[level-membership-for-gastroenterology-and-hepatology-category]
CHAPTER 52 Peptic Ulcer Disease
Decades of research focused on the role of acid secretion and the effects of stress, personality type, and genetics in the pathogenesis of ulcer disease. The discovery of histamine-2 (H2) receptors1 and development of H2-receptor antagonist drugs, and the subsequent development of proton pump inhibitor drugs led to major changes in the management of peptic ulcer disease. The discovery of H. pylori and its treatment led to dramatic changes in the prevalence and recurrence of peptic ulcer disease, transforming peptic ulcer from a chronic recurrent disease to a curable one.2 H. pylori infection remains an important cause of peptic ulceration in the developing world. In the developed world, the use of NSAIDs has emerged as a leading cause of peptic ulcer disease, especially in the aging population in whom these drugs are often prescribed. Through all of these developments, the role of acid and pepsin in the genesis and perpetuation of mucosal injury remains a unifying aspect of the pathogenesis of peptic ulcer disease.
EPIDEMIOLOGY
The epidemiology of peptic ulcer disease has undergone a remarkable change in the past century. The incidence of duodenal ulcer and gastric ulcer has declined in parallel with the decline in H. pylori prevalence, likely as a result of improved sanitary conditions and a safer food and water supply. The risk of developing peptic ulcer disease and the risk of dying from peptic ulcer disease increased in successive cohorts born between 1840 and 1890 and then declined thereafter.3 A peak in the incidence of gastric ulcer in the first half of the 19th century and a subsequent peak in the incidence of duodenal ulcer in the second half of the 19th century remain unexplained, although a number of theories have been proposed, among them the widespread adoption of smoking after the commercial manufacture of cigarettes in a setting of widespread H. pylori infection.
POPULATION-BASED STUDIES
In northern Sweden, a random sample of 1001 people underwent upper GI endoscopy after filling out symptom questionnaires.4 The prevalence of peptic ulcer disease in this sample was 4.1%, with 20 gastric ulcers and 21 duodenal ulcers. In a prospective Danish study of 2416 subjects, the 11-year cumulative incidence of peptic ulcer was 2.9%: 1.6% for duodenal ulcer, 1.3% for gastric ulcer, and 0.04% for combined ulcers.5 In countries with a high prevalence of H. pylori infection, the ratio between duodenal and gastric ulcers may be quite different. In a case-control study from Shanghai, China, in which the prevalence of H. pylori infection was 76%, recurrent or new peptic ulcers occurred in 3.6% of the population over 2 years and 85% of the ulcers were duodenal.6
TIME TRENDS
There has been a significant decline in mortality from peptic ulcer disease over time in most age groups.7,8 A notable exception is older adults, in whom peptic ulcer bleeding remains a life-threatening condition. A study based on the U.S. National Discharge survey reported that from 1992 to 1999, the annual rate of hospitalization for peptic ulcer disease declined 20%, from 205/100,000 population to 165/100,000 population. Mortality also declined 22%, from 7.7/100,000 to 6/100,000, respectively. Sales of acid-inhibitory drugs correlated with a decrease in peptic ulcer disease hospitalizations and mortality,9 although this correlation does not prove causality. The observed decline in ulcer-related mortality was probably related to improvements in the general health of the population and the availability of effective treatment for peptic ulcer disease.
In an analysis of the Canadian Institute of Health Information database, the prevalence of acute nonvariceal upper GI bleeding (largely ulcer-related bleeding) declined by 31%, from 77/100,000 population to 53/100,000 population over the 10-year period from 1993 to 2003.10 The need for surgical intervention also declined but the mortality rate remained unchanged. In the United Kingdom, overall hospital admission rates for peptic ulcer disease have also declined, as has mortality, but admission rates for peptic ulcer hemorrhage and perforation have increased.11 Peptic ulcer bleeding is most often seen in older adults, with 68% of patients presenting older than age 60, and 27% older than age 80.12
RISK FACTORS
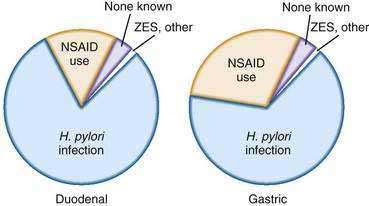
The principal risk factors of peptic ulcer disease are H. pylori infection and NSAID use (Fig. 52-1). However, some patients with peptic ulcer disease have neither of these risk factors.
HELICOBACTER PYLORI INFECTION (see Chapter 50)
H. pylori is a gram-negative bacillus that is uniquely adapted to life in the stomach. It is a major cause of peptic ulcer disease and accounts for a large proportion of peptic ulcers in countries where H. pylori infection is highly prevalent such as in Asia. In the United States and in western Europe, the original estimates that H. pylori infection was the cause of 90% or more of duodenal ulcers, and 60% or more of all gastric ulcers have been lowered by the declining prevalence of H. pylori infection.13 It is estimated that close to 70% of duodenal ulcers are related to H. pylori in Western populations. For example, in an analysis of patients who participated in H. pylori eradication trials in the United States, the proportion of patients with peptic ulcer disease and H. pylori infection was 73%.14 In Rochester, New York, the prevalence of H. pylori infection in patients with duodenal ulcer disease was 61%.15
ASPIRIN AND NONSTEROIDAL ANTI-INFLAMMATORY DRUGS (see also Chapter 51)
Aspirin is increasingly used in the prevention of cardiovascular disease.16,17 Aspirin and clopidogrel are frequently used in combination in patients who have had ischemic cardiac events or in patients who have recently had a stent placed in the coronary arteries.18 NSAIDs are used by approximately 11% of the U.S. population on a regular basis.19 This frequency is likely to increase as the population ages. Regular use of NSAIDs increases the odds of gastrointestinal bleeding five- to six-fold compared with persons not taking NSAIDs.20 Serious ulcer-related complications occur in 1% to 4% of NSAID users and NSAID-related complications are thought to account for 100,000 admissions every year.21 NSAID users who also take aspirin are at an especially high risk for complications. In a population-based study from Denmark, the odds ratio for GI bleeding if a person was taking low-dose aspirin was 2.6, and this risk increased to 5.6 in patients who were also taking an NSAID.22 In Spain, the death rate attributed to NSAID-aspirin use was 15.3/100,000. Up to one third of all NSAID-aspirin–related deaths in that study were attributable to low-dose aspirin use.23
H. pylori and NSAIDs may have a synergistic role in causing peptic ulcer disease. In a meta-analysis, the odds ratios for the development of a peptic ulcer in patients with H. pylori infection or NSAID use were 4.05 and 2.99, respectively, but the odds ratio increased significantly to 15.4 if both factors were present.24 The risk factors for peptic ulcer disease among patients taking NSAIDS and their risk ratios are listed in the next chapter (see Table 53-1).
OTHER ULCEROGENIC DRUGS
Deep ulcers and perforations of the stomach and duodenum have been described in cocaine and methamphetamine users, presumably due to mucosal ischemia.25 Bisphosphonates have also been associated with gastroduodenal ulceration,26 although esophageal injury with bisphosphonates in clinically more of a concern (see Chapter 45). There is little if any risk for peptic ulcer disease in patients taking glucocorticoids.27 In combination with NSAIDS, however, glucocorticoids increase the risk of peptic ulcer disease above the risk with NSAIDs alone.28
OTHER RISK FACTORS
Smoking has been implicated in the pathogenesis of peptic ulcer disease for decades, but its importance as a risk factor has declined after the discovery of H. pylori. A population-based study evaluated the risk factors for peptic ulcer disease in 2416 Danish adults who were interviewed between 1982 and 1994.29 As expected, H. pylori seropositivity was a significant risk factor for ulcer disease; smoking increased the risk of peptic ulcer only in H. pylori–infected subjects. A large body of literature suggests that smoking may predispose to peptic ulcer disease, but H. pylori infection remains a confounder that was not addressed in earlier studies. It is noteworthy that cigarette smoking does not increase the risk of recurrent ulceration once H. pylori has been eradicated, suggesting that smoking may only play a role in infected subjects.30
The role of alcohol remains uncertain. Alcoholic beverages stimulate gastric acid production. Moreover, direct application of high concentrations of alcohol to the gastric mucosa causes demonstrable mucosal injury. In the Danish study alluded to previously,29 intake of spirits increased the risk of peptic ulcer disease in H. pylori–infected patients.
