[level-membership-for-neurology-category]
Chapter 5 Peripheral Nerve Disorders
Anatomy
The spinal cord’s anterior horn cells form the motor neurons of the peripheral nerves – the PNS’ starting point. The peripheral nerves are the final link in the neuron chain that transmits motor commands from the brain through the spinal cord to muscles (Fig. 5-1). Nerve roots emerging from the anterior spinal cord mingle within the brachial or lumbosacral plexus to form the major peripheral nerves, such as the radial and femoral. Although peripheral nerves are quite long, especially in the legs, they faithfully conduct electrochemical impulses over considerable distances. Because myelin, the lipid-based sheath generated by Schwann cells, surrounds peripheral nerves and acts as insulation, the impulses are preserved.
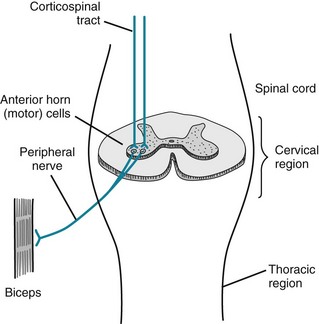
FIGURE 5-1 The corticospinal tracts, as discussed in Chapter 2, and as their name indicates, consist of upper motor neurons (UMNs) that travel from the motor cortex to the spinal cord. They synapse on the spinal cord’s anterior horn cells, which give rise to the lower motor neurons (LMNs). The LMNs join sensory fibers to form peripheral nerves.
When stimulated, motor nerves release packets of acetylcholine (ACh) from storage vesicles at the neuromuscular junction. The ACh packets traverse the junction and bind on to specific ACh receptors on the muscle end plate. The interaction between ACh and its receptors depolarizes the muscle membrane and initiates a muscle contraction (see Chapter 6). Neuromuscular transmission culminating in muscle depolarization is a discrete, quantitative action: ACh does not merely seep out of the presynaptic terminal as loose molecules and drift across the neuromuscular junction to trigger a muscle contraction.
Mononeuropathies
Disorders of single peripheral nerves, mononeuropathies, are characterized by flaccid paresis, DTR loss (areflexia), and reduced sensation, particularly for pain (hypalgesia [Greek, hypo, under + algos, pain] or analgesia [Greek, insensitivity to pain]) (Table 5-1). Paradoxically, mononeuropathies and other peripheral nerve injuries sometimes lead to spontaneously occurring sensations, paresthesias (Greek para, near + aisthesis, sensation) that may be painful, dysesthesias. Peripheral nerve injuries also convert stimuli that ordinarily do not cause pain, such as a light touch or cool air, into painful sensations, allodynia; exaggerate painful responses to mildly noxious stimuli, such as the point of a pin, hyperalgesia; or delay but then exaggerate and prolong pain from noxious stimuli, hyperpathia.
TABLE 5-1 Major Mononeuropathies
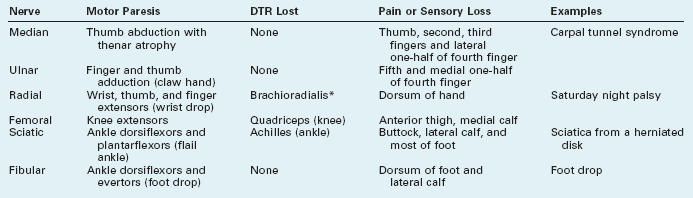
*Compression of the radial nerve in the spiral groove of the humerus spares the triceps deep tendon reflex (DTR).
Compression, especially of nerves protected only by overlying skin and subcutaneous tissue rather than by bone, viscera, or thick layers of fat, occurs frequently. People most susceptible are diabetics; those who have rapidly lost weight, thereby depleting nerves’ protective myelin covering; workers in certain occupations, such as watchmakers; and those who have remained in disjointed positions for long periods, often because of drug or alcohol abuse. One of the most common compressive mononeuropathies – “Saturday night palsy” – affects the radial nerve, which is vulnerable at the point where it winds around in the spiral groove of the humerus. Thus, people in alcohol-induced stupor who lean against their upper arm for several hours are apt to develop a wrist drop (Fig. 5-2). Foot drop, its lower-extremity counterpart, often results from common fibular nerve* damage from prolonged leg crossing compressing the nerve, lower-knee injuries traumatizing the nerve, or a constrictive lower-leg cast pushing against the nerve as it winds around the head of the fibula.
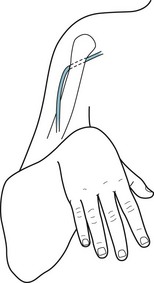
FIGURE 5-2 As the radial nerve winds around the humerus, it is vulnerable to compression and other forms of trauma. Radial nerve damage leads to the readily recognizable wrist drop that results from paresis of the extensor muscles of the wrist, finger, and thumb.
Carpal tunnel syndrome, the most common mononeuropathy, results from damage of the median nerve as it travels through the carpal tunnel of the wrist (Fig. 5-3, left). Forceful and repetitive wrist movements can traumatize the nerve in that confined passage. Meat and fish processing, certain assembly-line work, and carpentry are all closely associated with carpal tunnel syndrome; however, contrary to initial claims, word processing and other keyboarding actually have a weak association with the disorder. In another mechanism, fluid retention during pregnancy or menses entraps the median nerve in the carpal tunnel. Similarly, inflammatory tissue changes in the wrist from rheumatoid arthritis may compress the median nerve.
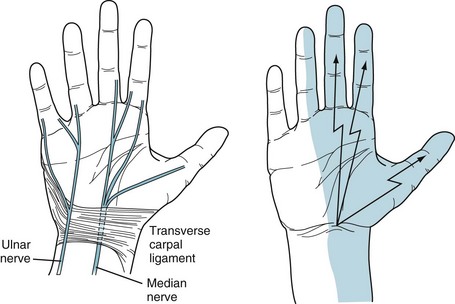
FIGURE 5-3 Left, The median nerve passes through the carpal tunnel, which is a relatively tight compartment. In it, the median nerve is vulnerable to repetitive movement and compression from fluid accumulation. The ulnar nerve, taking a different route, passes above and medial to the roof of the tunnel, the transverse carpal ligament. It thus escapes damage from most repetitive movements and fluid accumulation. Right, The usual sensory distribution of the median nerve encompasses the palm, thenar eminence (thumb base), thumb, and adjacent two fingers. In carpal tunnel syndrome, pain shoots distally from the wrist over this area. Physicians may elicit the Tinel sign, a reliable indication of carpal tunnel syndrome, by tapping the patient’s palmar wrist surface and finding that paresthesias emanate from the wrist and radiate in the median nerve distribution.
Whatever the mechanism, carpal tunnel syndrome causes paresthesias and pains that shoot from the wrist to the palm, thumb, and adjacent two or sometimes three fingers (Fig. 5-3, right). Symptoms worsen at night and awaken the victims, who shake their hands in an attempt to find relief. Neurologists test for the syndrome’s characteristic Tinel sign by percussing the wrist: The test is positive when the percussion generates electric sensations that shoot from the wrist into the palm and fingers.
In another example of upper-extremity nerve damage, pressure on the ulnar groove of the elbow (the “funny bone”) or cubital tunnel may damage the ulnar nerve. For instance, when individuals rest the weight of their arms on their elbows, the compression often injures the ulnar nerve. These individuals develop atrophy and weakness of their hand muscles (Fig. 5-4). The ulnar nerve damage also leads to loss of sensation of the fourth and fifth fingers and the medial surface of the hand.
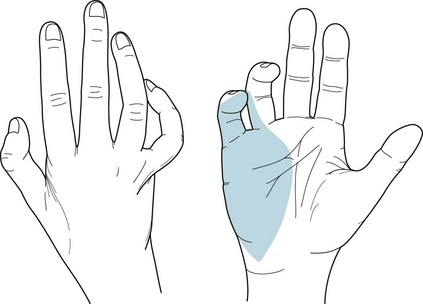
FIGURE 5-4 Left, With ulnar nerve injuries, the palmar view shows that intrinsic muscles of the hand, particularly those of the hypothenar eminence (fifth finger base), undergo atrophy. The fourth and fifth fingers are flexed and abducted. When raised, the hand and fingers assume the benediction sign. In addition, the medial two fingers and palm are anesthetic (numb). Right, Ulnar nerve injuries also produce a claw hand because of atrophy of the muscles between the thumb and adjacent finger (first dorsal interosseous and adductor pollicis), as well as of those of the hypothenar eminence.
