CHAPTER 75 PATHOPHYSIOLOGY OF MULTIPLE SCLEROSIS: DEMYELINATION AND AXONAL INJURY
The inflammatory demyelinating lesions of multiple sclerosis cause a range of axonal conduction abnormalities that, in turn, produce the major clinical features of the illness. The clinical effects of these lesions were previously thought to arise directly from the consequences of the demyelination alone, but more recent findings have substantially broadened this understanding. It is now recognized that (1) conduction abnormalities reflect a reorganization of axonal membrane currents after demyelination, (2) inflammation may have direct effects on axonal function, and (3) neuroaxonal degeneration plays a significant role in disease progression.
DEMYELINATION
Action potentials generated by inward sodium currents at the nodes of Ranvier propagate successfully along normal myelinated axons because of the insulating layer of myelin covering the internode. The thickness of the myelin reduces the leakage of current between the nodes by increasing the resistance of the membrane and, of more importance, by reducing its capacitance. These properties have the effect of reducing the leakage of current between nodes and thus reducing the current that needs to be generated at the node for secure conduction to occur. In normal myelinated axons, the action potential at an individual node generates three to seven times more current than required to discharge the next node along the axon; this is referred to as a safety factor of three to seven.1
It is now clear that the membrane of the adult myelinated axon has a complicated molecular structure (Fig. 75-1).2,3 Apart from sodium channels, which are clustered at the nodes and expressed at a much lower density in the internodal membrane, there are also a number of potassium channels, including so-called slow channels, located mainly at the nodes, and faster channels, located in the juxtaparanodal region. These serve to limit the excitability of the fiber.4 There are a number of other potassium channels, of which some are inhibited by adenosine triphosphate (ATP) and others are activated by intracellular calcium ions. The axonal membrane is also known to contain calcium channels, but these do not appear to contribute significantly to action potential generation or propagation, at least under normal conditions. The ionic balance across the axonal membrane is maintained by a number of ATP-dependent pumps, including the sodium and calcium pumps, and calcium homeostasis also depends on the sodium/calcium exchanger.
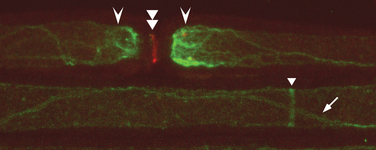
Demyelination reduces the safety factor for conduction, and conduction fails, at least initially, if an internode or more of myelin is removed (Fig. 75-2). The failure occurs partly because the local excitatory sodium current generated by the last node of Ranvier before the demyelinated segment is now dispersed across a much wider area of naked axonal membrane than just the nodal membrane. Even partial-thickness demyelination increases the effective capacitance of the axolemma as the thickness of the myelin is reduced, increasing the amount of local current necessary to depolarize the nodal membrane to its firing threshold and perhaps resulting in conduction block. Conduction is also likely to be blocked, or severely impaired, if the myelin loss occurs preferentially at the paranodes, which causes nodal widening and increases the capacitance of the “nodal” axolemma. Although conduction may later be restored (see later discussion), the density of sodium channels in the newly exposed axolemma is initially too low to support conduction along the demyelinated axolemma.5,6
(Reproduced from Smith KJ, Blakemore WF, McDonald WI: Central remyelination restores secure conduction. Nature 1979; 280:395-396. Reprinted by permission from Macmillan Publishers Ltd. Copyright 1979. http://www.nature.com/index.html.)
Effects of Temperature
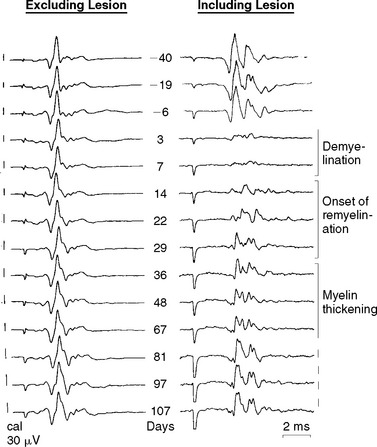
Small changes in temperature can have dramatic effects on conduction in demyelinated axons7–10 (Fig. 75-3), because the safety factor in demyelinated axons is typically reduced near to unity. The success of conduction in many demyelinated axons is therefore finely balanced, inasmuch as small changes can tip the safety factor either to just below unity, in which case conduction is blocked, or to just above unity, in which case conduction will succeed. Temperature changes affect the safety factor by altering the kinetics of the sodium channels.11 Warming shortens the action potential,12 thereby reducing the time that current is able to flow to depolarize the demyelinated region to its firing threshold, and cooling has the opposite effect.