With regard to diet, ulcer prevalence rates differ considerably in the north of India, where the principal cereal in the diet is wheat, and in the south, where rice is the predominant cereal. However, many other potential confounders were not accounted for in these populations.31 An association between the ingestion of spicy foods and peptic ulcer disease is weak, at best.32
Emotional stress was proposed as a major cause of ulcer disease or as a precipitant of ulcer complications,33 and much was written about its relationship to peptic ulcer disease prior to the description of H. pylori. Personality types and psychological profiles have been proposed to be linked to ulcer, but much of this literature is confounded by the lack of information on H. pylori infection. Well-documented descriptions of an increase in ulcer disease after natural calamities such as earthquakes suggest that emotional stress among those not physically injured may play a role in triggering overt manifestations of peptic ulcer disease, especially in individuals who may be otherwise predisposed to ulcer (e.g., patients infected with H. pylori).33,34
PATHOGENESIS
GASTRIC ACID SECRETION
Gastric acid and pepsin have long been considered the principal inciting agents in the pathogenesis of peptic ulcer disease.35 The control of gastric acid secretion in health and disease has been reviewed in detail36 and is discussed in Chapter 49.
HELICOBACTER PYLORI INFECTION (see also Chapters 50 and 51)
Most patients who have a chronic infection with H. pylori have a pan-gastritis in which the body and the antrum are equally involved. Gastritis results in inhibition of acid secretion, and various mechanisms have been proposed.37,38 These include direct inhibition of the parietal cell by lipopolysaccharides or toxins secreted by H. pylori or an indirect effect through stimulation of cytokines caused by the inflammation.1 Due to the decreased output of acid, these individuals with pan-gastritis do not usually develop ulcer disease related to H. pylori infection.39 The reduced acid production in H. pylori–infected individuals has been suggested as a factor that may protect against the development of reflux disease and its complications such as Barrett’s esophagus (see Chapters 43 and 44).
Another pattern of H. pylori infection affects approximately 10% to 20% of patients with chronic infection and consists of an antrum-predominant gastritis (see Chapters 50 and 51). In these individuals, through a series of steps not well understood but possibly involving reduced somatostatin concentrations in the antrum, basal and meal-stimulated acid secretion often increases. The increased acid output from the stomach results in increased acid delivery to the duodenum that can result in gastric metaplasia in the duodenal bulb. Some investigators believe that the metaplastic epithelium then becomes infected with H. pylori from the stomach, resulting in focal duodenitis, sometimes followed by ulcer formation. In contrast with patients with duodenal ulcer disease who often have increased acid secretion, patients with gastric ulcer typically have normal or decreased acid production, suggesting that the mechanism for the development of ulceration is a failure in the gastric mucosal protective mechanisms, described in more detail below. Acid antisecretory medication heals ulcers in patients with gastric ulcer and hypochlorhydria because acid changes the balance in favor of mucosal defense factors that restore mucosal integrity (see Chapter 53).
GASTRIC AND DUODENAL MUCOSAL DEFENSE MECHANISMS
As described in Chapter 49 the gastric mucosa has multiple defense mechanisms to protect it from digestion by acid and pepsin.40 These include the gastric surface epithelium,41–43 the mucus/phospholipid and bicarbonate barrier (mucus layer),44,45 epithelial cell renewal and regeneration,46,47 mucosal blood flow and the alkaline tide, and prostaglandin production.48–51 Prostaglandins also stimulate mucus, bicarbonate, and phospholipid production. Many prostaglandins also increase mucosal blood flow and stimulate epithelial regenerative processes. Inhibition of these protective effects by NSAIDs increases the likelihood of injury to the epithelium and decreases its ability to respond and regenerate.
Cyclooxygenase-1 (COX-1) and COX-2 are the enzymes responsible for the synthesis of prostaglandins. COX-1 is expressed in the stomach and is responsible for the maintenance of the integrity of gastric epithelium and the mucous barrier. COX-2 is not expressed in the healthy stomach but is rapidly expressed in response to the cytokines generated by inflammatory processes. Conventional NSAIDs such as ibuprofen inhibit the COX-1 and the COX-2 enzymes. It is believed that COX-1 inhibition reduces prostaglandin synthesis, which leads to a reduction in mucosal blood flow, hypoxia, and a reduction in mucosal defense. Neutrophil-endothelial interactions then occur as a result of the vascular disturbances and neutrophil activation. In experimental studies, COX-1 inhibition alone is not sufficient to cause ulceration.52 The inhibition of COX-1 up-regulates the expression of COX-2 that suppresses the neutrophil endothelial interaction that is stimulated by COX-1 inhibition. Inhibition of COX-1 and COX-2 enzymes is therefore important in the generation of gastric injury.
Some studies have suggested that a prostaglandin-independent mechanism may also contribute to damage by NSAIDs. One such mechanism is increased leukotriene production that occurs because arachidonic acid metabolism shifts to the alternative 5-lipooxygenase pathway when the COX-1 pathway is inhibited.40
The duodenum, like the stomach, secretes bicarbonate that neutralizes acid arriving into the duodenum.53 Decreased duodenal bicarbonate secretion has been reported in patients with duodenal ulcer.54 A protective role has also been postulated for pancreatic juice but the results have been conflicting.
HELICOBACTER PYLORI-NEGATIVE, NSAID-NEGATIVE ULCERS
As the prevalence of H. pylori infection declines in the United States, a growing proportion of patients with peptic ulcers who have no evidence of H. pylori infection and have no history of aspirin or NSAID use have emerged. Eradication of H. pylori generally cures peptic ulcer disease and prevents recurrence, but a small number of patients do not heal their ulcers or suffer an ulcer recurrence despite successful eradication of H. pylori. The exact prevalence of H. pylori–negative, NSAID-negative (idiopathic) peptic ulcers is unknown. The pathogenesis of H. pylori–negative, NSAID-negative (idiopathic) ulcers remains uncertain. One theory that has been proposed is that some idiopathic ulcers may occur from H. pylori colonization of the duodenum.55 These patients may test negative with conventional endoscopic tests for H. pylori because these tests focus on detecting H. pylori in gastric biopsies.
OTHER CAUSES OF ULCER DISEASE IN THE STOMACH AND DUODENUM
GASTRINOMA WITH OR WITHOUT MULTIPLE ENDOCRINE NEOPLASIA SYNDROME, TYPE I (see Chapter 32)
Gastrin-secreting tumors are an important cause of acid hypersecretion due to the sustained drive for acid secretion. This disorder may result in multiple ulcerations in the stomach or duodenum that are refractory to conventional treatment and are often associated with a chronic diarrhea. One fourth of all patients with Zollinger-Ellison syndrome have an autosomal dominant disorder characterized by pancreatic endocrine tumors, hyperparathyroidism, and pituitary adenomas.56
SYSTEMIC MASTOCYTOSIS (see Chapter 32)
This is an uncommon condition in which multiple ulcers may occur in the stomach or duodenum with infiltration of the mucosa with mast cells that can be recognized histologically.57 Secretion of histamine by the mast cells is thought to result in the excessive stimulation of acid production through the histamine receptor.
