CHAPTER 230 Pathophysiology of Surgical Nerve Disorders
Causes of Surgical Nerve Injuries
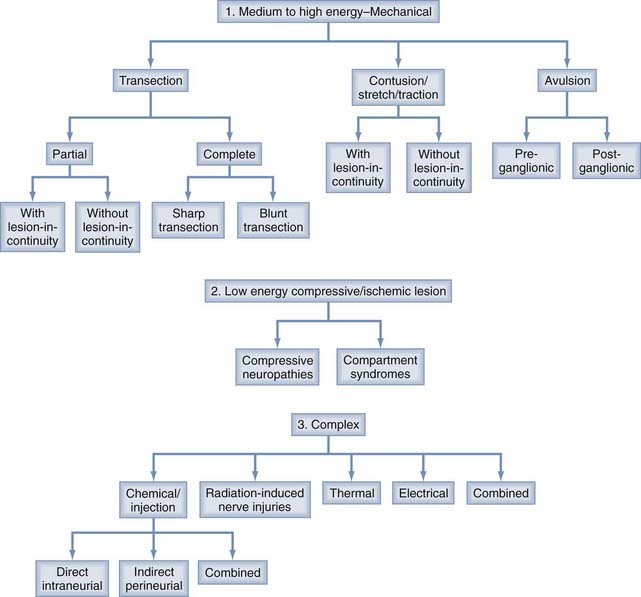
The different causes of PNS injury can generally be divided into three major categories based on the degree of biomechanical force exerted per surface area of the injured nerve and the complexity of the injury. Nerve injuries caused by transection, contusion, stretch, traction, and avulsion are generally sustained when medium- to high-energy force is applied directly or indirectly to nerves, whereas injuries such as compressive neuropathy tend to occur when nerves are subjected to chronic or repetitive low-energy force (Fig. 230-1, 1 and 2 and Table 230-1). Nerve injuries from injection and from radiation and thermal energy involve a rather heterogeneous combination of different injuring factors and can be grouped together as a complex group of nerve injuries (Fig. 230-1, 3).

Medium- to High-Energy Nerve Injuries
Transection
Soft tissue lacerations with objects such as knives, glass, propeller and fan blades, chain saws, auto metal, and surgical instruments may transect nerves in about 30% of cases.1 In the remaining 70% of these injuries, 20% leave the nerve in partial and 50% in complete (lesion in continuity) continuity.1,2 The extent of functional loss is determined by variable degrees of neurotmesis, axonotmesis, and neurapraxia sustained by the nerve,3 even when the nerve or a portion of it remains in continuity (Fig. 230-2B). The extent of functional loss varies from mild and incomplete to severe and total. If a nerve is partially transected, the injury to the fibers cut is, by definition, neurotmetic or Sunderland grade V. In contrast, the fibers not directly transected can have a variable degree of injury and be Sunderland grade II, III, or IV (see Table 230-1 for grading systems for nerve injuries).4 In humans, the partially transected portion of a nerve seldom regenerates spontaneously, but when it does, it is not sufficient to restore function and therefore needs microsurgical repair.5 Functional recovery in some of these patients can be attributed to reversal of neurapraxia or to regeneration in the bruised and stretched portion of the nerve rather than in the transected portion.
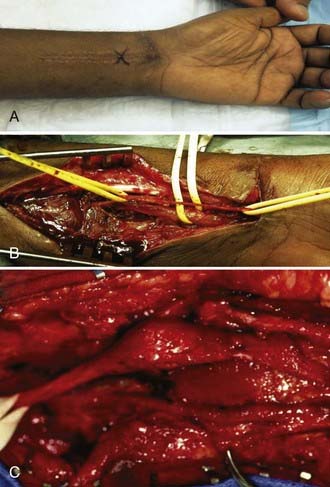
The physical appearance of a sharply transected nerve differs from that of a nerve that has sustained blunt transection changes over time. After sharp transection, the epineurium is cut cleanly, and there is minimal contusive change or hemorrhage in either stump. With time, the stumps of the cleanly cut nerve retract and become enveloped in scar. The amount of proximal neuroma and distal nerve stump scarring is much less than that formed in a more contusive or blunt transection. Blunt transection is associated with a ragged tear of the epineurium acutely and an irregular, longitudinal extent of damage to a segment of the nerve. Bruising and hemorrhage can extend for several centimeters up or down either stump. Retraction and proliferative scarring around the stumps are often more severe than that are seen with sharp transection.6
Stretch, Traction, and Contusion with or without Lesion in Continuity
Medium- to high-energy force applied to nerves can result in a combination of different types of serious nerve injury ranging from significant stretch and traction injuries to differing degrees of contusion.7 The perineurium of intact nerves is rich in elastin and collagen, which provide tensile strength.8 However, even 8% stretch leads to a disturbance in intraneural circulation and blood-nerve barrier function,9 whereas stretch beyond 10% to 20%, especially if applied acutely, results in structural failure.10 Such force can therefore occasionally distract a nerve and pull it apart totally or, more commonly, leave it in continuity but with considerable internal damage (Fig. 230-2C). If distracted by substantial force, the nerve becomes frayed, and both stumps are damaged over many centimeters. The retraction and scarring about both stumps are severe. If the nerve is left in continuity, as is more likely, the degree of intraneural damage is variable and may, on occasion, be manifested as a spectrum of nerve fiber changes, including neurapraxia, axonotmesis, and neurotmesis. A stretch mechanism is also responsible for segments of damage to nerves displaced by high-velocity missiles, especially with gunshot wounds.6,8
Traction force applied to nerves is commonly sufficient to tear apart the intraneural connective tissue structure, as well as disconnect axons.11 Such lesions are Sunderland grade IV and are neurotmetic despite physical continuity of the nerve.12 Less frequently, such force results in a more axonotmetic or Sunderland grade II or III lesion, and these lesions may have the potential for effective regeneration because of less disruption of connective tissue.4
Most nerve injuries leave the nerve in continuity, which can make determination of the degree of injury and prognostication of functional recovery quite difficult (Fig. 230-2C). Contusive lesions tend to leave the nerves in continuity. These lesions in continuity can be either focal or diffuse and may even be multifocal with intervening areas of seemingly intact nerve. In the diffuse subtype, which represents the majority of cases, the entire cross section of the nerve has a similar extent of internal damage.13 Clinical and electrophysiologic examination provides guidance about the extent of injury to the nerve fibers such that sparing of one or more fascicles may produce a partial neurological deficit with preserved but diminished nerve action potentials (NAPs) across the injury site.14,15
With the typical lesion in continuity, the nerve is acutely swollen, with extravasation of serum or blood; axons and their myelin coverings disintegrate, and there is disruption of the connective tissue elements16,17 (Fig. 230-3A). Wallerian degeneration occurs, and axonal and myelin debris is phagocytosed from both the injury site and more distal part of the nerve.18 The Schwann cells (SCs), basal lamina, and distal connective tissue elements survive and are well positioned and conducive for axonal outgrowth.19 Unfortunately, the endoneurial and perineurial elements at the injury site proliferate rapidly and lay down poorly structured collagen that interferes with organized and properly directed axonal regeneration.20 Because there is some retrograde damage proximal to the injury site with most nerve injuries, clusters of regenerating axons must first traverse this area of loss.21 These regenerating axons next encounter poorly restructured collagen at the injury site,22 which leads to further disorganization in their orientation and delay in the process of axonal regeneration (i.e., staggered axonal regeneration).23 Axons branch many times as they traverse the site of injury. Such axonal branching in humans may occur several hundred times.24 Other axons may be deflected into peripheral connective tissue layers at the injury site as well as distally. As a result, axons reaching the distal stump are thin, poorly myelinated, and therefore less likely to reach previous distal end-organs than with a more axonotmetic injury. Many serious lesions in continuity are therefore not capable of regeneration of sufficient quality to lead to recovery of useful distal function.2 Some of the complex interrelated factors that ultimately determine the success of axonal regeneration after nerve injury are outlined in Figure 230-3. In clinical practice, because it is difficult to discern the extent of internal damage after this type of injury, most lesions in continuity are monitored clinically and reevaluated at intervals for several months before surgical exploration25 (Table 230-2).
| PARTIAL INJURIES (INCOMPLETE LOSS WITH SIGNIFICANT DISTAL SPARING) |
|
Most patients improve with conservative treatment. Follow with serial clinical examination supplemented with electrophysiologic assessment
|
EMG, electromyographic; NAPs, nerve action potentials.