Polyneuropathies (Neuropathies)
Alternatively, neurologists divide neuropathies into sensory, motor, or mixed sensorimotor neuropathy. Patients with sensory neuropathy usually suffer predominantly or exclusively from numbness, paresthesias, or burning in their fingers and toes (i.e., stocking-glove hypalgesia: Fig. 5-5). The pain may reach intolerable proportions (see neuropathic pain, Chapter 14). When sensory neuropathy affects the feet, it may provoke leg movements, such as restless legs syndrome (see Chapter 17). Patients with motor neuropathy usually have distal limb weakness that impairs fine, skilled hand and finger movements, such as buttoning a shirt, or raising their feet when they walk, which causes a foot drop. Their neuropathy in chronic cases usually also leads to muscle atrophy and flaccidity. As with other LMN injuries, it diminishes DTRs (see Fig. 2-2C), first at the brachioradialis and Achilles’ and then at more proximal sites. Mixed sensorimotor neuropathy causes mixtures of those symptoms and signs.
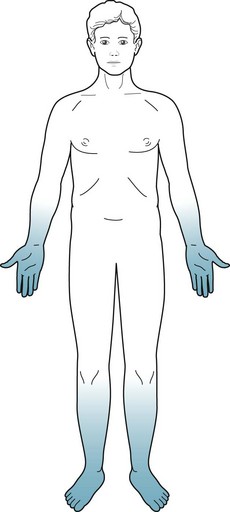
FIGURE 5-5 Patients with polyneuropathy lose pain and other sensations. The loss is symmetric, more severe distally than proximally, and more severe in the legs than arms. Neurologists term this pattern of sensory loss stocking-glove hypalgesia.
Neurologists who care for psychiatric patients may meaningfully divide neuropathies into those with and those without comorbid changes in mental status (Box 5-1).
Box 5-1
Important Causes of Neuropathy
Neuropathies Without Comorbid Mental Status Changes
Guillain–Barré Syndrome
When first affected, young and middle-aged adults feel paresthesias and numbness in the fingers and toes. Then they develop flaccid paresis of their feet and legs with characteristically absent knee and ankle DTRs. Weakness and areflexia, which soon become a much greater problem than numbness, ascend to involve the hands and arms. Many patients progress to respiratory insufficiency because of involvement of the phrenic and intercostal nerves, and require intubation for ventilation. If weakness ascends still further, patients develop cranial nerve involvement that may lead to dysphagia and other aspects of bulbar palsy (see Chapter 4). Additional involvement causes facial weakness and then sometimes even ocular immobility. Nevertheless, possibly because optic and acoustic nerves are protected by myelin generated by the CNS – not the PNS – patients continue to see and hear.
Even if the illness worsens to the point of total paralysis, patients usually remain conscious with a normal mental status – allowing for anxiety and depressive symptoms from enduring a life-threatening illness. Completely immobile and anarthric Guillain–Barré syndrome patients, typically with preserved consciousness and mental status, exist in a locked-in syndrome (see Chapter 11). Cerebrospinal fluid (CSF) exhibits an elevated protein concentration but with few white cells (i.e., the classic albumino-cytologic dissociation) (see Table 20-1).
The illness usually resolves almost completely within 3 months as the PNS myelin is regenerated. By way of treatment, plasmapheresis (plasma exchange), which extracts circulating inflammatory mediators, particularly autoantibodies, complement, and cytokines, reduces the severity and duration of the paresis. Alternatively, administration of intravenous human immunoglobulin, which “blocks” the antibodies at the neuromuscular junction, also restores patients’ strength. Steroids will not help, which is surprising because they are helpful in other inflammatory diseases of the nervous system, such as myasthenia gravis (see Chapter 6), multiple sclerosis (MS: see Chapter 15), and the chronic form of Guillain–Barré syndrome (chronic inflammatory demyelinating polyneuropathy).
Not only is Guillain–Barré syndrome a life-threatening illness, but it also epitomizes the distinction between PNS and CNS diseases. Although paraparesis or quadriparesis might be a feature common to PNS and CNS illnesses, different patterns of muscle weakness, changes in reflexes, and sensory distribution characterize PNS and CNS illnesses (Table 5-2). Also, in Guillain–Barré syndrome, as in most neuropathies other than diabetic neuropathy (see later), bladder, bowel, and sexual functions are preserved. In contrast, patients with spinal cord disease usually have incontinence and impotence at the onset of the injury.
TABLE 5-2 Differences between Central (CNS) and Peripheral Nervous System (PNS) Signs
| CNS | PNS | |
|---|---|---|
| Motor System | Upper motor neuron | Lower motor neuron |
| Paresis | Patterns* | Distal |
| Tone | Spastic† | Flaccid |
| Bulk | Normal | Atrophic |
| Fasciculations | No | Sometimes |
| Reflexes | ||
| Deep tendon reflexes | Hyperactive‡ | Hypoactive |
| Plantar | Babinski sign(s) | Absent |
| Sensory loss | Patterns* | Hands and feet |
*Examples: motor and sensory loss of one side or lower half of the body (e.g., hemiparesis or paraparesis), and hemisensory loss.
MS appears to be a partial exception to the rule that CNS demyelinating damage is permanent. In MS, episodes of demyelination of several CNS areas, including the optic nerves, partially or even completely resolve (see Chapter 15). However, the improvement results from resolution of myelin inflammation rather than myelin regeneration. When MS-induced demyelination eventually encompasses large areas of cerebral CNS myelin, it results in permanent quadriparesis and dementia.
Diabetes
Patients with diabetic neuropathy may also have autonomic nervous system involvement that causes gastrointestinal immobility, bladder muscle contraction, and sexual dysfunction. In fact, erectile dysfunction is occasionally the first or most disturbing symptom of diabetic autonomic neuropathy (see Chapter 16).
Toxic-Metabolic Disorders
Chronic, low-level intoxication by several other heavy metals causes polyneuropathy, dermatologic abnormality, and mental changes. In contrast, acute heavy metal poisoning typically leads to fatal gastrointestinal symptoms and cardiovascular collapse. Arsenic, which is tasteless and odorless, is a poison used in popular murder cases. With chronic, low-level, deliberate, accidental, or industrial intoxication, arsenic causes anorexia, malaise, and a distal neuropathy that might mimic Guillain–Barré syndrome. It also causes several characteristic dermatologic abnormalities: Mees lines on the fingernails (Fig. 5-6), hyperpigmentation, and hyperkeratosis.
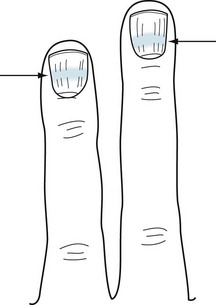
FIGURE 5-6 Mees lines, white bands (arrows) that stretch across the fingernails, characteristically indicate arsenic poisoning. In addition, poisoning by other heavy metals and trauma can cause them.
Mercury intoxication is more complex than arsenic poisoning. Individuals with mercury poisoning may develop a neuropathy and various CNS deficits, including cognitive impairment, ataxia, dysarthria, and visual field changes. Their gums accumulate a telltale dark line just below their teeth (Fig. 5-7).
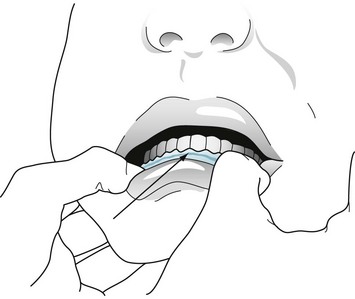
FIGURE 5-7 Chronic mercury poisoning causes a dark blue or black line (arrow) along the gum. Individuals with this sign usually also have neuropathy and central nervous system signs, such as ataxia and dysarthria.
Investigators at one time proposed that mercury-based dental amalgams (“fillings”) caused Alzheimer disease and other neurodegenerative illnesses either by dissolving in saliva and allowing mercury to enter the circulation or emitting a mercury vapor those individuals inhaled. In another inquiry, because ethyl mercury was a major component of the common vaccine preservative, thimerosal, investigators suspected that routine childhood immunizations caused autism (see Chapter 13). However, statistically powerful epidemiologic studies disproved both of those suspicions. In the case of vaccinations, the original “investigators” possibly engaged in fraud.
Neuropathies With Comorbid Mental Status Changes
Although most neuropathies, as described in the previous section, may be painful, incapacitating, or even devastating to the PNS, they generally do not cause mental changes in adults. For example, most people who are old, diabetic, on hemodialysis, or receiving chemotherapy remain intelligent, thoughtful, and competent even though beset by pain, sensory loss, and weakness. In contrast, only a few diseases (see Box 5-1) cause the combination of dementia and neuropathy, which would indicate both cerebral cortex and peripheral nerve damage. An analogous combination would be dementia and movement disorders, which would indicate cerebral cortex and basal ganglia damage (see Box 18-4).