MISCELLANEOUS DISORDERS
Ulcerations in the upper GI tract may be a manifestation of Crohn’s disease, which can cause ulcerations anywhere in the gastrointestinal tract (see Chapter 111). Associations between peptic ulcers and α1-antitrypsin deficiency, chronic lung disease, and chronic renal failure have been described. Many of these studies predate the routine evaluation for H. pylori in patients with peptic ulcer disease, and some of the observed associations may be questioned. For example, a recent study of α1-antitrypsin–deficient patients with duodenal ulcer found that all the patients were infected with H. pylori.58 Another study reported a higher than expected rate of ulcer recurrence in patients with renal failure following successful eradication of H. pylori.59
CLINICAL FEATURES
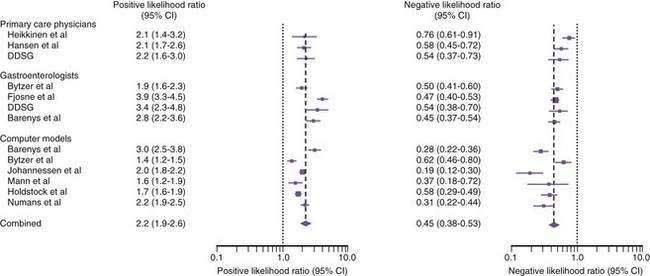
A systematic review evaluated, for the patient presenting with upper abdominal symptoms, the utility of the clinical examination and various computer models in predicting if peptic ulcer disease is present.60 Some studies evaluated the accuracy of gastroenterologists, some evaluated primary care physicians, and one evaluated both.61–66 The studies included 4684 patients of whom 802 (17%) had peptic ulcer disease (Fig. 52-2). The positive likelihood ratio of accurately diagnosing peptic ulcer by clinical features was 2.9 for gastroenterologists, which was somewhat better than the performance of primary care physicians (likelihood ratio, 2.2). The negative likelihood ratio for accurately excluding peptic ulcer by clinical features was similar in gastroenterologists and primary care physicians (0.62). Computer models for the prediction of peptic ulcer disease were also not highly accurate, with a positive likelihood ratio of 1.9 and a negative likelihood ratio of 0.34.
DIAGNOSIS
ENDOSCOPY
Endoscopy is the current reference standard for diagnosis of peptic ulcer disease. Its main limitation is its high cost in some countries such as the United States. The decision to perform endoscopy in a patient suspected of having peptic ulcer disease is based on a number of factors. Patients presenting with complications of peptic ulcer disease such as bleeding need endoscopic evaluation to allow an accurate diagnosis and for the administration of endoscopic therapy (see Chapters 19 and 53). The presence of “alarm” features such as weight loss or recurrent vomiting may prompt concern for malignancy (Table 52-1).
Table 52-1 Alarm Features in Patients with Suspected Peptic Ulcer Disease*
* These features should prompt an upper endoscopy and often other testing to establish a definitive diagnosis.
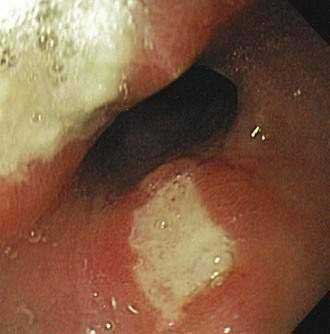
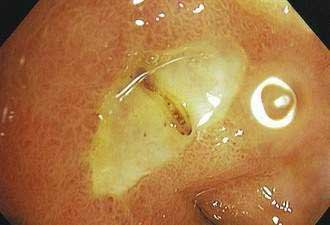
Although endoscopy is regarded as the standard for a diagnosis of peptic ulcer disease (Figs. 52-3 and 52-4), small ulcers may be missed at endoscopy.67 The proportion of ulcers that may be missed at endoscopy is uncertain because comparative studies of contrast radiography and endoscopy were performed many years ago and technologic innovations in the quality and resolution of endoscopes have improved the yield of endoscopy. Biopsies should be taken from the edges of a gastric ulcer because of the risk of malignancy. In patients with gastric ulcers it has been customary to repeat the endoscopy after approximately eight weeks of treatment to ensure that the ulcer has healed (see Chapter 53). A small number of chronic, nonhealing gastric ulcers can be identified as being malignant at the repeat endoscopy even though the initial biopsies had revealed no evidence of a malignancy. Although conflicting reports on the utility and cost-effectiveness of performing a repeat endoscopy after approximately eight weeks in patients with benign-appearing gastric ulcers have appeared in the literature, recent studies support the use of a repeat endoscopy in this setting.68,69 Up to 4% of apparently benign gastric ulcers at initial endoscopy can be found to be malignant at a subsequent examination, and cancers so detected are typically at an early stage with the possibility of a curative resection.
CONTRAST RADIOGRAPHY
Contrast radiography of the upper GI tract, also referred to as the barium meal or an upper GI series, can often demonstrate a peptic ulcer (Fig. 52-5). In historical studies, contrast radiography has performed as well as endoscopy in the diagnosis of ulcer disease when the contrast examinations are performed and interpreted by experts.67 Much has changed since then. Contrast radiography of the upper GI tract is now frequently performed by technicians, and the availability of personnel trained in the interpretation of barium studies has decreased. A further disadvantage to contrast radiography is radiation exposure, which can be substantial. Endoscopy offers the obvious advantage of allowing mucosal biopsy, which can be used to diagnose H. pylori infection and to rule out malignancy in chronic gastric ulcers. Contrast radiography therefore has limited utility in modern practice.
DIAGNOSTIC AND MANAGEMENT STRATEGIES FOR PATIENTS WITH SUSPECTED PEPTIC ULCER DISEASE
Upper abdominal symptoms that raise the question of peptic ulcer disease are common in clinical practice and account for 2% to 5% of visits in family practice.70 These symptoms, consisting of pain or discomfort in the upper abdomen are frequently referred to as dyspepsia. Due to the high cost of subjecting all dyspeptic individuals to endoscopy, noninvasive alternative strategies have been proposed as an initial step in the management of suspected peptic ulcer disease (Fig. 52-6).71 Economic models suggested that these noninvasive strategies could reduce the costs of managing peptic ulcer disease substantially.72 Subsequent clinical trials demonstrated the clinical efficacy of the approach and the initial strategy proposed was to perform noninvasive testing for infection, followed by antimicrobial treatment directed against H. pylori if the patient tested positive.73,74 This “test and treat” strategy offers a potentially curative treatment for patients who have H. pylori related ulcer disease and may cure a small proportion of patients with nonulcer dyspepsia. As the prevalence of H. pylori infection declines in Western populations, other empirical strategies can be considered. Because acid inhibition plays a significant role in the management of patients with upper GI symptoms, empirical treatment with a proton pump inhibitor has been proposed as an alternative to routine endoscopy. Cost-models have suggested that when the prevalence of H. pylori infection in the population falls to the 10% to 15% range, an empirical course of therapy with a proton pump inhibitor may be a reasonable initial strategy.75 A cost simulation model in the United States76 supported the empirical management of patients, as recommended in the American Gastroenterological Association dyspepsia guidelines (see Fig. 52-6).71 A randomized controlled clinical trial compared empirical testing and treatment for H. pylori with empirical proton pump inhibitor therapy and concluded that both were equally cost-effective in the initial management of dyspeptic patients, with the choice of which strategy to be taken left to a discussion between patient and physician.77
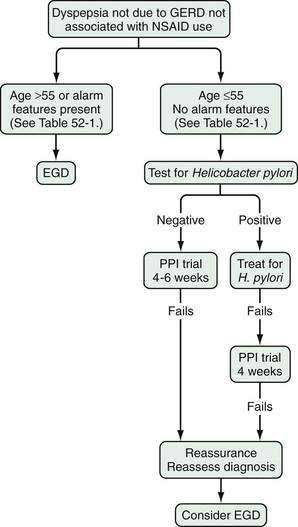
The incidence of upper GI malignancies rises with age, and thus current empirical management strategies are generally reserved for younger patients with upper abdominal symptoms. The age beyond which endoscopy should become routine is controversial and to a substantial degree depends on the epidemiology of gastric cancer in the population under consideration. In Western populations, upper GI cancer is rare in young individuals and therefore an age cutoff of 50 or 55 years is used.78 Older patients presenting with new-onset upper abdominal symptoms suggestive of peptic ulcer disease should be referred for endoscopy. Guidelines in the United Kingdom have not set an age cutoff for early endoscopy, and empirical therapy is preferred at all ages. In Asia and eastern Europe, an earlier age cutoff may be reasonable because the risk of gastric cancer is substantially higher than in Western nations and setting the age threshold too high may lead to a delayed diagnosis of a treatable cancer.79
The principal concern of any empirical treatment strategy for treating dyspepsia is that an underlying malignancy will be missed. To increase the odds of detecting an underlying malignancy, an age-based strategy has been proposed in which younger dyspeptic patients are treated empirically and older dyspeptic patients receive early endoscopy. Although there is some controversy about the age at which routine endoscopy for dyspepsia should begin, an age cutoff of 50 to 55 years is proposed in recent guidelines.80
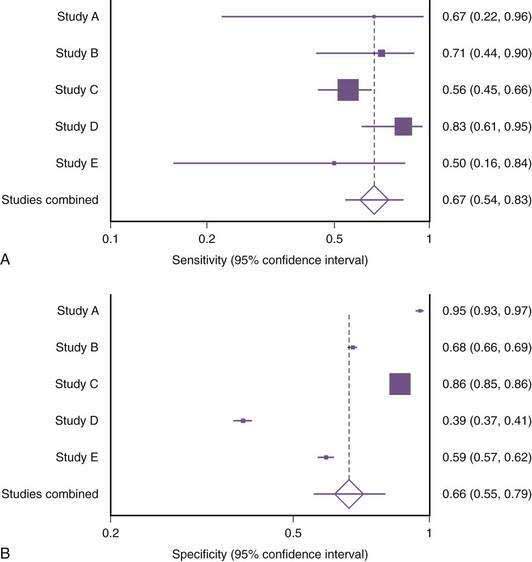
A systematic review and meta-analysis of alarm symptoms in predicting malignancy (as opposed to peptic ulcer and nonulcer dyspepsia) has raised questions about their accuracy.81 The sensitivity of alarm symptoms for serious underlying pathology in 15 studies including 57,363 patients ranged from 0% to 83%, with considerable heterogeneity between studies (Fig. 52-7). Specificity of alarm symptoms for malignancy varied from 40% to 83%. Although current management schemes still list alarm features as an indication for early endoscopy in patients with undiagnosed upper abdominal pain (see Fig. 52-4), further research is necessary in this area. The issue of evaluation of undiagnosed dyspepsia is also discussed in Chapter 13.
