Chapter 132 Perioperative Management of Severe Traumatic Brain Injury in Adults
Recent estimates of the magnitude of traumatic brain injury (TBI) suggest that each year approximately 1.7 million people sustain a head injury in the United States,1 with about 75% of patients having a concussion or a mild TBI.2 Based on data from the Centers for Disease Control, roughly 52,000 people die annually from TBI, while 275,000 people are hospitalized and nearly 1.4 million people are seen and discharged from an emergency room for TBI. TBI is a contributing factor in 30.5% of all injury-related deaths that occur in the United States, illustrating its importance as a public health problem. TBI is the leading cause of death under age 45 in the Western world.
The impact of brain trauma in the United States and on the country’s healthcare system is highlighted by estimates of direct medical expenditures and indirect costs (e.g., loss of productivity) attributable to TBI being upward of $60 billion in 2000.3 To help perform activities of daily living, 5.3 million Americans depend on long-term assistance, which further underscores the economic and social significance of TBI in today’s society.
Given these demographics, organized efforts to standardize the assessment and management of patients with TBI have resulted in the publication of Guidelines for the Management of Severe Traumatic Brain Injury, which is now in its third iteration.3a A recent cost-benefit analysis showed that widespread adherence and use of these guidelines would result in significant savings: $262 million in annual medical costs, $43 million in annual rehabilitation costs, and $3.84 billion in lifetime societal costs; 3607 lives would be spared annually from mortality due to head injury.4 Ultimately, evidence-based management of patients with severe TBI translates to better patient care, improved outcomes, and a decreased economic and societal burden imposed by TBI.
Initial Neurologic Assessment
Glasgow Coma Scale
The prehospital assessment of a patient’s neurologic condition serves as a guide to the appropriate triage of an individual following a TBI. When transported directly from the scene of injury to a trauma center, patients with severe TBI have lower mortality rates than those who are first taken to nontrauma centers.5 Furthermore, severely brain-injured patients who are admitted and treated at level I trauma centers have improved survival rates and experience better outcomes compared to matched counterparts treated at level II facilities.6
Over the years, many assessment schemes have been created to determine the neurologic status of head-injured patients, with the Glasgow Coma Scale (GCS) being the most commonly used tool to determine the severity of TBI (Table 132-1). Developed in 1974 by Teasdale and Jennett, the GCS gauges a patient’s level of consciousness through an assessment of eye opening, verbal response, and motor response.7 For patients who are mechanically ventilated by an endotracheal tube or via a tracheostomy, thus precluding the patient’s ability to communicate verbally, a GCS score of 1 point is given for the verbal response score with a “T” following to indicate the intubated status of the patient. There are instances in which accurately determining a GCS score is difficult or when the applicability of a score is of limited utility, as with the presence of concomitant facial trauma, ingestion of sedative drugs such as alcohol or barbiturates, or iatrogenic paralysis of patients for intubation. Moreover, physiologic derangements such as hypotension and hypoxia at the time of assessment hinder a patient’s neural response to stimuli; therefore, they represent important factors that may also lead to inaccurate GCS scores, as well as contributing to secondary brain injury.
| Best Response | Score |
|---|---|
| Eye Opening (E) | |
| Spontaneous | 4 |
| To speech | 3 |
| To pain | 2 |
| Not open | 1 |
| Verbal Response (V) | |
| Conversant | 5 |
| Confused | 4 |
| Nonsense | 3 |
| Sounds | 2 |
| Silence | 1 |
| Intubated | 1T |
| Motor Response (M) | |
| To command | 6 |
| To pain | |
| Localized | 5 |
| Withdrawal | 4 |
| Arm flexion | 3 |
| Arm extension | 2 |
| No response | 1 |
| GCS score = E + V + M (range 3 or 3T to 15) | |
From Teasdale G, Jennett B. Assessment of coma and impaired consciousness: A practical scale. Lancet 2:81-84, 1974.
Severe TBI is classified by total GCS scores of 3 to 8 points, while scores of 9 to 12 points indicate moderate TBI and those between 13 to 15 points denote mild TBI. GCS scores correlate significantly with outcome, with the motor component of the score being most reproducible and yielding the most prognostic information.8 Patients whose GCS scores decrease by 2 points or more between the field and the emergency department are more likely to require surgical intervention.9 Nearly 80% of patients with an initial hospital GCS score of 3 to 5 points have an eventual outcome of death, severe disability, or vegetative state, with a mortality rate of 65% seen in patients with an initial GCS score of 3 points.8,10,11 Among individuals with a GCS score of 3 points on presentation, only 13% have good functional outcomes at the 6-month mark.12 Age, pupil reactivity, radiologic findings on head computed tomography (CT) scan, and extent of extracranial injuries suffered during the trauma serve as additional independent predictors of patient outcome following severe TBI.13–15
Pupillary Examination
The pupillary examination is a critical component of a patient’s initial neurologic assessment following TBI. A dilated pupil that does not constrict in response to light indicates ipsilateral uncal herniation, until proved otherwise. Bilaterally dilated pupils can be seen with hypoxia, hypotension, bilateral oculomotor nerve dysfunction, or severe irreversible brain stem injury. A change in the pupillary examination is the most reliable indicator in determining the side with a mass lesion and carries an 80% positive predictive value. Moreover, pupillary size and reactivity are especially important in the assessment of trauma patients with an admission GCS score of 3 points, because those with bilaterally fixed and dilated pupils have a universally dismal prognosis, with mortality rates of upward of 100%.16–18
TBI Classification
Primary Brain Injury
Primary brain injury refers to damage suffered as a direct result of the initial traumatic event and may be caused by penetrating, blunt, or blast trauma. Bullets and other projectiles often create a significant amount of destruction to brain tissue along their track and can result in injury to brain parenchyma due to fragments of depressed skull. In addition, laceration of cerebral blood vessels can lead to subarachnoid hemorrhage and/or formation of intracerebral hematomas.19 Blunt trauma can result in brain injury because of transmission of force directly at the point of impact or at a point distant from impact due to the displaced brain rebounding against the inner surface of the skull, an event referred to as contrecoup injury.20 The primary injuries of blast TBI are only now becoming understood and represent a regrettable reality of modern warfare and civilian terrorism.
Primary brain injury can be broadly divided into focal and diffuse injuries (Table 132-2). The specific type of injury suffered depends on the nature of the impact during injury (contact or inertial loading), the resultant forces delivered during the impact (rotational, translational, or angular acceleration), and the magnitude and duration of the impact itself.
| Focal | Diffuse |
|---|---|
| Hematoma | Concussion |
| Epidural | Multifocal contusion |
| Subdural | Diffuse axonal injury |
| Intracerebral | |
| Contusion | |
| Concussion | |
| Laceration |
Focal injuries include traumatic intracranial hematomas and contusions. Based on data from the Traumatic Coma Data Bank (TCDB), diffuse injuries are more common and constitute upward of 60% of severe TBI, as they span a wide clinical spectrum ranging from concussion to diffuse axonal injury. Higher mortality rates have been seen with focal injuries,21 highlighting the necessity of expedient cranial imaging in evaluating TBI patients via CT scan, which can confirm the presence of a mass lesion and, in turn, prompt emergent surgical intervention if warranted.
Secondary Brain Injury
Secondary brain injury occurs when local phenomena within the skull or systemic factors cause further damage that manifests at the cellular level over the ensuing hours to days following the original insult. The reduction of mortality from severe TBI, even when adjusted for injury severity, age, and other admission prognostic parameters, from 50% to less than 25% over the past 30 years reflects the impact of preventing secondary brain injury specifically through avoiding and treating aggravating factors, such as hypotension, hypoxia, inadequate cerebral perfusion pressure (CPP), and intracranial hypertension.22 These unwanted physiologic perturbations result in cerebral ischemia, which can significantly compound the effects of the initial injury. The decrement in mortality and improvement in outcomes from TBI that has been achieved is largely attributable to the use of evidence-based protocols, which hold avoidance of cerebral ischemia—through intensive monitoring of neurophysiologic parameters and maintenance of adequate cerebral perfusion—as paramount.23,24 Knowing and understanding the pathophysiologic basis underlying cerebral ischemia and intracranial hypertension, the indications for monitoring, and the available treatments directed toward preventing or mitigating their effects are crucial to any neurosurgeon involved in the care of patients with severe TBI.
Cerebral Ischemia
Pathophysiology
At autopsy, 80% of patients who die after TBI have evidence of cerebral ischemia.25 Cerebral ischemia is defined as cerebral blood flow (CBF) that is inadequate to meet the metabolic demands of the brain. While the brain constitutes only 2% to 3% of the body’s weight, it consumes 25% of its oxygen and receives about 20% of its cardiac output. The brain is almost completely reliant on adequate blood flow: nearly 95% of the brain’s metabolism is oxidative without a significant oxygen storage capacity and limited glucose and glycogen reserves. Delivery of oxygen to the brain is contingent on the oxygen content of the blood and CBF; likewise, delivery of glucose and other substrates of metabolism are based on CBF. In turn, a discrete metabolic autoregulatory mechanism exists to couple CBF with the cerebral metabolic rate of oxygen (CMRO2) so that adequate perfusion exists to support local metabolic needs. This principle is illustrated by the Fick equation CMRO2 = CBF × AVDO2, where AVDO2 is the arteriovenous difference in oxygen (in milliliters per deciliter) and is measured by subtracting the jugular venous oxygen content from the systemic arterial oxygen content. Under physiologic conditions, changes in CMRO2 are paralleled by changes in CBF, with AVDO2 remaining constant. In states in which decreased CBF occurs, as in systemic hypotension or when aberrant cerebral pressure autoregulation exists, the brain increases its extraction of oxygen to avoid ischemia. Current evidence suggests that episodes of inadequate CBF, evidenced by low jugular venous oxygen saturation (SjvO2) values of less than 50%, portend bad outcomes, because they are associated with increased morbidity and mortality in patients with severe TBI.26
Two other important homeostatic mechanisms are involved in prevention of cerebral ischemia. The first mechanism indirectly links CBF to CPP via the partial pressure of carbon dioxide (PaCO2) within the arterial system.27 In this process, arteriolar dilation and constriction occurs secondary to changes in PaCO2 and concomitant changes in the pH within the perivascular space, with hypercapnia causing vasodilatation and hypocapnia resulting in vasoconstriction. The second mechanism, referred to as pressure autoregulation, similarly links CBF to CPP.
CBF and CPP
Pressure autoregulation is a fundamental concept in cerebrovascular physiology and is distinct from metabolic autoregulation. Pressure autoregulation allows a constant supply of oxygen and metabolic substrates to be delivered to the brain by maintaining a constant CBF over a range of CPPs. CPP is defined as the mean arterial pressure (MAP) minus the intracranial pressure (ICP). Under normal circumstances, CBF remains relatively constant over a range of CPPs between 50 and 150 mm Hg, ensuring adequate oxygen delivery through a dynamic system of arteriolar vasoconstriction and dilation. Hence, in situations of low perfusion pressures, arteriolar vasodilation occurs, resulting in decreased cerebral vascular resistance and, in turn, increased CBF to normal. On the contrary, when a state of high perfusion pressure exists, arteriolar constriction leads to a decrease in CBF toward normal levels. At CPPs outside the autoregulatory threshold, however, CBF is unable to be maintained within its normal parameters—an exceedingly low perfusion pressure results in arteriolar collapse and a consequent decrement in CBF; hyperemia occurs at high CPPs outside of the limit of autoregulation due to vessels reaching their capacity for vasoconstriction (Fig. 132-1).
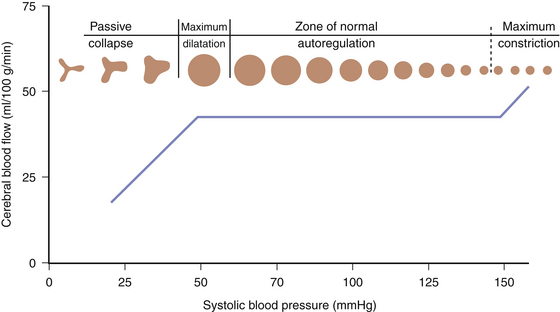
FIGURE 132-1 Cerebral pressure autoregulation.
(From White H, Venkatesh B. Cerebral perfusion pressure in neurotrauma: a review. Anesth Analg 107:979-988, 2008.)
Low CPP has been shown to be associated with higher mortality rates and increased morbidity among survivors following TBI,28–30 especially when systemic hypotension occurs concomitantly.31 Moreover, the normal response of cerebral arterioles to changes in CPP is frequently absent or impaired in 49% to 87% of patients after severe TBI.32–34 Aberrant pressure autoregulation at a range outside normal physiologic parameters may occur after TBI (Fig. 132-2),35,36 with significant consequences. Shifting the lower limit of autoregulation from 50 mm Hg to a supranormal range of 70 to 90 mm Hg could precipitate cerebral ischemia,29,37,38 which is especially noteworthy when considering that up to 60% of patients with severe brain injury experience a reduction in CBF during the first few hours following their trauma.39 Prolonged systemic hypotension or aggressive hyperventilation therapy would further exacerbate ischemia in vulnerable areas and could lead to subsequent cerebral edema. Conversely, autoregulation occurring at a lower-than-typical range of CPPs could result in malignant hyperemia or significant damage to the microcirculation, leading to secondary hemorrhages.40 Thus, absent or impaired cerebral pressure autoregulation represents a particularly significant risk factor for development of secondary brain injury, especially early in the postinjury period.41
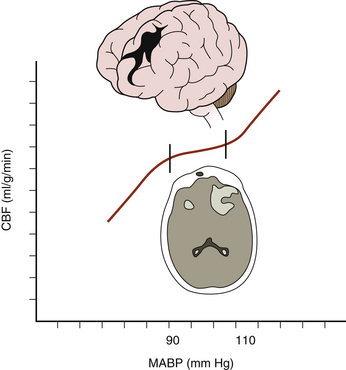
FIGURE 132-2 Aberrant cerebral pressure autoregulation after severe TBI.
(From Rangel-Castilla L, Gasco J, Nauta HJ, et al. Cerebral pressure autoregulation in traumatic brain injury. Neurosurg Focus 25:1-8, 2008.)
