105 Peritonitis and Intraabdominal Infection
 Pathogenesis
Pathogenesis
Host Defenses
The peritoneal cavity is a complex space lined with mesothelial cells in visceral and parietal layers. The healthy peritoneal cavity is quiescent immunologically but responds rapidly to bacterial contamination. Normally, about 50 to 100 mL of peritoneal fluid circulates freely among several potential and actual spaces within the peritoneal cavity.1 Net fluid movement is cephalad toward the diaphragm, facilitated by normal peristalsis, normal diaphragmatic excursions, splanchnic blood flow, and factors that maintain normal membrane permeability of the microcirculation. Conversely, ileus, mechanical ventilation, splanchnic hypoperfusion, and intraperitoneal inflammation can disrupt normal fluid movement and cause intraperitoneal fluid sequestration.
The three major intraperitoneal host defense mechanisms include clearance of bacteria by lymphatics, phagocytosis of bacteria by immune cells, and mechanical sequestration with abscess formation. A few phagocytic cells circulate as peritoneal macrophages, and opsonic proteins that facilitate phagocytosis are present. Experimentally, a small bacterial inoculum placed in the peritoneal cavity is cleared within a few minutes when the peritoneal fluid is absorbed by specialized lacunae on the undersurface of the diaphragm.1 The bacteria then pass into the central venous system via diaphragmatic and mediastinal lymphatics for disposition by systemic host defenses. When an infectious inoculum is introduced, a brisk inflammatory response attempts to localize the infection, leading to abscess formation rather than generalized peritonitis. Intraabdominal abscess formation is a source-containment process; mortality is lower for patients with abscess(es) than for patients with generalized peritonitis.2,3
With inflammatory injury, peritoneal mesothelial cells are denuded, exposing the underlying basement membrane. When platelets and fibrin come into contact with the basement membrane, fibrin polymerization occurs and produces a typical exudative rind on peritoneal surfaces. Fibrin and apoptotic neutrophils contribute to the formation of adhesions and the walls of abscesses. Normally the process is self-limited by up-regulation and/or activation of fibrinolytic factors such as plasminogen within the first week after mesothelial injury. If the insult is self-limited, peritoneal repair occurs within 3 to 5 days. Under local hypoxic conditions, the adhesions are invaded by fibroblasts, angiogenesis is up-regulated, and the adhesions become tenacious.4
Microbiology
Gastrointestinal perforation releases bacteria into the peritoneal cavity. Bacterial density within the gut lumen increases along the length of the gastrointestinal tract from the stomach to the colon. The bacteria must proliferate to cause infection, while local host defenses seek to prevent or contain the establishment of infection. In addition to the microbes present in peritoneal fluid, microbial colonization of peritoneal surfaces occurs rapidly after perforation or penetrating injury as a result of expression by the microorganisms of specific adherence factors. Enterobacteriaceae predominate within the first 4 hours but are superseded within 8 hours by members of the B. fragilis group. Adherent bacteria are difficult to eradicate by operative peritoneal lavage.5
Besides adherence factors, bacteria possess several other features that can enhance their virulence. Peptidoglycans and lipoteichoic acid in the cell walls of gram-positive bacteria, especially streptococci and staphylococci, stimulate a proinflammatory response. These organisms can elaborate exotoxins and proteases that cause tissue injury and promote the dissemination of the bacteria. Lipopolysaccharide (LPS) in the outer cell wall of gram-negative bacteria can interact with many cell types to stimulate an inflammatory response. As bacteria proliferate and the size of the inoculum increases, acidic bacterial metabolites can impair neutrophil function.6 Larger inocula can render antibiotics, particularly β-lactams, ineffective via a process called the inoculum effect.7 Additionally, bacteria demonstrate a quorum-sensing effect that maximizes survival and reproduction by altering their behavior based upon signaling pathways.8
Synergistic interactions, usually among members of the B. fragilis group and either facultative gram-negative bacilli or enterococci, can suppress local host defenses and promote bacterial survival and growth.1 B. fragilis produces a capsular polysaccharide antigen that suppresses complement activation and inhibits leukocyte recruitment and function.9 Anaerobic bacteria produce short-chain fatty acids that can impair the function of neutrophils. Facultative bacteria consume residual oxygen in the microenvironment, permitting the survival and proliferation of obligate anaerobes. Anaerobic bacteria lower the redox potential in the microenvironment, also favoring their growth. Aerobic and anaerobic bacteria can enhance the growth of other species by providing crucial nutrients or the producing enzymes that inactivate antibiotics.
In some respects, bacteria have evolved to take advantage of host defenses. As an example, bacterial adherence to colonocytes and bacterial growth is enhanced by physiologic concentrations of norepinephrine, which is secreted as part of the counter-regulatory response to stress, as well as being administered as an exogenous drug to promote arteriolar constriction and increase myocardial contractility.10
Peritonitis
Peritonitis can be classified as primary, secondary, or tertiary. Most critically ill patients with intraabdominal infection have secondary or tertiary peritonitis. The bacteriology characteristic of these classes of peritonitis is shown in Table 105-1.
| Primary (Monomicrobial) | Secondary (Polymicrobial) | Tertiary (Polymicrobial) |
|---|---|---|
| Escherichia coli | Bacteroides fragilis group | Acinetobacter spp. |
| Enterococcus spp. | Clostridium spp. | Enterobacter spp. |
| Klebsiella spp. | E. coli | Enterococcus spp. |
| Streptococcus pneumoniae | Klebsiella spp. | Pseudomonas spp. |
| Other anaerobes | Staphylococcus spp. | |
| Staphylococcus epidermidis | ||
| Streptococcus spp. | ||
| Candida spp. |
Device-associated peritonitis is a variant of primary peritonitis that also almost always is monomicrobial. The great majority of cases occur with chronic ambulatory peritoneal dialysis (CAPD) catheters, which become infected as often as once per year of dialysis.11 The most common pathogens are Staphylococcus aureus and species of Pseudomonas and Candida. Catheter removal is usually necessary to eradicate these infections, especially when caused by P. aeruginosa or Candida spp. Although rare, recurrent CAPD-related peritonitis due to methicillin-resistant S. aureus (MRSA) has been associated with the emergence of vancomycin-resistant strains after treatment with multiple courses of vancomycin.12
Secondary peritonitis follows perforation of a hollow gastrointestinal viscus. The vast majority of cases are community acquired. Appendicitis is the most common cause, and the polymicrobial bacterial flora typically are highly susceptible to antibiotics. Thorough microbiologic analysis of a carefully collected specimen of purulent peritoneal fluid from a patient with secondary peritonitis yields an average of five organisms. B. fragilis the most commonly isolated obligate anaerobe, and E. coli is the most commonly isolated facultative organism. Less common isolates include Enterococcus spp., Candida spp., Clostridium spp., and P. aeruginosa. These uncommon isolates do not need to be covered by the antibiotic regimen if the patient was previously healthy and does not have comorbid conditions that increase the risk for an adverse outcome. Early operative source control, combined with a short course of broad-spectrum antibiotics, are curative in more than 85% of all cases and more than 90% of appendicitis cases.13 Most cases of community-acquired peritonitis do not result in severe illness, and these cases seldom require care in an intensive care unit (ICU).
Tertiary peritonitis describes recurrent or persistent intraabdominal infection after failure of more than one source control procedure to control the infection.14–16 The flora usually include one or more strains of staphylococci (often methicillin-resistant S. epidermidis or MRSA) and Enterococcus spp., Candida spp., or Pseudomonas spp.17–19 It is debated whether tertiary peritonitis represents invasive infection or permissive colonization of the peritoneal cavity in the face of devastated host defenses. The notion that host defenses are compromised is supported by the observation that fluid collections are often poorly localized and serosanguineous rather than purulent. Cases of tertiary peritonitis are fortunately uncommon, but class I data regarding management are lacking.