COMPLICATIONS
HEMORRHAGE
Hemorrhage from a peptic ulcer occurs when the ulcer crater erodes a blood vessel. It is a major cause of morbidity in the United States with an annual cost that exceeds 2 billion dollars.82,83 GI hemorrhage is an important cause of morbidity and mortality in older adutls.84 The typical presentation is with melena or hematemesis although a small proportion of patients with bleeding may present with hematochezia. Bleeding peptic ulcers are discussed in more detail in Chapters 19 and 53. Strategies to prevent NSAID-related bleeding in patients at high risk have been developed85 and are also discussed in more detail in Chapter 53, as is the treatment of acute ulcers occurring in critically ill patients in intensive care units (stress ulcers).
PENETRATION AND PERFORATION86–93
As an ulcer deepens it can burrow into adjoining structures (penetration) or rupture into the peritoneal cavity (perforation). Ulcer-related penetration and perforation are less common than upper GI bleeding as a complication of peptic ulcer disease and are more common in older adults. Ulcers located posteriorally in the duodenum may penetrate the pancreas, whereas ulcers located more anteriorally located ulcers may penetrate the liver or biliary tract.88–90 There are no typical presenting signs that identify a penetrating ulcer, but pain radiating to the back is often cited as a symptom of a posterior penetrating ulcer. Computed tomography (CT) may be helpful in establishing a diagnosis.91
Free ulcer perforation with peritonitis is a medical emergency. The patient typically presents with severe generalized abdominal pain and signs of peritonitis (see Chapters 10 and 37). CT usually establishes the diagnosis.92 Perforation has a high mortality, particularly in older adults. A population-based study from three Danish counties reported that among 2061 patients hospitalized with peptic ulcer perforation, 38% were current NSAID users.93 The 30-day mortality associated with ulcer perforation in this population was 25% overall, and 35% among current NSAID users.
OBSTRUCTION
Chronic scarring, usually in the antrum of the stomach, can lead to a disorder characterized by recurrent vomiting and a narrowing of the H. pylori channel, which is usually visualized endoscopically or on contrast radiography. The symptoms may be insidious, manifesting as reflux disease that is difficult to control or dyspepsia until the narrowing becomes pronounced when vomiting and early satiety become prominent features. Benign pyloric stenosis due to recurrent peptic ulcer disease was a common condition in the past but is rather rare now. It is therefore important to rule out a neoplastic process, which is a more common cause of gastric outlet obstruction than benign pyloric stenosis due to ulcer disease.94
Aro P, Storskrubb T, Ronkainen J, et al. Peptic ulcer disease in a general adult population: The Kalixanda study: A random population-based study. Am J Epidemiol. 2006;163:1025-34. (Ref 4.)
Black J, Duncan W, Durant D. Definition and antagonism of histamine H2 receptors. Nature. 1972;236:365-90. (Ref 1.)
El-Omar EM. Mechanisms of increased acid secretion after eradication of Helicobacter pylori infection. Gut. 2006;55:144-6. (Ref 37.)
Laine L, Takeuchi K, Tarnawski A. Gastric mucosal defence and cytoprotection: Bench to bedside. Gastroenterology. 2008;135:41-60. (Ref 40.)
Lewis JD, Bilker WB, Brensinger C, et al. Hospitalization and mortality rates from peptic ulcer disease and GI bleeding in the 1990s: Relationship to sales of nonsteroidal anti-inflammatory drugs and acid suppression medications. Am J Gastroenterol. 2002;97:2540-9. (Ref 9.)
Marshall B, Warren J. Unidentified curved bacilli in the stomachs of patients with gastritis and peptic ulcer. Lancet. 1984;1:1311-15. (Ref 2.)
Moayyedi P, Talley N, Fennerty B, Vakil N. Can the clinical history distinguish between organic and functional dyspepsia? JAMA. 2006;295:1566-76. (Ref 60.)
Ofman JJ, Etchason J, Fullerton S, et al. Management strategies for Helicobacter pylori–seropositive patients with dyspepsia (clinical and economic consequences). Ann Intern Med. 1997;126:280-91. (Ref 72.)
Rosenstock S, Jorgensen T, Bonnevie O, Andersen L. Risk factors for peptic ulcer disease: A population based prospective cohort study comprising 2416 Danish adults. Gut. 2003;52:186-93. (Ref 29.)
Schubert M, Peura D. Control of gastric acid secretion in health and disease. Gastroenterology. 2008;134:1842-60. (Ref 36.)
Sonnenberg A. Time trends of ulcer mortality in non-European countries. Am J Gastroenterol. 2007;102:1101-7. (Ref 7.)
Spiegel BM, Vakil NB, Ofman JJ. Dyspepsia management in primary care (a decision analysis of competing strategies). Gastroenterology. 2002;122:1270-85. (Ref 75.)
Talley NJ, Vakil N. Practice Parameters Committee of the American College of Gastroenterology. Guidelines for the management of dyspepsia. Am J Gastroenterol. 2005;100:2324-37. (Ref 80.)
Targownik L, Nabalamba A. Trends in management and outcomes of acute nonvariceal upper gastrointestinal bleeding: 1993-2003. Clin Gastroenterol Hepatol. 2006;4:1459-66. (Ref 10.)
Vakil N, Moayyedi P, Fennerty BM, Talley N. Limited value of alarm features in the diagnosis of upper gastrointestinal malignancy: Systematic review and meta-analysis. Gastroenterology. 2006;131:390-401. (Ref 81.)
Wilcox C, Allison J, Benzuly K, et al. Consensus Development Conference on the Use of Nonsteroidal Anti-Inflammatory Agents, Including Cyclooxygenase-2 Enzyme Inhibitors and Aspirin. Clin Gastroenterol Hepatol. 2006;4:1082-9. (Ref 21.)