Based on the preceding information, CBF is assumed to have a linear relationship with CPP in managing patients with severe TBI. Other key factors that affect CBF include local factors such as physical compression of vessels by mass lesions or edematous brain, reduced cerebral metabolism of oxygen, and post-traumatic vasospasm.42,43 Nearly 33% of severely head-injured patients have CBF values near the ischemic threshold (less than 18 ml/dl) within the first 24 hours after primary injury,44,45 which is seen most often in individuals with diffuse cerebral edema or acute subdural hematomas.46–48 Low CBF values during the first 7 days after injury are associated with significantly higher mortality rates and increased morbidity.32,39,49–51
CPP thus serves as an important gauge of the adequacy of CBF and, in turn, must be monitored and maintained in patients with severe TBI. Through studies of brain tissue oxygen tension (PbtO2) and SjvO2 that correlate with unfavorable outcomes, some investigators have suggested a treatment and maintenance threshold CPP of 60 mm Hg,52,53 while other groups recommend a higher value of 70 mm Hg54,55 to avoid cerebral oxygen desaturation. Based on the results of cerebral microdialysis monitoring, others have discussed a lower CPP threshold of 50 mm Hg.56 In a prospective study in which a CPP-focused management strategy was used, Rosner et al.57 demonstrated a mean mortality rate of 21% in patients with severe TBI when CPP is maintained above 70 mm Hg, as compared to the 40% mortality rate in the TCDB series. Current practice guidelines dictate a threshold around 60 mm Hg due to concern over causing secondary brain injury in patients with aberrant pressure autoregulation, with further adjustments based on ancillary neurophysiologic monitoring of CBF, oxygenation, metabolism and the individual patient’s pressure autoregulatory status.58
Systemic Contributors to Ischemia: Hypotension and Hypoxia
Systemic hypotension and hypoxia have deleterious consequences on patient outcomes following TBI by contributing to secondary brain injury. Episodes of hypotension—defined as systolic blood pressure less than 90 mm Hg; the number of hypotensive events, inclusive of prehospital hypotensive events;13,59 and the increased total duration of such episodes—increase patients’ mortality and morbidity,60 with one study showing a 150% increased risk of mortality in patients with severe TBI who experienced at least one episode of hypotension.61
Similarly, hypoxia portends poor outcomes after TBI. Stochetti et al.62 noted that among severely brain-injured patients, those who had documented oxygen saturations of less than 60% had a mortality rate of 50%, with all survivors being severely disabled, in contrast to a mortality rate of 14.3% and a 4.8% rate of severe disability among nonhypoxic patients. As with hypotension, the number of hypoxic events, along with the duration of hypoxia,60 is predictive of worse outcomes. Establishing an airway early in the postinjury period is one important mechanism to prevent hypoxia; early intubation improves outcomes in patients with severe head injuries.63
Monitoring
Brain Oxygen Monitoring
Brain oxygen monitoring is a means of directly assessing the adequacy of oxygen delivery to brain tissue. Commonly employed modalities toward this goal include SjvO2 sampling via an internal jugular vein catheter directed toward the jugular bulb and direct brain tissue oxygen monitoring by a fiberoptic catheter. Episodes of jugular venous oxygen desaturation (SjvO2 less than 50%) are known to be associated with increased mortality and worse outcomes in patients after severe TBI.26,64 Low values of PbtO2 (less than 10-15 mm Hg) and the extent of their duration (more than 30 min) are also associated with high rates of mortality.65–67 The current guidelines for severe TBI management advocate treatment thresholds for SjvO2 and brain tissue oxygen tension at 50% and 15 mm Hg, respectively.68
Assessment of CBF via Other Modalities
Modalities aimed at directly measuring CBF include placement of thermal diffusion probes and noninvasive techniques such as transcranial Doppler sonography, xenon CT, nitrous oxide uptake, and positron emission tomography. Assessing the metabolic state of the brain via cerebral microdialysis gauges the adequacy of CBF.68 Finally, laser Doppler flowmetry and laser flow flowmetry, performed during surgery with the brain exposed, represent other technologies that measure CBF.69,70
Management Principles
Avoidance of secondary brain injury due to cerebral ischemia is a fundamental principle guiding care following severe head injury. The surgical team must ensure adequate CBF by keeping CPPs around 60 mm Hg, as well as avoiding systemic hypotension, hypoxia, and anemia. Maintenance of CPPs above 70 mm Hg via use of vasopressors and volume expansion has been shown to carry a significantly increased risk of developing acute respiratory distress syndrome, thus obviating the need to aggressively treat patients in such a manner.53,71
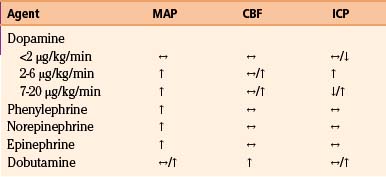
In patients with inadequate CPP, systemic causes of hypotension such as cardiac or spinal cord injury, tension pneumothorax, and bleeding must be ruled out. Intravascular volume must also be assessed via central venous pressure monitoring. Administration of intravenous isotonic fluids is a mainstay of management, but vasopressors are often necessary to maintain adequate CPP. The profiles of commonly used vasopressors are listed in Table 132-3. All listed agents are sympathomimetics, with phenylephrine being the most commonly used.72–75 Though these agents are of significant importance in the management of patients following severe head injury, their use may be associated with complications such as pulmonary edema and/or expansion of preexisting hematomas or contusions76 and therefore must be used with caution.
Delivery of oxygen to brain tissue is predicated on not only sufficient CBF but also oxygen content of the blood. As such, anemia can lead to cerebral ischemia. Despite evidence indicating that low hematocrit is associated with increased mortality77 and morbidity77a in patients with severe TBI, no consensus exists as to a particular threshold for transfusion of blood. While anemia is associated with worse outcomes, there is little support for the aggressive use of packed red blood cells to correct for anemia based on extant data: some studies have noted increasingly negative neurologic outcomes in patients with severe TBI who underwent transfusion,78 while other authors have shown no difference in mortality rates among patients treated under an aggressive transfusion protocol as opposed to those treated along a more restrictive one.79 Interestingly, studies have demonstrated that between 26% and 43% of patients with severe head injuries experience a paradoxical decrement in PbtO2 values in the setting of blood transfusion,80,81 with no effect seen on cerebral metabolism.81
Animal experiments, however, have shown that a hematocrit in the range of 30% to 33% provides an optimal balance between blood viscosity and oxygen-carrying capacity.82 Kee and Wood83 noted that brain tissue oxygen delivery begins to decrease at a hematocrit less than 33%, lending credence to the belief that maintenance of a hematocrit of approximately 30% to 33% is optimal.84 Most centers initiate transfusion of packed red blood cells at a hematocrit of less than 30%. As with all trauma patients, a hematocrit refractory to blood transfusion should prompt a search for extracranial bleeding sources.
Intracranial Hypertension
Pathophysiology
Intracranial contents include blood, brain, and cerebrospinal fluid (CSF). Under physiologic conditions, the intracranial vasculature contains 75 ml of blood, most of which is located within the venous system; CSF occupies a volume of approximately 75 ml and is primarily contained in the ventricles. The intracranial contents are enclosed within the rigid, nonexpansile skull. Based on the principle that the total volume available to intracranial contents is fixed, ICP is determined by the dynamic interplay among the individual components, as delineated by the Monro-Kellie doctrine. Therefore, an increase in an existing component—as seen with hydrocephalus or with post-traumatic cerebral edema—occurs with a concomitant decrease in another. The same concept holds true when a mass lesion occurs, because an additional component has been added to the intracranial space. In this way, in cases of hydrocephalus, post-traumatic cerebral edema, and even new mass lesion development, ICP may remain within normal limits through compensations of the intracranial vascular volume, CSF volume, and brain parenchyma (Fig. 132-3). Physiologic volume-buffering mechanisms exist through which intracranial vascular volume may reduce via increased arteriolar constriction and increased venous outflow, and CSF volume may decrease by being displaced into the thecal sac surrounding the spinal cord. It is estimated that in some instances upward of 100 to 150 ml of new intracranial volume can manifest without any significant increases in ICP, provided that the new mass does not present acutely.85 However, an uncompensated increase in one or more existing compartments or the presence of a new component results in an ICP higher than the normal range of 5 to 15 mm Hg.86,87
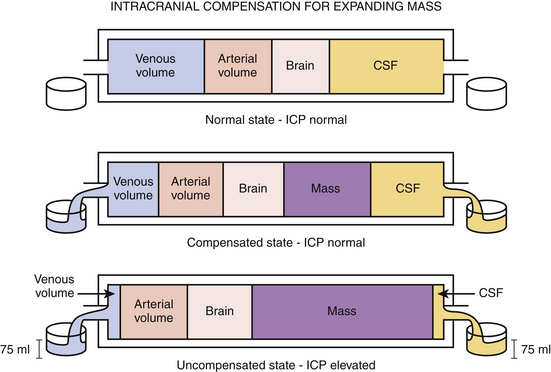
CSF normally circulates through the lateral ventricles, the interventricular foramen of Monro, the third ventricle, the cerebral sylvian aqueduct, and the fourth ventricle and out the foramina of Luschka and Magendie. It courses over the spinal cord and the brain and is eventually absorbed into the arachnoid granulations and superior sagittal sinus by means of passive transport. In situations in which there is no intracranial mass lesion, increased ICP following TBI occurs due to perturbations in CSF flow or CBV, as well as because of the development of vasogenic or cytotoxic edema.88 Breakdown products of blood secondary to traumatic subarachnoid hemorrhage preclude the passive absorption of CSF at the arachnoid granulations, which leads to an overall increase in CSF. Increase in CBV occurs when normal pressure and chemical autoregulatory mechanisms are compromised in severe TBI. Vasogenic and cytotoxic edema are the result of a disrupted blood–brain barrier and osmotic dysregulation in ischemic cells.
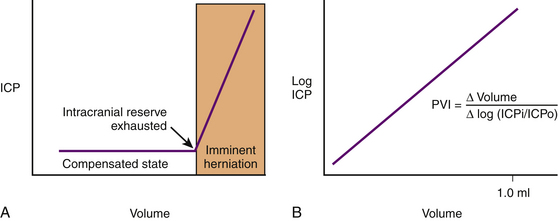
ICP rises exponentially when an uncompensated state exists. The shape of the pressure–volume curve reflects that as the ICP increases, the brain compliance decreases. The slope of the curve represents the intrinsic compliance of the brain without the influence of ICP. Marmarou et al.89 illustrated this by plotting ICP and volume on a semilogarithmic axis (thus removing the influence of ICP) and obtaining a linear relationship. The slope of the straight line is the pressure–volume index and is the amount of volume necessary to raise pressure 10-fold (Fig. 132-4).
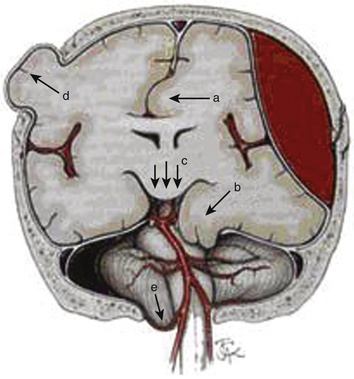
Elevated ICP, known as intracranial hypertension, leads to brain herniation and a decrement in the CPP (since CPP is defined as MAP-ICP). Herniation refers to an event in which brain parenchyma is displaced from its normal location through holes in the dura and skull. This may manifest as subfalcine herniation, uncal herniation, central downward herniation, external herniation of the brain through an open skull fracture, or tonsillar herniation (Fig. 132-5).
Monitoring
Intracranial hypertension is known to have a significant deleterious impact on patient outcome after severe TBI.10,59,90,91 The institution of modern neurocritical care protocols, predicated on monitoring of ICP and other important neurophysiologic parameters, has been credited with improved patient outcomes following severe TBI when compared to historic cohorts.92–94 The question of which patients benefit from ICP monitoring is based on risk stratification for developing intracranial hypertension. The presence of an abnormal head CT is one key factor in predicting which patients with severe brain injuries develop elevated ICPs. Narayan et al.95 found that the incidence of intracranial hypertension in this population is between 53% and 63%. Per findings of the same study and other investigations, individuals with a negative head CT scan on admission following severe TBI have a much lower risk of this occurring, with upper estimates in the range of 10% to 15%.14,96 Interestingly, among this particular subset of patients, the risk of developing intracranial hypertension was comparable to those with abnormal imaging findings if at least two of three risk factors (age over 40 years, unilateral or bilateral motor posturing, or systolic blood pressure less than 90 mm Hg) was present.95
Apart from their basic utility in guiding therapy involving the management of ICP and CPP, ICP monitors may also serve an important diagnostic purpose by indicating the development of worsening intracranial lesions, as a trend of progressively rising ICP values often correlates with the evolution of intracranial pathology.97
ICP Monitoring Devices, Indications, and Techniques of Usage
The Association for the Advancement of Medical Instrumentation has developed the American National Standard for Intracranial Pressure Monitoring Devices, which dictates performance requirements of currently available ICP monitoring devices. These parameters include the ability to (1) measure ICP readings from 0 to 100 mm Hg, (2) be accurate within 2 mm Hg in pressure ranges of 0 to 20 mm Hg, and (3) have a maximum error of 10% in pressure ranges of 20 to 100 mm Hg. Current technology allows for ICP measurement by external strain gauge transducers, microtip catheter strain gauge transducers, fiberoptic transducers, and coupling to a pneumatic transducer. There are many possible intracranial locations in which a particular ICP monitoring device may be placed, including within the ventricle, the brain parenchyma, the subdural space, the subarachnoid space, and epidurally. When considering which device to use, attention must be paid to accuracy, reliability, cost, and potential for patient morbidity. A ventricular catheter coupled to an external strain gauge transducer by fluid coupling is the gold standard.98
In the setting of severe TBI, placement of an external ventricular drain (EVD), or ventriculostomy, for ICP monitoring also allows for CSF diversion and is useful for patients believed to be at risk for development of intracranial hypertension or hydrocephalus. As such, it has a crucial role in guiding therapy, as well as serving as a therapeutic modality by reducing ICP on a sustained basis.99 The most common complications associated with the procedure are hemorrhage, infection, and malfunction. A recent meta-analysis estimated the risk of hemorrhage due to EVD placement as 5.7%, with only 0.61% of hemorrhages being clinically significant100; however, the significantly higher overall hemorrhage rates reported by other investigators highlight that obtaining postprocedural head CT scans may be a confounding factor.101 Lozier et al.’s102 review of ventriculostomy-associated infections (VAIs) cited a rate between 6% and 9%. Other important considerations involving VAIs include the significant association of CSF infections with increasing duration of EVD catheterization, the utility of administering prophylactic antibiotics prior to EVD placement, and the use of antimicrobial-impregnated catheters, which have been shown to reduce the risk of EVD-related infections.103 Based on the previously cited review of VAIs, prophylactic catheter exchange to reduce VAI rates is not efficacious. EVD malfunctions are usually attributable to obstruction in the fluid coupling mechanism by which the external strain gauge catheter transduces ICP, a complication estimated to occur in 2.5% of patients.104
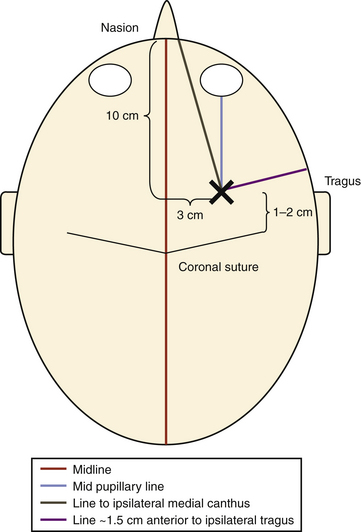
At our institution, right-sided EVDs are preferred to avoid the dominant hemisphere. The patient is maintained in a supine position with the head brought to the edge of the bed; the head of the bed is elevated to roughly 45 degrees. Hair clippers are used to widely shave the head on the same side of the EVD cannulation, and the head is prepared in the standard sterile fashion. Two lines are drawn on the scalp to establish Kocher’s point: one line in the midline from the nasion to a point 10 cm back and another from the previous point to a spot 3 cm lateral to it. This location is where the bur hole placement will occur and should be in the ipsilateral midpupillary line. Finally, two lines are drawn that guide the trajectory of ventricular cannulation: one line from Kocher’s point to the ipsilateral medial canthus of the eye and a second line from Kocher’s point to a point roughly 1.5 cm anterior to the ipsilateral tragus (Fig. 132-6). After administration of a local anesthetic containing epinephrine, a small linear skin incision is made down to bone and the periosteum is scraped off. A twist drill is used to open the cranium, bone fragments and dust are removed, and the dura and pia mater are pierced with a scalpel or sharp object. Using the demarcated lines as a guide, the trajectory at which the drill is used to create the bur hole is the same trajectory used for passing the ventricular catheter: aiming in the coronal plane toward the medial canthus of the ipsilateral eye and in the anteroposterior plane toward a point 1.5 cm anterior to the ipsilateral tragus. This trajectory is exceedingly important because it delineates the trajectory of the ventricular cannulation; if the bur hole is not created in an appropriate trajectory, the EVD cannot be optimally positioned due to its being limited by its bony opening, especially if the cranium is thick (Fig. 132-7). The ventricular catheter is passed to a depth of 6.25 cm using the previously described trajectory. The surgeon may alternatively pass the ventriculostomy using Dandy’s principle, which states that employing a trajectory directly perpendicular to the skull allows ventricular cannulation. Regardless, the ventricular catheter is never passed to a depth greater than 7 cm due to the risk of damaging the brain stem. Once the EVD is in place, it is tunneled through the skin away from the incision through a separate stab incision and is secured in place. A postprocedural head CT is then obtained.
There are cases in which ventricular cannulation can challenge even the most experienced of neurosurgeons. Our experience is to always use the landmarks and trajectory as outlined earlier, regardless of whether a mass lesion or significant shift is present. Most often, errant EVD passes are due to a lateral trajectory (Fig. 132-8), and the neurosurgeon must pay particular attention to avoiding an injury to the ipsilateral internal capsule. In cases in which the ventricle is believed to be cannulated with no egress of CSF, the neurosurgeon may consider attaching a small, plungerless syringe filled with sterile saline to the ventriculostomy catheter. If the ventricle is indeed cannulated, saline should flow from the syringe, through the catheter, and into the ventricle. If the neurosurgeon does not obtain CSF flow through the ventriculostomy after 3 passes, the EVD is left in place and a head CT scan is obtained to determine its position.
