6 Pharmacokinetics and Pharmacology of Drugs Used in Children
Pharmacokinetic Principles and Calculations
Pediatric Pharmacokinetic Considerations
Linking Pharmacokinetics with Pharmacodynamics
Developmental Changes of Specific Cytochromes
Central Nervous System Effects
The Drug Approval Process, the Package Insert, and Drug Labeling
Intermediate-Acting Nondepolarizing Relaxants
Long-Acting Nondepolarizing Relaxants
Antagonism of Muscle Relaxants
Relaxants in Special Situations
THE PHARMACOKINETICS AND PHARMACODYNAMICS of most medications, when used in children, especially neonates, differ from those in adults.1–11 Children exhibit different pharmacokinetics (PK) and pharmacodynamics (PD) from adults because of their immature renal and hepatic function, different body composition, altered protein binding, distinct disease spectrum, diverse behavior, and dissimilar receptor patterns.1,3,12–19 PK differences necessitate modification of the dose and the interval between doses to achieve the desired clinical response and to avoid toxicity.7,20–22 In addition, some medications may displace bilirubin from its protein binding sites and possibly predispose an infant to kernicterus.23–28 The capacity of the end organ, such as the heart or bronchial smooth muscle, to respond to medications may also differ in children compared with adults (PD effects). In this chapter we discuss basic pharmacologic principles as they relate to drugs commonly used by anesthesiologists.
Pharmacokinetic Principles and Calculations
Changes in drug concentrations within the body over time are referred to as pharmacokinetics. The principles and equations that describe these changes can be used to adjust drug doses rationally to achieve more effective drug concentrations at the site of action.29–33 The equations in this section are intended for general and practical use, whereas the more rigorous mathematical intricacies of PK are covered elsewhere.34–37
First-Order Kinetics
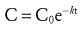 (Eq. 1)
(Eq. 1)where C is the concentration at time t, C0 is the starting concentration (a constant determined by the dose and distribution volume), and k is the elimination rate constant with units of time−1. First-order indicates that the exponent is raised to the first power (−kt in Equation 1). Second-order equations are those that are raised to the second power, such as e(z)2. First-order exponential equations, such as Equation 1, may be converted to the form of the equation of a straight line (y = mx + b) by taking the natural logarithm of both sides, after which they may be solved by linear regression.
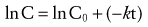 (Eq. 2)
(Eq. 2)Half-Life
Half-life can be determined by several methods. If concentration is converted to the natural logarithm of concentration and graphed versus time, as described in Equation 2, the slope of this graph is the elimination rate constant, k. For both accuracy and precision, at least three concentration-time points should be used to determine the slope, and they should be obtained over an interval during which the concentration decreases at least in half. In clinical practice, for infants and small children, however, k is often estimated from just two concentrations obtained during the terminal elimination phase. With multiple data points, the slope of ln C versus time may be calculated easily by least squares linear regression analysis. Half-life (T1/2) may be calculated from the elimination rate constant, k (time−1), as follows:
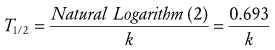 (Eq. 3)
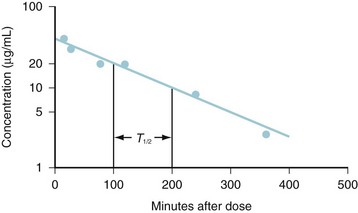
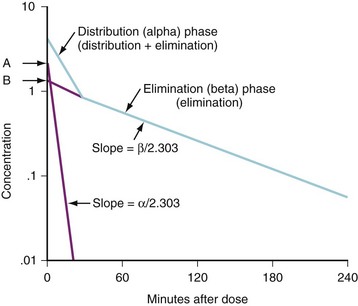
(Eq. 3)Graphic techniques may be used to determine half-life from a series of timed measurements of drug concentration. The concentration-time points should be graphed on semilogarithmic axes and used to determine the best fitting line either visually or by linear regression analysis. This approach is illustrated in Figure 6-1, in which the best-fitting line has been drawn to the concentration-points and crosses a concentration of 20 μg/mL at 100 minutes and a concentration of 10 μg/mL at 200 minutes. The concentration has decreased by one half in 100 minutes, so the half-life is 100 minutes. The elimination rate constant is (0.693/100) min−1 or 0.00693 min−1.
Elimination half-life is of no value in characterizing disposition of many intravenous (IV) anesthetic drugs during dosing periods relevant to anesthesia. A more useful concept is that of the context-sensitive half-time (CSHT) where “context” refers to the duration of the infusion. This is the time required for the plasma drug concentration to decrease by 50% after terminating the infusion.38 The CSHT is the same as the elimination half-life for a one-compartment model and does not change with the duration of the infusion. However, most drugs in anesthesia conform to multiple compartment models and the CSHTs are markedly different from their respective elimination half-lives.
CSHT may be independent of the duration of the infusion (e.g., remifentanil, 2.5 minutes); moderately affected (propofol, 12 minutes at 1 hour, 38 minutes at 8 hours); or display marked prolongation (e.g., fentanyl, 1 hour at 24 minutes, 8 hours at 280 minutes). This is a result of return of drug to plasma from peripheral compartments after stopping the infusion. Peripheral compartment sizes and clearances differ in children from adults and at termination of the infusion such that more or less drug remains in the body in children for any given plasma concentration compared with adults. The CSHT for propofol in children, for example, is greater than that in adults.39 The CSHT gives insight into the PK of a drug, but the parameter may not be clinically relevant; the percentage decrease in concentration required for recovery from the drug effect is not necessarily 50%.
First-Order Multiple-Compartment Kinetics
If drug concentrations are measured several times within the first 15 to 30 minutes after IV administration as well as during a more prolonged period, more than one rate of clearance is often present. This can be observed as a marked change in slope of a semilogarithmic graph of concentration versus time (Fig. 6-2). The number and nature of the compartments required to describe the clearance of a drug do not necessarily represent specific body fluids or tissues. When two first-order exponential equations are required to describe the clearance of drug from the circulation, the kinetics are described as first-order, two-compartment (e.g., central and peripheral compartments) that fit the following equation (Fig. 6-2)31:
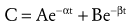 (Eq. 4)
(Eq. 4)Such two-compartment or biphasic kinetics are frequently observed after IV administration of drugs that rapidly distribute out of the central compartment of the circulation to a peripheral compartment.31 In such situations, the initial rapid decrease in concentration is referred to as the α or distribution phase and represents distribution to the peripheral (tissue) compartments in addition to drug elimination. The terminal (β) phase begins after the inflection point in the line when elimination starts to account for most of the change in drug concentration. To determine the initial change in concentration as a result of distribution (Fig. 6-2), the change in concentration that results from elimination must be subtracted from the total change in concentration. The slope of the line representing the difference between these two rates is the rate constant for distribution.
These parameters (A, B, α, β) have little connection with underlying physiology and an alternative parameterization is to use a central volume and three rate constants (k10, k12, k21) that describe drug distribution between compartments. Another common method is to use two volumes (central, V1; peripheral, V2) and two clearances (CL, Q). Q is the intercompartment clearance and the volume of distribution at steady state (Vdss) is the sum of V1 and V2. A more detailed mathematical discussion may be found elsewhere.31,40
Although many drugs demonstrate multiple-compartment kinetics, traditional studies of kinetics in neonates did not include enough samples immediately after dosing to determine more than one compartment. For clinical estimates of dose and dosing intervals, it is often not necessary to use multiple-compartment kinetics. To minimize cost, limit blood loss, and simplify PK calculations, dose adjustments are often based on only two plasma concentrations (peak and trough), and linear, single-compartment kinetics (such as that of gentamicin and vancomycin) is assumed. Because the elimination rate constant should be determined from the terminal elimination phase, it is important that peak concentrations of multiple-compartment drugs not be drawn prematurely, that is, during the initial distribution phase. If drawn too early, the concentrations will be greater than those during the terminal elimination phase (Fig. 6-2), which will overestimate the slope and the terminal elimination rate constant. Population modeling has improved analysis and interpretation of such data.41,42
Zero-Order Kinetics
The elimination of some drugs occurs with loss of a constant amount per time, rather than a constant fraction per time. Such rates are termed zero-order, and because e0 = 1, the change in the amount of drug in the body fits the following equation40:
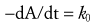 (Eq. 5)
(Eq. 5)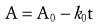 (Eq. 6)
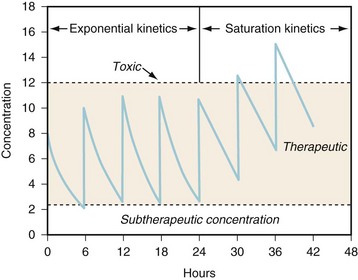
(Eq. 6)Zero-order (also known as Michaelis-Menten) kinetics may be designated saturation kinetics, because such processes occur when excess amounts of drug saturate the capacity of metabolic enzymes or transport systems. In this situation, only a constant amount of drug is metabolized or transported per unit of time. If kinetics are zero order, a graph of serum concentration versus time is linear on linear-linear axes and is curved when graphed on linear-logarithmic (i.e., semilogarithmic) axes. Clinically, first-order elimination may become zero order after administration of excessive doses or prolonged infusions or during dysfunction of the organ of elimination. Certain drugs administered to neonates exhibit zero-order kinetics at therapeutic doses and may accumulate to excessive concentrations, including thiopental, theophylline, caffeine, diazepam, furosemide, and phenytoin.43 Some drugs (e.g., phenytoin, ethyl alcohol) may exhibit mixed-order kinetics (i.e., first order at low concentrations and zero order after enzymes are saturated at higher concentrations). For these drugs, a small increment in dose may cause disproportionately large increments in serum concentrations (Fig. 6-3).
Apparent Volume of Distribution
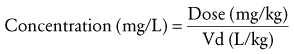 (Eq. 7)
(Eq. 7)If concentration is expressed with the unconventional units of milligrams per liter rather than micrograms per milliliter (which is equivalent), it is easier to balance the equation. This equation serves as the basis for most of the PK calculations because it is easily rearranged to solve for Vd and dose. It is also important to note that this equation represents the change in concentration after a rapidly administered IV dose of a drug whose elimination is great compared with its time for distribution. After a mini-infusion (e.g., of vancomycin or gentamicin), a more complex exponential equation may be required to account for drug elimination during the time of infusion.40 For neonates in whom drug elimination is relatively slow, only a small fraction of drug is eliminated during the time of infusion, and such adjustments can be omitted, whereas more complex equations may be needed in older children.
Knowledge of the apparent Vd is essential for dosage adjustments. Vd may be calculated by rearranging Equation 7.
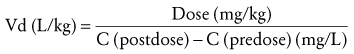 (Eq. 8)
(Eq. 8)Pharmacokinetic Example
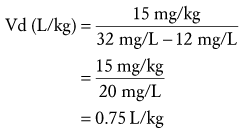
Step 1: Substituting the data into Equation 8, we calculate Vd.
Step 2: At steady-state, peak and trough concentrations reach the same levels after each dose. The time between the peak and trough concentrations is 10 hours, that is, 12 hours minus 1 hour infusion minus 1 hour to peak concentration. Half-life may be solved by rearranging Equation 2 to solve for k (elimination rate constant) and substituting the calculated k into Equation 3. In this case, the calculated elimination rate constant is 0.098 hour−1 and the corresponding half-life is 7.1 hours. However, a practical and clinically applicable “bedside” approach may be used without need for logarithmic calculations. For example, the plasma concentration decreased from 32 to 16 mg/L in one half-life and then from 16 to 12 mg/L in a fraction of the second half-life. At the end of the second half-life, the concentration would have decreased to 8 mg/L. Because 12 mg/L is the midpoint between the first and second half-lives, 1.5 half-lives have elapsed during the 10 hours between the peak and trough. Thus, if one assumes a linear decline, the half-life may be estimated as 6.67 hours (10 hours ÷ 1.5 half-lives). Note that the error between the actual half-life of 7.1 hours and the estimated half-life (6.67 hours) is a result of the linear assumptions of this calculation between half-lives. In fact, first-order elimination is a nonlinear process and concentration will actually decline from 32 mg/L to 22.6 mg/L during the first 50% of the first half-life rather than from 32 mg/L to 24 mg/L using this linear approach. The same occurs during subsequent half-lives. However, the small error associated with this method is often acceptable for rapid bedside estimates of PK parameters.
Step 3: A new dosage regimen must be calculated if the concentrations are unsatisfactory. Accordingly, one must decide on a desired peak and trough concentration. If, for example, the desired vancomycin peak and trough concentrations were 32 mg/L (20 to 40 mg/L) and 8 mg/L (5 to 10 mg/L), respectively, then Equation 8 may be rearranged to solve for the new dose.
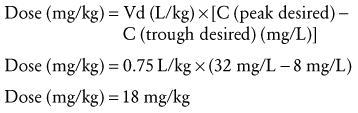 (Eq. 9)
(Eq. 9) hours) after the peak is reached (2 hours after beginning the dose infusion) will produce a trough concentration of 8 mg/L. The dose interval should be increased to 16 hours and the dose increased to 18 mg/kg.
hours) after the peak is reached (2 hours after beginning the dose infusion) will produce a trough concentration of 8 mg/L. The dose interval should be increased to 16 hours and the dose increased to 18 mg/kg.Repetitive Dosing and Drug Accumulation
When multiple doses are administered, the dose is usually repeated before complete elimination of the previous one. In this situation, peak and trough concentrations increase until a steady-state concentration (Css) is reached (Fig. 6-3). The average Css (AvgCss) can be calculated as follows32:
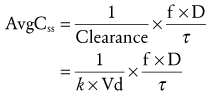 (Eq. 10)
(Eq. 10)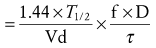 (Eq. 11)
(Eq. 11)In Equations 10 and 11, f is the fraction of the dose that is absorbed, D is the dose, τ is the dosing interval in the same units of time as the elimination half-life, k is the elimination rate constant, and 1.44 equals the reciprocal of 0.693 (see Equation 3). The magnitude of the average Css is directly proportional to the ratio of T1/2/τ and D.32
Steady State
Steady state occurs when the amount of drug removed from the body between doses equals the amount of the dose.33,37 Five half-lives are usually required for drug elimination and distribution among tissue and fluid compartments to reach equilibrium. When all tissues are at equilibrium (i.e., steady state), the peak and trough concentrations are the same after each dose. However, before this time, constant peak and trough concentrations after intermittent doses, or constant concentrations during drug infusions, do not prove that a steady state has been achieved because drug may still be entering and leaving deep tissue compartments. During continuous infusion, the fraction of steady-state concentration that has been reached can be calculated in terms of multiples of the drug’s half-life.32 After three half-lives, the concentration is 88% of that at steady state. When changing doses during chronic drug therapy, the concentration should usually not be rechecked until several half-lives have elapsed, unless elimination is impaired or signs of toxicity occur. Drug concentrations may not need to be checked if symptoms improve.
Loading Dose
If the time to reach a constant concentration by continuous or intermittent dosing is excessive, a loading dose may be used to reach plateau in the concentration more rapidly. This frequently is applied to initial treatment with digoxin, which has a 35- to 69-hour half-life in term neonates and an even longer half-life in preterm infants.44 Use of a loading dose increases the circulating concentration of drug earlier in the therapeutic course, but for the equilibration to reach a true steady-state still requires treatment for five or more half-lives. Loading doses must be used cautiously, because they increase the likelihood of drug toxicity, as has been observed with loading doses of digoxin.3,16,17,44
Dose calculations using a 1-compartment model (Eq. 9) may not be applicable to many anesthetic drugs that are characterized using multi-compartment models. The use of V1 results in a loading dose too high, while the use of Vdss results in a loading dose too low. Too high a dose may cause transient toxicity, although slowing the rate of administration may prevent excessive concentrations during the distributive phase.
The time to peak effect (Tpeak) is dependent on clearance and effect-site equilibration half-time (T1/2keo). At a submaximal dose, Tpeak is independent of dose. At supramaximal doses, maximal effect will occur earlier than Tpeak and persist for longer duration because of the shape of the response curve (see later discussion). The Tpeak concept has been used to calculate optimal initial bolus doses,45 because V1 and Vdss poorly reflect the required scaling factor. A new parameter, the volume of distribution at the time of peak effect-site concentration (Vpe) is used and is calculated.
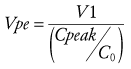 (Eq. 12)
(Eq. 12)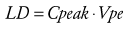 (Eq. 13)
(Eq. 13)Population Modeling
Pediatric anesthesiologists have embraced the population approach for investigating PK and PD. This approach, achieved through nonlinear mixed effects models, provides a means to study variability in drug responses among individuals representative of those in whom the drug will be used clinically. Traditional approaches to interpretation of time-concentration profiles relied on “rich” data from a small group of subjects. In contrast, mixed effects models can be used to analyze “sparse” (2 to 3 samples) data from a large number of subjects. Sampling times are not crucial for population methods and can be fit around clinical procedures or outpatient appointments. Sampling time-bands rather than exact times is equally effective and allows flexibility in children.45,46 Interpretation of truncated individual sets of data or missing data is also possible with this type of analysis, rendering it particularly useful for pediatric studies. Population modeling also allows pooling of data across studies to provide a single robust PK analysis rather than comparing separate smaller studies that are complicated by different methods and analyses.
Mixed effects models are “mixed” because they describe the data using a mixture of fixed and random effects. Fixed effects predict the average influence of a covariate, such as weight, as an explanation of some of the variability between subjects in a parameter like clearance. Random effects describe the remaining variability between subjects that are not predictable from the fixed effect average. Explanatory covariates (e.g., age, size, renal function, sex, temperature) can be introduced that explain the predictable part of the between-individual variability. Nonlinear regression is performed by an iterative process to find the curve of best fit.47,48
Pediatric Pharmacokinetic Considerations
Growth and development are two major aspects of children not readily apparent in adults. How these factors interact is not necessarily easy to determine from observations because they are quite highly correlated. Drug clearance, for example, may increase with weight, height, age, body surface area, and creatinine clearance. One approach is to standardize for size before incorporating a factor for maturation.49
Size
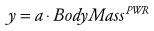 (Eq. 14)
(Eq. 14)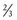 (i.e., body surface area) are at odds with those advocating a value of
(i.e., body surface area) are at odds with those advocating a value of 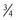 .