1. Black J, Duncan W, Durant D. Definition and antagonism of histamine H2 receptors. Nature. 1972;236:365-90.
2. Marshall B, Warren J. Unidentified curved bacilli in the stomachs of patients with gastritis and peptic ulcer. Lancet. 1984;1:1311-15.
3. Sonnenberg A. Causes underlying the birth-cohort phenomenon of peptic ulcer: Analysis of mortality data 1911-2000, England and Wales. Int J Epidemiol. 2006;35:1090-7.
4. Aro P, Storskrubb T, Ronkainen J, et al. Peptic ulcer disease in a general adult population: The Kalixanda study: A random population-based study. Am J Epidemiol. 2006;163:1025-34.
5. Rosenstock S, Jørgensen T, Bonnevie O, Andersen L. Risk factors for peptic ulcer disease: A population based prospective cohort study comprising 2416 Danish adults. Gut. 2003;52:186-93.
6. Wang JY, Liu SB, Chen SY, Dobson A. Risk factors for peptic ulcer in Shanghai. Int J Epidemiol. 1996;25:638-43.
7. Sonnenberg A. Time trends of ulcer mortality in non-European countries. Am J Gastroenterol. 2007;102:1101-7.
8. Sonnenberg A. Time trends of ulcer mortality in Europe. Gastroenterology. 2007;132:2320-7.
9. Lewis JD, Bilker WB, Brensinger C, et al. Hospitalization and mortality rates from peptic ulcer disease and GI bleeding in the 1990s: Relationship to sales of nonsteroidal anti-inflammatory drugs and acid suppression medications. Am J Gastroenterol. 2002;97:2540-9.
10. Targownik L, Nabalamba A. Trends in management and outcomes of acute nonvariceal upper gastrointestinal bleeding: 1993-2003. Clin Gastroenterol Hepatol. 2006;4:1459-66.
11. Higham J, Kang JY, Majeed A. recent trends in admissions and mortality due to peptic ulcer in England: increasing frequency of hemorrhage among older subjects. Gut. 2002;50:460-4.
12. Ohmann C, Imhof M, Ruppert C, et al. Time trends in the epidemiology of peptic ulcer bleeding. Scand J Gastroenterol. 2005;40:914-20.
13. Laine L, Hopkins RJ, Girardi LS. Has the impact of Helicobacter pylori therapy on ulcer recurrence in the United States been overstated? A meta-analysis of rigorously designed trials. Am J Gastroenterol. 1998;93:1409-15.
14. Ciociola AA, McSorley DJ, Turner K, et al. Helicobacter pylori infection rates in duodenal ulcer patients in the United States may be lower than previously estimated. Am J Gastroenterol. 1999;94:1834-40.
15. Jyotheeswaran S, Shah AN, Jin HO, et al. Prevalence of Helicobacter pylori in peptic ulcer patients in greater Rochester, NY: Is empirical triple therapy justified? Am J Gastroenterol. 1998;93:574-8.
16. Eidelman RS, Hebert PR, Weisman SM, Hennekens CH. An update on aspirin in the primary prevention of cardiovascular disease. Arch Intern Med. 2003;163:2006-10.
17. Pearson TA, Blair SN, Daniels SR, et al. AHA guidelines for primary prevention of cardiovascular disease and stroke: 2002 Update: Consensus Panel Guide to Comprehensive Risk Reduction for Adult Patients Without Coronary or Other Atherosclerotic Vascular Diseases. American Heart Association Science Advisory and Coordinating Committee. Circulation. 2002;106:388-91.
18. Bhatt D, Fox K, Hacke W, et al. Clopidogrel and aspirin versus aspirin alone for the prevention of atherothrombotic events. N Engl J Med. 2006;354:1706-17.
19. Kaufman D, Kelly J, Rosenberg L, et al. Recent patterns of medication use in the ambulatory adult population of the United States: The Slone survey. JAMA. 2002;287:337-44.
20. Garcia Rodriguez LA, Hernandez-Diaz S. Relative risk of upper gastrointestinal complications among users of acetaminophen and nonsteroidal anti-inflammatory drugs. Epidemiology. 2001;12:570-6.
21. Wilcox C, Allison J, Benzuly K, et al. Consensus Development Conference on the Use of Nonsteroidal Anti-Inflammatory Agents, Including Cyclooxygenase-2 Enzyme Inhibitors and Aspirin. Clin Gastroenterol Hepatol. 2006;4:1082-9.
22. Sørensen HT, Mellemkjaer L, Blot WJ, et al. Risk of upper gastrointestinal bleeding associated with use of low-dose aspirin. Am J Gastroenterol. 2000;95:2218-24.
23. Lanas A, Perez-Aisa MA, Feu F, et al. A nationwide study of mortality associated with hospital admission due to severe gastrointestinal events and those associated with nonsteroidal antiinflammatory drug use. Am J Gastroenterol. 2005;100:1685-93.
24. Papatheodoridis G, Sougioultzis S, Archimandritis A. Effects of Helicobacter pylori and nonsteroidal anti-inflammatory drugs on peptic ulcer disease: A systematic review. Clin Gastroenterol Hepatol. 2006;4:130-42.
25. Pecha RE, Prindiville T, Pecha BS, et al. Association of cocaine and methamphetamine use with giant gastroduodenal ulcers. Am J Gastroenterol. 1996;91:2523-7.
26. Lanza FL, Hunt RH, Thomson AB, et al. Endoscopic comparison of esophageal and gastroduodenal effects of risedronate and alendronate in postmenopausal women. Gastroenterology. 2000;119:631-8.
27. Conn H, Blitzer B. Nonassociation of adrenocorticosteroid therapy and peptic ulcer. N Engl J Med. 1976;294:473-9.
28. Piper J, Ray W, Daugherty J, Griffin M. Corticosteroid use and peptic ulcer disease: Role of non-steroidal anti-inflammatory drugs. Ann Intern Med. 1991;114:735-40.
29. Rosenstock S, Jorgensen T, Bonnevie O, Andersen L. Risk factors for peptic ulcer disease: A population based prospective cohort study comprising 2416 Danish adults. Gut. 2003;52:186-93.
30. Chan F. Dose smoking predispose to ulcer disease after the eradication of H. pylori?. Am J Gastroenterol. 1997;3:442.
31. Malhotra SL. A comparison of unrefined wheat and rice diets in the management of duodenal ulcer. Postgrad Med J. 1978;54:6.
32. Satyanarayana MN. Capsaicin and gastric ulcers. Crit Rev Food Sci Nutr. 2006;46:275-328.
33. Walker P, Luther J, Samloff IM, Feldman M. Life events stress and psychological factors in men with peptic ulcer disease. Gastroenterology. 1988;94:323-30.
34. Aoyama N, Kinoshita Y, Fujimoto S, et al. Peptic ulcers after the Hanshin-Awaji earthquake: Increased incidence of bleeding gastric ulcers. Am J Gastroenterol. 1998;93:311-16.
35. Schwartz K. Uber penetrrierende magen und jejunalgeschwure. Beitr Klin Chir. 1910;67:96-128.
36. Schubert M, Peura D. Control of gastric acid secretion in health and disease. Gastroenterology. 2008;134:1842-60.
37. El-Omar EM. Mechanisms of increased acid secretion after eradication of Helicobacter pylori infection. Gut. 2006;55:144-6.
38. Vakil N, Talley NJ, Stolte M, et al. Patterns of gastritis and the effect of eradicating Helicobacter pylori on gastro-oesophageal reflux disease in Western patients with non-ulcer dyspepsia. Aliment Pharmacol Ther. 2006;24:55-63.
39. El-Omar EM, Oien K, El-Nujumi A, et al. Helicobacter pylori infection and chronic gastric acid hyposecretion. Gastroenterology. 1997;113:15-24.
40. Laine L, Takeuchi K, Tarnawski A. Gastric mucosal defence and cytoprotection: Bench to bedside. Gastroenterology. 2008;135:41-60.
41. Oyaka J, Otaka M, Matsuhashi T, et al. Over-expression of 70-kDa heat shock protein confers protection against monochloramine-induced gastric mucosal cell injury. Life Sci. 2006;79:300-5.
