[level-membership-for-emergency-medicine-category]Chapter 82
Pericardial and Myocardial Disease
Pericardial Disease (Pericarditis)
Our knowledge of pericardial function and disease has increased greatly since Hippocrates described the pericardium in 460 BC as “a smooth tunic that envelops the heart and contains a small amount of fluid resembling urine.” Galen provided the first description of a pericardial effusion and performed the first pericardial resection.1 Lancisi first described the appearance of constrictive pericarditis at autopsy in 1728. Also in the 18th century, Laennec said, “There are few diseases attended by more variable symptoms and more difficult to diagnose than [pericarditis].”1 In 1935, Beck described the clinical presentation of cardiac tamponade, known as Beck‘s triad (hypotension, jugular venous distention, and muffled heart sounds).2 Despite the passage of time and the modern availability of many diagnostic tools, the diagnosis and treatment of pericardial disease still present a challenge.
Etiology
Each of the disorders listed in Box 82-1 can produce acute pericarditis, with or without pericardial effusion. In addition, most of these disorders can progress to cardiac tamponade or constrictive pericarditis.
Principles of Disease
Pericardial Anatomy and Physiology
The normal pericardium envelops the heart and attaches to the great vessels. It consists of parietal and visceral layers, with a narrow potential space between them. The visceral layer or epicardium adheres to the myocardium. It is separated from the parietal layer by a potential space. Each layer is 1 or 2 mm thick and is composed of elastic fibers. The position of the heart within the chest is stabilized by the attachment of the parietal pericardium to the sternum, the diaphragm, and the vertebral column. Its blood supply comes from the internal mammary artery, and its nerve supply from the phrenic nerve.3
Idiopathic Pericarditis
Diagnostic Strategies
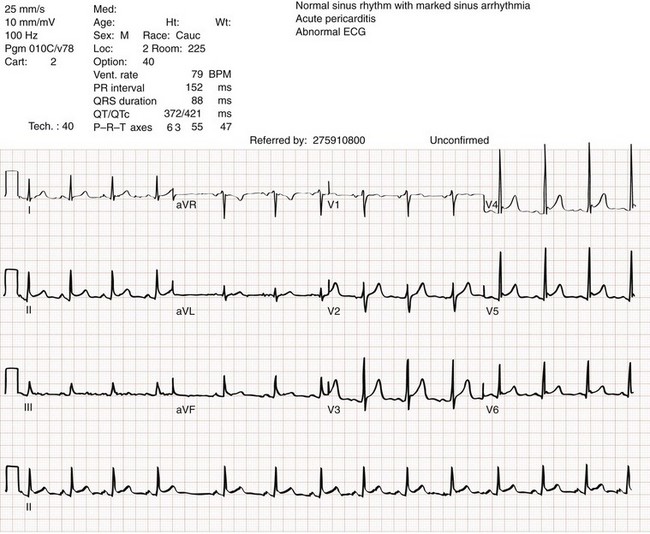
There is no single test that is diagnostic for pericarditis. The ECG is the most reliable diagnostic tool. It evolves through stages that occur over time. The first stage occurs in the first hours to days of illness. It includes diffuse ST segment elevation seen in leads I, II, III, aVL, aVF, and V2 through V6 and also reciprocal ST segment depression in aVR and V1. Most patients with acute pericarditis have concurrent PR segment depression (Fig. 82-1). In the next stages, the ST and PR segments normalize, but the T waves flatten, and then there is deep, symmetrical T wave inversion. At the last stage, the ECG reverts to normal, although the T wave inversions may become permanent.4
Management and Disposition
If a specific cause of pericarditis is found, therapy should be specific for that cause. Otherwise, therapy for acute pericarditis is symptomatic. Anti-inflammatory therapy will reduce pain, and a nonsteroidal anti-inflammatory drug (NSAID) is the treatment of first choice. Ibuprofen has the best side effect profile, but other NSAIDs should be equally effective. The patient will often report significant pain relief from the analgesic effect of the ibuprofen while in the ED, even before onset of the anti-inflammatory effect. A dose of 600 mg four times a day for 1 week is a good initial therapy. If the chosen NSAID is not effective within 1 week, a different class of NSAIDs should be tried, such as indomethacin 25 mg three times a day for 1 week. Colchicine (1.2 initially then 0.6 mg daily for up to 6 months) is effective for recurrent pericarditis, and steroid therapy (1 mg/kg daily) has shown mixed results.5,6
Complications
The clinical course of pericarditis is variable: 60% of patients have complete recovery within 1 week, and almost 80% have complete recovery within 3 weeks. Patients with fever, pericardial effusion, a subacute course, or failure of initial NSAID treatment have a worse prognosis. Eighteen percent of patients can have recurrent pericarditis that may require serial echocardiography to exclude effusion or tumor.6
Uremic Pericardial Disease
Clinical Features and Diagnostic Strategies
Patients with uremic pericarditis have chest pain, unexplained fever, and possibly a coarse friction rub. They also may have significant effusions. The ECG in uremic pericarditis is often normal because little epicardial inflammation occurs.7 In a dialysis patient, cardiac enlargement on chest radiograph in the absence of signs of volume overload or congestive heart failure (CHF) should prompt consideration of pericardial effusion. An echocardiogram will provide the definitive answer. Pericardiocentesis may be needed to exclude infection.
