Application of Pharmacokinetics in Pharmacotherapeutics
By applying knowledge of pharmacokinetics to drug therapy, we can help maximize beneficial effects and minimize harm. Recall that the intensity of the response to a drug is directly related to the concentration of the drug at its site of action. To maximize beneficial effects, a drug must achieve concentrations that are high enough to elicit desired responses; to minimize harm, we must avoid concentrations that are too high. This balance is achieved by selecting the most appropriate route, dosage, and dosing schedule.
Passage of Drugs Across Membranes
All four phases of pharmacokinetics—absorption, distribution, metabolism, and excretion—involve drug movement. To move throughout the body, drugs must cross membranes. Drugs must cross membranes to enter the blood from their site of administration. When in the blood, drugs must cross membranes to leave the vascular system and reach their sites of action. In addition, drugs must cross membranes to undergo metabolism and excretion. Accordingly, the factors that determine the passage of drugs across biologic membranes have a profound influence on all aspects of pharmacokinetics.
Biologic membranes are composed of layers of individual cells. The cells composing most membranes are very close to one another—so close, in fact, that drugs must usually pass through cells, rather than between them, to cross the membrane. Hence the ability of a drug to cross a biologic membrane is determined primarily by its ability to pass through single cells.
Three Ways to Cross a Cell Membrane
The three most important ways by which drugs cross cell membranes are (1) passage through channels or pores, (2) passage with the aid of a transport system, and (3) direct penetration of the membrane. Of the three, direct penetration of the membrane is most common.
Channels and Pores
Very few drugs cross membranes through channels or pores. The channels in membranes are extremely small and are specific for certain molecules. Consequently, only the smallest of compounds, such as potassium and sodium, can pass through these channels, and then only if the channel is the right one.
Transport Systems
Transport systems are carriers that can move drugs from one side of the cell membrane to the other side. All transport systems are selective. Whether a transporter will carry a particular drug depends on the drug’s structure.
Transport systems are an important means of drug transit. For example, certain orally administered drugs could not be absorbed unless there were transport systems to move them across the membranes that separate the lumen of the intestine from the blood. A number of drugs could not reach intracellular sites of action without a transport system to move them across the cell membrane. One transporter, known as P-glycoprotein (PGP) or multidrug transporter protein, deserves special mention. PGP is a transmembrane protein that transports a wide variety of drugs out of cells.
Direct Penetration of the Membrane
For most drugs, movement throughout the body is dependent on the ability to penetrate membranes directly because (1) most drugs are too large to pass through channels or pores, and (2) most drugs lack transport systems to help them cross all of the membranes that separate them from their sites of action, metabolism, and excretion.
A general rule in chemistry states that “like dissolves like.” Membranes are composed primarily of lipids; therefore, to directly penetrate membranes, a drug must be lipid soluble (lipophilic).
Certain kinds of molecules are not lipid soluble and therefore cannot penetrate membranes. This group consists of polar molecules and ions.
Polar Molecules
Polar molecules are molecules that have no net charge; however, they have an uneven distribution of electrical charge. That is, positive and negative charges within the molecule tend to congregate separately from one another. Water is the classic example. As depicted in Fig. 4.1, the electrons (negative charges) in the water molecule spend more time in the vicinity of the oxygen atom than in the vicinity of the two hydrogen atoms. As a result, the area around the oxygen atom tends to be negatively charged, whereas the area around the hydrogen atoms tends to be positively charged. In accord with the “like dissolves like” rule, polar molecules will dissolve in polar solvents (such as water) but not in nonpolar solvents (such as lipids).
Ions
Ions are defined as molecules that have a net electrical charge (either positive or negative). Except for very small molecules, ions are unable to cross membranes; therefore, they must become nonionized in order to cross from one side to the other. Many drugs are either weak organic acids or weak organic bases, which can exist in charged and uncharged forms. Whether a weak acid or base carries an electrical charge is determined by the pH of the surrounding medium. Acids tend to ionize in basic (alkaline) media, whereas bases tend to ionize in acidic media. Therefore drugs that are weak acids are best absorbed in an acidic environment such as gastric acid because they remain in a nonionized form. When aspirin molecules pass from the stomach into the small intestine, where the environment is relatively alkaline, more of the molecules change to their ionized form. As a result, absorption of aspirin from the intestine is impeded.
Ion Trapping (pH Partitioning)
Because the ionization of drugs is pH dependent, when the pH of the fluid on one side of a membrane differs from the pH of the fluid on the other side, drug molecules tend to accumulate on the side where the pH most favors their ionization. Accordingly, because acidic drugs tend to ionize in basic media, and because basic drugs tend to ionize in acidic media, when there is a pH gradient between two sides of a membrane, the following occur:
The process whereby a drug accumulates on the side of a membrane where the pH most favors its ionization is referred to as ion trapping or pH partitioning.
Absorption
Absorption is defined as the movement of a drug from its site of administration into the systemic circulation. The rate of absorption determines how soon effects will begin. The amount of absorption helps determine how intense effects will be. Two other terms associated with absorption are chemical equivalence and bioavailability. Drug preparations are considered chemically equivalent if they contain the same amount of the identical chemical compound (drug). Preparations are considered equal in bioavailability if the drug they contain is absorbed at the same rate and to the same extent. It is possible for two formulations of the same drug to be chemically equivalent while differing in bioavailability. The concept of bioavailability is discussed further in Chapter 6.
Factors Affecting Drug Absorption
The rate at which a drug undergoes absorption is influenced by the physical and chemical properties of the drug and by physiologic and anatomic factors at the absorption site.
Rate of Dissolution
Before a drug can be absorbed, it must first dissolve. Hence, the rate of dissolution helps determine the rate of absorption. Drugs in formulations that allow rapid dissolution have a faster onset than drugs formulated for slow dissolution.
Surface Area
The surface area available for absorption is a major determinant of the rate of absorption. When the surface area is larger, absorption is faster. For this reason, absorption of orally administered drugs is usually greater from the small intestine rather than from the stomach. (Recall that the small intestine, because of its lining of microvilli, has an extremely large surface area, whereas the surface area of the stomach is relatively small.)
Blood Flow
Drugs are absorbed most rapidly from sites where blood flow is high because blood containing a newly absorbed drug will be replaced rapidly by drug-free blood, thereby maintaining a large gradient between the concentration of drug outside the blood and the concentration of drug in the blood. The greater the concentration gradient, the more rapid absorption will be.
Lipid Solubility
As a rule, highly lipid-soluble drugs are absorbed more rapidly than drugs whose lipid solubility is low. This occurs because lipid-soluble drugs can readily cross the membranes that separate them from the blood, whereas drugs of low lipid solubility cannot.
pH Partitioning
pH partitioning can influence drug absorption. Absorption will be enhanced when the difference between the pH of plasma and the pH at the site of administration is such that drug molecules will have a greater tendency to be ionized in the plasma.
Characteristics of Commonly Used Routes of Administration
For each of the major routes of administration—oral (PO), intravenous (IV), intramuscular (IM), and subcutaneous (subQ)—the pattern of drug absorption (i.e., the rate and extent of absorption) is unique. Consequently, the route by which a drug is administered significantly affects both the onset and the intensity of effects. The distinguishing characteristics of the four major routes are summarized in Table 4.1. Additional routes of administration (e.g., topical, transdermal, inhaled) each have unique characteristics that are addressed throughout the book as we discuss specific drugs that employ them.
TABLE 4.1
Properties of Major Routes of Drug Administration
| Route | Barriers to Absorption | Absorption Pattern | Advantages | Disadvantages |
| PARENTERAL | ||||
| Intravenous (IV) | None (absorption is bypassed) | Instantaneous | ||
| Intramuscular (IM) | Capillary wall (easy to pass) | |||
| Subcutaneous (subQ) | Same as IM | Same as IM | Same as IM | Same as IM |
| ENTERAL | ||||
| Oral (PO) | Epithelial lining of gastrointestinal tract; capillary wall | Slow and variable | ||
Distribution
Distribution is defined as the movement of drugs from the systemic circulation to the site of drug action. Drug distribution is determined by three major factors: blood flow to tissues, the ability of a drug to exit the vascular system, and, to a lesser extent, the ability of a drug to enter cells.
Blood Flow to Tissues
In the first phase of distribution, drugs are carried by the blood to the tissues and organs of the body. The rate at which drugs are delivered to a particular tissue is determined by blood flow to that tissue. Because most tissues are well perfused, regional blood flow is rarely a limiting factor in drug distribution.
There are two pathologic conditions—abscesses and tumors—in which low regional blood flow can affect drug therapy. An abscess has no internal blood vessels; therefore, because abscesses lack a blood supply, antibiotics cannot reach the bacteria within. Accordingly, if drug therapy is to be effective, the abscess must usually be surgically drained.
Solid tumors have a limited blood supply. Although blood flow to the outer regions of tumors is relatively high, blood flow becomes progressively lower toward the core. As a result, it may not be possible to achieve high drug levels deep inside tumors. Limited blood flow is a major reason that solid tumors are resistant to drug therapy.
Exiting the Vascular System
After a drug has been delivered to an organ or tissue by blood circulation, the next step is to exit the vasculature. Because most drugs do not produce their effects within the blood, the ability to leave the vascular system is an important determinant of drug actions. Drugs in the vascular system leave the blood at capillary beds.
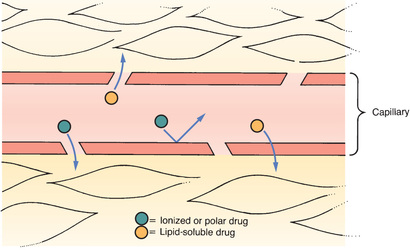
Typical Capillary Beds
Most capillary beds offer no resistance to the departure of drugs because, in most tissues, drugs can leave the vasculature simply by passing through pores in the capillary wall. Because drugs pass between capillary cells rather than through them, movement into the interstitial space is not impeded. The exit of drugs from a typical capillary bed is depicted in Fig. 4.2.
The Blood-Brain Barrier
The term blood-brain barrier (BBB) refers to the unique anatomy of capillaries in the central nervous system (CNS). As shown in Fig. 4.3, there are tight junctions between the cells that compose the walls of most capillaries in the CNS. These junctions are so tight that they prevent drug passage. Consequently, to leave the blood and reach sites of action within the brain, a drug must be able to pass through cells of the capillary wall. Only drugs that are lipid soluble or have a transport system can cross the BBB to a significant degree.
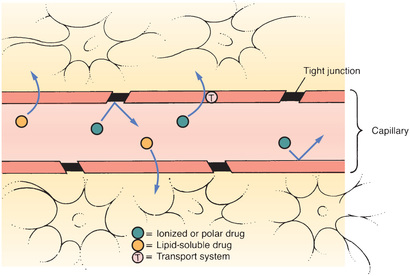
Recent evidence indicates that, in addition to tight junctions, the BBB has another protective component: PGP. As noted earlier, PGP is a transporter that pumps a variety of drugs out of cells. In capillaries of the CNS, PGP pumps drugs back into the blood and thereby limits their access to the brain.
