocity.9,21 Experimental data demonstrated that selective IKr inhibition produces a greater prolongation of APD and refractoriness in atria versus ventricles, at least at normal SR frequencies. Unfortunately, at fast driving frequencies, the relative role of IKr in atrial and ventricular repolarization diminishes so much that IKr inhibition is not able to prolong APD during the arrhythmia episodes. This property, known as reverse use-dependence, limits the clinical efficacy of these drugs to stop fast arrhythmias, particularly AF. Moreover, marked IKr inhibition produces excessive and inhomogeneous ventricular APD prolongation, results in TDR, and predisposes to EADs, which in turn can trigger TdP. Inhibition of IKr, and hence proarrhythmic risk, increases in the presence of bradycardia, hypokalemia, HF, or other IKr-inhibiting drugs. Indeed, many other drugs, including antihistamines, antipsychotics, antimicrobials, and diuretics, are able to markedly block Kr channels, thus prolonging the QT interval (drug-induced LQTS) and increasing the incidence of TdP and SCD. It has been proposed that prolongation of APD produced by pure IKr inhibitors is characterized by a set of four disturbances of repolarization including triangulation of the AP, reverse use-dependence, instability, and dispersion of APD whose magnitude would help to predict drug-induced proarrhythmia risk.22
HERG channel blockers gain access, from the intracellular side of the membrane, to a binding site located within the central cavity of the channel. The inner vestibule of HERG channels is larger than that of other K+ channels and presents two aromatic residues on each of the four subunits assembled to compose the conducting pore.23 These topological characteristics explain the marked pharmacologic promiscuity of HERG because they allow drugs with rather different chemical structures to establish interactions with the aromatic moieties by π electron stacking and block K+ efflux through the pore. Finally, it is worth mentioning that besides the “pure Kr blockers” many AADs, including amiodarone, quinidine, propafenone, flecainide, and ranolazine, also block HERG channels at therapeutic concentrations, an effect that could contribute to their antiarrhythmic properties particularly at the atrial level (i.e., amiodarone, ranolazine). However, unfortunately in some cases, HERG blockade can increase their ventricular proarrhythmic effects (quinidine).
In healthy human atrial and ventricular myocytes driven at SR frequencies, IKs seems to be small, and its pharmacologic inhibition does not alter APD.24 Importantly, the role of IKs in determining APD rises in prominence at increasing beating frequencies or under β-adrenergic stimulation, which causes channel accumulation in closed states near the open state. In addition, IKs helps to terminate the AP when the repolarization reserve is compromised (i.e., IKr decrease).25 Moreover, recent data demonstrated that chronic AF increases by 100% IKs density in myocytes from both the right and left atria.26 Mathematical models suggest that this chronic AF-induced remodeling of IKs density contributes to the atrial APD and refractoriness shortening produced by the arrhythmia, which in turn further enhances reentry, thus favoring AF maintenance. Moreover, there are data indicating that stable fast reentry sources (rotors) occur with significantly higher rotation frequencies, lower conduction velocities, and shorter AP in cells with prominent IKs.27 In addition, the frequency-dependent accumulation of IKs promotes post-repolarization refractoriness and fibrillatory conduction of waves emanating from rotors. Therefore, it seems reasonable to propose that IKs could be an interesting target for the treatment of AF. In fact, some AADs, such as propafenone, quinidine, and amiodarone, inhibit IKs—an effect that probably contributes to their antiarrhythmic effects.21 Unfortunately, selective inhibition of IKs would be difficult to achieve, because molecular determinants for channel block are similar to those required for HERG blockade and would not be devoid of ventricular proarrhythmic effects. Indeed, patients carrying mutations in KCNQ1 or KCNE1 genes, which encoded the α (Kv7.1) and β (minK) subunits that form the channels, present LQTS types 1 and 5, respectively. This finding suggests that excessive inhibition of IKs could also lead to a marked prolongation of ventricular APD, particularly under conditions of increased sympathetic tone that promote EADs and TdP.
Ito1 (4-aminopyridine sensitive component of the transient outward current) determines the amplitude of the phase 1 of early repolarization, and thus the height of the AP plateau, which in turn influences the activation of other currents involved in repolarization (mainly ICaL, IKr, and IKs).28 Variations of Ito1 strongly influence the shape of cardiac AP, intracellular Ca2+ transients, and cardiac contractility. Relative Ito1 density is much higher in atrial, Purkinje, epicardial, and midmyocardial cells than in endocardial cells. Ito1 densities are also reported to be higher in right than in left ventricular myocytes. Prominent Ito1 leads to the “spike and dome” morphology typical of epicardial (particularly right) and some atrial AP (Ito1 is also differentially expressed among atrial cells). HF and chronic AF reduce Ito1 density, this effect being more marked in the left than in the right atria. In regions of the myocardium exhibiting a prominent Ito1, marked accentuation of the AP notch results in a transmural voltage gradient, leading to coved ST-segment elevation diagnostic of Brugada syndrome (BrS).29 Because accentuation of the AP notch and loss of the dome could be secondary to either a decrease in inward currents (INa and ICaL) or an increase in outward potassium currents (mainly Ito1), sodium channel blockers such as procainamide, propafenone, flecainide, ajmaline, and disopyramide can be used to induce or unmask ST-segment elevation in patients with concealed J wave syndromes (BrS and early repolarizing syndromes).29 Conversely, quinidine, which is a potent Ito1 inhibitor, reduces the magnitude of the J wave and normalizes ST-segment elevation. The efficacy of quinidine in the prevention of BrS-associated arrhythmias is currently under evaluation.