.Support for a value of 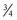 comes from investigations that show the log of BMR plotted against the log of body weight produces a straight line with a slope of
comes from investigations that show the log of BMR plotted against the log of body weight produces a straight line with a slope of 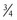 in all species studied, including humans. Fractal geometry mathematically explains this phenomenon. The
in all species studied, including humans. Fractal geometry mathematically explains this phenomenon. The 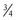 -power law for metabolic rates was derived from a general model that describes how essential materials are transported through space-filled fractal networks of branching tubes.50 A great many physiologic, structural, and time related variables scale predictably within and between species with weight (W) exponents (PWR) of
-power law for metabolic rates was derived from a general model that describes how essential materials are transported through space-filled fractal networks of branching tubes.50 A great many physiologic, structural, and time related variables scale predictably within and between species with weight (W) exponents (PWR) of 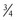 , 1, and
, 1, and 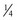 , respectively.51 These exponents have applicability to PK parameters, such as clearance (CL exponent of
, respectively.51 These exponents have applicability to PK parameters, such as clearance (CL exponent of 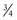 ), volume (V exponent of 1) and half-time (T1/2 exponent of
), volume (V exponent of 1) and half-time (T1/2 exponent of 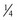 ).51 The factor for size (Fsize) for total drug clearance may be expressed:
).51 The factor for size (Fsize) for total drug clearance may be expressed:
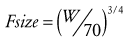 (Eq. 15)
(Eq. 15)Remifentanil clearance in children aged 1 month to 9 years is similar to adult rates when scaled using an allometric exponent of 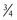 .52 Nonspecific blood esterases that metabolize remifentanil are mature at birth.53
.52 Nonspecific blood esterases that metabolize remifentanil are mature at birth.53
Maturation
Allometry alone is insufficient to predict clearance in neonates and infants from adult estimates for most drugs.54,55 The addition of a model describing maturation is required. The sigmoid hyperbolic or Hill model56 has been found useful for describing this maturation process (MF).
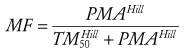 (Eq. 16)
(Eq. 16)The TM50 describes the maturation half-time, while the Hill coefficient relates to the slope of this maturation profile. Maturation of clearance begins before birth, suggesting that postmenstrual age (PMA) would be a better predictor of drug elimination than postnatal age.51 Figure 6-4 shows the maturation profile for dexmedetomidine, expressed as both the standard per-kilogram model and by using allometry. Clearance is immature in infancy. Clearance, expressed as per kilogram, is greatest at 2 years of age, decreasing subsequently with age. This “artifact of size” disappears with use of the allometric model.
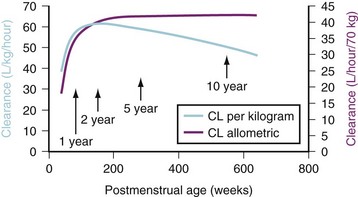
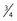 -power model. This maturation pattern is typical of many drugs cleared by the liver or kidneys.
-power model. This maturation pattern is typical of many drugs cleared by the liver or kidneys.Organ Function
Changes associated with normal growth and development can be distinguished from pathologic changes describing organ function.49 Morphine clearance is reduced in neonates because of immature glucuronide conjugation, but clearance was lower in critically ill neonates than healthier cohorts,57–59 possibly attributable to reduced hepatic function. The impact of organ function alteration may be concealed by another covariate. For example, positive pressure ventilation may be associated with reduced clearance. This effect may be attributable to a consequent reduced hepatic blood flow with a drug that has perfusion limited clearance (e.g., propofol, morphine).
Pharmacokinetic parameters (P) can be described in an individual as the product of size (Fsize), maturation (MF) and organ function (OF) influences, where Pstd is the parameter value in a standard size adult without pathologic changes in organ function49:
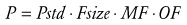 (Eq. 17)
(Eq. 17)Pharmacodynamic Models
Sigmoid Emax Model
The relation between drug concentration and effect may be described by the Hill equation or Emax model (see maturation model above)56:
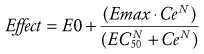 (Eq. 18)
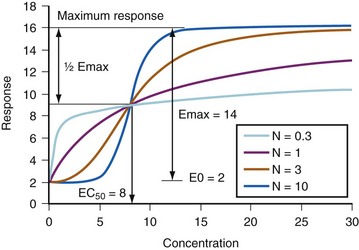
(Eq. 18)where E0 is the baseline response, Emax is the maximum effect change, Ce is the concentration in the effect compartment, EC50 is the concentration producing 50% Emax, and N is the Hill coefficient defining the steepness of the concentration-response curve (Fig. 6-5). Efficacy is the maximum response on a dose or concentration-response curve. EC50 can be considered a measure of potency relative to another drug, provided N and Emax for the two drugs are the same. A concentration-response relationship for acetaminophen has been described using this model. An EC50 of 9.8 mg/L, N = 1, and an Emax of 5.3 pain units (on a visual analog scale [VAS] of 0 to 10) was reported.60 Midazolam PD in adults have been similarly defined using electroencephalographic (EEG) responses.61,62
Quantal Effect Model
The potency of anesthetic vapors may be expressed by minimum alveolar concentration (MAC), and this is the concentration at which 50% of subjects move in response to a standard surgical stimulus. MAC appears, at first sight, to be similar to EC50, but is an expression of quantal response rather than magnitude of effect. There are two methods of estimating MAC. Responses can be recorded over the clinical dose range in a large number of subjects and logistic regression applied to estimate the relationship between dose and quantal effect; the MAC can then be interpolated. Large numbers of subjects may not be available, so an alternative is often used. The “up and down” method described by Dixon63,64 estimates only the MAC rather than the entire sigmoid curve. It usually involves a study of only one concentration in each subject and, in a sequence of subjects, each receives a concentration depending on the response of the previous subject; the concentration is either decreased if the previous subject did not respond or increased if they did. The MAC is calculated either as the mean concentration of equal numbers of responses and no-responses or is the mean concentration of pairs of “response–no response.”
Logistic Regression Model
When the pharmacologic effect is difficult to grade, then it may be useful to estimate the probability of achieving the effect as a function of plasma concentration. Effect measures, such as movement/no movement or rousable/nonrousable, are dichotomous. Logistic regression is commonly used to analyze such data and the interpolated EC50 value refers to the probability of response. For example, an EC50 of 0.52 mg/L for arousal after ketamine sedation in children has been estimated using this technique.65
Linking Pharmacokinetics with Pharmacodynamics
There may also be a delay as a result of transfer of the drug to the effect site (e.g., neuromuscular blockers), a lag time (e.g., diuretics), physiologic response (e.g., antipyresis), active metabolite (e.g., propacetamol), or synthesis of physiologic substances (e.g., warfarin). A plasma concentration-effect plot can form a hysteresis loop because of this delay in effect. Hull and Sheiner introduced the effect compartment concept for neuromuscular blockers.66,67 A single first-order parameter (T1/2keo) describes the equilibration half-time. This mathematical trick assumes that the concentration in the central compartment is the same as that in the effect compartment at equilibration, but that a time delay exists before drug reaches the effect compartment. The concentration in the effect compartment is used to describe the concentration-effect relationship.68
Adult T1/2keo values are well described (e.g., morphine, 16 minutes; fentanyl, 5 minutes; alfentanil, 1 minute; propofol, 3 minutes). This T1/2keo parameter is commonly incorporated into target controlled infusion pumps in order to achieve a rapid effect-site concentration. The adult midazolam T1/2keo of 5 minutes may be prolonged in the elderly, resulting in overdose if this is not recognized during dose titration.66
The T1/2keo for propofol in children has been described. As expected, a shorter T1/2keo with decreasing age based on size models has been described.67,69 Similar results have been demonstrated for sevoflurane and changes in the EEG.70 If the effect-site is targeted and peak effect (Tpeak) is anticipated to be later than it actually is because it was determined in a teenager or adult, this will result in excessive dose in a young child.
Drug Distribution
Protein Binding
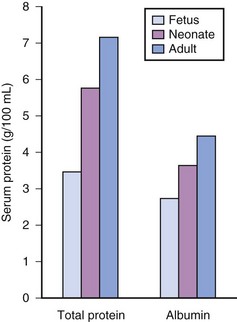
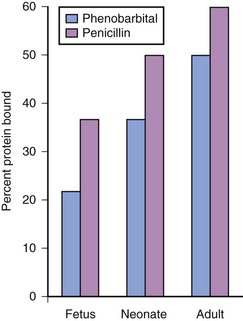
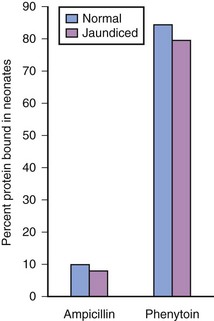
Acidic drugs (e.g., diazepam, barbiturates) tend to bind mainly to albumin while basic drugs (e.g., amide local anesthetic agents) bind to globulins, lipoproteins and glycoproteins. In general, plasma protein binding of many drugs is decreased in the neonate relative to the adult in part because of reduced total protein and albumin concentrations (Fig. 6-6).71 Many drugs that are highly protein bound in adults have less of an affinity for protein in neonates (E-Fig. 6-1).71–75 Reduced protein binding increases the free fraction of medications, thus providing more free medication and greater pharmacologic effect.1,3,12,14,17 This effect is particularly important for medications that are highly protein bound, because the reduced protein binding increases the free fraction of the medication to a greater extent than for low protein bound drugs. For example, phenytoin is 85% protein bound in healthy infants but only 80% in those who are jaundiced. This equates to a 33% increase in the free fraction of phenytoin when jaundice occurs (E-Fig. 6-2). Differences in protein binding may have considerable influence on the response to medications that are acidic and are, therefore, highly protein bound (e.g., phenytoin, salicylate, bupivacaine, barbiturates, antibiotics, theophylline, and diazepam).17 In addition, some medications, such as phenytoin, salicylate, sulfisoxazole, caffeine, ceftriaxone, diatrizoate (Hypaque), and sodium benzoate, compete with bilirubin for binding to albumin (see E-Fig. 6-2). If large amounts of bilirubin are displaced, particularly in the presence of hypoxemia and acidosis, which open the blood-brain barrier, kernicterus may result.24,25,72,75–77 Because these metabolic derangements often occur in sick neonates coming to surgery, special care must be taken when selecting medications for the anesthetic.77 Medications that are basic (e.g., lidocaine or alfentanil) are generally bound to plasma α1-acid glycoprotein; α1-acid glycoprotein concentrations in preterm and term infants are less than in older children and adults. Therefore, for a given dose, the free fraction of a drug is greater in preterm and term infants.78–80 Protein binding changes are important for the relatively unusual case of a drug that is more than 95% protein bound, with a high extraction ratio and a narrow therapeutic index, that is given parenterally (e.g., lidocaine administered IV), or a drug with a narrow therapeutic index that is given orally and has a very rapid T1/2keo (e.g., antiarrhythmic drugs; propafenone, verapamil).81


Maturational changes in tissue binding also affect drug distribution. Myocardial digoxin concentrations in infants are 6-fold greater than those in adults, despite similar serum concentrations. Erythrocyte/plasma concentration ratios of digoxin in infants are one-third smaller during loading digitalization than during maintenance digoxin therapy. These findings are consistent with a greater Vd of digoxin in infants and may explain, in part, the unusually large therapeutic doses needed in infants.82
Body Composition
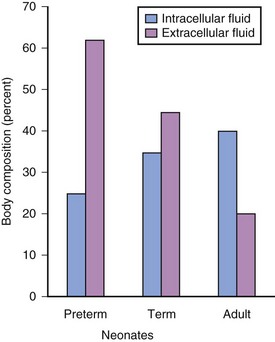
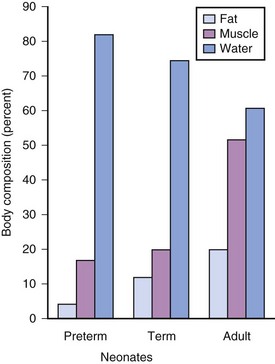
Preterm and term infants have a much greater proportion of body weight in the form of water than do older children and adults (Fig. 6-7).19 The net effect on water-soluble medications is a greater Vd in infants, which in turn increases the initial (loading) dose, based on weight, to achieve the desired target serum concentration and clinical response.1,3,14,83,84 Term neonates often require a greater loading dose (milligrams per kilogram) for some medications (e.g., digoxin, succinylcholine, and aminoglycoside antibiotics) than older children.83–87 However, neonates also tend to be sensitive to the respiratory, neurologic, and circulatory effects of many medications and therefore tend to be more responsive to these effects at reduced blood concentrations than are children and adults. Preterm infants are usually more sensitive than term neonates and in general require even smaller blood concentrations.1 On the other hand, dopamine may increase blood pressure and urine output in term neonates only at doses as large as 50 μg/kg/min. This dose, which would induce intense vasoconstriction in adults, suggests that neonates are less sensitive in their cardiovascular responsiveness.3,85,88–91 It is important to carefully titrate the doses of all medications that are administered to preterm and term infants to the desired response.
Compared with children and adolescents, preterm and term neonates have a smaller proportion of body weight in the form of fat and muscle mass; with growth, the proportion of body weight composed of these tissues increases (Fig. 6-8).* Therefore, medications that depend on their redistribution into muscle and fat for termination of their clinical effects likely have a larger initial peak blood concentration. These medications may also have a more sustained blood concentration because neonates have less tissue for redistribution of these medications. An incorrect dose may result in prolonged undesirable clinical effects (e.g., barbiturates and opioids may cause prolonged sedation and respiratory depression). The possible influence of small muscle mass on the response to muscle relaxants is exemplified by achieving neuromuscular blockade at smaller serum concentrations in infants.85
Absorption
Anesthetic drugs are mainly administered through the IV and inhalational routes, although premedication and postoperative pain relief is commonly administered enterally. Drug absorption after oral administration is slower in neonates than in children because of delayed gastric emptying (Fig. 6-9).
Adult enteral absorption rates may not be reached until 6 to 8 months after birth.94,95 Congenital malformations (e.g., duodenal atresia), co-administration of drugs (e.g., opioids), or disease characteristics (e.g., necrotizing enterocolitis) may further affect the variability in absorption. Delayed gastric emptying and reduced clearance may dictate reduced doses and frequency of repeated drug administration. For example, a mean steady state target paracetamol concentration greater than 10 mg/L at trough can be achieved by an oral dose of 25 mg/kg/day in preterm neonates at 30 weeks, 45 mg/kg/day at 34 weeks, and 60 mg/kg/day at 40 weeks PMA.96 Because gastric emptying is slow in preterm neonates, dosing may only be required twice a day.96 In contrast, the rectal administration of some drugs (e.g., thiopental, methohexital) is more rapid in neonates than adults. However, the interindividual absorption and relative bioavailability variability after rectal administration may be more extensive compared to oral administration, making rectal administration less suitable for repeated administration.97
The larger relative skin surface area, increased cutaneous perfusion, and thinner stratum corneum in neonates increase systemic exposure of topical drugs (e.g., corticosteroids, local anesthetic creams, antiseptics). Neonates have a greater tendency to form methemoglobin because of reduced methemoglobin reductase activity compared with older children. Furthermore, fetal hemoglobin is more readily oxidized compared with adult hemoglobin. Combined with an increased transcutaneous absorption, these have resulted in reluctance to apply repeat topical local anesthetics, such as EMLA (lidocaine-prilocaine) cream, in this age group.98 Similarly, cutaneous application of iodine antiseptics in neonates may result in transient hypothyroidism.
Metabolism and Excretion
Hepatic Metabolism
The liver is one of the most important organs involved in drug metabolism. Hepatic enzymatic drug metabolism usually converts the medication from a less polar state (lipid soluble) to a more polar, water-soluble compound (see later discussion). Although no categorical statement applies to all drugs and enzymes, the activities of most of these enzymes are reduced in neonates.3,4,16,20,22,87,99–104 Another important factor that influences hepatic degradation is hepatic blood flow. As the infant matures, a greater proportion of the cardiac output is delivered to the liver, therefore increasing drug delivery and potentially increasing drug metabolism. Some medications are extensively metabolized by the liver or other organs (e.g., the intestines or lungs) and are referred to as having high extraction ratios. This extensive metabolism produces a “first pass” effect in which a large proportion of an enteral dose is inactivated as it passes through the organ before reaching the systemic circulation. Metabolism via cytochrome P-450 in the intestinal wall may occur during drug absorption.105–107 Certain foods may induce or inhibit intestinal cytochromes, resulting in food–drug interactions.108 The concentrations of these enzymes in neonates are less than in older children. These enzymes may also be affected by diseases such as cystic fibrosis or celiac disease.109,110 Further metabolism may occur as the portal venous circulation from the small intestine passes through the liver before returning to the heart.105,107 In contrast, IV administration circulates drug to the liver or intestine for metabolism in proportion to the organ blood flow. Some of the drugs that exhibit extensive first-pass metabolism include propranolol, morphine, and midazolam.111–118
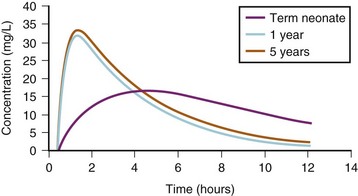
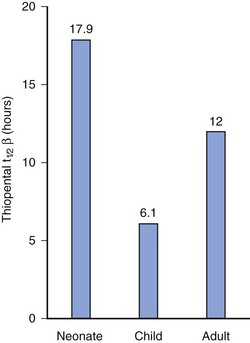
The opening or closing of a patent ductus may have profound effects on drug delivery to metabolizing organs in preterm infants.119,120 The ability to metabolize and conjugate medications improves considerably with age as a result of both increased enzyme activity and increased delivery of drug to the liver. Other factors influence the rate of hepatic maturation and metabolism (e.g., sepsis and malnutrition may slow maturation, whereas previous exposure to anticonvulsants, such as phenytoin or phenobarbital, may hasten maturation).3,89,99,100,104,121–125 The elimination half-lives of diazepam, thiopental, and phenobarbital are markedly increased in neonates compared with adults (i.e., the elimination half-life for thiopental in the neonate (17.9 hours) is almost three times that in children (6.1 hours) and 50% greater than that in adults (12 hours) (E-Fig. 6-3).12,74,126,127 In general, the half-lives of medications that are eliminated by the liver are prolonged in neonates, decreased in children 4 to 10 years of age, and reach adult values in adolescents, mirroring clearance changes with age (see Fig. 6-4).

Metabolism through biotransformation to more polar forms is required for many drugs before they can be eliminated. Two types of drug biotransformation can occur: Phase I and Phase II reactions. Phase I reactions transform the drug via oxidation, reduction, or hydrolysis. Phase II reactions transform the drug via conjugation reactions, such as glucuronidation, sulfation, and acetylation, into more polar forms.29,30 Although the liver is the primary site for biotransformation, other organs are also involved, including the lungs and kidneys. Hepatic drug metabolism activity appears as early as 9 to 22 weeks gestation, when fetal liver enzyme activity may vary from 2% to 36% of adult activity.128 It is inaccurate to generalize that the preterm neonate cannot metabolize drugs. Rather, the specific pathway(s) of drug metabolism must be considered.
Metabolism of many drugs involves the cytochrome P-450 (CYP) enzyme system. Multiple isoforms of the CYP enzyme system exist with different substrate specificities for different drugs.129–131 Induction and inhibition of these enzymes by different drugs and chemicals requires a thorough understanding of both the nomenclature of the CYP system, as well as the specific isoforms responsible for metabolism of the drugs used in pediatric anesthesia. There are both genetic and ethnic polymorphisms leading to clinically important differences in the capacity to metabolize drugs; these differences can make individual drug responses in some cases unpredictable.132–136 In the future it may be possible to tailor drug doses to the individual’s requirements by determining the child’s unique metabolic capacity.137,138
Cytochromes P-450: Phase I Reactions
CYPs are heme-containing proteins that provide most of the phase I drug metabolism for lipophilic compounds in the body.129 The generally accepted nomenclature of the cytochrome P-450 isozymes begins with CYP, and groups enzymes with more than 36% DNA homology into families designated with an Arabic number, followed by letters for the subfamily of closely related proteins (greater than 77% homology), followed by a number for the specific enzyme gene, such as CYP3A4.139,140 Isozymes that are important in human drug metabolism are found in the CYP1, CYP2, and CYP3 gene families. Table 6-1 outlines the CYP isozymes and their common substrates.