The BBB is not fully developed at birth. As a result, newborns have heightened sensitivity to medicines that act on the brain. Likewise, neonates are especially vulnerable to CNS toxicity.
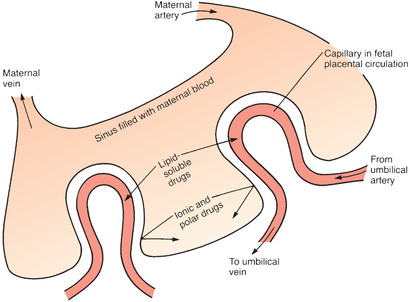
Placental Drug Transfer
The membranes of the placenta separate the maternal circulation from the fetal circulation (Fig. 4.4). However, the membranes of the placenta do NOT constitute an absolute barrier to the passage of drugs. The same factors that determine the movement of drugs across other membranes determine the movement of drugs across the placenta. Accordingly, lipid-soluble, nonionized compounds readily pass from the maternal bloodstream into the blood of the fetus. In contrast, compounds that are ionized, highly polar, or protein bound are largely excluded—as are drugs that are substrates for the PGP transporter that can pump a variety of drugs out of placental cells into the maternal blood.
Protein Binding
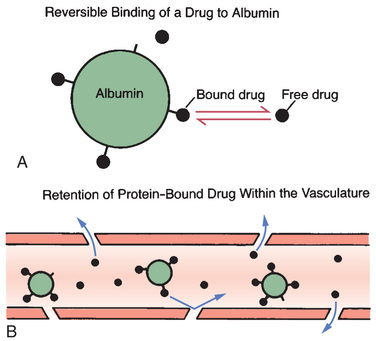
Drugs can form reversible bonds with various proteins in the body. Of all the proteins with which drugs can bind, plasma albumin is the most important. Like other proteins, albumin is a large molecule. Because of its size, albumin is too large to leave the bloodstream.
Fig. 4.5 depicts the binding of drug molecules to albumin. Note that the drug molecules are much smaller than albumin. As indicated by the two-way arrows, binding between albumin and drugs is reversible. Hence, drugs may be bound or unbound (free).
Even though a drug can bind albumin, only some molecules will be bound at any moment. The percentage of drug molecules that are bound is determined by the strength of the attraction between albumin and the drug. For example, the attraction between albumin and the anticoagulant warfarin is strong, causing nearly all (99%) of the warfarin molecules in plasma to be bound, leaving only 1% free. On the other hand, the attraction between the antibiotic gentamicin and albumin is relatively weak; less than 10% of the gentamicin molecules in plasma are bound, leaving more than 90% free.
An important consequence of protein binding is restriction of drug distribution. Because albumin is too large to leave the bloodstream, drug molecules that are bound to albumin cannot leave either (see Fig. 4.5B). As a result, bound molecules cannot reach their sites of action or undergo metabolism or excretion until the drug-protein bond is broken so that the drug is free to leave the circulation.
In addition to restricting drug distribution, protein binding can be a source of drug interactions. As suggested by Fig. 4.5A, each molecule of albumin has only a few sites to which drug molecules can bind. Because the number of binding sites is limited, drugs with the ability to bind albumin will compete with one another for those sites. As a result, one drug can displace another from albumin, causing the free concentration of the displaced drug to rise, thus increasing the intensity of drug responses. If plasma drug levels rise sufficiently, toxicity can result.
Entering Cells
Many drugs produce their effects by binding with receptors located on the external surface of the cell membrane; however, some drugs must enter cells to reach their sites of action, and practically all drugs must enter cells to undergo metabolism and excretion. The factors that determine the ability of a drug to cross cell membranes are the same factors that determine the passage of drugs across all other membranes, namely, lipid solubility, the presence of a transport system, or both.
Metabolism
Drug metabolism, also known as biotransformation, is defined as the enzymatic alteration of drug structure. Most drug metabolism takes place in the liver.
Hepatic Drug-Metabolizing Enzymes
Most drug metabolism that takes place in the liver is performed by the hepatic microsomal enzyme system, also known as the P450 system. The term P450 refers to cytochrome P450, a key component of this enzyme system.
It is important to appreciate that cytochrome P450 is not a single molecular entity, but rather a group of 12 closely related enzyme families. Three of the cytochrome P450 (CYP) families—designated CYP1, CYP2, and CYP3—metabolize drugs. The other nine families metabolize endogenous compounds (e.g., steroids, fatty acids). Each of the three P450 families that metabolize drugs is composed of multiple forms, each of which metabolizes only certain drugs. To identify the individual forms of cytochrome P450, designations such as CYP1A2, CYP2D6, and CYP3A4 are used to indicate specific members of the CYP1, CYP2, and CYP3 families, respectively.
Therapeutic Consequences of Drug Metabolism
Drug metabolism has six possible consequences of therapeutic significance:
Accelerated Renal Drug Excretion
The most important consequence of drug metabolism is promotion of renal drug excretion. The kidneys, which are the major organs of drug excretion, are unable to excrete drugs that are highly lipid soluble. Hence, by converting lipid-soluble drugs into more hydrophilic (water-soluble) forms, metabolic conversion can accelerate renal excretion of many agents.
Drug Inactivation
Drug metabolism can convert pharmacologically active compounds to inactive forms. This is the most common end result of drug metabolism.
Increased Therapeutic Action
Metabolism can increase the effectiveness of some drugs. For example, metabolism converts codeine into morphine. The analgesic activity of morphine is so much greater than that of codeine that formation of morphine may account for virtually all the pain relief that occurs after codeine administration.
Activation of Prodrugs
A prodrug is a compound that is pharmacologically inactive as administered and then undergoes conversion to its active form through metabolism. Prodrugs have several advantages; for example, a drug that cannot cross the BBB may be able to do so as a lipid-soluble prodrug that is converted to the active form in the CNS.
Increased or Decreased Toxicity
By converting drugs into inactive forms, metabolism can decrease toxicity. Conversely, metabolism can increase the potential for harm by converting relatively safe compounds into forms that are toxic. Increased toxicity is illustrated by the conversion of acetaminophen into a hepatotoxic metabolite. It is this product of metabolism, and not acetaminophen itself, that causes injury when acetaminophen is taken in overdose.
Special Considerations in Drug Metabolism
Several factors can influence the rate at which drugs are metabolized. These must be accounted for in drug therapy.
Age
The drug-metabolizing capacity of infants is limited. The liver does not develop its full capacity to metabolize drugs until about 1 year after birth. During the time before hepatic maturation, infants are especially sensitive to drugs, and care must be taken to avoid injury. Similarly, the ability of older adults to metabolize drugs is commonly decreased. Drug dosages may need to be reduced to prevent drug toxicity.
Induction and Inhibition of Drug-Metabolizing Enzymes
Drugs may be P450 substrates, P450 enzyme inducers, and P450 enzyme inhibitors. Drugs that are metabolized by P450 hepatic enzymes are substrates. Drugs that increase the rate of drug metabolism are inducers. Drugs that decrease the rate of drug metabolism are called inhibitors. Often a drug may have more than one property. For example, a drug may be both a substrate and an inducer.
Inducers act on the liver to stimulate enzyme synthesis. This process is known as induction. By increasing the rate of drug metabolism, the amount of active drug is decreased and plasma drug levels fall. If dosage adjustments are not made to accommodate for this, a drug may not achieve therapeutic levels.
Inhibitors act on the liver through a process known as inhibition. By slowing the rate of metabolism, inhibition can cause an increase in active drug accumulation. This can lead to an increase in adverse effects and toxicity.
First-Pass Effect
The term first-pass effect refers to the rapid hepatic inactivation of certain oral drugs. When drugs are absorbed from the gastrointestinal tract, they are carried directly to the liver through the hepatic portal vein before they enter the systemic circulation. If the capacity of the liver to metabolize a drug is extremely high, that drug can be completely inactivated on its first pass through the liver. As a result, no therapeutic effects can occur. To circumvent the first-pass effect, a drug that undergoes rapid hepatic metabolism is often administered parenterally. This permits the drug to temporarily bypass the liver, thereby allowing it to reach therapeutic levels in the systemic circulation before being metabolized.
Nutritional Status
Hepatic drug-metabolizing enzymes require a number of cofactors to function. In the malnourished patient, these cofactors may be deficient, causing drug metabolism to be compromised.
Competition Between Drugs
When two drugs are metabolized by the same metabolic pathway, they may compete with each other for metabolism and may thereby decrease the rate at which one or both agents are metabolized. If metabolism is depressed enough, a drug can accumulate to dangerous levels.
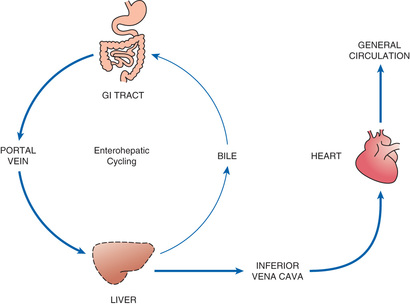
Enterohepatic Recirculation
As noted earlier and depicted in Fig. 4.6, enterohepatic recirculation is a repeating cycle in which a drug is transported from the liver into the duodenum (through the bile duct) and then back to the liver (through the portal blood). It is important to note, however, that only certain drugs are affected. Specifically, the process is limited to drugs that have undergone glucuronidation, a process that converts lipid-soluble drugs to water-soluble drugs by binding them to glucuronic acid. After glucuronidation, these drugs can enter the bile and then pass to the duodenum. In the intestine, some drugs can be hydrolyzed by intestinal beta-glucuronidase, an enzyme that breaks the bond between the original drug and the glucuronide moiety, thereby releasing the free drug. Because the free drug is more lipid soluble than the glucuronidated form, the free drug can undergo reabsorption across the intestinal wall, followed by transport back to the liver, where the cycle can start again. Because of enterohepatic recycling, drugs can remain in the body much longer than they otherwise would.
Excretion
Drug excretion is defined as the removal of drugs from the body. Drugs and their metabolites can exit the body in urine, bile, sweat, saliva, breast milk, and expired air. The most important organ for drug excretion is the kidney.
Renal Drug Excretion
The kidneys account for the majority of drug excretion. When the kidneys are healthy, they serve to limit the duration of action of many drugs. Conversely, if renal failure occurs, both the duration and intensity of drug responses may increase.
Steps in Renal Drug Excretion
Urinary excretion is the net result of three processes: (1) glomerular filtration, (2) passive tubular reabsorption, and (3) active tubular secretion.