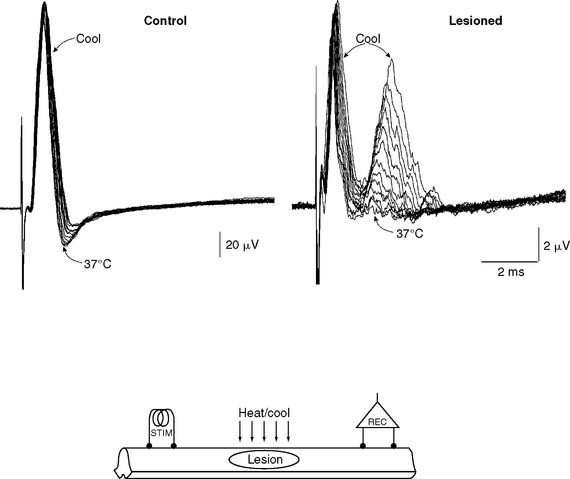
(Reproduced from Smith KJ: Conduction properties of central demyelinated and remyelinated axons, and their relation to symptom production in demyelinating disorders. Eye 1994; 8:224-237. Reprinted by permission from Macmillan Publishers Ltd. Copyright 1979. http://www.nature.com/eye/index.html.)
Even subtle changes in body temperature can profoundly alter the expression of symptoms, such as in the visual system (Uhthoff’s phenomenon).13 Thus, vision can improve simply by drinking a glass of cold water,14 whereas a hot shower or bath, or sunbathing, can aggravate disability significantly.15 Indeed, in earlier years, the deleterious effects of body warming underpinned the “hot bath test” for multiple sclerosis. The therapeutic benefits both of body cooling11,16 and of prolonging the action potential17,18 have been considered. Thus, the potassium channel-blocking drugs 4-aminopyridine and 3,4-diaminopyridine can prolong the action potential, favoring successful conduction in demyelinated axons.19 These drugs have been found to improve disability in clinical trials in multiple sclerosis,20 although it has also been suggested that this effect could arise from the potentiation of synaptic transmission.21 The widespread use of aminopyridines has been inhibited by the fact that they are markedly proconvulsant at doses that are not much higher than the therapeutic dose.22
Activity-Dependent Conduction Block
Apart from fixed conduction block, demyelinated axons with a safety factor just above unity have a prolonged refractory period for conduction through the lesion23 (Fig. 75-4) and can also exhibit intermittent conduction failure as a result of periods of sustained electrical activity. The rise in the intra-axonal sodium concentration with repetitive firing stimulates the activity of the electrogenic Na+/K+-ATPase (sodium pump) and leads to axonal hyperpolarization.24 Even a small hyperpolarization can effectively, and abruptly, block conduction in such demyelinated axons for periods of approximately 0.2 to 2 seconds.25 Conduction resumes as the intra-axonal sodium concentration recovers and pump activity falls, and this cycle may then be repeated, causing a continuous train of impulses to be divided into short bursts separated by silent periods. The intermittent failure of impulse transmission on sustained activation may contribute significantly to the fatigability experienced by many patients with multiple sclerosis: for example, those with spinal cord lesions whose gait becomes impaired on walking short distances, or those whose vision “fades” or becomes blurred on fixated gaze, particularly in bright surroundings.26,27 There is therefore a potential therapeutic role for partial blockade of Na+/K+-ATPase, and indeed some benefit with such a therapy has been demonstrated in a small number of patients.28
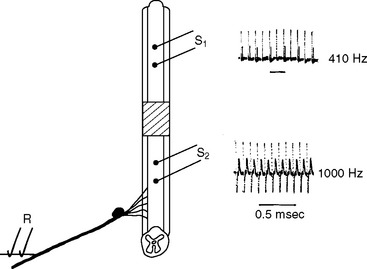
REMYELINATION
There is ample evidence that demyelinated axons can undergo repair by remyelination in multiple sclerosis29,30 and that remyelination is likely to play a major role in restoring conduction and in the remission of neurological deficits after relapses.31 New internodes are thinner and shorter than normal, but remyelinated axons can nonetheless conduct, even when invested with only thin new myelin sheaths composed of as few as five myelin lamellae.32 New nodes formed by remyelination show aggregations of sodium channel immunoreactivity,33 and, at least in the peripheral nervous system, the new nodes are excitable.34 Not only do axons repaired by remyelination conduct (see Fig. 75-2) but also remyelination restores the security5,35 and velocity of conduction to near-normal values regardless of whether the repair is effected primarily by oligodendrocytes,35 Schwann cells (even within the central nervous system),36,37 or transplanted olfactory ensheathing cells.38 Although the role of axonal impulse activity in promoting myelination and remyelination remains controversial, it is possible that because demyelinated axons often exhibit conduction block, the absence of electrical activity could contribute to the failure of remyelination in some multiple sclerosis lesions.39,40 Remyelination is likely to confer not only secure conduction to repaired axons but also protection from degeneration, and developing strategies to promote remyelination is an active area of research.