Other ICP monitoring devices are frequently used in centers, sometimes as an adjunctive means of monitoring ICP when they are placed in tandem with an EVD and at other times when slit-like ventricles preclude EVD placement. The scalp incision is made in the exact location as for a ventriculostomy, with a smaller drill bit and small stylet used to puncture the dura. When using microtip strain gauge or fiberoptic parenchymal monitors, the probe is advanced between 1 and 2 cm into the brain parenchyma. The microtip strain gauge probe must be tunneled through the skin via a separate stab incision and then be secured in place. However, the fiberoptic monitor can be held in place by a fixation bolt screwed into the skull. The reduced accuracy and reliability, increased propensity for malfunction, and high cost in comparison to EVD placement are factors that must be considered prior to using these alternative ICP monitors.
Management Principles
Current practice guidelines recommend that an ICP treatment threshold of 20 mm Hg be used,105 based on investigations showing that the incidence of morbidity and mortality among severely brain-injured patients is significantly associated with ICPs exceeding 20 mm Hg.59,95,106 The rationale behind treatment of intracranial hypertension is to prevent cerebral ischemia by ensuring adequate CPP and preventing brain herniation. ICP values, however, must be considered as part of the larger clinical picture, together with exam findings and imaging findings, such that established treatment thresholds are considered guidelines that do not preclude treatment at lower values, especially in light of herniation potentially occurring at ICP values less than 20 mm Hg.107
Several methods may be employed to decrease ICP. A stepwise, protocol-based approach to treatment of intracranial hypertension is best suited to prevent the utilization of an excessive number of modalities concomitantly. Maneuvers to optimize venous outflow from the intracranial vascular volume are first used, followed by pharmacotherapy and CSF diversion (Fig. 132-9).
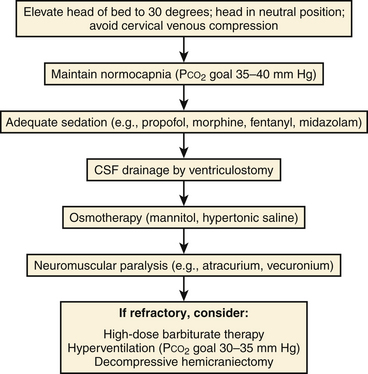
Positioning
Maintaining the head in a neutral position with elevation of the head of the bed to 30 degrees decreases the intracranial blood volume by maximizing venous return from the brain while preserving CPP and CBF.108–110 ICP reduction may thus be readily achieved without increasing the risk of cerebral ischemia.
Cervical Collar Removal
Of pertinence to patients with severe brain injuries is cervical spine clearance, and subsequent cervical collar removal, on ICP reduction. Studies have shown that patients involved in blunt trauma with a GCS score of 3 to 8 points have a higher incidence of cervical spine injuries as compared to other trauma patients,111,112 with rates of 10% to 14%.113,114 Recent investigations have demonstrated the efficacy of a negative helical cervical CT scan in cervical spine clearance among obtunded trauma patients without neurologic deficits that could be potentially explained by spinal cord pathology.115–118 Therefore, cervical spine magnetic resonance imaging (MRI) is unnecessary to remove external cervical orthoses designed for cervical immobilization, which are known to cause a significant and sustained elevation in baseline ICP.119–122 Hence, our institutional policy is that among patients with severe TBIs and no neurologic deficits that can be associated with injury to the spinal cord, cervical collars are expediently removed following a negative helical CT scan of the cervical spine.
Maintenance of Normocapnia
As previously mentioned, alterations in arterial PCO2 result in variations in arteriolar caliber with a consequent effect on CBF and ICP based on the phenomenon of CO2 reactivity. The physiologic mechanism by which the caliber of cerebral arterioles varies involves changes in the pH of perivascular tissue secondary to PaCO2 levels.27 In turn, hypoventilation increases PaCO2 and ICP, whereas hyperventilation reduces ICP at the expense of CBF. Maintenance of normocapnia (PaCO2 between 35 and 40 mm Hg) provides a balance between ensuring adequate CBF to avoid ischemia and preventing the development of intracranial hypertension associated with hypoventilation.
Sedation
A number of pharmacologic agents are used in patients with severe TBI, with the goal of treating pain and agitation and minimizing the effect of a multitude of noxious stimuli, which may contribute to elevations in blood pressure, body temperature, and ICP and resistance to controlled ventilation.123 In addition to using sedative agents in these patients, the surgeon often needs to determine their efficacy and titrate accordingly. Bispectral index monitoring is a useful tool to accomplish this, because it can provide objective information as to the degree of sedation in these critically injured patients.124 The effects of commonly used analgesic and sedative agents on MAP, CBF, and ICP are shown in Table 132-4.
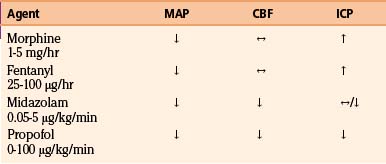
Propofol, a short-acting sedative anesthetic with a rapid onset of activity, is frequently used and has been shown to have superior effects on ICP reduction compared to narcotic-based agents.125 In addition, studies have demonstrated that propofol decreases cerebral metabolism and oxygen consumption, has anti-inflammatory properties, and is an antioxidant, leading to the possibility of its having a neuroprotective effect.126,127
Narcotics have also commonly been employed for providing sedation and analgesia, both alone and with other agents. Morphine sulfate has been shown to provide excellent analgesia but has poor sedative properties. Tachyphylaxis may be seen that can lead to a need for dose escalation and a potential for withdrawal when therapy is discontinued.123 Rapidly metabolized synthetic narcotics such as fentanyl, sufentanyl, and remifentanil are being used more often due to their brief duration of action. However, increases in ICP and decreases in MAP seen with fentanyl and sufentanyl usage mitigate their utility.128,129
Benzodiazepines have the untoward side effect profile of being neurodepressants and exhibiting long half-lives and thus are not considered ideal sedative agents in the brain-injured population. Midazolam, however, is a commonly employed agent in the management of patients with head injuries due to its short half-life compared to other benzodiazepines and its relatively minimal effect on hemodynamics, especially in comparison to propofol. Midazolam exhibits a similar safety and efficacy profile versus propofol,130,131 with a decrement in the cerebral metabolism and oxygen consumption seen with benzodiazepine administration being a potential additional benefit.
Dexmedetomidine, an alpha-2 agonist, is one of the newer agents to be studied and used in neurosurgical patients. Increased CPP and decreased ICP have been seen with its use, which, combined with a benign pharmokinetic profile,132 warrants further study for its use in patients with severe TBI.
Hyperosmolar Therapy
Osmotic Diuretics
Intravenous administration of osmotic diuretics has been used for several decades for reduction of ICP. Originally used by Wise and Chater in the 1950s,133 mannitol not only works to reduce ICP but also results in the improvement of MAP, CPP, and CBF.134 A randomized, controlled trial by Schwartz et al.135 comparing administration of mannitol and pentobarbital in patients with high ICP due to head injury showed superior ICP reduction, CPP preservation, and decreased mortality in those given mannitol. Similarly, improved ICP reduction and better CPP and SjvO2 values were seen in a subset of severe TBI patients who were given intravenous mannitol, in comparison to those treated with an EVD or hyperventilation alone.136
The effect of mannitol on ICP reduction is believed to be attributable to two separate factors, one involving its rheologic effect and the other pertaining to its osmotic effect. The rheologic properties of mannitol account for its ability to rapidly decrease ICP by mediating an expansion in the volume of plasma, which results in a reduction of the hematocrit and increased red blood cell deformability.137,138 The resultant decrease in blood viscosity leads to an increase in CBF and a subsequent decrease in ICP due to arteriolar vasoconstriction, as dictated by pressure autoregulation.139,140 Mannitol is able to reduce ICP over the longer term by creating an osmotic gradient by which water diffuses from the brain parenchyma into the intravascular compartment.141 Current guidelines support the administration of mannitol via bolus dosing of 0.25 to 1 g per kilogram of body weight.142
An important side effect to consider when administering mannitol is the potential for developing acute renal insufficiency secondary to acute tubular necrosis. Serum osmolarity of greater than 320 mOsm is a key risk factor for this to occur.143 Mannitol use may also lead to derangements in a patient’s intravascular volume status and concentrations of various plasma electrolytes; as such, blood urea nitrogen, creatinine, and sodium levels should be monitored closely. In addition, prolonged mannitol administration may result in a reversal of the osmotic gradient due to its crossing the blood–brain barrier and being taken up into edematous brain tissue, a scenario that is more likely if mannitol is administered as a continuous infusion.144
Studies have also shown that the use of furosemide (0.7 mg/kg) as an adjunctive ICP-lowering agent with mannitol elicits a more marked and sustained decrement in ICP.145 The usual intravenous dose is between 20 and 40 mg.
Hypertonic Saline
Hypertonic saline solutions are believed to act like mannitol and reduce ICP through both a rheologic and an osmotic effect.146 Reports of hypertonic saline’s use in concentrations ranging from 7.5% to 23.4% NaCl have shown it to be effective when given as boluses to brain-injured patients with intracranial hypertension refractory to mannitol administration.147–149 Giving hypertonic saline boluses to individuals with severe TBI may potentially be more efficacious when compared to mannitol in treating elevated ICPs, because they achieve a more robust decrement in ICP,150,151 with more patients responding to hypertonic saline as an ICP-lowering agent than to mannitol.150 Moreover, recent data have demonstrated it to be capable of eliciting improved CPP and PbtO2 values.152
The primary justification for using hypertonic saline solutions in lieu of mannitol resides in the belief that the intact blood–brain barrier is less permeable to saline than it is to mannitol. Therefore, putatively, hypertonic saline is a better, longer-lasting osmotic agent that does not carry the risk of inducing rebound intracranial hypertension that comes with administering mannitol (as described earlier). Potential complications seen with the use of hypertonic saline include developing central pontine myelinolysis in patients with preexisting chronic hyponatremia, triggering or aggravating electrolyte abnormalities, prompting seizures, causing pulmonary edema in patients with a history of cardiac or pulmonary disease, and possibly resulting in coagulopathy and phlebitis.146,153
Intracranial Hypertension Refractory to First-Line Treatment Modalities: Second-Line Treatments
Standard therapies in managing intracranial hypertension involve the use of the measures previously described in this section. When ICP is refractory despite maximal treatment, which occurs in upward of 25% of all patients with severe TBI,154–157 other methods must be employed. These second-line treatments include neuromuscular paralysis, hyperventilation, barbiturate-induced burst suppression, and decompressive craniectomy, all of which are associated with potentially significant morbidity.
Neuromuscular Paralysis
The physiologic rationale for using paralytic agents in the treatment of elevated ICP is predicated on their ability to reduce intrathoracic pressure, thus maximizing venous outflow and decreasing ICP. Pharmacologic paralysis prevents any muscle activity, in the form of posturing movements, shivering, or “bucking” the ventilator, from raising the respiratory muscle tone and triggering an increase in intrathoracic pressure.158 Hsiang et al.159 reviewed data from the TCDB and concluded that neuromuscular paralysis should only be employed for the management of refractory intracranial hypertension in patients with severe TBI. Early, routine, long-term use of such agents did not improve outcome and led to longer intensive care unit stays and higher rates of infectious complications, such as pneumonia and potentially sepsis.
If a neuromuscular blockade must be employed to address refractory intracranial hypertension in a patient with severe TBI, agents with a short half-life (e.g., vecuronium) are preferable since long-lasting pharmacotherapeutics preclude the patient’s participation in a neurologic examination.160,161
Hyperventilation
Hyperventilation therapy for the management of intracranial hypertension was introduced in the 1950s by Lundberg, a Swedish neurosurgeon, and his colleagues.162 The premise behind hyperventilating patients with high ICPs is based on the responsivity of arteriolar smooth muscle in the cerebral vasculature to changes in arterial CO2. CBF changes 2% to 3% for each millimeter of mercury change in PaCO2; thus, hypercarbia from hypoventilation causes reactive vasodilation and increased CBF, whereas hypocarbia from hyperventilation results in vasoconstriction and a decrement in CBF with a subsequent reduction in ICP.163 Even though pressure and metabolic autoregulation may be impaired after TBI, CO2 reactivity often remains intact.32,164
Though hyperventilation is an efficacious means of decreasing ICP, it does not have a role as a standard therapy in the management of patients with intracranial hypertension due to the potential of causing further brain injury secondary to ischemia. As noted earlier, studies using xenon CT39,165 and implantable thermodiffusion blood flow probes166 to measure CBF have demonstrated that patients with severe TBI have significant reductions in CBF in the first 24 hours after injury. Combined with evidence of cerebral ischemia seen in post mortem histologic analysis of patients with severe TBI, these data caution against aggressive hyperventilation in this patient population.25,167,168 In turn, worse clinical outcomes are seen among severely head-injured patients who are subjected to prophylactic hyperventilation during the first 5 days after the injury.169 Hence, hyperventilation should be avoided during the critical initial 24-hour postinjury period, and limiting the use of hyperventilation in patients with severe brain injuries could prevent iatrogenic cerebral ischemia from occurring and may potentially help improve neurologic recovery.170 Though the routine use of hyperventilation is not indicated in the management of intracranial hypertension in patients with severe TBI, it may be used as a short-term maneuver to reduce ICP in the event of acute neurologic deterioration and impending herniation while other more definitive measures are taken.
Barbiturate-Induced Burst Suppression
The ability of barbiturates to lower ICP has been known since the 1930s.171 The mechanism by which this reduction in ICP occurs is not entirely understood but likely involves mediating cerebral vasoconstriction,172 reversibly reducing CMRO2,173 and preventing the onset of numerous excitatory neurotoxic cascades. Studies investigating clinical outcomes in patients who received barbiturate therapy have been inconclusive. Though prophylactic use of barbiturates has no clinical benefit,135,174 Eisenberg et al.106 illustrated that in the setting of intractable intracranial hypertension, patients whose ICPs were responsive to barbiturate therapy had a significantly reduced mortality.
The administration of barbiturates such as pentobarbital is generally performed concomitant with continuous electroencephalogram monitoring. Maximal reductions in CBF and cerebral metabolism are seen when electrographic burst suppression of cerebral activity is achieved.175
A key side effect to consider prior to barbiturate administration is the potential to precipitate systemic hypotension. A meta-analysis by the Cochrane group noted that barbiturate infusion was associated with a fall in blood pressure in 25% of patients. In turn, the significant risk of causing hypotension—a factor known to be a harbinger of poor neurologic outcomes in patients with severe TBI—would offset any beneficial effect seen with ICP reduction.176 No clear improvement in outcome has been seen in studies involving the use of barbiturate therapy, which has led to the Brain Trauma Foundation stipulating that high-dose barbiturate therapy should only be instituted in cases of treating intracranial hypertension refractory to all other medical and surgical treatment alternatives.123 Furthermore, proper monitoring of cardiovascular status with a Swan-Ganz catheter and maintenance of hemodynamic stability are paramount if barbiturates are to be used.
Decompressive Craniectomy
Background
Surgical intervention to reduce ICP in the setting of severe TBI and medically refractory intracranial hypertension has experienced a resurgence in popularity over the past several years. Extrapolating from the Monro-Kellie doctrine, removing a portion of the cranial vault removes the volumetric constraint provided by the rigid, nonexpansile skull and thus provides a means of decreasing ICP when an uncompensated state exists. Recent data by Aarabi et al.177 supports this concept, as they showed a significant reduction in mean ICP from 24 to 14.6 mm Hg after decompressive surgery was performed in severe TBI patients with malignant intracranial hypertension.
The utility of decompressive craniectomy in the treatment of increased ICP was initially described in 1901 by Kocher.178 During the early 1900s, many other preeminent neurosurgeons provided further details about how to properly perform cranial decompressions. Horsley endorsed a technique in which a large portion of the skull was removed in the right temporal region with subsequent duraplasty.179 Cushing described a procedure in which subtemporal and suboccipital decompression was achieved after creating a 6 × 8 cm bone window, followed by performing dural incisions and subsequent closure of the temporalis muscle, fascia, galea, and scalp.180 The first large experience of decompressive craniectomy for treatment of malignant ICPs (known as therapeutic, or delayed, decompressive craniectomy) was reported in 1971 by Kjellberg and Prieto, who used a bifrontal approach.181 In their experience, only 22% of patients who underwent this procedure following TBI survived. Due to association with a high rate of complications and poor outcomes,182–184 performing delayed decompressive craniectomies for patients with severe TBI was nearly abandoned by neurosurgeons.
