CHAPTER 3 Pathophysiology of Varicose Veins
The World Health Organization defines varicose veins as ‘saccular dilatation of the veins which are often tortuous’.1 Further, this definition specifically excludes any tortuous veins associated with previous thrombophlebitis or arteriovenous connections or with venectasia.
Histochemical Physiology of Varicose Veins
Varicose veins differ from nonvaricose veins in physiologic function. This may occur in one or all of the histologic layers. Endothelial damage can occur in parts of a varicose vein,2 and has been noted both ultrastructurally and physiologically by a reduction in endothelial-mediated enhancement of norepinephrine (noradrenaline) induced vasoconstriction (Fig. 3.1).3,4
Characterization of the endothelin receptors in varicose veins compared with those in normal veins has shown decreased contraction to endothelin-1 in both varicose and saphenous veins of patients with primary varicosities. It may be that this observation will be associated with a decrease in the number of receptors.5
In most investigations the muscular layer has been found to be altered, with varicose veins having a considerable degree of smooth muscle hypertrophy and a 15% increase in muscle content compared with normal veins.6 This is thought to be a secondary response to venous hypertension. Other investigators have found that smooth muscle cells are capable of phagocytosis and decomposition of collagen fibers.7 Smooth muscle cells from varicose veins have been found to be less differentiated compared to normal veins and demonstrate increased synthetic capacity, greater proliferation and increased migration than smooth muscle cells found in normal veins.8 Thus, these cells may be part of the cellular basis for collagen breakdown. However, other investigators have noted a decrease in lactate dehydrogenase and creatine kinase activity in varicose versus normal veins and postulate that varicose vein weakness is due to a thinning or damaged muscular layer.9 This has been confirmed in a study of aging canine and human veins where a decrease in sympathetic innervation has been correlated with muscular layer thinning.10 In addition, the protein content of varicose veins (which is predominantly smooth muscle) is reduced.4 However, one research group has found no significant difference in the quantity of smooth muscle between normal and varicose veins.11
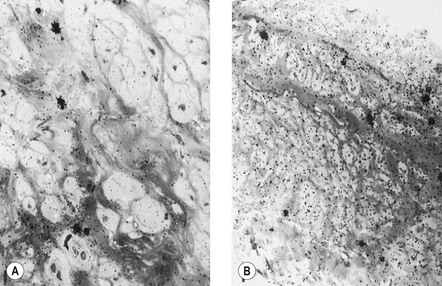
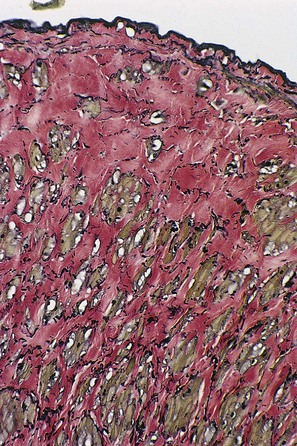
The adventitial layer has been noted to be altered in varicose veins. Some investigators have found that varicose veins have an extremely dense and compact fibrosis between the intima and adventitia, with a diminished and atrophied elastic network and a disorganized muscular layer (Fig. 3.2).12–15 Thickening and fibrillation of individual collagen fibers has also been noted.2,13,16,17 This translates to a reduced compliance that may lead to poor coaptation of venous valves and increased varicose vein wall stiffness. An in vivo measurement of venous elasticity in patients with normal, ‘high-risk’, and varicose veins confirmed reduced elasticity in both varicose and high-risk veins.18 In this study, individuals with high-risk veins were defined as having a family history of varicose veins, standing occupations, symptoms of venous disease and Doppler ultrasound reflux.
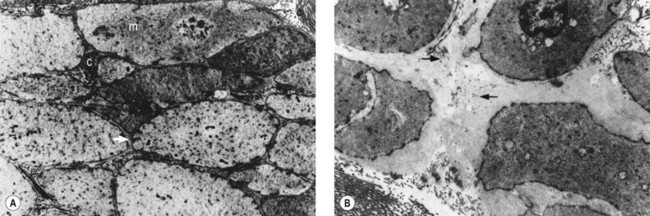
The described loss of tonicity of varicose veins is primarily the result of the loss of coordinated communication between smooth muscle cells. Electron microscopic studies of nonvaricose veins demonstrate the close approximation of smooth muscle cells. When veins become varicose, smooth muscle cells become vacuolated and are separated by collagen.12,13 With increasing varicose changes, intercellular collagen deposition accumulates and separates the smooth muscle cells, which then atrophy (Fig. 3.3). It is suggested that the resulting separation of smooth muscle cell hemidesmosomes causes inefficient smooth muscle contraction and increased venous distensibility.11,13,19 However, some varicose veins are capable of constricting in response to an infusion of dihydroergotamine. This venoconstriction is even more pronounced than that occurring in normal veins.20 The reason for this paradoxical effect is unknown, but a varicose vein appears to be a dysplastic vein characterized by malformations. Whether this is the result of continual high venous pressure or whether it is the primary etiologic event in the development of valvular incompetence is also unknown.
Elastin and collagen are known to play an important role in maintaining structural integrity of blood vessel walls. Normally when the wall is stretched, elastin generates a shortening force that opposes the traction exerted by the side branches and perivascular connective tissue and the lengthening force caused by pressure in the lumen. Type I collagen is believed to confer tensile strength to the vessel wall, whereas type III collagen may be involved in extensibility. In dilated and morphologically normal segments of varicose veins, type I collagen is present in a greater amount than type III. Furthermore, varicose veins contain more type I and type III collagen than do normal veins. It has been found that the elastin content is significantly reduced in dilated segments of varicose veins when compared with both normal veins and normal segments of varicose veins. Microscopically, the ratio of collagen to elastin appears to be significantly increased in the dilated segments of varicose veins. These findings tend to emphasize the important role of elastin in providing a retractile force that opposes development of dilation and tortuosity of the vein wall.21
Although collagen accumulation is thought to separate smooth muscle cells within the varicose vein wall, the collagen content of varicose veins is less than that in normal veins.6,22 The bulk of the varicose vein wall is made up of mucopolysaccharides and other ground substances. Varicose veins contain 67% more hexosamine (which comprises about 0.3% of normal vein dry material) than is found in normal veins.
Dysplasticity of the varicose vein wall may explain why varicose veins have an even greater susceptibility to pressure-induced distension than do nonvaricose veins. This anatomical–pathophysiologic correlation has been demonstrated by pharmacologic studies that show reduced maximal contraction of varicose veins compared with control veins.2,19 They have also been investigated with in vitro techniques measuring distensibility as a function of infused volumes of saline.23 However, some investigations have failed to discover a significant difference in the degree of intimal fibrosis between varicose and nonvaricose veins.24 Therefore, fibrosis of the vein wall alone is not totally responsible for the development of varicose veins.
In studying smooth muscle reactivity, the three main vasoconstrictor agents – norepinephrine, angiotensin II, and endothelin-1 – were compared. In diseased vein segments, a significant reduction in response to angiotensin II and norepinephrine was seen. Also, it was noted that there was reduction in response to endothelin-1. The reduction in angiotensin II affinity appeared at an early stage of varicose disease and supports the hypothesis that such an abnormality within the venous wall could play a role in the pathogenesis of primary varicose veins.25
Finally, a decrease in tocopherol concentration has been noted in varicose veins.26 A significant correlation also seems to exist between the inhibition of vessel wall tissue lipoperoxidation and their tocopherol concentration, independent of serum concentrations. This may be the result of the protective effect of blocking peroxidation of membrane-associated fatty acids by tocopherol and other antioxidants to prevent vein wall damage.27 It is clear that the dysplasticity of varicose veins correlates with the changes in their pharmacodynamics and histochemistry. Varicose veins have a demonstrated loss of contractility.28
Varicose veins are often complicated by local inflammation and thrombosis. This may be due to venous hypertension to an inherent histochemical abnormality in the varicose vein/endothelial wall. The formation of arachidonic acid-derived prostanoids was investigated in segments of varicose and nonvaricose veins. Venous production of prostacyclin was decreased, while that of thromboxane A2 and prostaglandin E2 was increased, in the varicose vein segments, regardless of whether they were macroscopically affected or unaffected.29 It is unknown whether this change in the cyclooxygenase pathway in the varicose vein wall is the cause or effect of its dysplasticity. In addition, histochemical examination discloses a marked increase in the activity of lysosomal enzymes,30 acid phosphatase, β-glucuronidase, and anaerobic isoenzymes (lactodehydrogenase) in primary varicose veins.31–33 These enzyme patterns suggest a decline in energy metabolism and an increase in cellular damage in the varicose veins. It has also been found that varicose veins accumulate and metabolize norepinephrine less efficiently than normal veins.34 Differences in expression and, probably more important, microscopic localization of matrix metalloproteinase (MMP) and tissue inhibitor of metalloproteinases between normal and varicose veins may explain the variability of disease between vein segments.35 MMP-2 has been found to cause relaxation of contracted vein segments which could lead to progressive venous dilatation, varicose vein formation and chronic venous insufficiency.36 Whether the abnormal level and/or action of MMP is the contributing factor or whether protracted increases in venous pressure lead to an increase in MMP expression is unknown.37 Therefore, both anatomical and biochemical abnormalities in the varicose vein wall contribute to its increased distensibility (Box 3.1).
Pathophysiology
Approximately 75% of the body’s total blood volume is contained within the peripheral venous system.38 The quantity of blood within the legs is a function of body position. When erect, 300 to 800 mL of extracellular and vascular fluids (the quantity varies according to the experimental method and the size of the subject measured) collects in the legs.39–41 This includes a 15% increase of blood volume.39 Thus, the venous system, especially in the legs, is an important component of the cardiovascular system’s circulatory reservoir. However, the arterial system plays an equally important role in cardiovascular adaptation to postural changes by virtue of changes in arterial resistance. In fact, studies have demonstrated that reflex changes in venous tone are not essential for this fluid shift.42
Venous blood pressure is determined by several factors. Among these are pressure generated by the heart, energy lost in the peripheral resistance of arterioles, hydrostatic gravitational forces, blood volume, anatomical composition of the venous wall, efficiency of one-way valves, vein wall distensibility (determined by hormonal, systemic alcohol and other factors), and contraction of venous smooth muscle as influenced by ambient temperature and sympathetic and parasympathetic nerve tone (Fig. 3.4).
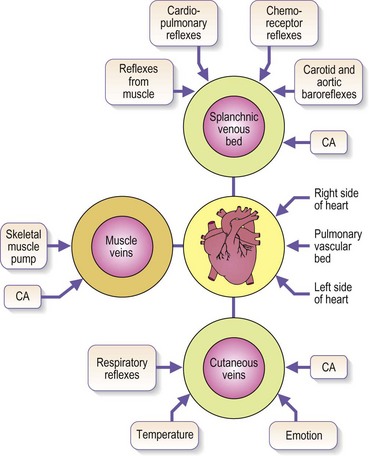
Although arterial pressure is one factor in the development of venous pressure, arterial hypertension has been noted to be associated with the development of varicose veins in some epidemiologic studies43 but not others.44 Curiously, atherosclerotic disease has been linked epidemiologically to varicose veins, although it might be two common conditions occurring concurrently.45,46 It is postulated that this coincidence may be related to an atherogenic risk profile, owing primarily to coexistent inactivity, obesity and hypertension.46 At rest, in the erect position, pressure in the saphenous vein is determined primarily by the height of the column of blood from the right atrium to the site of measurement (90 to 120 mmHg at the ankle) (Fig. 3.5).47,48 Contraction of calf muscles generates pressures of between 200 and 300 mm Hg.49–51
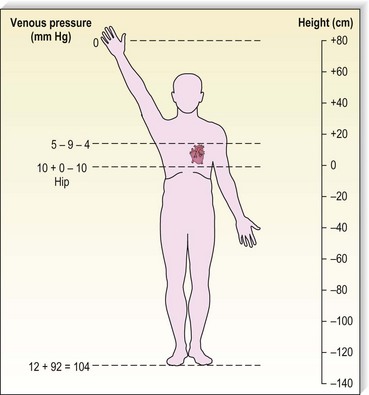
Figure 3.5 Venous pressure is that exerted by a column of blood from the heart to the location of measurement.
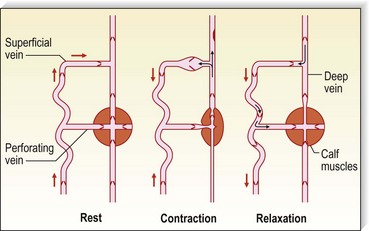
Pressure generated deep to the fascia, outside of muscles, is between 100 and 150 mmHg;51,52 however, with muscular activity, pressure in the normal saphenous vein at the level of the malleoli falls 45 to 68 mmHg below the resting level.53 It is reduced from 80 to 40 mmHg in the posterior tibial vein.54 Because of the one-way valves, blood flow is directed from the superficial venous system to the deep venous system through perforating vessels (see Fig. 1.4). This has been demonstrated visually by serial phlebography of the normal lower leg.55 The venous blood then flows towards the heart.
The venous pump in the foot is an important portion of the muscle pump of the lower leg. Weight bearing is usually necessary to propel blood up the leg. Bidirectional ultrasound velocity detector tracings of venous blood flow through the popliteal vein have demonstrated the importance of dorsiflexion of the foot when there is no weight bearing.56 Therefore, full flexion of the foot is important after sclerotherapy to maximize the efficacy of the lower extremity muscle pump.
Respiration produces alterations in intra-abdominal venous pressure. This ‘abdominal venous pump’ contributes to the flow of blood even when an individual is erect.41,57 Inspiration produces a rise in venous pressure in the external iliac vein, common iliac vein and inferior vena cava when measured in both the horizontal and erect positions (6.3 mmHg and 8.7 mmHg, respectively).57
In the supine position, blood flows evenly along all superficial and deep vessels towards the heart. It is propelled by the relatively small vis-à-tergo (force from behind) from the capillaries54 and the respiration-induced aspiration of blood into the abdominal and thoracic veins. In contrast to deep veins, superficial veins have smooth muscle in their walls. This allows contraction of these vessels in response to cold and to drugs such as dihydroergotamine58,59 and allows dilation in response to topical and systemic alcohol, estrogen and light physical trauma.54 As previously described, part of the pathophysiology of varicose veins may be a diminished response of such smooth muscle contraction.
