[level-membership-for-neurology-category]
11 Peripheral neuropathy
Introduction
Ability to differentiate between upper motor neurone (UMN) and LMN damage is fundamental to understanding PN (see Table 11.1). Cranial nerves represent a special type of peripheral nerve emanating from the brainstem. This raises an ‘old chestnut’, namely facial weakness of either UMN or LMN origin.
TABLE 11.1 Differentiation between upper motor neurone and lower motor neurone damage
| UMN | LMN | |
|---|---|---|
| Inspection | Limited or no loss of muscle bulkNo fasciculations | Localised or generalised muscle wastingFasciculations may be present |
| Tone | Increased muscle tone (clasp knife) | Decreased tone consistent with loss of bulk |
| Power | Decreased power in distribution of UMN (antigravity muscles) | Weakness of muscles innervated by damaged nerves |
| Reflexes | Increased (brisk) reflexes below level of damageUp going or splaying of toes | Decreased or absent reflexes in muscles denervatedToes down going |
| Sensation | Generally not affected | Decreased if sensory nerves also involved |
There are some conditions that may affect both UMN and LMN,1 such as motor neurone disease (often referred to as amyotrophic lateral sclerosis) or subacute combined degeneration, as may occur with vitamin B12 deficiency.2
What follows in this chapter is a discussion of the nature of nerve damage, causes of PN, diagnosis thereof, a look at some focal neuropathies, nutritionally induced neuropathies and some illnesses associated with PN. The aim is to help general practitioners become more involved in patient care and, hence, better enjoy the therapeutic partnerships with the consultant.
The Nature of Nerve Damage
Nerves transmit their messages via an electrical impulse from the nerve cell (the cell of the neurone) down the length of the ‘arm’ of the cell (the axon) to pass the message either to another nerve or the end target organ. The message is sent via the impulse causing release of the neurotransmitters that produce the desired effect, be it to stimulate or suppress a subsequent response, such as causing a muscle to contract or to stimulate another nerve (see Fig 11.1).
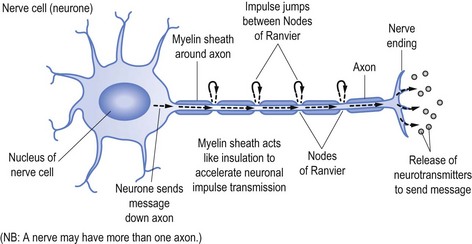
To speed up transmission the axons of the nerves are usually coated with myelin, which acts in a similar fashion to the plastic coating that insulates and allows unimpeded passage of electrical current down an electrical wire (see Fig 11.1). Along the myelinated nerve fibre the impulse ‘jumps’ between Nodes of Ranvier, breaks in the myelin.
In the same way the wire within the plastic transmits the electrical current, so too does the axon send the neuronal impulse. It follows that damage to the axon (analogous to the electrical wire) will reduce the amount of message able to be transmitted. In neurophysiological terms, axonal damage will reduce the amplitude of the impulse and hence the amplitude of the action potential generated,3 namely the size and strength of the message received rather than the speed of the transmission.