IKur is carried by Kv1.5 channels and is detected only in the human atria, but not in the ventricle. Therefore, IKur inhibitors would be expected to prolong atrial, but not ventricular APD and refractoriness.30 In myocytes from patients with SR, selective IKur inhibition shifts the plateau phase to more positive potentials at which the IKr is rapidly activated, so that mid to late repolarization is accelerated and APD is slightly shortened.30 This could explain why selective IKur inhibitors do not suppress or even induce AF in experimental models.8 Indeed, KCNA5 (the gene encoding Kv1.5) loss-of-function mutations can cause AF. Chronic AF reduces ICaL and markedly shortens atrial APD, so that IKur blockers increase the plateau potential over a range (below 0 mV) where the activation of IKr and IKs is less pronounced, and prolong atrial APD.30 Furthermore, mathematical models predict that IKur or Ito1 inhibition can induce rotor termination.31 However, chronic AF also reduces IKur, particularly in the right atria; this decrease suppresses the endogenous right-to-left gradient of the current.26 As a result, the relative contribution of IKur to repolarization in chronic AF can be small, decreasing the atrial sensitivity to IKur inhibitors.30 These findings raise serious doubts on the usefulness of “pure” IKur blockers, and in fact none of those currently under development have been successfully launched for therapeutics.32 Conversely, vernakalant, which combined blockade of IKur, Ito1, IKr, and IKACh, together with fast blockade of inactivated Na+ channels produces a high conversion rate to SR in patients with recent-onset AF.
IK1 contributes to the final phase of repolarization and stabilizes the RMP of atrial and ventricular myocytes.33 IK1 increase hyperpolarizes RMP, shortens APD, reduces spontaneous activity, and accelerates and stabilizes arrhythmia-maintaining rotors, playing a role in the genesis of AF and VF.31,33,34 IK1 exhibits differential biophysical properties in atrial and ventricular tissues. Moreover, experimental data suggest that the Kir2.1 channel is the major isoform underlying ventricular IK1, whereas relative contribution of Kir2.2 and 2.3 to atrial IK1 seems to be greater. IK1 increases in left and right atrial myocytes from patients with chronic AF, an augmentation that is greater in the left than in the right atria in patients with paroxysmal AF. Left-to-right ventricular differences in IK1 have been postulated to represent a mechanism for the preferential stabilization of high-frequency rotors in the LV.34 Moreover, patients carrying gain-of-function Kir2.1 mutations exhibit type 3 short QT syndrome characterized by a high incidence of VF and SCD. Furthermore, flecainide and propafenone at therapeutically relevant concentrations selectively increase IK1 current generated by Kir2.1 homotetramers, an effect that could contribute to their ventricular proarrhythmic effects. Overall, it seems that IK1 inhibitors could be an efficacious antifibrillatory strategy. Unfortunately, IK1 inhibition also results in proarrhythmia. In fact, patients carrying loss-of-function Kir2.1 mutations exhibit congenital LQTS type 7 (Andersen-Tawil syndrome) characterized by APD prolongation, diastolic depolarization, and increased propensity for EAD- and DAD-triggered arrhythmias.35 Moreover, the development of Kir2.1-based antiarrhythmics is hampered because Kir2.1 channels are expressed in many excitable tissues, which could lead to organ toxicity. More promising could be selective blockade of Kir2.3 over Kir2.1 channels for the treatment of AF. Kir2.3 channels are indeed preferentially inhibited by several drugs taking advantage of the low affinity of Kir2.3 channels for phosphatidylinositol 4,5-bisphosphate (PIP2), which critically determines the channel function.
Acetylcholine released from vagal nerve terminals activates IKACh. Because IKACh is exclusively present in the atria, it has been proposed as a target for development of atrial selective AADs. IKACh activation slows SAN pacemaker activity, produces a heterogeneous APD and ERP shortening, and hyperpolarizes RMP, increasing Na+ channel availability. These effects create a substrate for reentry, accelerate and stabilize high frequency rotors, and contribute to initiation and maintenance of AF.34 Some AADs inhibit IKACh via the blockade of muscarinic receptors (disopyramide) and may be useful in vagally mediated AF. Others inhibit IKACh via GTP binding proteins or the KACh channel itself (propafenone, dofetilide, and amiodarone). The new multichannel blocker dronedarone is 100-fold more potent in blocking IKACh than amiodarone, an effect that could contribute to its antiarrhythmic properties. Importantly, it has been demonstrated that chronic AF also reduces IKACh, reducing its relative role in atrial repolarization while increasing an IKACh component (IKAChC) with constitutive activity (active in the absence of muscarinic ligands).36 Ideally, an AAD should selectively block IKAChC (an atrial- and disease-specific therapeutic target) without affecting the IKACh that modulates SAN and AVN. IKAChC is inhibited by flecainide, AVE0118, and investigational AADs, but highly selective IKAChC blockers are not currently available.