Avulsion
Brachial plexus injury is a common disorder that is usually caused by a stretch mechanism. Stretch or traction injuries to the plexus most frequently result from extremes of movement at the shoulder joint, with or without actual dislocation or fracture of the humerus or the clavicle. With blunt or traction force, fracture of the scapula, ribs, cervical spine, or any combination of such fractures can also occur.26,27 A clavicular fracture seen with brachial plexus injury does not indicate that the injury was caused by the fracture but rather attests to the extensive force applied to the shoulder joint. On rare occasion, however, compressive upper trunk plexopathy may result in delayed fashion from bony callus generated from clavicular malunion28 (Fig. 230-4). Either upper or lower elements of the plexus may suffer the predominant injury, or with severe traction force, all elements may be involved in addition to the phrenic nerve and even subclavian vessels. All grades of damage are possible. Spinal nerves and roots can be avulsed from the spinal cord or more laterally from truncal or more distal outflow. The stretched elements may be left in continuity and have a mixture of neurapraxia and axonotmesis. A combination of neurapraxia, axonotmesis, and neurotmesis may coexist, but unfortunately, these mixed grades of injuries are more commonly severe in degree with significant neurotmetic components.
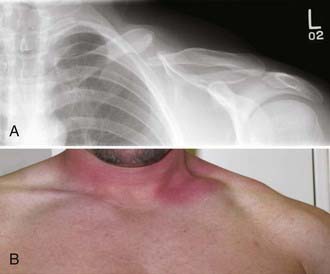
 years after the injury he started to notice paresthesia in his shoulder girdle emanating from the supraclavicular area and over the course of time became aware of progressive weakness in shoulder abduction. Physical examination confirmed a severe suprascapular neuropathy and mild weakness in deltoid function. Electromyography demonstrated evidence of denervation that was severe in the supraspinatus and infraspinatus and sparse, although active in the deltoid. A and B, Radiographic and clinical appearance of the nonunited midclavicular fracture with substantial callus formation. The patient underwent exploration and external neurolysis of the upper part of the trunk along with the suprascapular and posterior division branches from the trunk that were being impinged by the callus. The callus was resected widely and the fracture repaired by an orthopedic surgery colleague with a plate and lag screw. The patient eventually achieved excellent bony union of the site and progressive improvement in shoulder girdle muscle strength and function. This case illustrates delayed complication from a nonunited fracture with callus causing adjacent nerve compression.
years after the injury he started to notice paresthesia in his shoulder girdle emanating from the supraclavicular area and over the course of time became aware of progressive weakness in shoulder abduction. Physical examination confirmed a severe suprascapular neuropathy and mild weakness in deltoid function. Electromyography demonstrated evidence of denervation that was severe in the supraspinatus and infraspinatus and sparse, although active in the deltoid. A and B, Radiographic and clinical appearance of the nonunited midclavicular fracture with substantial callus formation. The patient underwent exploration and external neurolysis of the upper part of the trunk along with the suprascapular and posterior division branches from the trunk that were being impinged by the callus. The callus was resected widely and the fracture repaired by an orthopedic surgery colleague with a plate and lag screw. The patient eventually achieved excellent bony union of the site and progressive improvement in shoulder girdle muscle strength and function. This case illustrates delayed complication from a nonunited fracture with callus causing adjacent nerve compression.Some anatomic features of the brachial plexus may predispose it to traction or even rupture. After the roots penetrate dura, they become spinal nerves. The spinal nerves run in the gutters of the foramina in the vertebrae for which they are named. At this intraforaminal level, the nerves are relatively tethered by mesoneural-like connections to the gutters.29 The spinal nerves then angle inferiorly and appear between the scalenus anticus and scalenus medius muscles and thus gain entrance to the posterior triangle of the neck. Spinal nerves are often injured in a characteristic fashion just as they run off the lip of the gutter of the transverse process. Force here may distract a spinal nerve from the trunk and produce a rupture (Fig. 230-5). Alternatively, such force may produce severe intraneural damage. These result in lengthy lesions in continuity that not only involve the spinal nerves and trunks but may also extend into the divisions and rarely, even into the more distal infraclavicular elements. A common finding with severe stretch injury is to see cords pulled away from more proximal elements of the plexus, such as roots and trunks. Unfortunately, in these circumstances, intraneural damage to these proximal elements often extends close to, if not all the way to the spinal theca or cord.24
A key clinical feature of stretch injuries is that although some may improve, many do not and require operative reconstruction (see Fig. 230-3A). However, these type of injuries pose an especially difficult challenge because there may be no satisfactory operative solution for some of the most severe stretch injuries. If the major injury is neurotmetic, it can involve such a long segment of the nerve that the only operative method for replacing the resulting extensive neuroma is the use of lengthy grafts. The results of repair with such lengthy grafts are often poor, and they are especially prone to fail at the proximal levels, where many stretch injuries begin.
Low-Energy Compressive/Ischemic Nerve Injuries
Peripheral nerves, like other neural tissues, are critically dependent on blood flow. Because it is rarely possible to compress a nerve segment without simultaneously affecting its blood supply, the relative roles of ischemia and physical deformation in compression lesions remain unsettled.30 More recent evidence suggests that although ischemia may be primarily responsible for a mild type of rapidly reversible nerve lesion, direct mechanical distortion is the major factor underlying more severe, long-lasting forms of pressure palsy such as Saturday night palsy or tourniquet paralysis.31–33 Ischemia can produce a wide range of nerve fiber lesions and, if severe and prolonged, results in extensive axonal loss and wallerian degeneration.34 Studies on limb ischemia suggest that there is a critical period of approximately 8 hours after which irreversible nerve injury ensues.35
Compressive Neuropathies
The sequential pathology of nerve fiber injury is rather stereotyped and occurs regardless of the compressive agent except in minor forms of compression.36 Compression of nerve fibers appears to produce changes in myelinated nerve fibers that are unique to this mechanism.37,38 Such changes include alterations in paranodal myelination, axonal thinning, and segmental demyelination.39–42 Axonal injury and therefore wallerian degeneration result from more severe degrees of compression.