Transmission speed (or conduction time) is facilitated by the myelin sheath. It follows that damage to myelin, as occurs in demyelinating illnesses (such as Landry-Guillain-Barré syndrome, often referred to as GBS—omitting the name Landry, or multifocal motor neuropathy) will slow conduction time.4 In the same way damage to the plastic coating of the electrical wire will ultimately allow the wire to corrode and be damaged, so too will established demyelination allow subsequent axonal damage.5
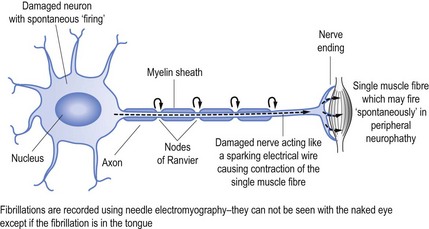
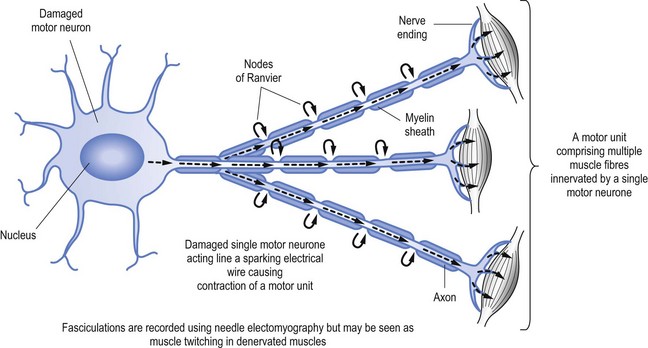
When nerves are damaged it may be akin to a short circuit, to continue the electrical wire analogy. Thus nerves may ‘fire’ spontaneously. This may result in the activation of a single muscle fibre (called fibrillation, see Fig 11.2) or a group of muscle fibres known as a motor unit, which is limited to muscle fibres innervation by a single motor nerve (called fasciculation, see Fig 11.3).
Causes of Nerve Damage
PN can be the result of either local damage or a more generalised process affecting a wider target (see Box 11.1).
Box 11.1 Treatable causes of peripheral neuropathy
Dietary deficiency neuropathies:
Heavy metals, such as lead or mercury poisoning
Neurotoxic drugs, such as nitrofurantoin
Landry-Guillain-Barré syndrome
The most common cause of local damage is consequent to pressure, which causes a ‘neurapraxia’ that damages the nerve at the site of the pressure. An example of this process is damage to the lateral popliteal nerve with resultant foot drop. The lateral popliteal nerve activates the tibialis anterior muscle that dorsiflexes the foot.6 Similarly, direct damage to the radial nerve as it transverses the humeral groove in the humerus bone will result in wrist drop due to deficits of wrist extension.7
Perhaps the best-known local nerve damage is carpal tunnel syndrome (CTS), which affects the median nerve. This causes weakness of the muscles innervated by the nerve plus specific sensory deficit referable to local median nerve damage (see Fig 11.4).
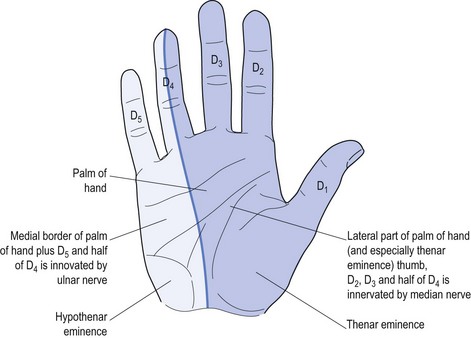
Only four intrinsic hand muscles are innervated by the median nerve.8 The remainder are innervated by the ulnar nerve. There is a mnemonic to remember these four muscles: LOAF (Lateral two lumbricals, Opponens pollicis, Abductor pollicis brevis and Flexor pollicis brevis).
Generalised PN results from a systemic problem that can be the result of a variety of toxins, for example drugs (e.g. vincristine)9 or infections, which may be the precipitant for conditions such as motor neuropathy (as may occur with infectious hepatitis)10. A variety of other environmental toxins, such as lead and other heavy metals,11 can also cause pure motor neuropathy.
Conditions such as Landry-Guillain-Barré syndrome (GBS, also known as idiopathic ascending inflammatory polyradiculopathy)12 and chronic inflammatory demyelinating neuropathy13 (CIDP) represent allergic responses to reputed infective agents causing demyelinating radiculopathy or neuropathy.12,13
Diagnosis of Nerve Damage
a History
Many patients with PN will start with the complaint of numbness, so clarification is important. Having confirmed that both doctor and patient understand the meaning relevant to the particular patient, the next step is to define the exact distribution of the sensory change(s). Specific nerve damage should cause a typical distribution of sensory change (see Fig 11.4) but often patients find it impossible to be specific. Often patients with CTS will report waking at night and state ‘my whole hand went to sleep’. They have trouble differentiating between palm and dorsum of the hand, and most will not recognise the anatomic ‘splint’ affecting the ring finger (see Fig 11.4). Some may report that the whole arm, meaning the whole upper limb, was affected and some may confuse the picture by stating that it started in the shoulder and passed to the hand, or started at the elbow.
