Lung growth is complex and modulated by multiple processes. Poor lung growth may result from maternal, fetal, placental, environmental, and/or genetic factors.
Lung Growth
The lung bud arises as an outgrowth of the foregut endoderm. Thereafter lung development occurs as a result of epithelial–mesenchyme interactions and branching morphogenesis (1). The stages and the timetable for human lung development are embryonic stage (3–7 weeks), pseudoglandular (5–17 weeks), canalicular (16–26 weeks), saccular (24–38 weeks), and alveolar (36 weeks to six months postnatal) (2). The lung is ready to function as an efficient gas exchange organ at birth as a result of the complex fetal lung developmental processes. The timing of different intrauterine insults will affect lung development in different ways, resulting in physiological consequences to the fetus and the newborn. This chapter will review the impact of common intrauterine exposures on fetal lung development. We will present current concepts from animal experiments and clinical studies.
Chorioamnionitis
Chorioamnionitis in humans is an ascending infection of genital organisms to the choriodecidual space or the chorioamnion space through the cervix. Organisms are thought to spread diffusely through the choriodecidual or the chorioamnion plane and then invade the amniotic cavity. However, a recent study using molecular microbiologic techniques in human placentae demonstrated that the initial event is a localized choriodecidual infection, which then invades the amniotic cavity, thereby infecting amniotic fluid and the fetus prior to diffuse choriodecidual inflammation (3). This sequence is consistent with experiments in the rhesus macaque demonstrating that localized choriodecidual infection with live Group B streptococci did not trigger preterm labor until the amniotic fluid was colonized. However, a transient choriodecidual infection can induce cytokine production in the amniotic fluid, which results in fetal lung inflammation without overt infection of amniotic fluid or preterm labor. Therefore animal models of chorioamnionitis resulting from injection of inflammatory agents or organisms into the amniotic fluid reproduce the pathology of chorioamnionitis. We will review experiments in which experimental animals were given intra-amniotic or intrauterine agonists/organisms. We will not review experiments with maternal intraperitoneal or intravascular injection of agonists because these models reflect maternal septicemia or bacteremia, which are rare events in human chorioamnionitis.
Animal Models of Chorioamnionitis
Models of chorioamnionitis have been described with intrauterine injection of agonists or live bacteria in the mouse and the rabbit. Chorioamnionitis can also be induced by intra-amniotic injection in the sheep using agonists including IL-1ß, IL-1α, lipopolysaccharide (LPS – ligand for TLR4), live Ureaplasma parvum, and live Candida albicans. In the Rhesus macaque, intra-amniotic injection of Group B streptococci, Ureaplasma parvum, IL-1ß, or TNF cause chorioamnionitis and preterm labor. In the sheep, intra-amniotic injection of PamCysK4 (ligand for TLR2) induced weak fetal lung inflammation, but poly I:C (TLR3 ligand) did not cause inflammation. Interestingly intra-amniotic injection of TNFα (4), IL-6 or IL-8 (5) did not induce lung inflammation in fetal sheep, and intra-amniotic injection of IL-6 or IL-8 did not induce preterm labor in the rhesus macaque (6). These experiments demonstrate relative specificity or potency of responses to different inflammatory agents.
Using broad-range microbe-specific PCR assays, the common organisms in the amniotic fluid of women with preterm labor and intact membranes or with preterm rupture of membranes were Ureaplasma, Mycoplasma, and Fusobacterium (7). The strongest evidence that Ureaplasma can cause preterm labor is from experiments in the rhesus macaque. Intra-amniotic injection of Ureaplasma parvum or the related organism Mycoplasma hominis induced chorioamnionitis, fetal inflammation, and preterm labor (8). In the sheep, Ureaplasma parvum is not cleared from the amniotic compartment after IA injection but does not cause preterm labor (9). In the amniotic fluid of sheep, Ureaplasmas have antigenic variations with changes in the multibanded antigen and extensive horizontal gene transfer resulting in hybrid forms of Ureaplasma serovars, implying unstable genotypes during the course of infection. These antigenic changes perhaps evolved as a mechanism to evade host immune system. Curiously, chorioamnionitis caused by Ureaplasma is of variable intensity in fetal sheep – ranging from minimal to severe, but without differences in the titer of the organism in amniotic fluid (9,10). Similarly in the human, Ureaplasma can be recovered without severe chorioamnionitis or progressive preterm labor. Candida spp. colonization/infection of the lower genital tract is fairly common in women. In addition, Candida spp. can also be recovered in amniotic fluid from women particularly with an indwelling intrauterine device and/or cervical cerclage. In contrast to Ureaplasma spp, intra-amniotic injection of a clinical isolate of live Candida albicans caused severe fetal inflammation and death in untreated sheep (11).
Chorioamnionitis and Maturation of the Fetal Lung
One of the striking and counterintuitive effects of intra-amniotic LPS or IL-1 in the fetal sheep is the increase in pulmonary surfactant lipids (Figure 10-2A) and surfactant-associated proteins A, B, and C (Figure 10-2B) (12,13). The increase in airway surfactant and a lung structural maturation results in improved lung mechanics in the preterm resulting in “clinical lung maturation” (Figure 10-2C). This effect of chorioamnionitis-induced lung maturation is more potent than that resulting from maternal administration of betamethasone alone in the sheep (Figure 10-3). Further, the combination of LPS and betamethasone caused the largest increase in lung volume (see following section on combined effects of chorioamnionitis and antenatal steroids). Intra-amniotic injection of LPS with tracheal ligation prevented the lung maturation (14,15), but fetal intratracheal infusion of LPS or IL-1 resulted in increased lung maturation, demonstrating the sufficiency of direct airway contact with LPS or IL-1 for lung maturation (16,17).