Adjuvants
Adjuvant substances act to decrease the bacterial inoculum necessary for infection. Adjuvants can increase virulence or interfere with host defenses and invariably are present to some degree in every patient with gastrointestinal perforation. Common adjuvants include ascites, blood, fibrin, bile, urine, chyle, pancreatic juice, and platelets.20 The most important adjuvant is blood. Hemoglobin, fibrin, and platelets all impair host defenses, and iron, which is essential for bacterial growth, also depresses phagocyte function. Fibrin promotes bacterial trapping and abscess formation and can sequester bacteria from neutrophils. Bile salts impair host defenses and are toxic to neutrophils.21 Pancreatic enzymes can be activated by bacterial infection, producing necrotic tissue that is an excellent culture medium.
 At-Risk Patient
At-Risk Patient
Fortunately, most patients with intraabdominal infection are not so sick as to require care in an ICU. In a population-based study of hospital discharges for peritonitis, severe sepsis developed in only 11% of cases (Table 105-2) but increased the mortality risk by 19-fold.2 Similarly, only about 15% of patients enrolled in clinical trials of antimicrobial therapy for secondary peritonitis have an APACHE II score above 15 points.22
TABLE 105-2 Risk Factors for Severe Sepsis in Patients with Intraabdominal Infections
| Parameter | Relative Risk | 95% Confidence Intervals |
|---|---|---|
| Age (Years) | ||
| <20 | 1.0 | |
| 20-39 | 1.4 | 0.8-2.5 |
| 40-59 | 3.2 | 1.8-5.6 |
| 60-79 | 4.6 | 2.6-8.0 |
| >79 | 6.5 | 4.7-11.8 |
| Site | ||
| Appendix | 1.0 | |
| Gallbladder | 2.7 | 1.9-3.8 |
| Colon | 3.9 | 2.6-5.8 |
| Stomach/duodenum | 6.9 | 4.6-10.3 |
| Small bowel | 9.0 | 6.1-13.4 |
| Extent | ||
| Localized | 1.0 | |
| Abscess | 1.2 | 0.8-1.8 |
| Diffuse | 1.5 | 1.1-1.9 |
| Comorbidities | ||
| Congestive heart failure | 1.2 | 1.0-1.6 |
| Stroke | 1.8 | 1.2-2.7 |
| Liver dysfunction | 2.0 | 1.4-2.8 |
| Renal dysfunction | 2.0 | 1.4-2.9 |
Data from Anaya and Nathens.2
Some patients with community-acquired secondary peritonitis have critical illness as a result of delayed presentation, immunosuppression, or extremes of age. However, most patients with critical illness have hospital-acquired peritonitis (Table 105-3). The leading causes of hospital-acquired peritonitis are gastrointestinal anastomotic dehiscence and splanchnic ischemia due to various causes including hypovolemia, distributive shock, atheroembolism, and thromboembolism. Hospital-acquired peritonitis is usually polymicrobial, and commonly cultured organisms include Enterococcus spp., Candida spp., Pseudomonas aeruginosa, and other antibiotic-resistant organisms such as MRSA.14,18,19
TABLE 105-3 Clinical Factors Predicting High-Risk Intraabdominal Infection
Data from Pieracci et al.13 and Solomkin et al.18
The frequency of intraabdominal infection encountered in a particular ICU is variable. Surgical ICUs that care for patients with multiple trauma or following emergency surgery are likely to have more cases of intraabdominal infection than medical ICUs. Surgical patients that have required an operation for source control or have a postoperative secondary nosocomial infection account for 25% to 40% of patients with severe sepsis. However, units with a low prevalence must be equally vigilant in their surveillance and assessment, because a missed intraabdominal infection is almost always fatal.23
When patients with intraabdominal infection are critically ill, mortality exceeds 25%. The risk of failure increases with increasing severity of illness, inadequate empirical antibiotic therapy, delayed surgical therapy, and failure of source control.14,18 Most clinical failures are not associated with multidrug-resistant pathogens, although some data suggest that resistant pathogens cause clinical failure in cases of postoperative peritonitis.24,25
 Spectrum of Disease Causing Critical Illness
Spectrum of Disease Causing Critical Illness
Abscess Of Solid Organs
Treatment of liver abscesses should be individualized. A source of origin should be sought and treated. Antibiotics are mandatory, and a prolonged course for more than 14 days may be necessary. If feasible, based upon the size and location of the abscess, percutaneous drainage always should be attempted.26,27 Operative drainage may be required for abscesses that cannot be drained percutaneously. Overall mortality rate is approximately 25% but is higher for patients with multiple abscesses that are too small to drain.28
Splenic abscesses are uncommon and are the result of hematogenous or local contamination. Hematologic sources include endocarditis, urinary tract infections, pneumonia, osteomyelitis, otitis, mastoiditis, and pelvic infections. Splenic abscesses have been reported with other systemic infections including typhoid, paratyphoid, malaria, and candidiasis. Direct extension from adjacent infections of the pancreas, retroperitoneum, subdiaphragmatic spaces, and diverticulitis can involve the spleen. Systemic disorders such as hemoglobinopathies or sickle cell disease, can cause splenic infarction. Devitalized splenic tissue resulting from trauma, infarction, or embolization can become infected and evolve into splenic abscesses.29
Acute Acalculous Cholecystitis
In contrast to cholecystitis due to gallstones, the etiology of acute acalculous cholecystitis is gallbladder ischemia; infection of the organ occurs secondarily.30 Although acute acalculous cholecystitis can complicate many illnesses, splanchnic hypoperfusion is the common feature. Risk factors in medical patients include congestive heart failure, diabetes mellitus, abdominal vasculitis, and malignancy (including after bone marrow transplantation). Acalculous cholecystitis is more common in surgical patients and can occur following burns, trauma, cardiopulmonary bypass, biliary instrumentation, and emergency aortic surgery.30
The diagnosis of acute acalculous cholecystitis can be challenging, and a high index of suspicion is required. Prompt diagnosis and therapy are necessary, as the disease can be fulminant. Necrosis of the gallbladder occurs in 50% of patients, and perforation of the gallbladder occurs in 20%. Fever and hyperbilirubinemia are common associated findings.29 Serum transaminase and alkaline phosphatase levels also may be elevated. When signs and symptoms can be localized to the right upper quadrant, the differential diagnosis includes gastroduodenal perforation, acute pancreatitis, right colonic ischemia, and acute hepatitis.
Bedside ultrasonography is favored for the diagnosis of acute acalculous cholecystitis; the most accurate diagnostic features are gallbladder wall thickness greater than 3.5 mm and presence of pericholecystic fluid. Computed tomography (CT) is equally accurate and can be utilized when there are no localizing findings and the patient is a candidate for intrahospital transport. Hepatobiliary scintigraphy is not useful to identify or exclude acute acalculous cholecystitis, owing to a high incidence of false-positive findings that result in part from a lack of dietary stimulus for gallbladder contraction. Coadministration of morphine sulfate, which increases biliary hydrostatic pressure, can promote filling of the gallbladder and increase diagnostic accuracy of hepatobiliary scintigraphy.31
Ischemic Colitis And Enteritis
Intestinal ischemia is a dangerous and relatively common complication of critical illness that can progress within hours to gangrene, perforation, and generalized peritonitis.32 The splanchnic circulation is especially vulnerable to low cardiac output, particularly when the cardiac index is less than 2 L/min/m2. Most cases are caused by nonocclusive ischemia; the origin is often multifactorial, including hypovolemia, shock, and administration of vasopressors. A number of other causes have been identified. Acquired protein C/protein S deficiency induces a hypercoagulable state that has been associated with mesenteric arterial and venous thrombosis. Chronic atrial fibrillation or dilated cardiomyopathy can lead to mesenteric arterial thromboembolism. Arteriography can cause cholesterol embolization from dislodgement of an atherosclerotic plaque. Intestinal obstruction also must be considered, and this diagnosis may not be obvious with partial or proximal obstructions.