Some additional considerations to be included in the history taking include: any identifiable causative factors; knowledge of any pre-existing diagnoses (such as diabetes discussed earlier); relevant family history (such Charcot-Marie-Tooth disease for hereditary neuropathies); exposure to medications; nutritional status (such as vegans not ingesting sufficient vitamin B12); excessive alcohol consumption (with direct alcohol toxicity or dietary deficiencies such as vitamin B1); and a comprehensive systems review (that may reveal conditions such as vasculitis, sarcoidosis or exposure to heavy metals such as lead or mercury, being recognised occupational hazards, or cytoxic or radiologic treatment for neoplasia).
b Examination
As suggested in Ch 5, O’Brien (2010) is an invaluable tool when evaluating the peripheral nervous system. It is highly recommended that anyone interested in the examination of the peripheral nervous system keep a copy handy, as it defines the relevant dermatomes and how to test specific radicular and nerve innervated muscles. Some examples will be provided in the discussion of classical presentations but this will not replace the value of this reference.
The ‘glove and stocking’ distribution of generalised PN usually starts in the distal end of the longest nerves, thus starting in the feet before involving more proximal areas such as the calf or thigh. It is easiest to start testing from the area of reduced sensation and move towards the region of increased perception. The patient finds it easier to identify when pain is first felt, rather than when it is diminished. The examiner should start testing at the periphery, be it the toes or fingers and move centrally towards the torso. If an abnormality is detected the testing should be repeated, ensuring that the patient is not watching the stimuli being applied. It is important to note if there is discrepancy between the two tests. If there is it should be repeated, marking the point at which change occurred using a pen and mapping the difference with repeated tests. If the distribution of the point of change is too great it provides ‘concrete’ evidence of non-organic disease (see Ch 5).
Much of what is included within this section on examination should be combined with Chapter 4 on the peripheral nervous system for a more comprehensive overview.
c Investigations
An understanding of the treatable causes of PN (see Box 11.1) will assist with appropriate tests to cast diagnostic light in some cases. These may obviate the need to refer the patient to a consultant. Most of the possible treatable causes can be explored by the general practitioner who can also prescribe appropriate treatment.
Focal Neuropathies
The most common focal PN is CTS,8 which reflects damage to the median nerve due to entrapment at the level of the carpal tunnel at the wrist. There should be an appropriate history (with dysaesthesia of the hand causing waking from sleep that often responds to shaking to return sensation). There should also be weakness of the median nerve innervated muscles (remember the mnemonic LOAF, as above), especially with weakness of abductor pollicis brevis and lateral two lumbricals (see Figs 11.5 and 11.6). There may be a sensory deficit (see Fig 11.4), which may not be as anatomically distinct as shown but, as a minimum, sensation in the thenar eminence should be less than that over the hypothenar eminence.