Nutritional Deficiencies
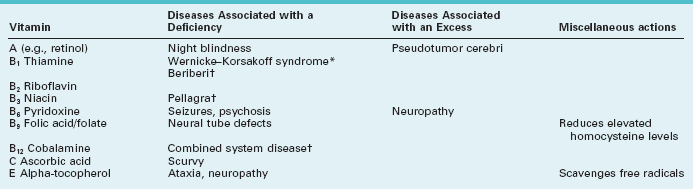
Deficiencies of thiamine (vitamin B1), niacin (nicotinic acid, B3), or vitamin B12 (cobalamine), each produce a predominantly sensory neuropathy accompanied by dementia or other mental status abnormality (Table 5-3). From a worldwide perspective, starvation has been the most common cause of deficiencies of vitamins, their carrier fats, minerals, and other nutrients. For example, beriberi was the starvation-induced neuropathy attributable to thiamine deficiency endemic in eastern Asia. In the United States, alcoholism, bariatric surgery, and malabsorption syndromes have replaced starvation as the most common causes of nutritional neuropathies. Curiously, few patients with anorexia nervosa or self-imposed extreme diets develop a neuropathy. Their protection may lie in a selective, possibly secret, intake of food or vitamins.
Bariatric surgery remains a unique example. After its rapid introduction and widespread acceptance, postoperative “micronutrient” deficiencies caused various neurologic illnesses. Thiamine, copper, and vitamins B12 and E deficiencies frequently caused neuropathy, but also occasionally encephalopathy or myelopathy (see Chapter 4), i.e., CNS problems. Routine postoperative administration of these micronutrients has prevented the problem.
Whatever the cause, thiamine deficiency generally leads to absent DTRs and loss of position sensation. In fact, until patients walk in the dark, when they must rely on position sense generated in the legs and feet, their deficits may remain asymptomatic. In the well-known Wernicke–Korsakoff syndrome, amnesia, dementia, and cerebellar degeneration accompany the neuropathy associated with alcoholism (see Chapter 7).
Among its many functions, vitamin B12 sustains both CNS and PNS myelin. Thus, B12 deficiency leads to the combination of CNS and PNS damage – combined system disease. Although its manifestations include a neuropathy, cognitive impairment and sensory loss reflecting demyelination of the posterior columns of the spinal cord (see Fig. 2-19B) predominate. Patients also develop a characteristic megaloblastic anemia. Most importantly, neurologists refer to combined system disease as a “correctable cause of dementia” because B12 injections can reverse the cognitive impairment as well as the other CNS and PNS manifestations. The usual causes of B12 deficiency include pernicious anemia, malabsorption, a pure vegetarian diet, or prolonged exposure to nitrous oxide, a gaseous dental anesthetic. (Nitrous oxide inactivates B12 by oxidizing its cobalt.)
The screening test for B12 deficiency consists of determining the serum B12 level. In equivocal cases, especially when cognitive impairment or spinal cord abnormalities are not accompanied by anemia, determining the serum homocysteine and methylmalonic acid levels can corroborate the diagnosis: in B12 deficiency, both homocysteine and methylmalonic acid levels rise to abnormally high levels (Fig. 5-8). Intrinsic factor antibodies, a classic finding in pernicious anemia, will be detectable in only about 60% of cases. The standard confirmatory test is the Schilling test.
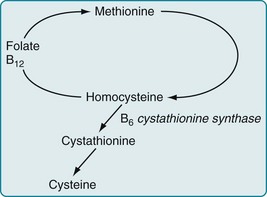
FIGURE 5-8 Vitamin B12, acting as a coenzyme, along with folate, facilitates the conversion of homocysteine to methionine. Other enzymes complete the cycle by converting methionine back to homocysteine. An absence of B12 leads to the accumulation of both methionine and homocysteine. Whatever the cause, an elevated homocysteine level is a risk factor for neural tube defects, cerebrovascular and cardiovascular disease, and other neurologic conditions.
In contrast to malnutrition causing neuropathy, excessive intake of certain vitamins causes neurologic problems. For example, although the normal adult daily requirement of vitamin B6 (pyridoxine) is only 2–4 mg daily, several food faddists who consumed several grams daily as part of a special diet developed a profound sensory neuropathy. Similarly, high vitamin A intake may cause pseudotumor cerebri (see Chapter 9) or induce fetal abnormalities (see Chapter 13).
Infectious Diseases
Several common organisms have a predilection for infecting the peripheral nerves and sparing the CNS. For example, herpes zoster infects a single nerve root or a branch of the trigeminal nerve, usually in people older than 65 years or those with an impaired immune system. An infection with herpes (Greek, herpes, spreading skin eruption) causes an ugly, red, painful, vesicular eruption (“shingles”) that may remain excruciating long after the skin infection has resolved (see postherpetic neuralgia, Chapter 14). As another example, leprosy (Hansen disease), infection with Mycobacterium leprae, causes anesthetic, hypopigmented patches of skin, anesthetic fingers and toes, and palpable nerves. It particularly affects the cool portions of the body, such as the nose, earlobes, and digits; however, depending on its variety, the infection strikes the ulnar or another large nerve either singly or along with others.
Acute Lyme disease typically produces multiple problems, such as arthritis, malaise, low-grade fever, cardiac arrhythmias, and a pathognomonic bull’s-eye-shaped expanding rash, erythema migrans (Greek, erythema, flush + migrans, move), surrounding the tick bite. In addition, Lyme disease frequently causes a facial nerve paresis, similar to Bell’s palsy, either unilaterally or bilaterally (see Fig. 4-15). Its PNS manifestations range from a mild neuropathy causing only paresthesias to a severe Guillain–Barré-like illness.
With CNS involvement, patients typically have headache, delirium (see Chapter 7), and other signs of meningitis or encephalitis. Their CSF may show a pleocytosis, elevated protein, decreased glucose concentrations, and Lyme antibodies. Serologic tests for Lyme disease remain unreliable. Another confusing aspect of the diagnosis is that patients may have a biologic false-positive test for syphilis because B. burgdorferi is a spirochete (see Chapter 7).
Numerous individuals and physicians attribute years of symptoms – cognitive impairment, weakness, fatigue, and arthralgias – after an attack of adequately treated Lyme disease to a persistent Lyme infection or disordered immunologic response to it. This condition, “chronic Lyme disease,” meets with skepticism in the neurologic community because it lacks consistent clinical criteria, pathology, and test results. Moreover, chronic Lyme disease symptoms do not respond to additional antibiotic treatment (see Chapters 6 and 7).
Inherited Metabolic Illnesses
Although numerous genetically determined illnesses cause neuropathy, two also cause psychosis.
Metachromatic leukodystrophy (MLD), an autosomal recessive illness carried on chromosome 22, derives its name from the colored granules (lipid sulfatides) that accumulate in the lysosomes of the brain, peripheral nerves, and many nonneurologic organs, such as the gallbladder, pancreas, and liver. Most importantly, MLD, like MS, causes a demyelination process in the CNS white matter (leukodystrophy) and, to a lesser extent, the PNS (see Chapter 15).
MLD symptoms usually first appear in infants and children, in whom the illness pursues a rapidly fatal course. In young adults, MLD presents with personality and behavioral changes, thought or mood disorder, and cognitive impairment. MLD-induced cognitive impairments typically progress slowly to dementia. Neurologists describe MLD-induced cognitive impairment as a “frontal dementia” because of its combination of personality, behavioral, and cognitive manifestations (see Chapter 7). Peripheral neuropathy and signs of CNS demyelination – spasticity and ataxia – follow and eventually overshadow the frontal dementia.
MLD is characterized by decreased activity of the lysosomal enzyme arylsulfatase A. Neurologists diagnose the illness by demonstrating a deficiency of this enzyme in leukocytes or cultured fibroblasts and the presence of metachromatic lipid material in biopsy specimens of peripheral nerves. In many cases, appropriate stains detect metachromatic lipid material in the urine. Autopsy specimens will show metachromatic material in cerebral tissue. As in MS, magnetic resonance imaging (MRI) shows demyelinated lesions in the brain (see Chapters 15 and 20). No treatment arrests the illness, but experimental treatments with bone marrow transplant and gene therapy hold promise.
Volatile Substance Exposure
In contrast, recreationally inhaling toluene, a component of spray paint and marker pens, predominantly damages CNS rather than PNS myelin. In single exposures, inhaling toluene produces an alcohol-like euphoria, but chronic overexposure, whether deliberate or accidental, may cause personality changes, psychosis, and cognitive impairment that can reach the severity of dementia. Toluene-induced dementia falls into the category of subcortical dementia, in which gait is impaired but language function is relatively preserved (see Chapter 7). Toluene may also cause ataxia, spasticity, and visual impairment. MRI can detect toluene-induced CNS demyelination (leukoencephalopathy, see Chapter 15). Thus, toluene exposure’s clinical findings and MRI abnormalities mimic those of MS.
Motor Neuron Disorders
Amyotrophic Lateral Sclerosis
The pathology of ALS, characteristically an absence of a cellular reaction surrounding degenerating motor neurons, weighs against inflammatory and infectious etiologies. Many ALS patients do respond, albeit modestly, to blocking glutamate, the excitatory neurotransmitter (see Chapter 21). Putting together these clues – the lack of a cellular response and a beneficial response to glutamate blocking – suggests that glutamate excitotoxicity leads to cell death from apoptosis (see Chapters 18 and 21).