Glomerular Filtration
Renal excretion begins at the glomerulus of the kidney tubule. As blood flows through the glomerular capillaries, fluids and small molecules—including drugs—are forced through the pores of the capillary wall. This process, called glomerular filtration, moves drugs from the blood into the tubular urine. Blood cells and large molecules (e.g., proteins) are too big to pass through the capillary pores and therefore do not undergo filtration. Because large molecules are not filtered, drugs bound to albumin remain in the blood.
Passive Tubular Reabsorption
As depicted in Fig. 4.7, the vessels that deliver blood to the glomerulus return to proximity with the renal tubule at a point distal to the glomerulus. At this distal site, drug concentrations in the blood are lower than drug concentrations in the tubule. This concentration gradient acts as a driving force to move drugs from the lumen of the tubule back into the blood. Because lipid-soluble drugs can readily cross the membranes that compose the tubular and vascular walls, drugs that are lipid soluble undergo passive reabsorption from the tubule back into the blood. In contrast, drugs that are not lipid soluble (ions and polar compounds) remain in the urine to be excreted.
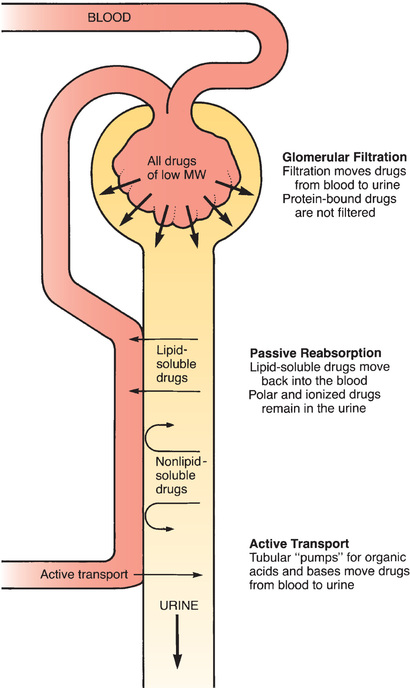
Active Tubular Secretion
There are active transport systems in the kidney tubules that pump drugs from the blood to the tubular urine. These pumps have a relatively high capacity and play a significant role in excreting certain compounds.
Factors That Modify Renal Drug Excretion
Renal drug excretion varies from patient to patient. Conditions such as chronic renal disease may cause profound alterations. Three other important factors to consider are pH-dependent ionization, competition for active tubular transport, and patient age.
pH-Dependent Ionization
The phenomenon of pH-dependent ionization can be used to accelerate renal excretion of drugs. Recall that passive tubular reabsorption is limited to lipid-soluble compounds. Because ions are not lipid soluble, drugs that are ionized at the pH of tubular urine will remain in the tubule and be excreted. Consequently, by manipulating urinary pH in such a way as to promote the ionization of a drug, we can decrease passive reabsorption back into the blood and can thereby hasten the drug’s elimination. This principle has been employed to promote the excretion of poisons as well as medications that have been taken in toxic doses.
Competition for Active Tubular Transport
Competition between drugs for active tubular transport can delay renal excretion, thereby prolonging effects. The active transport systems of the renal tubules can be envisioned as motor-driven revolving doors that carry drugs from the plasma into the renal tubules. These “revolving doors” can carry only a limited number of drug molecules per unit of time. Accordingly, if there are too many molecules present, some must wait their turn. Because of competition, if we administer two drugs at the same time, and if both drugs use the same transport system, excretion of each will be delayed by the presence of the other.
Age
The kidneys of newborns are not fully developed. Until their kidneys reach full capacity (a few months after birth), infants have a limited capacity to excrete drugs. This must be accounted for when medicating an infant.
In old age, renal function often declines. Older adults have smaller kidneys and fewer nephrons. The loss of nephrons results in decreased blood filtration. Additionally, vessel changes such as atherosclerosis reduce renal blood flow. As a result, renal excretion of drugs is decreased.
Nonrenal Routes of Drug Excretion
In most cases, excretion of drugs by nonrenal routes has minimal clinical significance. However, in certain situations, nonrenal excretion can have important therapeutic and toxicologic consequences.
Breast Milk
Some drugs taken by breast-feeding women can undergo excretion into milk. As a result, breastfeeding can expose the nursing infant to drugs. The factors that influence the appearance of drugs in breast milk are the same factors that determine the passage of drugs across membranes. Accordingly, lipid-soluble drugs have ready access to breast milk, whereas drugs that are polar, ionized, or protein bound cannot enter in significant amounts.
Other Nonrenal Routes of Excretion
The bile is an important route of excretion for certain drugs. Because bile is secreted into the small intestine, drugs that do not undergo enterohepatic recirculation leave the body in the feces.
The lungs are the major route by which volatile anesthetics are excreted. Alcohol is partially eliminated by this route.
Small amounts of drugs can appear in sweat and saliva. These routes have little therapeutic or toxicologic significance.
Time Course of Drug Responses
It is possible to regulate the time at which drug responses start, the time they are most intense, and the time they cease. Because the four pharmacokinetic processes—absorption, distribution, metabolism, and excretion—determine how much drug will be at its sites of action at any given time, these processes are the major determinants of the time course over which drug responses take place.
Plasma Drug Levels
In most cases, the time course of drug action bears a direct relationship to the concentration of a drug in the blood. Hence, before discussing the time course per se, we need to review several important concepts related to plasma drug levels.
Clinical Significance of Plasma Drug Levels
Providers frequently monitor plasma drug levels in efforts to regulate drug responses. When measurements indicate that drug levels are inappropriate, these levels can be adjusted up or down by changing dosage size, dosage timing, or both.
The practice of regulating plasma drug levels to control drug responses should seem a bit odd, given that (1) drug responses are related to drug concentrations at sites of action and (2) the site of action of most drugs is not in the blood. More often than not, it is a practical impossibility to measure drug concentrations at sites of action. Experience has shown that, for most drugs, there is a direct correlation between therapeutic and toxic responses and the amount of drug present in plasma. Therefore, although we cannot usually measure drug concentrations at sites of action, we can determine plasma drug concentrations that, in turn, are highly predictive of therapeutic and toxic responses. Accordingly, the dosing objective is commonly spoken of in terms of achieving a specific plasma level of a drug.
Two Plasma Drug Levels Defined
Two plasma drug levels are of special importance: (1) the minimum effective concentration, and (2) the toxic concentration. These levels are depicted in Fig. 4.8.
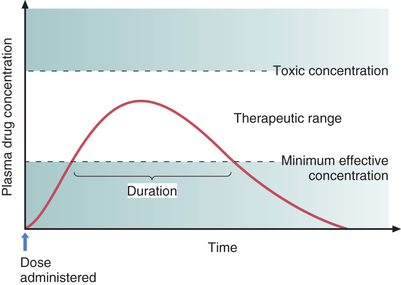
Minimum Effective Concentration
The minimum effective concentration (MEC) is defined as the plasma drug level below which therapeutic effects will not occur. Hence, to be of benefit, a drug must be present in concentrations at or above the MEC.
Toxic Concentration
Toxicity occurs when plasma drug levels climb too high. The plasma level at which toxic effects begin is termed the toxic concentration. Doses must be kept small enough so that the toxic concentration is not reached.
Therapeutic Range
As indicated in Fig. 4.8, there is a range of plasma drug levels, falling between the MEC and the toxic concentration, which is termed the therapeutic range. When plasma levels are within the therapeutic range, there is enough drug present to produce therapeutic responses but not so much that toxicity results. The objective of drug dosing is to maintain plasma drug levels within the therapeutic range.
The width of the therapeutic range is a major determinant of the ease with which a drug can be used safely. Drugs that have a narrow therapeutic range are difficult to administer safely. Conversely, drugs that have a wide therapeutic range can be administered safely with relative ease. The principle is the same as that of the therapeutic index discussed in Chapter 3. The therapeutic range is quantified, or measured, by the therapeutic index.
Understanding the concept of therapeutic range can facilitate patient care. Because drugs with a narrow therapeutic range are more dangerous than drugs with a wide therapeutic range, patients taking drugs with a narrow therapeutic range are the most likely to require intervention for drug-related complications. The provider who is aware of this fact can focus additional attention on monitoring these patients for signs and symptoms of toxicity.
Single-Dose Time Course
Fig. 4.8 shows how plasma drug levels change over time after a single dose of an oral medication. Drug levels rise as the medicine undergoes absorption. Drug levels then decline as metabolism and excretion eliminate the drug from the body.
Because responses cannot occur until plasma drug levels have reached the MEC, there is a latent period between drug administration and onset of effects. The extent of this delay is determined by the rate of absorption.
The duration of effects is determined largely by the combination of metabolism and excretion. As long as drug levels remain above the MEC, therapeutic responses will be maintained; when levels fall below the MEC, benefits will cease. Because metabolism and excretion are the processes most responsible for causing plasma drug levels to fall, these processes are the primary determinants of how long drug effects will persist.
Drug Half-Life
Before proceeding to the topic of multiple dosing, we need to discuss the concept of half-life. When a patient ceases drug use, the combination of metabolism and excretion will cause the amount of drug in the body to decline. The half-life of a drug is an index of just how rapidly that decline occurs for most drugs. (The concept of half-life does not apply to the elimination of all drugs. A few agents, most notably ethanol (alcohol), leave the body at a constant rate, regardless of how much is present. The implications of this kind of decline for ethanol are discussed in Chapter 31.
Drug half-life is defined as the time required for the amount of drug in the body to decrease by 50%. A few drugs have half-lives that are extremely short—on the order of minutes or less. In contrast, the half-lives of some drugs exceed 1 week.
Note that, in our definition of half-life, a percentage—not a specific amount—of drug is lost during one half-life. That is, the half-life does not specify, for example, that 2 g or 18 mg will leave the body in a given time. Rather, the half-life tells us that, no matter what the amount of drug in the body may be, half (50%) will leave during a specified period of time (the half-life). The actual amount of drug that is lost during one half-life depends on just how much drug is present: the more drug in the body, the larger the amount lost during one half-life.
The concept of half-life is best understood through an example. Morphine provides a good illustration. The half-life of morphine is approximately 3 hours. By definition, this means that body stores of morphine will decrease by 50% every 3 hours—regardless of how much morphine is in the body. If there are 50 mg of morphine in the body, 25 mg (50% of 50 mg) will be lost in 3 hours; if there are only 2 mg of morphine in the body, only 1 mg (50% of 2 mg) will be lost in 3 hours. Note that, in both cases, morphine levels drop by 50% during an interval of one half-life. However, the actual amount lost is larger when total body stores of the drug are higher.
The half-life of a drug determines the dosing interval (i.e., how much time separates each dose). For drugs with a short half-life, the dosing interval must be correspondingly short. If a long dosing interval is used, drug levels will fall below the MEC between doses, and therapeutic effects will be lost. Conversely, if a drug has a long half-life, a long time can separate doses without loss of benefits.