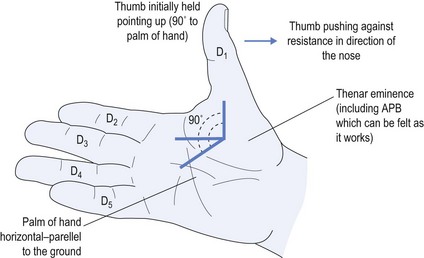
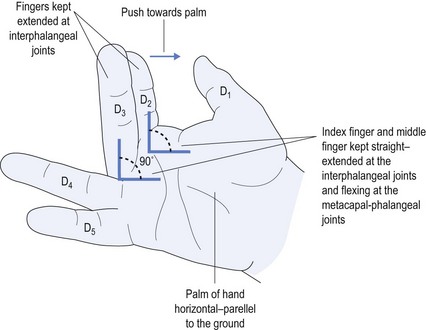
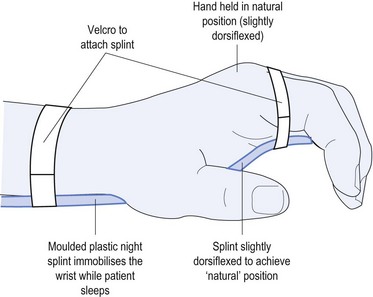
Causes of CTS include: using vibrating equipment, such as jack hammers; repeated pressure from using tools, such as a screwdriver with the palm pushing down on it; but most commonly from sleeping in such a position as to cause marked palmar flexion of the wrist thereby impeding the blood supply to the nerve. Before contemplating either neurophysiology or surgical referral, the most common effective treatment is to organise a physiotherapist to provide a moulded night splint made specifically to meet the anatomy of the patient (see Fig 11.7).
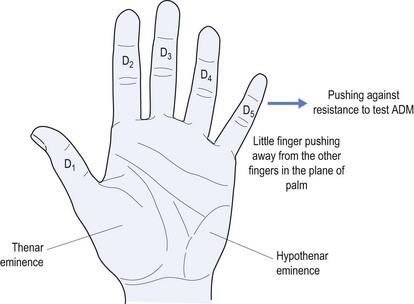
The next most common PN is an ulnar neuropathy14 at the elbow. This causes dysaesthesia along the ulnar border of the forearm, little finger (D5) and medial half of the ring finger (D4) (see Fig 11.4). It causes weakness of the ulnar-innervated muscles of the hand, typified by abductor digiti minimi that forces the little finger away from the rest of the fingers and away from the palm (see Fig 11.8). It also causes weakness of the medial two lumbricals (see Fig 11.9). Long-standing ulnar neuropathy may cause wasting of the first dorsal interosseus muscle best seen in the dorsum of the hand, the muscle between the thumb and index finger (D1 and D2).
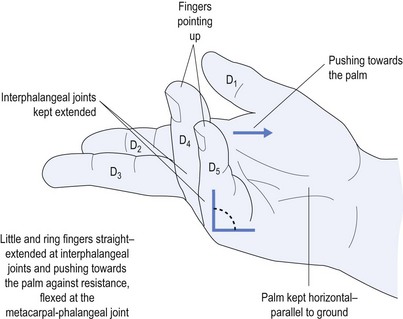
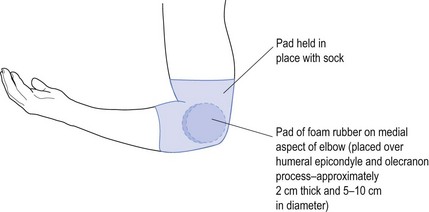
The main cause of ulnar neuropathy is direct trauma to the nerve as might occur when turning over in bed at night. This results in the whole body’s weight being supported by the elbow and hence the ulnar nerve as it crosses the elbow at the ulnar groove next to the humeral epicondyle. The way to treat this is to pad the elbow over the region. This includes the medial humeral epicondyle and olecranon of the ulnar bone. The easiest way to do this is to get a piece of foam rubber, approximately 2 cm thick and about 5–10 cm in diameter. This is placed in position and held either with a bandage or more simply using a large sports sock with the toe cut out so that it can be slipped up the forearm and over the elbow (see Fig 11.10). Such padding prevents repeated pressure upon the nerve that causes nerve damage (neuropraxia) by squashing the nerve against the hard bony surfaces while rolling over. The splint only needs to be worn at night while sleeping.
Nutritionally Induced Neuropathies
Various vitamin deficiencies are known to cause PN, such as B-group vitamins, possibly in association with alcohol abuse.15 The general practitioner should prescribe thiamine (B1) to all patients with suspected alcohol dependence. A proper history, including a dietary history, should be an integral part of the diagnostic process.