In up to 80% of cases, VT/VF occurs in the setting of CAD and HF. Attempts to prevent arrhythmic events and SCD with class I and III AADs in the setting of myocardial ischemia have failed. Activation of IKATP during acute myocardial ischemia results in shortening of the APD, accumulation of extracellular K+, membrane depolarization, and slow conduction velocity. These effects render the ischemic heart vulnerable to reentrant arrhythmias. It has been hypothesized that specific IKATP inhibitors prolong ventricular refractoriness in ischemic tissues with little or no effect on normal tissue, being truly ischemia-selective AADs.37 Conversely, the KATP opener nicorandil shortens the APD, reduces TDR, and suppresses EADs in patients with LQT1.38
Amiodarone and Dronedarone
Amiodarone is considered the epitome of class III AADs because, in chronic treatment, it prolongs APD and refractoriness in all cardiac tissues, an effect that is attributable to the blockade of several K+ channels (Ito1, IKur, IKr, IKs, IK1, and IKACh). However, as was mentioned before, amiodarone also inhibits peak INa (inhibits inactivated Na+ channels with fast kinetics [class I]), INaL, and ICaL (class IV), and antagonizes α-/β-adrenergic receptors (class II).1
Amiodarone presents a low proarrhythmic risk and is effective against supraventricular and ventricular arrhythmias caused by focal activity or reentry. It is the most effective AAD for preventing recurrences of AF, being the last-choice drug in patients with structural heart disease, and in the prophylaxis and treatment of life-threatening ventricular arrhythmias in patients with MI or congestive HF or after cardiac surgery, although it is being replaced by ICDs. Amiodarone decreases the incidence of SCD and is an alternative for patients who are not eligible for, or who do not have access to, ICD therapy for the prevention of SCD and to inhibit unpleasant ICD shocks.3,39 However, in patients with HF and LV ejection fraction of 35% or less, amiodarone has no favorable effect on survival.3
Calcium Channel Blockers: Class IV AADs
Nondihydropyridine Ca2+-channel blockers (e.g., diltiazem, verapamil) inhibit the ICaL and exhibit antiischemic and antihypertensive properties. These blockers stabilize the channel in its inactivated state and prolong its reactivation, so that their effects increase at fast rates (use-dependent block) and at depolarized membrane potentials (voltage-dependent block).1
Verapamil is a drug of choice in idiopathic outflow tract and fascicular VT and drug-induced TdP; it can be an alternative to β-blockers in CPVT and prevents VTs related to ischemia. However, class IV drugs have no effect on all-cause mortality in patients with prior MI and are contraindicated in congestive HF.3
Gap-Junction Coupling Enhancers
Connexins (Cx) are membrane proteins that assemble into hexameric hemichannels, also known as connexons. At the gap junctions, connexons allow electrical flow between cardiomyocytes and thus regulate intercellular coupling. Changes in Cx expression, location (lateralization), and function occur in many forms of heart disease (i.e., ischemia, hypertrophy, HF, AF) and contribute to arrhythmogenesis as they slow conduction velocity, cause heterogeneities in repolarization, and modulate automaticity. Cx40 has a critical role in mediating atrial conduction, whereas Cx43 seems to be the major ventricular form. Cellular uncoupling owing to dephosphorylation and redistribution of Cx43, together with increased fibrosis40 and decreased expression of Na+ channels, are implicated in conduction slowing during ischemia, increasing the risk of fatal ventricular arrhythmias. Rotigaptide increases gap junction conductance and prevents acute ischemic arrhythmias. However, it partially reverses the loss of Cx43 but does not restore normal conduction or prevent arrhythmias in the healing infarct border zone, such as after a prolonged period of gap junction remodeling.41 Thus, it seems that interventions that are effective in restoring cell-to-cell coupling under conditions of acute ischemia might not be effective in a substrate that has already undergone a prolonged period of gap junction remodeling. Gap junction–enhancing drugs can also reduce AF vulnerability in some models of AF (chronic mitral regurgitation and acute ischemia), but not in others (HF or atrial tachypacing).41 These findings suggest that the role of Cx in AF is disease specific and that gap junction enhancers may be effective only when conduction slowing is due to alterations in gap junctions.
Stretch-Activated Ion Channels
Chronic stretch causes heterogeneous conduction, prolongs refractoriness, and induces structural remodeling (dilatation, hypertrophy, fibrosis), which contributes to both atrial and ventricular arrhythmias. Recent evidence suggests that these electrophysiological effects can be explained via the activation of the nonselective cationic transient receptor potential TRPC6 channels.42 In experimental models, nonselective stretch-activated channel blockers (gadolinium and the tarantula peptide GsMtx-4) suppress AF inducibility.14 Nowadays, modulation of stretch-activated channels as an antiarrhythmic strategy is limited by the lack of effective drugs.
Modulation of Ion Channel Trafficking
Structural heart diseases and many mutations associated with channelopathies affect biogenesis, forward trafficking to the surface membrane subdomains, and degradation of cardiac ion channels.43 Recent evidence demonstrates that AADs inhibit ion conduction and modulate ion channel trafficking or surface density and that channel blockade and trafficking can be targeted independently.43,44 Some HERG channel blockers (cisapride, E4031, quinidine) can rescue defective HERG surface trafficking, whereas others (ketoconazole and fluoxetine) can decrease HERG surface density. Furthermore, there are drugs that do not block HERG channel conductance, but either decrease (pentamidine, probucol) or increase (fexofenadine) HERG surface density.45 Moreover, type 2 LQTS-associated mutant channels can be rescued to the plasma membrane with drugs (E4031, terfenadine, fexofenadine) that likely stabilize misfolded protein in the endoplasmic reticulum, although the drug effectiveness depends on the particular mutation present.45
Quinidine induces a stereospecific dose-, time- and subunit-dependent internalization of Kv1.5, concomitant with the classical pore block.43 The specific expression of Kv1.5 channels in human atria raises the possibility of designing drugs to specifically modulate Kv1.5 channel trafficking.43 Such drugs, in theory, can cause a rapid and reversible decrease in IKur and a prolongation of atrial APD terminating AF, with minimal risk of ventricular proarrhythmia. However, there is a need for a better understanding of trafficking mechanisms and how chronic decrease of channel expression contributes to arrhythmia-induced remodeling.