ADAPTIVE CHANGES IN THE EXPRESSION OF ION CHANNELS IN DEMYELINATION
The clustering of voltage-sensitive ion channels and other axolemmal proteins, such as the L1 cell adhesion molecule family members neurofascin and NrCAM, and 480/270kD ankyrin at nodes of Ranvier,41 requires contact with, or at least close proximity to, myelinating Schwann cells in the peripheral nervous system and with oligodendrocytes in the central nervous system.42–46 Because maintenance of ion channel distribution also seems to depend, at least in part, on the presence of compact myelin and the formation of normal, tight paranodal axoglial junctions,47–50 demyelination can produce changes in the patterns of expression of these ion channels. In fact, the restoration of conduction in demyelinated axons involves substantial remodeling of the axonal membrane at a molecular level. Immunocytochemical studies have revealed an increased expression of two different isoforms of voltagegated sodium channels, Nav 1.6 and Nav 1.2, along demyelinated axons.47,48,51 Both of these sodium channels, particularly Nav 1.6, can generate persistent sodium currents, which may be important in axonal degeneration (see later discussion). The Nav 1.8 channel, which is normally expressed only in adult spinal sensory and trigeminal neurons, has also been shown to occur aberrantly in Purkinje cells in multiple sclerosis and in experimental autoimmune encephalomyelitis and potentially to contribute to cerebellar dysfunction.52–55
On physiological examination, both a seemingly continuous distribution of sodium channels56 and, in contrast, an aggregation of sodium channels into “φ-nodes,”34 have been described. φ-Nodes appear to be the precursors of the new nodes of Ranvier formed during remyelination, and it may be significant that the initial observation of a continuous distribution of sodium channels was made in a lesion (diphtheria toxin) in which repair by remyelination is only slowly achieved. It is still not clear how sodium channels aggregate during remyelination to form the new nodes of Ranvier. The formation of the channel aggregations may be driven by the axon and may also depend on the adjacent myelinating cells.33,44,46,50,57,58
These adaptive changes can, in some axons, lead to the restoration of conduction, even in the absence of repair by remyelination. Factors favoring the restoration of conduction in a particular axon include a small diameter59,60 and either a short internode59,61,62 or a widened node before the demyelinated region. The first two of these factors characterize axons in the optic nerve, and they may contribute to the excellent recovery of vision that can follow optic neuritis. However, axons as large as 5.5μm in diameter have been shown to conduct in the absence of remyelination, which raises the possibility that most central axons might be able to conduct under ideal conditions. This possibility may help explain the presence in some patients of clinically silent demyelinating lesions in pathways that usually manifest in symptom production.63–67
The restoration of conduction in segmentally demyelinated axons was first demonstrated by a sophisticated examination of conduction in spinal root axons demyelinated by the intrathecal injection of diphtheria toxin56; nearly 20 years were to pass before there was proof that conduction could also occur in demyelinated central axons,32 although circumstantial evidence had accumulated.68–71 In the peripheral nervous system, conduction can resume as soon as 6 days after the induction of a demyelinating lesion, but in the central nervous system, longer intervals (e.g., 2 to 3 weeks) appear to be required. Once restored, conduction is continuous, or microsaltatory, rather than saltatory, and it is therefore much slower than normal and also less secure. The velocity of conduction along a demyelinated segment of rat ventral root axons is reduced from normal, to the range of 0.7 to 2.3m/second34,56 and similar values are likely to apply in the central nervous system. Even though this degree of slowing occurs across only the demyelinated portion of an axon, it is still sufficient to disperse and delay the compound action potential and, perhaps coupled with a reduction in the total number of axons conducting, to result in diagnostically valuable delays in the visual,72 somatosensory, and brainstem auditory evoked potentials. Slowing of conduction (as opposed to conduction block) generally causes few symptoms, although sensations dependent on the precise timing of impulses in different axons may be affected. For example, patients with unilateral optic neuritis may perceive the Pulfrich phenomenon (the tendency of a pendulum to appear to trace a circle when a neutral density filter is placed over one eye, because of delayed transmission of information in the filtered pathway) without the normal need of the filter,73 and auditory functions dependent on precisely coordinated information may be distorted by a unilateral lesion in the auditory pathway.
PLASTIC CHANGES
Recovery from a relapse of multiple sclerosis is also likely to involve “plastic” cortical changes; functional magnetic resonance imaging and other evidence support this view.74–81 Thus, to accomplish even minor tasks, patients with multiple sclerosis activate a larger part of the cortex than normal, including cortical areas not normally associated with the particular activity. Whether this increased cortical activation contributes to the sense of fatigue experienced by many patients remains unresolved.