Post–Myocardial Infarction Pericarditis
Dressler reported a syndrome of fever, pleuritis, leukocytosis, pericardial friction rub, and chest radiograph evidence of new pericardial or pleural effusions in post-MI patients.8 Frequent relapses and a high incidence of friction rubs led Dressler to describe this syndrome as a delayed complication of MI in contrast to the well-known syndrome of early post-MI pericarditis. The cause of late post-MI pericarditis (Dressler’s syndrome) may be immunologic, but the syndrome may also occur with pulmonary embolus and after pericardiotomy. Anticoagulants should be discontinued to reduce the risk of hemorrhage. Delayed post-MI pericarditis is treated with NSAIDs such as ibuprofen 600 mg four times per day or indomethacin 25 mg three times per day.
Neoplastic Pericardial Disease
Malignant pericardial tumors typically manifest late, which complicates diagnosis and treatment. Malignant involvement of the pericardium is observed in 3.4% of general autopsies and 2 to 31% of cancer autopsies. The most common causes are lung cancer (30%), breast cancer (23%), lymphoma (17%) and leukemia (9%). Primary malignancies of the pericardium are rare.9
Pericardial Effusion
Cardiac Tamponade
Ten percent of all patients with cancer develop cardiac tamponade.10 Cardiac tamponade should be suspected in patients with penetrating chest wounds. It is also common in patients with uremic pericarditis.
Cardiac tamponade is a physiologic continuum reflecting the amount of fluid, the rate of accumulation, and the nature of the heart. The three stages necessary for tamponade to develop are fluid filling the recesses of the parietal pericardium, fluid accumulating faster than the rate of the parietal pericardium’s ability to stretch, and accumulation that exceeds the body’s ability to increase blood volume to support right ventricle filling pressure. The final result is increased pericardial pressure, which causes decreased ventricle compliance and decreased flow of blood into the heart. The reduction of blood inflow into the right ventricle results in decreased stroke volume that leads to decreased cardiac output.10 The most important factor in the development of tamponade is the rate of fluid accumulation.
Symptoms and Signs
Cardiac tamponade symptoms are often nonspecific, but particularly include chest pain, cough, or dyspnea, any of which may be progressive and severe. The classic triad of cardiac tamponade signs described by Beck is hypotension, distended neck veins, and muffled heart sounds.11 These signs may not be present if tamponade develops quickly.
Diagnostic Strategies
The chest radiograph shows cardiomegaly only if there is a large accumulation of fluid (250 mL). The ECG classically shows decreased voltage or electrical alternans (Fig. 82-3), but the latter is rare. Echocardiography confirms the diagnosis when an effusion and paradoxical systolic wall motion are seen. Thermodilution catheters can also be diagnostic, showing equalization of right and left ventricular pressures.
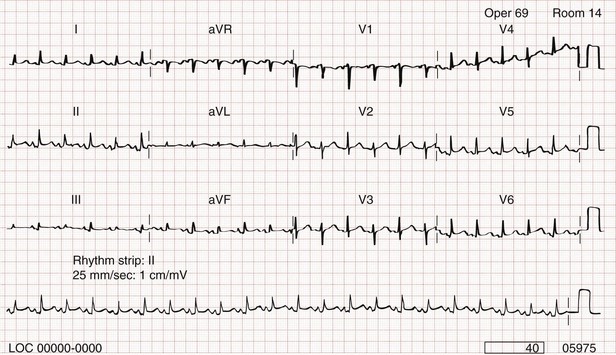
Figure 82-3 Electrocardiogram showing electrical alternans.
Management and Prognosis
Initial treatment includes volume augmentation to the right ventricle with intravenous fluids to increase the filling pressure to overcome the pericardial constriction. Pericardiocentesis or pericardial window is the treatment of choice. Enough fluid should be withdrawn to stabilize the patient. If tamponade recurs, pericardiocentesis may be repeated, or a drainage catheter may be left in the pericardial space. A pericardiectomy ultimately may be necessary. Cardiac tamponade has a high mortality that depends on the severity and nature of the underlying disease, the time course of onset, and the rapidity of diagnosis and intervention. Traumatic cardiac tamponade is discussed in Chapter 45.
Purulent Pericarditis
Tuberculous Pericarditis
Tuberculous pericarditis is estimated to occur in 1 or 2% of patients with pulmonary tuberculosis.12 In Africa, it is the most common cause of pericarditis. In countries in which tuberculosis is not a major health problem, tuberculous pericarditis is most common in patients who are socioeconomically deprived or immunodeficient. In many patients the chest radiograph shows an enlarged cardiac silhouette without a pulmonary infiltrate. Pericardial fluid aspirates reveal acid-fast bacilli by smear or culture (which may require 4-6 weeks to become positive) in approximately 50% of cases. Diagnostic workup should include assessment for human immunodeficiency virus (HIV). Patients with tuberculous pericarditis should be hospitalized and observed for evidence of cardiac tamponade. Triple-drug therapy should be started in the hospital and continued for at least 9 months. Patients with chronic pericardial effusions may benefit from oral prednisone therapy. The mortality rate is approximately 15% in HIV-negative patients and 20 to 35% in HIV-positive patients.12
Constrictive Pericarditis
Myocardial Diseases (Myocarditis)
Epidemiology and Etiology
Some degree of myocarditis is detected in almost 10% of routine autopsies but is often not recognized clinically. The overall incidence is unknown and probably underdiagnosed. The Coxsackie B virus was originally named as the causative organism, but the predominant agents in the 1990s were the adenoviruses, followed by parvovirus 19 and human herpesvirus 6 since 2000.14 In reality, any infectious agent can cause myocarditis (Box 82-2). The etiology varies by patient age and region. Cytomegalovirus and Toxoplasma gondii have emerged as potential causes of myocarditis in cardiac transplant patients. Of patients who have died of AIDS, 67% have myocarditis on autopsy. Worldwide, Chagas’ disease is a leading cause of myocarditis, especially in South America. Mortality from myocarditis has not changed since before 1990. The mortality rate is approximately 20% at 1 year and 50% at 5 years.15
Principles of Disease and Pathophysiology
Cardiac autoantibodies develop after myocarditis. There is also a higher concentration of immunoglobulin G (IgG) anti–β-myosin antibodies in patients with myocarditis and DCM than in controls. Because myocarditis is linked to the development of DCM, idiopathic DCM after myocarditis may be predominantly autoimmune in origin, resulting from either shared antigens or molecular mimicry.16 The amino acid sequences of the Coxsackie B virus and β-myosin heavy chain protein are similar. An immune response to the former yields damage to the latter (molecular mimicry).