Some drugs such as pentamidine and flecainide decrease and increase Kir2.1 channel expression in the membrane, respectively. Flecainide is also able to promote trafficking of some Kir2.1 loss-of-function mutants increasing the amplitude of the outward K+ current generated by the channels, if any. This effect could contribute to the empirically observed antiarrhythmic actions produced by flecainide in some patients with Andersen-Tawil syndrome.44
Targeting Intracellular Calcium Handling
Abnormal intracellular Ca2+ handling can promote both atrial and ventricular arrhythmias; it also has a key role in cardiac electrical and structural remodeling in patients with AF, HF, LV hypertrophy, and CAD.14,46–48 Moreover, CPVT associated with mutations in the sarcoplasmic reticulum Ca2+ release channel/ryanodine receptor (RyR2) and calsequestrin (CASQ2) genes destabilize the RyR2 channel complex and increase spontaneous Ca2+ release, which under certain conditions (e.g., during exercise or β-adrenergic stimulation) facilitates the development of DADs and triggered arrhythmias.20,46 Thus, intracellular Ca2+-handling proteins (Ca2+-ATPase [SERCA2a], phospholamban, RyR2, and its accessory protein calstabin 2, NCX, and CSQ), provide targets for developing new AADs.
Dysregulation of RyR2, characterized by improper SR Ca2+ release and increased diastolic Ca2+ leak is related to: (1) RyR2 hyperphosphorylation by protein kinase A (PKA) and Ca2+/calmodulin-dependent kinase II (CaMKII), which transiently dissociates calstabin 2 from the RyR2 complex increasing open channel probability; (2) increased sensitivity of RyR2 to activation by luminal Ca2+; and (3) reactive oxygen species generated in the diseased heart, which make the channel leaky.14,46–48 β-Adrenergic stimulation and renin-angiotensin-aldosterone system (RAAS) can promote triggered arrhythmias via PKA- and CaMKII-induced RyR2 hyperphosphorylation.46,47 β-Blockers prevent PKA- and CaMKII-mediated hyperphosphorylation of RyR2, depletion of calstabin 2, and SR Ca2+ leak being the only effective drugs in improving survival in CPVT.20 β-Blockers and verapamil act synergistically to prevent stress-induced increase in sarcoplasmic reticulum Ca2+ content by reducing heart rate, Ca2+ influx into the cell, and Ca2+ uptake into the sarcoplasmic reticulum.20 However, in patients with CPVT, cardiac events remain considerable even in this combination, so that alternative therapies are needed.
Carvedilol, a nonselective β- and α1-adrenergic antagonist, blocks several ion channels (IKur, IKr, ICaL, Ito1, and IKs) and exerts antioxidant and antiproliferative effects. It is the only β-blocker that suppresses sarcoplasmic reticulum Ca2+ release by directly reducing RyR2 mean open time, independently of its β- or α1-antagonism or antioxidant activities, which likely contributes to its antiarrhythmic effects in HF patients.20,46 Carvedilol analogues with minimal β-blocking activity retain the ability to suppress Ca2+ release and prevent CPVT without causing bradycardia. Interestingly, the combination of these analogues with selective β-blockers (metoprolol or bisoprolol) is more effective than each agent alone for preventing CPVT.
Stabilizers of the calstabin 2–RyR2 complex (Rycals) represent a new antiarrhythmic approach. Rycals (such as JTV519 and S44121) increase the affinity of calstabin-2 for PKA-hyperphosphorylated RyR2, reduce channel open probability and prevent arrhythmogenic diastolic Ca2+ leak without affecting normal RyR2.14,20,46,48 As a consequence, JTV519 prevents APD alternans and triggered ventricular arrhythmias and reduces Ca2+ waves in arrhythmogenic Purkinje fibers following MI. It also reduces ectopic activity and DADs in pulmonary vein cardiomyocytes, decreasing AF inducibility.47 Its putative efficacy in the treatment of AF and ventricular arrhythmias associated with HF and CPVT is currently being tested.
Flecainide and propafenone block the open state of RyR2, inhibit arrhythmogenic diastolic Ca2+ waves, and suppress DADs and triggered arrhythmias in experimental models and in patients with CPVT.20,47 In addition, their Na+ channel–blocking properties prevent DADs from reaching threshold potential and from triggering premature beats. Furthermore, β-blocking activity of propafenone can also contribute to its clinical efficacy in CPVT. As a result, class IC AADs can be an alternative to β-blockers for some patients with CPVT.20,47
Some RyR2 mutations can lead to an abnormally tight domain-domain interaction that results in an erroneous activation of the channel and diastolic Ca2+ leak.46 In experimental models, dantrolene stabilizes domain interactions within the RyR2 and prevents aberrant Ca2+ release and CPVT.20 The antioxidant edaravone also ameliorates the defective interdomain interaction of the RyR2 and prevents Ca2+ leak and LV remodeling during the development of HF.48
Even when strong evidence suggests that CaMKII control over Ca2+ handling might be more potent than that of PKA, CaMKII inhibitors might not represent a feasible target, because the ubiquitous role of CaMKII in cellular physiology can result in deleterious off-target side effects.48 In addition, some CaMKII inhibitors (KN-93) cannot discriminate between CaMKII and CaMKIV and inhibit voltage-gated K+ and Ca2+ channels. Therefore, to achieve an effective and safe cardiac effect, drugs with tissue and isoform-specific action (CaMKIIδ) are probably required. An alternative strategy is small peptides like autocamtide-2-related inhibitory peptide (AIP) that inhibit CaMKII activation by blocking calmodulin binding to CaMKII.