Recent series have shown more promising results. Gaab et al.185 reported their prospective results of the use of bifrontal or hemicraniectomy, in which they excluded patients older than 40 years and those with a GCS score of fewer than 5 points. They reported a 14% mortality rate and a 38% rate of “full restitution.” Polin et al.186 noted favorable outcomes in 60% of patients undergoing a modified Kjellberg procedure within 48 hours of injury with a GCS score between 3 and 8 points and intractable hypertension but with ICP of less than 40 mm Hg. However, these studies must be taken in perspective, because subsequent analysis has shown no evidence of the procedure achieving a decrement in the risk of death or disability following head injury in the adult population. Still, the same review highlighted the possibility of performing a decompressive craniectomy as a last-ditch rescue therapy in patients whose ICPs have failed to respond to maximal medical treatment.187
Importance
Decompressive craniectomy is an important intervention in the management of TBI. It is highly effective at reducing ICP and thus an important higher-treatment strategy for patients with medically refractory intracranial hypertension. A long-standing controversy in the management of severe TBI is whether early decompressive craniectomy is associated with improved long-term outcomes. The decompressive craniectomy trial (DECRA trial)188 was theoretically designed to address this question in the field; however, its numerous glaring flaws in study design and statistical analysis render its results irrelevant. In the DECRA trial, investigators from 15 centers across several continents randomized patients with diffuse brain injuries to early decompressive or medical management. However, the median ICP in the hours prior to randomization was 20 mm Hg, which raises the important question of whether the patients in the study truly had intracranial hypertension and whether the patients should have ever been considered for surgery. Furthermore, 27% of the patients randomized to surgery had bilateral nonreactive pupils, compared to only 12% of the patients in the medical group. This key discrepancy was statistically significant, and when accounting for this between-group difference, there was no difference in outcomes between patients in the decompressive craniectomy and medical management groups. Other criticisms of the trial include the investigators performing their analysis via an “intention-to-treat” design, despite an 18% crossover rate to surgery in the patient group initially randomized to medical management. Additional issues with the DECRA trial include managing ICPs for 15 minutes prior to randomization, changing the study design at the midpoint analysis instead of stopping the trial for futility, and enrolling in the study only 4% of screened patients over 7 years. The DECRA trial contains no data or valuable information to inform modern management of TBI and thus should be ignored by practitioners evaluating treatment options for severe TBI.
Adjunctive Therapy: Lumbar CSF Drainage
As previously mentioned, cannulation of the ventricular system with an EVD allows for ICP monitoring and is a first-line therapy utilized to lower ICPs in patients with severe TBI, because it provides a mechanism by which ICP reduction is achieved through CSF diversion. Lumbar CSF drainage as a means of treating intracranial hypertension was heretofore considered dangerous, as initially noted by Cushing, because of its risk of precipitating transtentorial or tonsillar herniation.189 Recent studies have posited that in cases of intracranial hypertension refractory to standard maneuvers directed toward lowering ICP, CSF drainage via placement of a lumbar drain is an effective means of achieving lasting ICP reduction, with a potentially acceptable risk of lethal outcome because of herniation at a rate of 6%. Improvement in neurophysiologic parameters such as PbtO2 and CBF, as well as a lower mortality rate compared to historic cohorts among TBI patients with malignant ICPs treated with lumbar CSF drainage, has been seen.190
Therapies to Avoid: Steroids
The majority of evidence to date indicates that administration of steroids does not affect outcome or lead to a reduction in ICP in patients with severe TBI. Individual trials involving the use of triamcinolone,191 tirilazad,192,193 dexamethasone,194 and methylprednisolone,195 as well as a large meta-analysis of glucocorticoid administration in the setting of TBI,196 have led to a level I recommendation by the Brain Trauma Foundation stating that the use of glucorticoids in patients with severe TBI is not supported.197 The use of steroids has been shown to be associated with increased mortality,195 with side effects including gastrointestinal bleeding and hyperglycemia.198
Management Paradigms
The goal of an ICP-based approach to the management of patients with severe TBI is to prevent intracranial hypertension and, in turn, avoid any deleterious effects on CPP and guard against the potential of brain herniation. As previously mentioned, an ICP treatment threshold of 20 mm Hg is recommended, with studies showing increased morbidity and mortality among individuals with severe brain trauma associated with ICPs that exceed such a level. A stepwise protocol in ICP management was illustrated in Fig. 132-9.
A CPP-based paradigm to severe TBI management involves focusing therapy on establishing and maintaining a defined CPP threshold, rather than managing ICP values. The concept behind this particular schema is that decrements in CPP, which occur as a result of increased ICPs or decreased MAPs, result in compensatory arteriolar vasodilation and subsequent increased CBVs, which lead to further increases in ICP and further compromise of CPP (Fig. 132-10). Establishing a CPP threshold from the outset thus would prevent such a vicious cycle from developing. The aforementioned work by Rosner et al.57 showing improved outcomes in patients whose CPP treatment threshold was 70 mm Hg has led to CPP preservation being a key component of TBI management. Current guidelines, however, call for a CPP threshold of around 60 mm Hg due to the significant risk of developing acute respiratory distress syndrome with aggressive treatment above 70 mm Hg.72
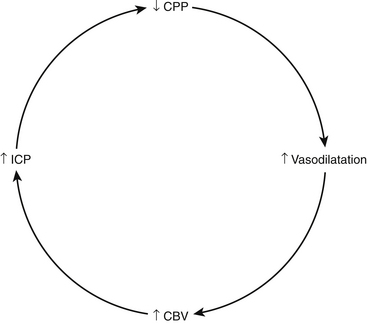
FIGURE 132-10 CPP-ICP relationship in preserved autoregulation.
(From Rosner MJ. Introduction to cerebral perfusion pressure management. Neurosurg Clin N Am 6:761-773, 1995.)
Novel Considerations in Severe TBI Management: The Utilization of a Brain–Tissue Oxygen-Directed Paradigm
Inadequate oxygen delivery to the brain is associated with higher mortality rates and worse outcomes in severe TBI patients. Several recent studies have shown reduced mortality rates,199–201 better overall outcomes,200 and an increased likelihood of a good outcome 6 months after the injury200,201 in patients treated with a PbtO2-directed paradigm (maintaining PbtO2 above either 20 or 25 mm Hg), in addition to adhering to standard ICP (less than 20 mm Hg) and CPP (more than 60 mm Hg) management protocols. While further investigation is warranted, the use of fiberoptic brain tissue monitors to ascertain the adequacy of CBF and guide therapy to prevent ischemia appears to be beneficial.
Systemic Medical Management
Electrolytes and Fluid Balance
Upward of 60% of patients have electrolyte abnormalities following TBI.202 Derangements in serum sodium levels are the most common and most pertinent from the standpoint of their role in maintaining neurologic homeostasis. Post-traumatic hyponatremia is associated with worse outcomes and longer hospital stays;203 thus, recognizing its etiology is crucial so that proper therapy may be directed accordingly. Hyponatremia commonly occurs in TBI patients as a consequence of the syndrome of inappropriate antidiuretic hormone (SIADH) or due to cerebral salt wasting. Management of SIADH includes fluid restriction and possible administration of demeclocycline. Patients with cerebral salt wasting often require sodium supplementation in the form of hypertonic saline or oral salt tablets. A key feature that distinguishes between these two entities is the patient’s fluid status: individuals with SIADH are euvolemic or hypervolemic, while those with cerebral salt wasting are generally hypovolemic. Regardless of etiology, slow correction of acute hyponatremia is a must to avoid iatrogenic central pontine myelinolysis.
Hypernatremia is also seen among patients who have suffered TBI. Central diabetes insipidus from pituitary stalk dysfunction often occurs with increased severity of brain injury and has been estimated to be the cause of hypernatremia in nearly 20% of cases, with significant implications on mortality.204 Administration of hypotonic fluids, desmopressin (DDAVP), and free water boluses may be considered in managing this problem.
Normal saline (0.9% NaCl) remains the standard choice of maintenance fluid in patients with head injury after initial resuscitation. As a rule, dextrose is excluded from the solution due to concern for causing hyperglycemia, which has been shown to worsen prognosis in patients with TBI.13,205 Hypotonic fluids are not an acceptable choice as a maintenance fluid because of their potential to exacerbate cerebral edema. Fluid choice, however, must be dictated by the clinical situation, as solutions of varying tonicity have to be used in cases of SIADH, cerebral salt wasting, and diabetes insipidus.
Glycemic Control
Elevated glucose levels are often seen among critically ill patients, regardless of whether they have a previous history of diabetes mellitus. Admission hyperglycemia has been demonstrated to correlate with increased risk of infection, longer lengths of stay in the intensive care unit and in the hospital, and higher mortality rates.206–209 Tight glycemic control and maintenance of blood glucose below 110 mg/dl has been shown to decrease morbidity and mortality in intensive care patients; thus, intensive insulin therapy is a fundamental principle in the management of the critically ill.210 Patients with severe head injuries commonly have supranormal glucose levels, likely secondary to a systemic surge in the sympathoadrenal axis and the resultant exaggerated catecholamine release.211 Hyperglycemia, both early and persistent, in patients with severe TBI has been shown to be associated with injury severity and portends a poor outcome.13,205 The potential pathophysiologic process involves the development of intracellular acidosis secondary to the conversion of glucose into lactate via anaerobic metabolism.205 Frequent glucose monitoring and aggressive glycemic control, starting with administration of sliding scale insulin, to maintain blood glucose levels below 130 mg/dl are standard parts of the severe TBI protocol at our institution.
Hyperpyrexia
Fever increases cerebral metabolism. Secondary ischemia may occur when metabolic autoregulation is uncoupled and CBF does not match CMRO2. Fever has been shown to be a significant predictor of increased mortality in patients with head injury60 and must be aggressively controlled with antipyretics and cooling blankets. Aggressively pursuing an infectious workup to determine the etiology of fever is of the utmost utility.
Altered Endocrine Function
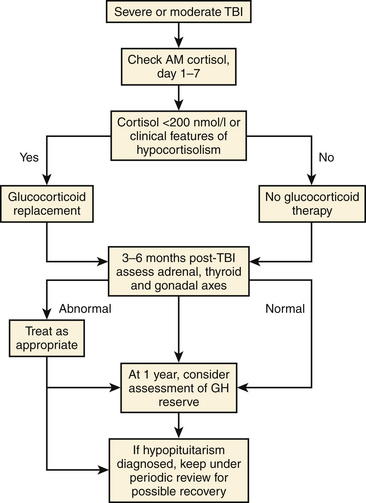
As mentioned previously, homeostatic mechanisms regulating water balance and electrolyte concentrations are frequently disturbed following TBI. Disorders of endocrine function secondary to damage to the anterior pituitary gland or the pituitary stalk result in aberrant circulating levels of hormones regulated by the anterior pituitary. Originally described in 1918,212 post-traumatic hypopituitarism was initially believed to be rare,213 but more recent studies have shown widespread neuroendocrine dysfunction in the acute post-TBI period.214,215 At our institution, a neuroendocrine panel is drawn on post-trauma days 3 and 7 in our severe TBI patients. Serum cortisol, insulin-like growth factor-1, free T4, thyroid stimulating hormone, estradiol and prolactin levels in females, and testosterone levels in males are checked. Of particular importance is adrenal insufficiency in the acute phase of TBI because of its potential to have life-threatening consequences in critically ill patients, manifesting as hyponatremia, hypotension, and hypoglycemia. Dramatic responses have been reported subsequent to glucocorticoid replacement in these individuals.216 Early post-traumatic pituitary hypopituitarism resolves in a significant proportion of patients, generally by 6 months after the injury.217
Post-traumatic hypopituitarism occurs in roughly 25% of long-term survivors of TBI,217 with growth hormone and gonadotropin secretion being most commonly affected. Anterior pituitary hormone dysfunction measured more than 12 months after the injury has been reported to be as high as 59% among patients with severe TBI.218 Other studies have noted that severe TBI, presence of higher ICP values, and longer intubation times are key factors associated with long-term prevalence of post-traumatic hypopituitarism.219
Interestingly, post-traumatic pituitary dysfunction can evolve: patients who are not affected in the acute, post-injury period can exhibit hormonal deficiencies later.217 Particular imaging findings, including CT evidence during the first 3 months after injury confirming the presence of brain swelling or a fracture of the cranial base or a MRI showing an empty sella within 12 months after the injury, are predictors of developing post-traumatic hypopituitarism.220
The presence of a neuroendocrine disorder is of tremendous clinical significance, not only from a general physiologic standpoint but also from a neurocognitive and neuropsychiatric standpoint.221,222 In the presence of a concomitant neurologic deficit, the deleterious effect of a dysfunctional hypothalamic–pituitary axis on a patient’s quality of life and capacity for rehabilitation confers significant morbidity. Improved cognition and mental well-being seen in patients with post-traumatic hypopituitarism after hormone replacement reinforce the importance of diagnosis and treatment of this disorder. One such algorithm is seen in Fig. 132-11.
Nutrition
Numerous studies over the past 25 years have highlighted the systemic hypermetabolic state induced in response to polytrauma and severe TBI. The overall state of catabolism is evidenced by patients’ supranormal caloric requirements in the acute, postinjury period: patients with severe head injuries expend upward of 140% to 160% of their basal rate, while those in barbiturate-induced burst suppression and patients who have undergone neuromuscular paralysis may require between 100% and 120% of normal resting levels.223
Nutritional support should be established as soon as possible, and patients should attain full caloric replacement by postinjury day 7.224 Severe undernutrition in the first 2 weeks after injury is associated with a much higher mortality rate as compared to the rate for patients who have had full caloric replacement by postinjury day 7.225 Protocols involving early initiation of feeding are correlated with decreased rates of infection and complications;226 regardless, to reach goal caloric replacement with the proper time afforded for advancement of parenteral or enteral nutrition, feeds should be started no later than 72 hours after injury.
The TBI literature is sparse with regards to research delineating the specific composition of feeds. To achieve an appropriate nitrogen balance, 15% of caloric intake should come from protein; otherwise, the appropriate amount of fats and carbohydrates constituting feeds has not been established among patients with severe brain injuries. The administration of glutamine, arginine, omega-3 fatty acids, nucleotides, and branch chain amino acids has shown promise in enhancing the immune response among critically ill patients,227–229 but the applicability of the results specifically in the TBI population is unknown.
Gastric, jejunal, and parenteral methods of feeding represent the three options available for nutritional support. Orogastric tubes are preferred to the nasogastric route in the presence of skull base fractures or sinus disease. Altered function of the lower esophageal sphincter and slowed gastric emptying are mitigating factors in gastric feeding, especially when weighing the risk of aspiration. Long-term feeding may require placement of a percutaneous endoscopic gastrostomy tube or jejunal tube. Although the enteral route decreases the risk of bacterial translocation and sepsis, parenteral nutrition may serve as a temporary route for nutritional support until gastroparesis resolves.
Infections
Higher rates of systemic infection are correlated with increased severity of brain trauma, as well as the presence of concomitant chest trauma.230 Upward of 70% of mechanically ventilated patients can develop pneumonia.231 The current recommendation of the Brain Trauma Foundation stipulates that administration of periprocedural antibiotics for intubation is warranted,232 based on evidence showing a decrement in the incidence of pneumonia with such a protocol.231 Unfortunately, prophylactic antibiosis during intubation in patients with severe TBI has not been shown to reduce mortality. Studies in which tracheostomy was performed within a week of injury similarly found no effect in reducing patient mortality in the severe TBI population, with no change in the rate of nosocomial pneumonia. The utility of early tracheostomy seems to lie in its reduction of the duration of mechanical ventilation in these critically ill patients.233 Caution is also warranted in considering prolonged antibiotic therapy in ventilator-dependent patients with severe TBI because of the potential to select for highly resistant bacterial species.