Except for the veterans, patients develop ALS at a median age of 66 years. Their first symptoms usually consist of weakness, atrophy, and subcutaneous muscular twitching ( fasciculations) – a sign of degenerating anterior horn cells – all in one arm or leg (Fig. 5-9). Surprisingly, even from these atrophic muscles, physicians can elicit brisk DTRs and Babinski signs – signs of upper motor degeneration – because damaged UMNs supply enough undamaged LMNs. The weakness, atrophy, and fasciculations spread asymmetrically to other limbs and also to the face, pharynx, and tongue. Dysarthria and dysphagia (bulbar palsy) eventually develop in most patients. When pseudobulbar palsy superimposes itself on bulbar palsy, patients’ speech becomes unintelligible and interrupted by “demonic” or “pathologic” laughing and crying, and their behavior falls into the category of involuntary emotional expression disorder (see Chapter 4). Despite their extensive paresis, they maintain control over their bladder and bowel function as well as over ocular muscle.
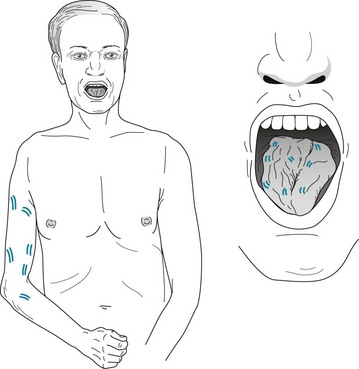
FIGURE 5-9 This elderly man with amyotrophic lateral sclerosis has typical asymmetric limb atrophy, paresis, and fasciculations. His tongue, which also has fasciculations, has undergone atrophy, as indicated by clefts and furrows.
Because ALS attacks UMNs and LMNs, it generally spares neurons involved in cognitive function. Except for approximately 10% of patients, ALS victims retain their cognitive and decisional capacity, remain tragically mentally competent, and have complete awareness of their plight. The small group usually has some clinical and pathological features of frontotemporal dementia, in which behavioral and emotional changes accompany cognitive impairment (see Chapter 7).
Poliomyelitis
In polio, as in ALS, oculomotor, bladder, bowel, and sexual functions are normal (see Chapters 12 and 16). Likewise, polio patients, no matter how devastating their illness, retain normal cognitive function. For example, Franklin Roosevelt, handicapped by polio-induced paraplegia, served as president of the United States.
Benign Fasciculations
Sometimes twitching, which mimics fasciculations, of the eyelid muscles (orbicularis oculi) creates annoying movements. Neurologists call them myokymia and blame lack of sleep, excessive caffeine, and other irritants. In a different situation – if the movements are bilateral, forceful enough to close the eyelids, or exceed a duration of 1 second – they may represent a facial dyskinesia, such as blepharospasm, hemifacial spasm, or tardive dyskinesia (see Chapter 18).
Spine Disease
Cervical spondylosis is the chronic age- and occupation-related degenerative condition, which neurologists loosely term “wear and tear,” where bony encroachment leads to stenosis of the vertebral foramina and spinal canal (Fig. 5-10). Stenosis of the neural foramina constricts cervical nerve roots, which causes neck pain with arm and hand paresis, atrophy, hypoactive DTRs, and fasciculations – signs of LMN injury. Cervical spondylosis may also create spinal canal stenosis that compresses the spinal cord to cause myelopathy with leg weakness, spasticity, hyperreflexia, and Babinski signs – signs of UMN injury.
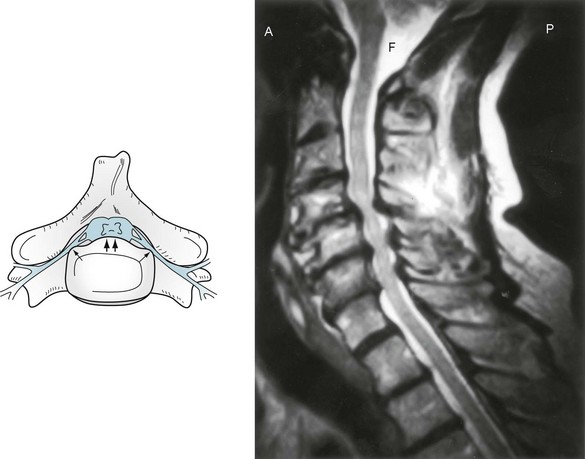
FIGURE 5-10 Left, In cervical spondylosis, bony proliferation damages upper and lower motor neurons. Intervertebral ridges of bone (double arrows) compress the cervical spinal cord. At the same time, narrowing of the foramina (single arrows) constricts cervical nerve roots. Right, Magnetic resonance imaging shows the lateral view of cervical spondylosis. The cerebrospinal fluid (CSF) in the foramen magnum (F) and surrounding the spinal cord is bright white. The spinal cord is gray. In the mid to low cervical spine, bony protrusions and hypertrophied ligaments compress the spinal cord and its surrounding CSF, giving the spinal cord a “washboard” appearance.
Lumbar spondylosis, the lower spine counterpart and frequent accompaniment of cervical spondylosis, produces lumbar nerve root compression and low back pain; however, because the spinal cord descends only to the first lumbar vertebra (see Fig. 16-1), lumbar spondylosis cannot cause spinal cord compression. Spondylolisthesis, which may accompany spondylosis, consists of the forward slip of adjacent lumbar vertebrae or of L5 on the sacrum. Lumbar spondylosis, with or without spondylolisthesis, commonly produces chronic buttock pain that radiates down the posterior aspect of the leg (sciatica). Its other symptoms are leg and feet fasciculations, paresis, atrophy, and knee or ankle areflexia. Sometimes patients with lumbar stenosis have symptoms of pain and weakness in their legs only when they walk (neurogenic claudication).
A ramification of cervical and lumbar spondylosis is that it causes chronic pain. It deprives people of work, mobility, and leisure activities. It sometimes contributes to depression and requires strong analgesics, perhaps opioids, as well as antidepressants (see Chapter 14). Carefully selected cases of severe spondylolisthesis and lumbar spondylosis with marked, symptomatic stenosis of the spinal canal will benefit from surgery.
More than 90% of lumbosacral disk herniations occur at either the L4–5 or L5–S1 intervertebral space. These interspaces are vulnerable because they bear the stress of the body’s weight on the lumbar spine curve. Herniated lumbar disks cause low back pain that radiates into the buttock and often down one or both legs, i.e., sciatica. They may also cause weakness of the ankle and foot muscles and loss of the ankle DTR, but infrequently the knee DTR. Large lumbar disk herniations may cause compression of all the lower lumbar and sacral nerve roots, which comprise the cauda equina (Fig. 5-11). Such herniated disks may produce the cauda equina syndrome: LMN paresis of one or both legs, perineal (“saddle area”) pain and anesthesia, incontinence, and sexual dysfunction.
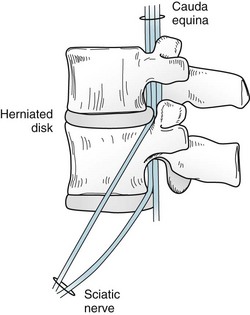
FIGURE 5-11 The cauda equina (Latin, horse’s tail) consists of the bundle of lumbar and sacral nerve roots in the lower spinal canal. The nerve roots leave the spinal canal through foramina. Herniated disks might compress the nerve roots in or near those narrow passages. Compressed nerve roots usually cause pain in the low back that radiates along the distribution of the sciatic nerve. Common movements that momentarily further herniate the disk, such as coughing, sneezing, or straining at stool, intensify the pain. Large herniated disks may compress the entire cauda equina.
Poor posture and obesity, as well as the causes of cervical herniated disks, predispose individuals to lumbar herniations. Coughing, sneezing, or elevating the straightened leg – because these maneuvers press the herniated disk more forcefully against the nerve root – characteristically increase buttock and leg pain (Fig. 5-12).
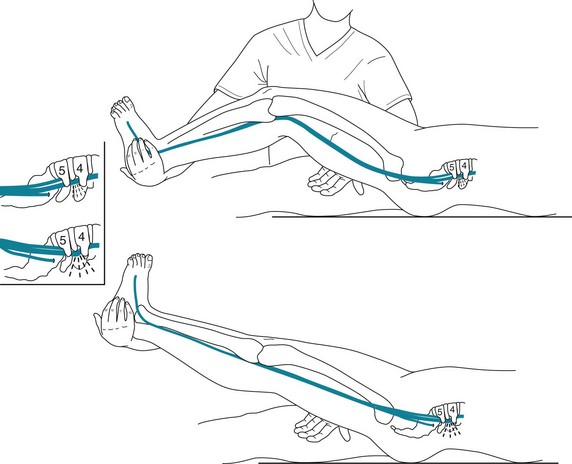
FIGURE 5-12 If a patient has a herniated lumbar intervertebral disk and a physician raises the patient’s straightened leg, the maneuver will probably cause low back pain to radiate to the buttocks and perhaps further down the leg (Lasègue sign). This position draws the nerve against the edge of a herniated disk, which leads to nerve compression and irritation.