Pharmacologic Treatment of Inherited Cardiac Arrhythmia Syndromes
During the past decade, there was an explosion of information linking mutations in genes encoding ion channel α- and accessory-subunits and cytoskeletal molecules to inherited cardiac arrhythmia syndromes, including short QT syndrome, LQTS, BrS, AF, idiopathic VF, and CPVT (Table 54-3).19,29,35,38,45 Identification of disease-associated genes provides important information on the molecular mechanisms of cardiac arrhythmias and opens the possibility for antiarrhythmic therapies based on genotype and clinical presentation. However, randomized trials have not been conducted because of the rarity of these conditions. Nevertheless, therapy alleviating the mutation consequences depends on the mutation class, although most AADs lack the desired selectivity for a given channel.
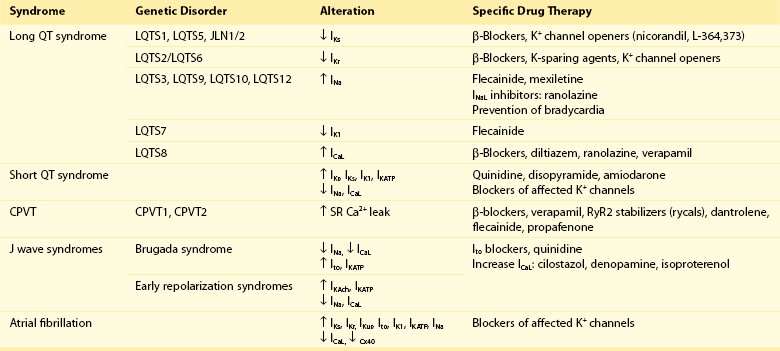
Target Cardiac Remodeling: Upstream Therapies
Cardiac arrhythmias produce changes (remodeling) in the electrophysiological (ion channel and gap junction function or expression, abnormal Ca2+ handling) and structural (hypertrophy, fibrosis, dilatation) properties of the myocardium often associated with stretch, oxidative stress, inflammation, or ischemia, creating a substrate for reentry and focal activity.13,14 Furthermore, aging and structural heart diseases also produce electrophysiological and structural remodeling that increases susceptibility to arrhythmias and makes arrhythmias more drug resistant.14,49 However, remodeling is a disease-specific process presenting important differences in signaling pathways and pharmacologic responses among comorbidities. Upstream therapies to prevent or delay myocardial remodeling are an attractive approach in the prophylaxis and treatment of arrhythmias associated with HF, hypertension, or MI.13,14,48,50,51 Potential drugs in this category include antiinflammatory agents, RAAS inhibitors (angiotensin-converting enzyme inhibitors [ACEIs], angiotensin II receptor antagonists [ARAs], aldosterone antagonists), omega-3 polyunsaturated fatty acids (PUFAs), statins, and antifibrotic agents.
Antiinflammatory Agents
Raised inflammatory markers (interleukin 6 and high-sensitivity C-reactive protein) are associated with a higher risk of atrial and ventricular arrhythmias in patients with HF or CAD.13,14,48 It has been hypothesized that inflammation contributes to the structural remodeling and might be a target for antiarrhythmic therapies. However, whether inflammation is an initiating event or a consequence in the development of arrhythmias remains controversial. Corticosteroids reduce the risk of postoperative AF, probably because inflammation has an important role in its pathogenesis,51 but their potential toxicity restricts their therapeutic value.
RAAS Inhibitors
ACEIs and ARAs inhibit angiotensin II AT1 receptor–mediated proarrhythmic effects including APD shortening, enhanced automaticity, hypokalemia, reduced cell coupling, pressure overload, abnormal Ca2+ handling, oxidative stress, neurohumoral activation, and structural remodeling (hypertrophy, fibrosis, inflammation).5,14,52
In primary prevention, ACEIs and ARAs reduce the risk of AF in patients with HF, hypertension, and LV hypertrophy, but not in patients with post–myocardial infarction.53 In secondary prevention, they exert a beneficial effect after cardioversion of persistent AF and in the prevention of paroxysmal AF, particularly in patients with significant underlying heart disease (e.g., LV dysfunction or hypertrophy) and when patients also received amiodarone.50,53 However, prospective randomized clinical trials question the value of ARAs in secondary prevention of AF.50 ACEIs decrease the risk of death following a recent MI by reducing cardiovascular mortality. In patients with congestive HF, ACEIs reduce all-cause mortality but not SCD.3
Aldosterone antagonists inhibit cardiac fibrosis, exert direct antiarrhythmic actions, and reduce the risk of hypokalemia; therefore, they represent a therapeutic option for patients with HF, LV hypertrophy, and AF. Indeed, preliminary data suggest that aldosterone inhibitors reduce the incidence of recent-onset AF, VT/VF, and SCD in patients with congestive HF.50
Statins
They exert multiple antiarrhythmic effects, including direct effects on cardiac ion channels, plaque stabilization, and antifibrotic, antiinflammatory, and antioxidant actions.54,55 In addition, they suppress triggered activity in canine pulmonary vein preparations. Studies of statins for primary or secondary prevention of AF do not support specific recommendations, except perhaps for primary prevention of postoperative AF.51 Statins do not reduce the risk of ventricular arrhythmias or of cardiac arrest and produce a modest benefit on SCD in patients with CAD treated with an ICD and in patients with nonischemic cardiomyopathy.3,56
Omega-3 Polyunsaturated Fatty Acids
They modulate ion channels and connexins, reduce fluctuations in Ca2+i, and exert antiinflammatory and antioxidant actions.57 However, there is no robust evidence to support their efficacy for primary or secondary prevention of AF or VT/VF.3,51,57
Antifibrotic Agents
A variety of stimuli (i.e., fast heart rates, pressure/volume overload, oxidative stress, inflammation, ischemia, cytokines, growth factors, neurohumoral activation) induce the proliferation of cardiac fibroblasts and its differentiation into myofibroblasts.58,59 Myofibroblasts when electrotonically coupled to cardiomyocytes cause slow conduction, ectopic activity, and electric remodeling based on paracrine interactions, which generate an arrhythmogenic substrate in fibrotic hearts. Cardiac fibrosis is the final result of multiple signaling pathways that can vary in different cardiac diseases, and many potential therapeutic antifibrotic targets have been identified, although its clinical relevance remains uncertain.