Invasive cerebral ICP monitoring confers a significant infection risk to the critically ill patient. Some studies have quoted rates of infection associated with placement of ICP monitoring devices as high as 27%.234 Interestingly, bacterial colonization of intraparenchymal monitors occurs at a higher rate than of EVDs.235 As previously mentioned, ventricular catheter exchange has not been found to be an efficacious method of reducing infection rates; in addition, prophylactic antibiotic use during ventricular catheter placement has not been recommended as a method of reducing infection. The key to avoiding infectious complications is predicated on using proper sterile technique when placing EVDs and other invasive intracerebral monitoring devices and, once the devices are placed, minimizing the frequency with which they are manipulated or flushed.
Pharmacologic Prophylaxis
Providing appropriate medical care to patients with severe TBI includes administration of pharmacologic agents to prevent potential complications. Gastric ulcers often form in patients after severe brain trauma, and in general, stress-induced ulceration of the gastric mucosa is a common complication among critically ill patients, with mortality rates of upward of 50% in patients requiring neurosurgical intensive care.236 Histamine type 2 (H2) antagonists or proton pump inhibitors are the most commonly used medicines for gastrointestinal prophylaxis, with recent data showing that both medications have a similar efficacy in preventing bleeding in the upper gastrointestinal tract in patients receiving mechanical ventilation.237 Given the relative cost effectiveness of H2 antagonists and the long-established effectiveness of these agents in the severe TBI population,238 the policy at our institution is to administer an H2 antagonist for gastric ulcer prophylaxis. Importantly, neutralizing gastric acid pH potentially places the patient at increased risk of developing nosocomial pneumonia; sucralfate, a medicine that strengthens gastric mucosa, may mitigate this risk.
The administration of anticonvulsants to prevent the development of post-traumatic seizures (PTSs) has been a mainstay in the management of brain trauma for the past several decades because, when not given pharmacologic prophylaxis, the incidence of clinically observed PTSs in the TBI population is relatively high: early PTS (those occurring within 7 days of injury) is estimated to occur in between 4% and 25% of patients, and late PTS (those occurring after postinjury day 7) happens in 9% to 42% of patients.240–242 Risk factors for PTS development include the presence of cortical contusions; depressed skull fracture; subdural, epidural, or intracranial hematoma; penetrating injury; seizure within 24 hours of injury; or initial GCS score of fewer than 10 points.242,243 Seizures increase ICP and CMRO2; alter blood pressure, CBF, and oxygen delivery; and cause excess neurotransmitter release. Temkin et al.244 demonstrated that administration of phenytoin to brain-injured patients resulted in a significant decrement in the incidence of early PTS but not late PTS. Carbamazepine240 and valproic acid245 were found to have similar effects on the development of PTSs. The current recommendation is that anticonvulsants be administered for 1 week after the injury due to the potential for untoward side effects of treatment, including neurobehavioral consequences, and because antiseizure prophylaxis has not been shown to achieve a reduction in the incidence of late PTS.246 If late seizures occur, they are to be managed identical to the manner in which a first-time seizure in any other patient population is addressed. A recently published study comparing phenytoin and levetiracetam demonstrated that the latter agent was similarly effective in the prevention of seizures among the study population, 90% of whom were severe TBI patients, and was associated with improved outcomes and a significantly better side-effect profile.247 Thus, studies involving novel therapeutics and use of electroencephalography to detect subclinical seizures may provide further guidance in our management of PTS.
Prevention of Venous Thromboembolism
Patients with severe head injuries constitute a high-risk group for developing venous thromboembolic events (VTEs).248 In the absence of prophylaxis, studies have estimated the risk of deep venous thrombosis (DVT) among severe TBI patients to be upward of 20%.249 A distinction must be drawn between clinically symptomatic DVTs and those detected by screening tests, as DVTs detected through screening usually are relegated to the calf and are not associated with adverse consequences to the patient.250 DVTs involving the proximal leg veins, on the other hand, place the patient at increased risk for developing pulmonary embolism, which has been estimated to occur in 0.38% of TBI patients during their acute hospital stay.251 The development of pulmonary embolisms has significant consequences on morbidity and mortality and important implications on the management of patients with TBI: the physician must weigh the safety of treating the thromboembolism with anticoagulant agents relative to the risk of progression of intracranial hemorrhage or may have to consider timing of anticoagulation as part of the patient’s postoperative management following craniotomy. Hence, prevention of VTE is of the utmost importance.
DVT prophylaxis can be accomplished via mechanical therapeutic modalities and by administration of pharmacologic agents. Mechanical therapies using compression stockings and intermittent pneumatic compression devices are efficacious in preventing DVTs.252–254 They are associated with lowered risk of bleeding-related complications as compared to pharmacologic agents, but confounding factors among TBI patients such as multisystem trauma, including lower extremity injuries, may limit the applicability of this prophylactic measure to all patients with severe TBI. Administration of low-dose heparin or low-molecular-weight heparin is an effective means of preventing VTE255 and has a higher efficacy than use of mechanical modalities alone.256,257 Pharmacologic prophylaxis, however, carries an increased risk of intracranial bleeding and should not be started in the perioperative setting. Though VTE prophylaxis was initiated in patients within 24 to 48 hours of admission in a number of studies, the exact time needed to establish a “safety window” following patients’ sustaining a TBI or after undergoing surgical intervention has not been defined. The policy at our institution to start upon admission VTE prophylaxis with intermittent pneumatic compression devices in all patients with severe TBI without extremity injuries precluding such an intervention, and pharmacologic prophylaxis is initiated 48 hours after either a CT scan demonstrates stable imaging findings or an operative intervention occurs.
Placement of a retrievable inferior vena cava (IVC) filter is another method to consider in preventing pulmonary embolism among trauma patients. Use of retrievable IVC filters also putatively spares patients from the untoward risks of anticoagulation associated with DVT treatment. Due to evidence of extremely low rates of filter retrieval, which are primarily attributable to poor patient follow-up but can involve technical issues, as well as development of significant thrombus within the IVC filter,258 we do not typically employ this modality in the management of patients with severe TBI.
TBI and the Elderly
The elderly comprise a growing percentage of the worldwide population. In the United States, with the advancing age of the baby-boomer generation, 87 million Americans are estimated to be over the age of 65 by 2050, representing more than 20% of the total U.S. population.259 Elderly TBI patients have a significantly higher mortality rate and experience greater morbidity than younger people;260 in addition, they exhibit a worse functional status at hospital discharge, a slower rate of advancement in functional measures while in TBI rehabilitation, and less overall improvement 1 year after the injury.261,262 The disproportionate morbidity and mortality associated with trauma in the elderly are attributable to many factors, some of which include decreases in physiologic reserve and the presence of comorbidities.263
Chronic oral anticoagulation with warfarin is employed in the management of many medical conditions whose prevalence is increased among the elderly, such as chronic atrial fibrillation and cardiomyopathies with low ejection fraction, and in patients with prosthetic heart valves.264,265 In turn, the number of people using warfarin has progressively increased,266 with more than 1 million Americans being prescribed warfarin, most of whom are older than 65 years.267 As a consequence, more anticoagulated, elderly people are at risk for trauma-related injuries. TBI among this particular patient population is especially significant, because it carries with it higher mortality rates and increased morbidity.268
Neuroprotective Interventions in TBI
The only “neuroprotective strategies” for secondary injury currently agreed upon target proven systemic and intracranial insults readily amenable to common therapeutic interventions (e.g., hypotension, hypoxemia, hypocapnia, hyperthermia, hyperglycemia, and intracranial hypertension). Therapeutic interventions being investigated include (1) therapies directed at increasing oxygen delivery and utilization, (2) hypothermia, and (3) decompressive surgical interventions for malignant edema and intraparenchymal mass lesions.
Aarabi B., Hesdorffer D.C., Ahn E.S., et al. Outcome following decompressive craniectomy for malignant swelling due to severe head injury. J Neurosurg. 2006;104:469-479.
Alderson P., Roberts I. Corticosteroids in acute traumatic brain injury: systematic review of randomised controlled trials. Br Med J. 1997;314:1855-1859.
Battison C., Andrews P.J., Graham C., et al. Randomized, controlled trial of the effect of a 20% mannitol solution and a 7.5% saline/6% dextran solution on increased intracranial pressure after brain injury. Crit Care Med. 2005;33:196-202.
Bondanelli M., Ambrosio M.R., Cavazzini L., et al. Anterior pituitary function may predict functional and cognitive outcome in patients with traumatic brain injury undergoing rehabilitation. J Neurotrauma. 2007;24:1687-1697.
Contant C.F., Valadka A.B., Gopinath S.P., et al. Adult respiratory distress syndrome: a complication of induced hypertension after severe head injury. J Neurosurg. 2001;95:560-568.
Eisenberg H., Frankowski R., Contant C., et al. High-dose barbiturate control of elevated intracranial pressure in patients with severe head injury. J Neurosurg. 1988;69:15-23.
Fieschi C., Battistini N., Beduschi A., et al. Regional cerebral blood flow and intraventricular pressure in acute head injuries. J Neurol Neurosurg Psychiatry. 1974;37:1378-1388.
Halloran L.G., Zfass A.M., Gayle W.E., et al. Prevention of acute gastrointestinal complications after severe head injury: a controlled trial of cimetidine prophylaxis. Am J Surg. 1980;139:44-48.
Hsiang J.K., Chesnut R.M., Crisp C.B., et al. Early, routine paralysis for intracranial pressure control in severe head injury: is it necessary? Crit Care Med. 1994;22:1471-1476.
Lam A.M., Winn H.R., Cullen B.F., et al. Hyperglycemia and neurological outcome in patients with head injury. J Neurosurg. 1991;75:545-551.
Lu J., Marmarou A., Choi S., et al. Mortality from traumatic brain injury. Acta Neurochir. 2005;95(suppl):281-285.
Marmarou A., Anderson R.L., Ward J.D., et al. Impact of ICP instability and hypotension on outcome in patients with severe head trauma. J Neurosurg. 1991;75:159-166.
Mendellow A.D., Teasdale G.M., Russell T., et al. Effect of mannitol on cerebral blood flow and cerebral perfusion pressure in human head injury. J Neurosurg. 1985;63:43-48.
Miller J.D., Sweet R.C., Narayan R., et al. Early insults to the injured brain. JAMA. 1978;240:439-442.
Muizelaar J.P., Marmarou A., Ward J.D., et al. Adverse effects of prolonged hyperventilation in patients with severe head injury: a randomized clinical trial. J Neurosurg. 1991;75:731-739.
Muizelaar J.P., van der Poel H.G., Li Z.C., et al. Pial arteriolar vessel diameter and CO2 reactivity during prolonged hyperventilation in the rabbit. J Neurosurg. 1988;69:923-927.
Narayan R.K., Greenberg R.P., Miller J.D., et al. Improved confidence of outcome prediction in severe head injury: a comparative analysis of the clinical examination, multimodality evoked potentials, CT scanning, and intracranial pressure. J Neurosurg. 1981;54:751-762.
Narayan R.K., Kishore P.R., Becker D.P., et al. Intracranial pressure: to monitor or not to monitor? A review of our experience with severe head injury. J Neurosurg. 1982;56:650-659.
Raichle M.E., Plum F. Hyperventilation and cerebral blood flow. Stroke. 1972;3:566-575.
Rapp R.P., Young B., Twyman D., et al. The favorable effect of early parenteral feeding on survival in head-injured patients. J Neurosurg. 1983;58:906-912.
Robertson C.S., Gopinath S.P., Goodman J.C., et al. SjvO2 monitoring in head-injured patients. J Neurotrauma. 1995;12:891-896.
Robertson C.S., Valadka A.B., Hannay H.J., et al. Prevention of secondary ischemic insults after severe head injury. Crit Care Med. 1999;27:2086-2095.
Rosner M.J., Rosner S.D., Johnson A.H. Cerebral perfusion pressure: management protocol and clinical results. J Neurosurg. 1995;83:949-962.
Stiefel M.F., Spiotta A., Gracias V.H., et al. Reduced mortality rate in patients with severe traumatic brain injury treated with brain tissue oxygen monitoring. J Neurosurg. 2005;103:805-811.
Temkin N.R., Dikmen S.S., Wilensky A.J., et al. A randomized, double-blind study of phenytoin for the prevention of post-traumatic seizures. N Engl J Med. 1990;323:497-502.
1. Faul M., Xu L., Wald M.M., et al. Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations and Deaths, 2002-2006. Atlanta, Georgia: Centers for Disease Control and Prevention, National Center for Injury Prevention and Control; 2010.
2. National Center for Injury Prevention and Control. Report to Congress on Mild Traumatic Brain Injury in the United States: Steps to Prevent a Serious Public Health Problem. Atlanta, GA: Centers for Disease Control and Prevention; 2003.
3. Finkelstein E., Corso P., Miller T., et al. The Incidence and Economic Burden of Injuries in the United States. New York: Oxford University Press; 2006.
3a. The Brain Trauma Foundation. The American Association of Neurological Surgeons and Congress of Neurological Surgeons. AANS/CNS Joint Section on Neurotrauma and Critical Care: Guidelines for the management of severe traumatic brain injury (3rd ed). J Neurotrauma. 2007;24(Suppl 1):S1-S95.
4. Faul M., Wald M.M., Rutland-Brown W., et al. Using a cost-benefit analysis to estimate outcomes of a clinical treatment guideline: testing the Brain Trauma Foundation guidelines for the treatment of severe traumatic brain injury. J Trauma. 2007;63:1271-1278.
5. Härtl R., Gerber L., Iacono L., et al. Direct transport within an organized state trauma system reduces mortality in patients with severe traumatic brain injury. J Trauma. 2006;60:1250-1256.
6. DuBose J.J., Browder T., Inaba K., et al. Effect of trauma center designation on outcome in patients with severe traumatic brain injury. Arch Surg. 2008;143:1213-1217.
7. Teasdale G., Jennett B. Assessment of coma and impaired consciousness: a practical scale. Lancet. 1974;2:81-84.
8. Choi S.C., Narayan R.K., Anderson R.L., et al. Enhanced specificity of prognosis in severe head injury. J Neurosurg. 1988;69:381-385.
9. Servadei F., Nasi M.T., Cremonini A.M., et al. Importance of a reliable admission Glasgow Coma Scale score for determining the need for evacuation of posttraumatic subdural hematomas: a prospective study of 65 patients. J Trauma. 1998;44:868-873.
10. Narayan R.K., Greenberg R.P., Miller J.D., et al. Improved confidence of outcome prediction in severe head injury: a comparative analysis of the clinical examination, multimodality evoked potentials, CT scanning, and intracranial pressure. J Neurosurg. 1981;54:751-762.
11. Fearnside M.R., Cook R.J., McDougall P., et al. The Westmead Head Injury Project outcome in severe head injury: a comparative analysis of pre-hospital, clinical and CT variables. Br J Neurosurg. 1993;7:267-279.
12. Chamoun R.B., Robertson C.S., Gopinath S.P. Outcome in patients with blunt head trauma and a Glasgow Coma Scale score of 3 at presentation. J Neurosurg. 2009;111:683-687.
13. Chesnut R.M., Marshall L.F., Klauber M.R., et al. The role of secondary brain injury in determining outcome from severe head injury. J Trauma. 1993;34:216-222.
14. Eisenberg H.M., Gary H.E.Jr., Aldrich E.F., et al. Initial CT findings in 753 patients with severe head injury. A report from the NIH Traumatic Coma Data Bank. J Neurosurg. 1990;73:688-698.
15. Signorini D.F., Andrews P.J., Jones P.A., et al. Predicting survival using simple clinical variables: a case study in traumatic brain injury. J Neurol Neurosurg Psychiatry. 1999;66:20-25.
16. Lieberman J.D., Pasquale M.D., Garcia R., et al. Use of admission Glasgow Coma Score, pupil size, and pupil reactivity to determine outcome for trauma patients. J Trauma. 2003;55:437-442.
17. Tien H.C., Cunha J.R., Wu S.N., et al. Do trauma patients with a Glasgow Coma Scale score of 3 and bilateral fixed and dilated pupils have any chance of survival? J Trauma. 2006;60:274-278.
18. Chaudhuri K., Malham G.M., Rosenfeld J.V. Survival of trauma patients with coma and bilateral fixed dilated pupils. Injury. 2009;40:28-32.