TABLE 6-1 Developmental Patterns and Activities for Important Cytochrome P-450 Enzymes (Phase I Reactions) in the Neonate
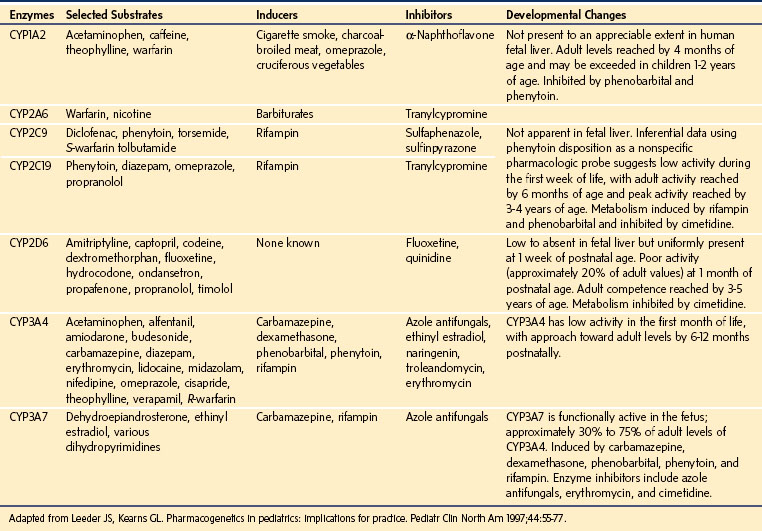
For many drugs, the reduced metabolism in neonates relates to reduced total quantities of CYP enzymes in the hepatic microsomes.141 Although the concentrations of CYP enzymes increase with gestational age, they may reach only 50% of adult values at term.141 In neonates, reduced CYP decreases clearance for many drugs, including theophylline, caffeine, diazepam, phenytoin, and phenobarbital.87,127,130,131,142–144 Although many isozymes are immature in the neonate, some CYP isozymes exhibit near-adult activity whereas others produce unique metabolic pathways in the neonatal period that invalidate broad generalizations about neonatal drug metabolism (see Table 6-1).
Developmental Changes of Specific Cytochromes
Cytochrome P-450 1A2 (CYP1A2) accounts for much of the metabolism of caffeine (1, 3, 7-trimethylxanthine)145,146 and theophylline (1,3-dimethylxanthine),147,148 which are methylxanthines frequently used to treat neonatal apnea and bradycardia. CYP1A2 activity is nearly absent in the fetal liver and remains minimal in the neonate.149 This limits N-3- and N-7-demethylation of caffeine in the neonatal period that prolongs elimination in preterm and term neonates.146,150 Elimination is through the immature renal system and consequent clearance is reduced. Adult levels of activity are reached between 4 and 6 months postnatally.151,152 A similar PK pattern of reduced metabolism at birth occurs with theophylline, in which CYP1A2 catalyzes 3-demethylation and 8-hydroxylation.147,148 Theophylline clearance reaches adult levels by 4 to 5 months, coincident with changes in CYP1A2 reflected in urine metabolite patterns.153
Other CYP enzymes that are reduced or absent in the fetus include CYP2D6 and CYP2C9.121,122,154 CYP2D6, which is involved in the metabolism of β-blockers, antiarrhythmics, antidepressants, antipsychotics, and codeine, is absent in the fetal liver and is eventually expressed postnatally (see Table 6-1).122,123 In contrast to the slow maturation of CYP1A2 and CYP2D6, CYP2C9, which are responsible for the metabolism of nonsteroidal antiinflammatory drugs (NSAIDs), warfarin, and phenytoin, have minimal activity antenatally121 and then develop rapidly postnatally.119,144
CYP3A is the most important cytochrome involved in drug metabolism, because of the broad range of drugs that it metabolizes and because it comprises the majority of adult human liver CYP (see Table 6-1).155 CYP3A is detectable during embryogenesis as early as 17 weeks, primarily in the form of CYP3A7,149 and reaches 75% of adult activity by 30 weeks gestation.122 In vivo, CYP3A activity appears to be mature at birth124; however, there is a poorly understood postnatal transition from the fetal CYP3A7 to the predominant adult isoform CYP3A4.156,157
Phase II Reactions
The other major route of drug metabolism, designated phase II reactions, involves synthetic or conjugation reactions that increase the hydrophilicity of molecules to facilitate renal elimination.29,30 The phase II enzymes include glucuronosyltransferase, sulfotransferase, N-acetyltransferase, glutathione S-transferase, and methyltransferase. The phase II enzymes also show developmental changes during infancy that influence drug clearance (Table 6-2).131,158–160
TABLE 6-2 Developmental Patterns for Important Conjugation (Phase II) Reactions in the Neonate
| Enzymes | Selected Substrates | Developmental Patterns |
|---|---|---|
| Uridine diphosphoglucuronyltransferase (UDP-GT) | Chloramphenicol, morphine, acetaminophen, valproic acid, lorazepam | Ontogeny is isoform specific. In general, adult activity is achieved by 6-18 months of age. May be induced by cigarette smoke and phenobarbital. |
| Sulfotransferase | Bile acids, acetaminophen, cholesterol, polyethylene, glycols, dopamine, chloramphenicol | Ontogeny seems to be more rapid than UDP-GT; however, it is substrate specific. Activity for some isoforms may exceed adult values during infancy and childhood, e.g., that responsible for acetaminophen metabolism. |
| N-Acetyltransferase 2 | Hydralazine, procainamide, clonazepam, caffeine, sulfamethoxazole | Some fetal activity present by 16 weeks. Virtually 100% of infants between birth and 2 months of age exhibit the slow metabolizer phenotype. Adult activity present by 1-3 years of age. |
Adapted from Leeder JS, Kearns GL. Pharmacogenetics in pediatrics: implications for practice. Pediatr Clin North Am 1997;44:55-77.
Most conjugation reactions have limited activity during fetal development.161 One of the most familiar synthetic reactions in young infants involves conjugation by uridine diphosphoglucuronosyltransferases (UGT). This enzyme system includes numerous isoforms and is also responsible for glucuronidation of endogenous compounds, such as bilirubin (by UGT1A1).161 As with the maturation of bilirubin conjugation, UGT activity is limited immediately postnatally and the different isoforms mature at different rates postnatally.162 Dosage adjustments are often needed to avoid toxicity in neonates from drugs that require conjugation by UGT for clearance. Experience with chloramphenicol in the 1960s illustrated this lesson when neonates received standard pediatric doses of chloramphenicol without understanding the immaturity of UGT and its role in the elimination of chloramphenicol. Infants accumulated high concentrations of chloramphenicol and developed fatal circulatory collapse, a condition known as the gray baby syndrome.163–165 Although the clearance of chloramphenicol is low during the neonatal period, appropriate dosage adjustments and monitoring allow safe treatment of preterm and term infants with chloramphenicol.166
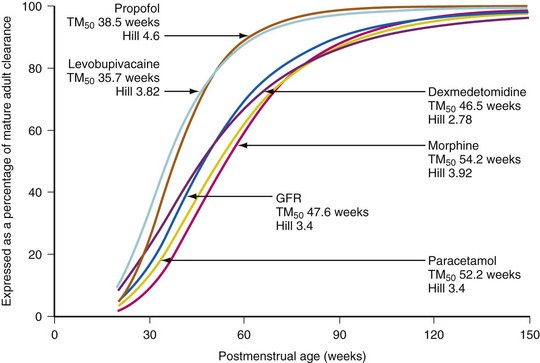
Morphine, acetaminophen, dexmedetomidine and lorazepam also undergo glucuronidation. The major steps in the metabolic disposition of morphine in children and adults is glucuronidation in the 3- and 6-position.111,167 The limited ability of neonates to metabolize morphine by glucuronidation necessitates dosage adjustment.58,168,169 Detailed studies have shown that morphine clearance,168,170 in particular 3- and 6-glucuronide formation, is limited at birth and increases with birth weight,169 gestational age,129 and postnatal age.58,167 In some studies, morphine clearance, expressed as per kilogram, approaches adult values by 1 month,58,171although others reported that the clearance does not reach adult values until at least 5 to 6 months.168,172 Overall, the maturation of glucuronosyltransferase enzymes varies among isoforms, but, in general, adult activity is reached by 6 to 18 months of age.140 Some of the confusion relating to maturation rates is attributable to the use of the per kilogram size model. The use of allometry with a maturation model has assisted understanding. The time courses of maturation of drug metabolism for morphine,57 acetaminophen,173 dexmedetomidine,174 and glomerular filtration rate175 (GFR) are strikingly similar (Fig. 6-10) with 50% of size-adjusted adult values being reached between 8 and 12 weeks (TM50) after full-term delivery. All three drugs are cleared predominantly by UGT that converts the parent compound into a water soluble metabolite that is excreted by the kidneys and the clearance maturation profiles of these drugs matches that of GFR maturation. Glucuronidation is also the major metabolic pathway of propofol metabolism, although multiple CYP isoenzymes, including CYP2B6, CYP2C9, or CYP2A6, contribute to its metabolism and cause a faster maturation profile than expected from glucuronide conjugation alone.176 A phase I reaction (CYP3A4) is the major enzyme system for oxidation of levobupivacaine and clearance through this pathway is faster than those associated with UGT maturation.57,173,175,177–180
In contrast to glucuronosyltransferase, the sulfotransferase enzyme system is well developed in the neonate, and for some compounds it may compensate for limited glucuronidation. In adults, the primary pathway for acetaminophen metabolism is glucuronidation, yet its half-life is only moderately prolonged in neonates compared with older infants and adults.181–183 This occurs partly because of the increased Vd in neonates (T1/2 α Vd/CL) and partly because the neonate forms more sulfate than glucuronide conjugate, leading to a greater percent of the dose excreted as the acetaminophen-sulfate conjugate.96,182–185 Unfortunately, this does not confer safety from hepatotoxicity. The toxic metabolite is created through the oxidative pathway mediated by CYP2E1.
Alterations in Biotransformation
Transition from the intrauterine to the extrauterine environment is associated with major changes in blood flow. There may also be an environmental trigger for the expression of some metabolic enzyme activities resulting in a slight increase in maturation rate above that predicted by postmenstrual age.176,179 Many biotransformation reactions, especially those involving certain forms of CYP, are inducible before birth through maternal exposure to drugs, cigarette smoke, or other inducing agents. Postnatally, biotransformation reactions may be induced through drug exposure (see Tables 6-1 and 6-2) and may be slowed by hypoxia, asphyxia, organ damage, and/or illness. The reduced thiopental clearance estimated from data when the drug was given to control neonatal seizures that resulted from hypoxic-ischemic insults, may not be applicable to healthy neonates undergoing anesthesia.186
Genotypic Variations in Drug Metabolism
Genetic variations can have impact on both PK and PD187 Single nucleotide changes or polymorphisms (SNPs) in the DNA sequence in CYP enzymes usually decrease but may also increase metabolic activity for a specific drug or drug substrate.188 Some of the explanation for variations in drug responses within large populations that are described as “biologic variation” likely relate to genetic differences in drug metabolism, receptor binding, and intracellular coupling to effector mechanisms. Following the recognition that certain individuals had exaggerated hypotensive responses to debrisoquine, the enzyme responsible for its metabolism, CYP2D6 became one of the first drug-metabolizing enzyme deficiencies identified.189–191 Earlier studies had shown that individuals might possess normal or reduced metabolic activity (poor metabolizers) for debrisoquine and sparteine.190,192 The frequency of poor metabolizers varied among ethnic groups, occurring in approximately 7% of Caucasians132 and in 0% to 1% of Chinese and Japanese.193
Codeine, is primarily a prodrug that undergoes metabolic activation by CYP2D6 O-demethylation to morphine.194 Without O-demethylation, codeine confers a small fraction of the analgesic molar potency of morphine, and much of its analgesic effect is likely contributed by a metabolite, codeine 6-glucuronide,195 although evidence suggests that up to 11% of codeine is also metabolized to hydrocodone.196 For the 2% to 10% of the population who are CYP2D6 poor metabolizers, codeine causes limited opioid effects.197,198 Gastrointestinal motility is affected only in extensive metabolizers, suggestive of a morphine-dependent mechanism of action.199 Individuals with duplicated active CYP2D6 genes are classified as ultra-extensive metabolizers.199a A 29-month-old previously healthy child experienced apnea resulting in brain injury, following a dose of codeine 2 days after an uneventful anesthetic for tonsillectomy. A genetic polymorphism leading to ultra-rapid metabolism of codeine into morphine, resulting in narcosis and apnea, was proposed.200
For other drugs, ranging from propranolol to warfarin to methotrexate, reduced metabolism through genetic polymorphisms of other enzymes lead to exaggerated effects when administered in conventional doses.127 Similar variations in activity have been reported for many of the CYPs involved in drug metabolism in humans.201 The potential importance of genotyping is demonstrated for patients scheduled to receive treatment with methotrexate; genotyping has become a routine part of the evaluation before treatment, for detection of reduced activity of thiopurine methyltransferase that may be lethal with conventional dosages.202,203 Genotyping has been proposed before drug treatment and as a guide to drug selection when specific SNPs have been correlated with adverse drug reactions or clinically significant alterations in metabolism.138 Although hundreds of possible SNPs have been identified that account for much of the variation in drug effects among individuals, specific SNPs of CYP2C9, CYP2C19, CYP2D6, CYP3A, and uridine diphosphate-glucuronosyltransferase 1A1 (UGT1A1) account for a sufficient number of adverse pharmacologic outcomes to warrant clinical testing when it becomes feasible.138 Until genotyping for many more abnormalities in drug metabolizing enzymes, receptors, and channels becomes a routine part of drug treatment, careful attention to a history of adverse drug reactions in the child and first-degree relatives, and careful attention to recording current reactions, are the best guides to detect clinically important variations in drug metabolism.
Extrahepatic Routes of Metabolic Clearance
Many drugs undergo metabolic clearance at extrahepatic sites. Remifentanil and atracurium are degraded by nonspecific esterases in tissues and erythrocytes. Clearance, expressed per kilogram, is increased in younger children,52,204–207 likely attributable to size, because clearance is similar when scaled to a 70-kg person using allometry.52 Nonspecific blood esterases that metabolize remifentanil are mature at birth.53
Ester local anesthetics are metabolized by plasma butyrylcholinesterase, which is thought to be reduced in neonates. The in vitro plasma half-life of 2-chloroprocaine in umbilical cord blood is twice that in maternal blood,208 but there are no in vivo studies of the effects of age on its metabolism. Succinylcholine clearance is increased in neonates when expressed as per kilogram, suggesting butyrylcholinesterase activity is mature at birth.209,210
Renal Excretion
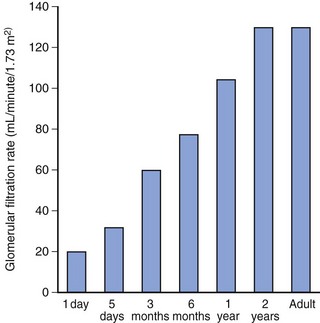
Renal function in preterm and term infants is less efficient than in adults, even after adjusting for the differences in body weight. This reduced efficiency is related to the combination of incomplete glomerular development, low perfusion pressure, and inadequate osmotic load to produce full countercurrent effects.211–216 However, glomerular filtration and tubular function both develop rapidly during the first few months of life,175 and are nearly mature by 20 weeks of age, and fully mature by 2 years of age (Figs. 6-10 and 6-11).212–216 For these reasons, drugs that are excreted primarily through glomerular filtration or tubular secretion, such as aminoglycoside and cephalosporin antibiotics, have a prolonged elimination half-life in neonates (E-Fig. 6-4).217–219

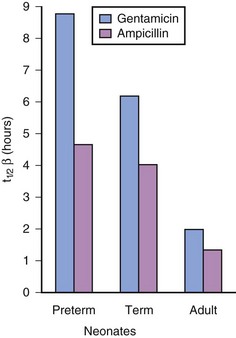
The PK and PD of the old muscle relaxant, curare, exemplify the complex interaction of increased Vd, smaller muscle mass, and decreased rate of excretion as a result of immaturity of glomerular filtration. The initial dose (per kilogram) of curare needed to achieve neuromuscular blockade is similar in infants and adults.85 In infants, however, this blockade is achieved at reduced serum concentrations compared with older children or adults, corresponding to differences in muscle mass and receptor immaturity. A larger Vd (total body water) accounts for the equivalent dose for each kilogram of body weight, and the reduced glomerular function in infants compared with older children or adults accounts in part for the longer duration of action.85 As in the case of drugs excreted by the liver, there is a triphasic developmental response to drugs excreted by the kidneys when expressed as per kilogram (see Fig. 6-4): a prolonged half-life in neonates (immature renal function), a shortened half-life in young children, and a greater elimination half-life in adolescents and adults (size related).
Reduced protein binding in neonates and preterm infants increases the free fraction of drugs delivered to the kidneys and liver for metabolism; however, reduced clearance results in a greater potential for toxicity.1,3,16,17 An important example is the immature clearance of bupivacaine, which resulted in high plasma concentrations that caused seizures in neonates given epidural infusions at rates greater than that at which it was metabolized.220
Central Nervous System Effects
Laboratory data have demonstrated the lethal dose in 50% of animals (LD50) for many medications to be significantly less in neonates than in adult animals.221,222 The sensitivity of human neonates to most of the sedatives, hypnotics, and opioids is clinically well known and may in part be related to increased brain permeability (immature blood-brain barrier or damage to the blood-brain barrier) for some medications.223–229 Laboratory studies have demonstrated greater brain concentrations of morphine and amobarbital in infant than in adult animals.230
However, respiratory depression, measured by carbon dioxide response curves or by arterial oxygen tension are similar from 2 to 570 days of age at the same morphine blood concentration.231 Altered PK may contribute to apparent morphine sensitivity in neonates. A reduced clearance and a reduced Vd in neonates will result in greater plasma concentrations in this age group compared with children given similar weight-scaled doses,232,233 and this greater concentration contributes more to respiratory depression, than does increased brain permeability.
Small molecules are thought to access fetal and neonatal brains more readily than in adults.234 Blood-brain barrier function improves gradually, possibly reaching maturity by full-term.234 Kernicterus, for example, is more common in preterm than in full-term neonates. In contrast to drugs bound to plasma proteins, unbound lipophilic drugs passively diffuse across the blood-brain barrier equilibrating very quickly. This may contribute to bupivacaine’s propensity for producing seizures in neonates. Decreased protein binding, as in the neonate, results in a greater proportion of unbound drug that is available for passive diffusion.