The onset of PN can be slow and subtle but damage can be profound, as in the case with B12 deficiency that can cause CNS and PN damage.16 Parenteral B12 injections may both arrest and allow some restorative benefits. Measurement of B12 and folate must be part of the patient assessment when considering PN. Folic acid is an essential component of B12 metabolism, but is often overlooked. Replacement with 5 mg folate per day is simple and effective.
Other nutritional conditions include pellagra,17 with its triad of gastrointestinal symptoms, dementia and PN. It is caused by nicotinic acid (niacin) deficiency. Some drugs, such as penicillamine or isoniazid, may cause B6 (pyridoxine) deficiency. B12 and B6 deficiency may cause elevation of homocysteine, which is implicated in stroke evolution due to hyperviscosity. Nutritional deficiencies may reflect malabsorption rather than poor dietary intake. Without a high index of suspicion, such causes may be overlooked.
Illness Associated with Peripheral Neuropathies
The most common diagnosis associated with diffuse PN is diabetes. This should be a fundamental consideration when diagnosing PN.18 Blood sugar level and glycosylated haemoglobin (HbA1c) should be ordered early in the evaluative process. Where there are haemoglobinopathies, such as thalassemia, fructosamine measurement may be preferable to HbA1c. Initial control of diet may be all that is required, but it has been personal preference to involve an endocrinologist early in the therapeutic process regarding diabetes and to advocate early introduction of insulin once PN has been confirmed. The variety of neuropathic complications with diabetes (such as painful diabetic amyotrophy) may necessitate a wider team approach and even use of immuno-modulating agents, such as IV immunoglobulin, but this is outside the scope of this review.
Neoplasia can cause PN via several routes, be it direct nerve damage (due to a space-occupying or pressure effect), as a consequence of cachexia or as a paraneoplastic expression, which can cause both small-fibre or large-fibre involvement. If in doubt, the general practitioner should look for a primary source using chest imaging and possibly chest and abdominal CT. Anti-neuronal antibody screening, such as anti-Hu together with other tumour markers—alpha-feta protein (AFP) or carcino-embryonic antigen (CEA)—may assist. Agents used to treat neoplasia (such as vincristine or thalidomide) may cause PN. Other medications may be associated with PN, such as statins—now used for more than just hyperlipidaemia—in stroke prevention for vessel-wall stabilisation and even for conditions such as dementia or multiple sclerosis.19
The cardiac medication amiodarone, the urinary antibiotic nitrofurantoin or the antiepileptic medication phenytoin (via its anti-folate effects) may all cause PN. Unless the general practitioner is vigilant, such iatrogenic causes may be overlooked. Medications may serve as a trigger to evoke conditions known to cause PN. One such condition is porphyria, with a pure motor neuropathy that may be provoked by use of barbiturates. Being a rare condition it is easily overlooked, but a detailed medication history may provoke its consideration.
Conclusion
Often general practitioners exclude themselves from the management of PN, but the above demonstrates what a valuable role they can play. As with all neurology, a good history is indispensable as is a clear understanding of what to look for and how to differentiate LMN from UMN lesions (see Table 11.1). The general practitioner may be the person who diagnoses the treatable PNs, having a high index of suspicion and having ordered the appropriate tests (see Box 11.1).
The most common PNs presenting to the general practitioner are CTS and ulnar neuropathy, both of which respond well to conservative treatment that might completely do away with the need for referral to a specialist (Figs 11.4–11.10). Only in those few cases that fail to respond would there be a need to seek neurophysiological diagnostic confirmation and consultant involvement. Where doubt exists as to whether the patient has CTS or ulnar neuropathy or a more widespread PN, early referral to a consultant is advisable.