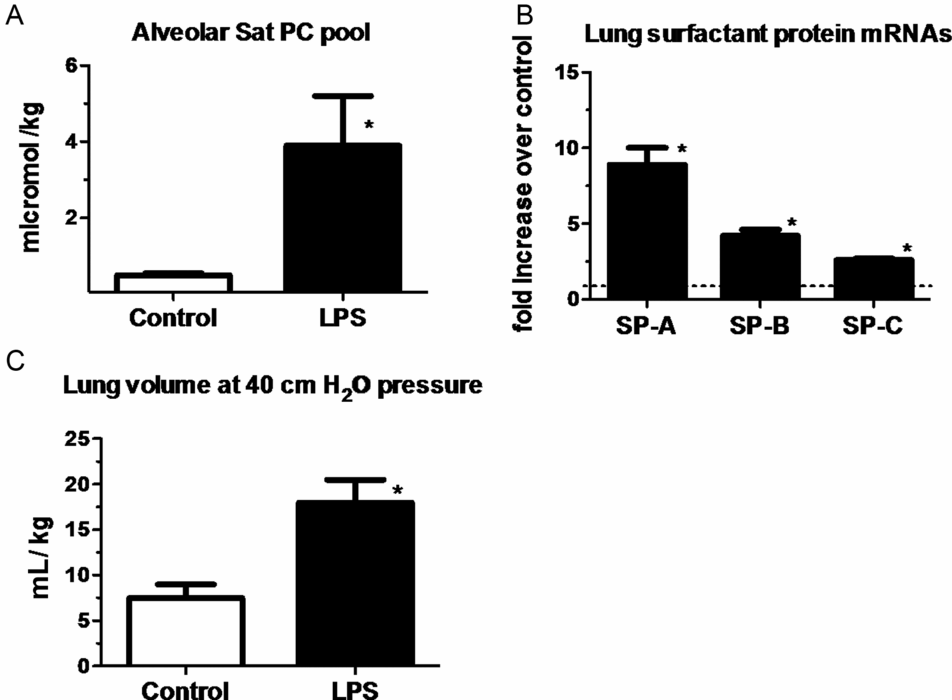
Measurements were done on lung and bronchoalveolar lavage fluid of fetal sheep after exposure to intra-amniotic LPS for 7 d (panels a & c) or 2 d (panel B). Exposure to LPS increased (A) surfactant lipid (saturated phosphatidylcholine) pool size normalized to body weight. (B) Induction of mRNAs for surfactant protein A, B, and C compared to control (value normalized to 1). (C) Lung volumes measured at 40 cmH20 pressure expressed relative to body weight. (*p < 0.05 vs. control)
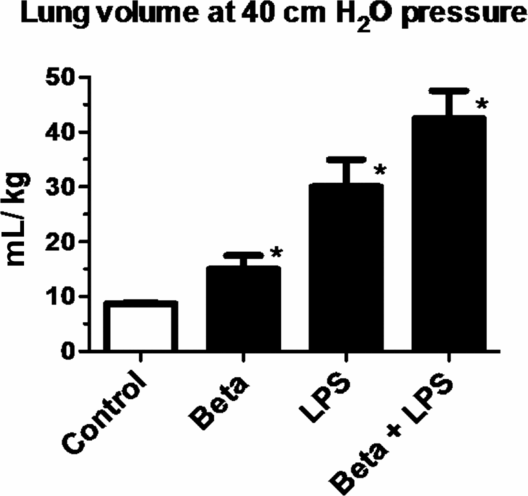
Pregnant ewes were given intramuscular betamethasone (Beta) 7 d before delivery or intra-amniotic LPS 14 d before delivery or the combined exposures of Beta + LPS. Fetuses delivered at 80% gestation had lung volumes measured at 40 cmH20 pressure at autopsy. Note the higher lung volumes induced by LPS compared to Beta. However, the most potent response was in animals receiving the combined exposures of LPS and Beta. (*p < 0.05 vs. control)
The precise signaling that results in fetal lung maturation is not known. However, intramuscular injection of an anti-CD18 antibody largely decreased the inflammatory cell influx into the lungs of fetal sheep and significantly decreased the lung maturation (18). Thus, inflammatory cell influx was critically required to mediate the clinical lung maturation induced by inflammatory agonist exposure in the sheep. Similarly, human IL-1 receptor antagonist decreased the IA LPS induced lung maturation (19). These experiments in the fetal sheep demonstrate that IL-1 signaling is central to the lung maturation induced by LPS. The numbers of inflammatory cells recruited to the lung correlated with the surfactant lipid pool size, suggesting that it will be difficult to separate the inflammatory effects of these agonists from the beneficial lung maturation effects. Similar changes of lung maturation with increases in surfactant proteins and type II cell numbers were found after exposure of fetal mouse or rabbit lungs to LPS or IL-1ß.
Chorioamnionitis – Inflammation/Injury of the Fetal Lung
Despite the beneficial lung maturation, intra-amniotic injection of pro-inflammatory agonists interferes with normal lung development. In the sheep, intra-amniotic LPS decreased alveolar numbers, caused thinning of the alveolar septae, and increased the size of the alveoli (Figure 10-4A) (20). An important feature of alveolarization is the expression of elastin that are the sites of secondary septation. Intra-amniotic LPS induced an aberrant and decreased expression of elastin foci (Figure 10-4B) (21). Because elastin deposition at the leading edge of alveolar septae is critical for secondary septation, these results are consistent with decreased alveolar septation in fetuses exposed to experimental chorioamnionitis. This inhibition of formation of terminal respiratory units also occurred in fetal mouse lung explants treated with LPS. In this explant model, LPS inhibited FGF-10 expression, which required macrophage NF-κB signaling (22). Intra-amniotic LPS in the preterm fetal sheep also inhibited several genes critical for vascular development, including VEGF-A, VEGFR2, and NOSIII (23). Intra-amniotic LPS caused smooth muscle hypertrophy in the resistance arterioles and adventitial fibroblast proliferation of the fetal sheep lung (Figure 10-4C,D) (23). These changes of vascular remodeling resulted in increased pulmonary vascular resistance (24).