42. Taupin D, Podolsky DK. Trefoil factors initiators of mucosal healing. Nat Rev Mol Cell Biol. 2003;4:721-32.
43. Bajaj-Elliott M, Fedeli P, Smith GV, et al. Modulation of host antimicrobial peptide (beta-defensins 1 and 2) expression during gastritis. Gut. 2002;51:356-61.
44. Flemström G. Active alkalinization by amphibian gastric fundic mucosa. Am J Physiol Endocrinol Metab Gastrointest Physiol. 1977;233:E1-12.
45. Atuma C, Strugala V, Allen A, et al. The adherent gastric mucus gel layer: Thickness and physical state in vivo. Am J Physiol Gastrointest Liver Physiol. 2001;280:G922-9.
46. Lacy ER, Ito S. Rapid epithelial restitution of the rat gastric mucosa after ethanol injury. Lab Invest. 1984;51:573-83.
47. Tarnawski A, Stachura J, Durbin T, et al. Increased expression of epidermal growth factor receptor during gastric ulcer healing in rats. Gastroenterology. 1992;102:695-8.
48. Yasue N, Guth PH. Role of exogenous acid and retransfusion in hemorrhagic shock-induced gastric lesions in the rat. Gastroenterology. 1988;94:1135-43.
49. Brzozowski T, Konturek PC, Konturek SJ, et al. Role of prostaglandins in gastroprotection and gastric adaptation. J Physiol Pharmacol. 2005;56:33-55.
50. Szabo S. Mechanisms of gastric mucosal injury and protection. J Clin Gastroenterol. 1991;13:S21-34.
51. Redfern JS, Feldman M. Role of endogenous prostaglandins in preventing gastrointestinal ulceration: induction of ulcers by antibodies to prostaglandins. Gastroenterology. 1989;96:596-605.
52. Odes H, Hogan D, Ballesteros M, et al. Human duodenal mucosal bicarbonate secretion. Evidence suggesting active transport under basal and stimulated conditions. Gastroenterology. 1990;98:867-72.
53. Isenberg JL, Selling JA, Hogan DL, et al. Impaired proximal duodenal mucosal bicarbonate secretion. New Engl J Med. 1987;316:374-8.
54. Tanaka A, Araki H, Hase S, et al. Inhibition of both COX-1 and COX-2 is required for development of gastric damage in response to nonsteroidal antiinflammatory drugs. J Physiol (Paris). 2001;95:21-7.
55. Pietroiusti A, Forlini A, Magrini A, et al. Isolated H. pylori duodenal colonization and idiopathic duodenal ulcers. Am J Gastroenterol. 2008;103:55-61.
56. Gibril F, Schumann M, Pace A, Jensen RT. Multiple endocrine neoplasia type 1 and Zollinger-Ellison syndrome: A prospective study of 107 cases and comparison with 1009 cases from the literature. Medicine (Baltimore). 2004;83:43-83.
57. Jensen RT. Gastrointestinal abnormalities and involvement in systemic mastocytosis. Hematol Oncol Clin North Am. 2000;14:579-623.
58. Elzouki AN, Tóth E, Florén CH, et al. Alpha1-antitrypsin deficiency may be a risk factor for duodenal ulcer in patients with Helicobacter pylori infection. Scand J Gastroenterol. 2000;35:32-5.
59. Tseng GY, Lin HJ, Fang CT, et al. Recurrence of peptic ulcer in uraemic and non-uraemic patients after Helicobacter pylori eradication: A 2-year study. Aliment Pharmacol Ther. 2007;26:925-33.
60. Moayyedi P, Talley N, Fennerty B, Vakil N. Can the clinical history distinguish between organic and functional dyspepsia? JAMA. 2006;295:1566-76.
61. Barenys M, Abad A, Pons JMV, et al. Scoring system has better discriminative value than Helicobacter pylori testing in patients with dyspepsia in a setting with a high prevalence of infection. Eur J Gastroenterol Hepatol. 2000;12:1275-82.
62. Ytxer P, Hansen J, Havelund T, et al. Predicting endoscopic diagnosis in the dyspeptic patient: The value of clinical judgement. Eur J Gastroenterol Hepatol. 1996;8:359-63.
63. Fjosne U, Kleveland P, Waldum H, et al. The clinical benefits of routine upper endoscopy. Scand J Gastroenterol. 1986;21:433-40.
64. Danish Dyspepsia Study Group. Value of the unaided clinical diagnosis in dyspeptic patients in primary care. Am J Gastroenterol. 2001;96:1417-21.
65. Hansen J, Bytzer P, Schaffalitzky de Muckadell O. Management of dyspeptic patients in primary care: Value of the unaided clinical diagnosis and of dyspepsia subgrouping. Scand J Gastroenterol. 1998;33:799-805.
66. Heikkinen M, Pikkarainen P, Takala J, et al. Etiology of dyspepsia: Four hundred unselected patients in general practice. Scand J Gastroenterol. 1995;30:519-23.
67. Rogers I, Sokhi G, Moule B, et al. Endoscopy and routine and double contrast barium meal in diagnosis of gastric and duodenal disorders. Lancet. 1976;1:901-2.
68. Hopper AN, Stephens MR, Lewis WG, et al. Relative value of repeat gastric ulcer surveillance gastroscopy in diagnosing gastric cancer. Gastric Cancer. 2006;9:217-22.
69. Bustamante M, Devesa F, Borghol A, et al. Accuracy of the initial endoscopic diagnosis in the discrimination of gastric ulcers: Is endoscopic follow-up study always needed? J Clin Gastroenterol. 2002;35:25-8.
70. Majumdar SR, Soumerai SB, Farraye FA, et al. Chronic acid-related disorders are common and underinvestigated. Am J Gastroenterol. 2003;98:2409-14.
71. Talley NJ. American Gastroenterological Association position statement: Evaluation of dyspepsia. Gastroenterology. 2005;129:1753-5.
72. Ofman JJ, Etchason J, Fullerton S, et al. Management strategies for Helicobacter pylori-seropositive patients with dyspepsia (clinical and economic consequences). Ann Intern Med. 1997;126:280-291.
73. Lassen AT, Pedersen FM, Bytzer P, et al. Helicobacter pylori test-and-eradicate versus prompt endoscopy for management of dyspeptic patients (a randomised trial). Lancet. 2000;356:455-60.
74. Lassen AT, Hallas J, Schaffalitzky de Muckadell OB. Helicobacter pylori test and eradicate versus prompt endoscopy for management of dyspeptic patients (6.7 year follow up of a randomised trial). Gut. 2004;53:1758-63.
75. Spiegel BM, Vakil NB, Ofman JJ. Dyspepsia management in primary care (a decision analysis of competing strategies). Gastroenterology. 2002;122:1270-85.
76. Barton PM, Moayyedi P, Talley NJ, et al. A second-order simulation model of the cost-effectiveness of managing dyspepsia in the United States. Med Decis Making. 2008;28:44-55.
77. Delaney BC, Qume M, Moayyedi P, et al. Helicobacter pylori test and treat versus proton pump inhibitor in initial management of dyspepsia in primary care: Multicentre randomised controlled trial (MRC-CUBE trial). BMJ. 2008;336:651-4.
78. Canga C3rd, Vakil N. Upper GI malignancy, uncomplicated dyspepsia, and the age threshold for early endoscopy. Am J Gastroenterol. 2002;97:600-3.
79. Sung JJ, Lao WC, Lai MS, et al. Incidence of gastroesophageal malignancy in patients with dyspepsia in Hong Kong: Implications for screening strategies. Gastrointest Endosc. 2001;54:454-8.
80. Talley NJ, Vakil N. Practice Parameters Committee of the American College of Gastroenterology. Guidelines for the management of dyspepsia. Am J Gastroenterol. 2005;100:2324-37.
81. Vakil N, Moayyedi P, Fennerty BM, Talley N. Limited value of alarm features in the diagnosis of upper gastrointestinal malignancy: Systematic review and meta-analysis. Gastroenterology. 2006;131:390-401.