Other AADs
Raised resting heart rate is a strong independent risk factor for cardiovascular events. The antianginal drug ivabradine, a selective If inhibitor, added on background therapy reduces the risk of cardiovascular death or hospitalization for worsening HF in patients with symptomatic HF and an LV ejection fraction of 35% or less with heart rate 70 beats/minute or greater.60 In addition, ivabradine reverses LV remodeling, suggesting that it prevents disease progression in these patients. Preliminary data suggest that ivabradine is a promising option in patients with inappropriate sinus tachycardia.
Unresolved Questions and Future Strategies
As mentioned, VT/VF associated with SCD preferentially occurs in the setting of myocardial ischemia, and it would be optimal to develop ischemia-selective AADs. Because class I and III AADs have failed to prevent ischemia-induced arrhythmias, antiarrhythmic strategy has been shifted to drugs commonly prescribed in patients with MI or HF that prevent plaque rupture and reduce myocardial ischemia, such as antianginal drugs (β-blockers, ivabradine, ranolazine), statins, RAAS inhibitors, and platelet antiaggregants, in an attempt to eliminate potential triggers for ventricular arrhythmias and, in the long-term, prevent the formation of myocardial scars and SCD. RAAS inhibitors and β-blockers might also limit the substrate for ventricular arrhythmias because they prevent LV remodeling after MI.3
Because AF is the most common arrhythmia, there are strategies for the development of atrial-selective AADs devoid of ventricular proarrhythmic effects based on the identification of atrial-specific targets. There are preliminary experimental data suggesting that several channels represent a possible atrial therapeutic target, including two-pore–domain K+ channels, Ca2+-activated nonselective cation channels, small-conductance Ca2+-activated K+ channels (SK1-3 channels are expressed in the atria), and transient receptor potential channels of the canonical (TRPC1/3/6) and melastin-related (TRPM3/4/6/7) subfamilies.9,14 However, because drugs used to target these currents lack specificity, the functional role of these channels in humans remains uncertain. In the same line, abnormal Ca2+ handling plays a major role in arrhythmogenesis, but the developed drugs do not have sufficient specificity.
References
1. Darbar, D. Standard antiarrhythmic drugs. In: Zipes D, Jalife J, eds. Cardiac electrophysiology: From cell to bedside. ed 5. Philadelphia: Saunders Elsevier; 2009:959–973.
2. Sheets, MF, Fozzard, HA, Lipkind, GM, et al. Sodium channel molecular conformations and antiarrhythmic drug affinity. Trends Cardiovasc Med. 2010; 20(1):16–21.
3. Das, MK, Zipes, DP. Antiarrhythmic and nonantiarrhythmic drugs for sudden cardiac death prevention. J Cardiovasc Pharmacol. 2010; 55(5):438–449.
4. Zipes, DP, Camm, AJ, Borggrefe, M, et al. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Circulation. 2006; 114(10):E385–E484.
5. Camm, AJ, Kirchhof, P, Lip, GY, et al. Guidelines for the management of atrial fibrillation: the Task Force for the Management of Atrial Fibrillation of the European Society of Cardiology (ESC). Eur Heart J. 2010; 31(19):2369–2429.
6. Fuster, V, Rydén, LE, Cannom, DS, et al. 2011 ACCF/AHA/HRS Focused Updates Incorporated Into the ACC/AHA/ESC 2006 Guidelines for the Management of Patients With Atrial Fibrillation: a Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines Developed in partnership with the European Society of Cardiology and in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. J Am Coll Cardiol. 2011; 57(11):e101–e198.
7. Hatem, SN, Coulombe, A, Balse, E. Specificities of atrial electrophysiology: Clues to a better understanding of cardiac function and the mechanisms of arrhythmias. J Mol Cell Cardiol. 2010; 48(1):90–95.
8. Burashnikov, A, Antzelevitch, C. Novel pharmacological targets for the rhythm control management of atrial fibrillation. Pharmacol Ther. 2011; 132(3):300–313.