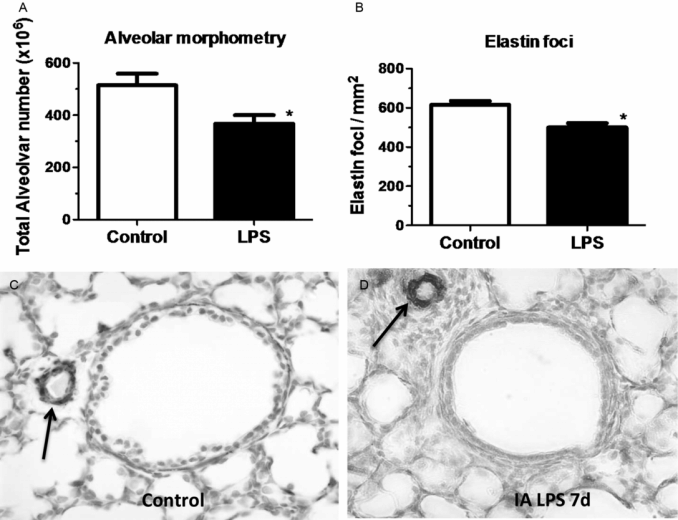
Pregnant sheep were given intra-amniotic LPS 7 d prior to preterm delivery at 80% gestation. Lung morphometry was performed using inflation-fixed fetal lung. Compared to control, LPS exposed lungs had (A) decreased alveolar numbers (B) decreased elastin foci. (C–D) Compared to control fetuses, LPS-exposed fetuses had increased pulmonary arteriolar α-smooth muscle actin staining (arrow showing brown staining) indicating media hypertrophy. Note the adventitial fibrocyte proliferation surrounding the arteriole in LPS exposed lung. (*p < 0.05 vs. control)
In the sheep, intra-amniotic injection of U. parvum increased lung surfactant pools and induced a clinical lung maturation, although less consistently than LPS or IL-1 (25). Intra-amniotic U. parvum also induced mild lung inflammation with mixed neutrophil and monocytic infiltration and low cytokine expression in the fetal lung. However, Ureaplasma caused a small decrease in elastic foci in secondary alveolar septae, impaired alveolar development, and increased smooth muscle around bronchioles and pulmonary artery/arterioles, similar to the changes caused by LPS. In contrast to what happens in the sheep, intra-amniotic Ureaplasma parvum (and the related organism Mycoplasma hominis) caused severe inflammation in the fetal rhesus macaque lung, characterized by increased neutrophils and macrophages and alveolar type II cell proliferation (8). These results suggest species differences in susceptibility to Ureaplasma.
In experiments simulating the effects of intrapartum exposure to inflammation, fetal lung inflammation induced changes similar to bronchopulmonary dysplasia (BPD) in transgenic mice. Human IL-1beta expressed in airway epithelial cells during the latter part of mouse gestation caused neutrophil and monocyte influx into the lung, respiratory insufficiency with increased postnatal mortality (26). IL-1beta disrupted alveolar septation with aberrant alpha-smooth muscle actin and elastin deposition in the septa of distal airspaces. IL-1beta also disrupted pulmonary vascular development.
Although antenatal infection causes aberrant pulmonary development, these changes are modest. For instance, the lung inflammation induced by chorioamnionitis in most experimental models is microscopic in nature and likely will not be visualized by X-ray imaging. Further, the delayed alveolar development apparent when fetuses are delivered seven days after intra-amniotic exposure to a pro-inflammatory agonist is no longer evident when fetuses are delivered twenty to thirty days later (27). These results imply that the fetus has a remarkable ability to repair the lung in utero. Understanding the mechanisms of lung repair following intra-uterine injury is clearly important.
Innate Immune Responses in the Fetal Lung Exposed to Chorioamnionitis
The fetal lung responds to chorioamnionitis with inflammation, induced “lung maturation,” and aberrant alveolar and vascular development. The central question is how the naïve immune system of the fetal lung responds to an intrauterine inflammatory challenge. The sentinel immune cell of the lung is the alveolar macrophage. In adult humans and animals, macrophages are located in the airspaces directly in contact with the alveolar hypophase. Fetuses do not normally have alveolar macrophages. In mice, macrophages can be detected in the lung interstitium from early gestation, while in other species, including nonhuman primates and sheep, very few mature macrophages are found in the fetal lung. In all species, mature alveolar macrophages begin populating the lung in large numbers postnatally with the onset of air breathing. Immature lung monocytes from preterm sheep have a minimal response (IL6 secretion) to an in vitro challenge to LPS and do not respond to TNFα (28). However, intra-amniotic LPS matures lung monocytes by stimulating GM-CSF and PU.1 expression in the fetal lung (Figure 10-5A,B). These monocytes migrate into the fetal alveolar spaces and respondvigorously to both LPS and TNFα in vitro (Figure 10-5C) (28). Thus exposure to a pro-inflammatory agonist in the amniotic fluid is a potent stimulus for maturation and responsiveness of monocytes in the lung.