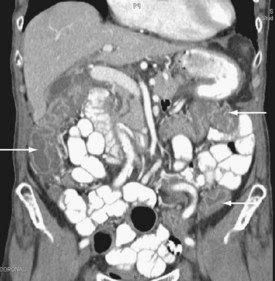
The pattern of injury with intestinal ischemia is variable depending on the mechanism, the presence of heart disease, and the status of the collateral circulation via the celiac and inferior mesenteric arteries. Large thrombi usually occlude the superior mesenteric artery where it narrows just distal to origin of the middle colic artery. The first 30 to 45 cm of small bowel and the left colon may be spared. Smaller emboli are more likely to infarct the small bowel and possibly the ascending colon (Figure 105-1); the distribution can be patchy. Nonocclusive ischemia classically occurs in watershed areas of the mesenteric circulation where collateral vessels bridge the two arterial distributions. A typical site is the splenic flexure of the colon at the watershed junction of the superior and the inferior mesenteric arteries. Although any segment of intestine can be affected by nonocclusive ischemia, the cecum (the point farthest from inferior mesenteric artery collaterals) and the left colon are most likely to be affected (Figure 105-2). The left colon is particularly vulnerable after abdominal aortic operations, especially when the inferior mesenteric artery has been ligated during the procedure.
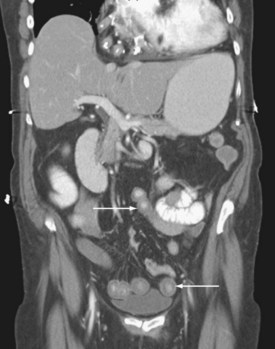
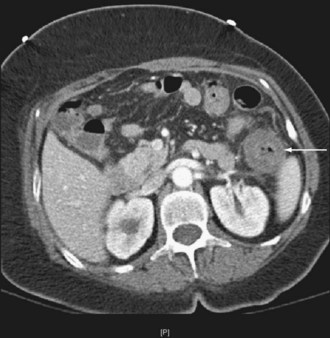
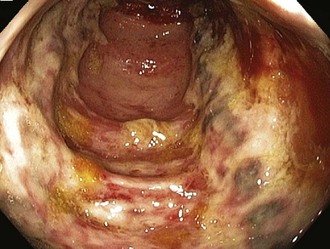
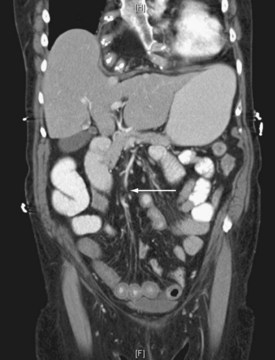
Given the propensity for left colonic involvement, lower endoscopy at the bedside is usually the first diagnostic modality to be employed, but a number of pitfalls should be recognized. Flexible sigmoidoscopy is simple and safe but may miss ischemia at or proximal to the splenic flexure; accordingly, colonoscopy is preferred (Figure 105-3). Endoscopy visualizes only the mucosa and can underestimate the extent of disease. CT and CT angiography are increasingly useful diagnostically and have largely supplanted use of formal arteriography (Figure 105-4).
Bedside diagnostic laparoscopy represents a new technique to identify intraabdominal pathology in an ICU setting, but reports to date are anecdotal. Laparoscopy should be considered when transfer of the patient to radiology or the operating room is considered unsafe or when routine radiologic examinations are inconclusive.33,34 Diagnostic testing should be foregone entirely in favor of immediate laparotomy if signs of peritonitis are present.
Surgical therapy is individualized based on the location and extent of intestinal compromise and the physiologic state of the patient. Infarcted bowel is resected, but bowel of questionable viability can be left for reinspection at a “second-look laparotomy” in 12 to 24 hours, particularly if a massive resection would otherwise be needed. Anastomosis can be performed in the stable patient without peritonitis or deferred in unstable patients by leaving the occluded ends of bowel in temporary discontinuity until the second-look procedure. Creation of a temporary ostomy is a third option but is performed with decreasing frequency. If there is confidence that a second-look procedure is unnecessary, the abdominal fascia can be closed. If reoperation is planned, or if bowel edema and distention are such that definitive closure would risk the development of intraabdominal hypertension and abdominal compartment syndrome, damage-control principles are utilized, and a temporary abdominal wall closure is performed. Methods for temporary closure include closure of skin only or closure with absorbable mesh, biological material, or plastic sheeting, usually in conjunction with a negative pressure system.35
Clostridium Difficile Colitis
The incidence and severity of Clostridium difficile infections (CDI) are increasing. CDI remains the most common nosocomial gastrointestinal infection, with significant morbidity and mortality. Early diagnosis and treatment are essential for a favorable outcome. A highly virulent and resistant strain, PCR ribotype 027, has been associated with recent outbreaks in North America and Europe characterized by increased incidence and a higher risk of death.36 The ICU patient is at increased risk for CDI, and in these patients the disease is more frequent, more severe, more refractory to medical therapy, and subject to higher rates of relapse.37 In one study, emergency colectomy was needed in 25% of patients with CDI requiring ICU admission. Other risk factors for CDI include preoperative antibiotic usage, uremia, burns, chronic obstructive pulmonary disease, cancer, abdominal surgery, cesarean section, antiperistaltic medications, proton pump inhibitors, ICU stay, prolonged hospital stay, chemotherapy, and postpyloric tube feeds.36,37
Clinical examination of patients with CDI may not demonstrate significant findings of peritonitis unless megacolon or perforation has developed. CT can demonstrate typical findings of colonic wall thickening, dilation, and the so-called accordion sign (thickened haustral fold and trapped contrast material, ascites, and/or pericolonic stranding; Figure 105-5). Measurement of stool toxin titers and endoscopy are useful diagnostic adjuncts, but definitive management in patients with evidence of refractory fulminant colitis should not be delayed as a consequence of waiting for the results of these tests. Routine cases of CDI are treated with oral or IV metronidazole. Severe disease is treated with oral vancomycin; administration of this antibiotic via the enteral route leads to high luminal concentrations of the drug owing to lack of absorption across the mucosa of the gut.