Drug Levels Produced With Repeated Doses
Multiple dosing leads to drug accumulation. When a patient takes a single dose of a drug, plasma levels simply go up and then come back down. In contrast, when a patient takes repeated doses of a drug, the process is more complex and results in drug accumulation. The factors that determine the rate and extent of accumulation are considered next.
The Process by Which Plateau Drug Levels Are Achieved
Administering repeated doses will cause a drug to build up in the body until a plateau (steady level) has been achieved. What causes drug levels to reach plateau? If a second dose of a drug is administered before all of the prior dose has been eliminated, total body stores of that drug will be higher after the second dose than after the initial dose. As succeeding doses are administered, drug levels will climb even higher. The drug will continue to accumulate until a state has been achieved in which the amount of drug eliminated between doses equals the amount administered. When the amount of drug eliminated between doses equals the dose administered, average drug levels will remain constant and plateau will have been reached (Fig. 4.9).
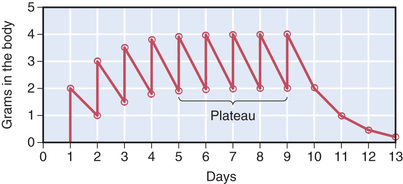
Time to Plateau
When a drug is administered repeatedly in the same dose, plateau will be reached in approximately four half-lives. For the hypothetical agent illustrated in Fig. 4.9, total body stores approached their peak near the beginning of day 5, or approximately 4 full days after treatment began. Because the half-life of this drug is 1 day, reaching plateau in 4 days is equivalent to reaching plateau in four half-lives.
As long as dosage remains constant, the time required to reach plateau is independent of dosage size. Put another way, the time required to reach plateau when giving repeated large doses of a particular drug is identical to the time required to reach plateau when giving repeated small doses of that drug. Referring to the drug in Fig. 4.9, just as it took four half-lives (4 days) to reach plateau when a dose of 2 g was administered daily, it would also take four half-lives to reach plateau if a dose of 4 g were administered daily. It is true that the height of the plateau would be greater if a 4-g dose were given, but the time required to reach plateau would not be altered by the increase in dosage. To confirm this statement, substitute a dose of 4 g in the previous exercise and see when plateau is reached.
Techniques for Reducing Fluctuations in Drug Levels
As illustrated in Fig. 4.9, when a drug is administered repeatedly, its level will fluctuate between doses. The highest level is referred to as the peak concentration, and the lowest level is referred to as the trough concentration. The acceptable height of the peaks and troughs will depend on the drug’s therapeutic range: the peaks must be kept below the toxic concentration, and the troughs must be kept above the MEC. If there is not much difference between the toxic concentration and the MEC, then fluctuations must be kept to a minimum.
Three techniques can be employed to reduce fluctuations in drug levels. One technique is to administer drugs by continuous infusion. With this procedure, plasma levels can be kept nearly constant. Another is to administer a depot preparation, which releases the drug slowly and steadily. The third is to reduce both the size of each dose and the dosing interval (keeping the total daily dose constant). For example, rather than giving the drug from Fig. 4.9 in 2-g doses once every 24 hours, we could give this drug in 1-g doses every 12 hours. With this altered dosing schedule, the total daily dose would remain unchanged, as would total body stores at plateau. However, instead of fluctuating over a range of 2 g between doses, levels would fluctuate over a range of 1 g.
Loading Doses Versus Maintenance Doses
As discussed previously, if we administer a drug in repeated doses of equal size, an interval equivalent to about four half-lives is required to achieve plateau. When plateau must be achieved more quickly, a large initial dose can be administered. This large initial dose is called a loading dose. After high drug levels have been established with a loading dose, plateau can be maintained by giving smaller doses. These smaller doses are referred to as maintenance doses.
The claim that use of a loading dose will shorten the time to plateau may appear to contradict an earlier statement, which said that the time to plateau is not affected by dosage size. However, there is no contradiction. For any specified dosage, it will always take about four half-lives to reach plateau. When a loading dose is administered followed by maintenance doses, the plateau is not reached for the loading dose. Rather, we have simply used the loading dose to rapidly produce a drug level equivalent to the plateau level for a smaller dose. To achieve plateau level for the loading dose, it would be necessary to either administer repeated doses equivalent to the loading dose for a period of four half-lives or administer a dose even larger than the original loading dose.
Decline From Plateau
When drug administration is discontinued, most (94%) of the drug in the body will be eliminated over an interval equal to about four half-lives. The time required for drugs to leave the body is important when toxicity develops. If a drug has a short half-life, body stores will decline rapidly, thereby making management of overdose less difficult. When an overdose of a drug with a long half-life occurs, however, toxic levels of the drug will remain in the body for a long time. Additional management may be needed in these instances.
Pharmacodynamics
Pharmacodynamics is the study of the biochemical and physiologic effects of drugs on the body and the molecular mechanisms by which those effects are produced. To participate rationally in achieving the therapeutic objective, an understanding of pharmacodynamics is essential.
Dose-Response Relationships
The dose-response relationship (i.e., the relationship between the size of an administered dose and the intensity of the response produced) is a fundamental concern in therapeutics. Dose-response relationships determine the minimal amount of drug needed to elicit a response, the maximal response a drug can elicit, and how much to increase the dosage to produce the desired increase in response.
Basic Features of the Dose-Response Relationship
The basic characteristics of dose-response relationships are illustrated in Fig. 4.10. Part A shows dose-response data plotted on linear coordinates. Part B shows the same data plotted on semilogarithmic coordinates (i.e., the scale on which dosage is plotted is logarithmic rather than linear). The most obvious and important characteristic revealed by these curves is that the dose-response relationship is graded. That is, as the dosage increases, the response becomes progressively larger. Because drug responses are graded, therapeutic effects can be adjusted to fit the needs of each patient by raising or lowering the dosage until a response of the desired intensity is achieved.
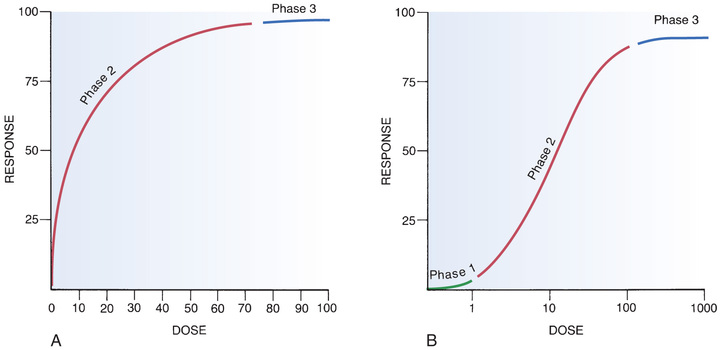
As indicated in Fig. 4.10, the dose-response relationship can be viewed as having three phases. Phase 1 (see Fig. 4.10B) occurs at low doses. The curve is flat during this phase because doses are too low to elicit a measurable response. During phase 2, an increase in dose elicits a corresponding increase in the response. This is the phase during which the dose-response relationship is graded. As the dose goes higher, eventually a point is reached where an increase in dose is unable to elicit a further increase in response. At this point, the curve flattens out into phase 3.
Maximal Efficacy and Relative Potency
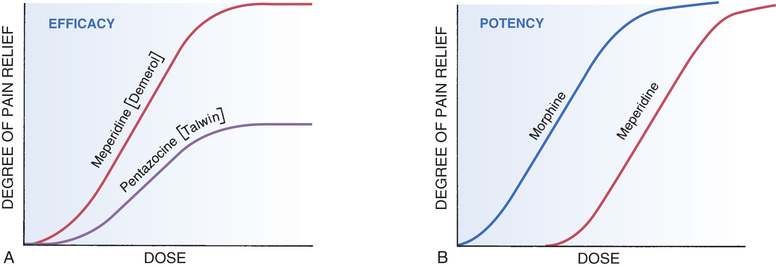
Dose-response curves reveal two characteristic properties of drugs: maximal efficacy and relative potency. Curves that reflect these properties are shown in Fig. 4.11.
Maximal Efficacy
Maximal efficacy is defined as the largest effect that a drug can produce. Maximal efficacy is indicated by the height of the dose-response curve.
The concept of maximal efficacy is illustrated by the dose-response curves for meperidine [Demerol] and pentazocine [Talwin], two morphine-like pain relievers (see Fig. 4.11A). As you can see, the curve for pentazocine levels off at a maximal height below that of the curve for meperidine. This tells us that the maximal degree of pain relief we can achieve with pentazocine is smaller than the maximal degree of pain relief we can achieve with meperidine. Put another way, no matter how much pentazocine we administer, we can never produce the degree of pain relief that we can with meperidine. Accordingly, we would say that meperidine has greater maximal efficacy than pentazocine.
Despite what intuition might tell us, a drug with very high maximal efficacy is not always more desirable than a drug with lower efficacy. Recall that we want to match the intensity of the response to the patient’s needs. This may be difficult to do with a drug that produces extremely intense responses. For example, certain diuretics (e.g., furosemide) have such high maximal efficacy that they can cause dehydration. If we only want to mobilize a modest volume of water, a diuretic with lower maximal efficacy (e.g., hydrochlorothiazide) would be preferred. Similarly, in a patient with a mild headache, we would not select a powerful analgesic (e.g., morphine) for relief. Rather, we would select an analgesic with lower maximal efficacy, such as aspirin.
Relative Potency
The term potency refers to the amount of drug we must give to elicit an effect. Potency is indicated by the relative position of the dose-response curve along the x (dose) axis.
The concept of potency is illustrated by the curves in Fig. 4.11B. These curves plot doses for two analgesics—morphine and meperidine—versus the degree of pain relief achieved. As you can see, for any particular degree of pain relief, the required dose of meperidine is larger than the required dose of morphine. Because morphine produces pain relief at lower doses than meperidine, we would say that morphine is more potent than meperidine. That is, a potent drug is one that produces its effects at low doses.
Potency is rarely an important characteristic of a drug. The only consequence of having greater potency is that a drug with greater potency can be given in smaller doses.
It is important to note that the potency of a drug implies nothing about its maximal efficacy! Potency and efficacy are completely independent qualities. Drug A can be more effective than drug B even though drug B may be more potent. Also, drugs A and B can be equally effective even though one may be more potent. As we saw in Fig. 4.11B, although meperidine happens to be less potent than morphine, the maximal degree of pain relief that we can achieve with these drugs is identical.
A final comment on the word potency is in order. In everyday parlance, people tend to use the word potent to express the pharmacologic concept of effectiveness. That is, when most people say, “This drug is very potent,” what they mean is, “This drug produces powerful effects.” They do not mean, “This drug produces its effects at low doses.” In pharmacology, we use the words potent and potency with the specific and appropriate terminology.
Drug-Receptor Interactions
Introduction to Drug Receptors
Drugs produce their effects by interacting with other chemicals. Receptors are the special chemical sites in the body that most drugs interact with to produce effects.