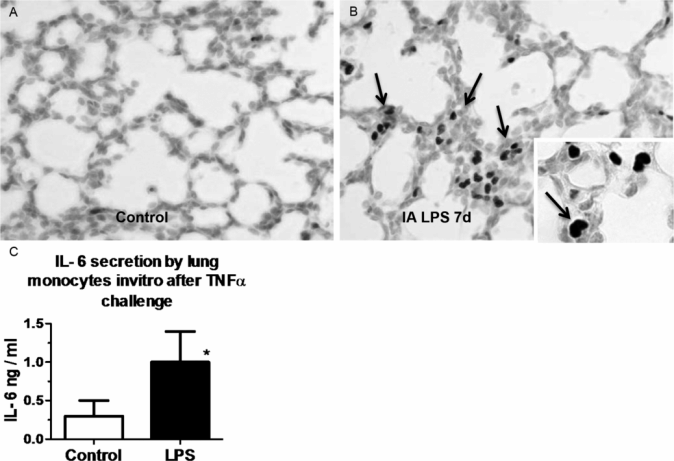
Pregnant sheep were given intra-amniotic LPS 7 d prior to preterm delivery at 80% gestation. (A,B) Immunohistology staining for PU.1 (a transcription factor necessary for maturation of monocyte) in fetal lung. Note increased monocyte PU.1 staining (arrow) after LPS exposure. (C) Lung monocytes were purified from fetuses and cultured in vitro in the presence of TNFα. Control preterm fetal lung monocyte responded with minimal IL-6 secretion, while fetuses exposed to intra-amniotic LPS responded with increased IL-6 secretion signifying a functional maturation of lung monocytes. (*p < 0.05 vs. control)
Intra-amniotic LPS also can cause an innate immune tolerance in the fetus. In adult animals and humans, endotoxin tolerance is the suppression of LPS signaling caused by a complex reprogramming of inflammatory responses. As part of endotoxin tolerance, pro-inflammatory cytokine expression is downregulated, while there is no change or an increase in the expression of anti-inflammatory genes, antimicrobial genes, and genes mediating phagocytosis. In the preterm fetal sheep, exposure to intra-amniotic LPS 2 d before delivery induces a robust expression of cytokines in the fetal lung. However, if the fetus is exposed to two intra-amniotic LPS injections of the same dose 7 d and 2 d prior to delivery, the fetal lung is refractory to the second LPS injection (Figure 10-6A) (29). Interestingly, both lung and blood monocytes are refractory to an in vitro challenge with LPS.
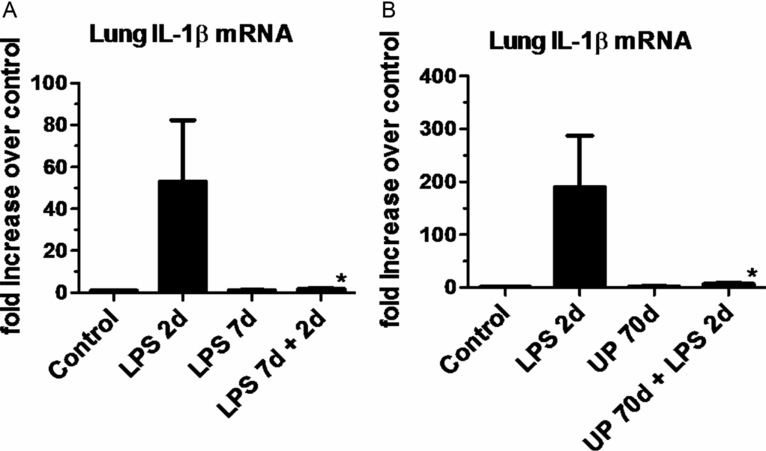
(a) Pregnant ewes were given intra-amniotic (IA) LPS either 2 d or 7 d or combined 7 d + 2 d prior to preterm delivery at 80% gestation. Quantitative rt-PCR for IL-1βmRNA was performed on whole-lung homogenates. Exposure to a single LPS injection increased IL-1βmRNA expression at 2 d with a decrease at 7 d. However, a prior exposure to IA LPS at 7 d blunted subsequent response to LPS injection at 2 d prior to delivery demonstrating endotoxin tolerance. (b) In a similar experimental schema pregnant ewes were given IA LPS injection 2 d or live Ureaplasma parvum (UP) 70 d or the combined exposure to Ureaplasma parvum 70 d + LPS 2 d prior to preterm delivery. Ureaplasma alone did not cause a significant induction of IL-1βmRNA expression but almost completely blunted LPS responsiveness, demonstrating a potent innate immune modulation (*p < 0.05 vs. LPS 2 d)
The phenomenon of innate immune tolerance is not restricted to exposure to LPS. Exposure to intra-amniotic Ureaplasma parvum almost completely abolished responsiveness of the fetal lung to LPS, implying a profound immune paralysis in the fetal lung induced by Ureaplasma exposure (Figure 10-6B) (30). The lung and blood monocytes from fetal sheep exposed to two injections of intra-amniotic LPS were also refractory to stimulation by a host of other TLR agonists, including PamCysK4 (TLR2), flagellin (TLR5), and CpG-DNA (TLR9) (31), demonstrating a cross tolerance to multiple TLRs. Other interactive phenomenon between antenatal LPS and postnatal inflammatory insults have also been reported. In rats, intra-amniotic LPS alone induced aberrant lung development and pulmonary hypertension. When these fetal rats exposed to LPS were exposed to postnatal moderate hyperoxia, the lung abnormalities were no longer evident. However, exposure to postnatal severe hyperoxia further enhanced the pulmonary abnormalities induced by antenatal LPS (32). Thus the interactive phenomena between different inflammatory insults can be complex and either exacerbate or reduce the lung injury response. Because innate immune tolerance is time dependent, it is not clear how these experimental phenomena will translate into clinical scenarios, where the timing of exposure to different inflammatory insults is not known. Although the precise mechanisms of innate immune tolerance are not known, the expression of negative regulator of Toll/IL-1 signaling, IRAK-M is increased in both the lung and blood monocytes (29).
Signaling Mediators Modulating Lung Development After Antenatal Insults
Intra-amniotic injection of IL-1 induced a large expression of IL-1ß, IL-8, GM-CSF, MCP-1, and the acute phase reactant serum amyloid A3 in the lungs of fetal sheep and fetal monkeys. Interestingly, the Th1 cytokines IFNγ, IL-12, type I interferon inducible genes CXCL9 (MIG) and CXCL10 (IP-10), and the Th2 cytokines IL-4 and IL-13 were not induced. IA LPS increased expression of additional genes inducible by the type I interferon signaling, including CXCL9 (MIG) and CXCL10 (IP-10) (33). In the fetal sheep, IA LPS increased lung expression of TLR4 and TLR2 mRNA expression but decreased TLR4 expression in the gut, demonstrating organ specific responses. Neither IA IL-1 nor LPS significantly increased expression of TNFα in the lung, and IA TNF did not induce chorioamnionitis or fetal lung inflammation (4). The counterregulatory cytokine IL-10 and IL-1 receptor antagonist are only modestly increased in the fetal lung exposed to IA IL-1 or LPS.