19. Guidelines for the management of penetrating brain injury. J Trauma. 2001;51(suppl 2):S1-S43.
20. Letarte P.. The Brain, Feliciano D.V., Mattox K.L., Moore E.E. Trauma, 6th ed., New York: McGraw-Hill, 2008. 397-416
21. Foulkes M., Eisenberg H.M., Jane J.A., et al. The Traumatic Coma Data Bank: design, methods, and baseline characteristics. J Neurosurg. 1991;75(suppl):S8-S13.
22. Lu J., Marmarou A., Choi S., et al. Mortality from traumatic brain injury. Acta Neurochir. 2005;95(suppl):281-285.
23. Ghajar J., Hariri R.J., Narayan R.K., et al. Survey of critical care management of comatose, head-injured patients in the United States. Crit Care Med. 1995;23:560-567.
24. Hesdorffer D., Ghajar J., Iacono L. Predictors of compliance with the evidence-based guidelines for traumatic brain injury care: a survey of United States trauma centers. J Trauma. 2002;52:1202-1209.
25. Graham D.I., Adams J.H. Ischaemic brain damage in fatal head injuries. Lancet. 1971;1:265-266.
26. Robertson C.S., Gopinath S.P., Goodman J.C., et al. SjvO2 monitoring in head-injured patients. J Neurotrauma. 1995;12:891-896.
27. Muizelaar J.P., van der Poel H.G., Li Z.C., et al. Pial arteriolar vessel diameter and CO2 reactivity during prolonged hyperventilation in the rabbit. J Neurosurg. 1988;69:923-927.
28. Clifton G.L., Miller E.R., Choi S.C., et al. Fluid thresholds and outcome from severe brain injury. Crit Care Med. 2002;30:739-745.
29. Juul N., Morris G.F., Marshall S.B., et al. Intracranial hypertension and cerebral perfusion pressure: influence on neurological deterioration and outcome in severe head injury. The Executive Committee of the International Selfotel Trial. J Neurosurg. 2000;92:1-6.
30. Struchen M.A., Hannay H.J., Contant C.F., et al. The relation between acute physiological variables and outcome on the Glasgow Outcome Scale and Disability Rating Scale following severe traumatic brain injury. J Neurotrauma. 2001;18:115-125.
31. Andrews P.J., Sleeman D.H., Statham P.F., et al. Predicting recovery in patients suffering from traumatic brain injury by using admission variables and physiological data: a comparison between decision tree analysis and logistic regression. J Neurosurg. 2002;97:326-336.
32. Fieschi C., Battistini N., Beduschi A., et al. Regional cerebral blood flow and intraventricular pressure in acute head injuries. J Neurol Neurosurg Psychiatry. 1974;37:1378-1388.
33. Bouma G.J., Muizelaar J.P. Cerebral blood flow, cerebral blood volume, and cerebrovascular reactivity after severe head injury. J Neurotrauma. 1992;9(suppl 1):S333-S348.
34. Hlatky R., Valadka A.B., Robertson C.S. Intracranial pressure response to induced hypertension: role of dynamic pressure autoregulation. Neurosurgery. 2005;57:917-923.
35. Bouma G.J., Muizelaar J.P. Relationship between cardiac output and cerebral blood flow in patients with intact and with impaired autoregulation. J Neurosurg. 1990;73:368-374.
36. McLaughlin M.R., Marion D.W. Cerebral blood flow and vasoresponsivity within and around cerebral contusions. J Neurosurg. 1996;85:871-876.
37. Gray W.J., Rosner M.J. Pressure–volume index. II: the effects of low cerebral perfusion pressure and autoregulation. J Neurosurg. 1987;67:377.
38. Rosner M.J. Introduction to cerebral perfusion pressure management. Neurosurg Clin N Am. 1995;6:761-773.
39. Bouma G.J., Muizelaar J.P., Stringer W.A., et al. Ultra-early evaluation of regional cerebral blood flow in severely head-injured patients using xenon-enhanced computerized tomography. J Neurosurg. 1992;77:360-368.
40. Simard J.M., Bellefleur M. Systemic arterial hypertension in head trauma. Am J Cardiol. 1989;63:32C-35C.
41. Rangel-Castilla L., Gasco J., Nauta H.J., et al. Cerebral pressure autoregulation in traumatic brain injury. Neurosurg Focus. 2008;25:1-8.
42. Obrist W.D., Langfitt T.W., Jaggi J.L., et al. Cerebral blood flow and metabolism in comatose patients with acute head injury: relationship to intracranial hypertension. J Neurosurg. 1984;61:241-253.
43. Weber M., Grolimund P., Seiler R.W. Evaluation of posttraumatic cerebral blood flow velocities by transcranial Doppler ultrasonography. Neurosurgery. 1990;27:106-112.
44. Bouma G.J., Muizelaar J.P., Handoh K., et al. Blood pressure and intracranial pressure–volume dynamics in severe head injury: relationship with cerebral blood flow. J Neurosurg. 1992;77:15-19.
45. Salvant J.B.Jr., Muizelaar J.P. Changes in cerebral blood flow and metabolism related to the presence of subdural hematoma. Neurosurgery. 1993;33:387-393.
46. Bouma G.J., Muizelaar J.P., Choi S.C., et al. Cerebral circulation and metabolism after severe traumatic brain injury: the elusive role of ischemia. J Neurosurg. 1991;5:685-693.
47. Jones T. Thresholds of focal cerebral ischemia in awake monkeys. J Neurosurg. 1981;54:773-782.
48. Schroder J.L., Muizelaar J.P., Kuta A.J., et al. Thresholds for cerebral ischemia after severe head injury: relationship with late CT findings and outcome. J Neurotrauma. 1996;13:17-23.
49. Robertson C.S., Contant C.F., Gokaslan Z.L., et al. Cerebral blood flow, arteriovenous oxygen difference, and outcome in head injured patients. J Neurol Neurosurg Psychiatry. 1992;55:594-603.
50. Jaggi J.L., Obrist W.D., Gennarelli T.A., et al. Relationship of early cerebral blood flow and metabolism to outcome in acute head injury. J Neurosurg. 1990;72:176-182.
51. Kelly D.F., Martin N.A., Kordestani R., et al. Cerebral blood flow as a predictor of outcome following traumatic brain injury. J Neurosurg. 1997;86:633-641.
52. Kiening K.L., Hartl R., Unterberg A.W., et al. Brain tissue pO2-monitoring in comatose patients: implications for therapy. Neurol Res. 1997;19:233-240.
53. Robertson C.S., Valadka A.B., Hannay H.J., et al. Prevention of secondary ischemic insults after severe head injury. Crit Care Med. 1999;27:2086-2095.
54. Chan K.H., Dearden N.M., Miller J.D., et al. Multimodality monitoring as a guide to treatment of intracranial hypertension after severe brain injury. Neurosurgery. 1994;32:547-552.
55. Chan K.H., Miller J.D., Dearden N.M., et al. The effect of changes in cerebral perfusion pressure upon middle cerebral artery blood flow velocity and jugular bulb venous oxygen saturation after severe brain injury. J Neurosurg. 1992;77:55-61.
56. Nordstrom C.H., Reinstrup P., Xu W., et al. Assessment of the lower limit for cerebral perfusion pressure in severe head injuries by bedside monitoring of regional energy metabolism. Anesthesiology. 2003;98:809-814.
57. Rosner M.J., Rosner S.D., Johnson A.H. Cerebral perfusion pressure: management protocol and clinical results. J Neurosurg. 1995;83:949-962.
58. Brain Trauma Foundation. The American Association of Neurological Surgeons and Congress of Neurological Surgeons. AANS/CNS Joint Section on Neurotrauma and Critical Care: guidelines for the management of severe traumatic brain injury (3rd ed). J Neurotrauma. 2007;24(suppl 1):S59-S64.
59. Marmarou A., Anderson R.L., Ward J.D., et al. Impact of ICP instability and hypotension on outcome in patients with severe head trauma. J Neurosurg. 1991;75:159-166.
60. Jones P.A., Andrews P.J.D., Midgely S., et al. Measuring the burden of secondary insults in head injured patients during intensive care. J Neurosurg Anesthesiol. 1994;6:4-14.
61. Miller J.D., Sweet R.C., Narayan R., et al. Early insults to the injured brain. JAMA. 1978;240:439-442.
62. Stochetti N., Furlan A., Volta F. Hypoxemia and arterial hypotension at the accident scene in head injury. J Trauma. 1996;40:764-767.
63. Winchel R.J., Hoyt D.B. Endotracheal intubation in the field improves survival in patients with severe head injury. Arch Surg. 1997;132:592-597.
64. Robertson C.S. Desaturation episodes after severe head injury: influence on outcome. Acta Neurochir (Wien) Suppl. 1993;59:98-101.
65. Valadka A.B., Gopinath S.P., Contant C.F., et al. Relationship of brain tissue PO2 to outcome after severe head injury. Crit Care Med. 1998;26:1576-1581.
66. Bardt T.F., Unterberg A.W., Hartl R., et al. Monitoring of brain tissue PO2 in traumatic brain injury: effect of cerebral hypoxia on outcome. Acta Neurochir Suppl. 1998;71:153-156.
67. van den Brink W.A., van Santbrink H., Steyerberg E.W., et al. Brain oxygen tension in severe head injury. Neurosurgery. 2000;46:868-878.
68. Brain Trauma Foundation. The American Association of Neurological Surgeons and Congress of Neurological Surgeons. AANS/CNS Joint Section on Neurotrauma and Critical Care: guidelines for the management of severe traumatic brain injury (3rd ed). J Neurotrauma. 2007;24(suppl 1):S65-S70.
69. Matz P.G., Pitts L. Monitoring in traumatic brain injury. Clin Neurosurg. 1997;44:267-294.
70. Alvarez del Castillo M. Monitoring neurologic patients in intensive care. Curr Opin Crit Care. 2001;7:49-60.
71. Contant C.F., Valadka A.B., Gopinath S.P., et al. Adult respiratory distress syndrome: a complication of induced hypertension after severe head injury. J Neurosurg. 2001;95:560-568.
72. von Essen C., Zervas N.T., Brown D.R., et al. Local cerebral blood flow in the dog during intravenous infusion of dopamine. Surg Neurol. 1980;13:181-188.
73. Myburgh J.A., Upton R.N., Grant C., et al. A comparison of the effects of norepinephrine, epinephrine, and dopamine on cerebral blood flow and oxygen utilisation. Acta Neurochir Suppl (Wien). 1998;71:19-21.
74. Berre J., De Backer D., Moraine J.J., et al. Effects of dobutamine and prostacyclin on cerebral blood flow velocity in septic patients. J Crit Care. 1994;9:1-6.
75. Robertson C.. Critical care management of traumatic brain injury, Winn H.R., editor. Youmans Neurological Surgery, 5th ed., Philadelphia: WB Saunders, 2004. 5103-5144
76. Kroppenstedt S.N., Kern M., Thomale U.W., et al. Effect of cerebral perfusion pressure on contusion volume following impact injury. J Neurosurg. 1999;90:520-526.
77. Alvarez M., Nava J.M., Rue M., et al. Mortality prediction in head trauma patients: performance of Glasgow Coma Score and general severity systems. Crit Care Med. 1998;26:142-148.
77a. Van Beek J.G., Mushkudiani N.A., Steyerberg E.W., et al. Prognostic value of admission laboratory parameters in traumatic brain injury: results from the IMPACT study. J Neurotrauma. 2007;24:315-328.
78. Carlson A.P., Schermer C.R., Lu S.W. Retrospective evaluation of anemia and transfusion in traumatic brain injury. J Trauma. 2006;61:567-571.
79. McIntyre L.A., Fergusson D.A., Hutchison J.S., et al. Effect of a liberal versus restrictive transfusion strategy on mortality in patients with moderate to severe head injury. Neurocrit Care. 2006;5:4-9.
80. Smith M.J., Stiefel M.F., Magge S., et al. Packed red blood cell transfusion increases local cerebral oxygenation. Crit Care Med. 2005;33:1104-1108.
81. Zygun D.A., Nortje J., Hutchinson P.J., et al. The effect of red blood cell transfusion on cerebral oxygenation and metabolism after severe traumatic brain injury. Crit Care Med. 2009;37:1074-1078.
82. Lee S.H., Heros R.C., Mullan J.C., et al. Optimum degree of hemodilution for brain protection in a canine model of focal cerebral ischemia. J Neurosurg. 1994;80:469-475.
83. Kee D.B., Wood J.H. Rheology of the cerebral circulation. Neurosurgery. 1984;15:125-131.
84. Andrews B.T. Head Injury Management. In: Andrews B.T., editor. Intensive Care in Neurosurgery. New York: Thieme, 2003. 125-136
85. Marion D.W. Pathophysiology and Treatment of Intracranial Hypertension. In: Andrews B.T., editor. Intensive Care in Neurosurgery. New York: Thieme, 2003. 47-54
86. Kellie G. On death from cold, and on congestions of the brain: an account of the appearances observed in the dissection of two of three individuals presumed to have perished in the storm of 3rd November 1821; with some reflections on the pathology of the brain. Trans Med Chir Soc Edinb. 1824;1:84-169.
87. Monro A. Observations Ion the Structure and Function of the Nervous System. Edinburgh: Creech & Johnson; 1823.
88. Marmarou A. Increased intracranial pressure in head injury and influence of blood volume. J Neurotrauma. 1992;9(suppl 1):S327-S332.
89. Marmarou A., Shulman K., Rosende R.M. A nonlinear analysis of the cerebrospinal fluid system and intracranial pressure dynamics. J Neurosurg. 1978;48:332-344.
90. Becker D.P., Miller J.D., Ward J.D., et al. The outcome from severe head injury with early diagnosis and intensive management. J Neurosurg. 1977;47:491-502.
91. Lundberg N., Troupp H., Lorin H. Continuous recording of the ventricular-fluid pressure in patients with severe acute traumatic brain injury. A preliminary report. J Neurosurg. 1965;22:581-590.
92. Fakhry S., Trask A., Waller M., et al. Management of brain-injured patients by evidence-based medicine protocol improves outcomes and decreases hospital charges. J Trauma. 2004;56:492-500.
93. Palmer S., Bader M., Qureshi A., et al. The impact of outcomes in a community hospital setting of using the AANS traumatic brain injury guidelines. J Trauma. 2001;50:657-662.
94. Patel H.C., Menon D.K., Tebbs S., et al. Specialist neurocritical care and outcome from head injury. Intensive Care Med. 2002;28:547-553.
95. Narayan R.K., Kishore P.R., Becker D.P., et al. Intracranial pressure: to monitor or not to monitor? A review of our experience with severe head injury. J Neurosurg. 1982;56:650-659.
96. Lobato R.D., Sarabia R., Rivas J.J., et al. Normal computerized tomography scans in severe head injury. Prognostic and clinical management implications. J Neurosurg. 1986;65:784-789.
97. Servadei F., Antonelli V., Giuliani G., et al. Evolving lesions in traumatic subarachnoid hemorrhage: prospective study of 110 patients with emphasis on the role of ICP monitoring. Acta Neurochir Suppl. 2002;81:81-82.
98. Brain Trauma Foundation. The American Association of Neurological Surgeons and Congress of Neurological Surgeons. AANS/CNS Joint Section on Neurotrauma and Critical Care: guidelines for the management of severe traumatic brain injury (3rd ed). J Neurotrauma. 2007;24(suppl 1):S45-S54.
99. Timofeev I., Dahyot-Fizelier C., Keong N., et al. Ventriculostomy for control of raised ICP in acute traumatic brain injury. Acta Neurochir Suppl. 2008;102:99-104.
100. Binz D.D., Toussaint L.G.3rd, Friedman J.A. Hemorrhagic complications of ventriculostomy placement: a meta-analysis. Neurocrit Care. 2009;10:253-256.
101. Gardner P.A., Engh J., Atteberry D., et al. Hemorrhage rates after external ventricular drain placement. J Neurosurg. 2009;110:1021-1025.
102. Lozier A.P., Sciacca R.R., Romagnoli M.F., et al. Ventriculostomy-related infections: a critical review of the literature. Neurosurgery. 2002;51:170-182.
103. Zabramski J.M., Whiting D., Darouiche R.O., et al. Efficacy of antimicrobial-impregnated external ventricular drain catheters: a prospective, randomized, controlled trial. J Neurosurg. 2003;98:725-730.
104. North B., Reilly P. Comparison among three methods of intracranial pressure recording. Neurosurgery. 1986;18:730-732.