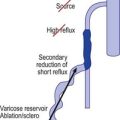
Regardless of its cause, chronic venous hypertension in the lower extremities causes an increase in venous diameter. This may lead to valvular insufficiency, which usually causes a reversal of blood flow from the deep veins into the superficial veins through incompetent perforating veins. This ‘private circulation’ may account for as much as 20% to 25% of the total femoral flow involved in a circular retrograde flow (Fig. 3.6).60,61
It has been found that prevalence of reflux in vein segments is correlated with signs of venous insufficiency, but in the general population, approximately 12% of limbs with no disease have reflux as detected by duplex ultrasound.62
Venous insufficiency has been correlated with standing occupations. A study that compared symptom-free vascular surgeons with normal individuals of nonmedical vocations showed that the superficial system was by far the most common site of venous incompetence in both groups. Vascular surgeons (standing occupation) showed a greater incidence of reflux than the controls. This was true even in subgroups in which reflux was seen in the superficial veins, as well as in those with reflux in the deep veins and perforating veins.63 In patients with symptoms of venous insufficiency, reflux in the great saphenous vein (GSV) territory is found in 85% of limbs, but only 68% of limbs show true saphenofemoral junction (SFJ) incompetence. Reflux is found in 20% of such patients in the small saphenous vein (SSV) territory, and strictly nonsaphenous origin of varicosities is found in 6%.64 One study demonstrated that 93% of the 10% of patients with nonsaphenous reflux in the study group were women with a mean of 3.2 pregnancies.65 This implied an association with female sex, hormones and/or number of pregnancies. Studies of causation of reflux focus on venous valves and vein walls. On one hand, an antiproteolytic milieu may favor the deposition of collagen and allow varicosities to develop;66 on the other hand, activation of monocytes and conversion to macrophages may cause weakening or destruction of valve segments.67
Direction of venous flow in varicose veins has been examined by McPheeters and Rice68 using fluoroscopy. Reversal of flow caused by incompetent perforator valves is beneficial during sclerotherapy. When a superficial varicosity is injected, its venous flow is forced distally to the smaller branching veins, where it is arrested (see Chapter 8).68 Thromboembolic disease is thereby prevented.
Superficial veins respond to increased pressure by dilating. Valvular incompetence occurs and varicosities appear.69 In addition, in muscular contraction, high compartmental pressure that normally occurs within the calf muscle pump is transmitted directly to the superficial veins and subcutaneous tissues drained by perforating veins.70,71 When this occurs, venous pressure in the cuticular venules may reach 100 mmHg in the erect position.54 This causes venular dilation over a broad area and may cause capillary dilation, increased permeability72–75 and a subsequent increase in the subcutaneous capillary bed through angiogenesis.73,76 This is expressed clinically as telangiectasia (venous blemishes). Histologically, cutaneous and subcutaneous hemosiderin deposition may also occur. This, in time, causes cutaneous pigmentation (see Chapter 2). However, some patients with chronic venous insufficiency are able to increase their venous blood flow through exercise.77 It is postulated that various factors (e.g. sympathetic tone, temperature, tissue metabolites) compensate venous hypertension to normalize cuticular blood flow. This finding demonstrates the complexity of the superficial venous system.
A special situation develops in the area of the medial malleolus. In this area, perforating veins are not surrounded by deep or superficial fascia. Therefore, any increase in deep venous pressure is transmitted directly through perforating veins to superficial connecting veins. This causes high cutaneous venous pressures and a transudation of extracellular fluid. This, in turn, leads to perivascular fibrin deposition, which has been blamed for decreased oxygenation of cutaneous and supporting tissues; this was thought to contribute to cutaneous ulceration (see Chapter 2).73,78,79 This theory has largely been discredited; the ability of any fibrin screen to prevent oxygen diffusion has never been proven.
The effect of temperature variations on the venous system is well studied.80,81 The cutaneous vasculature is intimately involved in thermoregulation. An increase in body core temperature results in cutaneous vasodilation. This does not occur as a result of relaxation of venous smooth muscle but because of the reduction in the vasoconstrictor impulses to the vein wall. Such vasodilation also occurs in varicose veins. Recently, strain gauge venous occlusion plethysmography has shown an increase in venous distensibility associated with temperature elevation.82 Similarly, alcohol ingestion may influence the development of varicose veins. Alcohol intake, like increased environmental temperature, causes cutaneous vasodilation. In an examination of 136 men with primary varicose veins greater than 4 mm in diameter, it was found that a significantly increased incidence of varicose veins occurred among men who consumed 4 oz (around 120 mL) of alcohol a day.83 Unfortunately, further experimental evaluations of this association have not been performed.
In summary, pathologic development of varicose veins can be divided into four broad categories, which may overlap and contribute to each other: increased deep venous pressure, primary valvular incompetence, secondary valvular incompetence and hereditary factors (such as vein wall weakness). All of these categories coexist and are influenced by temperature, alcohol, and hormonal and other vasodilatory stimuli (Box 3.2).
Increased Deep Venous Pressure
Most veins of the forearm and lower extremity remain competent even after maneuvers that induce venodilation and increase in blood flow, such as exercise hyperemia or postocclusion reactive hyperemia. However, veins with an inherent valvular weakness can be identified by reactive hyperemia in association with duplex flow analysis.84 The presence of femoropopliteal reflux is associated with clinical symptoms – it has been found in up to 15% of limbs having primary varicose veins. This is divided into those with superficial femoral venous reflux alone and those with isolated popliteal venous reflux.85
Proximal origin
Pelvic obstruction
Pelvic obstruction is an uncommon cause of varicose veins. Iliac vein compression syndrome is the phenomenon of compression of the left iliac vein by the left iliac artery overlying the fifth lumbar vertebra.86–90 This usually occurs in women, in whom it may be a cause of vulvar varicosities, but it has also been noted in men (see Chapter 5). Extravascular abdominal tumors, such as ovarian and uterine carcinoma or teratoma, may be causes of obstruction. More commonly, however, it is relative pelvic obstruction that provides a mechanism for impedance of return blood flow. Relative obstruction may occur in the third trimester of pregnancy, particularly during recumbency when the gravid uterus compresses the inferior vena cava against the lumbar spine and/or psoas muscles. Phlebographic studies have shown complete obstruction of the inferior vena cava at the confluence of the iliac veins in third-trimester pregnancies.91 Partial obstruction has been shown in earlier months of pregnancy. Some degree of compression is evident using phlebography, even in the left lateral decubitus position.
Increased intra-abdominal pressure
One popular hypothesis for the development of varicose veins is Western dietary and defecation habits that cause an increase in intra-abdominal pressure. A distended cecum or sigmoid colon, the result of constipation, may drag on the iliac veins and obstruct venous return from the legs.92 Population studies have demonstrated that a high-fiber diet is evacuated within an average of 35 hours.93 In contrast, a low-fiber diet has an average transit time of 77 hours. An intermediate diet has a stool transit time of 47 hours.
It is possible that the small increase in abdominal intravenous pressure caused by less than optimal bowel habits, when transmitted intravenously distally, gradually breaks down venous valves of leg veins.94,95 Evidence in support of this hypothesis is seen in populations of people who eat unprocessed high-fiber food. These persons are free of constipation and varicose veins.96,97 However, if this population’s diet is changed to low fiber, the incidence of varicose veins increases.92,98–101 In a diet that is intermediate between Western low-fiber and high-fiber diets, the prevalence of varicose veins is also found to be in an intermediate range.102
Defecatory straining induced by Western-style toilet seats has also been cited as a cause of varicose veins, in contrast to the African custom of squatting during defecation.99 However, venous pressures of subjects measured in both the sitting and squatting positions during defecation have not shown a significant difference.103 Venous flow has not been examined accurately in constipated and nonconstipated populations. Finally, there are other dietary factors besides fiber content that may explain the differences in prevalence of varicose veins. An increased incidence of varicose veins is found in populations of people who consume diets high in long-chain fatty acids, as opposed to diets high in short-chain fatty acids.104 Long-chain fatty acids have been shown in experimental systems to enhance blood coagulation and stimulate the development of blood clots.105–107 Clot lysis times were slower in the population group that consumed long-chain fatty acids.104 Accordingly, the type of dietary fatty acids consumed also may predispose the development of varicose veins. In addition, the Western diet has been found to be relatively deficient in vitamin E.108 It is hypothesized that the slight vitamin E deficiency, when aggravated by pregnancy, may predispose the vein wall to coagulation and fibrinolysis, thus causing the veins to become more sensitive to venous stasis and venous hypertension. Therefore, although it would seem prudent to recommend a high-fiber diet for several medical reasons, it remains an unproven treatment for the prevention of varicose veins.
An association between prostatic hypertrophy, inguinal hernia and varicose veins may be caused by straining at micturition with a resultant increase in intra-abdominal pressure.109
Another mechanism for increasing distal venous pressure by proximal obstruction is the practice of wearing girdles or tight-fitting clothing. A statistically significant excess of varicose veins was noted in women who wore corsets compared with women who wore less constrictive garments,110 although this finding was not confirmed by a subsequent study.111 However, a more recent study of 20 women aged 20 to 46, who wore ‘tight’ jeans (degree of compression was not measured), found that in 14 of these women there was an increase in subcutaneous pressure from 10 to 15 mmHg at rest to 30 mmHg when walking, as opposed to no change in pressure when wearing loose-fitting clothing. 112 Thus, the use of tight jeans can negatively affect venous return. A similarly increased incidence was noted in women who stand at work compared with those whose jobs entail more walking and sitting.45,47,110–116 However, this has not been confirmed universally.117,118
Leg crossing and sitting on chairs are two other potential mechanisms for producing a relative impedance in venous return. Habitual leg crossing is commonly thought to result in extravenous compression, but this has never been scientifically verified. A decreased incidence of varicose veins has been noted in population groups that do not sit in chairs.119,120 It is thought that sitting may produce some compression on the posterior thigh which produces a relative impedance to blood flow. Wright and Osborn121 have shown that the linear velocity of venous flow in the lower limbs in the recumbent position is reduced by half in the standing position and by two-thirds when sitting. Alexander120 found that the circumferential stress on the saphenous vein at the ankle was 2.54 times greater with chair sitting than with ground sitting. This may explain the increased incidence of varicose veins in men versus women in population groups in which only men sit on chairs and women sit on the floor. In this population study,119 varicose veins were present in 5.1% of men and only 0.1% of women. Finally, the practical implications regarding chair sitting concern those who travel for long periods in airplanes. Pulmonary thromboembolism has occurred in many people after prolonged air travel and has been termed economy class syndrome.122 Although pre-existing venous disease, dehydration and immobility are all contributing factors, chair sitting adds another insult to the venous system.
Most,43–45,115,116,123–129 but not all,111,125,130,131 studies have found that obesity is associated with the development of varicose veins. Careful examination of some of these epidemiologic studies shows that when the patient’s age is correlated with obesity, the statistical significance is eliminated.132 Obesity was especially correlated with the development of varicose veins in women when the varicosities occurred in unison with cutaneous changes indicative of venous stasis (see Chapter 2).129,133,134,135 This may be secondary to decreased exercise and associated medical problems specific to obesity, such as hypertension, diabetes, hypercholesterolemia and sensory impairment.136
Running has been demonstrated to raise the intra-abdominal pressure by 22 mmHg.137 This increase in abdominal pressure occurs because of a reflex tightening of abdominal muscles during running, which prevents the pelvis from tipping forwards during thigh flexion induced by contraction of the iliopsoas muscle group.138 Therefore, during strenuous leg exercise, elevated abdominal pressures may impede venous return. By way of comparison, a Valsalva maneuver was shown to elevate the intra-abdominal pressure by 50 mmHg or more.137 Strenuous exercise, particularly long-distance running, is often associated with prolonged increases in limb blood flow, which theoretically could overload the venous system and lead to progressive dilation.69 Usually, dilated veins that occur in this situation are normal and do not require treatment. Finally, it is commonly noted that occupations that require standing for prolonged periods have an increased incidence of varicose veins.139 This may be exacerbated by tall height, although this factor has not been supported by other studies.132
Saphenofemoral incompetence
Saphenofemoral impedance is rarely caused by anatomical abnormalities in the saphenofemoral triangle. When it occurs, pelvic tributary veins or accessory saphenous veins may converge in such a manner that flow to the femoral vein is impeded.140 Likewise, iliac venous incompetence caused by the congenital absence of venous valves, or by damage to the valves through thrombosis, may cause distal venous hypertension.
Our interest and focus on the venous valve dysfunction as a fundamental cause of distal venous hypertension began with unpublished observations using angioscopy. The angioscope provided a direct view of the internal architecture of saphenous veins. Patients taken to surgery who demonstrated preoperative reflux verified by duplex ultrasound showed a variety of pathologic lesions in the valves themselves. The first indication was a relative paucity of valves. The observation of a decrease in the number of GSV valves had already been reported by Cotton in 1961.141 Next, we encountered actual valve lesions. These observations were an extension of those reported by Hoshino et al,142 who classified valve damage in the saphenous vein into three categories ranging from stretched commissures to perforations and valve splitting.
From the preceding observations we suggest that the earliest valve defect is an increase in the commissural space, which allows reflux on the border of the vein. This may be one of the earliest causes of reflux in varicose veins. Later, thinning, elongation, stretching, splitting and tearing of the valves develop. The last stages are thickening, contraction, and possibly even adhesion between valves. These observations have been confirmed by Van Cleef et al.143 While we have proposed that this valve damage is acquired and causes axial reflux as well as outflow through check valves in perforating veins, others have proposed that the cause of primary venous insufficiency is an actual low number of valves in the saphenous system.144
The angioscopic observations could be confirmed by gross morphologic studies that, when extended to microscopy observations using monoclonal antibody labeling, have demonstrated monocytic infiltration into damaged venous valves.145 Others have found leukocytic infiltration into varicose veins and have called attention to the fact that the cells observed released vasoactive substances, including histamine, tryptase, prostaglandins, leukotrienes and cytokines.146 Observations in patients led to the conclusions that venous hypertension was related to leukocytic infiltration on the cranial surfaces of the venous valve and venous wall and that leukocytes there were greater in quantity than on the caudal portion of the valve leaflets and venous wall.
This inflammatory sequence occurred early during the phase of venous hypertension and progressed further after release of the occlusion. The model showed that venous occlusion with elevation of the hydrostatic pressure caused a highly injurious process for the surrounding tissues. It was accompanied by formation of microhemorrhages on the high-pressure side of the postcapillary venule and rolling and adhesion of leukocytes on the venular endothelium.147
Van Bemmelen et al148 produced a model of venous hypertension by creating arteriovenous fistulas in Wistar rats using microsurgical techniques. Valvular incompetence was seen as early as 1 day after creation of the arteriovenous fistula, and valvular structural changes were noticeable within 2 months of production of venous hypertension. Elongation of the cusps was observed. Separation and leakage of the cusps were encountered along the entire valvular free border, and, in later stages beyond 4 months, valve areas became difficult to recognize because commissures were lost and bulging of the valve sinus disappeared.