82. Viviane A, Barkun A. Estimates of costs of hospital stay for variceal and nonvariceal upper gastrointestinal bleeding in the United States. Value Health. 2008;11:1-3.
83. Alkhatib A, Abubakr S, Elkhatib F. An estimate of hospitalization cost for elderly patients with acute gastrointestinal bleeding in the USA. Dig Dis Sci. 2009;54:695. Epub 2008 Dec 3
84. Gralnek IM, Barkun AN, Bardou M. Management of acute bleeding from a peptic ulcer. N Engl J Med. 2008;359:928-37.
85. Wilcox CM, Allison J, Benzuly K, et al. Consensus development conference on the use of nonsteroidal anti-inflammatory agents including cyclooxygenase-2 enzyme inhibitors and aspirin. Clin Gastroenterol Hepatol. 2006;4:1082-9.
86. Taha A, Angerson WJ, Prasad R, et al. The incidence and early mortality after peptic ulcer perforation, and the use of low-dose aspirin and non-steroidal anti-inflammatory drugs. Aliment Pharmacol Ther. 2008;28:878-85.
87. West AB, Nolan N, O’Brian DS. Benign peptic ulcers penetrating pericardium and heart: clinicopathological features and factors favoring survival. Gastroenterology. 1988;94:1478-87.
88. Padda SS, Morales TG, Earnest DL. Liver penetration by a duodenal ulcer. Am J Gastroenterol. 1997;92:352-4.
89. Venkatesh KR, Halpern A, Riley LB. Penetrating gastric ulcer presenting as a subcapsular liver abscess. Am Surg. 2007;73:82-4.
90. Fournier D, Dey C, Hessler C. Gas in the gallbladder due to duodeno-cholecystic fistula: A rare complication of a penetrating duodenal ulcer. Sonographic findings with CT correlation. J Clin Ultrasound. 1994;22:506-9.
91. Madrazo BL, Halpert RD, Sandler MA, Pearlberg JL. Computed tomographic findings in penetrating peptic ulcer. Radiology. 1984;153:751-4.
92. Chen CH, Huang HS, Yang CC, Yeh YH. The features of perforated peptic ulcers in conventional computed tomography. Hepatogastroenterology. 2001;48:1393-6.
93. Thomsen RW, Riis A, Munk EM, et al. 30-day mortality after peptic ulcer perforation among users of newer selective COX-2 inhibitors and traditional NSAIDs: A population-based study. Am J Gastroenterol. 2006;101:2704-10.
94. Khullar SK, DiSario JA. Gastric outlet obstruction. Gastrointest Endosc Clin North Am. 1996;6:585-603.
[/level-membership-for-gastroenterology-and-hepatology-category][not-level-membership-for-gastroenterology-and-hepatology-category]
CHAPTER 52 Peptic Ulcer Disease
Decades of research focused on the role of acid secretion and the effects of stress, personality type, and genetics in the pathogenesis of ulcer disease. The discovery of histamine-2 (H2) receptors1 and development of H2-receptor antagonist drugs, and the subsequent development of proton pump inhibitor drugs led to major changes in the management of peptic ulcer disease. The discovery of H. pylori and its treatment led to dramatic changes in the prevalence and recurrence of peptic ulcer disease, transforming peptic ulcer from a chronic recurrent disease to a curable one.2 H. pylori infection remains an important cause of peptic ulceration in the developing world. In the developed world, the use of NSAIDs has emerged as a leading cause of peptic ulcer disease, especially in the aging population in whom these drugs are often prescribed. Through all of these developments, the role of acid and pepsin in the genesis and perpetuation of mucosal injury remains a unifying aspect of the pathogenesis of peptic ulcer disease.
EPIDEMIOLOGY
The epidemiology of peptic ulcer disease has undergone a remarkable change in the past century. The incidence of duodenal ulcer and gastric ulcer has declined in parallel with the decline in H. pylori prevalence, likely as a result of improved sanitary conditions and a safer food and water supply. The risk of developing peptic ulcer disease and the risk of dying from peptic ulcer disease increased in successive cohorts born between 1840 and 1890 and then declined thereafter.3 A peak in the incidence of gastric ulcer in the first half of the 19th century and a subsequent peak in the incidence of duodenal ulcer in the second half of the 19th century remain unexplained, although a number of theories have been proposed, among them the widespread adoption of smoking after the commercial manufacture of cigarettes in a setting of widespread H. pylori infection.
POPULATION-BASED STUDIES
In northern Sweden, a random sample of 1001 people underwent upper GI endoscopy after filling out symptom questionnaires.4 The prevalence of peptic ulcer disease in this sample was 4.1%, with 20 gastric ulcers and 21 duodenal ulcers. In a prospective Danish study of 2416 subjects, the 11-year cumulative incidence of peptic ulcer was 2.9%: 1.6% for duodenal ulcer, 1.3% for gastric ulcer, and 0.04% for combined ulcers.5 In countries with a high prevalence of H. pylori infection, the ratio between duodenal and gastric ulcers may be quite different. In a case-control study from Shanghai, China, in which the prevalence of H. pylori infection was 76%, recurrent or new peptic ulcers occurred in 3.6% of the population over 2 years and 85% of the ulcers were duodenal.6
TIME TRENDS
There has been a significant decline in mortality from peptic ulcer disease over time in most age groups.7,8 A notable exception is older adults, in whom peptic ulcer bleeding remains a life-threatening condition. A study based on the U.S. National Discharge survey reported that from 1992 to 1999, the annual rate of hospitalization for peptic ulcer disease declined 20%, from 205/100,000 population to 165/100,000 population. Mortality also declined 22%, from 7.7/100,000 to 6/100,000, respectively. Sales of acid-inhibitory drugs correlated with a decrease in peptic ulcer disease hospitalizations and mortality,9 although this correlation does not prove causality. The observed decline in ulcer-related mortality was probably related to improvements in the general health of the population and the availability of effective treatment for peptic ulcer disease.
In an analysis of the Canadian Institute of Health Information database, the prevalence of acute nonvariceal upper GI bleeding (largely ulcer-related bleeding) declined by 31%, from 77/100,000 population to 53/100,000 population over the 10-year period from 1993 to 2003.10 The need for surgical intervention also declined but the mortality rate remained unchanged. In the United Kingdom, overall hospital admission rates for peptic ulcer disease have also declined, as has mortality, but admission rates for peptic ulcer hemorrhage and perforation have increased.11 Peptic ulcer bleeding is most often seen in older adults, with 68% of patients presenting older than age 60, and 27% older than age 80.12
RISK FACTORS
The principal risk factors of peptic ulcer disease are H. pylori infection and NSAID use (Fig. 52-1). However, some patients with peptic ulcer disease have neither of these risk factors.