105. Brain Trauma Foundation. The American Association of Neurological Surgeons and Congress of Neurological Surgeons. AANS/CNS Joint Section on Neurotrauma and Critical Care: guidelines for the management of severe traumatic brain injury (3rd ed). J Neurotrauma. 2007;24(suppl 1):S55-S58.
106. Eisenberg H., Frankowski R., Contant C., et al. High-dose barbiturate control of elevated intracranial pressure in patients with severe head injury. J Neurosurg. 1988;69:15-23.
107. Andrews B.T., Chiles B.W., Olsen W.L., et al. The effect of intracerebral hematoma location on the risk of brain-stem compression and on clinical outcome. J Neurosurg. 1988;69:518-522.
108. Durward Q.J., Amacher A.L., Del Maestro R.F., et al. Cerebral and cardiovascular responses to changes in head elevation in patients with intracranial hypertension. J Neurosurg. 1983;59:938-944.
109. Feldman Z., Kanter M.J., Robertson C.S., et al. Effect of head elevation on intracranial pressure, cerebral perfusion pressure, and cerebral blood flow in head-injured patients. J Neurosurg. 1992;76:207-211.
110. Meixensberger J., Baunach S., Amschler J., et al. Influence of body position on tissue–pO2, cerebral perfusion pressure and intracranial pressure in patients with acute brain injury. Neurol Res. 1997;19:249-253.
111. Barba C.A., Taggert J., Morgan A.S., et al. A new cervical spine clearance protocol using computed tomography. J Trauma. 2001;51:652-656.
112. Chiu W.C., Haan J.M., Cushing B.M., et al. Ligamentous injuries of the cervical spine in unreliable blunt trauma patients: incidence, evaluation, and outcome. J Trauma. 2001;50:457-463.
113. Brohi K., Healy M., Fotheringham T., et al. Helical computed tomographic scanning for the evaluation of the cervical spine in the unconscious, intubated trauma patient. J Trauma. 2005;58:897-901.
114. Brooks R.A., Willett K.M. Evaluation of the Oxford protocol for total spinal clearance in the unconscious trauma patient. J Trauma. 2001;50:862-867.
115. Tomycz N.D., Chew B.G., Chang Y.F., et al. MRI is unnecessary to clear the cervical spine in obtunded/comatose trauma patients: the four-year experience of a level I trauma center. J Trauma. 2008;64:1258-1263.
116. Como J.J., Thompson M.A., Anderson J.S., et al. Is magnetic resonance imaging essential in clearing the cervical spine in obtunded patients with blunt trauma? J Trauma. 2007;63:544-549.
117. Schuster R., Waxman K., Sanchez B., et al. Magnetic resonance imaging is not needed to clear cervical spines in blunt trauma patients with normal computed tomographic results and no motor deficits. Arch Surg. 2005;140:762-766.
118. Hogan G.J., Mirvis S.E., Shanmuganathan K., et al. Exclusion of unstable cervical spine injury in obtunded patients with blunt trauma: is MR imaging needed when multi-detector row CT findings are normal? Radiology. 2005;237:106-113.
119. Davies G., Deakin C., Wilson A. The effect of a rigid collar on intracranial pressure. Injury. 1996;27:647-649.
120. Raphael J.H., Chotai M.B. Effects of the cervical collar on cerebrospinal fluid pressure. Anaesthesia. 1994;49:437-439.
121. Hunt K., Hallworth S., Smith M. The effects of rigid collar placement on intracranial and cerebral perfusion pressures. Anaesthesia. 2001;56:511-513.
122. Ferguson J., Mardel S.N., Beattie T.F., et al. Cervical collars: a potential risk to the head-injured patient. Injury. 1993;24:454-456.
123. Brain Trauma Foundation. The American Association of Neurological Surgeons and Congress of Neurological Surgeons. AANS/CNS Joint Section on Neurotrauma and Critical Care: guidelines for the management of severe traumatic brain injury (3rd ed). J Neurotrauma. 2007;24(suppl 1):S71-S76.
124. Deogaonkar A., Gupta R., DeGeorgia M., et al. Bispectral Index monitoring correlates with sedation scales in brain-injured patients. Crit Care Med. 2004;32:2403-2406.
125. Kelly P.F., Goodale D.B., Williams J., et al. Propofol in the treatment of moderate and severe head injury: a randomized, prospective double-blinded pilot trial. J Neurosurg. 1999;90:1042-1057.
126. Kotani Y., Shimazawa M., Yoshimura S., et al. The experimental and clinical pharmacology of propofol, an anesthetic agent with neuroprotective properties. CNS Neurosci Ther. 2008;14:95-106.
127. Adembri C., Venturi L., Pellegrini-Giampietro D.E. Neuroprotective effects of propofol in acute cerebral injury. CNS Drug Rev. 2007;13:333-351.
128. deNadal M., Ausina A., Sahuquillo J. Effects on intracranial pressure of fentanyl in severe head injury patients. Acta Neurochir. 1998;71:10-12.
129. Sperry R.T., Bailey P.L., Reichman M.V. Fentanyl and sufentanyl increase intracranial pressure in head trauma patients. Anesthesiology. 1992;77:416-420.
130. Sanchez-Izquierdo-Riera J.A., Caballero-Cubedo R.E., Perez-Vela J.L., et al. Propofol versus midazolam: safety and efficacy for sedating the severe trauma patient. Anesth Analg. 1998;86:1219-1224.
131. Ghori K.A., Harmon D.C., Elashaal A., et al. Effect of midazolam versus propofol sedation on markers of neurological injury and outcome after isolated severe head injury: a pilot study. Crit Care Resusc. 2007;9:166-171.
132. Aryan H.E., Box K.W., Ibrahim D., et al. Safety and efficacy of dexmedetomidine in neurosurgical patients. Brain Inj. 2006;20:791-798.
133. Wise B., Chater N. The value of hypertonic mannitol solution in decreasing brain mass and lowering cerebrospinal-fluid pressure. J Neurosurg. 1963;19:1038-1043.
134. Mendellow A.D., Teasdale G.M., Russell T., et al. Effect of mannitol on cerebral blood flow and cerebral perfusion pressure in human head injury. J Neurosurg. 1985;63:43-48.
135. Schwartz M.L., Tator C.H., Rowed D.W., et al. The University of Toronto head injury treatment study: a prospective, randomized comparison of pentobarbital and mannitol. Can J Neurol Sci. 1984;11:434-440.
136. Smith H.P., Kelly D.L.Jr., McWhorter J.M., et al. Comparison of mannitol regimens in patients with severe head injury undergoing intracranial monitoring. J Neurosurg. 1986;65:820-824.
137. Barry K.G., Berman A.R. Mannitol infusion. III: the acute effect of the intravenous infusion of mannitol on blood and plasma volumes. N Engl J Med. 1961;264:1085-1088.
138. Burke A., Quest D., Chien S., et al. The effects of mannitol on blood viscosity. J Neurosurg. 1981;55:550-553.
139. Muizelaar J.P., Wei E.P., Kontos H.A., et al. Mannitol causes compensatory cerebral vasoconstriction and vasodilatation to blood viscosity changes. J Neurosurg. 1983;59:822-828.
140. Muizelaar J.P., Lutz H.A.3rd, Becker D.P. Effect of mannitol on ICP and CBF and correlation with pressure autoregulation in severely head-injured patients. J Neurosurg. 1984;61:700-706.
141. Hartwell R.C., Sutton L.N. Mannitol, intracranial pressure, and vasogenic edema. Neurosurgery. 1993;32:444-450.
142. Brain Trauma Foundation. The American Association of Neurological Surgeons: the joint section on neurotrauma and critical care: guidelines for the management of severe head injury. J Neurotrauma. 2007;24:S14-S20.
143. Feig P.U., McCurdy D.K. The hypertonic state. N Engl J Med. 1977;297:1444-1454.
144. Kaufmann A.M., Cardoso E.R. Aggravation of vasogenic cerebral edema by multiple-dose mannitol. J Neurosurg. 1992;77:584-589.
145. Pollay M., Fullenwider C., Roberts P.A., et al. Effect of mannitol and furosemide on blood–brain osmotic gradient and intracranial pressure. J Neurosurg. 1983;59:945-950.
146. Ogden A.T., Mayer S.A., Connolly E.S.Jr. Hyperosmolar agents in neurosurgical practice: the evolving role of hypertonic saline. Neurosurgery. 2005;57:207-215.
147. Horn P., Munch E., Vajkoczy P., et al. Hypertonic saline solution for control of elevated intracranial pressure in patients with exhausted response to mannitol and barbiturates. Neurol Res. 1999;21:758-764.
148. Suarez J.I., Qureshi A.I., Bhardwaj A., et al. Treatment of refractory intracranial hypertension with 23.4% saline. Crit Care Med. 1998;26:1118-1122.
149. Worthley L.I., Cooper D.J., Jones N. Treatment of resistant intracranial hypertension with hypertonic saline: report of two cases. J Neurosurg. 1988;68:478-481.
150. Battison C., Andrews P.J., Graham C., et al. Randomized, controlled trial of the effect of a 20% mannitol solution and a 7.5% saline/6% dextran solution on increased intracranial pressure after brain injury. Crit Care Med. 2005;33:196-202.
151. Kerwin A.J., Schinco M.A., Tepas J.J.3rd, et al. The use of 23.4% hypertonic saline for the management of elevated intracranial pressure in patients with severe traumatic brain injury: a pilot study. J Trauma. 2009;67:277-282.
152. Rockswold G.L., Solid C.A., Paredes-Andrade E., et al. Hypertonic saline and its effect on intracranial pressure, cerebral perfusion pressure, and brain tissue oxygen. Neurosurgery. 2009;65:1035-1041.
153. Qureshi A.I., Suarez J.I. Use of hypertonic saline solutions in treatment of cerebral edema and intracranial hypertension. Crit Care Med. 2000;28:3301-3313.
154. Langfitt T.W., Gennarelli T.A. Can the outcome from head injury be improved? J Neurosurg. 1982;55:19-25.
155. Miller J.D., Butterworth J.F., Gudeman S.K., et al. Further experience in the management of severe head injury. J Neurosurg. 1981;54:289-299.
156. Nordby K.H., Nesbakken R. The effect of high dose barbiturate decompression after severe head injury: a controlled clinical trial. Acta Neurochir (Wien). 1984;72:157-166.
157. Saul T.G., Ducker T.B. Effect of intracranial pressure monitoring and aggressive treatment on mortality in severe head injury. J Neurosurg. 1982;56:498-503.
158. Shapiro H.M. Intracranial hypertension: therapeutic and anesthetic considerations. Anesthesiology. 1975;43:445-471.
159. Hsiang J.K., Chesnut R.M., Crisp C.B., et al. Early, routine paralysis for intracranial pressure control in severe head injury: is it necessary? Crit Care Med. 1994;22:1471-1476.
160. Rosa G., Sanfilippo M., Vilardi V., et al. Effects of vecuronium bromide on intracranial pressure and cerebral perfusion pressure: a preliminary report. Br J Anaesth. 1986;58:437-440.
161. Rosa G., Orfei P., Sanfilippo M., et al. The effects of atracurium besylate (Tracrium) on intracranial pressure and cerebral perfusion pressure. Anesth Analg. 1986;65:381-384.
162. Lundberg N., Kjallquist A., Bien C. Reduction of increased intracranial pressure by hyperventilation. Acta Psychiatr Scand. 1959;34:4-64.
163. Raichle M.E., Plum F. Hyperventilation and cerebral blood flow. Stroke. 1972;3:566-575.
164. Enevoldsen E.M., Jensen F.T. Autoregulation and CO2 responses of cerebral blood flow in patients with acute severe head injury. J Neurosurg. 1978;48:689-703.
165. Marion D.W., Darby J., Yonas H. Acute regional cerebral blood flow changes caused by severe head injuries. J Neurosurg. 1991;74:407-414.
166. Sioutos P.J., Orozco J.A., Carter L.P., et al. Continuous regional cerebral cortical blood flow monitoring in head-injured patients. Neurosurgery. 1995;36:943-949.
167. Graham D.I., Lawrence A.E., Adams J.H., et al. Brain damage in fatal non-missile head injury without high intracranial pressure. J Clin Pathol. 1998;41:34-37.
168. Ross D.T., Graham D.I., Adams J.H. Selective loss of neurons from the thalamic reticular nucleus following severe human head injury. J Neurotrauma. 1993;10:151-165.
169. Muizelaar J.P., Marmarou A., Ward J.D., et al. Adverse effects of prolonged hyperventilation in patients with severe head injury: a randomized clinical trial. J Neurosurg. 1991;75:731-739.
170. Brain Trauma Foundation. The American Association of Neurological Surgeons and Congress of Neurological Surgeons. AANS/CNS Joint Section on Neurotrauma and Critical Care: guidelines for the management of severe traumatic brain injury (3rd ed). J Neurotrauma. 2007;24(suppl 1):S87-S90.
171. Horsley J.S. The intracranial pressure during barbital narcosis. Lancet. 1937;1:141-143.
172. Ogura K., Takayasu M., Dacey R.G.Jr. Differential effects of pentobarbital on intracerebral arterioles and venules of rats in vitro. Neurosurgery. 1991;28:537-541.
173. Feustel P.J., Ingvar M.C., Severinghaus J.W. Cerebral oxygen availability and blood flow during middle cerebral artery occlusion: effects of pentobarbital. Stroke. 1991;12:858-863.
174. Ward J.D., Becker D.P., Miller J.D., et al. Failure of prophylactic barbiturate coma in the treatment of severe head injury. J Neurosurg. 1985;62:383-388.
175. Winer J.W., Rosenwasser R.H., Jimenez F. Electroencephalographic activity and serum and cerebrospinal fluid pentobarbital levels in determining the therapeutic end point during barbiturate coma. Neurosurgery. 1991;29:739-741.
176. Roberts I., Sydenham E. Barbiturates for acute traumatic brain injury. Cochrane Database of Systematic Reviews. 1999. Issue 3. Art. No.: CD000033. DOI: 10.1002/14651858.CD000033
177. Aarabi B., Hesdorffer D.C., Ahn E.S., et al. Outcome following decompressive craniectomy for malignant swelling due to severe head injury. J Neurosurg. 2006;104:469-479.
178. Kocher T. Die Therapie des Hirndruckes. In: Hölder A., editor. Hirnerschütterung, Hirndruck und chirurgische Eingriffe bei Hirnkrankheiten. Vienna: A Hölder; 1901:262-266.
179. Horsley V. On the technique of operations on the central nervous system. BMJ. 1906;2:411-423.
180. Cushing H. The establishment of cerebral hernia as a decompressive measure for inaccessible brain tumors; with the description of intramuscular methods of making the bone defect in temporal and occipital regions. Surg Gyn Obstet. 1905;1:297-314.
181. Kjellberg R.N., Prieto A.Jr. Bifrontal decompressive craniotomy for massive cerebral edema. J Neurosurg. 1971;34:488-493.
182. Cooper P.R., Rovit R.L., Ransohoff J. Hemicraniectomy in the treatment of acute subdural hematoma: a re-appraisal hemicraniectomy in the treatment of acute subdural hematoma: a re-appraisal. Surgical Neurology. 1976;5:25-28.
183. Cooper P.R., Hagler H., Clark W.K., et al. Enhancement of experimental cerebral edema after decompressive craniectomy: implications for the management of severe head injuries. Neurosurgery. 1979;4:296-300.
184. Cooper P.R., Hagler H., Clark W. Decompressive craniectomy, ICP and brain edema. In: Shulman K., Marmarou A., Miller J.D., Becker D.P., Hochwald G.M., Brock M. Intracranial pressure IV. Berlin: Springer-Verlag, 1980. 277-279
185. Gaab M.R., Rittierodt M., Lorenz M., et al. Traumatic brain swelling and operative decompression: a prospective investigation. Acta Neurochir Suppl (Wien). 1990;51:326-328.
186. Polin R.S., Shaffrey M.E., Bogaev C.A., et al. Decompressive bifrontal craniectomy in the treatment of severe refractory posttraumatic cerebral edema. Neurosurgery. 1997;41:84-92.