POSITIVE SYMPTOMS: AXONAL HYPEREXCITABILITY
The recovery processes in demyelinated axons can lead not only to the restoration of conduction but also to axonal hyperexcitability.82,83 Ectopic impulses can be generated in regular or bursting discharges, and they conduct away from their site of initiation in both directions along the axon.84,85 Discharges in sensory pathways may give rise to tingling sensations referred to the body parts normally innervated by those axons. Similar discharges are likely to underlie other paroxysmal phenomena, including trigeminal neuralgia, tonic spasms, hemifacial spasm, and possibly episodic dysarthria and limb ataxia. Positive phenomena can be enhanced in patients by measures that increase axonal excitability, such as hyperventilation, and treatment with bicarbonate or calcium chelators.83,86–88
It appears that spontaneous discharges can arise from the excitatory effects of inward sodium currents that can develop in the demyelinated axolemma,83,89 and sodium channel blockers such as carbamazepine and phenytoin are effective treatments for positive symptoms. Spontaneous discharges could also arise from inward potassium currents that can result from the accumulation of potassium ions in limited extracellular compartments around axons.90 Finally, it has been proposed that impulses can sometimes be “reflected” from sites of demyelination91–93; that is, an impulse propagating through a demyelinated site can induce the formation of a second impulse that travels back along the same axon in the opposite direction. A pair of reflecting sites could generate a train of apparently spontaneous impulses. Single, normally evoked impulses propagating through generator sites where any of these mechanisms of hyperexcitability have arisen could also trigger episodes of ectopic activity.25,91,94,95
Sensory axons are more prone to develop ectopic discharges than are motor axons, and, accordingly, positive sensory phenomena are more common in demyelinating disease than are their motor counterparts.96,97 These differences may occur partly because of the greater expression of persistent sodium currents along sensory axons.98 The application of tumor necrosis factor directly to the sciatic nerve has been found to evoke ectopic discharges in nociceptive axons, including unmyelinated axons.99 It is therefore possible that in inflammatory demyelinating diseases such as multiple sclerosis, tumor necrosis factor is involved in promoting the generation of ectopic discharges along demyelinated axons within inflammatory foci. Other inflammatory mediators may also favor ectopic activity. For example, exposure to nitric oxide is known to be conducive to persistent opening of sodium channels.100,101
Ephaptic Transmission between Axons
Ephaptic interactions involving electrical crosstalk between demyelinated axons can explain a number of clinical observations in which paroxysmal, positive phenomena affect anatomically adjacent axonal pathways.102–104 For example, tonic spasms may involve sensory symptoms along with contralateral limb contraction, as expected from a lateral spread of excitation from the spinothalamic to the corticospinal tract within the spinal cord. Hartmann102 described a patient in whom trigeminal neuralgia could be triggered by auditory stimuli and who had a pontine lesion involving the adjacent lateral lemniscus and trigeminal sensory pathway on magnetic resonance imaging. That said, ephaptic transmission has rarely been demonstrated electrophysiologically. Indeed, the best documented interaction occurs between normal and amyelinated (i.e., never myelinated, rather than demyelinated) axons in the spinal roots of the dystrophic mouse.105,106
The authors have occasionally observed massed synchronous discharges arising from the spinal cord in animals with experimental demyelinating lesions in the dorsal columns.107 The discharges take the form of repeated bouts of high-frequency bursts of compound action potentials, but their relevance to multiple sclerosis remains uncertain.
Mechanosensitivity
Demyelinated axons can become markedly mechanosensitive, so that even mild deformation can result in the generation of bursts of impulses or change the firing frequency of axons that are already spontaneously active.108 Stretch-sensitive ion channels, if they appear in the demyelinated membrane, might underlie mechanosensitivity, but this possibility remains speculative. It is reasonable to suppose that the mechanosensitivity of demyelinated axons in the cervical dorsal columns is responsible for Lhermitte’s phenomenon109–111 and for the perception of flashes of light (phosphenes) upon eye movements in patients with demyelinating lesions of the optic nerve.