9. Ravens, U. Antiarrhythmic therapy in atrial fibrillation. Pharmacol Ther. 2010; 128(1):129–145.
10. Zaza, A, Belardinelli, L, Shryock, JC. Pathophysiology and pharmacology of the cardiac “late sodium current. ”. Pharmacol Ther. 2008; 119(3):326–339.
11. Antzelevitch, C, Burashnikov, A, Sicouri, S, et al. Electrophysiologic basis for the antiarrhythmic actions of ranolazine. Heart Rhythm. 2011; 8(8):1281–1290.
12. Scirica, BM, Morrow, DA, Hod, H, et al. Effect of ranolazine, an antianginal agent with novel electrophysiological properties, on the incidence of arrhythmias in patients with non ST-segment elevation acute coronary syndrome: results from the Metabolic Efficiency With Ranolazine for Less Ischemia in Non ST-Elevation Acute Coronary Syndrome Thrombolysis in Myocardial Infarction 36 (MERLIN-TIMI 36) randomized controlled trial. Circulation. 2007; 116(15):1647–1652.
13. Schotten, U, Verheule, S, Kirchhof, P, et al. Pathophysiological mechanisms of atrial fibrillation: a translational appraisal. Physiol Rev. 2011; 91(1):265–325.
14. Wakili, R, Voigt, N, Kääb, S, et al. Recent advances in the molecular pathophysiology of atrial fibrillation. J Clin Invest. 2011; 121(8):2955–2968.
15. Vaquero, M, Calvo, D, Jalife, J. Cardiac fibrillation: from ion channels to rotors in the human heart. Heart Rhythm. 2008; 5(6):872–879.
16. Comtois, P, Sakabe, M, Vigmond, EJ, et al. Mechanisms of atrial fibrillation termination by rapidly unbinding Na+ channel blockers: insights from mathematical models and experimental correlates. Am J Physiol Heart Circ Physiol. 2008; 295(4):H1489–H1504.
17. López-Sendón, J, Swedberg, K, McMurray, J, et al. Expert consensus document on beta-adrenergic receptor blockers. Eur Heart J. 2004; 25(15):1341–1362.
18. Workman, AJ. Cardiac adrenergic control and atrial fibrillation. Naunyn Schmiedeberg Arch Pharmacol. 2010; 381(3):235–249.
19. Cerrone, M, Priori, SG. Genetics of sudden death: focus on inherited channelopathies. Eur Heart J. 2011; 32(17):2109–2118.
20. Werf, Cv, Zwinderman, AH, Wilde, AA. Therapeutic approach for patients with catecholaminergic polymorphic ventricular tachycardia: state of the art and future developments. Europace. 2012; 14(2):175–183.
21. Tamargo, J, Caballero, R, Gómez, R, et al. Pharmacology of cardiac potassium channels. Cardiovasc Res. 2004; 62(1):9–33.
22. Shah, RR, Hondeghem, LM. Refining detection of drug-induced proarrhythmia: QT interval and TRIaD. Heart Rhythm. 2005; 2(7):758–772.
23. Perry, M, Sanguinetti, M, Mitcheson, J. Revealing the structural basis of action of hERG potassium channel activators and blockers. J Physiol. 2010; 588(17):3157–3167.
24. Charpentier, F, Mérot, J, Loussouarn, G, et al. Delayed rectifier K+ currents and cardiac repolarization. J Mol Cell Cardiol. 2010; 48(1):37–44.
25. Varro, A, Baczkó, I. Cardiac repolarization reserve: a principle for understanding drug-related proarrhythmic risk. Br J Pharmacol. 2011; 164(1):14–36.
26. Caballero, R, de la Fuente, MG, Gómez, R, et al. In humans, chronic atrial fibrillation decreases the transient outward current and ultrarapid component of the delayed rectifier current differentially on each atria and increases the slow component of the delayed rectifier current in both. J Am Coll Cardiol. 2010; 55(21):2346–2354.
27. Muñoz, V, Grzeda, KR, Desplantez, T, et al. Adenoviral expression of IKs contributes to wavebreak and fibrillatory conduction in neonatal rat ventricular cardiomyocyte monolayers. Circ Res. 2007; 101(5):475–483.
28. Niwa, N, Nerbonne, JM. Molecular determinants of cardiac transient outward potassium current (Ito) expression and regulation. J Mol Cell Cardiol. 2010; 48(1):12–25.
29. Antzelevitch, C, Yan, GX. J wave syndromes. Heart Rhythm. 2010; 7(4):549–558.
30. Ravens, U, Wettwer, E. Ultra-rapid delayed rectifier channels: molecular basis and therapeutic implications. Cardiovasc Res. 2011; 89(4):776–785.
31. Pandit, SV, Berenfeld, O, Anumonwo, JM, et al. Ionic determinants of functional reentry in a 2-D model of human atrial cells during simulated chronic atrial fibrillation. Biophys J. 2005; 88(6):3806–3821.
32. Tamargo, J, Caballero, R, Gómez, R, et al. IKur/Kv1. 5 channel blockers for the treatment of atrial fibrillation. Expert Opin Investig Drugs. 2009; 18(4):399–416.
33. Anumonwo, JMB, Lopatin, AN. Cardiac strong inward rectifier potassium channels. J Mol Cell Cardiol. 2010; 48(1):45–54.
34. Jalife, J. Inward rectifier potassium channels control rotor frequency in ventricular fibrillation. Heart Rhythm. 2009; 6(11 Suppl):S44–S48.
35. Tristani-Firouzi, M, Etheridge, SP. Kir 2. 1 channelopathies: the Andersen-Tawil syndrome. Pflugers Arch. 2010; 460(2):289–294.