187. Sahuquillo J. Decompressive craniectomy for the treatment of refractory high intracranial pressure in traumatic brain injury. Cochrane Database of Systematic Reviews. 2006. Issue 1. Art. No.: CD003983. DOI: 10.1002/14651858.CD003983.pub2
188. Cooper D.J., Rosenfeld J.V., Murray L., et al. Decompressive craniectomy in diffuse traumatic brain injury. NEJM. 2011. Epub Ahead of Print
189. Cushing H. Some aspects of the pathological physiology of intracranial tumors. Boston Med Surg J. 1909;141:71-80.
190. Tuettenberg J., Czabanka M., Horn P., et al. Clinical evaluation of the safety and efficacy of lumbar cerebrospinal fluid drainage for the treatment of refractory increased intracranial pressure. J Neurosurg. 2009;110:1200-1208.
191. Grumme T., Baethmann A., Kolodziejczyk D., et al. Treatment of patients with severe head injury by triamcinolone: a prospective, controlled multicenter clinical trial of 396 cases. Res Exp Med. 1995;195:217-229.
192. Doppenberg E.M.R., Bullock R. Clinical neuro-protection trials in severe traumatic brain injury: lessons from previous studies. J Neurotrauma. 1997;14:71-80.
193. Marshall L.F., Maas A.L., Marshall S.B., et al. A multicenter trial on the efficacy of using tirilazad mesylate in cases of head injury. J Neurosurg. 1998;89:519-525.
194. Gaab M.R., Trost H.A., Alcantara A., et al. “Ultrahigh” dexamethasone in acute brain injury. Results from a prospective randomized double-blind multicenter trial (GUDHIS). German Ultrahigh Dexamethasone Head Injury Study Group. Zentralblatt Neurochirurgie. 1994;55:135-143.
195. Roberts I., Yates D., Sandercock P., et al. Effect of intravenous corticosteroids on death within 14 days in 10,008 adults with clinically significant head injury (MRC CRASH trial): randomized placebo controlled trial. Lancet. 2004;364:1321-1328.
196. Alderson P., Roberts I. Corticosteroids in acute traumatic brain injury: systematic review of randomised controlled trials. Br Med J. 1997;314:1855-1859.
197. Brain Trauma Foundation. The American Association of Neurological Surgeons and Congress of Neurological Surgeons. AANS/CNS Joint Section on Neurotrauma and Critical Care: guidelines for the management of severe traumatic brain injury (3rd ed). J Neurotrauma. 2007;24(suppl 1):S91-S95.
198. Lam A.M., Winn H.R., Cullen B.F., et al. Hyperglycemia and neurologic outcome in patients with head injury. J Neurosurg. 1985;62:806-810.
199. Stiefel M.F., Spiotta A., Gracias V.H., et al. Reduced mortality rate in patients with severe traumatic brain injury treated with brain tissue oxygen monitoring. J Neurosurg. 2005;103:805-811.
200. Narotam P.K., Morrison J.F., Nathoo N. Brain tissue oxygen monitoring in traumatic brain injury and major trauma: outcome analysis of a brain tissue oxygen-directed therapy. J Neurosurg. 2009;111:672-682.
201. Spiotta A.M., Stiefel M.F., Gracias V.H., et al. Brain tissue oxygen-directed management and outcome in patients with severe traumatic brain injury. J Neurosurg. 2010 Apr 23. [Epub ahead of print]
202. Piek J., Chesnut R.M., Marshall L.F., et al. Extracranial complications of severe head injury. J Neurosurg. 1992;77:901-907.
203. Moro N., Katayama Y., Igarashi T., et al. Hyponatremia in patients with traumatic brain injury: incidence, mechanism, and response to sodium supplementation or retention therapy with hydrocortisone. Surg Neurol. 2007;68:387-393.
204. Maggiore U., Picetti E., Antonucci E., et al. The relation between the incidence of hypernatremia and mortality in patients with severe traumatic brain injury. Crit Care. 2009;13:R110. [Epub]
205. Lam A.M., Winn H.R., Cullen B.F., et al. Hyperglycemia and neurological outcome in patients with head injury. J Neurosurg. 1991;75:545-551.
206. Laird A.M., Miller P.R., Kilgo P.D., et al. Relationship of early hyperglycemia to mortality in trauma patients. J Trauma. 2004;56:1058-1062.
207. Sung J., Bochicchio G.V., Joshi M., et al. Admission hyperglycemia is predictive of outcome in critically ill trauma patients. J Trauma. 2005;59:80-83.
208. Whitcomb B.W., Pradhan E.K., Pittas A.G., et al. Impact of admission hyperglycemia on hospital mortality in various intensive care unit populations. Crit Care Med. 2005;33:2772-2777.
209. Yendamuri S., Fulda G.J., Tinkoff G.H. Admission hyperglycemia as a prognostic indicator in trauma. J Trauma. 2003;55:33-38.
210. van den Berghe G., Wouters P., Weekers F., et al. Intensive insulin therapy in the critically ill patients. N Engl J Med. 2001;345:1359-1367.
211. Young B., Ott L., Dempsey R., et al. Relationship between admission hyperglycemia and neurologic outcome of severely brain-injured patients. Ann Surg. 1989;210:466-472.
212. Cyran E. Hypophysenschadigung durch Schadelbasisfraktur. Dtsch Med Wochenschr. 1918;44:261.
213. Escumilla R.F., Lisser H. Simmonds disease. J Clin Endocrinol. 1942;2:65-96.
214. Agha A., Rogers B., Mylotte D., et al. Neuroendocrine dysfunction in the acute phase of traumatic brain injury. Clin Endocrinol. 2004;60:584-591.
215. Tanriverdi F., Senyurek H., Unluhizarci, et al. High risk of hypopituitarism after traumatic brain injury: a prospective investigation of anterior pituitary function in the acute phase and 12 months after trauma. J Clin Endocrinol Metab. 2006;91:2105-2111.
216. Agha A., Sherlock M., Thompson C.J. Post traumatic hyponatraemia due to acute hypopituitarism. Q J Med. 2005;98:463-464.
217. Behan L.A., Phillips J., Thompson C.J., et al. Neuroendocrine disorders after traumatic brain injury. J Neurol Neurosurg Psychiatry. 2008;79:753-759.
218. Bondanelli M., De Marinis L., Ambrosio M.R., et al. Occurrence of pituitary dysfunction following traumatic brain injury. J Neurotrauma. 2004;21:685-696.
219. Klose M., Juul A., Poulsgaard L., et al. Prevalence and predictive factors of posttraumatic hypopituitarism. Clin Endocrinol. 2007;67:193-201.
220. Krahulik D., Zapletalova J., Frysak Z., et al. Dysfunction of hypothalamic–hypophyseal axis after traumatic brain injury in adults. J Neurosurg. 2009 Nov 20. [Epub ahead of print]
221. Popovic V. GH deficiency as the most common pituitary defect after TBI: clinical implications. Pituitary. 2005;8:239-243.
222. Bondanelli M., Ambrosio M.R., Cavazzini L., et al. Anterior pituitary function may predict functional and cognitive outcome in patients with traumatic brain injury undergoing rehabilitation. J Neurotrauma. 2007;24:1687-1697.
223. Clifton G.L., Robertson C.S., Choi S.C. Assessment of nutritional requirements of head-injured patients. J Neurosurg. 1986;64:895-901.
224. Brain Trauma Foundation. The American Association of Neurological Surgeons and Congress of Neurological Surgeons. AANS/CNS Joint Section on Neurotrauma and Critical Care: guidelines for the management of severe traumatic brain injury (3rd ed). J Neurotrauma. 2007;24(suppl 1):S77-S82.
225. Rapp R.P., Young B., Twyman D., et al. The favorable effect of early parenteral feeding on survival in head-injured patients. J Neurosurg. 1983;58:906-912.
226. Taylor S.J., Fettes S.B., Jewkes C., et al. Prospective, randomized, controlled trial to determine the effect of early enhanced enteral nutrition on clinical outcome in mechanically ventilated patients suffering head injury. Crit Care Med. 1999;27:2525-2531.
227. Garcia–de Lorenzo A.C., Ortiz-Leyba M., Planas J.C., et al. Parenteral administration of different amounts of branch chain amino acids in septic patients: clinical and metabolic aspects. Crit Care Med. 1997;25:418-424.
228. Huckleberry Y. Nutritional support and the surgical patient. Am J Health System Pharm. 2004;61:671-674.
229. Montejo J.C., Zarazaga A., Lopez-Martinez J., et al. Immunonutrition in the intensive care unit. A systematic review and consensus statement. Clin Nutr. 2003;22:221-233.
230. Helling T.S., Evans L.L., Fowler D.L., et al. Infectious complications in patients with severe head injury. J Trauma. 1988;28:1575-1577.
231. Sirvent J.M., Torres A., Mustafa E., et al. Protective effect of intravenously administered cefuroxime against nosocomial pneumonia in patients with structural coma. Am J Respir Crit Care Med. 1997;155:1729-1734.
232. Brain Trauma Foundation. The American Association of Neurological Surgeons and Congress of Neurological Surgeons. AANS/CNS Joint Section on Neurotrauma and Critical Care: guidelines for the management of severe traumatic brain injury (3rd ed). J Neurotrauma. 2007;24(suppl 1):S26-S31.
233. Bouderka M.A., Fakhir B., Bouaggad A., et al. Early tracheostomy versus prolonged endotracheal intubation in severe head injury. J Trauma. 2004;57:251-254.
234. Wyler A.R., Kelly W.A. Use of antibiotics with external ventriculostomies. J Neurosurg. 1972;37:185-187.
235. Brain Trauma Foundation American Association of Neurological Surgeons. Joint section on Neurotrauma and Critical Care: Recommendations for intracranial pressure monitoring technology. Management and Prognosis of Severe Traumatic Brain Injury. New York: Brain Trauma Foundation; 2000. 75-90
236. Lu W.Y., Rhoney D.H., Boling W.B., et al. A review of stress ulcer prophylaxis in the neurosurgical intensive care unit. Neurosurgery. 1997;41:416-425.
237. Ojiako K., Shingala H., Schorr C., et al. Famotidine versus pantoprazole for preventing bleeding in the upper gastrointestinal tract of critically ill patients receiving mechanical ventilation. Am J Crit Care. 2008;17:142-147.
238. Halloran L.G., Zfass A.M., Gayle W.E., et al. Prevention of acute gastrointestinal complications after severe head injury: a controlled trial of cimetidine prophylaxis. Am J Surg. 1980;139:44-48.
240. Glötzner F.L., Haubitz I., Miltner F., et al. Anfallsprophylaxe mit Carbamazepin nach schweren Schädelhirnverletzungen. Neurochirurgia. 1983;26:66-79.
241. Pechadre J.C., Lauxerois M., Colnet G., et al. Prevention de l’epelepsie posttraumatique tardive par phenytoine dans les traumatismes carniens graves: suivi durant 2 ans. Presse Med. 1991;20:841-845.
242. Temkin N.R., Dikmen S.S., Winn H.R. Management of head injury: posttraumatic seizures. Neurosurg Clin N Am. 1991;2:425-435.
243. Yablon S.A. Posttraumatic seizures. Arch Phys Med Rehabil. 1993;74:983-1001.
244. Temkin N.R., Dikmen S.S., Wilensky A.J., et al. A randomized, double-blind study of phenytoin for the prevention of post-traumatic seizures. N Engl J Med. 1990;323:497-502.
245. Temkin N.R., Dikmen S.S., Anderson G.D., et al. Valproate therapy for prevention of posttraumatic seizures: a randomized trial. J Neurosurg. 1999;91:593-600.
246. Brain Trauma Foundation. The American Association of Neurological Surgeons and Congress of Neurological Surgeons. AANS/CNS Joint Section on Neurotrauma and Critical Care: guidelines for the management of severe traumatic brain injury (3rd ed). J Neurotrauma. 2007;24(suppl 1):S83-S86.
247. Szaflarski J.P., Sangha K.S., Lindsell C.J., et al. Prospective, randomized, single-blinded comparative trial of intravenous levetiracetam versus phenytoin for seizure prophylaxis. Neurocrit Care. 2010;12:165-172.
248. Knudson M.M., Ikossi D.G., Khaw L., et al. Thromboembolism after trauma: an analysis of 1602 episodes from the American College of Surgeons National Trauma Data Bank. Ann Surg. 2004;240:490-496.
249. Kaufman H.H., Satterwhite T., McConnell B.J., et al. Deep vein thrombosis and pulmonary embolism in head-injured patients. Angiology. 1983;34:627-638.
250. Buller H.R., Agnelli, Hull R.D., et al. Antithrombotic therapy for venous thromboembolic disease: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest. 2004;126:401S-428S.
251. Page R.B., Spott M.A., Krishnamurthy S., et al. Head injury and pulmonary embolism: a retrospective report based on the Pennsylvania Trauma Outcomes study. Neurosurgery. 2004;54:143-148.
252. Black P.M., Baker M.F., Snook C.P. Experience with external pneumatic calf compression in neurology and neurosurgery. Neurosurgery. 1986;18:440-444.
253. Skillman J.J., Collins R.E., Coe N.P., et al. Prevention of deep vein thrombosis in neurosurgical patients: a controlled, randomized trial of external pneumatic compression boots. Surgery. 1978;83:354-358.
254. Turpie A.G., Hirsh J., Gent M., et al. Prevention of deep vein thrombosis in potential neurosurgical patients. A randomized trial comparing graduated compression stockings alone or graduated compression stockings plus intermittent compression with control. Arch Intern Med. 1989;149:679-681.
255. Iorio A., Agnelli G. Low-molecular-weight and unfractionated heparin for prevention of venous thromboembolism in neurosurgery. Arch Intern Med. 2000;160:2327-2332.
256. Agnelli G., Piovella F., Buoncristiani P., et al. Enoxaparin plus compression stocking compared with compression stocking alone in the prevention of venous thromboembolism after elective neurosurgery. N Engl J Med. 1998;339:80-85.
257. Nurmohamed M.T., van Riel A.M., Henkens C.M., et al. Low molecular weight heparin and compression stockings in the prevention of venous thromboembolism in neurosurgery. Thromb Haemost. 1996;75:233-238.
258. Karmy-Jones R., Jurkovich G.J., Velmahos G.C., et al. Practice patterns and outcomes of retrievable vena cava filters in trauma patients: an AAST multicenter study. J Trauma. 2007;62:17-24.
259. He W., Sengupta M., Velkoff V.A., et al. 65+ in the United States: 2005. In: U.S. Census Bureau, Current Population Reports [Internet]. Washington, DC: U.S. Government Printing Office; 2005:P23-209. Available from http://www.census.gov/prod/2006pubs/p23-209.pdf
260. Susman M., DiRusso S.M., Sullivan T., et al. Traumatic brain injury in the elderly: increased mortality and worse functional outcome at discharge despite lower injury severity. J Trauma. 2002;53:219-223.
261. Cifu D.X., Kreutzer J.S., Marwitz J.H., et al. Functional outcomes of older adults with traumatic brain injury: a prospective, multicenter analysis. Arch Phys Med Rehabil. 1996;77:883-888.
262. Livingston D.H., Lavery R.F., Mosenthal A.C., et al. Recovery at one year following isolated traumatic brain injury: a Western Trauma Association prospective multicenter trial. J Trauma. 2005;59:1298-1304.
263. Kauder D.R., Schwab C.W., Shapiro M.B.. Geriatric trauma: patterns, care, and outcomes, Moore E.E., Feliciano D.V., Mattox K.L. Trauma, 5th ed., New York: McGraw-Hill, 2004. 1041-1058
264. Wolf P.A., Abbott R.D., Kannel W.B. Atrial fibrillation: a major contributor to stroke in the elderly. The Framingham Study. Archives of Internal Medicine. 1987;147:1561-1564.
265. Fleming B. Head injury in patients using warfarin. Canadian Family Physician. 2001;47:727-728.
266. Stafford R.S., Singer D.E. National patterns of warfarin use in atrial fibrillation. Archives of Internal Medicine. 1996;156:2537-2541.
267. Fihn S.D., Callahan C.M., Martin D.C., et al. The risk for and severity of bleeding complications in elderly patients treated with warfarin. The National Consortium of Anticoagulation Clinics. Annals of Internal Medicine. 1996;124:970-979.
268. Grandhi R., Duane T.M., Dechert T., et al. Anticoagulation and the elderly head trauma patient. Am Surg. 2008;74:802-805.