1 Verma A, Bradley WG. Atypical motor neuron disease and related motor syndromes. Semin Neurol. 2001;21:177-187.
2 Reynolds E. Vitamin B12, folic acid, and the nervous system. Lancet Neurol. 2006;5:949-960.
3 Botteri M, Guarneri B. Electrophysiological tests in intensive care. Eur J Anaesthesiol suppl. 2008;42:174-180.
4 Vucic S, Black K, Chong PS, Cros D. Multifocal motor neuropathy with conduction block: distribution of demyelination and axonal degeneration. Clin Neurophysiol. 2007;118:124-130.
5 Mancardi G, Hart B, Roccatagliata L. Demyelination and axonal damage in a non-human primate model of multiple sclerosis. J Neurol Sci. 2001;184(1):41-49.
6 Stewart JD. Foot drop: where, why and what to do? Pract Neurol. 2008;8:158-169.
7 Lee YK, Kim YI, Choy WS. Radial nerve compression between the brachialis and brachioradialis muscles in a manual worker: a case report. J Hand Surg. 2006;31:744-746.
8 Bland JD. Carpal tunnel syndrome. British Medical J. 2007;335:343-346.
9 Hildebrand J. Neurological complications of medical anti-cancer therapies. Oncology Reviews. 2008;2:80-85.
10 de Freitas MR. Infectious neuropathy. Curr Opin Neurol. 2007;20:548-552.
11 Thomson RM, Parry GJ. Neuropathies associated with excessive exposure to lead. Muscle Nerve. 2006;6:732-741.
12 Winer J. Guillain-Barré syndrome. British Medical J. 2008;337:227-231.
13 Köller H, Kieseier BC, Jander S, Hartung HP. Chronic inflammatory demyelinating polyneuropathy. New England J Medicine. 2005;352:1343-1356.
14 Robertson C, Saratsiotis J. A review of compressive ulnar neuropathy at the elbow. J Manipulative Physiol Ther. 2005;28:345.
15 Koike H, Sobue G. Alcoholic neuropathy. Curr Opin Neurol. 2006;19:481-486.
16 Reynolds E. Vitamin B12, folic acid, and nervous system. Lancet Neurol. 2006;5:949-960.
17 Kumar N. Nutritional neuropathies. Neurol Clin. 2007;25:209-255.
18 Boulton AJ. Management of diabetic peripheral neuropathy. Clin Diab. 2005;23:9-19.
19 Chong PH, Boskovich A, Stevkovic N, Bartt RE. Statin-associated peripheral neuropathy: review of the literature. Pharmacotherapy. 2004;24:1194-1203.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
11 Peripheral neuropathy
Introduction
Ability to differentiate between upper motor neurone (UMN) and LMN damage is fundamental to understanding PN (see Table 11.1). Cranial nerves represent a special type of peripheral nerve emanating from the brainstem. This raises an ‘old chestnut’, namely facial weakness of either UMN or LMN origin.
TABLE 11.1 Differentiation between upper motor neurone and lower motor neurone damage
| UMN | LMN | |
|---|---|---|
| Inspection | Limited or no loss of muscle bulkNo fasciculations | Localised or generalised muscle wastingFasciculations may be present |
| Tone | Increased muscle tone (clasp knife) | Decreased tone consistent with loss of bulk |
| Power | Decreased power in distribution of UMN (antigravity muscles) | Weakness of muscles innervated by damaged nerves |
| Reflexes | Increased (brisk) reflexes below level of damageUp going or splaying of toes | Decreased or absent reflexes in muscles denervatedToes down going |
| Sensation | Generally not affected | Decreased if sensory nerves also involved |
There are some conditions that may affect both UMN and LMN,1 such as motor neurone disease (often referred to as amyotrophic lateral sclerosis) or subacute combined degeneration, as may occur with vitamin B12 deficiency.2
What follows in this chapter is a discussion of the nature of nerve damage, causes of PN, diagnosis thereof, a look at some focal neuropathies, nutritionally induced neuropathies and some illnesses associated with PN. The aim is to help general practitioners become more involved in patient care and, hence, better enjoy the therapeutic partnerships with the consultant.