THE ROLE OF INFLAMMATION
It is now accepted that the inflammatory response in multiple sclerosis plays a significant role in symptom production and clinical relapse.112 Clinical and magnetic resonance imaging studies have shown a correlation between inflammation in the optic nerve and visual loss in patients with acute optic neuritis,113 and administration of the antilymphocyte antibody alemtuzumab (Campath-1H) to patients with multiple sclerosis provokes acute exacerbations of previously expressed symptoms, presumably because conduction in damaged axons had been blocked.114
Inflammation may impair conduction indirectly by opening the blood-brain barrier, thereby exposing axons to potentially deleterious factors in the vasculature, or directly, as a result of the local action of soluble agents such as nitric oxide, cytokines, peptides, and antibodies produced during the immune response. Demyelinated axons have an inherently low safety factor for conduction and are therefore likely to be particularly susceptible to factors impairing conduction, even when the factors may exert only subtle effects on normal axons. Clinically, the time course of relapse and remission in multiple sclerosis tends to coincide with the onset and resolution of inflammatory activity within plaques, which supports the beliefs that inflammation contributes to the deficit and that recovery is associated with the restoration of conduction as inflammation subsides.113
Cytokines
Cytokines have been implicated as a cause of conduction block in some clinical studies, particularly in axons already compromised by disease processes. For example, the symptoms that occur after the administration of alemtuzumab are associated with a transient elevation of circulating proinflammatory cytokines,115 and they can be prevented by pretreatment with steroids. These neurological effects could be mediated by direct effects of cytokines on axonal ion channels and/or indirectly, because cytokines such as interleukin-1β and interferon γ are potent inducers of the inducible form of nitric oxide synthase (iNOS) in human astrocytes and also probably in microglia. It may therefore be clinically significant that interferon β inhibits both the expression of iNOS116 by astrocytes and the production of reactive oxygen species by monocytes from patients with relapsing-remitting multiple sclerosis.
Nitric Oxide
Levels of nitric oxide metabolites and of iNOS are increased in multiple sclerosis,117,118 and low micromolar concentrations of nitric oxide can block axonal conduction,119,120 particularly in demyelinated axons.119 The blockade is reversible, which raises the possibility that nitric oxide may be an important contributor to the reversible clinical deficits in the relapses and remissions of multiple sclerosis. The mechanisms underlying conduction block have not been established but may include direct effects of nitric oxide on ion channels,100,121–123 inhibition of mitochondrial respiration by nitric oxide,124,125 and effects on the levels of axonal cyclic guanosine monophosphate.126 If nitric oxide does impair conduction in inflammatory demyelinating disease, then agents that inhibit its production should ameliorate symptoms. However, this rationale may have unpredictable consequences in the context of an autoimmune demyelinating disease, in which nitric oxide may have protective as well as damaging effects. There have been more than 40 studies of the role of nitric oxide and its related compounds in experimental autoimmune encephalomyelitis, but there is little consensus regarding its effects or the mechanisms through which they are mediated.127
Antibodies
Antiganglioside antibodies have been identified in patients with progressive forms of multiple sclerosis.128,129 These antibodies may contribute to the neurological deficit because some (but not all) studies have revealed that they can have electrophysiological effects on peripheral axons.130
DISABILITY AND AXONAL DEGENERATION
It has long been believed that the persistence and progression of disability in multiple sclerosis can be explained by persistent conduction block in demyelinated axons caused by a failure of the repair mechanisms discussed earlier. However, it is now clear that axonal degeneration also plays an important and perhaps primary role.131–136 In pathological studies, there is prominent axonal degeneration at an early stage of multiple sclerosis,137,138 and in magnetic resonance studies, there is atrophy of the whole brain and regions that include the cerebellum, spinal cord and thalamus, as well as a reduction of the neuronal marker N-acetyl aspartate.131,133,139 Axonal injury is correlated with disability in both the pathological and magnetic resonance studies.