Clinical Features
Flulike complaints, including fever, fatigue, myalgias, vomiting, and diarrhea, are usually the first symptoms and signs of myocarditis. The most common presentation in children is dyspnea. In adults, it is dyspnea, chest pain, and dysrhythmias. Altered vital signs include fever, tachycardia, tachypnea, and, uncommonly, hypotension. Tachycardia disproportionate to the temperature or apparent toxicity may occur but is a nonspecific finding. No symptom or sign is sensitive or specific. Cardiac examination is often unremarkable. When chest pain or CHF occurs at initial presentation, there is a worse prognosis.17
Diagnostic Strategies
Endocardial biopsy, the historical gold standard, has variable sensitivity and specificity owing to sampling error when the tissue taken is different from the tissue involved (Fig. 82-4). Histologic criteria for myocarditis are present in only 5 to 30% of patients with clinically suspected myocarditis and up to half of patients with DCM. Molecular genetic probes, such as polymerase chain reaction assays, are used to supplement standard histologic analysis. In addition, polymerase chain reaction analysis of tracheal aspirates of intubated patients with myocarditis shows a correlation with endocardial biopsy. Because of the limitations of biopsy, magnetic imaging may become the diagnostic test of choice and the next gold standard.
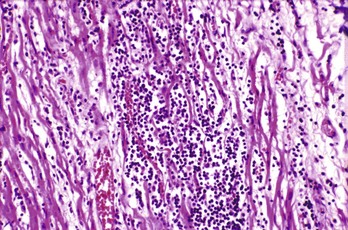
Management
Therapy is stage specific, given the three distinct phases of the disease. In the first phase, demonstration of replicating virus suggests that early antiviral agents, such as pleconaril or ribavirin, may be effective. Unfortunately, the diagnosis is often made after the initial viral phase. Agents active at the Coxsackievirus-adenovirus receptor present an intriguing theoretic approach.15
Multicenter trials of immunosuppressive therapy for the subacute phases of myocarditis have shown no benefit.15 Efforts to identify patient and treatment subsets in which immunosuppressive therapy may be beneficial are ongoing. High-dose gamma globulin therapy has been studied in a pediatric population. High-dose intravenous immunoglobulin may be associated with improved recovery of left ventricular function and better survival during the first year after presentation.
Chagas’ Disease
Chagas’ disease is one of the leading causes of myocardial disease in many countries in Latin America, particularly in Central America. Chagas’ disease is caused by the protozoan Trypanosoma cruzi with transmission by insect vectors.18
Approximately 75% of seropositive patients with Chagas’ disease never have symptoms. Systemic symptoms include fever, hepatic or splenic enlargement, and unilateral periorbital edema. Cardiac manifestations include angina-like chest pain, dysrhythmias, embolic episodes, heart failure, conduction abnormalities, multifocal ventricular premature contractions, and abnormal ST segment and T wave abnormalities. Ventricular tachycardia is common. Syncopal or near-syncopal episodes occur in nearly two thirds of patients.19
Chagas’ disease is treated with the antitrypanosomal drugs benznidazole (5 mg/kg/day divided into two daily doses for 60 days) and nifurtimox (15-20 mg/kg/day for patients younger than 10 years, 12.5-15 mg/kg/day for patients aged 10-18 years, and 8-10 mg/kg/day for patients older than 18 years, divided into four doses for 90 days). These medications are available from the Center for Disease Control and Prevention. Amiodarone may be useful to treat ventricular tachycardia. An angiotensin-converting enzyme inhibitor may be useful for CHF. Increased world attention, with elimination of the vector and improved blood donation screening, especially for platelet transfusions, is decreasing the incidence of this disease in Latin America.20
Trichinosis
The diagnosis is usually established with serologic studies or biopsy of any symptomatic muscle group. Treatment usually involves corticosteroids (1 mg/kg/day) together with anthelminthic drugs such as albendazole (400 mg twice a day for 14 days) and mebendazole (400 mg four times per day for 14 days).21
Diphtheria
Diphtheria presentation, diagnosis, and treatment are discussed in Chapter 129. Myocardial involvement is clinically evident in 10 to 25% of cases and is the major cause of death.22 Early signs of myocarditis are tachycardia and faint heart sounds. Cardiac enzymes are often elevated. Prolongation of the PR interval and ST-T wave abnormalities occur early in the course of the disease. Bundle branch block or complete heart block, when they occur, precede total circulatory collapse and are associated with a poor prognosis. Treatment with oral carnitine, a cofactor in the transport of fatty acids to mitochondria, is associated with a lower incidence of mortality, heart failure, and severe conduction blocks.22
Lyme Disease
Epidemiology and Clinical Features
Lyme disease is caused by infection with the spirochete Borrelia burgdorferi and is discussed in Chapter 141.134 Lyme disease–related carditis occurs weeks to months after the onset of erythema migrans. Cardiac complications occur in 4 to 10% of patients. Such complications most commonly is a conduction delay, usually a reversible effect on the AV node. Pericarditis and CHF also occur.