We can define a receptor as any functional macromolecule in a cell to which a drug binds to produce its effects. However, although the formal definition of a receptor encompasses all functional macromolecules, the term receptor is generally reserved for the body’s own receptors for hormones, neurotransmitters, and other regulatory molecules. The other macromolecules to which drugs bind, such as enzymes and ribosomes, can be thought of simply as target molecules, rather than as true receptors.
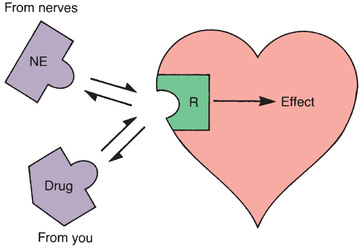
Binding of a drug to its receptor is usually reversible. Receptors are activated by interaction with other molecules (Fig. 4.12). Under physiologic conditions, endogenous compounds (neurotransmitters, hormones, other regulatory molecules) are the molecules that bind to receptors to produce a response. When a drug is the molecule that binds to a receptor, all that it can do is mimic or block the actions of endogenous regulatory molecules. By doing so, the drug will either increase or decrease the rate of the physiologic activity normally controlled by that receptor. Because drug action is limited to mimicking or blocking the body’s own regulatory molecules, drugs cannot give cells new functions. In other words, drugs cannot make the body do anything that it is not already capable of doing.2
Receptors and Selectivity of Drug Action
Selectivity, the ability to elicit only the response for which a drug is given, is a highly desirable characteristic of a drug, in that the more selective a drug is, the fewer side effects it will produce. Selective drug action is possible in large part because drugs act through specific receptors. There are receptors for each neurotransmitter (e.g., norepinephrine [NE], acetylcholine, dopamine); there are receptors for each hormone (e.g., progesterone, insulin, thyrotropin); and there are receptors for all of the other molecules the body uses to regulate physiologic processes (e.g., histamine, prostaglandins, leukotrienes). As a rule, each type of receptor participates in the regulation of just a few processes (Fig. 4.13). If a drug interacts with only one type of receptor, and if that receptor type regulates just a few processes, then the effects of the drug will be limited. Conversely, if a drug interacts with several different receptor types, then that drug is likely to elicit a wide variety of responses.
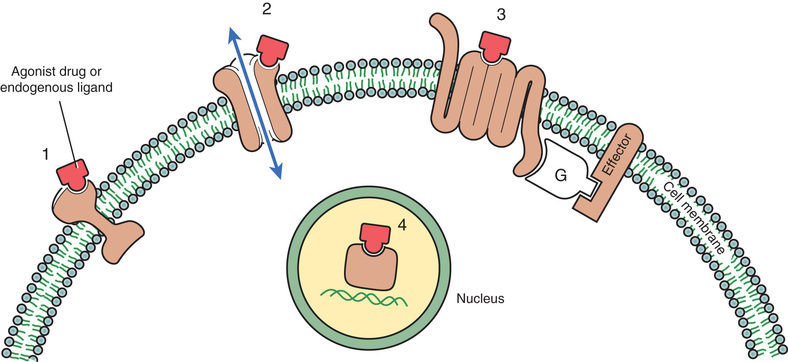
How can a drug interact with one receptor type and not with others? In some important ways, a receptor is analogous to a lock and a drug is analogous to a key for that lock: just as only keys with the proper profile can fit a particular lock, only those drugs with the proper size, shape, and physical properties can bind to a particular receptor (Fig. 4.14).
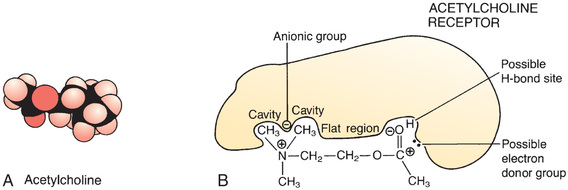
Theories of Drug-Receptor Interaction
In the discussion that follows, we consider two theories of drug-receptor interaction: (1) the simple occupancy theory and (2) the modified occupancy theory. These theories help explain dose-response relationships and the ability of drugs to mimic or block the actions of endogenous regulatory molecules.
Simple Occupancy Theory
The simple occupancy theory of drug-receptor interaction states that (1) the intensity of the response to a drug is proportional to the number of receptors occupied by that drug and (2) a maximal response will occur when all available receptors have been occupied. This relationship between receptor occupancy and the intensity of the response is depicted in Fig. 4.15.
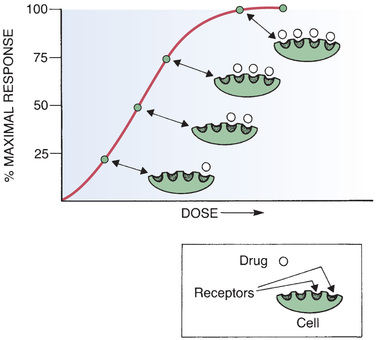
Although certain aspects of dose-response relationships can be explained by the simple occupancy theory, other important phenomena cannot. Specifically, there is nothing in this theory to explain why one drug should be more potent than another. In addition, this theory cannot explain how one drug can have higher maximal efficacy than another. That is, according to this theory, two drugs acting at the same receptor should produce the same maximal effect, provided that their dosages were high enough to produce 100% receptor occupancy. However, we have already seen this is not true. As illustrated in Fig. 4.11A, there is a dose of pentazocine above which no further increase in response can be elicited. Presumably, all receptors are occupied when the dose-response curve levels off. However, at 100% receptor occupancy, the response elicited by pentazocine is less than that elicited by meperidine. Simple occupancy theory cannot account for this difference.
Modified Occupancy Theory
The modified occupancy theory of drug-receptor interaction explains certain observations that cannot be accounted for with the simple occupancy theory. The modified theory ascribes two qualities to drugs: affinity and intrinsic activity. The term affinity refers to the strength of the attraction between a drug and its receptor. Intrinsic activity refers to the ability of a drug to activate the receptor after binding. Affinity and intrinsic activity are independent properties.
Affinity
As noted, the term affinity refers to the strength of the attraction between a drug and its receptor. Drugs with high affinity are strongly attracted to their receptors. Conversely, drugs with low affinity are weakly attracted.
The affinity of a drug for its receptor is reflected in its potency. Because they are strongly attracted to their receptors, drugs with high affinity can bind to their receptors when present in low concentrations. Because they bind to receptors at low concentrations, drugs with high affinity are effective in low doses. That is, drugs with high affinity are very potent. Conversely, drugs with low affinity must be present in high concentrations to bind to their receptors. Accordingly, these drugs are less potent.
Intrinsic Activity
The term intrinsic activity refers to the ability of a drug to activate a receptor upon binding. Drugs with high intrinsic activity cause intense receptor activation. Conversely, drugs with low intrinsic activity cause only slight activation.
The intrinsic activity of a drug is reflected in its maximal efficacy. Drugs with high intrinsic activity have high maximal efficacy. That is, by causing intense receptor activation, they are able to cause intense responses. Conversely, if intrinsic activity is low, maximal efficacy will be low as well.
It should be noted that, under the modified occupancy theory, the intensity of the response to a drug is still related to the number of receptors occupied. The wrinkle added by the modified theory is that intensity is also related to the ability of the drug to activate receptors after binding has occurred. Under the modified theory, two drugs can occupy the same number of receptors but produce effects of different intensity; the drug with greater intrinsic activity will produce the more intense response.
Agonists, Antagonists, and Partial Agonists
As previously noted, when drugs bind to receptors they can do one of two things: they can either mimic the action of endogenous regulatory molecules or they can block the action of endogenous regulatory molecules. Drugs that mimic the body’s own regulatory molecules are called agonists. Drugs that block the actions of endogenous regulators are called antagonists. Like agonists, partial agonists also mimic the actions of endogenous regulatory molecules, but they produce responses of intermediate intensity.
Agonists
Agonists are molecules that activate receptors. Because neurotransmitters, hormones, and other endogenous regulators activate the receptors to which they bind, all of these compounds are considered agonists. When drugs act as agonists, they simply bind to receptors and mimic the actions of the body’s own regulatory molecules. Dobutamine, for example, is a drug that mimics the action of NE at receptors on the heart, thereby causing heart rate and force of contraction to increase.
In terms of the modified occupancy theory, an agonist is a drug that has both affinity and high intrinsic activity. Affinity allows the agonist to bind to receptors, whereas intrinsic activity allows the bound agonist to activate or turn on receptor function.
Antagonists
Antagonists produce their effects by preventing receptor activation by endogenous regulatory molecules and drugs. Antagonists have virtually no effects of their own on receptor function.
In terms of the modified occupancy theory, an antagonist is a drug with affinity for a receptor but with no intrinsic activity. Affinity allows the antagonist to bind to receptors, but lack of intrinsic activity prevents the bound antagonist from causing receptor activation.
Although antagonists do not cause receptor activation, they most certainly do produce pharmacologic effects. Antagonists produce their effects by preventing the activation of receptors by endogenous regulatory molecules. Antihistamines, for example, are histamine receptor antagonists that suppress allergy symptoms by binding to receptors for histamine, thereby preventing activation of these receptors by histamine released in response to allergens.
It is important to note that the response to an antagonist is determined by how much agonist is present. Because antagonists act by preventing receptor activation, if there is no agonist present, administration of an antagonist will have no observable effect; the drug will bind to its receptors, but nothing will happen. On the other hand, if receptors are undergoing activation by agonists, administration of an antagonist will shut the process down, resulting in an observable response. An example is the use of the opioid antagonist naloxone, which is used to block opioid receptors in the event of an opioid overdose.
Antagonists can be subdivided into two major classes: (1) noncompetitive antagonists and (2) competitive antagonists. Most antagonists are competitive.
Noncompetitive (Insurmountable) Antagonists
Noncompetitive antagonists bind irreversibly to receptors. The effect of irreversible binding is equivalent to reducing the total number of receptors available for activation by an agonist. Because the intensity of the response to an agonist is proportional to the total number of receptors occupied, and because noncompetitive antagonists decrease the number of receptors available for activation, noncompetitive antagonists reduce the maximal response that an agonist can elicit. If sufficient antagonist is present, agonist effects will be blocked completely. Dose-response curves illustrating inhibition by a noncompetitive antagonist are shown in Fig. 4.16A.
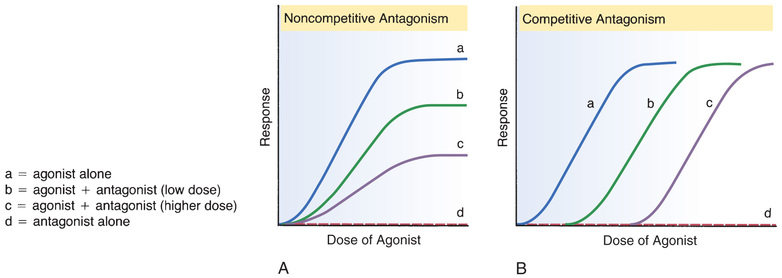
Because the binding of noncompetitive antagonists is irreversible, inhibition by these agents cannot be overcome, no matter how much agonist may be available.