We have pursued this line of investigation and have reproduced the human observations in the animal model.149–153
Another model of venous hypertension has been produced by Lalka.154 This model creates venous hypertension by ligation of the inferior vena cava, the common iliac veins and the common femoral veins. This preparation elevates rat hindlimb venous pressures compared with forelimb pressures. Myeloperoxidase assay indicates leukocyte trapping in hindleg tissues in the same way as it occurs in humans.155
The observations just mentioned suggest that valve damage in venous insufficiency is an acquired phenomenon related to leukocyte and endothelial interactions and an inflammatory reaction. This observation is not universally accepted. A study on 13 valve structures from varicose GSV showed an absence of lymphomonocyte infiltration in 85% and rare isolated ‘nonsignificant’ inflammatory cells in 15%.156 However, if this hypothesis is correct, pharmacologic intervention to block leukocyte adhesion, activation and subsequent valve damage may be a possibility.
Distal origin
Valvular incompetence
Unlike that described in the previous section, incompetence of the SFJ clearly produces distal retrograde flow into the GSV and thus produces distal venous hypertension (Fig. 3.7). The GSV then dilates, producing further distal valvular incompetence sequentially. Retrograde flow thus produced is channeled through the perforator veins back into the deep venous system. This produces a private circuit of blood flow from the femoral vein to the saphenous vein and back to the femoral vein through perforating veins.60 It has been estimated that the total volume of flow in this circuitous route may be 20% to 25% of the total limb blood flow during exercise.61 This paradoxical circulation can be maintained for a long time, but eventually the quantity of blood channeled by the perforator veins increases. As this happens, there is hypertrophy and dilation of the superficial veins, producing valvular incompetence and localized varicose veins.
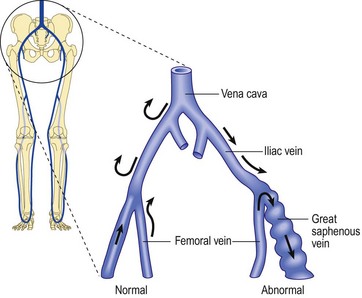
In addition to increasing superficial venous volume through perforator incompetence, retrograde flow produces an increase in acidity and potassium concentration with a decrease in venous oxygen concentration. These three factors promote vasodilatation to exacerbate venous stasis.157
Perforator incompetence in the lower part of the leg may occur from localized thrombosis in the vein following trauma. It is believed that localized thrombosis is usually masked by the local tissue injury. The valve cusps become involved by the thrombus and, after recanalization, remain functionless.158 Dodd and Cockett54 found, on examination of 54 legs with perforator incompetence, that the lower leg incompetent perforators were in communication with the soleal plexus of veins and, as such, were the channels most likely to be damaged as a result of thrombotic episodes in this region. In support of this concept, Fegan,159 Hobbs,160 Lofgren,161 and Beninson and Livingood162 have all pointed out that the treatment of varicosities may have no effect on superficial venous pressure. These investigators contend that treatment of perforating veins draining the ankle and lower calf area is important.70,110,160,163–165 The vessels may be either surgically ligated70,146–165 or sclerosed (see Chapters 9 and 10).160 Only then will retrograde flow under high pressure through the calf muscle pump be diverted upstream away from the skin. When this is done, a lowering of cuticular venous pressure and a decrease in dermal capillary pressure is accomplished. The excess transudation of fluid-producing edema and the associated decrease in tissue oxygenation and nutrition is halted. Quill and Fegan166 have demonstrated clearly the narrowing of the proximal SSV after obliteration of incompetent distal perforating veins in 9 out of 11 cases. This suggests that dilation and incompetence of the saphenous vein may be caused by distal reflux, as well as primary or irreversible abnormalities of the proximal venous wall. The finding has been confirmed through duplex examination of patients with cosmetic veins or primary varicose veins.
In a study of 500 lower limbs in ‘cosmetic’ patients, incompetence from below the knee extending upwards was found in 63.3%. However, only 9% were found to have perforator incompetence.167 An additional study of 167 consecutive patients with primary varicose veins demonstrated that 31% had incompetence of the GSV but no evidence of SFJ incompetence, and 24% of limbs had incompetence of the SSV without incompetence of the SFJ.168 Thus, perforator incompetence, although important as an etiologic factor, is not solely responsible for varicose vein development in all patients.
Duplex scanning has contributed to knowledge regarding valvular dysfunction produced by venous thrombosis. For the most part, deep valvular insufficiency comes from direct valve destruction rather than obstruction-induced venous pressure elevation and dilation of the vein wall. Destruction of the valve cusps occurs after venous thrombosis. It follows that the extent of venous valvular incompetence is related to the extent of the original deep venous thrombosis (DVT). One could extrapolate from these observations that valvular competence might be preserved if thrombus could be removed quickly by thrombolytic agents.169
Direct ambulatory venous pressure measurements and duplex examination have shown bidirectional flow through perforator veins. Exercise has been found to cause inward flow from the dilated superficial system and perforating veins into deep veins.170 The perforator vein here functions as a drainage pipe to limit cuticular venous hypertension. This explains the dilation of re-entry perforating veins, which drain the superficial reflux into the deep venous circulation and which become competent after superficial vein surgery.171–174 Compression distal to perforator veins in patients with venous disease demonstrated inward flow in 55 out of 56 perforator veins examined with duplex scanning.175 Thus, venous hypertension is related to both perforator incompetence and valvular dysfunction.
Venous obstruction
Venous obstruction may occur proximally or distally to varicose veins. An obstruction is typically produced by thrombus that may extend proximally and distally from its origin. The thrombus may also extend into communicating or perforating veins. Depending on the extent of thrombus, venous blood may be forced into the superficial veins in either a retrograde or lateral direction. Finally, it is interesting to speculate that the wearing of high-heeled shoes may lead to distal compression of the vein walls by leg muscles that are strained as a result of these shoes. When high-heeled shoes are worn, calf muscles are compressed and the gluteal muscles are used for walking. This secondary, logical factor, has been proposed previously but awaits experimental confirmation.176 At least one study has been unable to substantiate an effect of walking barefoot versus wearing high heels on leg volume.177
Arteriovenous anastomosis
Another important factor that may lead to increased venous pressure in cutaneous veins is the opening of arteriovenous communications. Arteriovenous anastomoses (AVAs) were suggested to be a component in the pathogenesis of varicose veins in 1949 by Pratt178 and by Piulachs et al179 in 1952. Pratt hypothesized that AVAs represented the failure of closure of femoral artery branches to the saphenous system.
A clue to the presence of AVAs is often provided by the presence of varicosities in an unusual anatomical location and in the absence of detectable abnormalities of the deep venous system.69 Physical examination often shows a lack of complete emptying of the varicose veins when the limb is elevated, during very rapid refilling of the varicose vein or venules with diascopy, during warmth over affected varicose veins, and in the rare presence of a bruit or thrill over these vessels.178 Arterial Doppler sounds are often demonstrated over varices, especially when they are associated with bright red venectasia. Between 60% and 80% of patients with congenital AVAs on the legs have associated varicose veins.180,181
AVAs have been demonstrated by direct operative microscopic dissection by Schalin182 and Gius.177 Indirect support of this theory has been provided by evaluating the oxygen content of varicose blood and skin temperature over varicose veins.183–187 These studies estimate that AVAs occur in 64% to 100% of patients with varicose veins. However, at least one study has found no difference in varicose vein blood oxygen levels.188 These authors postulated that oxygenation of venous blood is also related to metabolic activity, blood flow, blood volume in the varicosity, body position and intravenous pressure. This was confirmed indirectly by measuring skin oxygen levels in patients with and without varicose veins. In this study, no significant difference could be appreciated.189 An additional study of 39 patients with varicose veins failed to show pulsatile flow, or any significant change in mean venous O2, in comparison to controls.190 However, Pratt178 estimated that AVAs occur in 24% of varicose veins and in 50% of patients with recurrent varicose veins after surgical ligation and stripping.
Schalin191 has demonstrated AVAs with thermography (Fig. 3.8). He reviewed the role of arteriovenous shunting in the development of varicose veins and concluded that the recurrent and varying flow of arterial blood transmitted to veins over years causes the venous wall to distend. Schalin made his observations using operative microscopy and demonstrated that AVAs connect to varicose veins at the convex curves. Although AVAs are difficult to detect radiographically in association with varicose veins,192 rapid venous filling is commonly seen in arteriograms of limbs with severe venous stasis.
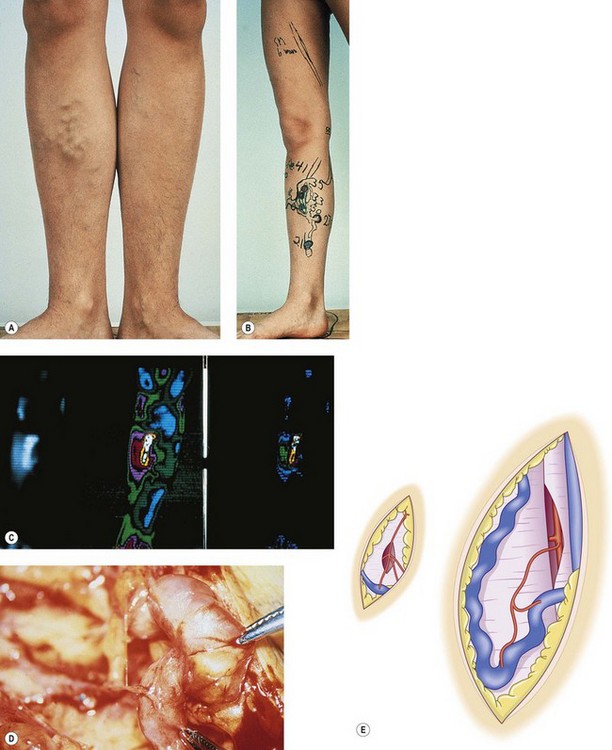
Opening of the AVA may occur through developmental or functional abnormalities of vasa vasorum of the venous wall. In one scenario, an association of excessive alcohol consumption in male patients with varicose veins was proposed to cause arteriolar dilation and new capillary formation, which may act like multiple AVAs.83 Alternatively, opening of the AVA may be caused by proximal venous hypertension breaking down the capillary barrier.192 Once dysfunction of the AVA is established, shunting of arterial blood directly into the venous system further increases venous dilation. Whether AVAs are the cause or the result of varicosities is still unknown. Despite this, common observations tend to support the concept of AVA-induced varicosities. Varicose veins may recur after anatomically correct sclerotherapy that obliterates all perforating veins. This may result from an AVA distal to the point of injection. Also, the bright red color of some leg telangiectasias may be caused by an underlying AVA. Finally, cutaneous ulceration after sclerotherapy treatment may also be related to injection of venules and their associated AVAs (see Chapter 8). Therefore, the AVA is important as either an etiologic or associated factor in the cause and treatment of varicose veins, but it is not the primary cause of all varicose veins.
Primary valvular incompetence
Primary valvular incompetence is a serious precursor of varicose veins because, by definition, such valves are permanently damaged, absent or incompetent. Thus, as originally suggested by William Harvey,193 an incompetent valve causes distension of the distal vein by gravitational back pressure and may produce a varicosity. Many factors may be responsible for the development of valvular incompetence, including developmental abnormalities and destruction of the venous valves. Direct venoscopic evaluation in 25 patients with varicose veins disclosed that the GSV was valveless from the SFJ to the upper calf, where the first normal valve appeared.194 Thus, regional differences occur in varicosities.
Congenital valvular agenesis is a very rare cause of varicose veins.195–199 Multiple case reports and a series of 14 patients have been described.195,200 Such patients have partial or complete absence of deep vein valves in the lower extremities. Familial occurrence in two pedigrees suggests a simple dominant mode of inheritance. Clinically, such patients are detected by development of venous insufficiency at an early age. Interestingly, signs and symptoms invariably occurred only after puberty. The most serious physical finding was cutaneous ulceration in the typical medial malleolar area. The most notable physical finding was ankle edema occurring during the day that was completely resolved at night or during rest. Another common sign was marked orthostatic hypotension from venous pooling in the legs. In these patients, wide variations in the number of valves were seen. They ranged from complete agenesis in both lower extremities to agenesis in only one leg or even partial agenesis. Therefore, although rarely reported, valvular agenesis may be found to some degree on careful examination of some patients. Its diagnosis is critical before performing injection sclerotherapy because a major complication of injecting veins without valves is a possible progression of venous fibrosis and thrombosis to the deep venous system. This could worsen venous insufficiency, even to the extent of jeopardizing the viability of the limb.
Scientific evaluation of the relative significance of valvular deficiency in relation to the development of varicose veins is not as clear as valvular agenesis as a cause for varicose veins. An autopsy study of 44 limbs disclosed four with varicose veins that had normal valves in the femoral vein and the SFJ. Valves were absent proximal to the SFJ in three of the four varicose vein limbs. This was statistically significant when compared with nonvaricose vein limbs.201
Another autopsy study demonstrated a lower number of venous valves in the left internal iliac vein compared with the right iliac vein. This could explain the relative increased incidence of left-sided varicose veins; the relative obstruction caused by the right iliac artery may be unimportant.202 Anatomical studies do not define the functional significance of venous valves. In a radiographic functional study of external iliac and femoral valves performed on 12 male volunteers with and without varicose veins, subjects with a family history of varicose veins were found not to have femoral or external iliac valves.203 Conversely, those men without a family history of varicose veins did have such valves. Another study of 54 normal adults and 19 children of patients with varicose veins confirmed these findings. Venous Doppler examination showed that incompetent iliofemoral valves were present in 16% of the normal adults and in 32% of the children of patients with varicose veins.204 These studies lend support to the theory of descending sequential valvular incompetence as a pathogenic mechanism for the development of varicose veins. Interestingly, a Nigerian study found that caucasians show a relative deficiency of venous valves relative to Africans.205 However, this cannot explain the lack of difference in the incidence of varicose veins between black and white people in America.
Despite the studies just mentioned, other studies unfortunately have failed to show a convincing association between the absence of iliofemoral valves and the presence of GSV incompetence.206 In addition, veins that have been reversed (thus rendering their valves incompetent) and used as arterial grafts fail to elongate or dilate. They certainly do not develop into varicose veins.207 Finally, at least one study demonstrated a competent saphenofemoral valve in up to 50% of GSVs with incompetent distal valves, thus suggesting that in these patients, reflux progresses distal to proximal.208,209 Therefore, primary valvular deficiency, with or without increased transmural pressure, is best thought of as a contributory factor and not as an absolute etiologic factor in all patients with varicose veins.