36. Dobrev, D, Friedrich, A, Voigt, N, et al. The G protein-gated potassium current IK,ACh is constitutively active in patients with chronic atrial fibrillation. Circulation. 2005; 112(24):3697–3706.
37. Billman, GE. The cardiac sarcolemmal ATP-sensitive potassium channel as a novel target for anti-arrhythmic therapy. Pharmacol Ther. 2008; 120(1):54–70.
38. Terzic, A, Alekseev, AE, Yamada, S, et al. Advances in cardiac ATP-sensitive K+ channelopathies from molecules to populations. Circ Arrhythm Electrophysiol. 2011; 4(4):577–585.
39. Piccini, JP, Berger, JS, O’Connor, CM. Amiodarone for the prevention of sudden cardiac death: a meta-analysis of randomized controlled trials. Eur Heart J. 2009; 30(10):1245–1253.
40. Severs, NJ, Bruce, AF, Dupont, E, et al. Remodelling of gap junctions and connexin expression in diseased myocardium. Cardiovasc Res. 2008; 80(1):9–19.
41. Dhein, S, Hagen, A, Jozwiak, J, et al. Improving cardiac gap junction communication as a new antiarrhythmic mechanism: the action of antiarrhythmic peptides. Naunyn Schmiedebergs Arch Pharmacol. 2010; 381(3):221–234.
42. Dyachenko, V, Husse, B, Rueckschloss, U, et al. Mechanical deformation of ventricular myocytes modulates both TRPC6 and Kir2. 3 channels. Cell Calcium. 2009; 45(1):38–54.
43. Schumacher, SM, Martens, JR. Ion channel trafficking: a new therapeutic horizon for atrial fibrillation. Heart Rhythm. 2010; 7(9):1309–1315.
44. Caballero, R, Dolz-Gaitón, P, Gómez, R, et al. Flecainide increases Kir2. 1 currents by interacting with cysteine 311, decreasing the polyamine-induced rectification. Proc Natl Acad Sci U S A. 2010; 107(35):15631–15636.
45. Sanguinetti, MC. HERG1 channelopathies. Pflugers Arch. 2010; 460(2):265–276.
46. Venetucci, L, Denegri, M, Napolitano, C, et al. Inherited calcium channelopathies in the pathophysiology of arrhythmias. Nat Rev Cardiol. 2012.
47. Dobrev, D, Voigt, N, Wehrens, XH. The ryanodine receptor channel as a molecular motif in atrial fibrillation: pathophysiological and therapeutic implications. Cardiovasc Res. 2011; 89(4):734–743.
48. Tamargo, J, López-Sendón, J. Novel therapeutic targets for the treatment of heart failure. Nat Rev Drug Discov. 2011; 10(7):536–555.
49. Michael, G, Xiao, L, Qi, XY, et al. Remodelling of cardiac repolarization: how homeostatic responses can lead to arrhythmogenesis. Cardiovasc Res. 2009; 81(3):491–499.
50. Savelieva, I, Kakouros, N, Kourliouros, A, et al. Upstream therapies for management of atrial fibrillation: review of clinical evidence and implications for European Society of Cardiology guidelines. Part I: primary prevention. Europace. 2011; 13(3):308–328.
51. Savelieva, I, Kakouros, N, Kourliouros, A, et al. Upstream therapies for management of atrial fibrillation: review of clinical evidence and implications for European Society of Cardiology guidelines. Part II: secondary prevention. Europace. 2011; 13(5):610–625.
52. Goette, A, Bukowska, A, Lendeckel, U. Non-ion channel blockers as anti-arrhythmic drugs (reversal of structural remodeling). Curr Opin Pharmacol. 2007; 7:219–224.
53. Schneider, MP, Hua, TA, Bohm, M, et al. Prevention of atrial fibrillation by renin-angiotensin system inhibition: a meta-analysis. J Am Coll Cardiol. 2010; 55(21):2299–2307.
54. Tamargo, J, Caballero, R, Gómez, R, et al. Lipid-lowering therapy with statins, a new approach to antiarrhythmic therapy. Pharmacol Ther. 2007; 114(1):107–126.
55. Thomas, G, Lerman, BB. Expanding the role of statins in postoperative atrial fibrillation. Heart Rhythm. 2012; 9(2):170–171.
56. Rahimi, K, Majoni, W, Merhi, A, et al. Effect of statins on ventricular tachyarrhythmia, cardiac arrest, and sudden cardiac death: a meta-analysis of published and unpublished evidence from randomized trials. Eur Heart J. 2012; 33(13):1571–1581.
57. Mozaffarian, D, Wu, JH. Omega-3 fatty acids and cardiovascular disease: effects on risk factors, molecular pathways, and clinical events. J Am Coll Cardiol. 2011; 58(20):2047–2067.
58. Rohr, S. Arrhythmogenic implications of fibroblast-myocyte interactions. Circ Arrhythm Electrophysiol. 2012; 5(2):442–452.
59. de Jong, S, van Veen, TA, van Rijen, HV, et al. Fibrosis and cardiac arrhythmias. J Cardiovasc Pharmacol. 2011; 57(6):630–638.
60. Tardif, JC, O’Meara, E, Komajda, M, et al. Effects of selective heart rate reduction with ivabradine on left ventricular remodelling and function: results from the SHIFT echocardiography substudy. SHIFT Investigators. Eur Heart J. 2011; 32(20):2507–2515.