Although noncompetitive antagonists bind irreversibly, this does not mean that their effects last forever. Cells are constantly breaking down old receptors and synthesizing new ones. Consequently, the effects of noncompetitive antagonists wear off as the receptors to which they are bound are replaced. Because the life cycle of a receptor can be relatively short, the effects of noncompetitive antagonists m ay subside in a few days. Still, this can be a long time for some functions; therefore, these agents are rarely used therapeutically.
Competitive (Surmountable) Antagonists
Competitive antagonists bind reversibly to receptors. As their name implies, competitive antagonists produce receptor blockade by competing with agonists for receptor binding. If an agonist and a competitive antagonist have equal affinity for a particular receptor, then the receptor will be occupied by whichever agent—agonist or antagonist—is present in the highest concentration. If there are more antagonist molecules present than agonist molecules, antagonist molecules will occupy the receptors and receptor activation will be blocked. Conversely, if agonist molecules outnumber the antagonists, receptors will be occupied mainly by the agonist and little inhibition will occur.
Because competitive antagonists bind reversibly to receptors, the inhibition they cause is surmountable. In the presence of sufficiently high amounts of agonist, agonist molecules will occupy all receptors and inhibition will be completely overcome. The dose-response curves shown in Fig. 4.16B illustrate the process of overcoming the effects of a competitive antagonist with large doses of an agonist.
Partial Agonists
A partial agonist is an agonist that has only moderate intrinsic activity. As a result, the maximal effect that a partial agonist can produce is lower than that of a full agonist. Pentazocine is an example of a partial agonist. As the curves in Fig. 4.11A indicate, the degree of pain relief that can be achieved with pentazocine is much lower than the relief that can be achieved with meperidine (a full agonist).
Partial agonists are interesting in that they can act as antagonists as well as agonists. For this reason, they are sometimes referred to as agonists-antagonists. For example, when pentazocine is administered by itself, it occupies opioid receptors and produces moderate relief of pain. In this situation, the drug is acting as an agonist. However, if a patient is already taking meperidine (a full agonist at opioid receptors) and is then given a large dose of pentazocine, pentazocine will occupy the opioid receptors and prevent their activation by meperidine. As a result, rather than experiencing the high degree of pain relief that meperidine can produce, the patient will experience only the limited relief that pentazocine can produce. In this situation, pentazocine is acting as both an agonist (producing moderate pain relief) and an antagonist (blocking the higher degree of relief that could have been achieved with meperidine by itself).
Regulation of Receptor Sensitivity
Receptors are dynamic components of the cell. In response to continuous activation or continuous inhibition, the number of receptors on the cell surface can change, as can their sensitivity to agonist molecules. For example, when the receptors of a cell are continually exposed to an agonist, the cell usually becomes less responsive. When this occurs, the cell is said to be desensitized or refractory, or to have undergone downregulation. Several mechanisms may be responsible, including destruction of receptors by the cell and modification of receptors such that they respond less fully. Continuous exposure to antagonists has the opposite effect, causing the cell to become hypersensitive (also referred to as supersensitive). One mechanism that can cause hypersensitivity is synthesis of more receptors.
Drug Responses That Do Not Involve Receptors
Although the effects of most drugs result from drug-receptor interactions, some drugs do not act through receptors. Rather, they act through simple physical or chemical interactions with other small molecules.
Common examples of these drugs include antacids, antiseptics, saline laxatives, and chelating agents. Antacids neutralize gastric acidity by direct chemical interaction with stomach acid. The antiseptic action of ethyl alcohol results from precipitating bacterial proteins. Magnesium sulfate, a powerful laxative, acts by retaining water in the intestinal lumen through an osmotic effect. Dimercaprol, a chelating agent, prevents toxicity from heavy metals (e.g., arsenic, mercury) by forming complexes with these compounds. All of these pharmacologic effects are the result of simple physical or chemical interactions, and not interactions with cellular receptors.
Interpatient Variability in Drug Responses
The dose required to produce a therapeutic response can vary substantially from patient to patient because people differ from one another. In this section we consider interpatient variation as a general issue. The specific kinds of differences that underlie variability in drug responses are discussed in Chapter 6.
To promote the therapeutic objective, you must be alert to interpatient variation in drug responses. Because of interpatient variation, it is not possible to predict exactly how an individual patient will respond to medication. The provider who appreciates the reality of interpatient variability will be better prepared to anticipate, evaluate, and respond appropriately to each patient’s therapeutic needs.
Fig. 4.17 illustrates an example of interpatient variability in response to a drug. Fig. 4.17A represents incremental increases in the milligram dose of drug to elevated gastric pH coupled with the number of patients who had a therapeutic response (pH of 5) at each dose. In Fig. 4.17B, these results are plotted on a frequency distribution curve. We can see from the curve that a wide range of doses is required to produce the desired response in all subjects. For some subjects, a dose of only 100 mg was sufficient to produce the target response. For other subjects, the therapeutic end point was not achieved until the dose totaled 240 mg.
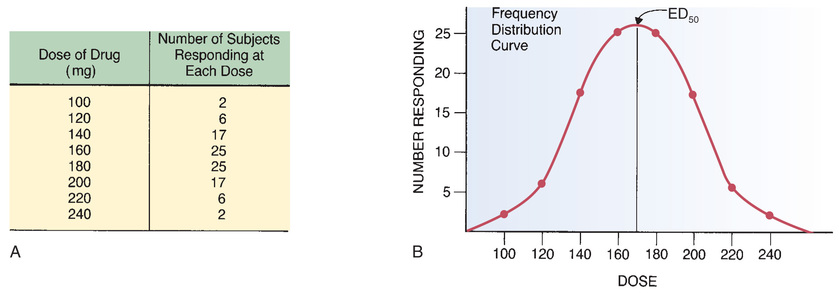
The ED50
The dose at the middle of the frequency distribution curve is termed the ED50 (see Fig. 4.17B). (ED50 is an abbreviation for average effective dose.) The ED50 is defined as the dose that is required to produce a defined therapeutic response in 50% of the population. In the case of the drug in our example, the ED50 was 170 mg—the dose needed to elevate gastric pH to a value of 5 in 50% of the 100 people tested.
The ED50 can be considered a standard dose and, as such, is frequently the dose selected for initial treatment. After evaluating a patient’s response to this standard dose, we can then adjust subsequent doses up or down to meet the patient’s needs.
Clinical Implications of Interpatient Variability
Interpatient variation has four important clinical consequences. As a provider you should be aware of these implications:
• The initial dose of a drug is necessarily an approximation. Subsequent doses may need to be fine-tuned based on the patient’s response.
• When given an average effective dose (ED50), some patients will be undertreated, whereas others will have received more drug than they need. Accordingly, when therapy is initiated with a dose equivalent to the ED50, it is especially important to evaluate the response. Patients who fail to respond may need an increase in dosage. Conversely, patients who show signs of toxicity will need a dosage reduction.
• Because drug responses are not completely predictable, you must monitor the patient’s response for both positive effects and adverse effects to determine whether too much or too little medication has been administered. In other words, dosage should be adjusted on the basis of the patient’s response and not just on the basis of clinical guidelines.
The Therapeutic Index
The therapeutic index is a measure of a drug’s safety. The therapeutic index is defined as the ratio of a drug’s LD50 to its ED50. (The LD50, or average lethal dose, is the dose that is lethal to 50% of the subjects treated.) A large (or high or wide) therapeutic index indicates that a drug is relatively safe. Conversely, a small (or low or narrow) therapeutic index indicates that a drug is relatively unsafe.
The concept of therapeutic index is illustrated by the frequency distribution curves in Fig. 4.18. Part A of the figure shows curves for therapeutic and lethal responses to drug X. Part B shows equivalent curves for drug Y. As you can see in Fig. 4.18A, the average lethal dose (100 mg) for drug X is much larger than the average therapeutic dose (10 mg). Because this drug’s lethal dose is much larger than its therapeutic dose, common sense tells us that the drug should be relatively safe. The safety of this drug is reflected in its high therapeutic index, which is 10. In contrast, drug Y is unsafe. As shown in Fig. 4.18B, the average lethal dose for drug Y (20 mg) is only twice the average therapeutic dose (10 mg). Hence, for drug Y, a dose only twice the ED50 could be lethal to 50% of those treated. Clearly, drug Y is not safe. This lack of safety is reflected in its low therapeutic index.
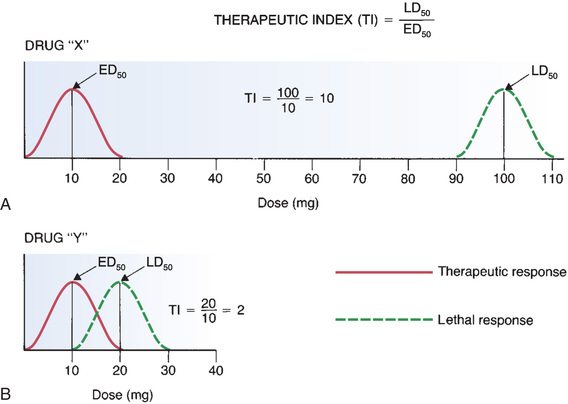
The curves for drug Y illustrate a phenomenon that is even more important than the therapeutic index. As you can see, there is overlap between the curve for therapeutic effects and the curve for lethal effects. This overlap tells us that the high doses needed to produce therapeutic effects in some people may be large enough to cause death in others. The message here is that, if a drug is to be truly safe, the highest dose required to produce therapeutic effects must be substantially lower than the lowest dose required to produce death.
Drug Interaction
Drug-Drug Interactions
Drug-drug interactions can occur whenever a patient takes two or more drugs. Some interactions are both intended and desired, as when we combine drugs to treat hypertension. In contrast, some interactions are both unintended and undesired.
Consequences of Drug-Drug Interactions
When two drugs interact, there are three possible outcomes: (1) one drug may intensify the effects of the other, (2) one drug may reduce the effects of the other, or (3) the combination may produce a new response not seen with either drug alone.
Intensification of Effects
When one drug intensifies, or potentiates, the effects of the other, this type of interaction is often termed potentiative. Potentiative interactions may be beneficial or detrimental.
Increased Therapeutic Effects
The interaction between sulbactam and ampicillin represents a beneficial potentiative interaction. When administered alone, ampicillin undergoes rapid inactivation by bacterial enzymes. Sulbactam inhibits those enzymes and thereby prolongs and intensifies ampicillin’s therapeutic effects.