The Nature of Nerve Damage
Nerves transmit their messages via an electrical impulse from the nerve cell (the cell of the neurone) down the length of the ‘arm’ of the cell (the axon) to pass the message either to another nerve or the end target organ. The message is sent via the impulse causing release of the neurotransmitters that produce the desired effect, be it to stimulate or suppress a subsequent response, such as causing a muscle to contract or to stimulate another nerve (see Fig 11.1).
To speed up transmission the axons of the nerves are usually coated with myelin, which acts in a similar fashion to the plastic coating that insulates and allows unimpeded passage of electrical current down an electrical wire (see Fig 11.1). Along the myelinated nerve fibre the impulse ‘jumps’ between Nodes of Ranvier, breaks in the myelin.
In the same way the wire within the plastic transmits the electrical current, so too does the axon send the neuronal impulse. It follows that damage to the axon (analogous to the electrical wire) will reduce the amount of message able to be transmitted. In neurophysiological terms, axonal damage will reduce the amplitude of the impulse and hence the amplitude of the action potential generated,3 namely the size and strength of the message received rather than the speed of the transmission.
Transmission speed (or conduction time) is facilitated by the myelin sheath. It follows that damage to myelin, as occurs in demyelinating illnesses (such as Landry-Guillain-Barré syndrome, often referred to as GBS—omitting the name Landry, or multifocal motor neuropathy) will slow conduction time.4 In the same way damage to the plastic coating of the electrical wire will ultimately allow the wire to corrode and be damaged, so too will established demyelination allow subsequent axonal damage.5
When nerves are damaged it may be akin to a short circuit, to continue the electrical wire analogy. Thus nerves may ‘fire’ spontaneously. This may result in the activation of a single muscle fibre (called fibrillation, see Fig 11.2) or a group of muscle fibres known as a motor unit, which is limited to muscle fibres innervation by a single motor nerve (called fasciculation, see Fig 11.3).
Causes of Nerve Damage
PN can be the result of either local damage or a more generalised process affecting a wider target (see Box 11.1).
Box 11.1 Treatable causes of peripheral neuropathy
Dietary deficiency neuropathies:
Heavy metals, such as lead or mercury poisoning
Neurotoxic drugs, such as nitrofurantoin
Landry-Guillain-Barré syndrome
The most common cause of local damage is consequent to pressure, which causes a ‘neurapraxia’ that damages the nerve at the site of the pressure. An example of this process is damage to the lateral popliteal nerve with resultant foot drop. The lateral popliteal nerve activates the tibialis anterior muscle that dorsiflexes the foot.6 Similarly, direct damage to the radial nerve as it transverses the humeral groove in the humerus bone will result in wrist drop due to deficits of wrist extension.7
Perhaps the best-known local nerve damage is carpal tunnel syndrome (CTS), which affects the median nerve. This causes weakness of the muscles innervated by the nerve plus specific sensory deficit referable to local median nerve damage (see Fig 11.4).
Only four intrinsic hand muscles are innervated by the median nerve.8 The remainder are innervated by the ulnar nerve. There is a mnemonic to remember these four muscles: LOAF (Lateral two lumbricals, Opponens pollicis, Abductor pollicis brevis and Flexor pollicis brevis).
Generalised PN results from a systemic problem that can be the result of a variety of toxins, for example drugs (e.g. vincristine)9 or infections, which may be the precipitant for conditions such as motor neuropathy (as may occur with infectious hepatitis)10. A variety of other environmental toxins, such as lead and other heavy metals,11 can also cause pure motor neuropathy.
Conditions such as Landry-Guillain-Barré syndrome (GBS, also known as idiopathic ascending inflammatory polyradiculopathy)12 and chronic inflammatory demyelinating neuropathy13 (CIDP) represent allergic responses to reputed infective agents causing demyelinating radiculopathy or neuropathy.12,13