The degree of recovery after compression or ischemic injury may be accurately predicted in some clinical situations. The characteristic Saturday night palsy results from compression of the radial nerve against the humerus. Total radial nerve palsy often results, but motor and sensory function is restored in the majority of patients without any need for surgical intervention. Most palsies associated with unconsciousness secondary to anesthesia and poor positioning or pressure during surgery, as well as those related to improper application of plaster casts, carry a good prognosis for spontaneous recovery.43 There are, however, important exceptions. Sometimes the compressive or crushing injury has been severe or prolonged enough to cause damage that is irreversible unless operative repair is undertaken. The brachial plexus and the ulnar, sciatic, and peroneal nerves are most commonly affected by these more severe compressive causes.44 Restoration of function after acute compression and ischemic injury may be uncertain in some circumstances. It may be difficult, for example, to predict the degree of recovery that follows evacuation of hematomas or relief of pseudoaneurysmal compression of such structures as the brachial plexus and the femoral or sciatic nerves.45 There are also circumstances in which two levels of compression may exist.46 A patient with cervical spondylolysis or relatively mild disk disease affecting a root or spinal nerve may be more likely to become symptomatic with an otherwise mild degree of compression of the median nerve at the wrist or ulnar nerve at the elbow. In these circumstances, multiple factors affect the outcome of peripheral nerve surgery, including the identity and level of the nerve involved, the age of the patient, the extent of precompression injury to the nerve, and the timing of corrective surgery.
Compartment Syndromes
Severe crushing injury, skeletal fracture with vascular compromise, and anticoagulant administration resulting in hemorrhage can lead to increased pressure within a fascial compartment. As a consequence, severe compression and ischemic damage to peripheral nerves and other soft tissues can result. A closed compartment syndrome with impending ischemic paralysis requires immediate decompression with properly placed and usually extensive longitudinal fasciotomies.47 Delay in treatment results in ischemic infarction of muscle, nerve, and other tissues, which leads to contractures and other crippling deformities.
Volkmann’s contracture is a serious example of ischemic compression caused by the development of compartment syndrome. There is injury to the brachial artery along with diffuse segmental damage to the median nerve and volar forearm muscles. The large median and sometimes radial nerve fibers serving motor and proprioceptive function are more severely involved than the smaller pain fibers. Electromyography may aid in diagnosis by showing temporary but repetitive and spontaneous motor discharges from muscles most distal to the injury site.48 Swelling of the forearm resulting in a painful paresthetic hand must alert the physician to an impending compartment syndrome long before more obvious signs of vascular compromise are apparent.
Complex Nerve Injuries
Electrical
Electrical injury induced by passage of a large current through a peripheral nerve usually results from accidental contact of the extremity with a high-tension wire.49 If the individual does not succumb because of respiratory or cardiac arrest, diffuse nerve and muscle damage results.50 Pathologic reports of peripheral nerve damage caused by this mechanism are sparse, and guidelines for treatment are controversial.51 Conservative management of the nerve injury itself and early orthopedic reconstruction of the extremity seem to be best.52 The prognosis with most low-voltage injuries is excellent, but it is quite variable with high-voltage injuries.52 Resection of a lengthy segment of damaged nerve plus repair with grafts is usually necessary. Histologically, the segment of the nerve is virtually replaced, first with necrosis and then with connective tissue reaction, including a severe degree of both perineurial and endoneurial scar tissue. The fascicular outline may be preserved, but intrafascicular damage and fibrosis can be severe enough to prevent any but fine and functionally fruitless axon regeneration.
Irradiation
Irradiation is a relatively rare cause of iatrogenic nerve injury when compared with injection (see the next section). It usually affects the brachial plexus, but the pelvic plexus can also be involved.53 Extensive scar formation in surrounding soft tissues and severe intraneural changes consisting of loss of myelin, axonal degeneration, and extensive endoneurial fibrosis often result.54
Injection
Injection injury is caused by insertion of a needle into or close to a nerve, and damage results from neurotoxic chemicals in the agent injected. The extent of damage depends not only on the agent injected but also on whether the needle and therefore the toxic agent were placed in or close to the nerve. There are cases in which some or all of the injury is caused by the damage done by needle placement itself. Experimentally, damage from injection seems to require placement of the agent either within the epineurium or, for more serious damage, at an intraneural locus, either intrafascicular or in the connective tissue layers between fascicles.55 In humans, however, about 10% of patients subsequently found to have an injection injury experience a delay of hours or even days before the onset of symptoms.56 This delayed onset suggests either a purely epineurial locus for deposition of the agent or placement of medication close to a nerve or in a tissue plane from which the agent can gravitate to and bathe the nerve.
The pathology of injection injury also depends on the injection site and the agent injected.57 The principal pathogenetic mechanism, however, is necrosis.58 With intraneural injection, acute edema and inflammatory changes, often with necrosis, take place and affect connective tissue elements, axons, and myelin.59 With time, connective tissue proliferation may occur and produce intraneural scarring, thereby thwarting effective axonal regeneration60 (see Fig. 230-3B). The blood-nerve barrier at both the perineurial and endoneurial capillary levels is severely disrupted,61 a finding that may occur despite preservation of fascicular structure (see Fig. 230-3A). After the first few days the injected segment is no longer swollen and may with time appear shrunken or even as a segment of nerve with normal diameter. On gross inspection with or without magnification, the nerve usually appears to have excellent physical continuity. Even intraneural dissection may reveal good fascicular continuity despite intrafascicular damage that is quite neurotmetic. Some agents injected into the epineurium or adjacent to nerve produce more proliferation of inflammatory tissue and scarring than at an intraneural locus, but the necrosis at the latter locus is especially damaging and difficult for the regenerative process to overcome spontaneously.
In the usual clinical setting, needle placement results in an electric-like shock down the extremity, followed by or concomitant with a severe radicular burning pain and paresthesias as the agent is injected. Acute symptoms are variously described but are generally severe. With delayed onset, which seems to occur in about 10% of patients with injection injuries, the symptoms are less dramatic but nonetheless bothersome.56 Such symptoms include a burning pain, paresthesias, and radiation of deep discomfort down the limbs and in the distribution of the involved nerve. It is worth mentioning that the most common neural injection sites are the sciatic nerve at the buttock level and the radial nerve in the lateral aspect of the upper part of the arm.56,62 Nonetheless, besides the sciatic and radial nerves, injection injuries involving almost every other major nerve in the body have been described, including injuries to the femoral and lateral femoral cutaneous nerves, as well as the ulnar and median nerves at the wrist, elbow, and upper arm levels.
Some of the biomechanical factors that affect the extent of nerve damage include the length of the needle used and the angle at which the needle penetrates soft tissues, as well as the force with which the injection is given.56,61 If the hub of the needle inverts the skin and underlying soft tissue with enough force during injection, the tip of the needle can penetrate to a level deeper than surmised. The injury may be compounded if the needle penetrates soft tissue at an angle toward nerve rather than at a right angle to a horizontal plane through the body. Other factors, such as movement of the patient as the injection is being made, can sometimes play a role. If the patient shrugs or jerks the shoulder, perhaps in anticipation of the pain connected with the event or the ability to see the approaching needle, an injection intended for the deltoid can injure the radial nerve. In part because of this possibility, the radial nerve at the midhumeral level is the second most frequent site of injection injury. Infusions or needles intended for veins can also inadvertently be placed in nerves. This is more likely to occur in the median or ulnar nerve, the former either at the wrist or elbow and the latter at the wrist. Infusion complications have, however, also involved the brachial plexus, the femoral nerve, and the posterior tibial nerve at the ankle.