There are probably numerous mechanisms of axonal degeneration. However, the magnitude of axonal degeneration in pathological studies is correlated with the intensity of the inflammatory response, which raises the possibility that some mediators of inflammation may have a damaging effect on adjacent axons. Experimentally sustained impulse activity at physiological frequencies has been shown to result in axonal degeneration, even in normal axons, if the activity occurs in conjunction with exposure to nitric oxide (Fig. 75-5).140 The combination of impulse activity and nitric oxide exposure may be expected to occur within inflammatory lesions; therefore, this mechanism could contribute to axonal degeneration in multiple sclerosis. In this respect, it is interesting that the smallest axons are the most susceptible to degeneration caused by impulse activity during nitric oxide exposure and that they are also the axons that are preferentially lost in multiple sclerosis lesions.141,142
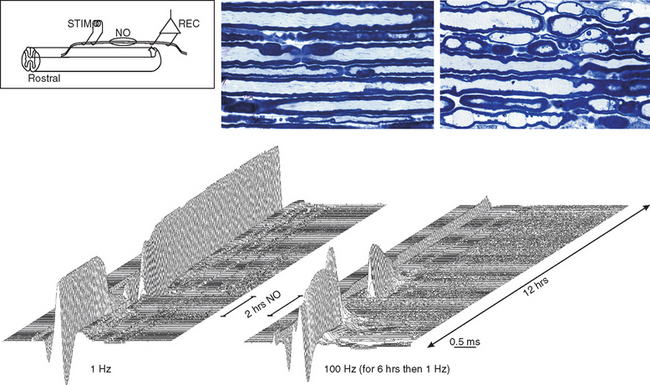
Figure 75-5 Records showing the vulnerability of nerve fibers to degeneration as a result of sustained impulse activity during exposure to nitric oxide (NO). Two series of averaged compound action potentials can be seen, recorded in parallel from two dorsal roots in an anesthetized rat, in the arrangement indicated (see inset). The data are shown in three-dimensional perspective, with the earliest records at the front and with a 2-minute interval between adjacent records; each plot therefore shows approximately 12 hours of recorded data. The left plot shows records obtained at continuous 1-Hz stimulation, whereas in the right plot, the nerve fibers were stimulated at 100 Hz for the first 6 hours of recording. On the left, the records were stable for the first 2.5 hours, but conduction block was imposed on nearly all the axons by a 2-hour exposure to nitric oxide. Such block has been described by Redford and colleagues,119 and it might help explain how inflammation can cause negative symptoms in multiple sclerosis (see text). The block was released on removal of nitric oxide, and the axons continued to conduct for the remaining 7.5 hours of the experiment. On the right, conduction was also blocked during exposure to nitric oxide, but recovery after washing was inconsistent and only temporary, so that within 2 hours, conduction had ceased in all axons. Histological examination of the roots at the end of the experiment revealed that all the axons exposed to nitric oxide in conjunction with stimulation at 100 Hz had undergone degeneration, whereas the root stimulated at only 1 Hz during the period of nitric oxide exposure was quite normal in appearance.
(Redrawn from Smith KJ, Kapoor R, Hall SM, et al: Electrically active axons degenerate when exposed to nitric oxide. Ann Neurol 2001; 49:470-476.)
The mechanisms responsible for the effects of nitric oxide are not yet certain, but because nitric oxide is known to inhibit mitochondrial energy production, and because demyelinated axons express more sodium channels, it seems reasonable to hypothesize that axonal degeneration might result from the combination of a reduced capacity for energy production as a result of nitric oxide exposure and an increased energy demand from sodium loading of the axon as a result of impulse activity.143 The consequent failure of Na+/K+-ATPase to maintain ionic homeostasis would result in an elevation of the intra-axonal sodium ion concentration, perhaps to a level capable of driving the sodium-calcium exchanger into reverse activity.144 This sequence of events could culminate in a pathological accumulation of calcium ions within the axon and the activation of intra-axonal degrading enzymes.
Demyelinated axons in multiple sclerosis or experimental autoimmune encephalomyelitis can also express immunoreactivity for subunits of the N-type calcium channel:145 If such channels are functional, they could add to an influx of calcium ions during impulse activity. Furthermore, there is increasing evidence of mitochondrial genetic dysfunction in multiple sclerosis,145a which might render subgroups of patients more vulnerable to axonal injury that depends on energy failure.
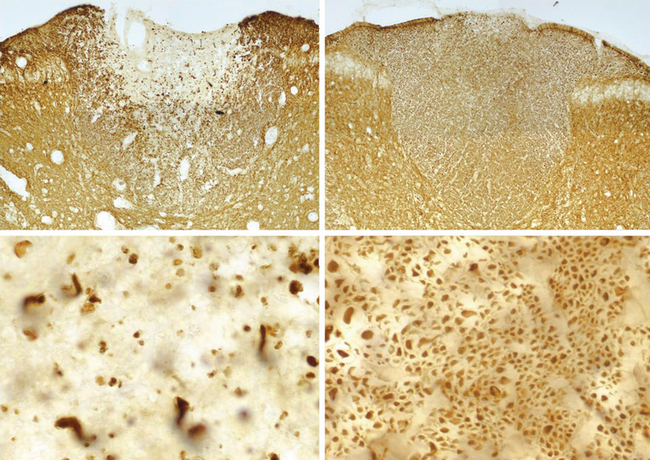
This hypothesis, which links inflammation, increased expression of sodium channels, metabolic failure, and ultimately calcium overloading of the axon, has received support from studies showing that sodium channel blockade with drugs such as phenytoin,146 flecainide (Fig. 75-6),147–150 and lamotrigine (unpublished observations) can indeed protect axons from degeneration, both on exposure to nitric oxide and in animals with experimental autoimmune encephalomyelitis and experimental autoimmune neuritis.