Light-microscope findings of the venous valve in varicose veins consist of multiple dystrophic changes. There is a proliferation of collagen fibers and smooth muscle cells, as well as distortion and tearing of elastic fibers in the cusp. This translates to intimal thickening and tortuosity (Fig. 3.9).210
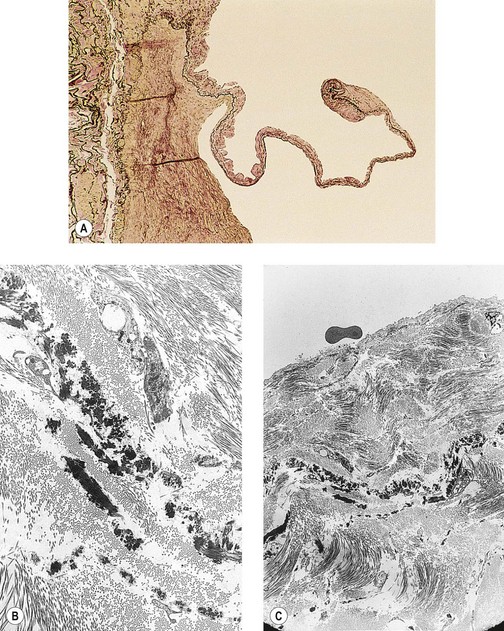
Common mechanisms for the destruction of these valves are DVT and thrombophlebitis. DVT is estimated to precede the development of varicose veins in as many as 25% of patients.211 Incompetence of the veins may also occur because of destruction of the valves by the inflammatory process of thrombophlebitis.212–214 In addition to spontaneous DVT, thrombophlebitis may occur as a result of chemical or mechanical trauma or inflammatory bowel disease. Thrombophlebitis may also occur postsurgically in association with various malignancies or as a result of hormonal elevations associated with birth control pills, the postpartum period, or even smoking.214
Finally, chronic venous dilation may in itself cause valvular fibrosis. Venous dilation increases tension on the cusps of the valves, which causes them to project into the lumen of the vein as rigid flanges (Fig. 3.10). The resultant turbulence of blood flow is thought to cause sclerosis and contraction of the valve as well as its eventual disappearance. This has been noted on histologic examination of varicose veins removed at surgery or postmortem operations.215
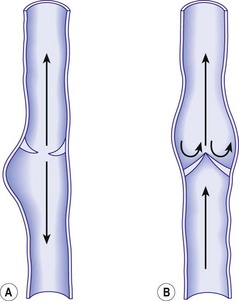
Secondary valvular incompetence
Secondary valvular incompetence is a common cause of varicose veins. In this situation the valves are normal but become incompetent because of dilation of the vein wall. Secondary valvular incompetence may occur as a result of destruction of the valvular system after DVT or because of the expansion in the diameter of the vein. This latter process may occur through an increase in venous volume, obstruction in venous return, or a hormonally induced increase in venous distensibility.216 Thrombotic destruction of venous valves usually begins in the venous sinuses, such as in the soleal sinuses. Here the thrombus may spread to the posterior tibial vein and subsequently into the ankle communicating veins.54 Organization and recanalization of the thrombus destroys the valves.54 An even more dangerous event occurs when the thrombus spreads proximally as a precursor to the development of pulmonary embolism.
Effects of pregnancy
Pregnancy is typically associated with secondary valvular incompetence. Many epidemiologic studies have found a significantly increased incidence of varicose veins in women who have been pregnant.46,115–117,131 However, some epidemiologic studies have failed to confirm this association when the effect of age is controlled.44,111 An additional study confirmed that the GSV diameter increases during the first pregnancy but does not increase further in subsequent pregnancies.217 Varices are often first noted during pregnancy and are exceedingly rare before puberty.218 Indeed, population studies have found that only 12% of women with varicose veins have never been pregnant.219 It has been suggested that, in addition to hormonal effects (discussed later), increased total blood flow in the iliac veins from the uterine and ovarian veins may explain the occurrence of varicose veins in early pregnancy.220 Pregnancy is accompanied by an increase in plasma volume to 149% of normal shortly before parturition.221 Blood flow through the uterine veins increases 4- to 16-fold in the first 2 months of pregnancy and doubles again during the 3rd month.222 Although uterine obstruction to venous flow and an increase in iliac blood volume and flow are measurable physiologic factors in pregnancy, it is obvious that factors other than venous congestion are important in the development of varicose veins.
Serial duplex scanning and air plethysmography determinations have revealed that women with no known venous disease can experience significant increases in common femoral vein and SFJ diameters without developing venous reflux. This is also true of women in pregnancy who have demonstrated prepregnancy venous obstruction.223
Femoral vein obstruction by the gravid uterus may lead to secondary valvular incompetence through an increase in proximal venous pressures. The obstructive effects of the uterus do not develop during pregnancy until the second and third trimesters.224 A sequential study of 50 gravid women through the first, second, and third trimesters using Doppler ultrasound demonstrated that femoral venous flow was obstructed in 72% of erect patients in the second trimester and in 86% of patients in the third trimester.225 An additional study of femoral blood flow in 61 pregnant patients demonstrated that the most significant decrease in femoral blood flow occurred when the head of the fetus became engaged during the latter part of the third trimester.226 However, a study of venous capacitance and outflow in pregnant women at term, 1 week, 6 weeks and 3 months after delivery demonstrated a decrease that persisted until 3 months after delivery.227 Thus, other factors affect venous function in pregnancy.
The effect of the gravid uterus on the impedance of femoral blood flow may be influenced by hereditary factors. Although some investigators have not found a consistent relationship between the presence or absence of varicose veins and obstruction to venous flow,225 others have measured venous pressure in the popliteal vein in pregnant patients in the third trimester and found a marked increase in venous pressure only in those women with varicose veins.228 Therefore both an increase in blood volume and an obstruction of venous return are responsible for the development of varicose veins in pregnancy. However, these two factors do not account for the development of smaller telangiectasias and venectasias; nor do these factors account for the development of varicose veins in the first trimester.
In pregnancy, hormonal factors are primarily responsible for venous dilation. As many as 70% to 80% of patients develop varicose veins during the first trimester, when the uterus is only slightly enlarged (Fig. 3.11). In the second trimester, 20% to 25% of patients develop varicose veins, and 1% to 5% of patients develop them in the third trimester.229–231
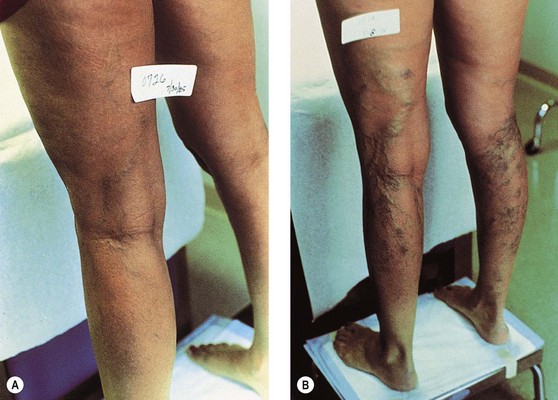
Varicose veins of the legs are first apparent as early as 6 weeks into gestation, a time when the uterus is not yet large enough to significantly impede venous return from the leg veins. In contrast, vulvar varices usually develop in the third trimester but may appear late in the first trimester.220,232 Furthermore, Mullane231 notes that symptoms of varicose veins can be the first sign of pregnancy and can occur even before the first missed menstrual period. This confirms the observations of many multiparous women and argues for a profound influence of progesterone on venous dilation and valvular insufficiency. Siegler233 states that 40% of all pregnant patients are affected and maintains that all women will develop varicosities if they have a sufficient number of pregnancies. Berg234 estimates that 30% of primigravidas and 60% of multigravidas suffer varicose changes in the veins during pregnancy. In support of this theory is Mullane’s patient, who developed varicose veins for the first time in her eleventh pregnancy.231 However, an evaluation of venous refilling times in primiparae and multiparae patients with varicose veins demonstrated no difference between the two groups.235,236 Therefore, the first pregnancy most likely represents the most important injurious factor, with each subsequent pregnancy producing minor deterioration of venous function.217,237
Venous distensibility has been found to increase in both forearm and calf veins with the progression of pregnancy, particularly after the 13 week.238 The increase in distensibility was greater in the calf than in the forearm and returned to normal by the eighth postpartum week.238
Incompetence of the saphenous veins may occur because of excessive dilation of the vein when there is sufficient separation of the valve cusps.213 Interestingly, retrospective studies have shown that 40% to 78% of patients note the development of varicosities during the second pregnancy rather than during the first.13,230,231 Rose and Ahmed13 postulate that veins that become subclinically varicose after the first pregnancy become clinically obvious after the second. As previously mentioned, this impression has been confirmed with photoplethysmography evaluation of venous refilling times.235
The pregnant state is associated with elevations of multiple hormones. Estrogen produces a relaxation of smooth muscle and softening of collagen fibers in general, which may explain the increased distensibility of veins.239,240 Increased distensibility of vein walls has been reported to occur as a result of estrogen therapy.241,242 McCausland230 believes that the progesterone level and, more importantly, the estrogen/progesterone ratio are primarily responsible for increased venous distensibility. Supporting this theory is the marked venous distensibility demonstrated in women who are given a synthetic progesterone.243 Furthermore, a study in 1946 reported that the administration of an estrogenic substance, Diovocylin (CIBA), to 27 pregnant patients with either varicose or telangiectatic veins, in weekly doses throughout pregnancy, actually produced an amelioration of the smaller and larger types of vein and a reduction of peripheral edema and subjective complaints in the majority of patients.244 This was confirmed in a subsequent study of 34 patients treated with 0.05 mg of oral ethinyl estradiol two to four times daily.245 Presumably, administration of this substance altered the progesterone/estrogen ratio. (No mention was made of feminization of male babies on follow-up examination.) An additional study has demonstrated a complete relief of symptoms in 13 of 15 patients with ‘angiectids’ (small, intradermal, raised, bluish mats of blood vessels) in pregnancy with diethylstilbestrol (E.R. Squibb) in doses ranging from 50 to 150 mg daily.246 In this regard, the degree of pain and disability caused by the angiectid was frequently related to the level of subnormal estrogen. Symptomless angiectids were correlated with low normal amounts of estrogen and progesterone. Therefore the hormonal factor responsible for the development or exacerbation of varicosities in pregnancy may very well be related to the estrogen–progesterone balance. Hormonal influences may also explain why varicose veins that develop in pregnancy resolve postpartum (Fig. 3.12).
One additional hypothesis to explain the development of varicose veins in pregnancy is the development of AVAs. Venous volume with correction for capillary filtration was found to be larger in pregnant women with varices than in pregnant women without varices or in nonpregnant women. This may indicate the presence of arteriovenous malformation.247
In pregnancy, two factors operate to dilate leg veins: increased venous distensibility from hormonal stimulation and increased venous pressure from obstruction by the gravid uterus and an increased blood volume. Sumner248 estimated that 80% to 90% of varicose veins tend to regress during the puerperium. He advises physicians to wait 6 to 12 weeks after delivery before performing surgery or sclerotherapy, to allow time for the varices to regress. This not only enhances treatment efficacy but also allows the physician to make a more accurate assessment of the true extent and severity of the disease.
The hormonal influence on the venous system extends beyond pregnancy. Studies have demonstrated that the increase in venous distensibility is different for different oral contraceptive agents. Oral contraceptives with a high progestogen component appear to increase venous capacitance and may induce venous stasis, whereas coagulability is partially enhanced by estrogen-dominant contraceptives.249 The effect of systemic estrogen and progesterone can be demonstrated with photoplethysmography, venous Doppler tensiometry and biomicroscopy. With these evaluations, women taking a low-dose oral contraceptive agent (0.020 mg ethinyl estradiol plus 0.150 mg desogestrel) undergo observable microcirculatory changes without clinical evidence of varicose or telangiectatic vein development. When women take a higher estrogen oral contraceptive (0.030 mg ethinyl estradiol plus 0.075 mg gestodene), significant capillaroscopic changes and a significant impairment of photoplethysmography occur, associated with clinical signs and symptoms such as orthostatic edema, itching, heaviness and slight pain in the lower limbs.250 A retrospective evaluation of 2295 patients who took various concentrations and types of oral contraceptives determined the existence of a significant relationship between the intensity of symptoms and functional symptomatology and the dosage of estrogen and progesterone.251 However, epidemiologic evaluations concerning the use of oral contraceptives demonstrate a trend but do not prove a statistically significant risk for their use in the development of varicose veins.115,252 In addition, alterations in leg vein distensibility were not found in the first half of pregnancy or during oral contraceptive therapy with estrogen-dominant ethyl adrianol (10 mg) by means of ascending phlebography.253
Finally, it has been noted by Gallagher254 in his practice of 20,000 patients that the incidence of minor asymptomatic varices decreases after menopause. This is correlated with a fall in estradiol production and plasma concentration. Therefore, neither estrogen nor progesterone seems to be an independent factor influencing venous capacitance: the proportional concentrations of either one may be more important.
Menstrual Cycle Effects
A change in venous distensibility occurs during the menstrual cycle,243,249 being higher during the luteal phase than during the follicular phase.249 This increase in distensibility correlates most closely with progesterone levels.243 However, studies on vein distensibility do not distinguish between relaxation of smooth muscle and alteration in the viscoelastic properties of the venous wall. Also, these studies do not exclude the possibility that the increased distensibility may result from the reduction in vasoconstrictor tone. Therefore it is difficult to make recommendations regarding the timing of sclerotherapy within the menstrual cycle or the necessity to discontinue oral contraceptives during sclerotherapy.
Recent studies measuring progesterone and estrogen receptors in varicose GSV from men and women demonstrate an increase in both estrogen and progesterone receptor-positive cells only in women with varicose veins. There was no difference between men with and without varicose veins and women without varicose veins.255,256 These gender differences may be hormone related or related to the presence of younger premenopausal females in the varicose vein patient group. Thus, women may be innately predisposed to developing varicose veins in addition to being susceptible to the systemic effects of these hormones.
Of more clinical concern is postpartum thrombophlebitis and its attendant dangers of pulmonary embolism and eventual development of the postphlebitic syndrome.257 The reported incidence of thrombophlebitis in pregnancy ranges from 0.35%, with a 0.085% incidence antepartum and 0.27% postpartum,258 up to 3.6% when mild cases are included in statistical analysis.259 Multiple factors are responsible for the increased incidence of thrombophlebitis in this group of patients. In late pregnancy, blood volume is increased, the uterus impedes venous return and elevated hormone levels produce an increased distensibility of veins with resulting valvular incompetence. In addition, multiple factors are present at childbirth, including increased clotting factors and a hormonal environment conducive to clotting, that appear to predispose this physiologic milieu to the development of thrombophlebitis.260,261 Therefore, graduated elastic support hosiery should be worn before, during and immediately after delivery.257
Constitutive Elements and Progression of Varicose Veins
Whatever the mechanism or the origin of the disease, fundamental lesions remain the same. All or several can be observed in a patient, and the description of the case uses them as bricks to build up the pathophysiologic model with which the therapeutic decision will be taken (Tables 3.1 and 3.2).