Although the deficit in neural function is generally caused by intraneural neuritis and scar tissue rather than extraneural scarring, some authors believe that external neurolysis for this complication can reverse the loss of function.63 We do not agree with this supposition; however, a lesion with partial loss of function and severe pain not responding to analgesics may be helped by internal neurolysis on a delayed basis. Occasional patients may have true causalgia after injection and can benefit from sympathectomy, especially if recurrent sympathetic blocks have provided temporary relief. The pain in most of these patients can be managed effectively with tricyclic antidepressants (such as amitriptyline) or anticonvulsant agents (such as gabapentin) because these agents have demonstrated good benefit in patients with neuropathic pain.64,65
If the nerve deficit is partial, expectant treatment is best, provided that the pain is not a severe problem, but if the deficit is complete after several months of observation, exploration becomes warranted. In this regard, if an injection injury to the sciatic nerve spares either the peroneal or tibial division but is complete in the other division, it is a complete lesion of one division, and this division may need resection and repair if function is to be regained.66 A reasonable management approach with injection injuries is to explore the nerve that shows little or no function after 12 to 16 weeks and attempt to evoke an NAP through the injury.56 Most lesions have a recordable NAP, but if no response is noted, the lesion must be resected and repaired in the hope of regaining function.
Neurobiology of Peripheral Nerve Injury, Regeneration, and Functional Recovery
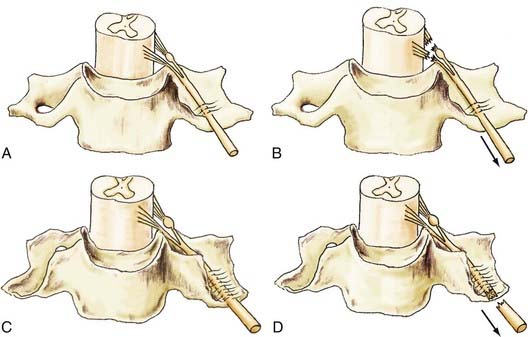
Nerve injuries trigger complex cell-molecular interactions that are essential for axonal regeneration and subsequently meaningful functional recovery of patients. Significant advances have been made in the technique of microsurgery of injured nerves, which often results in improved outcome for patients. However, recovery of function can be suboptimal in some patients despite the capacity of the PNS to regenerate axons, even after excellent microsurgical repair. This dichotomous observation has been studied experimentally by our group and others, and in this section of the chapter we discuss three key biochemical phenomena that have been shown to be responsible for suboptimal recovery of function, namely, chronic SC denervation, chronic neuronal axotomy, and misdirection of regenerating axons (Fig. 230-3B).
Regenerative Response after Nerve Injury and Its Role in Functional Outcome
Initial Phase of Axonal Regeneration
After nerve injury, the proximal and distal stumps of the injured nerve undergo structural and molecular changes in preparation for the process of axonal regeneration. The proximal stump undergoes dieback degeneration, which in human injuries with great force may be a centimeter or more. Each injured axon elaborates multiple daughter axons67,68 (Fig. 230-6A). Many of the daughter axons are pruned, and those that remain begin the process of elongation through the scar and attendant disorganization of the injury or repair site and then into the distal nerve stump. These constitute the regenerating units.21 This initial stage of axonal regeneration is sustained by the availability of both locally produced cytoskeletal material and neuronally produced and anterogradely transported cytoskeletal proteins such as actin and tubulin (Fig. 230-6A).68–71 Once the distal stump is reached, axon regeneration proceeds at a rate of 1 to 3 mm/day, with the rate corresponding to the slow rate of transport of cytoskeletal material. Further elongation and regeneration through the distal nerve stump are dependent on the growth-supportive milieu provided by the SCs of the distal nerve stumps. Lack of SC-laden endoneurial channels (Bands of Büngner) and oftentimes the presence of severe intraneural scarring result in misdirected regeneration and neuroma formation.8,72–74
Role of Schwann Cells and Regeneration-Associated Genes in Axonal Regeneration
The distal nerve stumps of severed nerves undergo Wallerian degeneration, a degenerative process named after Augustus Waller. We now understand that Wallerian degeneration is an essential preparatory stage of the process of axonal regeneration in which molecules that could be inhibitory to regeneration are eliminated. Schwann cells play a major role in this process by way of phagocytosis of axonal and myelin debris. They also secrete chemoattractive factors, such as interleukin-1 and monocyte chemoattractant protein-1. These factors recruit macrophages into the denervated distal nerve stumps, which contribute significantly to the phagocytosis of axon and myelin debris.71,75,76
Immediately after a nerve is injured, loss of axonal contact triggers SCs to proliferate and switch their phenotype from a myelinating to a nonmyelinating growth-supportive phenotype.70,71,77 mRNA expression of myelin-associated proteins such as P0 and myelin-associated glycoprotein are downregulated, and neurotrophins (e.g., nerve growth factor, brain-derived neurotrophic factor, glial-derived neurotrophic factor) and their receptors (e.g., p75, GFRA-1, GFRA-2), as well as adhesion molecules (e.g., neural cell adhesion molecule), are upregulated in preparation for the process of axonal regeneration70,74,78,79 (Fig. 230-6A). These upregulated genes are collectively called regeneration-associated genes (RAGs). The change in gene expression, as well as myelin and axonal degeneration and clearance are key features of the process of Wallerian degeneration.70,71,77
Likewise, neurons whose axons have been injured downregulate the mRNA of proteins required for neurotransmission and upregulate those for rebuilding their peripheral processes.70,80,81 Hence, actin, tubulin, and GAP-43 are upregulated immediately after injury70,80 (Fig. 230-6A). However, the upregulation of RAGs is not sustained in either the injured neurons or the SCs such that by 6 months in experimental animals, most of the upregulated mRNA is downregulated, thereby losing the growth-supportive environment for regenerating axons79,80 (Fig. 230-6B and C). As discussed in the following sections, the implication of the time-limited upregulation of RAGs is demonstrated by the progressive decline in the capacity of injured neurons to regenerate their axons and SCs to support regenerating axons as the duration of nerve regeneration is prolonged by the time and/or distance (Fig. 230-7C and D). Other factors that have effects on the process of axonal regeneration after nerve injury are listed in Figure 230-3.
Experimental Paradigms and Assessment of Axonal Regeneration after Nerve Injury and Surgical Repair
The experimental studies on nerve injury in animal and humans that were conducted in the early part of the 20th century concluded erroneously that poor functional recovery after nerve injury is due to irreversible denervation atrophy of muscle and its inability to accept innervation, especially after long periods.82,83 This conclusion was reached in a set of experimental studies of nerve injury and repair in rats in which regenerated nerve fibers in the distal nerve stumps were counted after nerve transection and surgical repair. The presence of countable regenerated nerve fibers in the distal nerve stumps led the authors to conclude that injured nerves do regenerate their fibers but that the regenerated nerve fibers could not reinnervate the target muscles because of their significant atrophy. This conclusion became quite popular and unfortunately is often repeated even in recent publications. We disagree with this concept for the following reasons: (1) the authors did not directly estimate the number of injured neurons that regenerated into the distal nerve stump and those that reinnervated denervated muscles, and (2) counting of nerve fibers overestimates the numbers of regenerated nerve fibers because many daughter axons arise from single regenerating nerve fibers in the proximal nerve stump and may be erroneously counted as representative of the individual nerve fibers in the proximal stump.