Increased Adverse Effects
The interaction between aspirin and warfarin represents a potentially detrimental potentiative interaction. Both aspirin and warfarin suppress formation of blood clots; aspirin does this through antiplatelet activity, and warfarin does this through anticoagulant activity. As a result, if aspirin and warfarin are taken concurrently, the risk for bleeding is significantly increased.
Reduction of Effects
Interactions that result in reduced drug effects are often termed inhibitory. As with potentiative interactions, inhibitory interactions can be beneficial or detrimental. Inhibitory interactions that reduce toxicity are beneficial. Conversely, inhibitory interactions that reduce therapeutic effects are detrimental.
Reduced Therapeutic Effects
The interaction between propranolol and albuterol represents a detrimental inhibitory interaction. Albuterol is taken by people with asthma to dilate the bronchi. Propranolol, a drug for cardiovascular disorders, can act in the lung to block the effects of albuterol. Hence, if propranolol and albuterol are taken together, propranolol can reduce albuterol’s therapeutic effects.
Reduced Adverse Effects
The use of naloxone to treat morphine overdose is an excellent example of a beneficial inhibitory interaction. When administered in excessive dosage, morphine can produce coma, profound respiratory depression, and eventual death. Naloxone blocks morphine’s actions and can completely reverse all symptoms of toxicity.
Creation of a Unique Response
Rarely, the combination of two drugs produces a new response not seen with either agent alone. To illustrate, let’s consider the combination of alcohol with disulfiram [Antabuse], a drug used to treat alcoholism. When alcohol and disulfiram are combined, a host of unpleasant and dangerous responses can result; however, these effects do not occur when disulfiram or alcohol is used alone.
Basic Mechanisms of Drug-Drug Interactions
Drugs can interact through four basic mechanisms: (1) direct chemical or physical interaction, (2) pharmacokinetic interaction, (3) pharmacodynamic interaction, and (4) combined toxicity.
Direct Chemical or Physical Interactions
Some drugs, because of their physical or chemical properties, can undergo direct interaction with other drugs. Direct physical and chemical interactions usually render both drugs inactive.
Direct interactions occur most commonly when drugs are combined in IV solutions. Frequently, the interaction produces a precipitate; however, direct drug interactions may not always leave visible evidence. Hence you cannot rely on simple inspection to reveal all direct interactions. Because drugs can interact in solution, it is essential to consider and verify drug incompatibilities when ordering medications.
The same kinds of interactions that can take place when drugs are mixed together in an IV solution can also occur when incompatible drugs are taken by other routes. However, because drugs are diluted in body water after administration, and because dilution decreases chemical interactions, significant interactions within the patient are much less likely than in IV solutions.
Pharmacokinetic Interactions
Drug interactions can affect all four of the basic pharmacokinetic processes. That is, when two drugs are taken together, one may alter the absorption, distribution, metabolism, or excretion of the other.
Altered Absorption
Drug absorption may be enhanced or reduced by drug interactions. There are several mechanisms by which one drug can alter the absorption of another.
• By elevating gastric pH, antacids can decrease the ionization of basic drugs in the stomach, increasing the ability of basic drugs to cross membranes and be absorbed. Antacids have the opposite effect on acidic drugs.
• Laxatives can reduce absorption of other oral drugs by accelerating their passage through the intestine.
• Drugs that depress peristalsis (e.g., morphine, atropine) prolong drug transit time in the intestine, thereby increasing the time for absorption.
• Drugs that induce vomiting can decrease absorption of oral drugs.
• Orally administered adsorbent drugs that do not undergo absorption (e.g., cholestyramine) can adsorb other drugs onto themselves, thereby preventing absorption of the other drugs into the blood.
• Drugs that reduce regional blood flow can reduce absorption of other drugs from that region. For example, when epinephrine is injected together with a local anesthetic, the epinephrine causes local vasoconstriction, thereby reducing regional blood flow and delaying absorption of the anesthetic.
Altered Distribution
There are two principal mechanisms by which one drug can alter the distribution of another: (1) competition for protein binding and (2) alteration of extracellular pH.
Competition for Protein Binding.
When two drugs bind to the same site on plasma albumin, coadministration of those drugs produces competition for binding. As a result, binding of one or both agents is reduced, causing plasma levels of free drug to rise. In theory, the increase in free drug can intensify effects. However, because the newly freed drug usually undergoes rapid elimination, the increase in plasma levels of free drug is rarely sustained or significant unless the patient has liver problems that interfere with drug metabolism or has renal problems that interfere with drug excretion.
Alteration of Extracellular pH.
A drug with the ability to change extracellular pH can alter the distribution of other drugs. For example, if a drug were to increase extracellular pH, that drug would increase the ionization of acidic drugs in extracellular fluids (i.e., plasma and interstitial fluid). As a result, acidic drugs would be drawn from within cells (where the pH was below that of the extracellular fluid) into the extracellular space. Hence, the alteration in pH would change drug distribution.
The ability of drugs to alter pH and thereby alter the distribution of other drugs can be put to practical use in the management of poisoning. For example, symptoms of aspirin toxicity can be reduced with sodium bicarbonate, a drug that elevates extracellular pH. By increasing the pH outside cells, bicarbonate causes aspirin to move from intracellular sites into the interstitial fluid and plasma, thereby minimizing injury to cells.
Altered Metabolism
Altered metabolism is one of the most important—and most complex—mechanisms by which drugs interact. Some drugs increase the metabolism of other drugs, and some drugs decrease the metabolism of other drugs. Drugs that increase the metabolism of other drugs do so by inducing synthesis of hepatic drug-metabolizing enzymes. Drugs that decrease the metabolism of other drugs do so by inhibiting those enzymes.
As discussed earlier in this chapter, the majority of drug metabolism is catalyzed by the cytochrome P450 enzymes, which are composed of a large number of isoenzyme families (CYP1, CYP2, and CYP3) that are further divided into specific forms. Five isoenzyme forms are responsible for the metabolism of most drugs: CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4. Table 4.2 lists major drugs that are metabolized by each isoenzyme and indicates drugs that can inhibit or induce those isoenzymes.
TABLE 4.2
Drugs That Are Important Substrates, Inhibitors, or Inducers of Specific CYP Isoenzymes
Induction of CYP Enzymes
In our discussion of metabolism earlier in the chapter, you learned about induction of CYP enzymes.
When it is essential that an inducing agent is taken with another medicine, dosage of the other medicine may need adjustment. For example, if a woman taking oral contraceptives were to begin taking phenobarbital, induction of drug metabolism by phenobarbital would accelerate metabolism of the contraceptive, thereby lowering its level. If drug metabolism were increased enough, protection against pregnancy would be lost. To maintain contraceptive efficacy, dosage of the contraceptive should be increased. Conversely, when a patient discontinues an inducing agent, dosages of other drugs may need to be lowered. If dosage is not reduced, drug levels may climb dangerously high as rates of hepatic metabolism decline to their baseline (noninduced) values.
Inhibition of CYP Enzymes
If drug A inhibits the metabolism of drug B, then levels of drug B will rise. The result may be beneficial or harmful. The interactions of ketoconazole (an antifungal drug) and cyclosporine (an expensive immunosuppressant) provide an interesting case in point. Ketoconazole inhibits CYP3A4, the CYP isoenzyme that metabolizes cyclosporine. If ketoconazole is combined with cyclosporine, the serum drug level of cyclosporine will rise. In this instance inhibition of CYP3A4 allows us to achieve therapeutic drug levels at lower doses, thereby greatly reducing the cost of treatment.
Although inhibition of drug metabolism can be beneficial, as a rule inhibition has undesirable results. That is, in most cases, when an inhibitor increases the level of another drug, the outcome is toxicity. Accordingly, when a patient is taking an inhibitor along with his or her other medicines, you should be alert for possible adverse effects. Unfortunately, because the number of possible interactions of this type is large, keeping track is a challenge. The safest practice is to check for drug interactions in one of the reliable software applications that are widely available.
Altered Renal Excretion
Drugs can alter all three phases of renal excretion: filtration, reabsorption, and active secretion. By doing so, one drug can alter the renal excretion of another. Glomerular filtration can be decreased by drugs that reduce cardiac output: a reduction in cardiac output decreases renal perfusion, which decreases drug filtration at the glomerulus, which in turn decreases the rate of drug excretion. By altering urinary pH, one drug can alter the ionization of another and thereby increase or decrease the extent to which that drug undergoes passive tubular reabsorption. Finally, competition between two drugs for active tubular secretion can decrease the renal excretion of both agents.
Interactions That Involve P-Glycoprotein
As discussed previously in this chapter, PGP is a transmembrane protein that transports a wide variety of drugs out of cells, including cells of the intestinal epithelium, placenta, BBB, liver, and kidney tubules. Like P450 isoenzymes, PGP is subject to induction and inhibition by drugs. In fact (and curiously), most of the drugs that induce or inhibit P450 have the same effect on PGP. Drugs that induce PGP can have the following effects on other drugs:
• Reduced absorption—by increasing drug export from cells of the intestinal epithelium into the intestinal lumen
• Reduced fetal drug exposure—by increasing drug export from placental cells into the maternal blood
• Reduced brain drug exposure—by increasing drug export from cells of brain capillaries into the blood
• Increased drug elimination—by increasing drug export from liver into the bile and from renal tubular cells into the urine
Drugs that inhibit PGP will have opposite effects.
Pharmacodynamic Interactions
By influencing pharmacodynamic processes, one drug can alter the effects of another. Pharmacodynamic interactions are of two basic types: (1) interactions in which the interacting drugs act at the same site and (2) interactions in which the interacting drugs act at separate sites. Pharmacodynamic interactions may be potentiative or inhibitory and can be of great clinical significance.
Interactions at the Same Receptor
Interactions that occur at the same receptor are almost always inhibitory. Inhibition occurs when an antagonist drug blocks access of an agonist drug to its receptor. There are many agonist-antagonist interactions of clinical importance. Some reduce therapeutic effects and are therefore undesirable. Others reduce toxicity and are of obvious benefit. The interaction between naloxone and morphine noted previously is an example of a beneficial inhibitory interaction: by blocking access of morphine to its receptors, naloxone can reverse all symptoms of morphine overdose.
Interactions Resulting From Actions at Separate Sites
Even though two drugs have different mechanisms of action and act at separate sites, if both drugs influence the same physiologic process, then one drug can alter responses produced by the other. Interactions resulting from effects produced at different sites may be potentiative or inhibitory.
The interaction between morphine and diazepam [Valium] illustrates a potentiative interaction resulting from concurrent use of drugs that act at separate sites. Morphine and diazepam are CNS depressants, but these drugs do not share the same mechanism of action. Hence, when these agents are administered together, the ability of each to depress CNS function reinforces the depressant effects of the other. This potentiative interaction can result in profound CNS depression.