In addition to passive diffusion, there are specific transport systems that mediate active transport. Pathologic CNS conditions can cause blood-brain barrier breakdown and alter these transport systems. Fentanyl is actively transported across the blood-brain barrier by a saturable ATP-dependent process; ATP-binding cassette proteins, such as P-glycoprotein, actively pump out opioids, such as fentanyl and morphine.235 P-glycoprotein modulation significantly influences opioid brain distribution and onset time, as well as the magnitude and duration of analgesic response.236 Modulation may occur during disease processes, fever, or in the presence of other drugs (e.g., verapamil, magnesium).235 Genetic polymorphisms that affect P-glycoprotein–related genes may explain differences in the sensitivity to CNS-active drugs.237
Incomplete myelination in infants may make it easier for drugs that are not particularly lipid soluble to enter the brain at a greater rate than if the blood-brain barrier were intact.223,224,230,238 When considering the use of any centrally acting medication in children younger than 1 year of age, and particularly those younger than 48 weeks postmenstrual age, one must balance the potential risks and benefits. Dosage must be carefully calculated and titrated to allow the lowest dose that provides the required patient response. Careful monitoring of vital signs is important, because prolonged effects or adverse clinical responses may occur in children of any age, but particularly in infants in whom CNS maturation may be incomplete.
Pharmacodynamics in Children
Children’s responses to drugs have much in common with the responses in adults.239 The perception that drug effects differ in children arises because the drugs have not been adequately studied in pediatric populations who have size and maturation related effects, as well as different diseases. Neonates and infants, however, often have altered PD. A series of examples are presented below.
The MAC for almost all anesthetic vapors is less in neonates than in infants, which is in turn greater than that observed in children and adults.240 MAC of isoflurane in preterm neonates less than 32 weeks gestation was 1.28%, and MAC in neonates 32-37 weeks gestation was 1.41%.241 This value rose to 1.87% by 6 months before decreasing again over childhood.241 The cause of these differences is uncertain and may relate to maturation changes in cerebral blood flow, γ-aminobutyric acid (GABA) class A receptor numbers, or developmental shifts in the regulation of chloride transporters.
Neonates have an increased sensitivity to the effects of neuromuscular blocking drugs (NMBDs).85 The reason for this is unknown but it is consistent with the observation that there is a threefold reduction in the release of acetylcholine from the infant rat phrenic nerve as well as a relatively reduced muscle mass.242–245 The increased Vd, however, means that a single NMBD dose (calculated as mg/kg) in the neonate results in blockade at a reduced plasma concentration while reduced clearance prolongs the duration of effect.
Cardiac calcium stores in the endoplasmic reticulum are reduced in the neonatal heart because of immaturity. Exogenous calcium has greater impact on contractility in this age group than in older children or adults. There are some data to suggest greater sensitivity to warfarin in children, but the mechanism is not determined.246 Amide local anesthetic agents induce shorter block duration and require a larger weight-scaled dose to achieve similar dermatomal levels, when given by subarachnoid block to infants. This may be due, in part, to myelination, spacing of nodes of Ranvier and length of nerve exposed, increased relative volume of CSF, as well as other size factors. There is an age-dependent expression of intestinal motilin receptors and the modulation of gastric antral contractions in neonates. Prokinetic agents may not be useful in very preterm infants, partially useful in older preterm infants, and useful in full-term infants. Similarly, bronchodilators in infants are less effective because of the paucity of bronchial smooth muscle that can cause bronchospasm.
Measurement of Pharmacodynamic Endpoints
Outcome measures are more difficult to assess in neonates and infants than in children or adults. Measurement techniques, disease and pathology differences, inhomogeneous groups, recruitment issues, ethical considerations, and endpoint definitions for establishing efficacy and safety often confuse data interpretation.247
Common effects measured include anesthesia depth, pain responses, depth of sedation, and intensity of neuromuscular blockade. A common effect-measure used to assess depth of anesthesia is the EEG or a modification of detected EEG signals (spectral edge frequency, bispectral index [BIS], entropy). Physiologic studies in adults and children indicate that EEG-derived anesthesia depth monitors can provide an imprecise and drug-dependent measure of arousal. Although the outputs from these monitors do not closely represent any true physiologic entity, they can be used as guides for anesthesia, and in so doing, may improve outcomes in adults. In older children the physiology, anatomy, and clinical observations indicate the performance of the monitors may be similar to that in adults. In infants, however, their use cannot yet be supported in theory or in practice.248,249 During anesthesia, the EEG in infants is fundamentally different from the EEG in older children; there remains a need for specific neonate-derived algorithms if EEG-derived anesthesia depth monitors are to be used in neonates.250,251 Examples of problems with BIS monitoring in infants and young children include the observations that BIS numbers paradoxically increase when sevoflurane concentrations exceed 3%, there is often a difference between the right and left side of the brain, equivalent MAC concentrations produce different BIS values with each agent, and values in children tend to be greater than values in adults at equivalent MAC concentrations.252–256
The Children’s Hospital of Wisconsin Sedation Scale has been used to investigate ketamine in the emergency department.65,257 However, despite the use of such scales in procedural pain or sedation studies, few behavioral scales have been adequately validated in this setting and inter-observer variability can be substantial.258–260 Most scores are validated for the acute, procedural setting and perform less well for subacute or chronic pain or stress.
The Target Concentration Approach
The goal of treatment is the target effect. A PD model is used to predict the target concentration given a target effect. Population estimates for the PD model parameters and covariate information are used to predict typical PD values in a specific patient. Population estimates of PK model parameter estimates and covariate information are then used to predict typical PK values in a typical patient. For example, a dexmedetomidine steady-state target concentration of 0.6 μg/L may be achieved with an infusion of 0.33 μg/kg/hr in a neonate, 0.51 μg/kg/hr in a 1-year-old, and 0.47 μg/kg/hr in an 8-year-old.180 This target concentration strategy is a powerful tool for determining clinical dose.261 Monitoring of serum drug concentrations and Bayesian forecasting may be used to improve dosing in individual patients.
Defining Target Concentration
A target concentration of 10 μg/L may be used for morphine analgesia. Observations in children after cardiac surgery found that steady state serum concentrations greater than 20 μg/L resulted in hypercarbia (Paco2 greater than 55 mm Hg) and depressed CO2 response curve slopes. During washout, morphine concentrations more than 15 μg/L resulted in hypercarbia in 46% of children, whereas concentrations less than 15 μg/L were associated with hypercarbia in 13% of children. No age-related differences in respiratory effect were seen in these studies at the same serum morphine concentration.231 Observation or self-reporting pain scales are used as part of the feedback loop for dose incremental changes.
The target concentration may vary, depending on the desired target effect. The target concentration for ketamine analgesia (0.25 mg/L) is quite different from that of anesthesia (2 mg/L), and BIS monitoring would be totally useless because ketamine causes central excitation thereby increasing bispectral monitoring numbers.262–265
Drug Interactions
There are many common examples of drug interactions that increase or decrease responses mediated through either PK or PD routes. Phenobarbitone induces CYP3A4 metabolism, increasing ketamine requirements for radiologic sedation.266 An increase in the T1/2keo of d-tubocurarine with increasing inspired halothane concentrations has been demonstrated.267 Halothane is a negative inotrope268 and reduces skeletal muscle blood flow,269 so it seems reasonable to interpret changes in T1/2keo as a result of changes in organ blood flow. Inhalation anesthetic agents can also prolong the duration of block and this affect is agent specific. When compared with halothane, sevoflurane potentiates the effects of vecuronium to a greater extent. When compared with balanced anesthesia, sevoflurane and halothane decrease the dose requirements of vecuronium by 60% and 40%, respectively.270
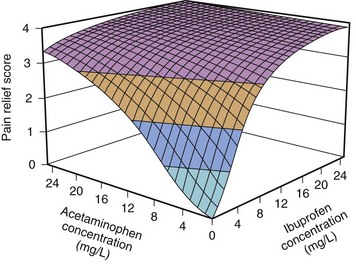
Anesthetic drug interactions traditionally have been characterized using isobolographic analysis or multiple logistic regression. Minto proposed a model based on response-surface methodology.271 Computer simulations based on interactions at the effect-site predicted that the maximally synergistic three-drug combination (midazolam, propofol, and alfentanil) tripled the duration of effect compared with propofol alone. The response surface for ibuprofen and acetaminophen is shown in Figure 6-12. The addition of acetaminophen to ibuprofen improved analgesia when the dose of ibuprofen was less that 100 mg (5 mg/kg) in a 5-year-old child.272 Response surfaces can describe anesthetic interactions, even those between agonists, partial agonists, competitive antagonists, and inverse agonists.271
Synergism between propofol and alfentanil,273 as well as propofol and remifentanil,274 has also been demonstrated using response-surface methodology. Remifentanil alone had no appreciable effect on response to shaking and shouting or response to laryngoscopy while propofol could ablate both responses. Modest remifentanil concentrations dramatically reduced the concentrations of propofol required to ablate both responses.275 When comparing the different combinations of midazolam, propofol, and alfentanil, the responses varied markedly at each endpoint assessed and could not be predicted from the responses observed with each individual agent.276 Similar response-surface methodology has been used to investigate the combined administration of sevoflurane and alfentanil,273 and remifentanil and propofol,274 on ventilation. These combinations have a strikingly synergistic effect on respiration, resulting in severe respiratory depression in adults. These synergistic associations can be extended to pediatric sedation techniques. It is little wonder that the use of three or more sedating medications compared with one or two medications is strongly associated with adverse outcomes.274
The Drug Approval Process, the Package Insert, and Drug Labeling
One area of concern has been the general lack of approval of many medications for populations of pediatric patients. This is particularly ironic because most of the changes in legislation pertaining to pharmaceuticals have been a result of adverse events in infants and children. The 1938 Federal Food, Drug, and Cosmetic Act277 replaced the original Federal Food and Drugs Act of 1906 (Wiley Act)278 because nearly 100 individuals, mostly children, were poisoned by diethylene glycol (an antifreeze analogue for vehicles) that had been added to an elixir of sulfanilamide. This new legislation prohibited the addition of poisonous substances (unless they were demonstrated to be safe in low concentrations) and instituted other measures to protect the consumer. The next major piece of legislation was the Kefauver-Harris Amendments, which were passed in 1962 as a result of the thalidomide catastrophe.279 This legislation strengthened the safety standards by requiring the drug company to demonstrate effectiveness before marketing. The U.S. Food and Drug Administration (FDA) then allowed drugs to be marketed to adults as “safe and effective,” but now the drug label was required to indicate that “safety and effectiveness had not been established in children” because no trials in children had been carried out. This had an enormous negative impact on drug development for children and led Shirkey to coin the now common expression “therapeutic orphans” when referring to drug development for children.280
Until the late 1990s, nearly 80% of approved medications contained language within the drug label (package insert) that excluded children of varying ages. The majority of the drugs used in the operating room (OR) and the intensive care unit (ICU) today have similar language.281 Common examples of disclaimers for drugs used in our daily practice include those for bupivacaine (“Until further experience is gained in children younger than 12 years, administration of Sensorcaine [bupivacaine HCl] injection is not recommended”)282 and for fentanyl (“It should not be administered to children 2 years of age or younger because safety in this age group has not yet been established”).283 Such disclaimers are placed in the package insert because the contents of the package insert must, by law, be based on “adequate, well controlled studies involving children.”279,284–286 Any use of a drug that is not specifically described in the package insert is considered “unapproved” or “off label.” The reason for the lack of labeling for children is that the appropriate controlled clinical trials were never supported by industry and the FDA did not have the legislative power to force the pharmaceutical companies to perform pediatric studies.287 In 1994, the FDA passed a new interpretation of the original Food, Drug, and Cosmetic Act277,285 that allowed manufacturers to review the published medical literature and submit these data to the FDA to support revised pediatric labeling.286 This did result in additional changes in the drug label for approximately 100 medications. Unfortunately, for drugs that are no longer under patent protection, there was no financial incentive to force the issue, so many drugs remain unlabeled for children despite the many papers published describing their safe use in children of all ages.
During the early stages of the AIDS epidemic, there was great pressure placed on the FDA to reduce the time for the drug approval process. New legislation was passed to raise funds to pay for additional consultants and experts to help the FDA with this process (The Prescription Drug User Fee Act [PDUFA]).288 This legislation was renewed in 1997, 2002, 2007, and was reaffirmed for the fourth time in July 2012 (PDUFA V). The monies from these fees (~ $1,170,000 per full application) greatly reduced the time from a New Drug Application until a drug reaches market, and the most recent iteration has expanded funding to support marketing safety and pharmacoepidemiology activities, as well as increased inspection of non-USA based pharmaceutical manufacturing facilities. Approximately 55 feasibility and in-depth studies were launched under PDUFA IV.289
Additional changes at the FDA occurred in the late 1990s when The Food and Drug Administration Modernization Act290 and The Final Rule were passed.291 Tacked onto this legislation was the Better Pharmaceutical Act for Children, which granted 6 months’ patent extension in exchange for pediatric studies of drugs that were still patent protected. This was later replaced with the Best Pharmaceutical Act for Children (BPCA) in 2002, which earmarked money for the National Institutes of Health (NIH) to support study of drugs no longer patent protected.292 This was subsequently challenged as giving excessive legal power to the FDA, but further legislation reinstituted the legal power to the FDA to now require drug companies to conduct research in children if the drug would have use in children (The Pediatric Research Equity Act).293 Legislation passed in 2007 that renewed PDUFA IV was a much larger bill entitled the Food and Drug Administration Amendments Act of 2007. This bill renewed PDUFA IV, the Medical Device User Fee and Modernization Act, and BPCA.294 Of importance to researchers was the new requirement for registration of all clinical trials with an archive of thousands of trials that is easily searchable for clinicians as well as the public.295 Many journals now will not publish clinical pharmaceutical trials that have not been registered. Since the first legislation for children passed in 1998, there has been an explosion of pediatric drug trials (more than 600 requested or carried out from 1997 until 2011) and new drug labels have been created for nearly 400 drugs. Unfortunately, the definition of a pediatric study is still somewhat unclear and the money that was supposed to be earmarked to the NIH for generic drug trials has not been fully provided, so deficiencies in labeling for older drugs still persist.
It is important for clinicians to understand that, despite language on the label regarding use in children, they are perfectly within their medical and legal rights to use these drugs in children. “Unapproved use does not imply an improper use and certainly does not imply an illegal use.”296,297 The use of a drug in a child is the decision of the individual physician and may be based on the available literature, despite the fact that formal FDA approval and labeling have not been achieved.284,285 The Committee on Drugs of the American Academy of Pediatrics is very clear on this issue: “Lack of approval for a specific use should not prevent physicians from prescribing an available drug in the best interest of their patients.”296,297
Inhalation Anesthetic Agents
Physicochemical Properties
The potent synthesized inhaled anesthetics are ether anesthetics based on either a methyl ethyl (enflurane, isoflurane, and desflurane) or a methyl isopropyl (sevoflurane) polyhalogenated ether skeleton (Table 6-3). The single exception in chemical structure is halothane, which is a polyhalogenated alkane. Of the methyl ethyl ether anesthetics, isoflurane and enflurane are identical in chemical structure but are stereoisomers. Desflurane differs from isoflurane in the single atomic substitution of a fluoride for a chlorine atom on the α-carbon of isoflurane. Sevoflurane differs from isoflurane in the substitution of a trifluoromethyl group for the chlorine atom. Although the general chemical structures of the four ether inhalational agents are similar, the single atomic substitutions confer substantially different physicochemical and pharmacologic properties that are described below and contrasted to the properties of halothane (see Table 6-3). All of these agents are liquids at room temperature and pressure.
TABLE 6-3 Pharmacology of Inhaled Anesthetics
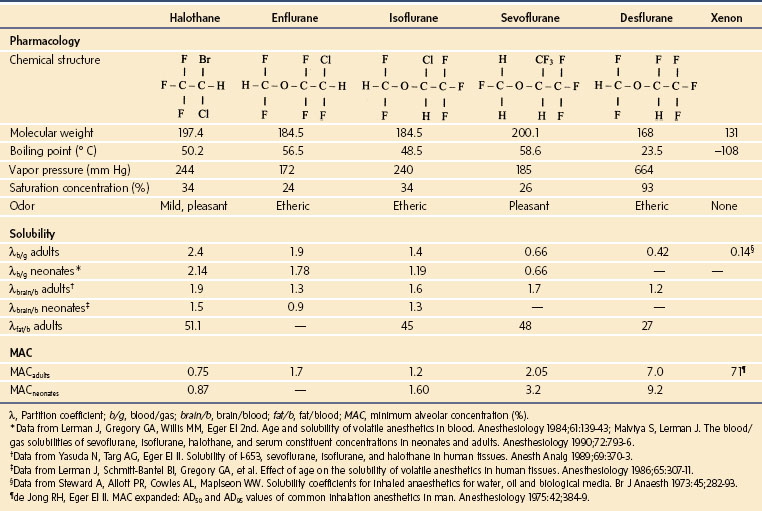
Pharmacokinetics of Inhaled Anesthetics
The rate of increase or equilibration of the partial pressures of alveolar to inspired anesthetic (also known as the wash-in) is a function of the rate of delivery of anesthetic to, and uptake from, the lungs. Six factors determine the wash-in of inhalational anesthetics (Table 6-4)298: the first three determine the delivery of anesthetics to the lungs, and the second three determine their rate of removal (uptake) from the lungs. The wash-in, defined as the ratio of the alveolar to inspired anesthetic partial pressures (Fa/Fi, or fractional alveolar to fractional inspired partial pressures), increases from zero to a value of unity (1), when the inspired and alveolar partial pressures have equilibrated (Fig. 6-13).299 Although not shown in the figure, the wash-in of xenon is exceedingly rapid based on its physicochemical properties (see Table 6-3).300 For the Fa/Fi to increase toward equilibration, the rate of delivery of anesthetic to the lungs must substantially exceed its uptake from the lungs.
TABLE 6-4 Determinants of the Wash-In of Inhalational Agents
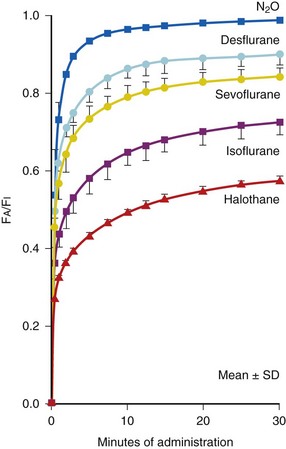
Salanitre and Rackow first demonstrated that the rate of increase of Fa/Fi of halothane in infants and children was more rapid than in adults (Fig. 6-14).301 Subsequent studies substantiated similar relationships for isoflurane, enflurane, and nitrous oxide.302,303 The more rapid rate of increase of Fa/Fi in neonates compared with adults has been attributed to four factors (Table 6-5); the order in the table reflects their relative contributions to the rapid wash-in. Based on these factors and the solubilities of sevoflurane and desflurane, we speculate that the wash-in curves of sevoflurane and desflurane in neonates and infants will be comparable to those in adults. This amounts to a safety factor for these anesthetics that was not previously afforded with halothane.