Other Causes of Myocarditis
The cardiac manifestations of AIDS are diverse and cause death in at least 6% of patients with HIV (HIV is discussed in Chapter 132). The prevalence of left ventricular dysfunction in adult AIDS patients is approximately 20%. Myocarditis is described in approximately 46% of AIDS patients undergoing postmortem examination. Cardiac involvement increases as the disease worsens. HIV treatment may also lead to cardiac toxicity. Pentamidine can cause torsades de pointes ventricular tachycardia. Zidovudine and dideoxyinosine also can lead to cardiac dysfunction.
Cardiomyopathies and Specific Heart Muscle Disease
Cardiomyopathies are a heterogeneous group of diseases associated with mechanical or electrical dysfunction.24 They usually exhibit inappropriate ventricular hypertrophy or dilation and have a variety of causes. It is becoming increasingly evident that many of these diseases result from genetic mutations. The diseases included are listed in Box 82-3.
A variety of pathologic processes may initiate myocyte injury. When injury occurs, common pathophysiologic pathways are activated. These pathways involve neurohumoral factors, immune factors, and cytokines that cause myocyte dysfunction and subsequent remodeling. At the organ level the remodeling is hypertrophy or dilation. There is also an increase in interstitial fibrosis, which impairs ventricular filling; this leads to increased metabolic demands on the myocardium. At a cellular level, the pathophysiologic derangement may be the troponin complex, intracellular concentration of calcium, myocardial subproteins, or the sarcomere. These lesions lead to alteration in the cardiac muscle’s ability to contract, which eventually leads to clinical pathology.25 There is also change in the cardiac microvascular circulation that is an independent predictor of morbidity and mortality.26 Although traditionally defined by organ level pathology, the classification of cardiomyopathies may evolve into a unified theory that shows all types of cardiomyopathy to be variations of a common genetic, anatomic, and humoral pathophysiologic process.
Mutations of genes for myocardial protein components lead to molecular changes in the myocardium that eventually lead to clinical disease. Mutations have been found for abnormalities in cytoskeleton force transmission, impaired force production, and cell junction regulation.27 The exact correlation between genotype, phenotype, and clinical presentation is unknown, as is the point when molecular level changes transition from compensatory to pathologic.
Dilated Cardiomyopathy
DCM is a spectrum of disorders that have in common a dilated and failing heart. The incidence of DCM is estimated to be 1 case per 2500 adults and 0.57 cases per 100,000 children. The true incidence is probably underestimated because many asymptomatic cases remain undiagnosed. DCM is estimated to affect 5.5 million adults and 500,000 children in the United States, costing $10 billion annually.28 Many cases previously thought to be of unknown cause may be secondary to infection. Myocarditis is the most common cause of DCM in children.28 It is now known that 30 to 40% of DCM cases have a genetic cause, with more than 35 gene mutations discovered, primarily coding for cytoskeleton or sarcomere proteins.29
Pathophysiology
There are both primary and secondary causes for DCM. Possible pathophysiologic causes include myocardial inflammation mediated by cytokines, macrophages, and natural killer cells; local inflammation caused by the release of cytokines by infiltrating lymphocytes; direct reaction of antibodies with receptors on myocardial muscle; toxins, such as ethanol, impairing myocardial biochemical processes; and loss or dysfunction of myocardial matrix proteins.30 Myocardial damage initiates a vicious cycle of hypertrophic cell death that increases the burden of the remaining cells; this leads to signaling, more work, and cell death.31
Diagnostic Strategies
As many as one third of patients may have a genetic defect as the underlining cause. Obtaining a family history and genetic counseling may be indicated.32
Management and Disposition
Angiotensin-converting enzyme inhibitors reduce morbidity and mortality. Other afterload-reducing agents, such as isosorbide dinitrate and hydralazine, also prolong survival in patients with heart failure. Spironolactone and the angiotensin receptor-blocking agents also prolong survival. Implantable defibrillators improve survival and should be considered in patients with an ejection fraction less than 30% or symptoms for more than 9 months. Short-term statin therapy may help.33 Antidysrhythmics are not usually effective.
Hypertrophic Cardiomyopathy
Principles of Disease
Anatomy: HCM is a disease involving abnormalities of heart muscle at anatomic, cellular, and genetic levels. Specifically, it is a genetic disease of sarcomere proteins. The defining anatomic feature of HCM is a hypertrophied left ventricle in the absence of another cause of left ventricular hypertrophy. The thickening is usually asymmetrical and involves the septum more than the free ventricular wall. The extent of hypertrophy at any given site can vary greatly and bears significantly on the manifestation of the disease. The dimensions of the left ventricular and right ventricular cavities are small or normal. Atrial dilation is another feature.