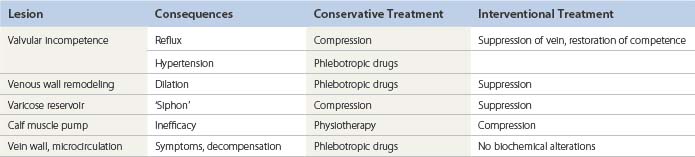
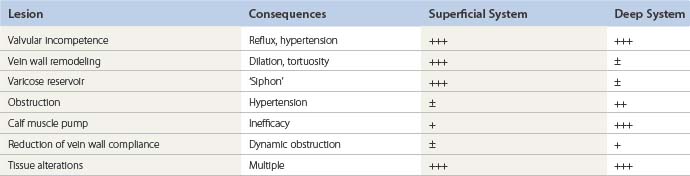
Although each individual lesion has its proper treatment, the relative importance of its proximal position, distal (ascending theory) versus proximal (descending theory), for the development of valvular incompetence can lead to different treatment approaches. Contrary to the classic descending approach (crossectomy), it has been shown that in patients with a large varicose reservoir and a reflux of the GSV, ablation of the varicose reservoir by phlebectomy can correct the saphenous reflux in more than 66% of cases and provide satisfactory midterm (4 years) clinical results.262 These findings are consistent with the evaluation of systematic color-flow duplex scan images showing the pattern of varicose veins in a cross-sectional study of untreated patients, which showed a predominance of early lesions in tributaries rather than in the saphenous trunk.263 It seems that the progression of varicose veins can be ascending, descending or a combination of both directions, either in different subsets of the disease or even in the same patient, consecutively or simultaneously. As this natural history issue of varicose veins is likely to strongly influence the long-term results of the treatment, follow-up studies in treated patients, and also in untreated subjects from the general population, are clearly needed.
Heredity
Although development of varicose veins can usually be ascribed to one of the previously mentioned pathologic states, postmortem examination may not disclose the apparent source of the high-pressure leak from the deep to the superficial system.264 Therefore, other inherent factors such as vein wall weakness, increased primary valvular dysfunction or agenesis, and other genetic factors, may enhance the development of varicose veins.
In an extensive study in France, 134 families were examined. Of these, 67 were patients with varicose veins plus their parents, and 67 were controls without varicose veins plus their parents. A total of 402 subjects were examined. The results demonstrated the prominent role of heredity in the development of varicose veins (P <0.001). For the children, the risk of developing varicose veins was 90% when both parents were afflicted. When only one parent was affected, the risk of developing varicose veins was 25% for males and 62% for females. The overall risk of varicose vein development was 20% when neither parent was affected by varicosities.265
A familial tendency toward the development of varicose veins has been described in many population groups.114–116,132,135,266–270 This may also be demonstrated by the development over time of varicose veins bilaterally when patients with unilateral varicose and telangiectatic veins are followed for 10 years.269 A limited study of 50 patients with varicose veins in Great Britain disclosed a simple dominant type of inheritance.271 Only 28% of patients had no family history of varicose veins. In Scandinavia, questionnaires completed by 124 women with varicose veins disclosed a 72% prevalence of varicose veins of an autosomal type in the women’s siblings.267 Of these cases, 28% were of a recessive pattern. Troisier and Le Bayon272 examined 154 families with 514 descendants. They found that if both parents had varicose veins, 85% of children had evidence of varicose veins, whereas 27% of the children were affected if neither parent had varicose veins and 41% of the children were affected if one parent had varicosities. These authors concluded that the inheritance of varicose disease is recessive. However, some studies have not found a significant familial tendency.118,273
A first twin study found that 75% of 12 monozygotic pairs were concordant with regard to varicose veins versus 52% of 25 dizygotic same-sexed pairs.274 But the definite proof regarding the influence of heredity came in 2005 from a large study of 2060 unselected pairs of female twins aged from 18 to 80 years,275 which showed that the concordance rate for varicose veins phenotype was significantly (P < 0.001) higher for monozygotic (67%) than for dizygotic twins (45%). In the same study, a significant linkage of varicose veins to a marker of the FOXC2 region of the chromosome 16 was found, suggesting FOXC2, a gene already known for its role in the embryogenesis of the lymphatic system, is implicated in the development of varicose veins. Although this study provides a clear demonstration of the role of genetic factors in the pathogenesis of varicose veins, it also demonstrates obviously that environmental factors play an important role as well, since the concordance rate in monozygous twins is far from 100%. Other studies have found more of a multifactorial inheritance. In a detailed study from Sweden of 250 probands of patients with varicose veins requiring treatment, the overall frequency of varicose veins in female relatives was 43%, compared with 19% in male relatives.276
The absence of venous valves in the external iliac and femoral veins has been shown to be a marker of varicose veins in a limited radiographic study of 12 male volunteers, some with and some without varicose veins,203 and in a venous Doppler study of 54 patients with varicose veins.277 In addition, a simple dominant mode of inheritance has been reported in 14 patients with congenital partial or total absence of venous valves of the leg.184,278 Thus, this genetic predisposition may be the result of multiple factors and the subsequent development of varicose veins may depend on one or more occupational or hormonal factors.
Congenitally weak or nonfunctioning venous valves may be an initiating factor in the altered venous hemodynamics that lead to the formation of varicose veins.279 The argument against this theory is that valvular cusps consist of a fibrous tissue core covered by endothelium. This structure has been demonstrated to be extremely strong in experimental models.280 In fact, it has been estimated experimentally that valves will not rupture at the physiologic pressures to which a valve might be subjected during life. Therefore it is more likely that alterations in the vein wall, and not valve strength, are responsible for the development of incompetence.
Rose and Ahmed13 postulated that an inherited alteration in vein wall collagen and/or elastin, or an increase in vein wall collagen deposition with separation of smooth muscle cells (as previously described), is a major etiologic precursor to the development of varicose veins. They reasoned that increased venous pressure should lead to hypertrophy of the vein wall as demonstrated in arterialized venous bypass coronary grafts280–284 and not the dilation associated with varicose veins. Accordingly, dilation of varicose veins, at times only in certain areas of the vein, must be caused by a vein wall defect – not merely by the presence of high venous pressure. A generalized increase in venous distensibility was found in superficial forearm and hand veins in patients with a saphenous varicosity as compared with patients without varicosities.285,286 Abnormal distensibility curves were found to be similar regardless of the age and sex of the patient. This may be related to a reported decrease in collagen content in the saphenous veins of patients with varicosities, which occurs even in vein segments that are not varicose.22,32 The decrease in venous distensibility may also be related to a constitutional decrease in venous α-adrenergic receptor responsiveness in patients with varicosities. Patients with varicose veins require significantly higher doses of norepinephrine for vasoconstriction than do control subjects. This finding applies to both varicose and normal veins in the same individual.287 The neural regulatory network in the saphenous vein also consists of acetylcholinergic and peptidergic neurons, as well as both circulating and endothelium-derived vasoactive substances.288 Thus, neural and hormonal factors are important regulators of venous distensibility.
The decreased collagen content in varicose vein walls has been related physically to its viscoelastic properties, with varicose veins breaking at lower pressures than normal veins.289 This may occur in certain patients from a genetic defect affecting the biosynthesis of certain collagen types. One example is type 4 Ehlers-Danlos syndrome (vascular type), in which patients have a deficiency in collagen 3, normally present in the skin, arteries and gut. These patients also frequently have varicose veins.290 In addition, patients with previous hernia surgery also have an increased incidence of varicose veins suggesting that ligamentous laxity may be a risk factor.135 However, this theory does not explain why correction of proximal valvular dysfunction with a tourniquet can correct abnormal venous pressures distally if, indeed, it is the vein wall that is abnormally distensible.291
Generalized dystrophic changes in the vein wall were confirmed histologically through biopsy of normal dorsal foot veins in 97.3% of patients with varicose veins.292 The generalization of venous wall changes from superficial to deep veins was also demonstrated.22 The authors speculate that this generalized trend may allow improvement of sclerotherapy techniques by choosing a stronger sclerosing agent when a peripheral venous biopsy demonstrates severe dystrophy (see Chapter 9).
Another interesting relationship is the recently described association of varicose veins with the ABO blood group system. Numerous studies have demonstrated a relationship between blood groups of the ABO system and DVT of the lower limbs.293–296 These studies indicate an increased incidence of DVT in patients with blood type O, particularly when associated with pregnancy or the use of oral contraceptive agents.296 However, one study found an increased incidence of thromboembolism in people with blood group A.297 A study of 569 French men and women showed the risk of varicose veins in patients with type A blood to be double that of patients with all other blood groups.298 Varicose veins were defined as the presence, in the standing position, of a permanent dilation of at least one leg vein with a diameter of 3 mm or more with reflux. The risk of varicose veins persisted after adjustment for age, sex, paternal or maternal history and a personal history of DVT. No association was found between Rhesus factor and varicose veins. Therefore, a patient’s blood group may be taken into account when assessing the need for prophylactic treatment of varicose veins or assessing the risk of postoperative venous thrombosis.
Recent studies on varicose and normal veins using gene expression profiling based on cDNA microarray analysis suggest that pathways associated with fibrosis and wound healing may be altered in varicose veins.299 Whether the upregulated varicose vein genes are a sequel to the changes in the varicose vein wall rather than a primary contributing factor to varicose pathogenesis awaits additional study.
Aging
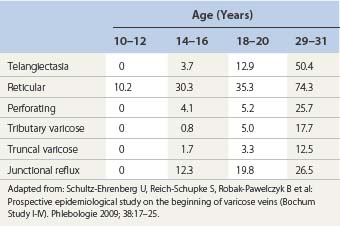
The incidence of varicose veins increases with age (Table 3.3);300 therefore, vein wall damage should also be more pronounced in the veins of older patients. Superficial venous reflux also sed a marked increase with age.62,301 An autopsy study of the popliteal vein in 127 persons demonstrated diffuse changes, with an increase in connective tissue in the media, which became most pronounced in the fifth decade and are progressed thereafter. This is associated with the loss of muscle cells in the media.302 The finding correlated with an abnormality in the physical property of axial tension testing in 93 specimens of saphenous veins from 22 patients harvested during coronary bypass surgery.303 However, one study of 31 normal veins and 41 varicose veins in patients and autopsy samples ranging in age from 25 to 92 years failed to disclose an age-related difference.304 The latter study concluded that varicose veins were a predetermined disease unrelated to aging effects.
1 Prerovsky I. Disease of the veins, internal communication. MHO-PA: World Health Organization; 1964.
2 Thulesius O, Ugaily-Thulesius L, Gjores JE, Neglen P. The varicose saphenous vein: functional and ultrastructural studies with special reference to smooth muscle. Phlebology. 1988;3:89.
3 Thulesius O, Said S, Shuhaiber H, et al. Endothelial mediated enhancement of noradrenaline induced vasoconstriction in normal and varicose veins. Clin Physiol. 1991;11:153.
4 Lowell RC, Gloviczki P, Miller VM. In vitro evaluation of endothelial and smooth muscle function of primary varicose veins. J Vasc Surg. 1992;16:679.
5 Svejcar J, Prerovsky I, Linhart J, Kruml J. Biochemical differences in the composition of primary varicose veins. Am Heart J. 1964;67:572.
6 Barber DA, Wang X, Gloviczki P, Miller VM. Characterization of endothelin receptors in human varicose veins. J Vasc Surg. 1997;26:61.
7 Jurukova Z, Milenkov C. Ultrastructural evidence for collagen degradation in the walls of varicose veins. Exp Mol Path. 1982;37:37.
8 Xiao Y, Huang Z, Lin Y, Wang S. In vitro differences between smooth muscle cells derived from varicose veins and normal veins. J Vasc Surg. 2009;50:1149.
9 Garcia-Rospide V, Penafiel-Marfil R, Moreno-Padilla F, et al. Enzymatic and isoenzymatic study of varicose veins. Phlebology. 1991;6:187.
10 Azevedo I, Albino Teixeira A, Osswald W. Changes induced by ageing and denervation in the canine saphenous vein: a comparison with the human varicose vein. In: Vanhoutte PM, editor. Return circulation and norepinephrine: an update. Paris: John Libbey Eurotext, 1991.
11 Lees TA, Mantle D, Lambert D. Analysis of the smooth muscle content of the normal and varicose vein wall via analytical electrophoresis. In: Raymond-Martimbeau P, Prescott R, Zummo M, editors. Phlébologie ’92. Paris: John Libbey Eurotext, 1992.
12 Bouissou H, Julian M, Piraggi M, Louge L. Vein morphology. Phlebology. 1988;3(Suppl 1):1.
13 Rose SS, Ahmed A. Some thoughts on the aetiology of varicose veins. Cardiovasc Surg. 1986;27:534.
14 Acsady G, Lengyel I. Modifications between histomorphological and pathobiochemical changes leading to primary varicosis. In: Davy A, Stemmer R, editors. Phlébologie ’89. Montrouge, France: John Libbey Eurotext, 1989.
15 Andreotti L, Cammelli D. Connective tissue in varicose veins. Angiology. 1979;30:798.
16 Zwillenberg LO, Laszt L, Willenberg H. Die feinstruktur der venenwand bei varikose. Angiologica. 1971;8:318.
17 Gandhi RH, Irizarry E, Nackman GB, et al. Analysis of the connective tissue matrix and proteolytic activity of primary varicose veins. J Vasc Surg. 1993;18:814.
18 Clarke H, Smith SR, Vasdekis SN, et al. Role of venous elasticity in the development of varicose veins. Br J Surg. 1989;76:577.
19 Merlen JF, Curri SB, Saout J, et al. Le devenir histologique d’une veine sclerosee. Phlebologie. 1978;31:17.
20 Partsch H, Mostbeck A. Constriction of varicose veins and improvement of venous pumping by dihydroergotamine. Vasa. 1985;14:74.
21 Chello M, Mastroroberto P, Zofrea S, Marchese AR. Analysis of collagen and elastin content in primary varicose veins: letter to the editor. J Vasc Surg. 1994;24:490.
22 Fuchs U, Petter O. Phlebosclerosis of the stem veins in varicosis. Phlebo Proktol. 1990;19:120.
23 Zsoter T, Moore S, Keon W. Venous distensibility in patients with varicosities: in vitro studies. J Appl Physiol. 1967;22:505.