Fulminant CDI with perforation, toxic megacolon, severe ileus, hypotension, or refractory septicemia occurs in approximately 3% to 8% of patients. Patients who have a history of inflammatory bowel disease, recent surgery, prior treatment with IV immunoglobulin, vasopressor requirements, leukocytosis, or increased blood lactate concentration should have early surgical consultation. Mortality was notably decreased in patients who had no more than 6 days of medical treatment prior to operation, supporting consideration of early subtotal colectomy.36–39
Acute Pancreatitis
Gallstones and alcohol together account for about 80% of cases of pancreatitis, with the remainder due to trauma, upper abdominal surgery, or cardiopulmonary bypass. Approximately 85% of cases are self-limited and have a good prognosis. The remaining 15% of cases account for most of the morbidity and all of the mortality. Infection is the most common complication and can lead to multiple organ dysfunction syndrome and death. Mortality risk is associated with large IV fluid requirement, acidosis, and hypocalcemia.40
Pancreatic infections include infected pseudocysts, discrete pancreatic abscesses, or infected pancreatic necrosis. The last is a poorly localized process that affects the retroperitoneal fat as well as the pancreas itself. Infection can develop as early as 5 days after the onset of acute pancreatitis, with peak incidence at day 14. Almost any common organism can cause infection, including staphylococci, enteric gram-negative bacilli, obligate anaerobes, P. aeruginosa, and Candida spp. Assuming appropriate microbial susceptibility, imipenem, meropenem, doripenem, or a fluoroquinolone plus metronidazole are recommended empirical antimicrobial agents, based on kinetic studies of drug accumulation in normal pancreas or pancreatic juice. Fluconazole achieves adequate concentrations in pancreatic tissue, whereas aminoglycosides do not.41
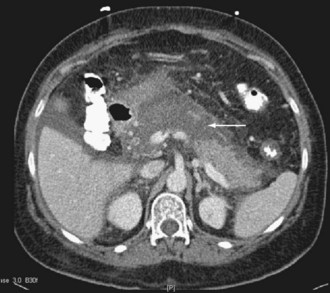
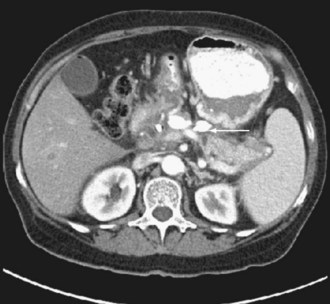
Many aspects of the prevention and management of pancreatic infection are controversial, including the role of antibiotic prophylaxis, diagnostic methods, and techniques and timing of surgical drainage and débridement. Antibiotic prophylaxis of severe pancreatitis with imipenem is employed by many physicians and surgeons, but this practice is not supported by class I data and has been associated with an increased risk of fungal infection.42 Regardless of the severity of illness, all patients with pancreatitis should undergo biliary ultrasonography in search of gallstones. Pancreatic protocol CT is the best study to define anatomic severity and infection. The study should be performed with contrast infusion and thin cuts through the region of the pancreas to assess for viability and presence of devitalized tissues (Figure 105-6). When peripancreatic infection is suspected, CT-guided fine-needle aspiration can be performed to obtain material for culture.
Source control is essential for patients with established pancreatic infection. Newer techniques with acceptable outcomes and lower morbidity have begun to replace traditional open drainage and surgical necrosectomy. These techniques include minimally invasive endoscopic, radiologic, and laparoscopic approaches. Endoscopic ultrasonography as a guide to drainage and laparoscopic pancreatic necrosectomy has demonstrated success in managing pancreatitis necrosis (Figure 105-7).43,44 Recent results from the Dutch Pancreatitis Study Group demonstrated a significant reduction in major complications (new-onset multiple-organ failure, incisional hernias, and new-onset diabetes) following the step-up approach for managing infected pancreatic necrosis. This approach consists of percutaneous drainage followed (if necessary) by minimally invasive retroperitoneal necrosectomy.45 Improvements in resuscitation and operative management of infectious complications have reduced the mortality rate to about 20%, half that of an earlier era.
 Diagnosis
Diagnosis
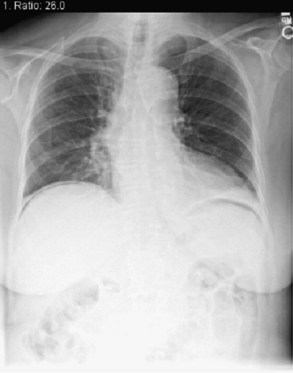
Although good-quality plain abdominal radiographs are difficult to obtain at the bedside, pneumoperitoneum (Figure 105-8), intestinal obstruction, or signs of intestinal ischemia may be found. Pneumoperitoneum can be an innocuous finding in mechanically ventilated patients and for as long as 7 days after abdominal operations.46,47 Plain radiography can be augmented by water-soluble contrast injection of drains, fistulas, or sinuses to define the anatomy of complex infections or monitor resolution after drainage.
CT with oral and IV contrast is the primary radiologic imaging tool for the abdomen and pelvis.48–50 CT signs of intraabdominal infection include extraluminal gas (Figure 105-9), free fluid, contrast extravasation, fat stranding, and presence of a contrast-enhancing rim that is characteristic of abscess (Figure 105-10). CT is now the test of choice for diagnosis of intestinal obstruction. Intramural gas may be identified when mesenteric ischemia is present; occasionally, a thrombus is seen in a visceral vessel (see Figure 105-4). CT-guided percutaneous drainage of abscesses and intraabdominal fluid collections is the treatment of choice for source control in the absence of generalized peritonitis or disruption of visceral structures.26,27,50
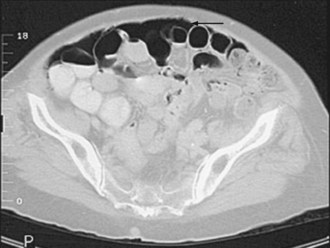
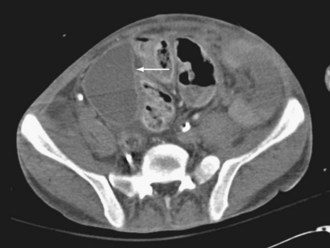
Although CT is an excellent diagnostic modality, obtaining an adequate study in critically ill patients can be problematic. When patients are hemodynamically unstable or dependent on a high level of mechanical ventilatory support, transport out of the ICU can be risky.51 Iodinated contrast agents can precipitate or aggravate renal dysfunction. If the need for CT can be anticipated 24 hours in advance, pretreatment with N-acetylcysteine and IV sodium bicarbonate can limit the risk of contrast-induced nephropathy.52
 Principles of Management
Principles of Management
Source Control
Source control for complicated intraabdominal infections remains the most important component of successful treatment. Consensus guidelines aim to implement source control interventions within the first 6 hours of management.53,54
The elements of definitive source control include removal of infected or nonviable material, closure or control of perforations, and reduction of peritoneal contamination by bacteria and toxins. Multiple staged operative procedures may be needed. Failure to obtain adequate source control reportedly occurs in 10% to 25% of cases of intraabdominal infection, depending on the severity and complexity of the infection.14,54
Abscess
Abscesses are characterized by low oxygen tension, poor antibiotic penetration, and impaired leukocyte function. Small abscesses may resolve with antibiotics alone. Source control with percutaneous drainage is the treatment of choice for most abscesses, provided adequate drainage is possible and no débridement or repair of anatomic structures is necessary.12,53 Percutaneous decompression of abscesses is successful and produces rapid clinical improvement in 85% of cases.26,27 Formal operative intervention should be performed without delay if clinical improvement does not occur promptly following drainage.
Peritoneal Toilet
Once source control has been achieved, additional intraoperative measures to cleanse the peritoneal cavity of microscopic infection, including irrigation with fluid or antibiotic solutions and débridement of peritoneal surfaces, are ineffective and may be deleterious.55–57 Bacteria adhere to mesothelial cells on the serosal surfaces in peritonitis, rendering them resistant to removal by passive irrigation.57–59 Moreover, animal studies suggest that irrigation fluids disseminate infection by hindering the normal immunologic function of the peritoneum. Irrigation with antibiotic solutions is of no benefit if parenteral antibiotics are administered.4 High-volume lavage and pulse irrigation used in fecal and purulent peritonitis may be of benefit but also can increase the risk of fistula formation.57–59 Closed-suction drains do not prevent recurrent fluid collections and are ineffective at draining the peritoneal cavity.