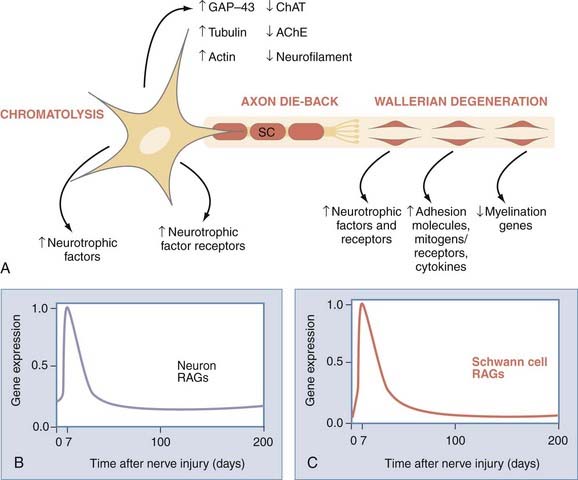
We carried out several experimental studies in which we studied the process of axonal regeneration after immediate and delayed repair of nerve injury. Furthermore, we used quantitative methods of counting the motoneurons that regenerated their axons into distal nerve stumps and of counting the number of reinnervated motor units in the target muscles to assess the capacity of motoneurons to regenerate their axons and to reinnervate muscle. Using a cross-suture technique in rats, which allows independent study of the effects of delayed reinnervation of the distal nerve stump (termed chronic or prolonged denervation) and delayed neuronal regeneration to their targets (termed chronic or prolonged axotomy), we studied the number of regenerated motoneurons and reinnervated motor units. Figure 230-7 illustrates our surgical paradigm and methods of assessment of axonal regeneration and muscle reinnervation after nerve injury and repair.
Chronic Schwann Cell Denervation
Provision of the growth-supportive environment by SCs is intimately related to loss and timely reestablishment of axonal contact with the cells.84 Schwann cells whose reinnervation by regenerating axons is delayed are said to be “chronically denervated.” The deleterious effect of chronic denervation on SCs worsens as the duration of chronic denervation is prolonged.84,85 In our experiments, the number of motoneurons that were back-labeled with fluororuby dye applied to the distal nerve stump 10 mm from the cross-suture site was reduced after chronic denervation of more than 6 months, to less than 10% of the number that regenerated after immediate suture of the nerve stumps (Fig. 230-7). There was excellent correspondence between the proportion of motoneurons that regenerated their axons into the chronically denervated nerve stump and the proportion of freshly axotomized motoneurons that regenerated and reinnervated the denervated muscle after 4 to 6 months85 (Fig. 230-7). The reduced number of motoneurons that regenerate their axons through the chronically denervated SCs not only reinnervated the denervated muscle fibers but also reinnervated up to three times the number of muscle fibers that they normally do. Because this enlargement of the reinnervated motor units constitutes the maximum sprouting capacity of the motoneurons,84,85 it follows that it is not the inability of chronically denervated muscle fibers to accept reinnervation that limits functional recovery after chronic nerve injuries.71,84,85 A clinical correlate of our experimental cross-suture paradigm is injury to large nerve stumps in which SCs of the distal nerve stumps are left chronically denervated because of the slow rate of regenerating axons. The longer it takes for regenerating axons to reinnervate SCs in the distal nerve stumps, the more prolonged the period of chronic denervation during which SCs do not have contact with axons and the greater the likelihood that their capacity to support regenerating axons is impaired70,84,85 (Fig. 230-8D; also see Fig. 230-7D). We believe that this is partly why reinnervation and thereby functional recovery in muscle targets located close to regenerating motoneurons are better than in more distally placed muscle targets.
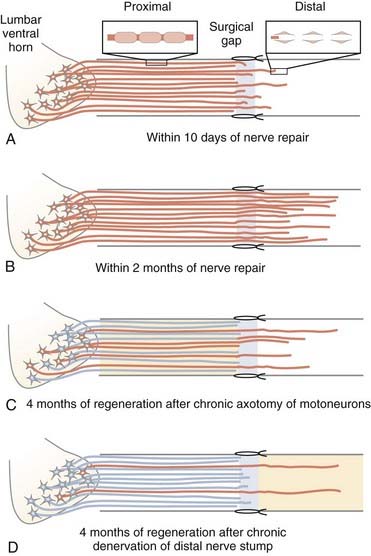
FIGURE 230-8 Factors that limit functional recovery after nerve injury and surgical repair. Even after microsurgical repair of injured nerves, a surgical gap is left between the proximal and distal nerve stumps. Schwann cells divide, multiply, and form the bands of Büngner that line the endoneurial tubes to support, guide, and finally myelinate the regenerating axons. A, Within the disarray of the extracellular matrix provided by disorganized proliferation of scar tissue and absence of the defined structures of the connective tissue nerve sheaths at the repair site, axons grow out from the proximal nerve stump and often emit multiple sprouts (not shown) that grow toward and into the distal nerve stump, where in turn the growing axons are guided by Schwann cells lining the endoneurial tubes. The “staggered axon regeneration” across the suture site proceeds slowly as indicated by the relatively small number of axons that enter the distal nerve stumps during early regeneration. B, After immediate nerve repair, all axons may regenerate across the suture site and reinnervate the distal nerve stump (successful regeneration denoted by an orange neuron/axon). The multiple regenerating branches in the distal nerve stumps are gradually withdrawn after connections are made with denervated muscles. The process of withdrawal of axons is also slow and takes years to complete.73 C, If nerve repair is delayed or in the clinically frequent circumstance of regeneration of axons over long distances to denervated target connections, the regenerative capacity of the chronically axotomized neurons progressively declines such that fewer axons regenerate successfully into the distal stump (denoted by orange) and remake functional connections (not shown). D, Regeneration through the long-term chronically denervated Schwann cells also declines progressively because the atrophic Schwann cells are less able to support axon regeneration. However, once axons do regenerate, the Schwann cells support their growth and remyelinate the axons.84
Chronic Neuronal Axotomy
Injured neurons whose axons may or may not be regenerating but have not made target connections are said to be subjected to “chronic axotomy.”86–88 For example, after brachial plexus injury, the injured neurons have to regenerate over long distance before they can reinnervate some of the denervated muscles and are thereby being subjected to chronic axotomy. Using our outcome measures described earlier, we found that the number of motoneurons that regenerated their axons fell progressively as a function of prolonged duration of chronic axotomy to approximately 37% of those that regenerated without the effect of chronic axotomy88,89 (see Figs. 230-7C and 230-8C). This reduced capacity to regenerate after chronic axotomy is significant, especially when combined with the deleterious effect of chronic denervation, both of which occur concurrently, particularly after injuries to large nerve trunks such as the brachial plexus.
Staggered Axon Regeneration and Misdirection of Regenerating Axons
One of the key prerequisites for successful functional recovery is that regenerating axons regenerate into the correct endoneurial tubes that direct them back to their original target organs. However, we found that regenerating axons encounter a significant delay at the injury site and traverse the injury site into the distal nerve stumps in a staggered fashion (Fig. 230-8A; i.e., staggered axonal regeneration). A factor largely responsible for both misdirection of regenerating axons and their delay in arriving at the injury or repair site is the disorganized proliferation of scar tissue seen at that level, especially in human and other primate species. Indeed, crossing of regenerating axons occurs slowly before the entry of regenerating axons into the distal nerve stump. It is only once the axons enter the distal stumps that they regenerate at the slow rate of transport of 1 to 3 mm/day (Fig. 230-8B). After immediate nerve repair, axon regeneration proceeds and all neurons regenerate their axons to reach their targets. However, there is a degree of misdirection of the regenerating axons such that a proportion of both motor and sensory axons fail to regenerate into their appropriate motor and sensory pathways.22,90 The proportion of neurons that regenerate into inappropriate pathways may be as high as 30%. We believe that this misdirection of regenerating injured axons plays an important role in reducing functional recovery after nerve injuries.