Glutamate Excitotoxicity
Glutamate is likely to be present in active multiple sclerosis lesions after its liberation by activated microglia and leukocytes151 and perhaps by axons,144 and its levels may be amplified still further within inflammatory foci by the action of proinflammatory cytokines, such as interleukin-1β and tumor necrosis factor α, which have been reported to inhibit the glutamate uptake activity of astrocytes in vitro by a mechanism that involves nitric oxide. Glutamate-mediated excitotoxicity acting through Ca2+-permeable α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA)/kainate receptors may contribute to the death of oligodendrocytes (and neurons) in multiple sclerosis, and indeed the blockade of AMPA/kainate receptors has been found to reduce both axon and oligodendrocyte loss in experimental allergic encephalomyelitis, with a corresponding amelioration of clinical outcome.152,153 Furthermore, excitotoxic damage to oligodendrocyte-like cells is prevented by agents that elevate cyclic adenosine monophosphate levels, such as rolipram, which has been shown to be protective in experimental allergic encephalomyelitis.154,155 The precise mechanisms of injury to neurons and to oligodendrocytes may differ, inasmuch as oligodendrocytes appear to possess only AMPA/kainate receptors, whereas neurons possess N-methyl-D-aspartate receptors as well. The situation is less clear for axons, although there is emerging evidence that they do express novel glutamate receptors after demyelination. Finally, research suggests that axonal degeneration in experimental allergic encephalomyelitis may be influenced by the activity of cannabinoid receptors, acting in part through their effects on glutaminergic systems.156
Cortical Lesions and Neuronal Loss in Gray Matter
The lesions of multiple sclerosis are not restricted to the white matter, and different forms of cortical lesions are known to occur.157,158 Their contribution to the clinical features of multiple sclerosis is a subject of increasing interest. Magnetic resonance imaging and spectroscopy have demonstrated that atrophy and lowered N-acetyl aspartate levels are present in cortical and deep gray matter areas.159–163 This work, which is in keeping with the results of histopathological studies, indicates that there is a significant component of degeneration of neuronal cell bodies in multiple sclerosis, not only in advanced secondary progressive disease but also in early relapsing disease. Gray matter neuroaxonal pathology may therefore be an important factor in the development of disability. The mechanisms of damage to neuronal cell bodies probably differ to some extent from those causing axonal degeneration, and it may be relevant that cortical lesions in multiple sclerosis contain a smaller leukocytic infiltrate and more microglial activity than do white matter lesions.
Bechtold DA, Smith KJ. Sodium-mediated axonal degeneration in inflammatory demyelinating disease. J Neurol Sci. 2005;233:27-35.
Burke D, Kiernan MC, Bostock H. Excitability of human axons. Clin Neurophysiol. 2001;112:1575-1585.
Craner MJ, Newcombe J, Black JA, et al. Molecular changes in neurons in multiple sclerosis: altered axonal expression of Nav1.2 and Nav1.6 sodium channels and Na+/Ca2+ exchanger. Proc Natl Acad Sci U S A. 2004;101:8168-8173.
Smith KJ, Felts PA, Kapoor R. Axonal hyperexcitability: mechanisms and role in symptom production in demyelinating diseases. Neuroscientist. 1997;3:237-246.
Smith KJ, McDonald I, Miller D, et al. The pathophysiology of multiple sclerosis. In: Compston A, Confavreux C, Lassman H, et al, editors. McAlpine’s Multiple Sclerosis. London: Churchill Livingstone; 2005:601-659.
1 Tasaki I. Nervous Transmission. Springfield, IL: Charles C Thomas, 1953.
2 Arroyo EJ, Scherer SS. On the molecular architecture of myelinated fibers. Histochemistry. 2000;113:1-18.
3 Kazarinova-Noyes K, Shrager P. Molecular constituents of the node of Ranvier. Mol Neurobiol. 2002;26:167-182.
4 Reid G, Scholz A, Bostock H, et al. Human axons contain at least five types of voltage-dependent potassium channel. J Physiol (Lond). 1999;518:681-696.
5 Smith KJ, Hall SM. Nerve conduction during peripheral demyelination and remyelination. J Neurol Sci. 1980;48:201-219.
6 Waxman SG, Ritchie JM. Molecular dissection of the myelinated axon. Ann Neurol. 1993;33:121-136.
7 Davis FA, Schauf CL, Reed BJ, et al. Experimental studies of the effects of extrinsic factors on conduction in normal and demyelinated nerve. J Neurol Neurosurg Psychiatry. 1975;39:442-448.
8 Rasminsky M. The effects of temperature on conduction in demyelinated single nerve fibers. Arch Neurol. 1973;28:287-292.
9 Sears TA, Bostock H, Sheratt M. The pathophysiology of demyelination and its implications for the symptomatic treatment of multiple sclerosis. Neurology. 1978;28:21-26.