Alao AO, Soderberg MG, Geller G. Psychiatric symptoms in acute intermittent porphyria. Resident Staff Physician. 2004;50:28–32.
Albert SM, Rabkin JG, Del Bene ML, et al. Wish to die in end-stage ALS. Neurology. 2005;65:68–74.
Armon C, Argoff CE, Samuels J, et al. Assessment: Use of epidural steroid injections to treat radicular lumbosacral pain. Neurology. 2007;68:723–729.
Aubourg P, Adamsbaum C, Lavallard-Rousseau MC, et al. A two-year trial of oleic and erucic acids (“Lorenzo’s oil”) as treatment for adrenomyeloneuropathy. N Engl J Med. 1993;329:745–752.
Black DN, Taber KH, Hurley RA. Metachromatic leukodystrophy: A model for the study of psychosis. J Neuropsychiatry Clin Neurosci. 2003;15:289–293.
Clarkson TW, Magos L, Myers GL. The toxicology of mercury – current exposures and clinical manifestations. N Engl J Med. 2003;349:1731–1736.
England J, Gronseth G, Franklin G, et al. Practice parameter: Evaluation of distal symmetric polyneuropathy: Role of laboratory and genetic testing (an evidence-based review). Neurology. 2009;72:185–192.
Estrov Y, Scaglia F, Bodamer OA. Psychiatric symptoms of inherited metabolic disease. J Inherit Metab Dis. 2000;23:2–6.
Eyeson J, House I, Yang YH, et al. Relationship between mercury levels in blood and urine and complaints of chronic mercury toxicity from amalgam restorations. Br Dent J. 2010;208:E7.
Fang F, Valdimarsdottir U, Furst CJ, et al. Suicide among patients with amyotrophic lateral sclerosis. Brain. 2008;131:2729–2733.
Feder HM, Johnson B, O’Connell S, et al. A critical appraisal of “chronic Lyme disease.”. N Engl J Med. 2007;357:1422–1430.
Filley CM, Kleinschmidt-DeMasters BK. Toxic leukoencephalopathies. N Engl J Med. 2001;345:425–432.
Filley CM, Halliday W, Kleinschmidt-DeMasters BK. The effects of toluene on the central nervous system. J Neuropathol Exp Neurol. 2004;63:1–12.
Gauthier A, Vignola A, Cavlo A, et al. A longitudinal study on quality of life and depression in ALS patient–caregiver couples. Neurology. 2007;68:923–926.
Gonzalez-Arriaza HL, Bostwick JM. Acute porphyrias. Am J Psychiatry. 2003;160:450–458.
Herskovitz S, Scelsa SN, Schaumburg HH. Peripheral Neuropathies in Clinical Practice. New York: Oxford University Press; 2010.
Hyde TM, Ziegler JC, Weinberger DR. Psychiatric disturbances in metachromatic leukodystrophy. Arch Neurol. 1992;49:401–406.
Isbister G, Kiernan MC. Neurotoxic marine poisoning. Lancet Neurol. 2005;4:219–228.
Katz JN, Simmons BP. Carpal tunnel syndrome. N Engl J Med. 2002;346:1807–1812.
Koffman BM, Greenfield LJ, Ali P. Neurologic complications after surgery for obesity. Muscle Nerve. 2006;33:166–176.
Kumperscak HG, Plesnicar BK, Zalar B, et al. Adult metachromatic leukodystrophy: A new mutation in the schizophrenia-like phenotype with early neurologic signs. Psychiatr Genet. 2007;17:85–91.
Miller RG, Jackson CE, Kararskis EJ. Practice parameter update: The care of the patient with amyotrophic lateral sclerosis: Drug, nutritional, and respiratory therapies: Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2009;73:1218–1226.
Peul WC, Houwelingen HCV, Hout WBVD. Surgery versus prolonged conservative treatment for sciatica. N Engl J Med. 2007;356:2245–2256.
Phukan J, Pender NP, Hardiman O. Cognitive impairment in amyotrophic lateral sclerosis. Lancet Neurol. 2007;6:994–1003.
Quecedo E, Sanmartin O, Febrer MI, et al. Mees’ lines: A clue to the diagnosis of arsenic poisoning. Arch Dermatol. 1996;132:349–350.
Research Advisory Committee on Gulf War Veterans’ Illnesses. U. S. Department of Veterans Affairs. Gulf War Illness and the Health of Gulf War Veterans. http://sph.bu.edu/insider/racreport.
Rowland LP, Shneider NA. Amyotrophic lateral sclerosis. N Engl J Med. 2001;344:1688–1700.
Saito M, Kumano H, Yoshiuchi K, et al. Symptoms profile of multiple chemical sensitivity in actual life. Psychosom Med. 2005;67:318–325.
Schaumburg HH, Albers JW. Pseudoneurotoxic disease. Neurology. 2005;65:22–26.
Sharief MK, Priddin J, Delamont RS, et al. Neurophysiologic analysis of neuromuscular symptoms in UK Gulf War veterans: A controlled study. Neurology. 2002;59:1518–1525.
Simpson DM, Kitch D, Evans SR, et al. HIV neuropathy natural history cohort study: Assessment measures and risk factors. Neurology. 2006;66:1679–1687.
Staudenmayer H, Selner JC, Buhr MP. Double-blind provocations chamber challenges in 20 patients presenting with “multiple chemical sensitivity. Regul Toxicol Pharmacol. 1993;18:44–53.
Strong MJ, Lomen-Hoerth C, Caselli RJ, et al. Cognitive impairment, frontotemporal dementia, and the motor neuron disease. Ann Neurol. 2003;54(Suppl 5):S20–S23.
Triebig G, Nasterlack M, Hacke W, et al. Neuropsychiatric symptoms in active construction painters with chronic solvent exposure. Neurotoxicology. 2000;21:791–794.
Yuki N, Hartung HP. Guillain-Barré syndrome. N Engl J Med. 2012;366:2294–2304.
Chapter 5 Questions and Answers
3. A 24-year-old woman has the sudden onset of low back pain with inability to dorsiflex and evert her right ankle. She also has mild weakness of ankle inversion. Raising her straightened right leg produces back pain that radiates down the lateral leg. Sensation is diminished on the dorsum of her right foot. Her DTRs remain normally reactive. Which is the most likely lesion?
4. A 54-year-old man with pulmonary carcinoma has had 2 weeks of mid thoracic back pain. He describes the sudden onset of abnormal sensation in his legs and difficulty walking. He has weakness of both legs, which are areflexic, and hypalgesia from the toes to the umbilicus. What process is evolving?
5. After recovering consciousness, while still sitting on a toilet, a 27-year-old drug addict is unable to walk. He has paresis of the knee flexor (hamstring) muscles and all ankle and toe muscles. His knee DTRs are normal, but his ankle DTRs and plantar reflexes are absent. Sensation is absent below the knees. Where is the lesion(s)?
7. A 30-year-old computer programmer describes painful tingling in both of her palms and first three fingers on both hands that often wakes her from sleep. In addition, she reports frequently dropping small objects. She has mild paresis of her thumb (thenar) abduction and opposition muscles. Percussion of her wrists recreates the paresthesias. DTRs are normal. What is the cause?
12. After losing a fistfight, a 17-year-old man finds that he cannot walk or feel anything below his waist. Although he has total inability of his legs, they have retained normally active DTRs and flexor plantar responses. He has no response to noxious (pinprick) stimulation below his umbilicus, but sensation of position, vibration, and temperature is preserved. Where is the lesion?
14. A 34-year-old man with chronic low back pain experienced an exacerbation while raking leaves. He has difficulty walking and pain that radiates from the low back down the left posterior thigh to the lateral ankle. He has paresis of plantar flexion of the left ankle and an absent ankle DTR. He has an area of hypalgesia along the left lateral foot. What is the etiology of his condition?
15. A 62-year-old man has the onset over 3 months of weakness of both arms and then the left leg. He is alert and oriented but has dysarthria. His jaw jerk is hyperactive and his gag reflex is absent. The tongue is atrophic and has fasciculations. The muscles of his arms and left leg also have atrophy and fasciculations. All DTRs are hyperactive, and Babinski signs are present. Sensation is intact. What is the etiology of his illness?
a. He has a classic case of ALS with both bulbar and pseudobulbar palsy and wasting of his limb and tongue muscles. Although fasciculations are commonplace and usually benign, when they occur in multiple limbs and are associated with weakness, muscle atrophy, and hyperactive DTRs, they indicate ALS. The corticobulbar and corticospinal tracts, as well as the brainstem nuclei and spinal anterior horn cells, are all involved. He typically has normal ocular movements and mental faculties. A syrinx would have a characteristic “suspended sensory loss” (see Fig. 2-18) and absent upper extremity DTRs. Both multiple sclerosis and multiple strokes would have exclusively upper motor neuron signs.