Open Abdomen
Open abdomen techniques are used in selected patients with diffuse peritonitis, inadequate primary source control, intestinal ischemia and discontinuity, abdominal compartment syndrome, or necrotizing infections of the anterior abdominal wall.60,61 Open abdomen is a component of the damage-control strategy used in unstable patients with massive trauma. In cases of peritonitis, goals of damage control include reassessment of intestinal viability, decompression of the abdomen, and access for peritoneal toilet. Open abdomen management has disadvantages including promotion of excessive losses of fluid and protein, ileus, fistula formation, and ventral hernias. Neither planned re-laparotomy nor open abdomen management offer a survival benefit as compared with on-demand re-laparotomy.62
Many variations in open abdomen techniques have been reported. Most often, a negative-pressure system is utilized with a fenestrated nonadherent material to cover the bowel, suction drains to aspirate fluid above this layer, and an airtight adherent outer drape to maintain a vacuum and prevent evisceration until adhesions form (Figures 105-11 and 105-12). Nasogastric and urinary catheter decompression are maintained, and parenteral nutritional support is provided to prevent bowel distention. At reexploration, the abdomen is lavaged, and loculated fluid collections are evacuated. After achieving resuscitation and adequate source control, aggressive diuresis should begin to reverse edema and facilitate closure of the abdomen. In a substantial percentage of patients, the fascia cannot be closed primarily; absorbable mesh or biological grafts, skin grafts, and enteral nutrition allow healing to occur, and ventral hernias are repaired electively when all acute problems have resolved, but no earlier than 3 to 6 months after the acute episode.
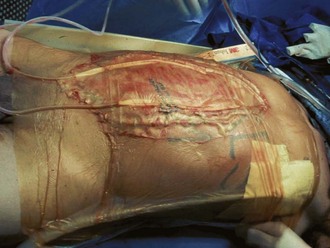
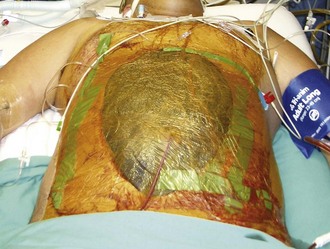
Antibiotic Therapy
Optimal antibiotic therapy for intraabdominal infection requires an agent or combination of agents active against gut-derived facultative enteric gram-negative bacilli as well as obligate anaerobes.12,13 Initial antibiotic therapy should be empirical, because the source is not always known.
A 2005 Cochrane review of 40 studies with 5094 patients comparing 16 different antibiotic regimens for empirical first-line therapy demonstrated equivalent efficacy and made no specific recommendations based on class I evidence.21 Given this equivalence (Table 105-4), the selection of a regimen should be based on considerations of cost, availability, ease of administration, susceptibilities, and the risk of toxicity, including allergy to β-lactam agents.12,63
TABLE 105-4 Antimicrobial Agent Regimens for Therapy of Serious Intraabdominal Infections
| Single Agents |
Data from Solomkin et al.18 and Mazuski et al.19
Evidence-based guidelines for selection of antimicrobial therapy for high-risk patients with intraabdominal infections have been formulated by both the Surgical Infection Society and the Infectious Diseases Society of America.12 The most commonly accepted empirical treatment regimens for complicated intraabdominal infections include extended-range β-lactam/β-lactamase agents such as piperacillin/tazobactam, carbapenems such as imipenem-cilastatin, meropenem, and ertapenem, or a third- or fourth-generation cephalosporin plus metronidazole.
In hospital-acquired peritonitis, local antimicrobial resistance patterns should be considered. Although mortality appears to be higher when an Enterococcus spp. is isolated from polymicrobial intraabdominal infections, there is no evidence that antienterococcal therapy improves outcome.12,24,64 Combination therapy directed against a specific pathogen (e.g., double-drug coverage of P. aeruginosa) has no demonstrated benefit in sepsis and can worsen outcomes. Newer agents including the glycylcycline antibiotic, tigecycline; the carbapenems, ertapenem and doripenem; and the fluoroquinolone, moxifloxacin also have equivalency in the treatment of complicated intraabdominal infections and provide new options in dealing with the problems of emerging bacterial resistance.65–70
Fungal species are common components of the normal intestinal flora, and fungi are common isolates from peritoneal fluid during operations for perforated viscera. Treatment of fungal isolates in an otherwise immunocompetent patient has not been found to improve survival.71 Because fungal colonization usually precedes invasive infection in surgical patients, some experts advocate systemic antifungal treatment when fungal species are recovered from peritoneal fluid.12,72,73 Empirical antifungal therapy is warranted with isolation of fungi from two or more normally sterile sites, from the bloodstream of critically ill patients with fungal abscesses, and in immunosuppressed patients.2,3,12,74 Fungal species cultured from an abscess or peritoneal fluid in a profoundly immunosuppressed patient also should be treated.2,3,12 In the critically ill patient, initial antifungal therapy with an echinocandin (caspofungin, micafungin, anidulafungin) instead of a triazole is recommended.12,71
Shorter courses of antimicrobial therapy and modification of agents once susceptibilities are obtained are safe in patients with adequate source control and will limit intraabdominal infections caused by multidrug-resistant pathogens.75–77 When source control is adequate, generalized peritonitis can be treated with antibiotics for as few as 3 to 5 days.77 The duration of therapy should typically be limited to 7 to 10 days. In hospital-acquired peritonitis, the duration of therapy is less defined and is based upon clinical parameters such as fever, abdominal pain, leukocytosis, and return of gastrointestinal function. Persistent sepsis should raise the possibility of inadequate source control, other nosocomial infections, or tertiary peritonitis. Rather than broadening antibiotic coverage or continuing the current regimen, a complete diagnostic reevaluation with physical examination, cultures, and imaging is indicated to identify any source of ongoing infection.
The concept of deescalation, where empirical broad-spectrum antibiotics are replaced by targeted narrower-spectrum agents once susceptibilities are obtained, is safe and should help reduce the risk of emergence of antibiotic-resistant isolates. While evidence-based recommendations are unequivocal, compliance with deescalation is poor.76 When intestinal function returns, oral antibiotics with good bioavailability can be administered to complete the course of therapy.12,78
The role of antibiotics for tertiary peritonitis is even less well defined. There is little proven benefit to empirical antibiotics, and most bacterial isolates tend to be resistant to standard regimens. Empirical coverage of isolated Enterococcus spp. and Candida spp. has been well studied and is without clearly defined benefit. Such coverage is recommended for complicated, nosocomial infections in immunosuppressed patients, critically ill patients, and patients with valvular heart disease or implanted prosthetic materials.12 Antibiotics for tertiary peritonitis should be of narrow spectrum and administered briefly; anti-anaerobic therapy is probably unnecessary.
 Complications
Complications
Enterocutaneous Fistula
Fistula formation is a dreaded complication of peritoneal inflammation and bowel injury. More than 80% of fistulas occur postoperatively, whereas fistulas that arise primarily from infection or irradiated bowel are rare.79,80 A fistula can be an occult source of sepsis before drainage to the skin makes the diagnosis obvious. A fistula can contain an abscess cavity along its tract or exist internally as a connection between two intraabdominal structures.
When a fistula develops, initial care should be supportive. Therapy is directed at appropriate antibiotic therapy if signs of secondary infection are present, along with bowel rest, skin care, and parenteral nutritional support. Administration of octreotide may reduce fistula output, minimize losses of fluid, electrolytes, and proteins, and facilitate spontaneous closure.81,82 Spontaneous closure with nonoperative therapy occurs in 30% to 50% of fistulas, usually within 3 to 4 weeks.80
 Mortality
Mortality
Significant progress in the management of peritonitis and intraabdominal infection has been made over the past century, but mortality remains about 25% for critically ill patients.3 Age and severity of illness at the time of presentation are more important predictors of mortality than the site of infection within the abdomen.3 Failure of the initial source control procedure is more likely to result in death than infection caused by a multidrug-resistant pathogen.