Some candidate genes associated with lung injury have been assessed. Caveolins (Cavs) are implicated as major modulators of lung injury and remodeling by multiple signaling pathways by virtue of their strategic positioning in the lipid rafts of plasma membranes. Intra-amniotic LPS decreased the expression of Cav-1 in the preterm fetal lung (34). The decreased expression of Cav-1 was associated with the activation of the Smad2/3, Stat 3, a-SMase/ceramide pathways, and increased expression of HO-1 (34). Consistent with activation of Smads, intra-amniotic LPS increased lung TGF-ß1 mRNA and protein expression (35). However, the lung expression of CTGF – a key pro-fibrotic protein induced by TGF-ß, decreased on exposure to intra-amniotic LPS (35). Metallo-proteinases regulate the breakdown of extracellular matrix in the lung. The expression of MMP-9 was increased in sheep given intra-amniotic LPS (36) and in transgenic mice that overexpress IL-1ß in the lung (37). Sonic Hedgehog (Shh) signaling is a major pathway directing lung development. Intra-amniotic LPS also decreased Shh mRNA levels and Gli1 protein expression in the fetal sheep (21). In a fetal mouse lung explant system, abnormal saccular lung development induced by LPS was mediated by NFκB activation in the macrophage, resulting in IL-1ß secretion (22). The link between inflammation and abnormal lung development was FGF-10 in this mouse lung explant system, a finding that is consistent with reduced FGF-10 in the tracheal aspirates of preterm infants developing chronic lung disease (38). In a recent study, vitamin D administration prenatally and postnatally reversed intra-amniotic LPS-induced aberrant alveolar and pulmonary vascular development in rats (39). Clearly, chorioamnionitis directly and indirectly changes multiple pathways that have been linked to lung growth and development.
Vascular endothelial growth factor (VEGF) signaling also is important in fetal lung development. Women with preeclampsia have increased amniotic fluid levels of soluble VEGF receptor 1, an increased risk of delivering infants with intrauterine growth restriction, and an increased the risk for chronic lung disease (40). Intra-amniotic injection of soluble VEGF receptor 1 in rats inhibits alveolar development and causes pulmonary hypertension postnatally (41). Hypoxia and HIF-1 are well-known regulators of VEGF expression (42). Pulmonary expression of HIF-1alpha, HIF-2alpha, and the downstream target of their regulation, VEGF mRNA, is impaired following RDS in neonatal lambs (43). Furthermore, high postnatal HIF-1 levels increased VEGF levels and stimulated pulmonary angiogenic factors in a preterm baboon model of chronic lung disease (44). Infants dying of chronic lung disease had lower levels of VEGF and other angiogenic factors in tracheal aspirates (45) consistent with inhibited expression of angiogenic proteins in a baboon model of chronic lung disease and fetal sheep exposed to chorioamnionitis (23,46). These studies underscore the tight linkage between pulmonary vascular development and fetal lung growth.
Human Studies of Chorioamnionitis
Chorioamnionitis in pregnant women is diagnosed clinically or after histological examination of the placenta. Thus it is often difficult to compare rates between centers or between different populations. In one U.S. study of over 2.2 million births, the prevalence of chorioamnionitis was 9.7 per 1,000 live births with odds ratio for neonatal mortality of 1.72 (95% CI 1.2 to 2.45) (47). The association of respiratory diseases including BPD following chorioamnionitis is complex. Early reports suggested that chorioamnionitis was associated with decreased rates of RDS and increased rates of developing BPD. While a systemic review did show an association between chorioamnionitis and development of BPD, the authors concluded that there was “strong evidence of publication bias which suggests potential overestimation of the measure of association between chorioamnionitis and BPD” (48). Been and colleagues have recently reported that severity of RDS may be greater in chorioamnionitis where there is fetal involvement compared to when there is not (49). Furthermore, responses of surfactant may vary after exposure to chorioamnionitis, especially when there is fetal inflammation (50). It is thus very likely that respiratory outcomes may depend on a number of factors associated with exposure to chorioamnionitis, including timing, fetal involvement, underlying cause of the infective process, and so on. Chorioamnionitis observed in infants born preterm may be associated with adverse longer-term respiratory outcomes. Jones and colleagues classified 95 preterm born infants according to histologic examination of the placenta (51). They observed lower maximal expiratory flows at forty weeks postconceptional age but interestingly noted that this outcome was mainly due to females being affected. In the Boston prospective study of 1,096 preterm subjects, those born at less than thirty-three weeks gestation with chorioamnionitis had the greatest risk of wheezing, and physicians diagnosed asthma up to the follow-up age of 2.2 years (52). Although chorioamnionitis certainly affects lung development, further work is required to address the specific population at highest risk of future risk of respiratory compromise.
Ureaplasma and Bronchopulmonary Dysplasia (BPD)
Ureaplasma is the most commonly isolated organism from the uterine cavity of women who deliver prematurely. The organism readily transfers from the mother to the fetus and has been shown to cause chorioamnionitis and significant neonatal morbidity. Macrolide treatment of pregnant women presenting in preterm labor did not alter newborn outcomes (53,54). Several meta-analyses found an association between pulmonary presence of Ureaplasma and subsequent development of BPD (55–57). The most recent report gave odds ratios for the association between the presence of pulmonary Ureaplasma and subsequent development of BPD – oxygen dependency for at least twenty-eight days of age of 3.04 (95% CI 2.41, 3.83) or oxygen dependency at least thirty-six weeks postconceptional age of 2.22 (95% CI 1.42, 3.47) (57).
Disappointingly, initial studies using erythromycin to determine if eradication of Ureaplasma in preterm infants decreased rates of BPD were unsuccessful but were markedly underpowered. More recent studies are encouraging, although they should be considered as proof of principle studies (58,59). In the first, Ballard and colleagues, reported decreased duration of mechanical ventilation for infants treated with up to six weeks of azithromycin when compared to placebo. In the other study, Ozdemir et al. treated 74 Ureaplasma culture-positive preterm infants with clarithromycin or placebo and noted that Ureaplasma was eradicated in 68.5% of the cases. Rates of BPD decreased to 2.9% in the treated group compared to 36.4% in the placebo group (p < 0.001). Partial eradication suggests either inadequate treatment or antibiotic resistance to the macrolide.