TABLE 6-5 Determinants of the Rapid Wash-In of Inhalational Agents in Infants Compared to Adults
Factors Affecting Delivery of Anesthetics to the Lungs
Inspired Concentration
The effect of the inspired concentration on the Fa/Fi of anesthetics relates only to those that are administered in high concentrations (i.e., nitrous oxide). The greater the FI of nitrous oxide, the more rapid the increase of Fa/Fi.304 This effect, known as the concentration effect (second gas effect), depends on both a concentrating effect and an increase in alveolar ventilation that results from an increased uptake of nitrous oxide.298 Hence, agents that depend on alveolar ventilation for their wash-in (i.e., the more soluble anesthetics) will have a more rapid wash-in when administered with nitrous oxide. This effect diminishes as the solubility of the anesthetic decreases and as time passes.305–307 Although the rate of increase of Fa/Fi of nitrous oxide in adults is rapid, it is even more rapid in neonates and infants.301,303
Alveolar Ventilation and Functional Residual Capacity
The ratio of alveolar ventilation to functional residual capacity (Va/FRC) is the primary determinant of the rate of the delivery of anesthetic to the lungs. The greater the Va/FRC ratio, the more rapid the Fa/Fi (E-Fig. 6-5). However, the ratio does not affect all anesthetics similarly: more soluble anesthetics (i.e., halothane) are affected to a greater extent than are less soluble anesthetics (i.e., sevoflurane and desflurane). The reason for this differential effect of Va/FRC relates to the relative delivery of anesthetic to and uptake from the lungs: in the case of soluble anesthetics (halothane), uptake from the lungs is substantial, thereby limiting the increase in Fa/Fi to the delivery of anesthetic to the lungs. Hence, changes in the alveolar ventilation will directly affect the Fa/Fi of anesthetics in proportion to their blood solubilities. In terms of age-related effects of Fa/Fi, the Va/FRC ratio accounts for most of the differences between the Fa/Fi of anesthetics in neonates and adults (see Table 6-5). The Va/FRC ratio is approximately 5 : 1 in neonates compared with only 1.5 : 1 in adults. The greater Va/FRC ratio in neonates may be attributed to the threefold greater metabolic rate and, therefore, threefold greater alveolar ventilation in neonates compared with adults. This is true for both spontaneous and controlled ventilation, depending on the settings used during controlled ventilation.

Factors Affecting the Uptake (Removal) of Anesthetics from the Lungs
Cardiac Output
The rate of increase in Fa/Fi is inversely related to changes in cardiac output: that is, the smaller the cardiac output, the more rapid the increase in Fa/Fi and vice versa (E-Fig. 6-6). As cardiac output diminishes, removal of anesthetic from the lungs diminishes and the rate of equilibration of Fa/Fi increases. Thus, a child in heart failure who receives an inhalational induction may achieve greater anesthetic concentrations in the lungs more rapidly than expected compared with a child with a normal cardiac output. This very serious problem may be exacerbated if the “overpressure” technique has been used, because it may cause an acute anesthetic-induced myocardial depression and decompensation of cardiac output. Conversely, in a high cardiac output state (as in the case of anxiety), the greater blood flow through the lungs removes anesthetic from the alveoli, thus reducing the alveolar partial pressure of anesthetic, which slows the rate of equilibration of Fa/Fi. The impact of changes in cardiac output on Fa/Fi depends, in part, on the solubility of the anesthetic: the more soluble the anesthetic (e.g., halothane), the greater the effect of changes in cardiac output on Fa/Fi and the less soluble the anesthetic (sevoflurane and desflurane), the less the effect.298 This is another safety feature of the less soluble anesthetics.

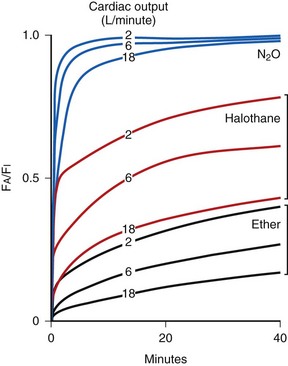
Solubility
The rate of increase of Fa/Fi of inhalational anesthetics, which varies inversely with the solubility of the anesthetic in blood, follows the order: nitrous oxide > desflurane > sevoflurane > isoflurane > enflurane > halothane > methoxyflurane (see Table 6-3 and Fig. 6-13).298,299 Although the solubilities of nitrous oxide and desflurane are similar, the rate of increase in Fa/Fi of nitrous oxide is more rapid than that after desflurane because of the concentration effect from administering 70% nitrous oxide. After a stepwise change in the inspired partial pressure of less soluble anesthetics, the alveolar partial pressure equilibrates rapidly with the new inspired partial pressure. Because the washout of these anesthetics is equally rapid (see later discussion), the alveolar partial pressure can be adjusted to previous values rapidly by decreasing the inspired partial pressure. Thus, anesthetic depth can be controlled more rapidly with a less soluble (i.e., desflurane or sevoflurane) than with a more soluble inhalational anesthetic (i.e., halothane).
The blood solubility of xenon is one-third that of nitrous oxide and desflurane (see Table 6-3). Although the tissue solubility of xenon has not been determined, its low blood solubility would suggest that its wash-in is the most rapid of the inhalational anesthetics.
Age is an important determinant of the solubility of inhalational anesthetics in blood. The blood solubilities of halothane, isoflurane, enflurane, and methoxyflurane are 18% less in neonates than they are in adults (E-Fig. 6-7; see also Table 6-3).308 Serum cholesterol and proteins (including albumin) account for these age-related differences in blood solubilities.308,309 In contrast, the blood solubility of the less soluble anesthetic sevoflurane is similar in neonates and adults.309 Factors that do not appear to significantly affect the blood solubility of most anesthetics include age-related differences in hemoglobin, serum concentration of α1-acid glycoprotein, and prematurity.308,309

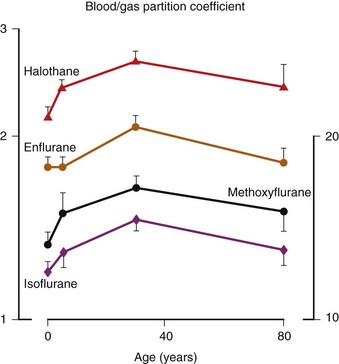
The tissue/gas solubilities of the inhalational anesthetics in the VRG in neonates are approximately one-half those in adults (E-Fig. 6-8).310 The reduced tissue solubilities of halothane, isoflurane, enflurane, and methoxyflurane in neonates are attributable to two differences in the composition of tissues: (1) greater water content and (2) decreased protein and lipid concentrations. In terms of the uptake and distribution of anesthetics in tissues, the tissue/blood solubilities determine the speed of equilibration of anesthetics in tissues. The reduced tissue solubilities of inhalational anesthetics decrease the time for partial pressure equilibration of anesthetics (see time constant discussion, later). Although the partial pressures of inhalational agents in tissues cannot easily be measured in vivo, they may be estimated by their concentrations in the exhaled or alveolar gases. The solubilities of the inhalational anesthetics in the brain of adults vary approximately 50% from desflurane to halothane (see Table 6-3). In the case of neonates, the reduced tissue solubilities of inhalational anesthetics speed the rate of increase in Fa/Fi compared with the rates in adults. In the cases of sevoflurane and desflurane, their respectively low but similar blood solubilities and likely similar tissues solubilities in neonates and adults offer a safety factor in neonates compared with adults, because tissue equilibration of these relatively insoluble inhalational anesthetics should be similar in both age groups. In contrast, the reduced tissue solubility of halothane in neonates likely results in a more rapid and unexpected anesthetic effect compared with the time course in adults.

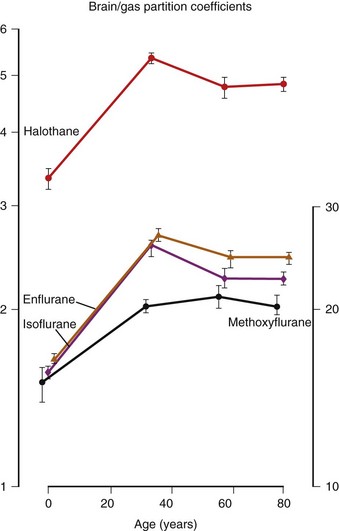
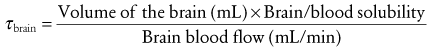
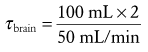
Knowing that three time constants achieve 95% equilibration, and four time constants achieve 98%, then the time to 95% equilibration is 12 minutes, and to 98% equilibration is 16 minutes. If the brain/blood solubility ratio were halved to 1.0, as it might be in the case of the neonate, then the time to 95% equilibration would decrease by 50% to 8 minutes. Thus, the time to equilibration of anesthetic partial pressure within the brain of the neonate would be approximately one half that of the adult but still requires 8 minutes! This holds true for the more soluble anesthetics, such as halothane, whose tissue solubility in neonates is diminished compared with adults310 but not for the less soluble anesthetics, such as desflurane and sevoflurane, whose tissue solubilities may be similar in neonates and adults.
Whereas the PK of inhalational anesthetics during the first 15 to 20 minutes depends primarily on the characteristics of the VRG, the PK during the subsequent 20 to 200 minutes depends primarily on those of the muscle group.298 The solubility of inhalational anesthetics in skeletal muscle varies directly with age in a logarithmic relationship.310 Thus, the lower solubility of inhalational anesthetics in the muscle of neonates and the smaller muscle mass speed the increase in Fa/Fi during this period compared with that in adults. This effect of age on the solubility of anesthetics in muscle has been attributed to age-dependent increases in protein concentration (i.e., muscle bulk) during the first 5 decades of life and in fat content during the subsequent 3 decades of life.310 Overall, the reduced solubility combined with the reduced muscle mass in neonates (and infants) decreases uptake by the muscle group, leaving the Fa/Fi to equilibrate more rapidly in neonates compared with adults.
The net effect of these differences between neonates and adults is to speed the equilibration of anesthetic partial pressures in alveoli and tissues, and thereby speed the rate of increase in Fa/Fi in neonates compared with adults.301,302 This is particularly true for the more soluble anesthetics, such as halothane. However, the difference in the rate of wash-in of less soluble anesthetics, such as sevoflurane and desflurane, between neonates and adults may be diminished.
Second Gas Effect
When two anesthetics are administered simultaneously, the wash-in of the anesthetic administered in a small concentration may be increased if the uptake of the second anesthetic is relatively large.298 Nitrous oxide is the only anesthetic for which the uptake may be relatively large compared with that of the potent inhalational anesthetics, as described earlier. There is evidence, however, that casts doubt on the clinical relevance of the second gas effect.311,312 These data suggest that the concentrating effect, if it exists in humans at all, is a small and weak effect. This may be of minor importance in any regard, as induction of anesthesia may be hastened by using the overpressure technique, which refers to administering much greater inspired concentrations of inhalational anesthetic than the target goal.
Induction
The rate of wash-in of inhalational anesthetics into the lungs varies inversely with their solubilities in blood. Although anesthetics that are less soluble (e.g., sevoflurane and desflurane) wash into the lungs more rapidly than more soluble anesthetics (e.g., halothane), the more rapid increase in Fa/Fi of less soluble anesthetics is offset by their greater MAC (see Table 6-3). To ensure that induction of anesthesia is as rapid with less soluble anesthetics as it is with more soluble anesthetics, two criteria must be satisfied. First, the inspired concentration of the less soluble anesthetic must be increased in greater increments (based on the relative MAC values and wash-in profile, where MAC is defined as the minimum alveolar [or end-tidal or end-expiratory] anesthetic concentration at which 50% of subjects do not move in response to a noxious stimulus) than the more soluble anesthetic and, second, the maximum inspired concentration of the less soluble anesthetic must provide an alveolar concentration that is equipotent with that of the more soluble anesthetic. Theoretically, the overpressure technique should provide rapid and similar rates of induction of anesthesia with anesthetics of differing solubilities. However, if the maximum inspired anesthetic concentrations from the vaporizers preclude the delivery of equipotent concentrations or if airway irritability (as in the case of coughing and breath holding) interrupts the smooth delivery of anesthetic, induction of anesthesia will not be comparable.
To illustrate this, contrast the rate of equilibration of Fa/Fi for sevoflurane and halothane during the first few minutes of induction of anesthesia (see Fig. 6-13). Data on the wash-in of inhalational anesthetics in adults is used because comparable data for children are not available. In adults, Fa/Fi for halothane reaches 0.35 in the first few minutes of anesthesia. Given the previous data from Salanitre and Rackow,301 Fa/Fi in children should increase more rapidly than that in adults, reaching perhaps 0.45 in the same time frame. For the maximum inspired concentration of halothane of 5%, the alveolar or end-tidal concentration achieved in the first few minutes in a child is 0.45 × 5% = 2.25%. With a MAC for halothane in children of ~1.0%, this is equivalent to 2.25%/1% or 2.25 MAC multiples. Contrast this to the wash-in of sevoflurane during the same time frame. The Fa/Fi for sevoflurane reaches 0.5. Because of its low blood solubility, the Fa/Fi for sevoflurane in children is likely to be similar to that in adults. With a vaporizer that delivers a maximum sevoflurane inspired concentration of 8%, the alveolar or end-tidal concentration in children achieved in the first few minutes is 0.5 × 8% = 4%. Given that the MAC for sevoflurane in children is 2.5%, 4% sevoflurane is equivalent to 4%/2.5% or 1.6 MAC multiples. This number of MAC multiples for sevoflurane is approximately 25% less than that achieved with halothane. This model will result in even greater differences between halothane and sevoflurane if the sevoflurane vaporizer is limited to 5% or 7%, as it is in some countries.
These two examples illustrate several extremely important features of the pharmacology of sevoflurane that distinguish it from halothane. First, it may be difficult to rapidly achieve a deep level of anesthesia with sevoflurane in children (as was previously achieved with halothane) when sevoflurane is the sole anesthetic. Hence, inserting an IV catheter or performing laryngoscopy or bronchoscopy immediately after induction of anesthesia with sevoflurane may result in a physiologic or motor (withdrawal) response, even if the inspired concentration remains at 8%. We caution against decreasing the inspired concentration of sevoflurane (and nitrous oxide) as soon as the eyelash reflex is lost or the child appears to have lost consciousness, because a deep level of anesthesia has not been achieved (despite theoretical fears of seizure-like activity, see below). In such cases, supplemental IV anesthetics may be required to rapidly deepen the level of anesthesia. Second, these examples illustrate an important safety feature of sevoflurane. With the current vaporizer design, excessive concentrations of sevoflurane cannot be administered to neonates and infants because their large MAC values more than offset their reduced solubilities. These insights contribute to the cardiovascular safety profile of sevoflurane and may help to explain why the morbidity and mortality associated with sevoflurane in children appear to be less than are associated with halothane.313
Control of Anesthetic Depth
During spontaneous respirations, as the partial pressure of inhaled anesthetics increases, alveolar ventilation decreases, thereby limiting both the wash-in of anesthetics and the depth of anesthesia achieved (E-Fig. 6-9, A).314 This negative-feedback response is a protective mechanism that permits the safe use of inspired concentrations of inhalational anesthetics that are severalfold greater than MAC (overpressure technique) during spontaneous respirations. Excessive depth of anesthesia cannot normally be achieved during spontaneous respirations (irrespective of the inspired concentrations of anesthetics, even if multiple anesthetics are administered simultaneously), because of the negative-feedback effect such anesthetic concentrations have to depress minute ventilation. As alveolar ventilation decreases and the wash-in of anesthetics slows, the uptake of anesthetic by blood slows and the delivery of anesthetics to the VRG slows. When the partial pressure of anesthetics in the VRG exceeds that in blood, anesthetics move along their partial pressure gradients from the VRG into blood and other tissues, thus decreasing the depth of anesthesia. As the depth of anesthesia decreases, alveolar ventilation again increases and uptake of anesthetic from the alveoli resumes. Thus, spontaneous ventilation protects against an anesthetic overdose by virtue of its negative feedback effect on respirations.

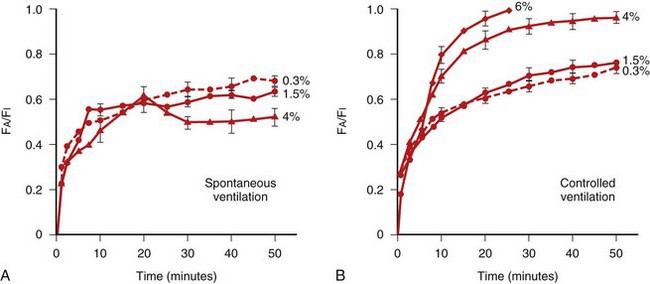
In contrast to the negative-feedback loop of spontaneous ventilation, the positive-feedback effect of controlled ventilation relentlessly delivers inhaled anesthetic to the alveoli, increasing Fa/Fi, but at the same time diminishing cardiac output (E-Fig. 6-9, B).314 The decrease in cardiac output limits the uptake of anesthetic from the alveoli, further increasing Fa/Fi. Hence, as cardiac output decreases, Fa/Fi increases and the depth of anesthesia increases, thereby further decreasing cardiac output. For a specific minute ventilation in a neonate, the speed at which cardiovascular collapse occurs is reflected in part by the maximum number of MAC-multiples the vaporizer can deliver (Table 6-6). This is a positive-feedback loop. This response creates a downward spiral that may result in death, if it is not interrupted.
TABLE 6-6 Minimum Alveolar Concentration (MAC) Multiples for a Neonate Allowed by Current Vaporizers
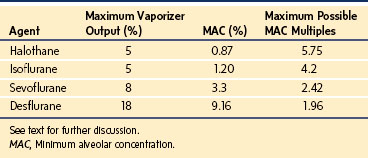

In a model of uptake and distribution, dogs who breathed spontaneously received up to 4% inspired concentrations of halothane314; the anesthetic was tolerated without cardiovascular collapse because the negative feedback response of respiratory depression prevented excessive concentrations of anesthetic from depressing the VRG. In contrast, when ventilation was controlled, cardiovascular collapse occurred at inspired concentrations 4% or more, and some dogs succumbed. High concentrations of halothane (i.e., as those used in the overpressure technique) are commonly administered during inhalational inductions, either as stepwise increases in inspired concentrations or as a single-breath large concentration. These large concentrations are tolerated provided that spontaneous respiration is maintained. If, however, ventilation is controlled, then the negative respiratory feedback loop is bypassed and cardiovascular collapse becomes possible. This is of particular concern in neonates and small infants who are more susceptible to the cardiodepressant effects of inhaled agents.298
Shunts
Two types of shunts exist in the lungs and heart: left-to-right or right-to-left. Left-to-right shunts refer to conditions in which blood recirculates through the lungs (usually via an intracardiac defect, such as a ventricular septal defect). In contrast, right-to-left shunts refer to conditions in which venous blood returning to the heart bypasses the lungs as in an intracardiac shunt (cyanotic heart disease) or intrapulmonary shunt (pneumonia or an endobronchial intubation). The effects of shunts on the rate of increase in Fa/Fi are poorly understood. In general, left-to-right shunts do not significantly affect the PK of inhalational anesthetics (they may affect IV medications), provided cardiac output remains unchanged. In contrast, right-to-left shunts can significantly delay the equilibration of Fa/Fi of inhalational anesthetics. The magnitude of the delay with a right-to-left shunt depends on the solubility of the anesthetic: the wash-in of less soluble anesthetics is delayed to a greater extent than that of the more soluble anesthetics.298 These effects are independent of the location of the shunt: intracardiac or intrapulmonary.