Organ dysfunction is present to some degree in every patient dying with intraabdominal infection. Early recognition of organ dysfunction as a sign of persistent intraabdominal infection offers an opportunity to intervene while the process is still reversible.83–85 The key elements of management to minimize mortality include early recognition of the problem, rapid resuscitation, timely and correct performance of source control procedures, and administration of appropriate broad-spectrum antibiotics.
Administration of glucocorticoids in patients with septic shock has not demonstrated a survival benefit or clinical improvement of sepsis and increases the risk of superinfection.86 Judicious transfusion of red blood cell concentrates may be of benefit as an adjunct in sepsis, but additional data are needed for confirmation.87 The concept of very tight control of serum glucose concentration in the management of the critically ill patient with sepsis has been controversial, but recent results indicate that this practice increases complications and mortality and should be avoided.88–91 One recent trial demonstrated that therapy with drotrecogin alfa (recombinant human activated protein C) improves survival among patients with severe sepsis and a high risk of death (APACHE II score ≥ 25 points or dysfunction of at least two organs), but follow-up studies failed to confirm this finding.92–94 Surgical patients had more significant bleeding complications during the 96-hour infusion period, but most were treated successfully. At the time of this writing, a second multicentric pivotal trial of drotrecogin alfa (activated) is in progress, and results should be available soon. Based on currently available evidence, therapy with drotrecogin alfa (activated) should be considered for patients with severe abdominal sepsis associated with a high risk of death.62,93,94
Key Points
Mazuski J, Solomkin J. Intraabdominal infections. Surg Clin N Am. 2009;89:421-437.
Bradley JS, et al. Diagnosis and management of complicated intra-abdominal infection in adults and children: guidelines by the Surgical Infection Society and the Infectious Diseases Society of America. Surg Infect. 2010;11:79-109.
Wong PF, Gilliam AD, Kumar S, Shenfine J, O’Dair GN, Leaper DJ. Antibiotic regimens for secondary peritonitis of gastrointestinal origin in adults. Cochrane Database Syst Rev. 2005;2:1-78.
Rivera-Sanfeliz G. Percutaneous abdominal abscess drainage: a historical perspective. AJR Am J Roentgenol. 2008;191:642-643.
Sailhamer EA, Carson K, Chang Y, et al. Fulminant Clostridium difficile colitis: patterns of care and predictors of mortality. Arch Surg. 2009;144:433-439.
van Santvoort HC, Besselink MG, Bakker OJ, et alDutch Pancreatitis Study Group. A step-up approach or open necrosectomy for necrotizing pancreatitis. N Engl J Med. 2010;362:1491-1502.
Dellinger RP, Levy MM, Carlet JM, et al. Surviving Sepsis Campaign: International guidelines for management of severe sepsis and septic shock: 2008. Intensive Care Med. 2008;34:17-60.
Blot SI, Vandewoude KH, De Waele JJ. Candida peritonitis. Curr Opin Crit Care. 2007;13:195-199.
De Waele JJ, Ravyts M, Depdt P, Blot SI, Decruyenaere J, Vogelaers D. De-escalation after empirical meropenem treatment in the intensive care unit: fiction or reality? J Crit Care. 2010 Jan 13. Epub ahead of print
Finfer S, Chittock DR, Su SY, et alNICE-SUGAR Study Investigators. Intensive versus conventional glucose control in critically ill patients. N Engl J Med. 2009;360:1283-1297. Epub 2009 Mar 24
Payen D, Sablotzki A, Barie PS, et al. International integrated database for the evaluation of severe sepsis and drotrecogin alfa (activated) therapy: analysis of efficacy and safety data in a large surgical cohort. Surgery. 2006;140:726-739.
1 Solomkin JS, Wittman DW, West MA, Barie PS. Intraabdominal infections. In: Schwartz SI, Shires GT, Spencer FC, et al, editors. Principles of Surgery. 7th ed. New York: McGraw-Hill; 1999:1515-1550.
2 Anaya DA, Nathens AB. Risk factors for severe sepsis in secondary peritonitis. Surg Infect (Larchmt). 2003;4:355-362.
3 Barie PS, Hydo LJ, Eachempati SR. Longitudinal outcomes of intraabdominal infection complicated by critical illness. Surg Infect (Larchmt). 2004;5(4):365-373.
4 Molinas CR, Campo R, Elkelani OA, et al. Role of hypoxia inducible factors 1alpha and 2alpha in basal adhesion formation and in carbon dioxide pneumoperitoneum-enhanced adhesion formation after laparoscopic surgery in transgenic mice. Fertil Steril. 2003;80(Suppl 2):795-802.
5 Platell C, Papadimitriou JM, Hall JC. The influence of lavage on peritonitis. J Am Coll Surg. 2000;191:672-680.
6 Grinstein S, Furuya W, Nasmith PE, Rotstein OD. Na+H+ exchange and the regulation of intracellular pH in polymorphonuclear leukocytes. Comp Biochem Physiol A. 1988;90:543-549.
7 Queenan AM, Foleno B, Gownley C, et al. Effects of inoculum and beta-lactamase activity in AmpC- and extended-spectrum beta-lactamase (ESBL)-producing Escherichia coli and Klebsiella pneumoniae clinical isolates tested by using NCCLS ESBL methodology. J Clin Microbiol. 2004;42:269-275.
8 March JC, Bentley WE. Quorum sensing and bacterial cross-talk in biotechnology. Curr Opin Biotechnol. 2004;15:495-502.
9 Patrick S, Lutton DA, Crockard AD. Immune reactions to Bacteroides fragilis populations with three different types of capsule in a model of infection. Microbiology. 1995;141:1969-1976.
10 Alverdy JC, Laughlin RS, Wu L. Influence of the critically ill state on host-pathogen interactions within the intestine: Gut-derived sepsis redefined. Crit Care Med. 2003;31:598-607.
11 Borg D, Shetty A, Williams D, Faber MD. Fivefold reduction in peritonitis using a multifaceted continuous quality initiative program. Adv Perit Dial. 2003;19:202-205.
12 O’Riordan A, Abraham KA, Ho JK, Walshe JJ. Vancomycin-resistant peritonitis associated with peritoneal dialysis: A cause for concern. Isr J Med Sci. 2002;171:42-43.
13 Pieracci FM, Barie PS. Management of Severe Sepsis of Abdominal Origin. Scand J Surg. 2007;96:184-196.
14 Mazuski J, Solomkin J. Intraabdominal Infections. Surg Clin N Am. 2009;89:421-437.
15 Malangoni MA. Evaluation and management of tertiary peritonitis. Am Surg. 2000;66:157-161.
16 Evans HL, Raymond DP, Pelletier SJ, et al. Tertiary peritonitis (recurrent diffuse or localized disease) is not an independent predictor of mortality in surgical patients with intraabdominal infection. Surg Infect. 2001;2:255-263.
17 Fierobe L, Decre D, Muller C, et al. Methicillin-resistant Staphylococcus aureus as a causative agent of post-operative intra-abdominal infection: Relation to nasal colonization. Clin Infect Dis. 1999;29:1231-1238.
18 Solomkin JS, Mazuski JE, Bradley JS, et al. Diagnosis and management of complicated intra-abdominal infection in adults and children: guidelines by the Surgical Infection Society and the Infectious Diseases Society of America. Surg Infect. 2010;11(1):79-109.
19 Mazuski JE. Antimicrobial treatment for intra-abdominal infections. Expert Opin Pharmacother. 2008;17:2933-2945.
20 Farthmann EH, Schoffel U. Epidemiology and pathophysiology of intraabdominal infections (IAI). Infection. 1998;26:329-334.
21 Schneierson SS, Amsterdam D, Perlman E. Enhancement of intraperitoneal staphylococcal virulence for mice with different bile salts. Nature. 1961;190:829-830.