24 Leu HJ, Vogt M, Pfrunder H. Morphological alterations of nonvaricose and varicose veins. Basic Res Cardiol. 1979;74:435.
25 Rizzi A, Quaglio D, Vasquez G, et al. Effects of vasoactive agents in healthy and diseased human saphenous veins. J Vasc Surg. 1998;28:855.
26 Deby C, Hariton C, Pincemail J, Coget J. Decreased tocopherol concentration of varicose veins associated with a decrease in antilipoperoxidant activity without similar changes in plasma. Phlebology. 1989;4:113.
27 Kontos HA, Hess ML. Oxygen radicals and vascular damage. Adv Exp Med Biol. 1983;161:365.
28 Felix W. Pharmakologische Beeinflussbarkeit kapazitativer Venen. In: Schneider KW, editor. Die venose Insuffizienz, Wittstock. East Germany: Baden-Baden, 1972.
29 Biagi G, Lapilli A, Zendron R, et al. Prostanoid production in varicose veins: evidence for decreased prostacyclin with increased thromboxane A2 and prostaglandin E2 formation. Angiology. 1988;39:1036.
30 Kreysel HW, Nissen HP, Enghofer E. A possible role of lysosomal enzymes in the pathogenesis of varicosis and the reduction in their serum activity by venostasis. Vasa. 1983;12:377.
31 Haardt B. Histological comparison of the enzyme profiles of healthy veins and varicose veins. Phlebology. 1986;39:921.
32 Svejcar J, Prerovsky I, Linhart J, et al. Content of collagen, elastin, and hexosamine in primary varicose veins. Clin Sci. 1963;24:325.
33 Niebes P. Biochemical studies of varicosis. Monogr Stand Cardioangiological Methods. 1977;4:233.
34 Branco D, Osswald W. The influence of Ruscus extract on the uptake and metabolism of noradrenaline in the normal and varicose vein. Phlebology. 1988;3(Suppl 1):33.
35 Woodside KJ, Hu M, Burke A, et al. Morphologic characteristics of varicose veins: possible role of metalloproteinases. J Vasc Surg. 2003;38:162.
36 Raffetto JD, Ross RL, Khailil RA. Matrix metalloproteinase 2-induced venous dilation via hyperpolarization and activation of K+ channels: relevance to varicose vein formation. J Vasc Surg. 2007;45:373.
37 Raffetto JD, Qiao X, Koledova VV, Khalil RA. Prolonged increases in vein wall tension increase matrix metalloproteinase and decrease constriction in rat vena cava: Potential implications in varicose veins. J Vasc Surg. 2008;48:447.
38 Mellander S. Operative studies on the adrenergic neuro-hormonal control of resistance and capacitance blood vessels in the cat. Acta Physiol Scand. 1960;50:5.
39 Waterfield RL. The effect of posture on the volume of the leg. J Physiol. 1931;72:121.
40 Ludbrook J, Loughlin J. Regulation of volume in postarteriolar vessels of the lower limb. Am Heart J. 1964;67:493.
41 Rushmer RF. Effects of posture. In Rushmer RF, editor: Cardiovascular dynamics, 4th ed, Philadelphia: WB Saunders, 1976.
42 Samueloff SL, Browse NL, Shepherd JT. Response of capacity vessels in human limbs to head-up tilt and suction on lower body. J Appl Physiol. 1966;21:47.
43 Reinis Z, Pokorny J, Bazika V, et al. Incidence of venous diseases of veins of the lower extremities in the Czech population. Vnitr Lek. 1983;29:105.
44 Abramson JH, Hopp C, Epstein LM. The epidemiology of varicose veins: a survey in western Jerusalem. J Epidemiol Community Health. 1981;35:213.
45 Ducimetiere P, Richard JL, Pequignot G, Warnet JM. Varicose veins: a risk factor for atherosclerotic disease in middle-aged men? Int J Epidemiol. 1981;10:329.
46 Brand FN, Dannenberg AL, Abbott RD, Kannel WB. The epidemiology of varicose veins: the Framingham study. Am J Prev Med. 1988;4:96.
47 Pollack AA, Taylor BE, Myers TT, Wood EH. The effect of exercise and body position on the venous pressure at the ankle in patients having venous valvular defects. J Clin Invest. 1949;28:559.
48 Pollack AA, Wood EH. Venous pressure in the saphenous vein at the ankle in man during exercise and changes in posture. J Appl Physiol. 1949;1:649.
49 Barcroft H, Dornhorst AC. The blood flow through the human calf during rhythmic exercise. J Physiol (Lond). 1949;107:402.
50 Wells HS, Youmans JB, Miller DG. Tissue pressure, intracutaneous, subcutaneous, and intramuscular, as related to venous pressure, capillary filtration, and other factors. J Clin Invest. 1938;17:489.
51 Ludbrook J. The musculo-venous pumps of the human lower limb. Am Heart J. 1966;71:635.
52 Hojensgard IC, Sturup H. Static and dynamic pressures in superficial and deep veins of the lower extremity in man. Acta Physiol Scand. 1953;27:49.
53 Arnoldi CC. Venous pressure in the leg of healthy human subjects at rest and during muscular exercise in the nearly erect position. Acta Chir Scand. 1965;130:570.
54 Dodd H, Cockett FB, editors. The pathology and surgery of the veins of the lower limb, 2nd ed, Edinburgh: Churchill Livingstone, 1976.
55 Almen T, Nylander G. Serial phlebography of the normal lower leg during muscular contraction and relaxation. Acta Radiol. 1962;57:264.
56 Dicker JWJr. A study of venous return from the leg during exercise. Phlebology. 1989;4:185.
57 Fegan WG, Milliken JC, Fitzgerald DE. Abdominal venous pump. Arch Surg. 1966;92:44.
58 Lange L, Echt M. Comparative studies on drugs which increase venous tone using nor-adrenaline, ethyl-andrianol, dihydroergotamine, and horsechestnut extract. Fortschr Med. 1972;90:1161.
59 Mellander S, Nordenfelt L. Comparative effects of dihydroergotamine and noradrenaline on resistance, exchange, and capacitance functions in the peripheral circulation. Clin Sci. 1970;39:183.
60 Bjordal R. Simultaneous pressure and flow recordings in varicose veins of the lower extremities. Acta Chir Scand. 1970;136:309.
61 Strandness DEJr, Thiele BL. Selected topics in venous disorders. New York: Futura; 1981.
62 Evans CJ, Allan PL, Lee AJ, et al. Prevalence of venous reflux in the general population on duplex scanning: the Edinburgh vein study. J Vasc Surg. 1998;28:767.
63 Labropoulos N, Delis KT, Nicolaides AN. Venous reflux in symptom-free vascular surgeons. J Vasc Surg. 1995;22:150.
64 Guex JJ, Hiltbrand B, Bayon JM, et al. Anatomical patterns in varicose vein disease. Phlebology. 1995;10:94.
65 Labropoulos N, Tiongson J, Pryor L, et al. Nonsaphenous superficial vein reflux. J Vasc Surg. 2001;34:872.
66 Parra JR, Cambria RA, Hower CD, et al. Tissue inhibitor of metalloproteinase-1 is increased in the saphenofemoral junction of patients with varices in the leg. J Vasc Surg. 1998;28:665.
67 Ono T, Bergan JJ, Schmid-Schönbein GW, Takase S. Monocyte infiltration into venous valves. J Vasc Surg. 1998;27:158.
68 McPheeters HO, Rice CO. Varicose veins: the circulation and direction of venous flow. Surg Gynecol Obstet. 1929;49:29.
69 Farber EM, Bates EE. Pathologic physiology of stasis dermatitis. Arch Dermatol. 1954;70:653.
70 Negus D, Friedgood A. The effective management of venous ulceration. Br J Surg. 1983;70:623.
71 Cockett FB, Jones BE. The ankle blow-out syndrome: a new approach to the varicose ulcer problem. Lancet. 1953;261:17.
72 Landis EM. Factors controlling the movement of fluid through the human capillary wall. Yale J Biol Med. 1933;5:201.
73 Burnand KG, Clemenson G, Whimster I, et al. The effect of sustained venous hypertension on the skin capillaries of the canine hind limb. Br J Surg. 1982;69:41.
74 Ryan TJ, Wilkinson DS. Diseases of the veins: venous ulcers. In Rook A, Wilkinson DS, Ebling FJG, editors: Textbook of dermatology, 3rd ed, London: Blackwell Scientific, 1979.
75 Ryan TJ. Diseases of the skin: management of varicose ulcers and eczema. Br Med J. 1974;1:192.
76 [cited in]. Whimster I, Dodd H, Cockett FB, editors. The pathology and surgery of the veins of the lower limb, 2nd ed, Edinburgh: Churchill Livingstone, 1976.
77 Peters K, Sindrup JH, Petersen LJ, et al. Lower leg subcutaneous blood flow during walking and passive dependency in chronic venous insufficiency. Br J Dermatol. 1991;124:177.
78 Falanga V, Moosa HH, Nemeth AJ, et al. Dermal pericapillary fibrin in venous disease and venous ulceration. Arch Dermatol. 1987;123:620.
79 Lotti T, Fabbri P, Panconesi E. The pathogenesis of venous ulcers. J Am Acad Dermatol. 1987;16:877.
80 Ludbrook J, Loughlin J. Regulation of volume in postarteriolar vessels of the lower limb. Am Heart J. 1964;67:493.
81 Shepherd JT, Vanhoutte PM. Role of the venous system in circulatory control. Mayo Clin Proc. 1978;53:247.
82 Boccalon H, Ginestet MC. Influence of temperature variations on venous return: clinical observations. Phlebology. 1988;3(Suppl 1):47.
83 Tanyol H, Menduke H. Alcohol as a possible etiologic agent in varicose veins: a study of drinking habits in 136 patients with varicose veins and in 70 control patients. Angiology. 1961;12:382.
84 Andersson J, Thurin A, Thulesius O. Valvular function of peripheral veins after hyperemic dilation. J Vasc Surg. 1996;23:611.
85 Sakurai T, Matsushita M, Nishikimi N, Nimura Y. Hemodynamic assessment of femoropopliteal venous reflux in patients with primary varicose veins. J Vasc Surg. 1997;26:260.
86 May R, Thurner J. Ein Gefäβsporn in der Vena iliaca communis sinistra als Urasche der überwiegend linksseitigen Beckeneven-thrombosen. Z Kreislaufforsch. 1956;45:912.
87 Murray JD, Brennan FJ, Hall LD, et al. Left iliac vein occlusion: its clinical spectrum. Ann Vasc Surg. 2000;14:510.
88 Nazarian GK, Bjarnason H, Dietz CAJr, et al. Iliofemoral venous stenoses: effectiveness of treatment with metallic endovascular stents. Radiology. 1996;200:193.
89 Berger A, Jaffe JW, York TN. Iliac compression syndrome treated with stent placement. J Vasc Surg. 1995;21:510.
90 Kibbe MR, Ujiki M, Goodwin AL, et al. Iliac vein compression in an asymptomatic patient population. J Vasc Surg. 2004;39:937.
91 Kerr MG, Scott DB, Samuel E. Studies of the inferior vena cava in late pregnancy. Br Med J. 1964;1:532.
92 Cleave TL. On the causation of varicose veins and their prevention and arrest by natural means. Bristol: Wright; 1960.
93 Cambell GC, Cleave TL. Diverticular disease of colon. Br Med J. 1968;3:741.
94 Beaglehole R. Epidemiology of varicose veins. World J Surg. 1986;10:898.
95 Burkitt DP. Varicose veins: facts and fantasy. Arch Surg. 1976;111:1327.
96 Foote RR. Varicose veins. St Louis: CV Mosby; 1949.
97 Pirner F. Der varikose Symptomen-komplex mit seien folgen und nachkrakheiten unter besonerer bereichsichtigung der fehlerquellen. In: Diagnostik und therapie. Stuttgart: Enke; 1957.
98 Dodd H. The cause, prevention, and arrest of varicose veins. Lancet. 1964;2:809.
99 Burkitt DP. Varicose veins, deep vein thrombosis, and haemorrhoids: epidemiology and suggested etiology. Br Med J. 1972;2:556.
100 Stamler J. Epidemiology as an investigative method for the study of human atherosclerosis. J Natl Med Assoc. 1958;50:161.
101 Brodribb AJM, Humphreys DM. Diverticular disease: three studies. Part I: relation to other disorders of fibre intake. Br Med J. 1976;6:424.
102 Richardson JB, Dixon M. Varicose veins in tropical Africa. Lancet. 1977;309:791.
103 Martin A, Odling-Smee A. Pressure changes in varicose veins. Lancet. 1976;307:768.
104 Malhotra SL. An epidemiological study of varicose veins in Indian railroad workers from the south and north of India, with special reference to the causation and prevention of varicose veins. Int J Epidemiol. 1982;1:117.
105 Connor WE, Hoak JG, Warner EA. Massive thrombosis produced by fatty acid infusion. J Clin Invest. 1963;42:860.
106 Connor WE, Poole JCF. The effect of fatty acids on the formation of thrombi. Q J Exp Physiol. 1961;46:1.
107 Poole JCF. Effective diet and lipidemia on coagulation and thrombosis. Fed Proc. 21(4), 1962. pt 2:20
108 Melet JJ. La place de l’alimentation parmi les facteurs de rísque des maladies veineuses. Phlebologie. 1981;34:469.
109 Abramson JH, Gofin J, Hopp C, et al. The epidemiology of inguinal hernia: a survey in western Jerusalem. J Epidemiol Community Health. 1978;32:59.
110 Schilling RSF, Walford J. Varicose veins in women cotton workers: an epidemiological study in England and Egypt. Br Med J. 1969;2:591.
111 Guberan E, Widmer LK, Glaus L, et al. Causative factors of varicose veins: myths and facts. Vasa. 1973;2:115.
112 Pereira GJM, de Fatima GM, Marcolino BD. The effect of wearing jeans on venous circulation during activities. Int Angiol. 2005;24:174.
113 Askar O, Emara A. Varicose veins and occupation. J Egypt Med Assoc. 1970;53:341.
114 Stvrtinova V, Kolesar J, Wimmer G. Prevalence of varicose veins of the lower limbs in the women working at a department store. Int Angiol. 1991;10:2.
115 Reinis Z, Pokorny J, Bazika V, et al. Incidence of venous diseases of the veins of the lower extremities in the Czech population. Vnitr Lek. 1983;29:105.