20. Pat, a 25-year-old anxious medical student who is in psychotherapy, describes fasciculations in the limb muscles, calf cramps at night, and muscle aches during the day. A classmate found that Pat’s strength was normal and that no muscle was atrophic; however, all DTRs were brisk. Pat’s father, who had been a house painter, had developed arm muscle weakness and fasciculations before he died of pulmonary failure. Which is the most likely cause of Pat’s fasciculations?
26–36. Which conditions are associated with fasciculations? (Yes or No)
26-No; 27-No; 28-Yes; 29-Yes; 30-Yes; 31-Yes; 32-No; 33-Yes; 34-Yes; 35-Yes; 36-Yes.
d. He has most likely swallowed a common anticholinesterase-based insecticide. Most of them block neuromuscular transmission (see Chapter 6), which causes an acute generalized flaccid paralysis accompanied by fasciculations. In addition, the increased parasympathetic activity causes miosis and bradycardia. The bradycardia and other manifestations of excessive parasympathetic activity can be reversed by atropine.
c. The transverse carpal ligament, which underlies the skin of the palmar (flexor) surface of the wrist, forms the roof of the carpal tunnel. The median nerve passes through the carpal tunnel, but the ulnar nerve passes above and medial to it (see Fig. 5-3).
42. His hyperactive DTRs and Babinski signs indicate that he sustained CNS rather than PNS damage.
43. Although his quadriparesis might have several explanations, the oculomotor paresis and apnea indicate a brainstem injury. The lesion spared his cerebral functions, such as mentation and vision, as well as his upper brainstem functions, such as blinking. He exists in the well-known “locked-in syndrome” (see Chapter 11). Physicians should not misdiagnose him as being comatose, demented, or vegetative.
44. Of course, localization makes a difference. Neurologists often say, “Location, location, location … is destiny.” With lesion confined to the brainstem, as in this case, intellectual function is preserved. On the other hand, extensive cerebral damage causes dementia.
45. Although computed tomography (CT) might be performed to detect or exclude a cerebral lesion, only a MRI would be sensitive enough to detect his brainstem lesion. An electroencephalogram in this case would be a valuable test because, with his cerebral hemispheres intact, it would show a relatively normal electrical pattern. Visual-evoked responses would determine the integrity of the entire visual system, which is not part of the brainstem. Brainstem auditory-evoked responses would determine the integrity of the auditory circuits, which are predominantly based in the brainstem. Positron emission tomography and single photon emission CT may also be helpful (see Chapter 20).
47. Typically follows an upper respiratory tract infection
50. Internuclear ophthalmoplegia
52. Cognitive impairment early in the course of the illness
54. A demyelinating polyneuropathy
56. Leads to pseudobulbar palsy
59. A monophasic illness that typically peaks in several weeks and last several months
60. Sexual dysfunction can be the only or primary persistent deficit
46-b; 47-b; 48-a; 49-c; 50-a; 51-c; 52-d; 53-b; 54-b; 55-a; 56-a; 57-b; 58-d; 59-b; 60-a.
71. The wife of a homicidal neurologist enters psychotherapy because of several months of fatigue and painful paresthesias. She also describes numbness in a stocking-glove distribution, darkening of her skin, and the appearance of white lines across her nails. In addition to a general medical evaluation, which specific test should be performed?
78. A 35-year-old man with AIDS reports developing painful burning sensations of his feet. Although he has no significant weakness, he has lost his ankle DTRs. He has no history of diabetes, use of medications, exposure to chemicals, or other illness. Which of the following is least likely to be present?
87. Two physician vacationers in a Caribbean restaurant had, they felt, a wonderful fish dinner of shrimp, barracuda, and, for dessert, rum ice cream to celebrate the end of a fantastic visit. However, severely painful abdominal cramps, unremitting nausea, protracted vomiting, and brief but intense diarrhea awoke both of them later that night. After they self-medicated with antibiotics, antiemetics, and antidiarrheals, and caught several hours of sleep, the pair of physicians improved enough to catch their flight back to their urban teaching hospital. Never fully recovering their strength, 2 days later they began to have weakness, clumsiness, and numbness of the fingers, hands, and feet. A striking feature of their sensory loss was that all cold objects felt burning hot to the touch. Moreover, cold drinks seemed to burn their mouth and throat. A neurologic examination showed generalized mild weakness, hypoactive DTRs, and diminished sensation to pin and light touch in their distal extremities. They also displayed a Romberg sign. Given the obvious diagnosis of food poisoning, which is the most likely agent?
89. A community hospital transferred a 23-year-old waitress for Guillain–Barré syndrome after she developed areflexic quadriparesis. Neurologists at the tertiary center requested a psychiatry consultation because the patient demanded narcotics for abdominal pain, which they explained would not constitute a symptom of Guillain–Barré syndrome. The psychiatrist found her disoriented and inattentive. When alert, she insisted on narcotics. She had a low-grade fever but otherwise her vital signs were normal. Her oxygen saturation, blood glucose, and electrolytes were normal. Her urine tests were normal, but the urine darkened in the sunlight of the laboratory. Which test would probably clinch the diagnosis?
90. A 45-year-old physician slipped and fell on his buttocks. As soon as he hit the pavement, he had excruciating low lumbar pain. On attempting to arise, he found that both legs were weak. In the emergency room, a neurologist confirmed that he had paraparesis and was unable to elicit DTRs or plantar reflexes. Not only did the neurologist detect a distended bladder, he found that the patient had no perception of it and that he also had perineal (“saddle”) anesthesia. What is the most likely cause of the pain and neurologic deficits?
*Anatomists have renamed the peroneal nerves and muscles because of their similarity in sound to perineum. Their new name, which this book has adopted, is fibular nerves and muscles.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 5 Peripheral Nerve Disorders
Anatomy
The spinal cord’s anterior horn cells form the motor neurons of the peripheral nerves – the PNS’ starting point. The peripheral nerves are the final link in the neuron chain that transmits motor commands from the brain through the spinal cord to muscles (Fig. 5-1). Nerve roots emerging from the anterior spinal cord mingle within the brachial or lumbosacral plexus to form the major peripheral nerves, such as the radial and femoral. Although peripheral nerves are quite long, especially in the legs, they faithfully conduct electrochemical impulses over considerable distances. Because myelin, the lipid-based sheath generated by Schwann cells, surrounds peripheral nerves and acts as insulation, the impulses are preserved.
FIGURE 5-1 The corticospinal tracts, as discussed in Chapter 2, and as their name indicates, consist of upper motor neurons (UMNs) that travel from the motor cortex to the spinal cord. They synapse on the spinal cord’s anterior horn cells, which give rise to the lower motor neurons (LMNs). The LMNs join sensory fibers to form peripheral nerves.
When stimulated, motor nerves release packets of acetylcholine (ACh) from storage vesicles at the neuromuscular junction. The ACh packets traverse the junction and bind on to specific ACh receptors on the muscle end plate. The interaction between ACh and its receptors depolarizes the muscle membrane and initiates a muscle contraction (see Chapter 6). Neuromuscular transmission culminating in muscle depolarization is a discrete, quantitative action: ACh does not merely seep out of the presynaptic terminal as loose molecules and drift across the neuromuscular junction to trigger a muscle contraction.
Mononeuropathies
Disorders of single peripheral nerves, mononeuropathies, are characterized by flaccid paresis, DTR loss (areflexia), and reduced sensation, particularly for pain (hypalgesia [Greek, hypo, under + algos, pain] or analgesia [Greek, insensitivity to pain]) (Table 5-1). Paradoxically, mononeuropathies and other peripheral nerve injuries sometimes lead to spontaneously occurring sensations, paresthesias (Greek para, near + aisthesis, sensation) that may be painful, dysesthesias. Peripheral nerve injuries also convert stimuli that ordinarily do not cause pain, such as a light touch or cool air, into painful sensations, allodynia; exaggerate painful responses to mildly noxious stimuli, such as the point of a pin, hyperalgesia; or delay but then exaggerate and prolong pain from noxious stimuli, hyperpathia.
TABLE 5-1 Major Mononeuropathies
*Compression of the radial nerve in the spiral groove of the humerus spares the triceps deep tendon reflex (DTR).
Compression, especially of nerves protected only by overlying skin and subcutaneous tissue rather than by bone, viscera, or thick layers of fat, occurs frequently. People most susceptible are diabetics; those who have rapidly lost weight, thereby depleting nerves’ protective myelin covering; workers in certain occupations, such as watchmakers; and those who have remained in disjointed positions for long periods, often because of drug or alcohol abuse. One of the most common compressive mononeuropathies – “Saturday night palsy” – affects the radial nerve, which is vulnerable at the point where it winds around in the spiral groove of the humerus. Thus, people in alcohol-induced stupor who lean against their upper arm for several hours are apt to develop a wrist drop (Fig. 5-2). Foot drop, its lower-extremity counterpart, often results from common fibular nerve* damage from prolonged leg crossing compressing the nerve, lower-knee injuries traumatizing the nerve, or a constrictive lower-leg cast pushing against the nerve as it winds around the head of the fibula.