To understand the effects of right-to-left shunts on the PK of inhalational anesthetics, it is useful to consider a simplified model of a right-to-left shunt using the lung to mimic the shunt. In this model, each lung is represented by one alveolus and each is perfused by one pulmonary artery (Fig. 6-15). When a tracheal tube is positioned with its tip at the mid-trachea level (Fig. 6-15, A), ventilation is divided equally between both alveoli, thereby yielding equivalent anesthetic partial pressures in both pulmonary veins (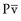 = 1). However, if the tip of the tracheal tube is advanced into the right main-stem bronchus (equivalent to a right-to-left shunt) (Fig. 6-15, B), all of the ventilation is delivered to one alveoli, that is, the ventilation to that alveoli is doubled, whereas ventilation to the nonventilated lung is zero. Under these conditions, total ventilation remains unchanged. For the remainder of this discussion, it is important to recognize that with a right-to-left shunt, the end-tidal and blood anesthetic partial pressures will differ, the magnitude of which will depend on the solubility of the anesthetic.
= 1). However, if the tip of the tracheal tube is advanced into the right main-stem bronchus (equivalent to a right-to-left shunt) (Fig. 6-15, B), all of the ventilation is delivered to one alveoli, that is, the ventilation to that alveoli is doubled, whereas ventilation to the nonventilated lung is zero. Under these conditions, total ventilation remains unchanged. For the remainder of this discussion, it is important to recognize that with a right-to-left shunt, the end-tidal and blood anesthetic partial pressures will differ, the magnitude of which will depend on the solubility of the anesthetic.
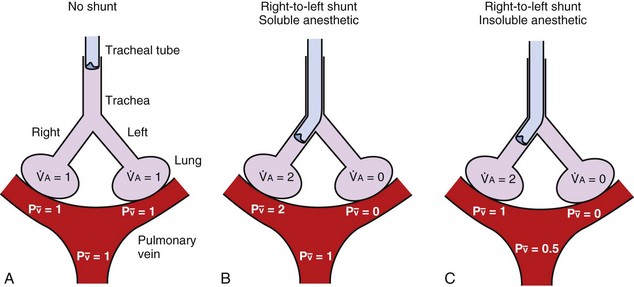
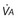 ) to both lungs, and normocapnia. B, The effect of a right-to-left shunt (via an endobronchial intubation) with a more soluble anesthetic (e.g., halothane). Ventilation and therefore normocapnia are maintained, and hypoxic pulmonary vasoconstriction is negligible. In this case, the increased ventilation to the ventilated lung speeds the increase in F
) to both lungs, and normocapnia. B, The effect of a right-to-left shunt (via an endobronchial intubation) with a more soluble anesthetic (e.g., halothane). Ventilation and therefore normocapnia are maintained, and hypoxic pulmonary vasoconstriction is negligible. In this case, the increased ventilation to the ventilated lung speeds the increase in F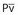 = 1) are similar to those in A. C, The effect of a shunt with a less soluble anesthetic (e.g., desflurane or sevoflurane). Because the increase in alveolar ventilation does not increase F
= 1) are similar to those in A. C, The effect of a shunt with a less soluble anesthetic (e.g., desflurane or sevoflurane). Because the increase in alveolar ventilation does not increase F = 0.5).
= 0.5).When a more soluble anesthetic (e.g., halothane) is administered through a tracheal tube that is positioned in the right main-stem bronchus (to model a right-to-left shunt), doubling the ventilation to that lung speeds the increase in Fa/Fi (effect of changes in ventilation on the wash-in of soluble anesthetics) (E-Fig. 6-5) such that it compensates, for the most part, for the absence of ventilation to the contralateral lung (Fig. 6-15, B).298 The more soluble the anesthetic, the closer the partial pressure of anesthetic in the combined pulmonary vein that drains both the ventilated and nonventilated lungs approximates the partial pressure from delivering the anesthetic to lungs without a right-to-left shunt. The net effect of a right-to-left shunt on the Fa/Fi of a more soluble inhalational anesthetic is thus negligible.

In contrast, when a less soluble anesthetic (e.g., sevoflurane or desflurane) is administered in the presence of such a right-to-left shunt, doubling the ventilation to the lung minimally increases the Fa/Fi, because ventilation has a limited effect on the speed of increase of Fa/Fi of less soluble anesthetics (see E-Fig. 6-5 and Fig. 6-15, C).298 Consequently, the increase in Fa/Fi in the ventilated lung is insufficient to offset the absence of anesthetic in the blood draining the nonventilated lung. The net effect is to almost halve the anesthetic partial pressure in the combined pulmonary vein. The less soluble the anesthetic, the greater the discrepancy between the anesthetic partial pressure in the pulmonary vein that drains the ventilated and nonventilated lungs and the partial pressure when the tube is in the trachea. The overall effect of a right-to-left shunt is to slow induction of anesthesia or even limit the depth of anesthesia that can be achieved with less soluble anesthetics.315,316

Few studies have documented the clinical importance of such a shunt.315,316 Clinical situations that have posed challenges with these shunts include children with right-to-left cardiac shunts and infants with chronic lung disease. In the past, very soluble anesthetics, such as methoxyflurane (blood/gas solubility of 12), would have been unaffected by a right-to-left shunt. In the case of halothane, the most soluble anesthetic currently available, anesthesia remained quite effective in children with right-to-left shunts even though the ratio of arterial to inspired partial pressures lagged behind the ratio when the shunt was closed.316 The most likely explanation was that the 5% inspired concentration of halothane from the vaporizer permitted a 5 × MAC overpressure effect. Although the ratios of the arterial to inspired partial pressures of sevoflurane and desflurane have not been measured in children with right-to-left shunts, we expect they will pose even greater difficulties than halothane, particularly with their limited overpressure effect (i.e., the maximal inspired concentrations of the vaporizers are limited to 3 × MAC or less [see Table 6-6]). Our experience suggests that when we use these less soluble anesthetics in such circumstances (e.g., bronchoscopy for a bronchial foreign body), IV anesthetics are needed to achieve an adequate depth of anesthesia in infants and younger children.
Washout and Emergence
The washout of inhalational anesthetics follows an exponential decay (the inverse of the wash-in curves, see Fig. 6-13) and during emergence, this is achieved by setting the inspired concentration to zero.299 The order of the washout (and speed of emergence) of the inhalational anesthetics parallels their blood solubilities: desflurane > sevoflurane > isoflurane > halothane > methoxyflurane (see Table 6-3).299,317 For most inhalational anesthetics, metabolism does not contribute substantively to the washout. Halothane is the one exception; its washout is as rapid as that of isoflurane, likely because its metabolism is 15- to 20-fold greater than that of isoflurane (see later discussion). The order of the washout of anesthetics in children should be similar to that in adults whereas the washout in neonates and infants is likely to be more rapid than that in adults for the same reasons the rate of wash-in is more rapid (see Table 6-5). Further studies are required to substantiate this notion.
Although some advocate switching inhalational anesthetics to a less soluble agent toward the end of surgery for economy and to facilitate a rapid emergence, there is a dearth of data to support such a practice in children. In fact, it has been suggested that switching from isoflurane to desflurane 30 minutes before the end of anesthesia in adults does not speed emergence.318
A number of other strategies have been used to speed emergence and recovery from anesthesia. In adults, discontinuing nitrous oxide accelerates the washout of and emergence from inhalational anesthesia.319 Most recently, charcoal filters added to anesthesia breathing circuits adsorb anesthetics and have been shown to speed emergence.320 Hypercapnic hyperventilation with a charcoal filter to adsorb the inhaled anesthetic has been shown to speed emergence from isoflurane, sevoflurane, and desflurane anesthesia in adults by about 60%.321,322
When comparing the speed of recovery after anesthesia, the results are heavily influenced by the study design. Studies in which the anesthetic concentration is maintained at a fixed MAC multiple until the end of surgery usually demonstrate a pattern of recovery that parallels the solubilities of the anesthetics in blood, at least during the early recovery period: halothane > isoflurane > sevoflurane > desflurane (see earlier).323–328
However, in the clinical setting, the inspired concentrations of inhalational anesthetics are usually tapered toward the end of surgery and this attenuates these differences. Second, differences in the speed of recovery among anesthetics parallels the duration of anesthesia; for example, differences will be less for brief surgery and greater for surgery of greater duration.324,329–331 Third, failure to prevent or treat pain before emergence will trigger a much more rapid and stormy emergence after less soluble than after more soluble anesthetics.324,327 A more sophisticated approach to the washout of inhalational anesthetics is to use the CSHT, which is a measure of the time to decrease the anesthetic partial pressure by 50%. Using a computer model and PK data from adults, the CSHTs of the potent inhalational anesthetics enflurane, isoflurane, sevoflurane, and desflurane were similar (less than 5 minutes) and were unaffected by the duration of the anesthetic.332 The 80% decrement times were similar for desflurane and sevoflurane (less than 8 minutes) whereas those for isoflurane and enflurane were greater (30 and 35 minutes, respectively). However, after 6 hours of simulated anesthesia, the 90% decrement times differed substantially: 14 minutes for desflurane, 65 minutes for sevoflurane, 86 minutes for isoflurane, and 100 minutes for enflurane. These data suggest that the early recovery (up to 80% decrement in partial pressure) after inhalational anesthesia is similar among these four anesthetics (although sevoflurane and desflurane are more rapid), but after 6 hours (i.e., prolonged anesthesia) 90% decrement is achieved much more rapidly with desflurane than with the remainder.
In animal models, recovery of motor function (an indicator of more complete recovery than simple measurement of expired anesthetic concentrations) parallels the wash-out of inhalational anesthetics from fastest to slowest: desflurane < sevoflurane < isoflurane < halothane.333 Notably, the time to recover increases in parallel with the duration of anesthesia.333 In pediatric studies in which the recovery times after two or more anesthetics were compared, the end-tidal concentrations of the anesthetics were maintained at approximately 1 MAC until the conclusion of surgery, after which the anesthetics were abruptly discontinued.323,324,334 In this paradigm, the rates of recovery paralleled the rates of wash-out, which in turn paralleled the solubilities of the inhalational anesthetics, including xenon and desflurane.335 In clinical practice, however, anesthetic concentrations are gradually tapered as the end of surgery approaches. This practice may attenuate the differences in the rates of recovery among inhalational anesthetics.
Pharmacodynamics of Inhaled Anesthetics
Minimum Alveolar Concentration
MAC is defined as the minimum alveolar (or end-tidal or end-expiratory) anesthetic concentration at which 50% of patients do not move in response to a noxious stimulus. The classic stimulus for MAC in humans is skin incision; throughout the remainder of this chapter, MAC will refer to this stimulus. MAC has also been determined in response to other stimuli, including tracheal intubation, insertion of a laryngeal mask airway, tracheal extubation, and awake responsiveness. The difference in the potency (or MAC) of inhalational anesthetics varies inversely with the lipid solubility of these anesthetics; that is, as the lipid solubility decreases, the potency decreases in parallel (i.e., MAC increases) (see Table 6-3).
In children, MAC varies significantly with age. For example, the MAC of halothane increases as age decreases, reaching a maximum value in infants 1 to 6 months of age (1.20 ± 0.06%) and then decreases by about 30% to (0.87 ± 0.03%) in full-term neonates.336,337 Similar relationships hold true for isoflurane and desflurane (Figs. 6-16 and E-Fig. 6-10).338,339 However, the relationship for sevoflurane differs from that of the other inhalational anesthetics in that the MAC of sevoflurane does not increase steadily as age decreases (Fig. 6-17).340 In fact, the MAC of sevoflurane in neonates and infants younger than 6 months of age is 3.3%, whereas in older infants and children, it is 2.5%.340–342 The explanation for this different relationship for sevoflurane remains unclear.
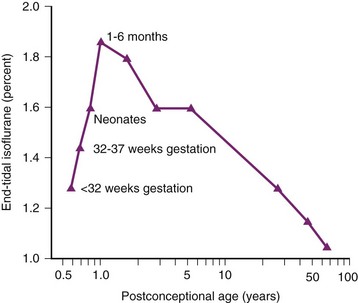
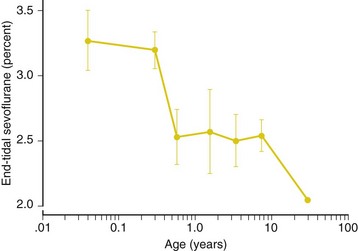

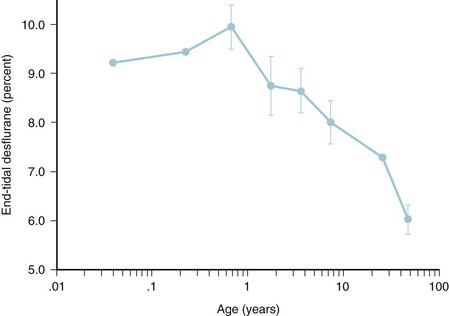
The MAC of inhalational anesthetics in preterm neonates has been determined only for isoflurane (see Fig. 6-16). The (mean ± SD) MAC of isoflurane in preterm neonates younger than 32 weeks gestation (1.28 ± 0.17%) is 10% less than it is in neonates of 32 to 37 weeks gestational age (1.41 ± 0.18%), which in turn is 12% less than it is in full-term neonates (1.60 ± 0.03%).241 The etiology of these age-dependent changes in MAC remains elusive. Several possible causes have been proposed, including maturational changes in the CNS and neurohumoral factors, but none of them have been confirmed.
Other factors are known to affect MAC. The melanocortin-1 receptor gene has been shown to affect the MAC of desflurane. That is, 90% of adults who were either homozygous or heterozygous for mutations of this gene (i.e., redheads) required 20% more anesthesia than those who had no mutations (brunettes).343 A similar relationship should hold true for children with these mutations. Hypothermia decreases MAC. In children 4 to 10 years, the MAC of isoflurane decreases 5% per degree Celsius.344
Cerebral palsy and severe cognitive impairment reduce the MAC of halothane approximately 25% compared with healthy children.345 Although tetanic stimulation was used to elicit a pain response, this stimulus underestimates the MAC compared with skin incision.346 Nonetheless, the MAC of halothane in healthy children was similar to published data with skin incision.337 Chronic anticonvulsant therapy decreased the MAC of halothane in handicapped children by 15%, compared with those without anticonvulsants, from 0.71% ± 0.10% to 0.62% ± 0.03%.345 Several factors may account for the decrease in MAC in children with cognitive impairment, including central sensory impairment, increased pain threshold or insensitivity, and a disequilibrium of inhibitory and excitatory regulatory neurons within the spinal cord in these children.347,348 Although the acute administration of barbiturates and benzodiazepines decreases MAC,349,350 chronic administration of similar medications does not.351 The effects of specific anticonvulsants, such as valproic acid and phenytoin, on the MAC of inhalational anesthetics in children remain unclear.
The MAC for nitrous oxide has been estimated to be 104% in adults352; comparable data do not exist in children. The additivity of MAC fractions of inhalational anesthetics (as well as nitrous oxide) is well established. For example, the total anesthetic delivered when 0.5 MAC of one agent and 0.5 MAC of a second agent are administered is 1 MAC. In adults, the concept of additivity has been confirmed for all inhalational anesthetics, including sevoflurane and desflurane, in combination with nitrous oxide.353,354 In children, the concept of MAC additivity holds true when nitrous oxide is combined with halothane or isoflurane355,356 but not when nitrous oxide is combined with sevoflurane or desflurane.219,220,238 Nitrous oxide 60% decreases the MAC of sevoflurane only 20% and that of desflurane 26% (Table 6-7).339,340,357 The MAC response to tracheal intubation during sevoflurane anesthesia in children is also attenuated with nitrous oxide.358 The explanation for the differential additivity effect of nitrous oxide in children remains unclear.
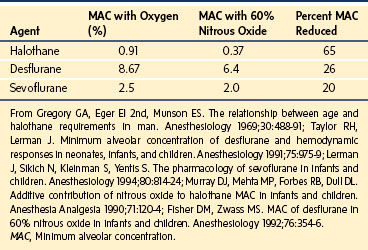
The MAC for xenon in middle-aged adults is about 70%.359,360 The MAC of xenon in children has not been determined.
The MAC responses to stimuli other than skin incision have also been determined in children (Table 6-8). The MAC for tracheal intubation is 10% to 50% greater than the MAC for skin incision for halothane,361,362 enflurane,363 and sevoflurane,358,364,365 whereas the MAC for tracheal extubation is 10% to 25% less than the MAC for skin incision for isoflurane,366 desflurane,367 and sevoflurane.368,369 The MAC values for laryngeal mask airway insertion and removal during sevoflurane differ by 15%.362,370 The MAC response to tracheal intubation during sevoflurane anesthesia in children is attenuated in the presence of adjuvants, such as clonidine (E-Table 6-1).365 Conflicting evidence exists regarding the relative potencies of stereoisomers or enantiomers of chiral inhalational anesthetics.371–373 Studies in animals suggest that the (+)S optical enantiomer may be more potent than the (−)R enantiomer, as evidenced by its ability to enhance potassium conductance in neurons.371,372 In adults, the (−)R enantiomer of isoflurane was nominally (17%) more potent than the (+)S enantiomer.373
| MAC (%) | References | |
|---|---|---|
| Tracheal intubation | Halothane: 1.33 | 361 |
| Enflurane: 2.93 | 363 | |
| Sevoflurane: 2.69, 2.66, 2.83 | 342, 358, 364 | |
| Tracheal extubation | Isoflurane: 1.4 | 366 |
| Sevoflurane: 1.70, 2.3 | 368, 369 | |
| Desflurane: 7.7 | 367 | |
| LMA insertion | Sevoflurane: 2.0 | 364 |
| LMA extubation | Sevoflurane 1.84 | 370 |
| Desflurane (with 1-1.3 μg/kg Fentanyl) 3.56% | 1909 | |
| Tracheal intubation/skin incision ratio* | Halothane, enflurane, sevoflurane: 1.33 | Calculated from MAC data |
| MAC awake | Sevoflurane: 0.66 (2-5 years) and 0.43 (5-12 years) |
1910 |
LMA, Laryngeal mask airway; MAC, minimum alveolar concentration.
*Calculated using the above MAC data.