In addition to their anti-infective activities, macrolides are attractive as they have potent anti-inflammatory activities. Two studies have reported the potential role of azithromycin in targeting pulmonary inflammation in infants at risk of developing BPD. In the first, Ballard and colleagues treated 220 preterm infants for up to six weeks with azithromycin or placebo but did not note any differences in BPD or death between the two groups (60). In their subanalyses they noted decreased BPD rates in the Ureaplasma colonized infants (73% vs. 94%, p = 0.03). In the other study, Gharehbaghi and colleagues, treated 108 preterm infants with two weeks of azithromycin and noted decreased rates of BPD in the treated group (25% vs. 43%, p = 0.4) (61). These studies are encouraging, but adequately powered studies are required to assess if indeed targeting Ureaplasma and/or pulmonary inflammation can indeed alter the causal pathway in the development of BPD.
Chorioamnionitis and Antenatal Glucocorticoids: Effects on Fetal Lung Development
After the pioneering studies of Liggins (62) and the subsequent meta-analysis by Crowley (63), antenatal maternal corticosteroid treatment became a standard clinical practice for women at risk for preterm birth. A majority of very low-birth-weight deliveries also are exposed to infection or inflammation. Thus the fetus at high risk for preterm delivery is often exposed to both chorioamnionitis and maternal glucocorticoids. The common scenario is likely a fetus exposed to chorioamnionitis initially who subsequently is exposed to maternal glucocorticoids. However it is also possible that a pregnant woman who received betamethasone will subsequently develop rupture of membrane and consequently chorioamnionitis. The combined effects of these two exposures is of great biologic interest because betamethasone is a potent anti-inflammatory agent while chorioamnionitis causes fetal inflammation. Studies in fetal sheep have evaluated the pulmonary effects of fetal exposure to chorioamnionitis and maternal betamethasone with differing orders of exposures. If intra-amniotic LPS and maternal betamethasone were given simultaneously to pregnant ewes, there was an initial suppression of fetal lung inflammation induced by LPS. Curiously, there was an unexpected amplification of pulmonary inflammation at later time points. Thus maternal betamethasone had an initial innate immune suppressive effect as expected, but the later amplification of innate immune response was not anticipated (64). Despite the modulation of inflammatory response, the physiologic effect of the combined exposure was more airway surfactant production and improved lung mechanics compared to either exposure alone (Figure 10-3). When maternal betamethasone is administered before intra-amniotic LPS in the ewes, lung inflammation was suppressed (65). Interestingly, betamethasone treatment after LPS did not counteract inflammation but enhanced lung maturation. Thus the order of exposures of intra-amniotic LPS or maternal betamethasone had large effects on fetal lung inflammation and maturation. Extrapolation of these experimental results would support the clinical practice of giving maternal betamethasone to women with suspected chorioamnionitis because the net physiological effect of the combined exposure is for the fetal lung to be more “clinically mature.”
Fetal Growth Restriction, Maternal Nutrition, and Lung Development
Intra-uterine growth restriction (IUGR) is a relatively common complication of pregnancy. Various terms including IUGR, fetal growth restriction, small for gestational age, and low birth weight are often confused. IUGR can be constitutional or nonconstitutional and is often described as symmetrical or nonsymmetrical. The IUGR, defined by both birth weight and gestation may have a pathological basis, which may occur due to fetal, maternal, or placental conditions with additional environmental or genetic influences (66). Its origin is likely to involve deficiencies of delivery of nutrition and/or oxygen. The resultant effect on growth trajectories will depend on when these insults occur during the different stages of lung growth.
A number of different fetal exposures can result in IUGR. These include hypertensive disorders in the mother, maternal smoking and poor nutrition, multiple pregnancies, uterine malformations, and fetal intra-uterine infections. From a physiological perspective, IUGR is caused by decreased fetal perfusion due to increased placental resistance or due to fetal abnormal somatic growth. Decreased tissue perfusion causes hypoxemia, decreased delivery of nutrients, and increased cortisol concentrations, all of which can affect lung development. Fetal growth restriction is independently associated with an increased risk of BPD in preterm infants (67). Calorie or protein restriction in multiple species or placental blood restriction to fetuses are established models of fetal growth restriction. IUGR is associated with decrease lung weight and total DNA content, decreased maturation of type II cells and subsequent surfactant maturation, decreased alveoli numbers, and thickened airway walls in animal models.
A mechanism by which IUGR may interfere with lung development is via PPAR gamma, a transcription factor known to regulate epigenetic changes, particularly of chromatin-modifying enzymes. In a rat model of IUGR, maternal supplementation of docosahexaenoic acid (DHA) increased PPAR gamma levels and restored aberrant fetal lung development (68). Another mechanism of aberrant lung development during IUGR is via retinoic acid signaling. The retinoids act on their receptors RARs and RXRs and modulate alveolar formation, branching morphogenesis and surfactant protein synthesis (69). In a rat model, fetal growth restriction induced by a low-calorie maternal diet during pregnancy induced alveolar hypoplasia that was reversible by postnatal vitamin A supplementation (70). Administration of vitamin A can reduce lung injury and BPD in sheep and humans (71,72).