22 Wong PF, Gilliam AD, Kumar S, Shenfine J, O’Dair GN, Leaper DJ. Antibiotic regimens for secondary peritonitis of gastrointestinal origin in adults. Cochrane Database Syst Rev. 2005;2:1-78.
23 Gajic O, Urrurtia LA, Sewani H, et al. Acute abdomen in the medical intensive care unit. Crit Care Med. 2002;30:1187-1190.
24 Sitges-Serra A, Lopez MJ, Girvent M, et al. Postoperative enterococcal infection after treatment of complicated intraabdominal sepsis. Br J Surg. 2000;89:361-367.
25 Montravers P, Gauzit R, Muller C, et al. Emergence of antibiotic-resistant bacteria in cases of peritonitis after intraabdominal surgery affects the efficacy of empirical antibiotic therapy. Clin Infect Dis. 1996;23:486-494.
26 Men S, Akhan O, Koroglu M. Percutaneous drainage of abdominal abscess. Eur J Radiol. 2002;43:204-218.
27 Rivera-Sanfeliz G. Percutaneous abdominal abscess drainage: a historical perspective. AJR Am J Roentgenol. 2008 Sep;191(3):642-643.
28 Huang CJ, Pitt HA, Lipsett PA, et al. Pyogenic hepatic abscess: Changing trends over 42 years. Ann Surg. 1996;223:600-607.
29 Phillips GS, Radosevich MD, Lipsett PA. Splenic abscess: Another look at an old disease. Arch Surg. 1997;132:1331-1335.
30 Barie PS, Eachempati SR. Acute acalculous cholecystitis. Curr Gastroenterol Rep. 2003;5:302-309.
31 Flancbaum L, Choban PS. Use of morphine cholescintigraphy in the diagnosis of acute cholecystitis in critically ill patients. Intensive Care Med. 1995;21:120-124.
32 Scharff JR, Longo WE, Vartanian SM, et al. Ischemic colitis: Spectrum of disease and outcome. Surgery. 2003;134:624-629.
33 Branicki FJ. Abdominal emergencies: Diagnostic and therapeutic laparoscopy. Surg Infect. 2002;3:269-282.
34 Peris A, Matano S, Manca G, et al. Bedside diagnostic laparoscopy to diagnose intraabdominal pathology in the intensive care unit. Crit Care. 2009;13(1):R25.
35 Boele van Hensbroek P, Wind J, Dijkgraaf MG, Busch OR, Carel Goslings J. Temporary closure of the open abdomen: a systematic review on delayed primary fascial closure in patients with an open abdomen. World J Surg. 2009 Feb;33(2):199-207.
36 Jaber MR, Olafsson S, Fung WL, Reeves ME. Clinical review of the management of fulminant clostridium difficile infection. Am J Gastroenterol. 2008 Dec;103(12):3195-3203.
37 Sailhamer EA, Carson K, Chang Y, et al. Fulminant Clostridium difficile colitis: Patterns of care and predictors of mortality. Arch Surg. 2009;144:433-439.
38 Chan S, Kelly M, Helme S, et al. Outcomes following colectomy for Clostridium difficile colitis. Int J Surg. 2008;7:78-81.
39 Butala P, Divino CM. Surgical aspects of fulminant Clostridium difficile colitis. Am J Surg. 2010 Apr 19.
40 Eachempati SR, Hydo LJ, Barie PS. Severity scoring for prognostication in patients with severe acute pancreatitis: Comparative analysis of the Ranson score and the APACHE III score. Arch Surg. 2002;137:730-736.
41 Bassi C, Pederzoli P, Vesentini S, et al. Behavior of antibiotics during human necrotizing pancreatitis. Antimicrob Agents Chemother. 1994;38:830-836.
42 Maravi-Poma E, Gener J, Alvarez-Lerma F, et al. The Spanish Group for the Study of Septic Complications in Severe Acute Pancreatitis. Early antibiotic treatment (prophylaxis) of septic complications in severe acute necrotizing pancreatitis: A prospective, randomized, multicenter study comparing two regimens with imipenem-cilastatin. Intensive Care Med. 2003;29:1974-1980.
43 Seewald S, Ang TL, Teng KC, Soehendra N. EUS-guided drainage of pancreatic pseudocysts, abscesses and infected necrosis. Dig Endosc. 2009;21(Suppl 1):S61-S65.
44 Babu BI, Siriwardena AK. Current status of minimally invasive necrosectomy for post-inflammatory pancreatic necrosis. HPB (Oxford). 2009;11:96-102.
45 Dutch Pancreatitis Study Groupvan Santvoort HC, Besselink MG, Bakker OJ, et al. A step-up approach or open necrosectomy for necrotizing pancreatitis. N Engl J Med. 2010 Apr 22;362(16):1491-1502.
46 Canivet JL, Yans T, Piret S, Frere P, Beguin Y. Barotrauma-induced tension pneumoperitoneum. Acta Anaesthesiol Belg. 2003;54:233-236.
47 Gayer G, Hertz M, Zissin R. Postoperative pneumoperitoneum: Prevalence, duration, and possible significance. Semin Ultrasound CT MR. 2004;25:286-289.
48 Rozycki GS, Tremblay L, Feliciano DV, et al. Three hundred consecutive emergent celiotomies in general surgery patients: Influence of advanced diagnostic imaging techniques and procedures on diagnosis. Ann Surg. 2002;235:681-688.
49 Velmahos GC, Kamel E, Berne TV, et al. Abdominal computed tomography for the diagnosis of intra-abdominal sepsis in critically injured patients. Arch Surg. 1999;134:831-836.
50 Lohrmann C, Ghanem N, Pache G, Makowiec F, Kotter E, Langer M. CT in acute perforated sigmoid diverticulitis. Eur J Radiol. 2005;56:78-83.
51 Szem JW, Hydo LJ, Fischer E, et al. High-risk intrahospital transport of critically ill patients: Safety and outcome of the necessary “road trip.”. Crit Care Med. 1995;23:1660-1666.
52 Brown JR, Block CA, Malenka DJ, et al. Sodium bicarbonate plus N-acetylcysteine prophylaxis: a meta-analysis. JACC Cardiovasc Interv. 2009 Nov;2(11):1116-1124.
53 Stijn Blot S, De Waele JJ. Critical issues in the clinical management of complicated intra-abdominal infections [review]. Drugs. 2005;65:1611-1620.
54 Dellinger RP, Levy MM, Carlet JM, et al. Surviving Sepsis Campaign: International guidelines for management of severe sepsis and septic shock: 2008. Intensive Care Med. 2008;34:17-60.
55 Qadan M, Dajani D, Dickinson A, Polk HCJr. Meta-analysis of the effect of peritoneal lavage on survival in experimental peritonitis. Br J Surg. 2010 Feb;97(2):151-159.
56 Polk HC, Fry DE. Radical surgical debridement in the treatment for established peritonitis: The results of a prospective randomized clinical trial. Ann Surg. 1980;192:350-355.
57 Edmiston CEJr, Goheen MP, Kornhall S, et al. Fecal peritonitis: Microbial adherence to serosal mesothelium and resistance to peritoneal lavage. World J Surg. 1990;14:176-183.
58 Moussavian MR, Richter S, Kollmar O, Schuld J, Schilling MK. Staged lavage versus single high-volume lavage in the treatment of feculent/purulent peritonitis: a matched pair analysis. Langenbecks Arch Surg. 2009 Mar;394(2):215-220.
59 Sugimoto K, Hirata M, Takishima T, et al. Mechanically assisted intra-operative peritoneal lavage for generalized peritonitis as a result of perforation of the upper part of the gastrointestinal tract. J Am Coll Surg. 1994;179:443-448.