FIGURE 5-2 As the radial nerve winds around the humerus, it is vulnerable to compression and other forms of trauma. Radial nerve damage leads to the readily recognizable wrist drop that results from paresis of the extensor muscles of the wrist, finger, and thumb.
Carpal tunnel syndrome, the most common mononeuropathy, results from damage of the median nerve as it travels through the carpal tunnel of the wrist (Fig. 5-3, left). Forceful and repetitive wrist movements can traumatize the nerve in that confined passage. Meat and fish processing, certain assembly-line work, and carpentry are all closely associated with carpal tunnel syndrome; however, contrary to initial claims, word processing and other keyboarding actually have a weak association with the disorder. In another mechanism, fluid retention during pregnancy or menses entraps the median nerve in the carpal tunnel. Similarly, inflammatory tissue changes in the wrist from rheumatoid arthritis may compress the median nerve.
FIGURE 5-3 Left, The median nerve passes through the carpal tunnel, which is a relatively tight compartment. In it, the median nerve is vulnerable to repetitive movement and compression from fluid accumulation. The ulnar nerve, taking a different route, passes above and medial to the roof of the tunnel, the transverse carpal ligament. It thus escapes damage from most repetitive movements and fluid accumulation. Right, The usual sensory distribution of the median nerve encompasses the palm, thenar eminence (thumb base), thumb, and adjacent two fingers. In carpal tunnel syndrome, pain shoots distally from the wrist over this area. Physicians may elicit the Tinel sign, a reliable indication of carpal tunnel syndrome, by tapping the patient’s palmar wrist surface and finding that paresthesias emanate from the wrist and radiate in the median nerve distribution.
Whatever the mechanism, carpal tunnel syndrome causes paresthesias and pains that shoot from the wrist to the palm, thumb, and adjacent two or sometimes three fingers (Fig. 5-3, right). Symptoms worsen at night and awaken the victims, who shake their hands in an attempt to find relief. Neurologists test for the syndrome’s characteristic Tinel sign by percussing the wrist: The test is positive when the percussion generates electric sensations that shoot from the wrist into the palm and fingers.
In another example of upper-extremity nerve damage, pressure on the ulnar groove of the elbow (the “funny bone”) or cubital tunnel may damage the ulnar nerve. For instance, when individuals rest the weight of their arms on their elbows, the compression often injures the ulnar nerve. These individuals develop atrophy and weakness of their hand muscles (Fig. 5-4). The ulnar nerve damage also leads to loss of sensation of the fourth and fifth fingers and the medial surface of the hand.
FIGURE 5-4 Left, With ulnar nerve injuries, the palmar view shows that intrinsic muscles of the hand, particularly those of the hypothenar eminence (fifth finger base), undergo atrophy. The fourth and fifth fingers are flexed and abducted. When raised, the hand and fingers assume the benediction sign. In addition, the medial two fingers and palm are anesthetic (numb). Right, Ulnar nerve injuries also produce a claw hand because of atrophy of the muscles between the thumb and adjacent finger (first dorsal interosseous and adductor pollicis), as well as of those of the hypothenar eminence.
Polyneuropathies (Neuropathies)
Alternatively, neurologists divide neuropathies into sensory, motor, or mixed sensorimotor neuropathy. Patients with sensory neuropathy usually suffer predominantly or exclusively from numbness, paresthesias, or burning in their fingers and toes (i.e., stocking-glove hypalgesia: Fig. 5-5). The pain may reach intolerable proportions (see neuropathic pain, Chapter 14). When sensory neuropathy affects the feet, it may provoke leg movements, such as restless legs syndrome (see Chapter 17). Patients with motor neuropathy usually have distal limb weakness that impairs fine, skilled hand and finger movements, such as buttoning a shirt, or raising their feet when they walk, which causes a foot drop. Their neuropathy in chronic cases usually also leads to muscle atrophy and flaccidity. As with other LMN injuries, it diminishes DTRs (see Fig. 2-2C), first at the brachioradialis and Achilles’ and then at more proximal sites. Mixed sensorimotor neuropathy causes mixtures of those symptoms and signs.
FIGURE 5-5 Patients with polyneuropathy lose pain and other sensations. The loss is symmetric, more severe distally than proximally, and more severe in the legs than arms. Neurologists term this pattern of sensory loss stocking-glove hypalgesia.
Neurologists who care for psychiatric patients may meaningfully divide neuropathies into those with and those without comorbid changes in mental status (Box 5-1).
Box 5-1
Important Causes of Neuropathy
Neuropathies Without Comorbid Mental Status Changes
Guillain–Barré Syndrome
When first affected, young and middle-aged adults feel paresthesias and numbness in the fingers and toes. Then they develop flaccid paresis of their feet and legs with characteristically absent knee and ankle DTRs. Weakness and areflexia, which soon become a much greater problem than numbness, ascend to involve the hands and arms. Many patients progress to respiratory insufficiency because of involvement of the phrenic and intercostal nerves, and require intubation for ventilation. If weakness ascends still further, patients develop cranial nerve involvement that may lead to dysphagia and other aspects of bulbar palsy (see Chapter 4). Additional involvement causes facial weakness and then sometimes even ocular immobility. Nevertheless, possibly because optic and acoustic nerves are protected by myelin generated by the CNS – not the PNS – patients continue to see and hear.
Even if the illness worsens to the point of total paralysis, patients usually remain conscious with a normal mental status – allowing for anxiety and depressive symptoms from enduring a life-threatening illness. Completely immobile and anarthric Guillain–Barré syndrome patients, typically with preserved consciousness and mental status, exist in a locked-in syndrome (see Chapter 11). Cerebrospinal fluid (CSF) exhibits an elevated protein concentration but with few white cells (i.e., the classic albumino-cytologic dissociation) (see Table 20-1).
The illness usually resolves almost completely within 3 months as the PNS myelin is regenerated. By way of treatment, plasmapheresis (plasma exchange), which extracts circulating inflammatory mediators, particularly autoantibodies, complement, and cytokines, reduces the severity and duration of the paresis. Alternatively, administration of intravenous human immunoglobulin, which “blocks” the antibodies at the neuromuscular junction, also restores patients’ strength. Steroids will not help, which is surprising because they are helpful in other inflammatory diseases of the nervous system, such as myasthenia gravis (see Chapter 6), multiple sclerosis (MS: see Chapter 15), and the chronic form of Guillain–Barré syndrome (chronic inflammatory demyelinating polyneuropathy).
Not only is Guillain–Barré syndrome a life-threatening illness, but it also epitomizes the distinction between PNS and CNS diseases. Although paraparesis or quadriparesis might be a feature common to PNS and CNS illnesses, different patterns of muscle weakness, changes in reflexes, and sensory distribution characterize PNS and CNS illnesses (Table 5-2). Also, in Guillain–Barré syndrome, as in most neuropathies other than diabetic neuropathy (see later), bladder, bowel, and sexual functions are preserved. In contrast, patients with spinal cord disease usually have incontinence and impotence at the onset of the injury.
TABLE 5-2 Differences between Central (CNS) and Peripheral Nervous System (PNS) Signs
| CNS | PNS | |
|---|---|---|
| Motor System | Upper motor neuron | Lower motor neuron |
| Paresis | Patterns* | Distal |
| Tone | Spastic† | Flaccid |
| Bulk | Normal | Atrophic |
| Fasciculations | No | Sometimes |
| Reflexes | ||
| Deep tendon reflexes | Hyperactive‡ | Hypoactive |
| Plantar | Babinski sign(s) | Absent |
| Sensory loss | Patterns* | Hands and feet |
*Examples: motor and sensory loss of one side or lower half of the body (e.g., hemiparesis or paraparesis), and hemisensory loss.
MS appears to be a partial exception to the rule that CNS demyelinating damage is permanent. In MS, episodes of demyelination of several CNS areas, including the optic nerves, partially or even completely resolve (see Chapter 15). However, the improvement results from resolution of myelin inflammation rather than myelin regeneration. When MS-induced demyelination eventually encompasses large areas of cerebral CNS myelin, it results in permanent quadriparesis and dementia.
Diabetes
Patients with diabetic neuropathy may also have autonomic nervous system involvement that causes gastrointestinal immobility, bladder muscle contraction, and sexual dysfunction. In fact, erectile dysfunction is occasionally the first or most disturbing symptom of diabetic autonomic neuropathy (see Chapter 16).
Toxic-Metabolic Disorders
Chronic, low-level intoxication by several other heavy metals causes polyneuropathy, dermatologic abnormality, and mental changes. In contrast, acute heavy metal poisoning typically leads to fatal gastrointestinal symptoms and cardiovascular collapse. Arsenic, which is tasteless and odorless, is a poison used in popular murder cases. With chronic, low-level, deliberate, accidental, or industrial intoxication, arsenic causes anorexia, malaise, and a distal neuropathy that might mimic Guillain–Barré syndrome. It also causes several characteristic dermatologic abnormalities: Mees lines on the fingernails (Fig. 5-6), hyperpigmentation, and hyperkeratosis.