The interaction between two diuretics—hydrochlorothiazide and spironolactone—illustrates how the effects of a drug acting at one site can counteract the effects of a second drug acting at a different site. Hydrochlorothiazide acts on the distal convoluted tubule of the nephron to increase excretion of potassium. Acting at a different site in the kidney, spironolactone works to decrease renal excretion of potassium. Consequently, when these two drugs are administered together, the potassium-sparing effects of spironolactone tend to balance the potassium-wasting effects of hydrochlorothiazide, leaving renal excretion of potassium at about the same level it would have been had no drugs been given at all.
Combined Toxicity
If drug A and drug B are both toxic to the same organ, then taking them together will cause more injury than if they were not combined. For example, when we treat tuberculosis with isoniazid and rifampin, both of which are hepatotoxic, the potential to cause liver injury is greater than it would be if we used just one of the drugs. As a rule, drugs with overlapping toxicity are not used together. Unfortunately, when treating tuberculosis, the combination is essential.
Clinical Significance of Drug-Drug Interactions
Clearly, drug interactions have the potential to affect the outcome of therapy. As a result of drug-drug interactions, the intensity of responses may be increased or reduced. Interactions that increase therapeutic effects or reduce toxicity are desirable. Conversely, interactions that reduce therapeutic effects or increase toxicity are detrimental.
Interactions are especially important for drugs that have a narrow therapeutic range. For these agents, an interaction that produces a modest increase in drug levels can cause toxicity. Conversely, an interaction that produces a modest decrease in drug levels can cause therapeutic failure.
Although a large number of important interactions have been documented, many more are yet to be identified. Therefore, if a patient develops unusual symptoms, it is wise to suspect that a drug interaction may be the cause—especially because yet another drug might be given to control the new symptoms.
Minimizing Adverse Drug-Drug Interactions
We can minimize adverse interactions in several ways. The most obvious is to minimize the number of drugs a patient receives. A second and equally important way to avoid detrimental interactions is to take a thorough drug history. A history that identifies all drugs the patient is taking, including illicit and over-the-counter preparations, allows the provider to adjust the regimen accordingly. Additional measures for reducing adverse interactions include adjusting the dosage when an inducer of metabolism is added to or deleted from the regimen, adjusting the timing of administration to minimize interference with absorption, monitoring for early signs of toxicity when combinations of toxic agents cannot be avoided, and being especially vigilant when the patient is taking a drug with a narrow therapeutic range.
Drug-Food Interactions
Drug-food interactions are both important and poorly understood. They are important because they can result in toxicity or therapeutic failure. They are poorly understood because research has been largely lacking.
Effects of Food on Drug Absorption
Decreased Absorption
Food frequently decreases the rate of drug absorption and occasionally decreases the extent of absorption. Reducing the rate of absorption merely delays the onset of effects; peak effects are not lowered. In contrast, reducing the extent of absorption reduces the intensity of peak responses.
The interaction between calcium-containing foods and tetracycline antibiotics is a classic example of food reducing drug absorption. Tetracyclines bind with calcium to form an insoluble and nonabsorbable complex. Hence, if tetracyclines are administered with milk products or calcium supplements, absorption is reduced and antibacterial effects may be lost.
High-fiber foods can reduce absorption of some drugs. For example, absorption of digoxin [Lanoxin], used for cardiac disorders, is reduced significantly by wheat bran, rolled oats, and sunflower seeds. This reduced absorption can result in therapeutic failure.
Increased Absorption
With some drugs, food increases the extent of absorption. When this occurs, peak effects are heightened. For example, a high-calorie meal more than doubles the absorption of saquinavir [Invirase], a drug for HIV infection. If saquinavir is taken without food, absorption may be insufficient for antiviral activity.
Effects of Food on Drug Metabolism: The Grapefruit Juice Effect
Grapefruit juice can inhibit the metabolism of certain drugs, thereby raising their blood levels. Four compounds have been identified as the responsible agents: two furanocoumarins (bergapten and 6′,7′-dihydroxybergamottin) and two flavonoids (naringin and naringenin). This effect is not seen with other citrus juices, including orange juice.
Grapefruit juice raises drug levels mainly by inhibiting CYP3A4 metabolism. CYP3A4 is an isoenzyme of cytochrome P450 found in the liver and the intestinal wall. Inhibition of the intestinal isoenzyme is much greater than inhibition of the liver isoenzyme. By inhibiting CYP3A4, grapefruit juice decreases the intestinal metabolism of many drugs (Table 4.3) and thereby increases the amount available for absorption. As a result, blood levels of these drugs rise, causing peak effects to be more intense. Because inhibition of CYP3A4 in the liver is minimal, grapefruit juice does not usually affect metabolism of drugs after they have been absorbed. Importantly, grapefruit juice has little or no effect on drugs administered intravenously. Why? Because, with IV administration, intestinal metabolism is not involved. Inhibition of CYP3A4 is dose dependent; the more grapefruit juice the patient drinks, the greater the inhibition.
TABLE 4.3
Some Drugs Whose Levels Can Be Increased by Grapefruit Juice
| Drug | Indications | Potential Consequences of Increased Drug Levels |
| Dihydropyridine CCBs: amlodipine, felodipine, nicardipine, nifedipine, nimodipine, nisoldipine | Hypertension; angina pectoris | Toxicity: flushing, headache, tachycardia, hypotension |
| Nondihydropyridine CCBs: diltiazem, verapamil | Hypertension; angina pectoris | Toxicity: bradycardia, AV heart block, hypotension, constipation |
| Statins: lovastatin, simvastatin (minimal effect on atorvastatin, fluvastatin, pravastatin, or rosuvastatin) | Cholesterol reduction | Toxicity: headache, GI disturbances, liver and muscle toxicity |
| Amiodarone | Cardiac dysrhythmias | Toxicity |
| Caffeine | Prevents sleepiness | Toxicity: restlessness, insomnia, convulsions, tachycardia |
| Carbamazepine | Seizures; bipolar disorder | Toxicity: ataxia, drowsiness, nausea, vomiting, tremor |
| Buspirone | Anxiety | Drowsiness, dysphoria |
| Triazolam | Anxiety; insomnia | Increased sedation |
| Midazolam | Induction of anesthesia; conscious sedation | Increased sedation |
| Saquinavir | HIV infection | Increased therapeutic effect |
| Cyclosporine | Prevents rejection of organ transplants | Increased therapeutic effects; if levels rise too high, renal and hepatic toxicity will occur |
| Sirolimus and tacrolimus | Prevent rejection of organ transplants | Toxicity |
| SSRIs: fluoxetine, fluvoxamine, sertraline | Depression | Toxicity: serotonin syndrome |
| Pimozide | Tourette syndrome | Toxicity: QT prolongation resulting in a life-threatening ventricular dysrhythmia |
| Praziquantel | Schistosomiasis | Toxicity |
| Dextromethorphan | Cough | Toxicity |
| Sildenafil | Erectile dysfunction | Toxicity |
Inhibition of CYP3A4 persists after grapefruit juice is consumed, so the problem cannot be easily resolved by scheduling medication hours after drinking the juice. In fact, when grapefruit juice is consumed on a regular basis, inhibition can persist up to 3 days after the last drink.
The effects of grapefruit juice vary considerably among patients because levels of CYP3A4 show great individual variation. In patients with very little CYP3A4, inhibition by grapefruit juice may be sufficient to stop metabolism completely. As a result, large increases in drug levels may occur. Conversely, in patients with an abundance of CYP3A4, metabolism may continue more or less normally, despite inhibition by grapefruit juice.
The effects of grapefruit juice also vary considerably among CYP3A4 substrates. In one study, coadministration of grapefruit juice produced a 406% increase in blood levels of the calcium channel blocker felodipine [Plendil]. For some other drugs metabolized by these isoenzymes, the effects are negligible.
The clinical consequences of inhibition may be good or bad. As indicated in Table 4.3, by elevating levels of certain drugs, grapefruit juice can increase the risk for serious toxicity. On the other hand, by increasing levels of two drugs—saquinavir and cyclosporine—grapefruit juice can intensify therapeutic effects to promote positive outcomes.
What should providers do if the drugs they are prescribing can be affected by grapefruit juice? Unless a predictable effect is known or desirable, prudence dictates advising patients to avoid grapefruit juice entirely.
Effects of Food on Drug Toxicity
Drug-food interactions sometimes increase toxicity. The most dramatic example is the interaction between monoamine oxidase (MAO) inhibitors and foods rich in tyramine (e.g., aged cheeses, yeast extracts, Chianti wine). If an MAO inhibitor is combined with these foods, blood pressure can rise to a life-threatening level. To avoid disaster, patients taking MAO inhibitors must be warned about the consequences of consuming tyramine-rich foods and must be given a list of foods to strictly avoid (see Chapter 25). Other drug-food combinations that can increase toxicity include the following:
• Theophylline (an asthma medicine) plus caffeine, which can result in excessive CNS excitation
• Potassium-sparing diuretics (e.g., spironolactone) plus salt substitutes, which can result in dangerously high potassium levels
• Aluminum-containing antacids (e.g., Maalox) plus citrus beverages (e.g., orange juice), which can result in excessive absorption of aluminum
Effects of Food on Drug Action
Although most drug-food interactions concern drug absorption or drug metabolism, food may also (rarely) have a direct effect on drug action. For example, foods rich in vitamin K (e.g., broccoli, Brussels sprouts, cabbage) can reduce the effects of warfarin, an anticoagulant. This occurs because warfarin inhibits vitamin K–dependent clotting factors. Accordingly, when vitamin K is more abundant, warfarin is less able to inhibit the clotting factors, and therapeutic effects decline.
Timing of Drug Administration With Respect to Meals
Administration of drugs at the appropriate time with respect to meals is an important part of drug therapy. As discussed, the absorption of some drugs can be significantly decreased by food, and hence these drugs should be administered on an empty stomach (i.e., at least 1 hour before or 2 hours after a meal). Conversely, the absorption of other drugs can be increased by food, and hence these drugs should be administered with meals.
Many drugs cause stomach upset when taken without food. If food does not significantly reduce their absorption, then these drugs should be administered with meals. However, if food does reduce their absorption, then we have a difficult choice: we can administer them with food and thereby reduce stomach upset, but also reduce absorption—or, we can administer them without food and thereby improve absorption, but also increase stomach upset. Unfortunately, the correct choice is not obvious. The best solution, when possible, may be to select an alternative drug that doesn’t upset the stomach.
Drug-Supplement Interactions
Dietary supplements (herbal medicines and other nonconventional remedies) create the potential for frequent and significant interactions with conventional drugs. Of greatest concern are interactions that reduce beneficial responses to conventional drugs and interactions that increase toxicity. These interactions occur through the same pharmacokinetic and pharmacodynamic mechanisms by which conventional drugs interact with each other. Unfortunately, reliable information about dietary supplements is largely lacking, including information on interactions with conventional agents. Interactions that have been well documented are discussed as appropriate throughout this text.