60 Lamme B, Boermeester MA, Belt EJ, et al. Mortality and morbidity of planned relaparotomy versus relaparotomy on demand for secondary peritonitis. Br J Surg. 2004;91:1046-1054.
61 Lamme B, Boermeester MA, Reitsma JB, et al. Meta-analysis of relaparotomy for secondary peritonitis. Br J Surg. 2002;89:1516-1524.
62 Pieracci FM, Barie PS. Intra-abdominal infections. Curr Opin Crit Care. 2007;13:440-449.
63 Weigelt JA. Empiric treatment options in the management of complicated intra-abdominal infections. Cleve Clin J Med. 2007;74:29-37.
64 Barie PS, Christou NV, Dellinger EP, et al. Pathogenicity of the enterococcus in surgical infections. Ann Surg. 1990;212:155-159.
65 Lucasti C, Jasovich A, Umeh O, Jiang J, Kaniga K, Friedland I. Efficacy and tolerability of IV doripenem versus meropenem in adults with complicated intra-abdominal infection: a phase III, prospective, multicenter, randomized, double-blind, noninferiority study. Clin Ther. 2008 May;30(5):868-883.
66 Eagye KJ, Kuti JL, Nicolau DP. Evaluating empiric treatment options for secondary peritonitis using pharmacodynamic profiling. Surg Infect. 2007;8:215-226.
67 Oliva ME, Rekha A, Yellin A, Pasternak J, Campos M, Rose GM, et al301 Study Group. A multicenter trial of the efficacy and safety of tigecycline versus imipenem/cilastatin in patients with complicated intra-abdominal infections. BMC Infect Dis. 2005 Oct 19;5:88.
68 Towfigh S, Pasternak J, Poirier A, Leister H, Babinchak T. A multicentre, open-label, randomised comparative study of tigecycline versus ceftriaxone sodium plus metronidazole for the treatment of hospitalised subjects with complicated intra-abdominal infections. Clin Microbiol Infect. 2009 Nov 20.
69 Malangoni MA, Song J, Herrington J, et al. Randomized controlled trial of moxifloxacin compared with piperacillin-tazobactam and amoxicillin-clavulanate for the treatment of complicated intra-abdominal infections. Ann Surg. 2006;244:204-211.
70 Weiss G, Reimnitz P, Hampel B, Muehlhofer E, Lippert H, AIDA Study Group. Moxifloxacin for the treatment of patients with complicated intra-abdominal infections (the AIDA Study). J Chemother. 2009 Apr;21(2):170-180.
71 Blot SI, Vandewoude KH, De Waele JJ. Candida peritonitis. Curr Opin Crit Care. 2007;13:195-199.
72 Dupont H, Paugam-Burtz C, Muller-Serieys C, et al. Predictive factors of mortality due to polymicrobial peritonitis with Candida isolation in peritoneal fluid in critically ill patients. Arch Surg. 2002;137:1341-1346.
73 Bujik SL, Gyssens IC, Mouton JW, et al. Pharmacokinetics of sequential intravenous and enteral fluconazole in critically ill surgical patients with invasive mycoses and compromised gastrointestinal function. Intensive Care Med. 2000;27:115-121.
74 Dupont H, Bourichon A, Paugam-Burtz C, et al. Can yeast isolation in peritoneal fluid be predicted in intensive care unit patients with peritonitis? Crit Care Med. 2003;31:752-757.
75 Wittman DH, Schein M. Let us shorten antibiotic prophylaxis and therapy in surgery. Am J Surg. 1996;172:26S-32S.
76 De Waele JJ, Ravyts M, Depuydt P, Blot SI, Decruyenaere J, Vogelaers D. De-escalation after empirical meropenem treatment in the intensive care unit: Fiction or reality? J Crit Care. 2010 Jan 13. Epub ahead of print
77 Basoli A, Chirletti P, Cirino E, et al. A prospective, double-blind, multicenter, randomized trial comparing ertapenem 3 vs. 5 days in community-acquired intraabdominal infection. J Gastrointest Surg. 2008;12:592-600.
78 De Marie S, Vanden Bergh MF, Bujik SL, et al. Bioavailability of ciprofloxacin after multiple enteral and intravenous doses in ICU patients with severe gram-negative intraabdominal infections. Intensive Care Med. 1998;24:343-346.
79 Sitges-Serra A, Jaurrieta E, Sitges-Crues A. Management of postoperative enterocutaneous fistulas: The role of parenteral nutrition and surgery. Br J Surg. 1989;69:147-150.
80 Rose D, Yarborough MF, Canizaro PC, Lowry SF. One hundred and fourteen fistulas of the gastrointestinal tract treated with total parenteral nutrition. Surg Gynecol Obstet. 1986;163:345-350.
81 Sites-Serra A, Guirao X, Pereira JA, et al. Treatment of gastrointestinal fistulas with sandostatin. Digestion. 1993;54(suppl 1):38-40.
82 Leandros E, Antonkis PT, Albanopoulos K, et al. Somatostatin versus octreotide in the treatment of patients with gastrointestinal and pancreatic fistulas. Can J Gastroenterol. 2004;18:303-306.
83 Sugerman HJ, Bloomfield GL, Saggi BW. Multisystem organ failure secondary to increased intraabdominal pressure. Infection. 1999;27:61-66.
84 Paugam-Burtz C, Dupont H, Marmuse JP, et al. Daily organ-system failure for diagnosis of persistent intra-abdominal sepsis after postoperative peritonitis. Intensive Care Med. 2002;28:594-598.
85 Fry DE, Pearlstein DE, Fulton RL, Polk HC. Multiple system organ failure: The role of uncontrolled infection. Arch Surg. 1980;115:136-140.
86 CORTICUS Study GroupSprung CL, Annane D, Keh D, et al. Hydrocortisone therapy for patients with septic shock. N Engl J Med. 2008 Jan 10;358(2):111-124.
87 Hill GE, Frawley WH, Griffith KE, et al. Allogeneic blood transfusion increases the risk of postoperative bacterial infection: A meta-analysis. J Trauma. 2003;54:908-914.
88 van den Berghe G, Wouters P, Weekers P, et al. Intensive insulin therapy in critically ill patients. N Engl J Med. 2001;345:1359-1367.
89 Arabi YM, Dabbagh OC, Tamim HM. Intensive versus conventional insulin therapy: a randomized controlled trial in medical and surgical critically ill patients. Crit Care Med. 2008 Dec;36(12):3190-3197.
90 Wiener RS, Wiener DC, Larson RJ. Benefits and risks of tight glucose control in critically ill adults: a meta-analysis. JAMA. 2008 Aug 27;300(8):933-944.
91 NICE-SUGAR Study InvestigatorsFinfer S, Chittock DR, Su SY, et al. Intensive versus conventional glucose control in critically ill patients. N Engl J Med. 2009;360(13):1283-1297.
92 Barie PS, Williams MD, McCollam JS, et alfor the PROWESS Surgical Evaluation committee. Benefit/risk profile of drotrecogin alfa (activated) in surgical patients with severe sepsis. Am J Surg. 2004;188:212-220.
93 Barie PS, Hydo LJ, Shou J, Eachempati SR. Efficacy and safety of drotrecogin alfa (activated) for the therapy of surgical patients with severe sepsis. Surg Infect. 2006;7(Suppl 2):S77-S80.
94 Payen D, Sablotzki A, Barie PS, et al. International integrated database for the evaluation of severe sepsis and drotrecogin alfa (activated) therapy: Analysis of efficacy and safety data in a large surgical cohort. Surgery. 2006;140:726-739.