116 Sadick NS. Predisposing factors of varicose and telangiectatic leg veins. J Dermatol Surg Oncol. 1992;18:883.
117 Maffei FH, Magaldi C, Pinho SZ, et al. Varicose veins and chronic venous insufficiency in Brazil: prevalence among 1755 inhabitants of a country town. Int J Epidemiol. 1986;15:210.
118 Weddell JM. Varicose veins pilot survey, 1966. Br J Prev Soc Med. 1969;23:179.
119 Stanhope JM. Varicose veins in a population of lowland New Guinea. Int J Epidemiol. 1975;4:221.
120 Alexander CJ. Chair-sitting and varicose veins. Lancet. 1972;299:822.
121 Wright HP, Osborn SB. Effect of posture on venous velocity. Br Heart J. 1952;14:325.
122 Symington IS, Stack BH. Pulmonary thromboembolism after travel. Br J Dis Chest. 1977;71:138.
123 Myers TT. Varicose veins. In Allen EV, Barker NW, Hines EA, editors: Peripheral vascular diseases, 3rd ed, Philadelphia: Saunders, 1962.
124 Leipnitz G, Kiesewetter H, Waldhausen P, et al. Prevalence of venous disease in the population: first results from a prospective study carried out in greater Aachen. In: Davy A, Stemmer R, editors. Phlébologie ’89. Montrouge, France: John Libbey Eurotext, 1989.
125 Beaglehold R, Salmond CE, Prior IAM. Varicose veins in New Zealand: prevalence and severity. N Z Med J. 1976;84:396.
126 Fowkes FGR. Prevalence and risk factors for chronic venous insufficiency. Acta Phlebol. 2000;1:69.
127 Iannuzzi A, Panico S, Ciardullo AV, et al. Varicose veins of the lower limbs and venous capacitance in postmenopausal women: relationship with obesity. J Vasc Surg. 2002;36:965.
128 Danielsson G, Eklof B, Grandinetti A, Kistner RL. The influence of obesity on chronic venous disease. Vasc Endovasc Surg. 2002;36:271.
129 Canonico S, Gallo C, Paolisso G, et al. Prevalence of varicose veins in an Italian elderly population. Angiology. 1998;49:129.
130 Widmer LK. Peripheral venous disorders: prevalence and socio-medical importance: observations in 4529 apparently healthy persons, Basle study III, Berne. Switzerland: Hans Huber; 1978.
131 Coughlin LB, Gandy R, Rosser S, de Cossart L. Factors associated with varicose veins in pregnant women. Phlebology. 2002;16:167.
132 Hirai M, Nakayama R. Prevalence and risk factors of varicose veins in Japanese women. Angiolology. 1990;3:228.
133 Ludbrook J. Obesity and varicose veins. Surg Gynecol Obstet. 1964;118:843.
134 Seidell JC, Bakx JC, De Boer E, et al. Fat distribution of overweight persons in relation to morbidity and subjective health. Int J Obes. 1985;9:363.
135 Criqui MH, Denenberg JO, Bergan J. Risk factors for chronic venous disease: The San Diego population study. J Vasc Surg. 2007;46:331.
136 Padburg F, Cerveira JJ, Lal BK, et al. Does severe venous insufficiency have a different etiology in the morbidly obese? Is it venous? J Vasc Surg. 2003;37:79.
137 Stegall HF. Muscle pumping in the dependent leg. Circ Res. 1966;19:180.
138 Floyd WF, Silver PHS. Electromyographic study of patterns of activity of the anterior abdominal muscles in man. J Anat. 1950;84:132.
139 Stewart AM, Webb JW, Hewitt D. Social medicine studies based on civilian medical board records. II. Physical and occupational characteristics of men with varicose conditions. Br J Prev Med. 1955;9:26.
140 Kriessmann A. Klinische pathophysiologie des venosen Ruckstroms. In: Ay RE, Kriessmann A, editors. Periphere Venendruckmessung. Stuttgart: Thieme, 1978.
141 Cotton LT. Varicose veins: gross anatomy and development. Br J Surg. 1961;48:589.
142 Hoshino S, Satakawa H, Iwaya F, et al. External valvuloplasty under preoperative angioscopic control. Phlebologie. 1993;46:521.
143 Van Cleef JF, Desvaux P, Hugentobler JP, et al. Étude endoscopique des reflux valvulaires sapheniens. J Mal Vasc. 1992;17:113.
144 Sales CM, Rosenthal D, Petrillo KA, et al. The valvular apparatus in venous insufficiency: a problem of quantity? Ann Vasc Surg. 1998;12:153.
145 Ono T, Bergan JJ, Schmid-Schönbein GW, Takase S. Monocyte infiltration into venous valves. J Vasc Surg. 1998;27:158.
146 Yamada T, Tomita S, Mori M, et al. Increased mast cell infiltration in varicose veins of the lower limbs: a possible role in the development of varices. Surgery. 1996;119:494.
147 Takase S, Lerond L, Bergan JJ, Schmid-Schönbein GW. The inflammatory reaction during venous hypertension in the rat. Microcirculation. 2000;7:41.
148 Van Bemmelen SP, Hoynck van Papendrecht AA, Hodde KC, Klopper PJ. A study of valve incompetence that developed in an experimental model of venous hypertension. Arch Surg. 1986;121:1048.
149 Takase S, Pascarella L, Bergan JJ, Schmid-Schönbein GW. Hypertension-induced venous valve remodeling. J Vasc Surg. 2004;39:1329.
150 Takase S, Pascarella L, Lerond L, et al. Venous hypertension, inflammation and valve remodeling. Eur J Vasc Endovasc Surg. 2004;28:484.
151 Takase S, Lerond L, Bergan JJ, Schmid-Schönbein GW. The inflammatory reaction during venous hypertension in the rat. Microcirculation. 2000;7:41.
152 Takase S, Lerond L, Bergan JJ, Schmid-Schönbein GW. Enhancement of reperfusion injury by elevation of microvascular pressures. Am J Physiol Heart Circ Physiol. 2002;282:H1387.
153 Pascarella L, Schmid-Schönbein GW, Bergan J. An animal model of venous hypertension: the role of inflammation in venous valve failure. J Vasc Surg. 2005;41:303.
154 Hahn TL, Unthank JL, Lalka SG. Increased hindlimb leukocyte concentration in a chronic rodent model of venous hypertension. J Surg Res. 1999;81:38.
155 Thomas PRS, Nash GB, Dormandy JA. White cell accumulation in the dependent legs of patients with venous hypertension: a possible mechanism for trophic changes in the skin. Br Med J. 1988;296:1693.
156 Botta G, Belcastro M, Mancini S, et al. Both subterminal greater saphenous vein valve and wall are not affected by lymphomonocyte infiltration in primary chronic venous insufficiency: a preliminary report. Acta Phlebol. 2001;2:1.
157 Haddy FJ, Scott JB. Metabolic factors in peripheral circulatory regulation. Fed Proc. 1975;34:2006.
158 Fegan G. Varicose veins: compression sclerotherapy. London: William Heinemann; 1967.
159 Fegan WG. Continuous uninterrupted compression technique of injecting varicose veins. Proc R Soc Med. 1960;53:837.
160 Hobbs JT. The problem of the post-thrombotic syndrome. Postgrad Med J. 1973;Aug Suppl:48.
161 Lofgren EP. Leg ulcers: symptom of an underlying disorder. Postgrad Med. 1984;76:51.
162 Beninson J, Livingood CS. Stasis dermatitis. In: Demis DJ, editor. Clinical dermatology, vol 2. Philadelphia: Harper & Row; 1985.
163 Cockett FB, Thomas ML. The iliac compression syndrome. Br J Surg. 1965;52:816.
164 Kwaan JHM, Jones RN, Connolly JE. Simplified technique for the management of refractory varicose ulcers. Surgery. 1976;80:743.
165 Lim LT, Michuda M, Bergan JJ. Therapy of peripheral vascular ulcers: surgical management. Angiology. 1978;29:654.
166 Quill RD, Fegan WG. Reversibility of femorosaphenous reflux. Br J Surg. 1971;55:389.
167 Thibault P, Bray A, Wlodarczyk J, Lewis W. Cosmetic leg veins: evaluation using duplex venous imaging. J Dermatol Surg Oncol. 1990;16:612.
168 Abu-Own A, Scurr JH, Coleridge Smith PD. Saphenous vein reflux without sapheno-femoral or sapheno-popliteal junction incompetence. Presented at the Sixth Annual Meeting of the American Venous Forum, Maui, 1994.
169 Van Ramhorst B, van Bemmelen P, Hoeneveld H, Eilekboom BC. The development of valvular incompetence after deep vein thrombosis: a follow-up study with duplex scanning. J Vasc Surg. 1994;20:1059.
170 Bjordal RI. Circulation patterns in incompetent perforating veins of the calf in venous dysfunction. In: May R, Partsch H, Staubesand J, editors. Perforating veins. Munich: Urban & Schwarzenberg, 1981.
171 Bergan JJ. Venous insufficiency and perforating veins. Br J Surg. 1998;85:721.
172 Proebstle TM, Weisel G, Paepcke U, et al. Light reflection rheography and clinical course of patients with advanced venous disease before and after endoscopic subfascial division of perforating veins. Dermatol Surg. 1998;24:771.
173 Labropoulos N, Tassiopoulos AK, Kang SS, et al. Prevalence of deep venous reflux in patients with primary superficial vein incompetence. J Vasc Surg. 2000;32:663.
174 Labropoulos N, Tassiopoulos AK, Bhatti AF, Leon L. Development of reflux in the perforator veins in limbs with primary venous disease. J Vasc Surg. 2006;43:558.
175 McMullin GM, Coleridge Smith PD, Scurr JH. Which way does blood flow in the perforating veins of the leg? Phlebology. 1991;6:127.
176 Lake M, Pratt GH, Wright IS. Arteriosclerosis and varicose veins: occupational activities and other factors. JAMA. 1942;119:696.
177 Lindemayr H, Santler R. Der Einfluss der Schuhabasastzhohe auf den Venendruk des Beines. Wien Klin Wochenschr. 1979;91:496.
178 Pratt GH. Arterial varices: a syndrome. Am J Surg. 1949;77:456.
179 Piulachs P, Vidal-Barraquer F, Biel JM. Considerations pathogeniques sur les varices de la grossesse. Lyon Chir. 1952;47:263.
180 Szilagyi DE, Smith RF, Elliott JP, Hageman JH. Congenital arteriovenous anomalies of the limbs. Arch Surg. 1976;111:423.
181 Coursley G, Ivins JC, Barker NW. Congenital arteriovenous fistulas in the extremities: an analysis of sixty-nine cases. Angiology. 1956;7:201.
182 Schalin L. Reevaluation of incompetent perforating veins: a review of all the facts and observations interpreted on account of a controversial opinion and our own results. In: Tese M, Dormandy JA, editors. Superficial and deep venous diseases of the lower limbs. Torino, Italy: Edizioni Panminerva Medica, 1984.
183 Gius JA. Arteriovenous anastomoses and varicose veins. Arch Surg. 1960;81:299.
184 Schroth R. Venose sauerstoffsattigung bei varicen. Arch Klin Chir. 1962;300:419.
185 Haeger KHM, Bergman L. Skin temperature of normal and varicose legs and some reflections on the etiology of varicose veins. Angiology. 1963;14:473.
186 Piulachs P, Vidal-Barraquer F. Pathogenic study of varicose veins. Angiology. 1953;4:59.
187 Baron HC, Cassaro S. The role of arteriovenous shunts in the pathogenesis of varicose veins. J Vasc Surg. 1986;4:124.
188 McEwan AJ, McArdle CS. Effect of hydroxyethylrutosides on blood oxygen levels and venous insufficiency symptoms in varicose veins. Br Med J. 1971;2:138.
189 Clyne CA, Ramsden WH, Chant AD, Webster JH. Oxygen tension of the skin of the gaiter area of limbs with venous disease. Br J Surg. 1985;72:644.
190 Murphy MA, Hands L. Is arteriovenous shunting involved in the development of varicosities? A study of the intraluminal pressure and oxygen content in varicose veins. Phlebology. 2008;23:137.
191 Schalin L. Role of arteriovenous shunting in the development of varicose veins. In: Eklof B, Gjores JE, Thulesius O, Bergqvist D, editors. Controversies in the management of venous disorders. London: Butterworth, 1989.
192 Haimovici H, Steinman C, Caplan LH. Role of arteriovenous anastomoses in vascular diseases of the lower extremity. Ann Surg. 1966;164:990.
193 Bylebyl JJ. Harvey on the valves in the veins. Bull Hist Med. 1983;3:351.
194 Gradman WS, Segalowitz J, Grandfest W. Venoscopy in varicose vein surgery: initial experience. Phlebology. 1993;8:145.
195 Lindvall N, Lodin A. Congenital absence of venous valves. Acta Chir Scand. 1962;124:310.
196 Luke JC. The diagnosis of chronic enlargement of the leg: with description of a new syndrome. Surg Gynecol Obstet. 1941;73:472.
197 Norman AG. The significance of a venous cough impulse in the short saphenous vein. Angiology. 1956;7:523.
198 Ludin A. Congenital absence of valves in the veins. In: van der Molen HR, editor. Progres clinques et therapeutiques dans le domaine de la phlebologie. Apeldoorn, Netherlands: Stenvert, 1970.
199 Lodin A. Congenital absence of valves in the deep veins of the leg: a factor in venous insufficiency. Acta Derm Venerol. 1961;62:1.
200 Lodin A, Lindvall N, Gentele H. Congenital absence of venous valves as a cause of leg ulcers. Acta Chir Scand. 1958–1959;116:256.
201 Chadwick SJD, Clowes T, Dudley HAF. The relationship of varicose veins to the absence of venous valves. Br J Surg. 1984;71:222.
202 Villavicencio JL. Personal communication, 1989.
203 Ludbrook J, Beale G. Femoral vein valves in relation to varicose veins. Lancet. 1962;279:79.
204 Reagan B, Folse R. Lower limb venous dynamics in normal persons and children of patients with varicose veins. Surg Gynecol Obstet. 1971;132:15.
205 Geelhoed GW, Burkitt DP. Varicose veins: a reappraisal from a global perspective. South Med J. 1991;84:1131.
206 Basmajian JV. The distribution of valves in the femoral, external iliac, and common iliac veins and their relationship to varicose veins. Surg Gynecol Obstet. 1952;95:537.
207 LiCalzi LK, Stansel HCJr. Failure of autogenous reversed saphenous vein femoropopliteal grafting: pathophysiology and prevention. Surgery. 1982;91:352.
208 Fassiadis N, Holdstock JM, Whiteley MS. The saphenofemoral valve: gate keep turned into rear guard. Phlebology. 2002;17:29.