Longer-term effects of IUGR are of great interest as an initial study in the UK reported increased cardiovascular disease in adults who had growth restriction during pregnancy and during the first year of life (73). Placental insufficiency may accelerate maturation of the lungs as indicated by increased lecithin/sphingomyelin ratios in amniotic fluid (74). However, it is unclear if IUGR is associated with increased or decreased rates of respiratory distress syndrome in infancy. There is increasing evidence that growth restriction is associated with decreased lung function in childhood and beyond. In adulthood, Lawlor and colleagues in a meta-analyses of eight studies of adults found that birth weight was associated with approximately 48 mL (95% CI 26 to 70) increase in FEV1 for each kg increase in birth weight (75). Catch-up growth during childhood may improve the outlook (76), but caution is required as increased weight gain during infancy may worsen lung function (77). IUGR may be caused by fetal, maternal, or placental factors. Increasing evidence suggests that IUGR is associated with increased long-term morbidity. The challenge is to prevent IUGR where possible by health education factors, including decreasing antenatal smoking and optimal maternal nutrition.
[level-membership-for-neonatal-and-perinatal-medicine-category]
Effects of Prenatal Exposure to Toxins on Fetal Lung Development
Environmental pollutants are well recognized to cause major lung structure and function changes. In particular, antenatal smoking has received much attention. The effects of nicotine, the major toxin in cigarettes, on fetal lung development are complex. The animal experiments and clinical studies point to morphological effects that translate to altered lung function after the fetus is born. Although the rate of smoking during pregnancy has decreased in the past decade in the United States, about 12% of all pregnant mothers reported smoking cigarettes (78). Maternal smoking causes harmful effects on the fetus due to fetal hypoxemia induced by carboxyhemoglobin and growth restriction in addition to direct fetal effects of the components of cigarette smoke. Nicotine readily crosses the placenta and is likely to be a prime mediator of the toxic effects of smoking on the fetal lungs. Nicotine causes DNA damage and promotes oxidant injury.
Nicotine has direct effects on both the conducting airways and the parenchyma but also appears to alter the epigenome resulting in lifelong pulmonary consequences. Maternal cigarette smoke exposure in rats results in fewer numbers of alveoli in exposed pups (79). In fetal monkeys, prenatal exposure to nicotine also causes lung hypoplasia and increases collagen deposition around large airways and vessels (80). Prenatal nicotine exposure of lambs and monkeys causes proximal airway obstruction (81,82). Apart from the detrimental effects of nicotine on fetal lungs, nicotine stimulated branching morphogenesis and increased expression of surfactant proteins in embryonic mouse lungs cultured in vitro and increased surfactant producing alveolar type II cells in monkeys (80,83).
Consistent with these experimental results, human studies have also suggested a decreased risk for respiratory distress syndrome in infants exposed to maternal smoking (84). However, infants born to mothers who smoke during pregnancy are at greater risk of sudden infant death syndrome (SIDS), decreased lung function at birth and into adulthood, increased risk of respiratory symptoms, increased diagnosis of asthma, and increased risk of bronchial hyper-responsiveness. One study showed increased inner airway wall thickening in infants who died from SIDS for mothers who smoked 20 cigarettes or more during pregnancy when compared to those who did not (85). Clinical studies have also demonstrated more restriction in pulmonary airflow in preterm infants compared with term infants (86, 87). A more recent prospective study of a twenty-one-year follow-up of 2,409 young adults measured decreased lung spirometry (FEV1 and FEF25–75%) in the young adults born to mothers who smoked during pregnancy (88). Another recent study evaluated lung function of infants whose mothers smoked cigarettes during pregnancy. Predictably, compared to healthy term infants, lung function of infants born to smokers was decreased at birth, and these infants also had increased wheezing reports at one year of age (89). This effect was particularly pronounced in mothers with homozygous mutant alleles in the α5 nicotinic acetylcholine receptor. Remarkably, daily vitamin C supplementation (500 mg) of smoking mothers starting in the second trimester rescued decreased lung function at birth and the incidence of wheezing at one year of age. Presumably, the salutary effect of vitamin C was due to its antioxidant effect. Thus, the effects of antenatal smoking are long lasting but do provide an opportunity to intervene either by smoking cessation or by vitamin C supplementation to decrease the long-term consequences of antenatal exposure to a noxious substance.
The fetal lung may also be affected by exposure to drugs. Antidepressant, particularly selective serotonin reuptake inhibitor (SSRI), usage has increased greatly during pregnancy. In a recent meta-analysis, the use of antidepressants during pregnancy was associated with an increased the risk for persistent pulmonary hypertension of the newborn if the drug was used in the third trimester (2.50, 1.32 to 4.73; P = 0.005) but not the first trimester (odds ratio 1.23, 95% confidence interval 0.58 to 2.60; P = 0.58) (90). Because the disease is not very common, the overall risk for neonatal pulmonary hypertension with maternal SSRI use is small when considered against the potential benefits.
Another area receiving increasing scrutiny is the exposure to environmental pollutions such as carbon particles and gases such as nitrous oxide and sulphur oxide particularly from charcoal and kerosene used for cooking and heating. Increased risk of stillbirth, decreased birth weight, and prematurity are associated with increased antenatal exposure. In one study, birth weight was decreased by 110 g for coal, 107 g for kerosene, and 78 g for biomass fuels when compared to “cleaner” fuels. Low birth weight was also increased with use of kerosene and biomass fuels (91). An increased risk of neonatal death is strongly associated with household use of coal (OR 18.54; 95% CI: 6.31–54.45) and perhaps with kerosene (OR 2.30; 95% CI: 0.95–5.55). The effects of pollution may indirectly affect lung growth and development in the newborn, and further work is needed to assess if any of the effects directly affect lung growth and development of the fetus.
Fetal Breathing, Fetal Lung Fluid, Amniotic Fluid, and Lung Development
For optimal lung growth and development, adequate intra- and extrathoracic fluid, adequate intrathoracic space, pulmonary blood flow, and fetal breathing are required. Abnormalities of the thoracic cage or muscle disorders may also affect lung growth and development. The elegant experiments by Desai and Wigglesworth showed that transaction of the phrenic nerve results in hypoplastic lungs suggesting that breathing movements are essential to lung development (92). In humans, breathing movements are observed from eleven weeks gestation and occur approximately 30% of the time in the third trimester. Breathing movements appear to be essential to maintain an adequate fetal lung fluid volume. The rhythmic movements are necessary to promote mitogenic growth factor activity. Attempts have been made to use breathing movements and patterns to predict outcomes, but their clinical use are limited by their episodic nature.