Conclusion
A tremendous amount of progress has been made in our understanding of the microanatomy, pathophysiology, and microsurgical management of injured nerves. These advances have improved the quality of care provided to patients sustaining nerve injuries and often result in better functional recovery. However, the other groups of patients who fail to recover good function despite excellent microsurgical care pose a challenge to the nerve surgeon. The upregulation of RAGs is short-lived such that there appears to be a window of opportunity during which SCs provide a growth-supportive environment and the injured neurons can regenerate their axons. This time-limited upregulation of RAGs and the slow rate of axonal regeneration result in progressive loss of neurotrophic support for the injured neurons and their regenerating axons, and hence chronic SC denervation and chronic neuronal axotomy ensue.70,71,88 Therefore, two possible approaches to combat the gap between the timing of upregulation of RAGs and the slow rate of regeneration will be to (1) accelerate the rate of axonal regeneration or (2) sustain the neurotrophic environment for regenerating axons for longer periods. Brief (1 hour) low-frequency electrical stimulation has been shown to accelerate axonal regeneration by modulating the expression of RAGs.22,90,91 Likewise, the beneficial effects of the application of neurotrophins on axonal regeneration have been demonstrated, either exogenously74,86 or by the use of genetically engineered lentivirus to stimulate the production of neurotrophins.92 Reactivation of chronically denervated SCs by exposing them to cytokines such as transforming growth factor-β has been shown to promote nerve regeneration,93 and transplantation of stem cells into the distal nerve stumps also shows very promising results with regard to nerve regeneration.94 Extensive research has also been done in the design of enhanced nerve guidance channels or conduits that will not only eliminate the need for autografts but also allow positive modulation of the growth-permissive environment for axons traversing the guidance channels into the distal nerve stumps.95,96 Perhaps the ultimate solution to improve functional recovery in the subset of patients who currently do not do well with microsurgical repair is a combination of several neurobiologic approaches to overcome the challenge of the limited time window for optimal nerve regeneration.
Bisby MA, Tetzlaff W. Changes in cytoskeletal protein synthesis following axon injury and during axon regeneration. Mol Neurobiol. 1992;6:107-123.
Boyd JG, Gordon T. Glial cell line–derived neurotrophic factor and brain-derived neurotrophic factor sustain the axonal regeneration of chronically axotomized motoneurons in vivo. Exp Neurol. 2003;183:610-619.
Boyd JG, Gordon T. A dose-dependent facilitation and inhibition of peripheral nerve regeneration by brain-derived neurotrophic factor. Eur J Neurosci. 2002;15:613-626.
Brushart TM, Hoffman PN, Royall RM, et al. Electrical stimulation promotes motoneuron regeneration without increasing its speed or conditioning the neuron. J Neurosci. 2002;22:6631-6638.
Cajal SR. Degeneration and Regeneration of the Nervous System [translated by R. M. May]. New York: Oxford University Press; 1928.
Fu SY, Gordon T. The cellular and molecular basis of peripheral nerve regeneration. Mol Neurobiol. 1997;14:67-116.
Fu SY, Gordon T. Contributing factors to poor functional recovery after delayed nerve repair: prolonged denervation. J Neurosci. 1995;15:3886-3895.
Furey MJ, Midha R, Xu QG, et al. Prolonged target deprivation reduces the capacity of injured motoneurons to regenerate. Neurosurgery. 2007;60:723-732.
Gentili F, Hudson AR, Midha R. Peripheral nerve injuries: types, causes, and grading. In: Wilkins RH, Rengachary SS, editors. Neurosurgery. New York: McGraw-Hill; 1996:3105-3114.
Gutmann E, Young JZ. The re-innervation of muscle after various periods of atrophy. J Anat. 1944;78:15-44.
Hoke A, Gordon T, Zochodne DW, et al. A decline in glial cell-line–derived neurotrophic factor expression is associated with impaired regeneration after long-term Schwann cell denervation. Exp Neurol. 2002;173:77-85.
Kline DG, Hudson AR. Acute injuries of peripheral nerves. In: Youmans J, editor. Neurological Surgery. Philadelphia: WB Saunders, 1990.
Lundborg G. Nerve regeneration. In: Lundborg G, editor. Nerve Injury and Repair. London: Churchill Livingstone; 1988:149-195.
Mackinnon SE, Dellon AL. Surgery of the Peripheral Nerve. New York: Thieme Medical; 1988.
Martini R. Expression and functional roles of neural cell surface molecules and extracellular matrix components during development and regeneration of peripheral nerves. J Neurocytol. 1994;23:1-28.
Midha R, Kline DG. Evaluation of the neuroma in continuity. In: Omer GE, Spinner M, Van Beek AL, editors. Management of Peripheral Nerve Problems. 2nd ed. Philadelphia: WB Saunders; 1998:319-327.
Rudge P, Ochoa J, Gilliatt RW. Acute peripheral nerve compression in the baboon. J Neurol Sci. 1974;23:403-420.
Seddon HJ. Three types of nerve injury. Brain. 1943;66:238-288.
Sulaiman OAR, Boyd JG, Gordon T. Axonal regeneration in the peripheral system of mammals. In: Kettenmann H, Ransom BR, editors. Neuroglia. 2nd ed. Oxford: Oxford University Press; 2005:454-466.
Sulaiman OAR, Gordon T. Transforming growth factor-beta and forskolin attenuate the adverse effects of long-term Schwann cell denervation on peripheral nerve regeneration in vivo. Glia. 2002;37:206-218.
Sulaiman OAR, Gordon T. Effects of short- and long-term Schwann cell denervation on peripheral nerve regeneration, myelination, and size. Glia. 2000;32:234-246.
Sunderland S. Nerve and Nerve Injuries. Edinburgh: Livingstone; 1978.
Toft PB, Fugleholm K, Schmalbruch H. Axonal branching following crush lesions of peripheral nerves of rat. Muscle Nerve. 1988;11:880-889.
Waller A. Experiments on the section of the glossopharyngeal and hypoglossal nerves of the frog, and observations of the alterations produced thereby in the structure of their primitive fibres. Philos Trans R Soc Lond. 1850;140:423-429.
You S, Petrov T, Chung PH, et al. The expression of the low affinity nerve growth factor receptor in long-term denervated Schwann cells. Glia. 1997;20:87-100.
1 Kline DG. Physiological and clinical factors contributing to the timing of nerve repair. Clin Neurosurg. 1977;24:425-455.
2 Sunderland S. Nerve and Nerve Injuries. Baltimore: Williams & Wilkins; 1968.
3 Seddon HJ. Three types of nerve injury. Brain. 1943;66:238-288.
4 Sunderland S. A classification of peripheral nerve injuries producing loss of function. Brain. 1951;74:491-516.
5 Hudson AR, Hunter D. Timing of peripheral nerve repair: important local neuropathological factors. Clin Neurosurg. 1977;24:391-405.
6 Ducker TB. Pathophysiology of peripheral nerve trauma. In: Omer GE, Spinner M, editors. Management of Peripheral Nerve Problems. Philadelphia: WB Saunders, 1980.
7 Gentili F, Hudson AR, Midha R. Peripheral nerve injuries: types, causes, and grading. In: Wilkins RH, Rengachary SS, editors. Neurosurgery. New York: McGraw-Hill; 1996:3105-3114.
8 Sunderland S. Nerve and Nerve Injuries. Edinburgh: Livingstone; 1978.
9 Lundborg G, Rydevik B. Effects of stretching the tibial nerve of the rabbit. A preliminary study of the intraneural circulation and the barrier function of the perineurium. J Bone Joint Surg Br. 1973;55:390-401.