HELICOBACTER PYLORI INFECTION (see Chapter 50)
H. pylori is a gram-negative bacillus that is uniquely adapted to life in the stomach. It is a major cause of peptic ulcer disease and accounts for a large proportion of peptic ulcers in countries where H. pylori infection is highly prevalent such as in Asia. In the United States and in western Europe, the original estimates that H. pylori infection was the cause of 90% or more of duodenal ulcers, and 60% or more of all gastric ulcers have been lowered by the declining prevalence of H. pylori infection.13 It is estimated that close to 70% of duodenal ulcers are related to H. pylori in Western populations. For example, in an analysis of patients who participated in H. pylori eradication trials in the United States, the proportion of patients with peptic ulcer disease and H. pylori infection was 73%.14 In Rochester, New York, the prevalence of H. pylori infection in patients with duodenal ulcer disease was 61%.15
ASPIRIN AND NONSTEROIDAL ANTI-INFLAMMATORY DRUGS (see also Chapter 51)
Aspirin is increasingly used in the prevention of cardiovascular disease.16,17 Aspirin and clopidogrel are frequently used in combination in patients who have had ischemic cardiac events or in patients who have recently had a stent placed in the coronary arteries.18 NSAIDs are used by approximately 11% of the U.S. population on a regular basis.19 This frequency is likely to increase as the population ages. Regular use of NSAIDs increases the odds of gastrointestinal bleeding five- to six-fold compared with persons not taking NSAIDs.20 Serious ulcer-related complications occur in 1% to 4% of NSAID users and NSAID-related complications are thought to account for 100,000 admissions every year.21 NSAID users who also take aspirin are at an especially high risk for complications. In a population-based study from Denmark, the odds ratio for GI bleeding if a person was taking low-dose aspirin was 2.6, and this risk increased to 5.6 in patients who were also taking an NSAID.22 In Spain, the death rate attributed to NSAID-aspirin use was 15.3/100,000. Up to one third of all NSAID-aspirin–related deaths in that study were attributable to low-dose aspirin use.23
H. pylori and NSAIDs may have a synergistic role in causing peptic ulcer disease. In a meta-analysis, the odds ratios for the development of a peptic ulcer in patients with H. pylori infection or NSAID use were 4.05 and 2.99, respectively, but the odds ratio increased significantly to 15.4 if both factors were present.24 The risk factors for peptic ulcer disease among patients taking NSAIDS and their risk ratios are listed in the next chapter (see Table 53-1).
OTHER ULCEROGENIC DRUGS
Deep ulcers and perforations of the stomach and duodenum have been described in cocaine and methamphetamine users, presumably due to mucosal ischemia.25 Bisphosphonates have also been associated with gastroduodenal ulceration,26 although esophageal injury with bisphosphonates in clinically more of a concern (see Chapter 45). There is little if any risk for peptic ulcer disease in patients taking glucocorticoids.27 In combination with NSAIDS, however, glucocorticoids increase the risk of peptic ulcer disease above the risk with NSAIDs alone.28
OTHER RISK FACTORS
Smoking has been implicated in the pathogenesis of peptic ulcer disease for decades, but its importance as a risk factor has declined after the discovery of H. pylori. A population-based study evaluated the risk factors for peptic ulcer disease in 2416 Danish adults who were interviewed between 1982 and 1994.29 As expected, H. pylori seropositivity was a significant risk factor for ulcer disease; smoking increased the risk of peptic ulcer only in H. pylori–infected subjects. A large body of literature suggests that smoking may predispose to peptic ulcer disease, but H. pylori infection remains a confounder that was not addressed in earlier studies. It is noteworthy that cigarette smoking does not increase the risk of recurrent ulceration once H. pylori has been eradicated, suggesting that smoking may only play a role in infected subjects.30
The role of alcohol remains uncertain. Alcoholic beverages stimulate gastric acid production. Moreover, direct application of high concentrations of alcohol to the gastric mucosa causes demonstrable mucosal injury. In the Danish study alluded to previously,29 intake of spirits increased the risk of peptic ulcer disease in H. pylori–infected patients.
With regard to diet, ulcer prevalence rates differ considerably in the north of India, where the principal cereal in the diet is wheat, and in the south, where rice is the predominant cereal. However, many other potential confounders were not accounted for in these populations.31 An association between the ingestion of spicy foods and peptic ulcer disease is weak, at best.32
Emotional stress was proposed as a major cause of ulcer disease or as a precipitant of ulcer complications,33 and much was written about its relationship to peptic ulcer disease prior to the description of H. pylori. Personality types and psychological profiles have been proposed to be linked to ulcer, but much of this literature is confounded by the lack of information on H. pylori infection. Well-documented descriptions of an increase in ulcer disease after natural calamities such as earthquakes suggest that emotional stress among those not physically injured may play a role in triggering overt manifestations of peptic ulcer disease, especially in individuals who may be otherwise predisposed to ulcer (e.g., patients infected with H. pylori).33,34
PATHOGENESIS
GASTRIC ACID SECRETION
Gastric acid and pepsin have long been considered the principal inciting agents in the pathogenesis of peptic ulcer disease.35 The control of gastric acid secretion in health and disease has been reviewed in detail36 and is discussed in Chapter 49.
HELICOBACTER PYLORI INFECTION (see also Chapters 50 and 51)
Most patients who have a chronic infection with H. pylori have a pan-gastritis in which the body and the antrum are equally involved. Gastritis results in inhibition of acid secretion, and various mechanisms have been proposed.37,38 These include direct inhibition of the parietal cell by lipopolysaccharides or toxins secreted by H. pylori or an indirect effect through stimulation of cytokines caused by the inflammation.1 Due to the decreased output of acid, these individuals with pan-gastritis do not usually develop ulcer disease related to H. pylori infection.39 The reduced acid production in H. pylori–infected individuals has been suggested as a factor that may protect against the development of reflux disease and its complications such as Barrett’s esophagus (see Chapters 43 and 44).
Another pattern of H. pylori infection affects approximately 10% to 20% of patients with chronic infection and consists of an antrum-predominant gastritis (see Chapters 50 and 51). In these individuals, through a series of steps not well understood but possibly involving reduced somatostatin concentrations in the antrum, basal and meal-stimulated acid secretion often increases. The increased acid output from the stomach results in increased acid delivery to the duodenum that can result in gastric metaplasia in the duodenal bulb. Some investigators believe that the metaplastic epithelium then becomes infected with H. pylori from the stomach, resulting in focal duodenitis, sometimes followed by ulcer formation. In contrast with patients with duodenal ulcer disease who often have increased acid secretion, patients with gastric ulcer typically have normal or decreased acid production, suggesting that the mechanism for the development of ulceration is a failure in the gastric mucosal protective mechanisms, described in more detail below. Acid antisecretory medication heals ulcers in patients with gastric ulcer and hypochlorhydria because acid changes the balance in favor of mucosal defense factors that restore mucosal integrity (see Chapter 53).
GASTRIC AND DUODENAL MUCOSAL DEFENSE MECHANISMS
As described in Chapter 49 the gastric mucosa has multiple defense mechanisms to protect it from digestion by acid and pepsin.40 These include the gastric surface epithelium,41–43 the mucus/phospholipid and bicarbonate barrier (mucus layer),44,45 epithelial cell renewal and regeneration,46,47 mucosal blood flow and the alkaline tide, and prostaglandin production.48–51 Prostaglandins also stimulate mucus, bicarbonate, and phospholipid production. Many prostaglandins also increase mucosal blood flow and stimulate epithelial regenerative processes. Inhibition of these protective effects by NSAIDs increases the likelihood of injury to the epithelium and decreases its ability to respond and regenerate.
Cyclooxygenase-1 (COX-1) and COX-2 are the enzymes responsible for the synthesis of prostaglandins. COX-1 is expressed in the stomach and is responsible for the maintenance of the integrity of gastric epithelium and the mucous barrier. COX-2 is not expressed in the healthy stomach but is rapidly expressed in response to the cytokines generated by inflammatory processes. Conventional NSAIDs such as ibuprofen inhibit the COX-1 and the COX-2 enzymes. It is believed that COX-1 inhibition reduces prostaglandin synthesis, which leads to a reduction in mucosal blood flow, hypoxia, and a reduction in mucosal defense. Neutrophil-endothelial interactions then occur as a result of the vascular disturbances and neutrophil activation. In experimental studies, COX-1 inhibition alone is not sufficient to cause ulceration.52 The inhibition of COX-1 up-regulates the expression of COX-2 that suppresses the neutrophil endothelial interaction that is stimulated by COX-1 inhibition. Inhibition of COX-1 and COX-2 enzymes is therefore important in the generation of gastric injury.
Some studies have suggested that a prostaglandin-independent mechanism may also contribute to damage by NSAIDs. One such mechanism is increased leukotriene production that occurs because arachidonic acid metabolism shifts to the alternative 5-lipooxygenase pathway when the COX-1 pathway is inhibited.40
The duodenum, like the stomach, secretes bicarbonate that neutralizes acid arriving into the duodenum.53 Decreased duodenal bicarbonate secretion has been reported in patients with duodenal ulcer.54 A protective role has also been postulated for pancreatic juice but the results have been conflicting.