The importance of a satisfactory fetal lung fluid volume was shown by Moessinger et al., who observed that when one lobe in fetal lambs was allowed to drain freely, marked lung hypoplasia occurred (93). In contrast, ligation of the contralateral lobe resulted in lung hyperplasia. Fetal lung fluid is formed by epithelial cells that actively transport chloride ions from the interstitium to the lumen. In the near term fetal lung, the fluid production rate is 4 to 5 mL/kg/hr (94). Assuming a 3 to 4 kg fetus, the daily production of fetal lung fluid is therefore about 400 mL/day. Fetal lung fluid flows intermittently up the trachea with fetal breathing movements, and some of this fluid is swallowed while the rest mixes with the amniotic fluid. The pressure in the fetal trachea exceeds that in the amniotic fluid by about 2 mm Hg, demonstrating a resistance to outflow that maintains the fetal lung fluid volume. Although the presence of fetal lung fluid is essential for normal lung development, its clearance is equally essential for normal neonatal respiratory adaptation. Fluid clearance after birth results from active sodium transport via the ENaC sodium channel, which can be blocked with amiloride (95). In experimental models, tracheal occlusion in late gestation can reverse much of the pulmonary hypoplasia resulting from diaphragmatic hernia, but the occlusion induces a decrease in type II cell number and causes surfactant deficiency (96).
Decreased amniotic fluid or oligohydramnios complicates about 10% of pregnancies (97). Although a number of factors are responsible for amniotic fluid volume, fetal urine is a significant contributor to amniotic fluid volume during later stages of pregnancy. Severe pulmonary hypoplasia associated with renal agenesis (Potter’s syndrome), and prolonged oligohydramnios is characterized by a decrease in lung size and cell number together with narrow airways, a delay of epithelial differentiation, and surfactant deficiency (98). The importance of oligohydramnios was shown in study of 5,228 women with oligohydramnios during the third trimester (99). When compared to 20,912 unaffected pregnancies, the offspring of the oligohydramnios group had increased rates of respiratory failure and increased rates of hospitalization.
Another important cause of decreased amniotic fluid is early preterm prelabor rupture of membranes (pPROM), often occurring during the second trimester. Relatively short-term oligohydramnios caused by ruptured membranes in the sixteenth to twenty-eighth week of gestation can also result in pulmonary hypoplasia; the severity in general correlates with the length of the oligohydramnios (100). Williams and colleagues reported their two-year follow-up of fifteen survivors whose mothers had ruptured their membranes before twenty-five weeks of completed gestation (101). Surprisingly they did not note a difference in BPD between the groups, but those infants with pPROM had increased length of respiratory support and increased hospital stay. Encouragingly, these affected infants did not have neurological deficits, but they did have increased postdischarge hospital readmissions for respiratory illness.
Pulmonary hypoplasia can also result from either a restriction of lung growth or the absence of fetal breathing (102). Any reduction of the chest cavity by a mass, effusion, or external compression can impact lung growth. Lung hypoplasia can be minimal or very severe. Infants with congenital diaphragmatic hernia have more severe hypoplasia on the ipsilateral side than on the contralateral side, although the contralateral lung may be hypoplastic (103). The lungs have decreased acinar units as well as delayed epithelial maturation with an associated surfactant deficiency.
Lung growth is dependent on the coordinated interaction between the intra- and extrathoracic volumes, adequate fetal lung volume and fluid production, and breathing movements. Disturbance of any of these factors will disrupt normal development of the fetal lung.
Knowledge gaps and the future: Although the concept of origin of cardiovascular and respiratory diseases in adults due to perturbations in early infancy is well known, the fetal origins hypothesis is still relatively new. We have reviewed a number of fetal insults including infection, inflammation, mechanical perturbations, and toxin exposure to the fetus as determinants of aberrant lung growth. Much of these observations are still at an early descriptive stage. Focused research using animal models and targeted clinical studies will be needed to unravel mechanisms of the pulmonary insult. A major area that desperately needs further study is the understanding of developmental ontogeny of the innate immune system of the fetus. How does the fetal immune system recognize and respond to its environmental challenge remains a mystery. The clinical correlate of this question is: Does the patterning of fetal immune system by exposure to infectious/inflammatory insult lead to lasting effects, and if so, what are the pathways? Pulmonary diseases such as reactive airway disease particularly resulting from prenatal exposures likely are mediated by aberrant lung growth and aberrant immune modulation. Understanding the relative contributions of these pathways will be important to devise targeted therapies. Similar questions can be asked about how mechanical stretch mediates lung development in the fetus. At present, treatment with antimicrobials alone have not shown to substantively improve perinatal outcomes even for women with chorioamnionitis. In the future, it may be important to devise anti-inflammatory immunomodulatory therapies as adjunct to antimicrobials to optimize the perinatal outcomes including optimizing pulmonary outcomes for the fetus. Vitamin C supplementation for women who are not able to cease smoking during pregnancy might be an inexpensive but effective treatment to improve pulmonary outcomes of the fetus.
Summary
A multitude of exposures can affect fetal lung development. Understanding the physiology and basic mechanisms of fetal lung development may allow us to intervene in the future. For example, imaging studies are being used to predict the degree of lung hypoplasia in high-risk pregnancies. Unfortunately, antenatal prediction of pulmonary hypoplasia is still far from accurate. However, more sophisticated imaging modalities in the future can improve the antenatal prediction of pulmonary hypoplasia. Currently, the long-term benefits of fetal tracheal occlusion therapy to reduce pulmonary hypoplasia resulting from diaphragmatic hernia have not been proven, and this therapy remains investigative. Finally, public health measures to reduce fetal exposure to toxins that adversely affect lung development need to be intensified.
References
[/level-membership-for-neonatal-and-perinatal-medicine-category][not-level-membership-for-neonatal-and-perinatal-medicine-category]
[/not-level-membership-for-neonatal-and-perinatal-medicine-category]