Histologically, individual muscle cells are hypertrophied, with a disorganized, characteristic whorled pattern.34 Sarcomere disarray is the histologic hallmark. Abnormal fibrous tissue is often found in the left ventricle, and the scarring mimics a healed MI.
Pathophysiology: HCM is an autosomal dominant disease caused by mutations in genes that encode for sarcomere contractile proteins. Ten genes encoding for cardiac sarcomere proteins and more than 200 different mutations of these genes have been identified. Half the mutations involve three genes: those for β-myosin heavy chain (which constitutes 30% of myocardial protein), myosin-binding protein C, and troponin T.
Clinical Features
Although HCM occurs at all ages, the average age at diagnosis is 30 to 40 years. Approximately 2% of cases are diagnosed in children younger than age 5 years, and 7% are diagnosed before 10 years of age. HCM gained notoriety after the press coverage of the sudden deaths of several young athletes. There is advocacy for large-scale ECG screening of young athletes, but no studies demonstrate an appropriate strategy for effective screening. The presentation of HCM varies widely, and it may be discovered by screening relatives of patients who have HCM.34
Management
Nitroglycerin, the traditional initial ED management for chest pain, is avoided in HCM-associated chest pain because it decreases ventricular volume. Amiodarone is the drug of choice for treatment of ventricular dysrhythmias in HCM. Amiodarone may also control atrial fibrillation. Automatic implantable cardioverter defibrillators are indicated for patients with sudden death or a history of a sudden death risk factor.35 Updated American Heart Association guidelines do not recommend prophylactic antibiotics in patients with HCM.36
Restrictive Cardiomyopathy
The most common cause of RCM worldwide is tropical endomyocardial fibrosis, which is endemic to India, Africa, and Latin America. Symptoms include an initial viral-like illness followed by persistent fever, malaise, and the development of severe right-sided heart failure.37
Peripartum Cardiomyopathy
Peripartum cardiomyopathy (PPCM) is uncommon and represents less than 1% of the cardiovascular problems associated with pregnancy. PPCM is a form of DCM with symptoms and signs of heart failure that appear initially during the last 3 months of pregnancy or the first 5 months postpartum. The diagnosis is made if there is no previous heart disease and no other cause for the heart failure.38
Etiology and Epidemiology
The cause of PPCM is unknown. Risk factors include myocarditis, excessive use of tocolytics, preeclampsia, advanced maternal age, multiparity, twins, and genetic predisposition. The incidence is estimated to be 1 case of PPCM per 13,000 to 15,000 live births.39
Management and Disposition
Treatment of PPCM includes limitation of physical activity, beta-blockers, alteration of preload with nitrates and diuretics, increase in ventricular contractility, and afterload reduction.40 Angiotensin-converting enzyme inhibitors are teratogenic and should be avoided, if possible, in pregnancy. Hydralazine and labetalol are effective choices.
Mortality for PPCM in the United States is approximately 2%.39 Half of the survivors have complete or near-complete recovery of cardiac function within the first 6 months. Patients who do not recover completely show either continuous clinical deterioration or persistent left ventricular dysfunction. Subsequent pregnancies may be associated with a high risk of maternal mortality. In the ED, patients with signs of hemodynamic instability or failure to maintain oxygenation should be admitted for treatment and fetal monitoring.
Takotsubo Cardiomyopathy
Takotsubo cardiomyopathy (TCM), also known as stress cardiomyopathy, broken heart syndrome, or tako-tsubo cardiomyopathy, was first reported from Japan in 1991.41 In Japanese, tako-tsubo means octopus trap. The term was used because the cardiac abnormality observed in this disease resembles that of the device used to catch octopi.42 There have been hundreds of cases reported in the medical literature since that time (Box 82-4). The pathophysiology of TCM is a sudden temporary myocardial weakening. The exact mechanism is unknown. Speculated causes include stress hormones, microvascular spasm, focal myocarditis, and cellular level muscle changes.
Clinical Features and Diagnostic Strategies
The clinical presentation is usually chest pain, although dyspnea occurs, especially on exertion. Almost 90% of cases involve women.43 There is uniformly a significant stressful incident that precedes the onset of symptoms. ECG is often consistent with an anterior MI. There are transient Q waves and ST elevation. Later, the ECG shows T wave inversion and a prolonged QT interval. Cardiac enzymes are elevated slightly and resolve to normal levels quickly. Coronary artery angiography shows no—or very little—disease. The diagnosis is made by left ventricular angiography or echocardiography that shows a ballooning of the apex during the acute phase of the disease. The involved area does not match a coronary artery anatomic distribution.