FIGURE 5-6 Mees lines, white bands (arrows) that stretch across the fingernails, characteristically indicate arsenic poisoning. In addition, poisoning by other heavy metals and trauma can cause them.
Mercury intoxication is more complex than arsenic poisoning. Individuals with mercury poisoning may develop a neuropathy and various CNS deficits, including cognitive impairment, ataxia, dysarthria, and visual field changes. Their gums accumulate a telltale dark line just below their teeth (Fig. 5-7).
FIGURE 5-7 Chronic mercury poisoning causes a dark blue or black line (arrow) along the gum. Individuals with this sign usually also have neuropathy and central nervous system signs, such as ataxia and dysarthria.
Investigators at one time proposed that mercury-based dental amalgams (“fillings”) caused Alzheimer disease and other neurodegenerative illnesses either by dissolving in saliva and allowing mercury to enter the circulation or emitting a mercury vapor those individuals inhaled. In another inquiry, because ethyl mercury was a major component of the common vaccine preservative, thimerosal, investigators suspected that routine childhood immunizations caused autism (see Chapter 13). However, statistically powerful epidemiologic studies disproved both of those suspicions. In the case of vaccinations, the original “investigators” possibly engaged in fraud.
Neuropathies With Comorbid Mental Status Changes
Although most neuropathies, as described in the previous section, may be painful, incapacitating, or even devastating to the PNS, they generally do not cause mental changes in adults. For example, most people who are old, diabetic, on hemodialysis, or receiving chemotherapy remain intelligent, thoughtful, and competent even though beset by pain, sensory loss, and weakness. In contrast, only a few diseases (see Box 5-1) cause the combination of dementia and neuropathy, which would indicate both cerebral cortex and peripheral nerve damage. An analogous combination would be dementia and movement disorders, which would indicate cerebral cortex and basal ganglia damage (see Box 18-4).
Nutritional Deficiencies
Deficiencies of thiamine (vitamin B1), niacin (nicotinic acid, B3), or vitamin B12 (cobalamine), each produce a predominantly sensory neuropathy accompanied by dementia or other mental status abnormality (Table 5-3). From a worldwide perspective, starvation has been the most common cause of deficiencies of vitamins, their carrier fats, minerals, and other nutrients. For example, beriberi was the starvation-induced neuropathy attributable to thiamine deficiency endemic in eastern Asia. In the United States, alcoholism, bariatric surgery, and malabsorption syndromes have replaced starvation as the most common causes of nutritional neuropathies. Curiously, few patients with anorexia nervosa or self-imposed extreme diets develop a neuropathy. Their protection may lie in a selective, possibly secret, intake of food or vitamins.
Bariatric surgery remains a unique example. After its rapid introduction and widespread acceptance, postoperative “micronutrient” deficiencies caused various neurologic illnesses. Thiamine, copper, and vitamins B12 and E deficiencies frequently caused neuropathy, but also occasionally encephalopathy or myelopathy (see Chapter 4), i.e., CNS problems. Routine postoperative administration of these micronutrients has prevented the problem.
Whatever the cause, thiamine deficiency generally leads to absent DTRs and loss of position sensation. In fact, until patients walk in the dark, when they must rely on position sense generated in the legs and feet, their deficits may remain asymptomatic. In the well-known Wernicke–Korsakoff syndrome, amnesia, dementia, and cerebellar degeneration accompany the neuropathy associated with alcoholism (see Chapter 7).
Among its many functions, vitamin B12 sustains both CNS and PNS myelin. Thus, B12 deficiency leads to the combination of CNS and PNS damage – combined system disease. Although its manifestations include a neuropathy, cognitive impairment and sensory loss reflecting demyelination of the posterior columns of the spinal cord (see Fig. 2-19B) predominate. Patients also develop a characteristic megaloblastic anemia. Most importantly, neurologists refer to combined system disease as a “correctable cause of dementia” because B12 injections can reverse the cognitive impairment as well as the other CNS and PNS manifestations. The usual causes of B12 deficiency include pernicious anemia, malabsorption, a pure vegetarian diet, or prolonged exposure to nitrous oxide, a gaseous dental anesthetic. (Nitrous oxide inactivates B12 by oxidizing its cobalt.)
The screening test for B12 deficiency consists of determining the serum B12 level. In equivocal cases, especially when cognitive impairment or spinal cord abnormalities are not accompanied by anemia, determining the serum homocysteine and methylmalonic acid levels can corroborate the diagnosis: in B12 deficiency, both homocysteine and methylmalonic acid levels rise to abnormally high levels (Fig. 5-8). Intrinsic factor antibodies, a classic finding in pernicious anemia, will be detectable in only about 60% of cases. The standard confirmatory test is the Schilling test.
FIGURE 5-8 Vitamin B12, acting as a coenzyme, along with folate, facilitates the conversion of homocysteine to methionine. Other enzymes complete the cycle by converting methionine back to homocysteine. An absence of B12 leads to the accumulation of both methionine and homocysteine. Whatever the cause, an elevated homocysteine level is a risk factor for neural tube defects, cerebrovascular and cardiovascular disease, and other neurologic conditions.
In contrast to malnutrition causing neuropathy, excessive intake of certain vitamins causes neurologic problems. For example, although the normal adult daily requirement of vitamin B6 (pyridoxine) is only 2–4 mg daily, several food faddists who consumed several grams daily as part of a special diet developed a profound sensory neuropathy. Similarly, high vitamin A intake may cause pseudotumor cerebri (see Chapter 9) or induce fetal abnormalities (see Chapter 13).
Infectious Diseases
Several common organisms have a predilection for infecting the peripheral nerves and sparing the CNS. For example, herpes zoster infects a single nerve root or a branch of the trigeminal nerve, usually in people older than 65 years or those with an impaired immune system. An infection with herpes (Greek, herpes, spreading skin eruption) causes an ugly, red, painful, vesicular eruption (“shingles”) that may remain excruciating long after the skin infection has resolved (see postherpetic neuralgia, Chapter 14). As another example, leprosy (Hansen disease), infection with Mycobacterium leprae, causes anesthetic, hypopigmented patches of skin, anesthetic fingers and toes, and palpable nerves. It particularly affects the cool portions of the body, such as the nose, earlobes, and digits; however, depending on its variety, the infection strikes the ulnar or another large nerve either singly or along with others.
Acute Lyme disease typically produces multiple problems, such as arthritis, malaise, low-grade fever, cardiac arrhythmias, and a pathognomonic bull’s-eye-shaped expanding rash, erythema migrans (Greek, erythema, flush + migrans, move), surrounding the tick bite. In addition, Lyme disease frequently causes a facial nerve paresis, similar to Bell’s palsy, either unilaterally or bilaterally (see Fig. 4-15). Its PNS manifestations range from a mild neuropathy causing only paresthesias to a severe Guillain–Barré-like illness.
With CNS involvement, patients typically have headache, delirium (see Chapter 7), and other signs of meningitis or encephalitis. Their CSF may show a pleocytosis, elevated protein, decreased glucose concentrations, and Lyme antibodies. Serologic tests for Lyme disease remain unreliable. Another confusing aspect of the diagnosis is that patients may have a biologic false-positive test for syphilis because B. burgdorferi is a spirochete (see Chapter 7).
Numerous individuals and physicians attribute years of symptoms – cognitive impairment, weakness, fatigue, and arthralgias – after an attack of adequately treated Lyme disease to a persistent Lyme infection or disordered immunologic response to it. This condition, “chronic Lyme disease,” meets with skepticism in the neurologic community because it lacks consistent clinical criteria, pathology, and test results. Moreover, chronic Lyme disease symptoms do not respond to additional antibiotic treatment (see Chapters 6 and 7).
Inherited Metabolic Illnesses
Although numerous genetically determined illnesses cause neuropathy, two also cause psychosis.
Metachromatic leukodystrophy (MLD), an autosomal recessive illness carried on chromosome 22, derives its name from the colored granules (lipid sulfatides) that accumulate in the lysosomes of the brain, peripheral nerves, and many nonneurologic organs, such as the gallbladder, pancreas, and liver. Most importantly, MLD, like MS, causes a demyelination process in the CNS white matter (leukodystrophy) and, to a lesser extent, the PNS (see Chapter 15).
MLD symptoms usually first appear in infants and children, in whom the illness pursues a rapidly fatal course. In young adults, MLD presents with personality and behavioral changes, thought or mood disorder, and cognitive impairment. MLD-induced cognitive impairments typically progress slowly to dementia. Neurologists describe MLD-induced cognitive impairment as a “frontal dementia” because of its combination of personality, behavioral, and cognitive manifestations (see Chapter 7). Peripheral neuropathy and signs of CNS demyelination – spasticity and ataxia – follow and eventually overshadow the frontal dementia.
[/not-level-membership-for-neurology-category]