10 Liu CT, Benda CE, Lewey FH. Tensile strength of human nerves: experimental physiological and histological study. Arch Neurol Psychiatry. 1948;59:322-336.
11 Speed JS, Knight RA. Peripheral nerve injuries. In: Campbell’s Operative Orthopedics. St. Louis: C.V. Mosby; 1956:947-1014.
12 Highet J. Effects of stretch on peripheral nerve. Br J Surg. 1942;30:355-369.
13 Kline DG, Hudson AR. Acute injuries of peripheral nerves. In: Youmans J, editor. Neurological Surgery. Philadelphia: WB Saunders, 1990.
14 Tiel RL, Happel LTJr, Kline DG. Nerve action potential recording method and equipment. Neurosurgery. 1996;39:103-108.
15 Seddon HJ. Surgical Disorders of the Peripheral Nerves. Baltimore: Williams & Wilkins; 1972.
16 Seddon HJ. Peripheral Nerve Injuries: Special Report Series No. 282. London: HMSO: Medical Research Council; 1954.
17 Zachary RB, Raof R. Lesions in continuity. Spec Rep Ser Med Res Counc (G B). 1954;282:57-81.
18 Waller A. Experiments on the section of the glossopharyngeal and hypoglossal nerves of the frog, and observations of the alterations produced thereby in the structure of their primitive fibres. Philos Trans R Soc Lond. 1850;140:423-429.
19 Martini R. Expression and functional roles of neural cell surface molecules and extracellular matrix components during development and regeneration of peripheral nerves. J Neurocytol. 1994;23:1-28.
20 Siironen J, Sandberg M, Vuorinen V, et al. Expression of type I and III collagens and fibronectin after transection of rat sciatic nerve. Reinnervation compared with denervation. Lab Invest. 1992;67:80-87.
21 Morris JH, Hudson AR, Weddell G. A study of degeneration and regeneration in the divided rat sciatic nerve based on electron microscopy. II. The development of the “regenerating unit.”. Z Zellforsch Mikrosk Anat. 1972;124:103-130.
22 Al-Majed AA, Brushart TM, Gordon T. Electrical stimulation accelerates and increases expression of BDNF and trkB mRNA in regenerating rat femoral motoneurons. Eur J Neurosci. 2000;12:4381-4390.
23 Kline DG. Macroscopic and microscopic concomitants of nerve repair. Clin Neurosurg. 1979;26:582-606.
24 Woodhall B, Nulsen FE, White JC, et al. Neurosurgical Implications. Peripheral Nerve Regeneration. Washington, D.C.: Veterans Administration Monograph; 1957.
25 Midha R, Kline DG. Evaluation of the neuroma in continuity. In: Omer GE, Spinner M, Van Beek AL, editors. Management of Peripheral Nerve Problems. 2nd ed. Philadelphia: WB Saunders; 1998:319-327.
26 Midha R. Epidemiology of brachial plexus injuries in a multitrauma population. Neurosurgery. 1997;40:1182-1188.
27 Noble J, Munro CA, Prasad VS, et al. Analysis of upper and lower extremity peripheral nerve injuries in a population of patients with multiple injuries. J Trauma. 1998;45:116-122.
28 Sunderland S. Nerve Injuries and Their Repair. A Critical Appraisal. Melbourne: Churchill Livingstone; 1991.
29 Bowden REM, Abdullah S, Gooding MR. Anatomy of the cervical spine, membranes, spinal cord, nerve roots, and brachial plexus, in brain. In: Wilkinson MF, editor. Cervical Spondylosis and Other Disorders of the Cervical Spine. Philadelphia: WB Saunders, 1967.
30 Denny-Brown D, Brenner C. Paralysis of nerve induced by direct pressure and tourniquet. Arch Neurol Psychiatry. 1944;51:1-26.
31 Weisl H, Osbourne GV. The pathological changes in rats’ nerves subject to moderate compression. J Bone Joint Surg Br. 1964;46:297-306.
32 Eames RA, Lange LS. Clinical and pathological study of ischaemic neuropathy. J Neurol Neurosurg Psychiatry. 1967;30:215-226.
33 Williams IR, Jefferson D, Gilliatt RW. Acute nerve compression during limb ischaemia—an experimental study. J Neurol Sci. 1980;46:199-207.
34 Lundborg G. Nerve regeneration. In: Lundborg G, editor. Nerve Injury and Repair. London: Churchill Livingstone; 1988:149-195.
35 Lundborg G. Ischemic nerve injury. Experimental studies on intraneural microvascular pathophysiology and nerve function in a limb subjected to temporary circulatory arrest. Scand J Plast Reconstr Surg Suppl. 1970;6:3-113.
36 Aguayo AJ. Neuropathy due to compression and entrapment. In: Dyck PJ, Thomas PK, Lambert EH, editors. Peripheral Neuropathy. Philadelphia: WB Saunders; 1975:688-713.
37 Aguayo A, Nair CP, Midgley R. Experimental progressive compression neuropathy in the rabbit. Histologic and electrophysiologic studies. Arch Neurol. 1971;24:358-364.
38 Ochoa J, Marotte L. The nature of the nerve lesion caused by chronic entrapment in the guinea-pig. J Neurol Sci. 1973;19:491-495.
39 Fullerton PM, Gilliatt RW, Lascelles RG, et al. The relation between fibre diameter and internodal length in chronic neuropathy. Proc Physiol Soc. 1965;19-20:26P-28P.
40 Fullerton PM, Gilliatt RW. Median and ulnar neuropathy in the guinea-pig. J Neurol Neurosurg Psychiatry. 1967;30:393-402.
41 Fullerton PM, Gilliatt RW. Pressure neuropathy in the hind foot of the guinea-pig. J Neurol Neurosurg Psychiatry. 1967;30:18-25.
42 Rudge P, Ochoa J, Gilliatt RW. Acute peripheral nerve compression in the baboon. J Neurol Sci. 1974;23:403-420.
43 Mackinnon SE, Dellon AL. Surgery of the Peripheral Nerve. New York: Thieme Medical; 1988.
44 Parks BJ. Postoperative peripheral neuropathies. Surgery. 1973;74:348-357.
45 Gilden DH, Eisner J. Lumbar plexopathy caused by disseminated intravascular coagulation. JAMA. 1977;237:2846-2847.
46 Upton AR, McComas AJ. The double crush in nerve entrapment syndromes. Lancet. 1973;2:359-362.
47 Spinner M. Injuries to the Major Branches of the Peripheral Nerve of the Forearm. Philadelphia: WB Saunders; 1978.
48 Kimura J. Electrodiagnosis in Diseases of Nerve and Muscles: Principles and Practice. Philadelphia: FA Davis; 1983.
49 DiVincenti FC, Moncrief JA, Pruitt BAJr. Electrical injuries: a review of 65 cases. J Trauma. 1969;9:497-507.
50 Aita JA. Neurologic manifestations of electrical injury. Nebr State Med J. 1965;50:530-533.
51 Fischer H. Pathological effects and sequelae of electrical accidents. Electrical burns (secondary accidents, renal manifestations, sequelae). J Occup Med. 1965;7:564-571.
52 Grube BJ, Heimbach DM, Engrav LH, et al. Neurologic consequences of electrical burns. J Trauma. 1990;30:254-258.
53 Gilbert H, Kagen AR. Radiation Damage to the Nervous System. New York: Raven Press; 1980.
54 Match RM. Radiation-induced brachial plexus paralysis. Arch Surg. 1975;110:384-386.
55 Gentili F, Hudson AR, Hunter D. Clinical and experimental aspects of injection injuries of peripheral nerves. Can J Neurol Sci. 1980;7:143-151.