Ion Channelopathies
Specific Heart Muscle Diseases
Massive amyloid deposition results in an increased cardiac weight and the diastolic dysfunction of RCM. CHF occurs in most cases of cardiac amyloidosis. Treatment involves standard CHF and antidysrhythmic therapy, although the dysrhythmias in amyloid heart disease are often refractory to treatment. The prognosis is poor, with death often resulting from progressive heart failure within 1 year of symptom onset.44
Connective Tissue Disorders and Disease of the Myocardium
Primary myocardial involvement is a major complication of diffuse scleroderma and develops as scleroderma worsens. Estimates of the frequency of myocardial involvement in scleroderma vary widely. The clinical presentation includes CHF, angina, and dysrhythmias. Pericardial disease can also occur. Although prognosis is poor, azathioprine may be a beneficial adjunct to steroid therapy.45
Sudden Death
• Age younger than 20 years—myocarditis 22% and HCM 22%





References
1. Spodick, DH. Medical history of the pericardium. Am J Cardiol. 1970;26:447.
2. Beck, CS. Two cardiac compression triads. JAMA. 1935;104:714.
3. Troughton, R, Asher, C, Klein, A. Pericarditis. Lancet. 2004;363:717.
4. Lange, RA, Hills, D. Acute pericarditis. N Engl J Med. 2004;351:2195.
5. Lotrionte, M, et al. International collaborative systematic review of controlled clinical trials on pharmacologic treatment for acute pericarditis and its recurrences. Am Heart J. 2010;160:662.
6. Imazio, M, et al. Indicators of poor prognosis of acute pericarditis. Circulation. 2007;115:2739.
7. Gunukula, S, Spodick, D. Pericardial disease in renal patients. Semin Nephrol. 2001;21:52.
8. Dressler, W. A post-myocardial infarction syndrome. JAMA. 1956;160:1379.
9. Retter, A. Pericardial disease in the oncology patient. Heart Dis. 2002;4:387.
10. Spodick, DH. Acute cardiac tamponade. N Engl J Med. 2003;349:684.
11. Roy, CL, Minot, MA, Brookhart, MA, Choudhry, NK. Does this patient with pericardial effusion have cardiac tamponade? JAMA. 2007;297:1810.
12. Mayosi, BM, Burgess, LJ, Doubell, AF. Tuberculous pericarditis. Circulation. 2005;112:3608.
13. Goldstein, J. Cardiac tamponade, constrictive pericarditis and restrictive cardiomyopathy. Curr Probl Cardiol. 2004;29:503.
14. Cooper, L. Myocarditis. N Engl J Med. 2009;360:1526.
15. Magnani, JV, Dec, W. Myocarditis. Current trends in diagnosis and treatment. Circulation. 2006;113:876.
16. Cunningham, MW. T cell mimicry in inflammatory heart disease. Mol Immunol. 2004;40:1121.
17. Marholdt, H, et al. Presentation, patterns of myocardial damage, and clinical course of viral myocarditis. Circulation. 2006;114:1581.
18. Moncayo, A, Ortiz Yanine, MI. An update on Chagas disease (human American trypanosomiasis). Ann Trop Med Parasitol. 2006;100:663.
19. Bern, C, et al. Evaluation and treatment of Chagas disease in the United States. JAMA. 2007;298:2171.
20. Rassi, A, Jr., Rassi, A, Marin-Neto, JA. Chagas disease. Lancet. 2010;375:1388.
21. Kennedy, E, et al. Trichinellosis surveillance—United States, 2002-2007. MMWR Surveill Summ. 2009;58:SS9.
22. Vitek, C. Diphtheria. Curr Top Microbiol Immunol. 2006;304:71.
23. Steere, AC. Lyme disease. N Engl J Med. 2001;345:115.
24. Maron, BJ, et al. Contemporary definitions and classifications of the cardiomyopathies: An American Heart Association scientific statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care Outcomes and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2007;113:1807.
25. Gomes, A, Potter, J. Molecular and cellular aspects of troponin cardiomyopathies. Ann N Y Acad Sci. 2004;1015:214.
26. Camici, PG, Crea, F. Coronary microvascular dysfunction. N Engl J Med. 2007;356:830.
27. Thiene, G, Corrado, D, Basso, C. Cardiomyopathies: Is it time for a molecular classification? Eur Heart J. 2004;25:1772.
28. Towbin, JA, et al. Incidence, causes and outcomes of dilated cardiomyopathy in children. JAMA. 2006;296:1867.
29. Jefferies, J, Towbin, J. Dilated cardiomyopathy. Lancet. 2010;375:752.
30. Maisch, B, Richter, A, Sandmoller, A. Inflammatory dilated cardiomyopathy (DCM). Herz. 2005;30:535.
31. Katz, A. Pathophysiology of heart failure: Identifying targets for pharmaceutical therapy. Med Clin North Am. 2003;87:303.
32. Howlett, JG, et al. Canadian Cardiovascular Society Consensus Conference guidelines on heart failure, update 2009: Diagnosis and management of right sided heart failure, myocarditis, device therapy and recent important clinical trials. Can J Cardiol. 2009;25:85.
33. Dickinson, MG, et al. Statin use was associated with reduced mortality in both ischemic and nonischemic cardiomyopathy and in patients with implantable defibrillators: Mortality data and mechanistic insights from the Sudden Cardiac Death in Heart Failure Trial (SCD-HeFT). Am Heart J. 2007;153:573.
34. Maron, BJ, et al. Recommendations and considerations related to preparticipation screening for cardiovascular abnormalities in competitive athletes: 2007 update: A scientific statement from the American Heart Association Council on Nutrition, Physical Activity, and Metabolism: Endorsed by the American College of Cardiology Foundation. Circulation. 2007;115:1643.
35. Maron, BJ, Spirito, P, Shen, WK. Implantable cardioverter-defibrillators and prevention of sudden cardiac death in hypertrophic cardiomyopathy. JAMA. 2007;298:405.