E-TABLE 6-1 Effects of Adjunctive Therapies on MAC of Sevoflurane
| Adjunct | MAC Sevoflurane | References |
|---|---|---|
| MAC tracheal intubation in oxygen | 2.69, 2.66%, 3.2% | 342, 358, 365 |
| MAC tracheal intubation and 60% N2O | 1.57% (41% decrease), 2.4% (25% decrease) | 358, 365 |
| MAC tracheal intubation and oral clonidine (4 μg/kg) | 40% decrease | 365 |
| MAC tracheal intubation and clonidine (4 μg/kg) and 60% N2O | 56% decrease | 365 |
MAC, Minimum alveolar concentration.
Central Nervous System
All potent inhalational anesthetics depress the central nervous system (CNS), as evidenced by dose-dependent decreases in the cerebral vascular resistance and the cerebral metabolic rate for oxygen (CMRO2). The decrease in vascular resistance causes a reciprocal increase in cerebral blood flow (CBF) that begins at approximately 0.6 MAC.374 The extent of the increase in CBF, however, depends on the inhalational anesthetics: halothane > enflurane ~ desflurane > isoflurane > sevoflurane.374–377 In adults, the cerebral vasculature remains responsive to CO2 under general anesthesia but decreases with increasing MAC, disappearing at 1.5 MAC in the case of desflurane.378 The net effect of inhalational anesthetics is a dose-dependent increase in the ratio of the CBF to CMRO2.
The effects of inhalational anesthetics on the CNS in children have not been fully elucidated. Autoregulation of CBF does not appear to vary with age in children up to 1.5 MAC sevoflurane.379,380 CBF velocity in children varies directly with the end-tidal CO2 during halothane and isoflurane anesthesia.381 CBF velocity increases as the concentrations of halothane382 and desflurane383 increase. Compared with halothane, however, sevoflurane does not increase CBF velocity, suggesting it may be the preferred anesthetic.384 Based on current evidence, sevoflurane and isoflurane remain the preferred inhalational anesthetics for neuroanesthesia in children at low MAC values (less than 1 MAC) and in the presence of mild hyperventilation (see Chapter 24).
In children, the EEG activity during halothane anesthesia differs substantially from that of sevoflurane. In the case of halothane, the EEG is characterized by slow waves superimposed on fast rhythms (α and β waves), whereas in the case of sevoflurane, the EEG is characterized by mainly sharp slow waves.385 Furthermore, the shift of power of the EEG from low (1 to 4 Hz) to medium frequencies (8 to 30 Hz) is greater for halothane than it is for sevoflurane. The clinical relevance of these EEG differences remains unclear at this time, but may explain in part the inconsistencies reported with processed EEG monitoring in children anesthetized with various inhalation anesthetics (see later discussion).
Enflurane was the first inhalational anesthetic to be associated with seizure activity both clinically and electroencephalographically in humans.386,387 High concentrations of enflurane and a respiratory alkalosis were associated with seizure activity.
Both myoclonic movement of the extremities and transient spike and wave complexes on EEG have been reported during sevoflurane anesthesia in children.385,388–391 Diffuse spike and wave complexes were noted on EEG at 5% and 7% inspired concentrations in two children with histories of epilepsy.389 In a third child without a history of seizures, a 30-second burst of spike and wave complexes was recorded during sevoflurane anesthesia, but identified only after the event resolved.392 The EEG data were analyzed in children who were premedicated with midazolam and then anesthetized with halothane or sevoflurane. There was no evidence of seizure activity.385 Cortical epileptiform EEG activity has been reported during 1 to 2 MAC sevoflurane in adults with one episode of seizures that occurred with a pCO2 of 34 mm Hg.393 This prompted some to recommend limiting the depth of anesthesia with sevoflurane to minimize epileptiform EEG activity or even clinically evident seizure activity.394 However, the co-administration of other anesthetic medications, such as midazolam, nitrous oxide, and opioids, may attenuate epileptiform EEG activity.393 Furthermore, the notion of limiting the depth of sevoflurane anesthesia by reducing the concentration of sevoflurane as soon as the eyelash reflex is lost has not been shown to benefit children, and may actually increase the risk of awareness (see later discussion).
Awareness during inhalational anesthesia has been reported to occur with an incidence of 0.2% to 1.2% during sevoflurane anesthesia in children,395–397 which exceeds that in adults. The reason(s) for this discrepancy remains unclear. These studies do suggest that children who experience awareness do not develop long-term sequelae.398 Concerns over awareness during anesthesia has increased interest in the use of processed EEG monitoring in children.399 Processed EEG monitoring during anesthesia has been evaluated using a number of EEG signals and techniques, processing signals in a “black box” and displaying a single numerical result. The most widely studied monitor to date is the BIS monitor, which displays the value on a scale between 0 and 100 (see Chapter 51). In adults, BIS readings below 60 are thought to be associated with a small risk of awareness and recall, whereas readings greater than 70 are associated with a large risk for awareness. The BIS value has been studied in children to a limited degree to date, but its validity and role in the anesthetic management of infants and children under 5 years of age remain in question. One concern is that the EEG algorithm in current BIS software is derived from adult EEG data, not from children. Despite use of an algorithm derived from adults, BIS measurements in children appear, for the most part, to track the depth of anesthesia, and these values correlate with the end-tidal concentration of the anesthetic. However, several unexplained curiosities have been reported. The BIS readings for halothane in children exceed those for isoflurane, desflurane, and sevoflurane at equipotent anesthetic concentrations.255,256,400 This has been attributed to the differential effects of anesthetics on the EEG. The BIS readings for a specific sevoflurane concentration decreases with increasing ages.252,255,401 The validity of processed EEG monitoring in children younger than 5 years has not been established. In children, the BIS values actually increased between 3% and 4% with sevoflurane, a paradoxical response to date unexplained.401 A further concern is the enormous interindividual variability in processed EEG monitoring, making it difficult to define thresholds for awareness or lack thereof.401 Interestingly, spontaneous ventilation increases the variability in the BIS readings.402 The issue is further complicated by the fact that ketamine, nitrous oxide, and opioids do not depress the EEG in a dose-dependent manner. Discrepant BIS readings have been reported from the right and left sides of the brain403 as well as in the prone position.404 The BIS readings in children with cognitive impairment are reported to be 28 points less than those in unimpaired children at the same anesthetic concentration, although the authors failed to account for the reduced MAC in the impaired group.405 Had they done so, the BIS readings may have been identical. The aggregate of this evidence undermines the validity of processed EEG monitoring in children.
Cardiovascular System
Inhalational anesthetics (with the exception of xenon) affect the cardiovascular system either directly (by depressing myocardial contractility, altering the conduction system, or by dilating the peripheral vasculature) or indirectly (by affecting the balance of parasympathetic and sympathetic nervous systems and neurohumoral, renal, or reflex responses). The cardiovascular responses to inhalational anesthetics in children are further complicated by maturational changes in the cardiovascular system and its responsiveness to these anesthetics. When all of these developmental changes are taken into consideration there is a reduced margin of safety between adequate anesthesia and severe cardiopulmonary depression in infants and children compared with adults. The salient features of the immature cardiovascular system in infants and children at both the macroscopic and microscopic levels have been reviewed.406
Assessment of cardiovascular variables in infants and children presents a challenge for clinicians. Although blood pressure (BP) and electrocardiography are standard monitors of hemodynamics in infants and children of all ages, measures of cardiac output and myocardial contractility are much more difficult to quantitate accurately. Two-dimensional echocardiography and impedance cardiometry have been used to estimate cardiac output and myocardial contractility in infants and children,407–410 although the echocardiographic measurements are subject to variability depending on the preload and afterload. Load-independent derived echocardiographic variables (stress-velocity and stress-shortening indices) have improved the accuracy of echocardiographic estimates of myocardial function and are used with increasing frequency.411 Transesophageal echocardiography is used much more frequently in children, although its use is limited to children with congenital heart disease undergoing cardiac surgery.
In children, several factors determine the BP responses to inhalational anesthetics, including the particular anesthetic studied, the dose, the presence of a premedication, the level of preoperative anxiety, and the systemic pressure measured: systolic, diastolic, or mean. Most studies demonstrate modest, dose-dependent decreases in BP with all of the inhalational anesthetics, although the magnitudes of the changes vary. In a direct comparison of sevoflurane and halothane, systolic BP decreased 7.5% at 1 MAC sevoflurane and 12.5% at 1 MAC halothane, but returned to awake values at 1.5 MAC with both anesthetics.411 In children older than 1 year of age, systolic BP decreased 0% to 11% at 1 MAC sevoflurane, and 22% to 28% at 1 MAC desflurane, compared with awake values.339,340 At 1 MAC, mean BP in children decreased 15% to 25% with isoflurane and sevoflurane.408,410 All of the inhalational anesthetics (in concentrations up to 1.5 MAC) modestly depress the systemic BP in children.
Myocardial contractility decreases to a greater extent during halothane (up to 1.5 MAC) than during isoflurane or sevoflurane anesthesia, as evidenced by decreases in cardiac output and ejection fraction in healthy children.409,410,412 Cardiac index decreased to similar extents with halothane and sevoflurane at 1 and 2 MAC: 10% at 1 MAC and 20% to 35% at 2 MAC.410 Ejection fraction decreased 30% at 0.5 and 1.5 MAC halothane compared with awake values, but is unchanged at equipotent concentrations of isoflurane.408 The addition of nitrous oxide to halothane or isoflurane in infants and small children depresses myocardial function to a similar extent as equipotent anesthetic concentrations of halothane or isoflurane in oxygen.412 In children, halothane decreases myocardial contractility in a dose-dependent manner and to a greater extent than the ether anesthetics. Isoflurane and sevoflurane decrease myocardial contractility to a lesser extent than halothane and are preferred for children with limited cardiovascular reserves. IV atropine restores the decrease in myocardial function, in part, associated with halothane anesthesia,412–415 whereas IV balanced salt solution restores the decrease in myocardial function associated with isoflurane anesthesia.409
The mechanism by which inhalational anesthetics depress myocardial function remains controversial. Studies in both animal and human myocardial cells suggest that halothane, isoflurane, and sevoflurane directly depress myocardial contractility by decreasing intracellular Ca2+ flux. Inhalational anesthetics decrease the Ca2+ flux by their action on the calcium channels themselves, ion exchange pumps, and the sarcoplasmic reticulum.406 Evidence suggests that inhalational anesthetics attenuate contractility of ventricular myocytes via voltage-dependent L-type calcium channels (which are responsible for release of large amounts of calcium from the sarcoplasmic reticulum).416,417
That neonates and infants are more sensitive to the depressant actions of inhalational anesthetics than are older children is supported by experimental evidence of maturational differences between neonatal and adult rat, rabbit, and feline myocardium.417–420 Structural differences that may account, in part, for the changes in myocardial sensitivity to inhalational anesthetics with age, include a reduction in contractile elements, immature sarcoplasmic reticulum, and functional differences in calcium sensitivity of the contractile elements, calcium channels, and the sodium-calcium pump in the neonatal myocardium.406,416–418,420–423 The determinants of Ca2+ homeostasis in neonatal ventricular myocardial cells depend on transsarcolemmic Ca2+ flux to a far greater extent than on the sarcoplasmic reticulum.406 This is based on a growing body of experimental evidence that includes the finding that the concentration of the Na+-Ca2+ exchange protein in the neonatal myocardium, a protein that regulates transsarcolemmic flux of Ca2+, exceeds that in adult cells by 2.5-fold and that its concentration decreases with age as the concentration of L-type voltage-dependent calcium channels increases.418 Furthermore, halothane reversibly inhibits the Na+-Ca2+ exchange protein in immature myocardial cells.418 The sarcoplasmic reticulum is poorly developed in neonatal myocardial cells, and this finding weighs heavily against the sarcoplasmic reticulum being the major source of Ca2+ required for myocardial contractility. Further research is required before the contribution of each aspect of Ca2+ homeostasis to myocardial contractility in the neonate can be confirmed.
Ever since the introduction of halothane into clinical practice, clinicians have been aware of a greater incidence of hypotension and bradycardia in neonates who were anesthetized with halothane than in adults. However, this was not the case when equipotent concentrations (approximately 1 MAC) of halothane337,414 were administered to neonates and older infants 1 to 6 months of age. Subsequent studies demonstrated that isoflurane,424 sevoflurane,340 and desflurane339 all decreased systolic pressure in neonates to similar extents as in older infants 1 to 6 months of age. Interestingly, systolic BP decreased 30% in response to 1 MAC sevoflurane in neonates and infants 1 to 6 months of age, which was substantially greater than the 5% decrease in older infants and children up to 12 years of age.340 In the case of desflurane, systolic BP decreased 30% in response to 1 MAC desflurane across all age groups.339 These data suggest that systolic BP decreases up to 30% in response to 1 MAC of all inhalational anesthetics in infants and children, and that caution should be exercised when administering these anesthetics to infants and children who are at risk for hemodynamic instability, or in whom greater concentrations of inhaled anesthetics are required. On the basis of echocardiographic determinations, cardiac output and ejection fraction decrease in a dose-dependent fashion from awake to 1.5 MAC halothane and isoflurane in neonates and infants.407 In a comparison of sevoflurane and halothane up to 1.5 MAC in infants, sevoflurane maintained cardiac index but decreased BP and systemic vascular resistance.425 Myocardial contractility decreased in a dose-dependent manner with halothane as well as with sevoflurane, although the decrease with the former exceeded that with the latter.
The baroreceptor reflex response is also depressed in infants with either halothane426 or isoflurane,427 albeit to a greater extent with halothane. In view of the greater incidence of hypotension in neonates and infants than older children, an intact baroreceptor reflex could offset in part, the cardiovascular consequences. However, inhalational anesthetics blunt this response, leaving the infant vulnerable to the direct cardiodepressant actions of the anesthetics. Prophylactic anticholinergics augment the cardiac output by increasing the heart rate.
Two studies evaluated the effects of inhalational anesthesia on the hemodynamics of children with congenital heart disease undergoing cardiac surgery.428,429 Sevoflurane maintained cardiac index and heart rate with less hypotension and negative inotropic effect than halothane. Isoflurane maintained cardiac index and ejection fraction, increased heart rate, and caused less depression of mean arterial pressure than halothane.428 Sevoflurane was also associated with fewer episodes of severe hypotension and reduced need for vasopressors and chronotropes during emergence than halothane.429
Inhalational anesthetics also vary in their effect on cardiac rhythm. Halothane slows the heart rate, in some cases leading to junctional rhythms, bradycardia, and asystole. This response is dose dependent. Three mechanisms have been proposed to explain the genesis of halothane-associated dysrhythmias: a direct effect on the sinoatrial node, a vagal effect, or an imbalance in the parasympathetic and sympathetic tone. It has also been suggested that the etiology of the bradycardia during halothane anesthesia may be a withdrawal of sympathetic tone. Bradycardia is particularly marked in the neonate, presumably because parasympathetic influences predominate over the sparse sympathetic innervation of the myocardium in this age group. Junctional rhythms are also common during halothane anesthesia. Atrial or ventricular ectopic beats are rare, except in the presence of hypercapnia.430 In infants and children anesthetized with halothane, 10 μg/kg atropine increases heart rate by 50% or more and promotes sinus rhythm.431 This dose of atropine also increases BP in infants and children 2 years of age and older.
Halothane also sensitizes the myocardium to catecholamines, particularly in the presence of hypercapnia and “light anesthesia.”430 Halothane decreases the threshold for ventricular extrasystoles during epinephrine administration threefold.432–434 In contrast, isoflurane, desflurane, and sevoflurane maintain or increase heart rate during the early induction period of anesthesia,* although a slowing of the heart rate has been reported during sevoflurane anesthesia. When bradycardia occurs in an anesthetized child, hypoxia must be considered first before other causes, such as a direct drug effect (i.e., high concentration of halothane). Isoflurane, desflurane, and sevoflurane do not sensitize the myocardium to catecholamines to the same extent as does halothane, and ventricular arrhythmias are rare.432,433,438 The mechanism by which the sinus node controls automaticity is incompletely understood but may include K+ currents, hyperpolarization-activated current, and T and L forms of Ca2+ currents.406 Moreover, developmental changes in these channels likely account, in part, for the differential effects of inhalational anesthetics on heart rate with age.421
Recent concerns of a relationship between inhalational anesthetics and prolonged QT interval that progressed to induce cardiac arrest or torsades de pointes have emerged. Although the ether inhalational anesthetics prolong the QTc interval (with greater than 500 milliseconds being abnormal),439,440 this alone appears to be insufficient to induce torsades de pointes. Torsades de pointes also requires the transmural dispersion of repolarization. This is defined as the variability in the rate of repolarization across the myocardium, from epi- to endocardium. The rate of repolarization may be estimated by the interval in the peak to end of the T wave on a 12-lead electrocardiogram. Evidence has demonstrated that the risk of torsades de pointes during sevoflurane anesthesia is minimal because the transmural dispersion of repolarization is limited.441
Paroxysmal increases in BP (both systolic and diastolic pressures) and heart rate have been reported in adults after a rapid increase in the inspired concentration of isoflurane or desflurane.442 This occurs as a result of a massive sympathetic response, mediated by norepinephrine and/or epinephrine, that culminates in tachycardia and hypertension.443,444 Further increases in the inspired concentration of the inciting anesthetic while attempting to attenuate the tachycardia and hypertension are ineffective and may perpetuate or augment the response. In order to restore vital signs to normal, the inciting anesthetic should be discontinued and replaced with another anesthetic. Repetitive small increases (1%) in the inspired concentration of the putative anesthetic produces transient but attenuated catecholamine bursts and cardiovascular responses compared with larger increases in concentration.445,446 Fentanyl (2 μg/kg), esmolol, and clonidine have all been shown to be effective in preventing, attenuating, or eliminating these responses.447–449 The origin of these responses is unknown, although the rapidity of the response points to the lung.450 Others, however, dispute this notion, contending that two sites must be responsible for triggering the sympathetic discharge; the lung and the VRG,451 with the latter mediating the greater response.451,452 Neuroexcitatory responses have not been reported in children with isoflurane, desflurane, or sevoflurane.453
Xenon offers an immense advantage over the ether-based inhalational anesthetics in that it maintains circulatory stability. This anesthetic may have a niche role when anesthetizing infants and children with congenital and acquired heart disease. However, the effects of xenon on the hearts in children have not been studied. Although the MAC of xenon in adults is very large (see Table 6-3), MAC for xenon in children may be even greater. If the MAC exceeds the adult values, this may limit not only the dose that may be administered to children (e.g., much less than 1 MAC) but it may limit the inspired concentration of oxygen as well. Notwithstanding the MAC of xenon in children, this anesthetic will certainly be much less effective in children with cyanotic congenital heart disease, as it is the least soluble anesthetic and its large MAC restricts the MAC multiple that may be administered.