56 Midha R, Guha A, Gentili F, et al. Peripheral nerve injection injury. In: Omer GE, Spinner M, Van Beek AL, editors. Management of Peripheral Nerve Problems. 2nd ed. Philadelphia: WB Saunders; 1999:406-413.
57 Broadbent TR, Odom GL, Woodhall B. Peripheral nerve injuries from administration of penicillin; report of four clinical cases. JAMA. 1949;140:1008-1010.
58 Kolb LC, Gray SJ. Peripheral neuritis as a complication of penicillin therapy. JAMA. 1946;132:323-326.
59 Mackinnon SE, Hudson AR, Gentili F, et al. Peripheral nerve injection injury with steroid agents. Plast Reconstr Surg. 1982;69:482-490.
60 Pizzolato P, Mannheimei W. Histopathologic Effects of Local Anesthetic Drugs and Related Substances. Springfield, IL: Charles C Thomas; 1961.
61 Gentili F, Hudson AR, Hunter D, et al. Nerve injection injury with local anesthetic agents: a light and electron microscopic, fluorescent microscopic, and horseradish peroxidase study. Neurosurgery. 1980;6:263-272.
62 Villarejo FJ, Pascual AM. Injection injury of the sciatic nerve (370 cases). Childs Nerv Syst. 1993;9:229-232.
63 Matson DD. Early neurolysis in the treatment of injury of the peripheral nerves due to faulty injection of antibiotics. N Engl J Med. 1950;242:973-975.
64 Merren MD. Gabapentin for treatment of pain and tremor: a large case series. South Med J. 1998;91:739-744.
65 Kingery WS. A critical review of controlled clinical trials for peripheral neuropathic pain and complex regional pain syndromes. Pain. 1997;73:123-139.
66 Kline DG, Kim D, Midha R, et al. Management and results of sciatic nerve injuries: a 24-year experience. J Neurosurg. 1998;89:13-23.
67 Cajal SR. Degeneration and Regeneration of the Nervous System [translated by R. M. May]. New York: Oxford University Press; 1928.
68 Erturk A, Hellal F, Enes J, et al. Disorganized microtubules underlie the formation of retraction bulbs and the failure of axonal regeneration. J Neurosci. 2007;27:9169-9180.
69 McQuarrie IG. Effect of conditioning lesion on axonal sprout formation at nodes of Ranvier. J Comp Neurol. 1985;231:239-249.
70 Fu SY, Gordon T. The cellular and molecular basis of peripheral nerve regeneration. Mol Neurobiol. 1997;14:67-116.
71 Sulaiman OAR, Boyd JG, Gordon T. Axonal regeneration in the peripheral system of mammals. In: Kettenmann H, Ransom BR, editors. Neuroglia. 2nd ed. Oxford: Oxford University Press; 2005:454-466.
72 Aitken JT, Sharman M, Young JZ. Maturation of peripheral nerve fibres with various peripheral connections. J Anat. 1947;81:1-22.
73 Toft PB, Fugleholm K, Schmalbruch H. Axonal branching following crush lesions of peripheral nerves of rat. Muscle Nerve. 1988;11:880-889.
74 Navarro X, Vivo M, Valero-Cabre A. Neural plasticity after peripheral nerve injury and regeneration. Prog Neurobiol. 2007;82:163-201.
75 Sulaiman OAR, Gordon T. Cellular and molecular interactions after peripheral and central injury. Biomed Rev. 2003;14:51-62.
76 Hoke A, Mi R. In search of novel treatments for peripheral neuropathies and nerve regeneration. Discov Med. 2007;7:109-112.
77 Scherer SS, Salzer JL. Axon-Schwann cell interactions during peripheral nerve degeneration and regeneration. In: Jessen KR, Richardson WD, editors. Glial Cell Development: Basic Principles and Clinical Relevance. Oxford, UK: Bios Scientific; 1996:169-196.
78 Hoke A, Gordon T, Zochodne DW, et al. A decline in glial cell-line–derived neurotrophic factor expression is associated with impaired regeneration after long-term Schwann cell denervation. Exp Neurol. 2002;173:77-85.
79 You S, Petrov T, Chung PH, et al. The expression of the low affinity nerve growth factor receptor in long-term denervated Schwann cells. Glia. 1997;20:87-100.
80 Bisby MA, Tetzlaff W. Changes in cytoskeletal protein synthesis following axon injury and during axon regeneration. Mol Neurobiol. 1992;6:107-123.
81 Boyd JG, Gordon T. Neurotrophic factors and their receptors in axonal regeneration and functional recovery after peripheral nerve injury. Mol Neurobiol. 2003;27:277-324.
82 Gutmann E. Effect of delay of innervation on recovery of muscle after nerve lesions. J Neurophysiol. 1948;11:279-294.
83 Gutmann E, Young JZ. The re-innervation of muscle after various periods of atrophy. J Anat. 1944;78:15-44.
84 Sulaiman OAR, Gordon T. Effects of short- and long-term Schwann cell denervation on peripheral nerve regeneration, myelination, and size. Glia. 2000;32:234-246.
85 Fu SY, Gordon T. Contributing factors to poor functional recovery after delayed nerve repair: prolonged denervation. J Neurosci. 1995;15:3886-3895.
86 Boyd JG, Gordon T. Glial cell line–derived neurotrophic factor and brain-derived neurotrophic factor sustain the axonal regeneration of chronically axotomized motoneurons in vivo. Exp Neurol. 2003;183:610-619.
87 Furey MJ, Midha R, Xu QG, et al. Prolonged target deprivation reduces the capacity of injured motoneurons to regenerate. Neurosurgery. 2007;60:723-732.
88 Fu SY, Gordon T. Contributing factors to poor functional recovery after delayed nerve repair: prolonged axotomy. J Neurosci. 1995;15:3876-3885.
89 Boyd JG, Gordon T. A dose-dependent facilitation and inhibition of peripheral nerve regeneration by brain-derived neurotrophic factor. Eur J Neurosci. 2002;15:613-626.
90 Al-Majed AA, Neumann CM, Brushart TM, et al. Brief electrical stimulation promotes the speed and accuracy of motor axonal regeneration. J Neurosci. 2000;20:2602-2608.
91 Brushart TM, Hoffman PN, Royall RM, et al. Electrical stimulation promotes motoneuron regeneration without increasing its speed or conditioning the neuron. J Neurosci. 2002;22:6631-6638.
92 Kwon BK, Liu J, Lam C, et al. Brain-derived neurotrophic factor gene transfer with adeno-associated viral and lentiviral vectors prevents rubrospinal neuronal atrophy and stimulates regeneration-associated gene expression after acute cervical spinal cord injury. Spine. 2007;32:1164-1173.
93 Sulaiman OAR, Gordon T. Transforming growth factor-beta and forskolin attenuate the adverse effects of long-term Schwann cell denervation on peripheral nerve regeneration in vivo. Glia. 2002;37:206-218.
94 Heine W, Conant K, Griffin JW, et al. Transplanted neural stem cells promote axonal regeneration through chronically denervated peripheral nerves. Exp Neurol. 2004;189:231-240.
95 Midha R. Emerging techniques for nerve repair: nerve transfers and nerve guidance tubes. Clin Neurosurg. 2006;53:185-190.
96 Pfister LA, Papaloizos M, Merkle HP, et al. Nerve conduits and growth factor delivery in peripheral nerve repair. J Peripher Nerv Syst. 2007;12:65-82.