36. Wilson, W, et al. Prevention of infective endocarditis: Guidelines from the American Heart Association: A guideline from the American Heart Association Rheumatic Fever, Endocarditis, and Kawasaki Disease Committee, Council on Cardiovascular Disease in the Young, and the Council on Clinical Cardiology, Council on Cardiovascular Surgery and Anesthesia, and the Quality of Care and Outcomes Research Interdisciplinary Working Group. Circulation. 2007;116:1736.
37. Nihoyannopoulos, P, Dawson, D. Restrictive cardiomyopathies. Eur J Echocardiogr. 2009;10:iii23.
38. Abboud, J, Murad, Y, Chen-Scarabelli, C, Saravolatz, L, Scarabelli, TM. Peripartum cardiomyopathy: A comprehensive review. Int J Cardiol. 2007;118:295.
39. Mielniczuk, LM, et al. Frequency of peripartum cardiomyopathy. Am J Cardiol. 2006;97:1765.
40. Sliwa, K, Fett, J, Elkayam, U. Peripartum cardiomyopathy. Lancet. 2006;368:687.
41. Dote, K, et al. Myocardial stunning due to simultaneous multivessel coronary spasms: A review of 5 cases. J Cardiol. 1991;21:203.
42. Bielecka-Dabrowa, A, et al. Takotsubo cardiomyopathy—the current state of knowledge. Int J Cardiol. 2010;142:120.
43. Dorfman, T, Iskandrian, A. Takutsubo cardiomyopathty: State-of-the-art review. J Nucl Cardiol. 2009;16:122.
44. Shah, KB, Inoue, Y, Mehra, MR. Amyloidosis and the heart. Arch Int Med. 2006;166:1805.
45. Leyngold, I, Baughman, K, Kasper, E, Ardehali, H. Comparison of survival among patients with connective tissue disease and cardiomyopathy (systemic sclerosis, systemic lupus erythematosus, and undifferentiated disease). Am J Cardiol. 2007;100:513.
[/level-membership-for-emergency-medicine-category][not-level-membership-for-emergency-medicine-category]Chapter 82
Pericardial and Myocardial Disease
Pericardial Disease (Pericarditis)
Our knowledge of pericardial function and disease has increased greatly since Hippocrates described the pericardium in 460 BC as “a smooth tunic that envelops the heart and contains a small amount of fluid resembling urine.” Galen provided the first description of a pericardial effusion and performed the first pericardial resection.1 Lancisi first described the appearance of constrictive pericarditis at autopsy in 1728. Also in the 18th century, Laennec said, “There are few diseases attended by more variable symptoms and more difficult to diagnose than [pericarditis].”1 In 1935, Beck described the clinical presentation of cardiac tamponade, known as Beck‘s triad (hypotension, jugular venous distention, and muffled heart sounds).2 Despite the passage of time and the modern availability of many diagnostic tools, the diagnosis and treatment of pericardial disease still present a challenge.
Etiology
Each of the disorders listed in Box 82-1 can produce acute pericarditis, with or without pericardial effusion. In addition, most of these disorders can progress to cardiac tamponade or constrictive pericarditis.
Principles of Disease
Pericardial Anatomy and Physiology
The normal pericardium envelops the heart and attaches to the great vessels. It consists of parietal and visceral layers, with a narrow potential space between them. The visceral layer or epicardium adheres to the myocardium. It is separated from the parietal layer by a potential space. Each layer is 1 or 2 mm thick and is composed of elastic fibers. The position of the heart within the chest is stabilized by the attachment of the parietal pericardium to the sternum, the diaphragm, and the vertebral column. Its blood supply comes from the internal mammary artery, and its nerve supply from the phrenic nerve.3
Idiopathic Pericarditis
Diagnostic Strategies
There is no single test that is diagnostic for pericarditis. The ECG is the most reliable diagnostic tool. It evolves through stages that occur over time. The first stage occurs in the first hours to days of illness. It includes diffuse ST segment elevation seen in leads I, II, III, aVL, aVF, and V2 through V6 and also reciprocal ST segment depression in aVR and V1. Most patients with acute pericarditis have concurrent PR segment depression (Fig. 82-1). In the next stages, the ST and PR segments normalize, but the T waves flatten, and then there is deep, symmetrical T wave inversion. At the last stage, the ECG reverts to normal, although the T wave inversions may become permanent.4
Management and Disposition
If a specific cause of pericarditis is found, therapy should be specific for that cause. Otherwise, therapy for acute pericarditis is symptomatic. Anti-inflammatory therapy will reduce pain, and a nonsteroidal anti-inflammatory drug (NSAID) is the treatment of first choice. Ibuprofen has the best side effect profile, but other NSAIDs should be equally effective. The patient will often report significant pain relief from the analgesic effect of the ibuprofen while in the ED, even before onset of the anti-inflammatory effect. A dose of 600 mg four times a day for 1 week is a good initial therapy. If the chosen NSAID is not effective within 1 week, a different class of NSAIDs should be tried, such as indomethacin 25 mg three times a day for 1 week. Colchicine (1.2 initially then 0.6 mg daily for up to 6 months) is effective for recurrent pericarditis, and steroid therapy (1 mg/kg daily) has shown mixed results.5,6
Complications
The clinical course of pericarditis is variable: 60% of patients have complete recovery within 1 week, and almost 80% have complete recovery within 3 weeks. Patients with fever, pericardial effusion, a subacute course, or failure of initial NSAID treatment have a worse prognosis. Eighteen percent of patients can have recurrent pericarditis that may require serial echocardiography to exclude effusion or tumor.6
Uremic Pericardial Disease
Clinical Features and Diagnostic Strategies
Patients with uremic pericarditis have chest pain, unexplained fever, and possibly a coarse friction rub. They also may have significant effusions. The ECG in uremic pericarditis is often normal because little epicardial inflammation occurs.7 In a dialysis patient, cardiac enlargement on chest radiograph in the absence of signs of volume overload or congestive heart failure (CHF) should prompt consideration of pericardial effusion. An echocardiogram will provide the definitive answer. Pericardiocentesis may be needed to exclude infection.