Respiratory System
During spontaneous ventilation, both tidal volume and respiratory rate vary with the specific anesthetic, depth of anesthesia, and nociception. The increased respiratory rate during inhalational anesthesia in children has been attributed to sensitization of the stretch receptors within the lung as well as possible central effects. Inhalational anesthetics significantly affect respiration in infants and children in a dose-dependent fashion via effects on the respiratory center, chest wall muscles, and reflex responses. Halothane depresses minute ventilation by decreasing tidal volume and attenuating the response to carbon dioxide.454–457 This depression is offset, in part, by an increase in the respiratory rate.454,457 These ventilatory responses to halothane are age dependent; minute ventilation in infants decreases to a greater extent than in children.456 In infants and young children anesthetized with halothane, intercostal muscle activity is attenuated before the diaphragm.454,458 This effect is most pronounced in preterm and full-term neonates and infants, and when a tracheal tube is used in place of a laryngeal mask airway.459 The movement of the chest and abdomen during the respiratory cycle is one of synchronized protrusion of the abdomen and collapse of the chest wall during inspiration (with some intercostal indrawing) and indrawing of the abdomen and flattening of the chest wall during expiration. This is commonly referred to as the “rocking horse” movement of the chest (similar to the phenomenon that occurs during upper airway obstruction in infants and children). This results in loss of FRC and is of particular concern in infants younger than 2 years of age who have decreased type I muscle fibers in both the diaphragm and intercostal muscles (see Fig. 12-11). This explains why infants fatigue easily and why positive end-expiratory pressure is useful in this age group. Isoflurane, enflurane, sevoflurane, and desflurane also depress ventilatory drive, decrease tidal volume, and attenuate the response to carbon dioxide.455,457,460–466 The increase in respiratory frequency that follows respiratory depression (decreased tidal volume) may not restore minute ventilation to preanesthetic levels.
Sevoflurane depresses respiration to a similar extent as halothane up to 1.4 MAC but depresses respiration to a greater extent at concentrations greater than 1.4 MAC.460 This results from a direct effect of sevoflurane on respiratory frequency.463 Although respiratory effort may decrease rapidly during sevoflurane or desflurane anesthesia, their low blood solubilities and rapid wash-out profiles in part ensure that this is a self-limiting phenomenon during spontaneous respirations. Sevoflurane decreases the tone of the intercostal muscles to a lesser extent than halothane.458,462 The compensatory changes in respiratory rate differ among the anesthetics; respiratory rate increases at 1.4 MAC or more with halothane, is unchanged with isoflurane, but decreases at 1.4 MAC or less with sevoflurane and enflurane.455,457 This inadequate compensatory response to respiratory depression with sevoflurane and enflurane suggests that when children are anesthetized with those anesthetics, spontaneous ventilation must be monitored carefully to avoid hypopnea or apnea. Sevoflurane maintains or decreases airway resistance in children with normal airways, and those with asthma or a recent upper respiratory tract infection. Insertion of a tracheal tube during sevoflurane anesthesia increases airway resistance without sequelae.467,468
Desflurane depresses respiration in children during spontaneous respiration at greater than 1 MAC by decreasing tidal volume.466 However, desflurane does increase airway resistance in children with asthma and respiratory tract infections to a greater extent than sevoflurane.468 Desflurane is probably best avoided in these children.
Studies of the upper airway in children anesthetized with inhalational anesthesia have sought to explain the pathophysiology of airway obstruction. Sevoflurane at 1 MAC causes more upper airway obstruction than halothane.469 In escalating doses between 0.5 and 1.5 MAC, sevoflurane decreased the cross-sectional area of the airway by one third, predominantly in the anteroposterior dimension.470 This effect primarily results from pharyngeal wall collapse, which can be easily offset with positive end-expiratory pressure (see Chapter 12).
Renal System
Inhalational anesthetics are metabolized in vivo by the CYP isozyme system to varying extents (Table 6-9). Metabolism of inhalational anesthetics may release both inorganic and organic fluoride moieties.471 It is the inorganic fluoride that is released from these ether anesthetics that has stimulated interest in renal dysfunction after inhalational anesthesia.
| Inhalational Agent | Percent Metabolized | Reference |
|---|---|---|
| Methoxyflurane | 50 | 1911 |
| Halothane | 20 | 1912 |
| Sevoflurane | 5 | 486 |
| Enflurane | 2.4 | 1913 |
| Isoflurane | 0.2 | 1914 |
| Desflurane | 0.02 | 1915 |
Isoflurane and desflurane undergo limited metabolism in vivo, resulting in very small plasma concentrations of inorganic fluoride, even after 131 MAC hours of isoflurane.472 In contrast, halothane is metabolized to a substantially greater extent but releases most of the fluoride in an organic form, trifluoroacetate. Trifluoroacetate has, however, been linked to halothane hepatitis (see later discussion). The metabolism of enflurane, sevoflurane, and methoxyflurane yields greater plasma concentrations of inorganic fluoride than isoflurane. The metabolism of sevoflurane yields both inorganic and organic fluoride moieties.473 The organic form, hexafluoroisopropanol, is rapidly conjugated and excreted by the kidneys473 and poses no threat to humans. Peak plasma concentrations of inorganic fluoride after exposure to inhalational anesthetics follow a similar order to that in Table 6-9: methoxyflurane > sevoflurane > enflurane > isoflurane > halothane ~ = desflurane.474–478 In the case of methoxyflurane, two metabolites are produced: inorganic fluoride and oxalic acid. Both were implicated in the pathogenesis of renal dysfunction, although, clinically, the renal injury was more consistently associated with inorganic fluoride.479 Subsequent studies demonstrated that subclinical nephrotoxicity occurred after more than 2.5 MAC hours of methoxyflurane, provided the plasma concentration of inorganic fluoride exceeded 50 µmol/L; nephrotoxicity occurred after more than 5 MAC hours if the concentration of inorganic fluoride exceeded 90 µmol/L.480 These clinical concerns led to the voluntary withdrawal of methoxyflurane from clinical practice.
In contrast to adults, renal dysfunction was not a feature after methoxyflurane anesthesia in children. The peak plasma concentrations of inorganic fluoride in children anesthetized with methoxyflurane were significantly less than in adults after an equivalent anesthetic exposure.481 The reduced plasma concentrations of fluoride in children were attributed to several causes, including a decreased metabolism of methoxyflurane, greater uptake of fluoride by bone, increased excretion of fluoride ions, or a reduced renal sensitivity to fluoride.481
That the plasma concentrations of inorganic fluoride in children who were anesthetized with sevoflurane were similar to or greater than those after enflurane482–484 raised concerns about possible renal dysfunction after prolonged exposure. However, inorganic fluoride concentrations after relatively brief anesthetics in children are similar to those in adults: less than 20 µmol/L after about 1 MAC hour, which decreases to less than 10 µmol/L by 4 hours after discontinuation of anesthesia.465 Nonetheless, the peak plasma concentrations of inorganic fluoride paralleled the MAC hour exposure to sevoflurane in both children and adults.484 Concerns regarding the risk of renal dysfunction after sevoflurane were heightened after reports that the peak plasma concentration of inorganic fluoride in some adults exceeded the purported threshold for nephrotoxicity (50 µmol/L).485 Despite the high plasma concentrations of inorganic fluoride after sevoflurane anesthesia, there was no evidence of renal dysfunction. These reports, together with a dearth of evidence in the toxicology literature, suggested that fluoride-mediated nephrotoxicity may be independent of the plasma concentration of inorganic fluoride.
Kharasch and colleagues postulated that inhalational anesthetic-induced nephrotoxicity might be anesthetic specific. They determined that the primary isozyme responsible for the degradation of enflurane, isoflurane, sevoflurane, and methoxyflurane anesthetics was CYP450 2E1,471,486–488 with secondary isozymes including CYP450 2A6 and 3A.489 Subsequently, they reported large quantities of CYP450 2E1 not only within the liver, but also within the kidneys.486 They also noted that the affinity of renal CYP450 2E1 for methoxyflurane was fivefold greater than it was for sevoflurane.489 This provided further evidence that the renal dysfunction after ether inhalational anesthetics resulted from the local production of inorganic fluoride within the renal medulla rather than extrarenal production, and that certain anesthetics were more prone to release of inorganic fluoride than others (e.g., methoxyflurane much more so than sevoflurane). Because CYP450 2E1 has a greater affinity for methoxyflurane than sevoflurane, we now understand why renal dysfunction occurs after methoxyflurane and not after sevoflurane.489 The lack of an association between sevoflurane, plasma inorganic fluoride concentration, and renal dysfunction in children and adults supports this new understanding of the mechanism of renal dysfunction after inhalational anesthesia. Consequently, the risk of renal dysfunction after sevoflurane is independent of the duration of exposure to sevoflurane. Sevoflurane does not pose any greater risk for perioperative renal disease than other maintenance anesthetics.490 A second theoretical cause of sevoflurane-associated renal dysfunction is compound A, a product of alkaline hydrolysis of sevoflurane in the presence of carbon dioxide absorbents (see later discussion).
Hepatic System
In vivo metabolism of inhalational anesthetics varies with age, increasing to adult values within the first 2 years of life. The developmental changes in metabolism may be attributed to several factors, including reduced activity of the hepatic microsomal enzymes, reduced fat stores, and more rapid elimination of inhalational anesthetics in infants and children compared with adults. Halothane, isoflurane, enflurane, sevoflurane, and desflurane have all been associated with postoperative liver dysfunction and/or liver failure in adults, although the relationship between sevoflurane and desflurane and liver dysfunction remains tenuous.491–494 In children, halothane and sevoflurane have been associated with transient hepatic dysfunction.495–498 Indeed, several pediatric cases of transient postoperative liver failure and one case of fulminant hepatic failure and death have been attributed to “halothane hepatitis” that was confirmed serologically with antibodies to halothane-altered hepatic cell membrane antigens.495 The exact mechanism of the hepatic dysfunction after halothane exposure remains unclear, although some clinicians have speculated that it is caused by an immunologic response to a metabolite of halothane. This putative toxic metabolite, a trifluoroacetyl halide compound, is produced during oxidative metabolism of halothane. It is thought that this compound induces an immunologic response in the liver by binding covalently to hepatic microsomal proteins, thereby forming an immunologically active hapten. A subsequent exposure to halothane then incites an immunologic response in the liver.499 Hepatic enzymes may also be induced by previous administration of drugs, such as barbiturates, phenytoin, and rifampin. Although some have admonished clinicians for administering repeat anesthetics with halothane in children, it is our opinion that, in view of the millions of uneventful repeat halothane anesthetics in infants and children worldwide, insufficient evidence exists to support such an admonition.
Clinical Effects
Induction Techniques
Although the physicochemical characteristics of the ether series of anesthetics would predict that anesthesia could be induced smoothly and more rapidly with these agents than with halothane,308,310 this has not proved to be the case. All of the ether anesthetics (except sevoflurane) irritate the upper airway in children, resulting in a high incidence of breath holding, coughing, salivation, excitement, laryngospasm, and hemoglobin–oxygen desaturation.435,500–509 Clinical studies with desflurane in children demonstrated a high incidence of breath holding, laryngospasm, and desaturation during inhalational induction (approximately 50%).435,509 As a result, a “black box” warning was issued against the use of desflurane for induction of anesthesia in infants and children. Before the introduction of desflurane, some advocated inducing anesthesia in children with isoflurane.502–506 For example, it has been suggested that the quality and speed of induction of anesthesia with isoflurane in oxygen in infants and children is similar to that with halothane.503 However, airway reflexes were commonly triggered with isoflurane despite the use of a number of strategies to attenuate them,507,509,510 including slowly increasing the inspired concentration, and scented masks. Given the smooth induction characteristics, economy, and availability of sevoflurane, there are no reasons to consider other anesthetics for induction of anesthesia in infants and children.
In contrast to the noxious effects of the methyl ethyl ether series of anesthetics on the airway, sevoflurane does not irritate the upper airway and is well tolerated when administered by mask to infants and children at any concentration.* The introduction of sevoflurane has challenged and displaced halothane as the induction agent of choice in children in most countries. The incidences of coughing, breath holding, laryngospasm, and hemoglobin–oxygen desaturation during induction with sevoflurane, whether by slow incremental increases in concentration or a single breath, are similar to those that occur during halothane (Table 6-10). The observation that the airway reflex responses are infrequent after a single-breath induction with 8% sevoflurane or 5% halothane casts doubt on the adage that high concentrations of inhalational anesthetics trigger airway reflex responses.515–517 In fact, the induction is so smooth with sevoflurane that adjuvants, such as a premedication, concurrent use of nitrous oxide, or other strategies to prevent airway reflex responses, are unnecessary.Induction of anesthesia with xenon has not been studied in children. In adults, inhalational induction with xenon at equi-MAC with sevoflurane resulted in a more rapid induction with stability of respirations with xenon.518 These data suggest that xenon may be an excellent induction anesthetic in children, provided its characteristics are upheld in well-designed clinical trials.
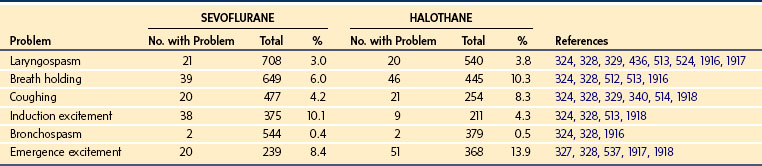
There is no single ideal approach to induce anesthesia by inhalation for all children. However, we advocate empowering children as much as possible to minimize fear. After appropriate preoperative preparation (involving premedication or parental presence or distraction techniques), the child is seated on the bed and encouraged to breathe through a face mask scented with a favorite flavor (to disguise the plastic odor of the mask) that is held over the nose and mouth. A fresh gas composed of 70% nitrous oxide in oxygen is breathed (while the pop-off value is completely open for 1 to 2 minutes). As soon as the child becomes “silly” or ceases to respond verbally, 8% sevoflurane is administered. Induction of anesthesia with halothane was performed with stepwise increments in the inspired concentration of 0.5% to 1.0% every three to four breaths until an adequate depth of anesthesia was achieved. This slow increase in the inspired concentration of halothane was thought to attenuate the incidence of airway reflex responses, although this is not evidence based. In fact, when a single breath vital capacity induction was performed in children older than 6 years of age with 5% halothane, the incidence of airway reflex responses was surprisingly small.515 Initially, sevoflurane was also administered in slow increments of 1% to 1.5% until 8%, but this caused transient agitation and involuntary movement of the extremities, frequently and sometimes violently, particularly in adolescents.340 This was attributed to an exaggerated excitement phase. To minimize the excitement phase, we recommend that the inspired concentration of sevoflurane be increased as rapidly as possible, from 0% to 8% in a single-step increase.515 This is based on a study of single-breath induction with 8% sevoflurane and 5% halothane in which the incidence of involuntary movement and need for restraint was significantly less with sevoflurane than it was with halothane.515 Recently, induction of anesthesia with 12% sevoflurane was reported to be more rapid than with 8%.519 This is not a surprising finding, although 12% sevoflurane vaporizers should not be used in clinical practice because inspired concentrations of sevoflurane (in a laboratory setting) of 11% in oxygen and 10% in nitrous oxide support combustion.
Although some studies report more rapid induction of anesthesia with sevoflurane than with halothane, others have not. This inconsistency in the relative speed of induction reflects differences in study design that likely failed to take advantage of the 8% sevoflurane vaporizer and the differences in MAC. For children who are unable to perform a single-breath vital capacity induction, a rapid increase in the inspired concentration of sevoflurane has been a very effective alternative, with results comparable to the single-breath technique.329,513–515
Sevoflurane does not trigger airway reflex responses either alone or in combination with other agents, such as nitrous oxide. One study suggested that sevoflurane is the least irritating to the airway of all the inhalational anesthetics.520 Previous studies suggested that airway irritability and excitement during sevoflurane anesthesia were similar whether nitrous oxide was present or absent, although others have disputed these findings.515,521 The lack of effect of nitrous oxide on the speed of induction in the single-breath study was attributed to its concentration-reducing effect on sevoflurane.515
Although halothane has been the preferred agent for induction of anesthesia because of its lack of airway irritability, arrhythmias occur more frequently during halothane than during anesthesia with the ether anesthetics. Halothane-induced arrhythmias occur more frequently during spontaneous ventilation and in association with high levels of circulating catecholamines. Most of these arrhythmias are unifocal or multifocal premature ventricular beats, nodal rhythm, bigeminy, or supraventricular arrhythmias.330,430,508 Despite their appearance, most of these arrhythmias are benign and preserve the blood pressure; ventricular tachycardia, however, may cause hypotension. Management of arrhythmias during halothane anesthesia includes inflating the lungs with large tidal volumes, hyperventilation to decrease the arterial carbon dioxide tension, increasing the concentration of halothane if there is evidence of “light” anesthesia (i.e., sweating, hypertension), and substituting another inhalational anesthetic for halothane.430,522 Lidocaine has no role in the treatment of these arrhythmias because the myocardium is not intrinsically irritable. Moreover, a rapid IV bolus of lidocaine (2 mg/kg) may cause profound bradycardia.523 Arrhythmias are rare during anesthesia with the ether series of anesthetics, but when they occur they are usually nodal in origin.* Arrhythmias during anesthesia with the ether anesthetics are usually self-limiting, resolving spontaneously or with parenteral administration of an anticholinergic. If the arrhythmias persist, then a cardiology consultation should be sought, particularly in a child with a history of congenital heart disease. Both IV and inhalational anesthetics have been used for induction and maintenance of anesthesia in children with congenital heart disease. (See cardiovascular section earlier and Chapter 21 for a more detailed discussion.)
Once an adequate depth of anesthesia has been achieved, it is prudent to maintain spontaneous respirations with the maximal inspired concentration of sevoflurane that is tolerated until IV access has been achieved. If hypopnea or apnea occurs, then ventilation should be assisted while avoiding hypocapnia. The reason to recommend this practice is to prevent awareness from occurring in the early induction period.395,396 Discontinuing or decreasing the inspired concentration of nitrous oxide or sevoflurane individually or together may predispose to awareness, particularly if the child is stimulated at a light plane of anesthesia. This may occur in situations where anesthesia is induced in one location (induction room) and the child is then transferred to another (operating room) without continuously supplying sevoflurane. Appreciating the limited solubility of nitrous oxide and sevoflurane will help to understand how rapidly these anesthetics egress from the body, particularly after a brief exposure. Once IV access is established, some practitioners administer propofol or another IV anesthetic before discontinuing the nitrous oxide to facilitate insertion of a laryngeal mask airway or tracheal intubation. The concentration of sevoflurane can then be decreased.
Emergence
Emergence or recovery has been arbitrarily divided into early (extubation, eye opening, following commands) and late (drinking, discharge time from postanesthesia care unit or hospital). Although most studies have demonstrated a more rapid early recovery after less soluble anesthetics,326–328,331,334 few have demonstrated a more rapid late recovery.324,334,524,525
The speed of emergence and recovery from anesthesia are discussed above. The incidence of complications, such as airway reflex responses and vomiting during emergence from anesthesia, after mask anesthesia or tracheal intubation, are similar with most inhalational agents.* However, the incidence of airway responses of any severity after desflurane were significantly greater than after isoflurane. Moreover, the incidence of airway adverse responses after removing a laryngeal mask airway (LMA) deep during desflurane anesthesia was significantly greater than was the incidence after removal of an LMA following recovery from desflurane (awake) or after isoflurane anesthesia.527