209 Labropoulous N, Giannoukas AD, Delis K, et al. Where does venous reflux start? J Vasc Surg. 1997;26:736.
210 Obitsu Y, Ishimaru S, Furukuva K, Yoshihama I. Histopathological studies of the valves of varicose veins. Phlebology. 1990;5:245.
211 Gjores JE. The incidence of venous thrombosis and its sequelae in certain districts of Sweden. Acta Chir Scand. 1956;206(Suppl):11.
212 Miller RP, Sparrow TD. The pathogenesis, diagnosis and treatment of varicose veins and varicose ulcers. NC Med J. 1948;9:574.
213 Lofgren KA. Varicose veins: their symptoms, complications, and management. Postgrad Med. 1979;65:131.
214 Beninson J. Thrombophlebitis. In: Demis DJ, editor. Clinical dermatology. Philadelphia: Harper & Row, 1986.
215 Cotton LT. Varicose veins: gross anatomy and development. Br J Surg. 1961;48:589.
216 Lee AJ, Evans CJ, Hau CM, et al. Pregnancy, oral contraception, hormone replacement therapy, and the occurrence of varicose veins: Edinburgh vein study. Phlebology. 1999;14:111.
217 Morrison N, Salles-Cunha S, Neuhardt D. Relationship between number of pregnancies and great saphenous vein diameter. Int Angiol. 2009;28:34.
218 Tolins SH. Treatment of varicose veins: an update. Am J Surg. 1983;145:248.
219 Henry M, Corless C. The incidence of varicose veins in Ireland. Phlebology. 1989;4:133.
220 Nabatoff RA. Varicose veins of pregnancy. JAMA. 1960;174:1712.
221 Diem K, editor. Documenta Geigy: Scientific tables. Basle: Ciba-Geigy, 1962.
222 Barrow DW. Clinical management of varicose veins, 2nd ed. New York: Paul B Hoeber; 1957.
223 Cordts PRO, Gawley TS. Anatomic and physiologic changes in lower extremity venous hemodynamics associated with pregnancy. J Vasc Surg. 1996;24:763.
224 McLennan CE. Antecubital and femoral venous pressures in normal and toxemic pregnancy. Am J Obstet Gynecol. 1943;45:568.
225 Ikard RW, Ueland K, Folse R. Lower limb venous dynamics in pregnant women. Surg Gynecol Obstet. 1971;132:483.
226 Wright HP, Osborn SB, Edmonds DG. Changes in the rate of flow of venous blood in the leg during pregnancy, measured with radioactive sodium. Surg Gynecol Obstet. 1950;90:481.
227 Skudder PAJr, Farrington DT, Weld E, Putman C. Venous dysfunction of late pregnancy persists after delivery. J Cardiovasc Surg. 1990;31:748.
228 Veal JR, Hussey HH. Venous circulation in lower extremities during pregnancy. Surg Gynecol Obstet. 1941;72:841.
229 Tournay R, Wallois P. Les varices de la grossesse et leur traitment principalement par les injections sclerosantes, expansion. Paris: Scient Franc; 1948.
230 McCausland AM. Varicose veins in pregnancy. Cal West Med. 1939;50:258.
231 Mullane DJ. Varicose veins in pregnancy. Am J Obstet Gynecol. 1952;63:620.
232 Dodd H, Wright HP. Vulval varicose veins in pregnancy. Br Med J. 1959;1:831.
233 Siegler J. The treatment of varicose veins in pregnancy. Am J Surg. 1939;44:403.
234 Berg D. First results with Ruscus extract in the treatment of pregnancy-related varicose veins. In: Vanhoutte PM, editor. Return circulation and norepinephrine: an update. Paris: John Libbey Eurotext, 1991.
235 Sohn CH, Karl C, Zelihic N. Untersuchungen zum Einfluss der Schwangerschaftsanzahl auf das Venensystem. Phlebologie. 1991;20:41.
236 Struckmann JR, Meiland H, Bagi P, Juul-Jorgensen B. Venous muscle pump function during pregnancy: assessment of ambulatory strain-gauge plethysmography. Acta Obstet Gynecol Scand. 1990;69:209.
237 Pemble L. Reversibility of pregnancy-induced changes in the superficial veins of the lower extremities. Phlebology. 2007;22:60.
238 Barwin BN, Roddie IC. Venous distensibility during pregnancy determined by graded venous congestion. Am J Obstet Gynecol. 1976;125:921.
239 Wahl LM. Hormonal regulation of macrophage collagenase activity. Biochem Biophys Res Commun. 1977;74:838.
240 Woolley DE. On the sequential changes in levels of oestradiol and progesterone during pregnancy and parturition and collagenolytic activity. In: Piez KA, Eddi AH, editors. Extracellular matrix biochemistry. New York: Elsevier Science, 1984.
241 Reynolds RM, Foster I. Peripheral vascular action of estrogen, observed in the ear of the rabbit. J Pharmacol Exp Ther. 1940;68:173.
242 Goodrich SM, Wood JE. Peripheral venous distensibility and velocity of venous blood flow during pregnancy or during oral contraceptive therapy. Am J Obstet Gynecol. 1964;90:740.
243 McCausland AM, Holmes F, Trotter ADJr. Venous distensibility during the menstrual cycle. Am J Obstet Gynecol. 1963;86:640.
244 Marazita AJD. The action of hormones on varicose veins in pregnancy. Med Rec. 1946;159:422.
245 McPheeters HO. The value of estrogen therapy in the treatment of varicose veins complicating pregnancy. Lancet. 1949;69:2.
246 Fried PH, Perilstein PK, Wagner FBJr. Saphenous varicosities vs. angiectids; consideration of so-called varicose veins of the lower extremities complicating pregnancy. Obstet Gynecol. 1953;2:418.
247 Sandstrom B, Bjerle P. Calf blood flow and venous capacity during late pregnancy in women with varices. Acta Obstet Gynecol Scand. 1974;53:355.
248 Sumner DS. Venous dynamics: varicosities. Clin Obstet Gynecol. 1981;24:743.
249 Fawer R, Dettling A, Weihs D, et al. Effect of menstrual cycle, oral contraception, and pregnancy on forearm blood flow, venous distensibility, and clotting factors. Eur J Clin Pharmacol. 1978;13:251.
250 Allegra C, Carlizza A, Mari A. Estroprogestins and microcirculation. In: Raymond-Martimbeau P, Prescott R, Zummo M, editors. Phlébologie ’92. Paris: John Libbey Eurotext, 1992.
251 Vin F, Allaert FA, Levardon M. Influence of estrogens and progesterone on the venous system of the lower limbs in women. J Dermatol Surg Oncol. 1992;18:888.
252 Durand JL, Bressler R. Clinical pharmacology of the steroidal oral contraceptives. Adv Intern Med. 1979;24:97.
253 Dandstrom B, Lowegren L. Phlebographic studies of the leg veins in the first half of pregnancy. Acta Obstet Gynecol Scand. 1970;49:375.
254 Gallagher PG. Major contributing role of sclerotherapy in the treatment of varicose veins. Vasc Surg. 1986;20:139.
255 Krasinski Z, Oszkinis G, Kotwicka M, et al. Ocena komorek miesniowych gladkich z receptorami dla hormonow plciowych w zylach prawidlowych I zylakach. Prezeglad Flebologiczny. 2005;13:61.
256 Mashiah A, Berman V, Thole HH, et al. Estrogen and progesterone receptors in normal and varicose saphenous veins. Cardiovas Surg. 1999;7:327.
257 Harridge WH. The treatment of primary varicose veins. Surg Clin North Am. 1960;40:191.
258 Husni EA, Pena LI, Lenhert AE. Thrombophlebitis in pregnancy. Am J Obstet Gynecol. 1967;97:901.
259 Quattlebaum FW, Hodgson JF. The surgical treatment of varicose veins in pregnancy. Surg Gynecol Obstet. 1952;95:336.
260 Pechet L, Alexander B. Increased clotting factors in pregnancy. N Engl J Med. 1961;265:1093.
261 Wood JE. Oral contraceptives, pregnancy, and the veins. Circulation. 1968;38:627.
262 Pittaluga P, Chastanet S, Rea B, Barbe R. Midterm results of the surgical treatment of varices by phlebectomy with conservation of a refluxing saphenous vein. J Vasc Surg. 2009;50:107.
263 Labropoulos N, Kokkosis AA, Spentzouris G, et al. The distribution and significance of varicosities in the saphenous trunks. J Vasc Surg. 2010;51:96.
264 Thompson H. The surgical anatomy of the superficial and perforating veins of the lower limb. Ann R Coll Surg Engl. 1979;61:198.
265 Cornu-Thenard A, Boivin P, Baud JM, et al. Importance of the familial factor in varicose disease. J Dermatol Surg Oncol. 1994;20:318.
266 Von Curtius F. Untersuchungen uber das menschliche Venensystem. Deutsches Arch Klin Med. 1928;162:194.
267 Arnoldi C. The heredity of venous insufficiency. Dan Med Bull. 1958;5:169.
268 Almgren B, Eriksson I. Primary deep venous incompetence in limbs with varicose veins. Acta Chir Scand. 1989;155:455.
269 Arenander E, Lindhagen A. The evolution of varicose veins studied in a material of initially unilateral varices. Vasa. 1978;7:180.
270 Carpentier PH, Maricq HR, Biro C, et al. Prevalence, risk factors, and clinical patterns of chronic venous disorders of lower limbs: a population-based study in France. J Vasc Surg. 2004;40:650.
271 Ottley C. Heredity and varicose veins. Br Med J. 1934;1:528.
272 Troisier J, Le Bayon. Etude génétique des varices. Ann de Med. 1937;41:30.
273 King ESJ. The genesis of varicose veins. Aust N Z J Surg. 1950;20:126.
274 Niermann H. Zwillingsdermatologie. Berlin: Springer-Verlag; 1964.
275 Ng MY, Andrew T, Spector TD, Jeffery S. (representing the Lymphoedema Consortium): Linkage to the FOXC2 region of chromosome 16 for varicose veins in otherwise healthy, unselected sibling pairs. J Med Genet. 2005;42:235.
276 Gundersen J, Hauge M. Hereditary factors in venous insufficiency. Angiology. 1969;20:346.
277 Folse R. The influence of femoral vein dynamics on the development of varicose veins. Surgery. 1970;68:974.
278 Almgren B. Non-thrombotic deep venous incompetence with special reference to anatomic, haemodynamic and therapeutic aspects. Phlebology. 1990;5:255.
279 Baron HC. Varicose veins, Consultant May 1983, p 108.
280 Ackroyd JS, Pattison M, Browse NL. A study of the mechanical properties of fresh and preserved human femoral vein wall and valve cusps. Br J Surg. 1985;72:117.
281 Campbell PA, McGeachie JK, Prendergast FJ. Vein grafts for arterial repair: their success and reasons for failure. Ann R Coll Surg Engl. 1981;4:257.
282 Szilagyi DE, Elliot JP, Hageman JH, et al. Biological fate of autologous vein implants as arterial substitutes. Ann Surg. 1973;178:232.
283 Fuchs JCA, Michener JS, Hagen PO. Postoperative changes in autologous vein grafts. Ann Surg. 1978;188:1.
284 Brody WR, Kosek JC, Angell WW. Changes in vein grafts following aorto-coronary bypass induced by pressure and ischaemia. J Thorac Cardiovasc Surg. 1972;64:847.
285 Zsoter T, Cronin RFP. Venous distensibility in patients with varicose veins. Can Med Assoc J. 1966;4:1293.
286 Eiriksson E, Dahn I. Plethysmographic studies of venous distensibility in patients with varicose veins. Acta Chir Scand Suppl. 1968;398:19.
287 Blöchl-Daum B, Schuller-Petrovic S, Wolzt M, et al. Primary defect in alpha-adrenergic responsiveness in patients with varicose veins. Clin Pharmacol Ther. 1991;49:49.
288 Herbst WM, Eberle KP, Ozen Y, Hornstein OP. The innervation of the great saphenous vein: an immunohistological study with special regard to regulatory peptides. Vasa. 1992;21:253.
289 Psaila JV, Melhuish J. Viscoelastic properties and collagen content of the long saphenous vein in normal and varicose veins. Br J Surg. 1989;76:37.
290 Gertsch P, Loup PW, Lochman A, Anani P. Changing patterns in the vascular form of Ehlers-Danlos syndrome. Arch Surg. 1986;121:1061.
291 Ludbrook J. Valvular defect in primary varicose veins: cause or effect? Lancet. 1963;282:1289.
292 Corcos L, Peruzzi GP, Romeo V, et al. Peripheral venous biopsy: significance, limitations, indications and clinical applications. Phlebology. 1989;4:271.
293 Jick H, Porter J. Thrombophlebitis of the lower extremities and the ABO type. Arch Intern Med. 1978;138:1566.
294 Robinson WN, Roisenberg I. Venous thromboembolism and the ABO blood groups in a Brazilian population. Hum Genet. 1980;55:129.
295 Talbot S, Wakley EJ, Ryrie D, Langman MJ. ABO blood groups and venous thromboembolic disease. Lancet. 1970;295:1257.
296 Jick H, Westerholm B, Vessey MP, et al. Venous thromboembolic disease and the ABO blood type. Lancet. 1969;293:539.
297 Vessay MP, Mann JI. Female sex hormones and thrombosis. Br Med Bull. 1978;34:157.
298 Cornu-Thenard A, Dab W, De Vincenzi I, Valty J. Relationship between blood groups (ABO) and varicose veins of the lower limbs: a case-control study. Phlebology. 1989;4:37.
299 Lee S, Lee W, Choe Y, et al. Gene expression profiles in varicose veins using complementary DNA microarray. Dermatol Surg. 2005;31:391.
300 Schultz-Ehrenberg U, Reich-Schupke S, Robak-Pawelczyk B, et al. Prospective epidemiological study on the beginning of varicose veins (Bochum Study I-IV). Phlebologie. 2009;38:17.
301 Maurins U, Hoffmann BH, Losch C, et al. Distribution and prevalence of reflux in the superficial and deep venous system in the general population – results from the Bonn vein study, Germany. J Vasc Surg. 2008;48:680.
302 Lev M, Saphir O. Endophlebohypertrophy and phlebosclerosis. Arch Pathol Lab Med. 154, 1951.
303 Donovan DL, Schmidt SP, Townshend SP, et al. Material and structural characterization of human saphenous veins. J Vasc Surg. 1990;12:531.
304 Bouissou H, Julian M, Pieraggi MT, et al. Structure of healthy and varicose veins. In: Vanhoutte PM, editor. Return circulation and norepinephrine: an update. Paris: John Libbey Eurotext, 1991.