Post–Myocardial Infarction Pericarditis
Dressler reported a syndrome of fever, pleuritis, leukocytosis, pericardial friction rub, and chest radiograph evidence of new pericardial or pleural effusions in post-MI patients.8 Frequent relapses and a high incidence of friction rubs led Dressler to describe this syndrome as a delayed complication of MI in contrast to the well-known syndrome of early post-MI pericarditis. The cause of late post-MI pericarditis (Dressler’s syndrome) may be immunologic, but the syndrome may also occur with pulmonary embolus and after pericardiotomy. Anticoagulants should be discontinued to reduce the risk of hemorrhage. Delayed post-MI pericarditis is treated with NSAIDs such as ibuprofen 600 mg four times per day or indomethacin 25 mg three times per day.
Neoplastic Pericardial Disease
Malignant pericardial tumors typically manifest late, which complicates diagnosis and treatment. Malignant involvement of the pericardium is observed in 3.4% of general autopsies and 2 to 31% of cancer autopsies. The most common causes are lung cancer (30%), breast cancer (23%), lymphoma (17%) and leukemia (9%). Primary malignancies of the pericardium are rare.9
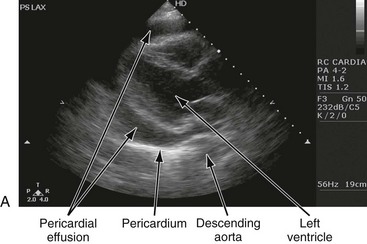
Pericardial Effusion
Cardiac Tamponade
Ten percent of all patients with cancer develop cardiac tamponade.10 Cardiac tamponade should be suspected in patients with penetrating chest wounds. It is also common in patients with uremic pericarditis.
Cardiac tamponade is a physiologic continuum reflecting the amount of fluid, the rate of accumulation, and the nature of the heart. The three stages necessary for tamponade to develop are fluid filling the recesses of the parietal pericardium, fluid accumulating faster than the rate of the parietal pericardium’s ability to stretch, and accumulation that exceeds the body’s ability to increase blood volume to support right ventricle filling pressure. The final result is increased pericardial pressure, which causes decreased ventricle compliance and decreased flow of blood into the heart. The reduction of blood inflow into the right ventricle results in decreased stroke volume that leads to decreased cardiac output.10 The most important factor in the development of tamponade is the rate of fluid accumulation.
Symptoms and Signs
Cardiac tamponade symptoms are often nonspecific, but particularly include chest pain, cough, or dyspnea, any of which may be progressive and severe. The classic triad of cardiac tamponade signs described by Beck is hypotension, distended neck veins, and muffled heart sounds.11 These signs may not be present if tamponade develops quickly.
Diagnostic Strategies
The chest radiograph shows cardiomegaly only if there is a large accumulation of fluid (250 mL). The ECG classically shows decreased voltage or electrical alternans (Fig. 82-3), but the latter is rare. Echocardiography confirms the diagnosis when an effusion and paradoxical systolic wall motion are seen. Thermodilution catheters can also be diagnostic, showing equalization of right and left ventricular pressures.
Figure 82-3 Electrocardiogram showing electrical alternans.
Management and Prognosis
Initial treatment includes volume augmentation to the right ventricle with intravenous fluids to increase the filling pressure to overcome the pericardial constriction. Pericardiocentesis or pericardial window is the treatment of choice. Enough fluid should be withdrawn to stabilize the patient. If tamponade recurs, pericardiocentesis may be repeated, or a drainage catheter may be left in the pericardial space. A pericardiectomy ultimately may be necessary. Cardiac tamponade has a high mortality that depends on the severity and nature of the underlying disease, the time course of onset, and the rapidity of diagnosis and intervention. Traumatic cardiac tamponade is discussed in Chapter 45.
Purulent Pericarditis
Tuberculous Pericarditis
Tuberculous pericarditis is estimated to occur in 1 or 2% of patients with pulmonary tuberculosis.12 In Africa, it is the most common cause of pericarditis. In countries in which tuberculosis is not a major health problem, tuberculous pericarditis is most common in patients who are socioeconomically deprived or immunodeficient. In many patients the chest radiograph shows an enlarged cardiac silhouette without a pulmonary infiltrate. Pericardial fluid aspirates reveal acid-fast bacilli by smear or culture (which may require 4-6 weeks to become positive) in approximately 50% of cases. Diagnostic workup should include assessment for human immunodeficiency virus (HIV). Patients with tuberculous pericarditis should be hospitalized and observed for evidence of cardiac tamponade. Triple-drug therapy should be started in the hospital and continued for at least 9 months. Patients with chronic pericardial effusions may benefit from oral prednisone therapy. The mortality rate is approximately 15% in HIV-negative patients and 20 to 35% in HIV-positive patients.12
[/not-level-membership-for-emergency-medicine-category]