10 Smith KJ. Conduction properties of central demyelinated and remyelinated axons, and their relation to symptom production in demyelinating disorders. Eye. 1994;8:224-237.
11 Davis FA, Schauf CL. Approaches to the development of pharmacological interventions in multiple sclerosis. Adv Neurol. 1981;31:505-510.
12 Paintal AS. The influence of diameter of medullated nerve fibres of cats on the rising and falling phases of the spike and its recovery. J Physiol (Lond). 1966;184:791-811.
13 Uhthoff W. Untersuchungen über die bei der multiplen Herd-sklerose vorkommenden Augenstörungen. Arch Psychiatr Nervenkr. 1890;21:55-116.
14 McDonald WI. The pathophysiology of multiple sclerosis. In: McDonald WI, Silberberg DH, editors. Multiple Sclerosis. London: Butterworth; 1986:112-133.
15 Waxman SG, Geschwind N. Major morbidity related to hyperthermia in multiple sclerosis. Ann Neurol. 1983;13:348.
16 Waxman SG, Utzschneider DA, Kocsis JD. Enhancement of action potential conduction following demyelination: experimental approaches to restoration of function in multiple sclerosis and spinal cord injury. Prog Brain Res. 1994;100:233-243.
17 Sherratt RM, Bostock H, Sears TA. Effects of 4-aminopyridine on normal and demyelinated mammalian nerve fibres. Nature. 1980;283:570-572.
18 Targ EF, Kocsis JD. 4-Aminopyridine leads to restoration of conduction in demyelinated rat sciatic nerve. Brain Res. 1985;328:358-361.
19 Bowe CM, Kocsis JD, Targ EF, et al. Physiological effects of 4-aminopyridine on demyelinated mammalian motor and sensory fibers. Ann Neurol. 1987;22:264-268.
20 Bever CTJr. 4-Aminopyridine: use in multiple sclerosis. CNS Drug Rev. 1995;1:261-279.
21 Smith KJ, Felts PA, John GR. Effects of 4-aminopyridine on demyelinated axons, synapses and muscle tension. Brain. 2000;123(Pt 1):171-184.
22 Blight AR, Toombs JP, Bauer MS, et al. The effects of 4-aminopyridine on neurological deficits in chronic cases of traumatic spinal cord injury in dogs: a phase I clinical trial. J Neurotrauma. 1991;8:103-119.
23 McDonald WI, Sears TA. The effects of experimental demyelination on conduction in the central nervous system. Brain. 1970;93:583-598.
24 Bostock H, Grafe P. Activity-dependent excitability changes in normal and demyelinated rat spinal root axons. J Physiol (Lond). 1985;365:239-257.
25 Felts PA, Kapoor R, Smith KJ. A mechanism for ectopic firing in central demyelinated axons. Brain. 1995;118(Pt 5):1225-1231.
26 Smith KJ, McDonald I, Miller D, et al. The pathophysiology of multiple sclerosis. In: Compston A, Confavreux C, Lassman H, et al, editors. McAlpine’s Multiple Sclerosis. London: Churchill Livingstone; 2005:601-659.
27 Waxman SG. Clinicopathological correlations in multiple sclerosis and related diseases. Adv Neurol. 1981;31:169-182.
28 Kaji R, Happel L, Sumner AJ. Effect of digitalis on clinical symptoms and conduction variables in patients with multiple sclerosis. Ann Neurol. 1990;28:582-584.
29 Prineas JW, Barnard RO, Kwon EE, et al. Multiple sclerosis: remyelination of nascent lesions. Ann Neurol. 1993;33:137-151.
30 Prineas JW, Kwon EE, Sharer LR, et al. Massive early remyelination in acute multiple sclerosis [Abstract]. Neurology. 1987;37(Suppl):109.
31 Jeffery ND, Blakemore WF. Locomotor deficits induced by experimental spinal cord demyelination are abolished by spontaneous remyelination. Brain. 1997;120(Pt 1):27-37.
32 Felts PA, Baker TA, Smith KJ. Conduction in segmentally demyelinated mammalian central axons. J Neurosci. 1997;17:7267-7277.
33 Novakovic SD, Deerinck TJ, Levinson SR, et al. Clusters of axonal Na+ channels adjacent to remyelinating Schwann cells. J Neurocytol. 1996;25:403-412.
34 Smith KJ, Bostock H, Hall SM. Saltatory conduction precedes remyelination in axons demyelinated with lysophosphatidyl choline. J Neurol Sci. 1982;54:13-31.
35 Smith KJ, Blakemore WF, McDonald WI. The restoration of conduction by central remyelination. Brain. 1981;104:383-404.
36 Felts PA, Smith KJ. Schwann cell remyelination of demyelinated central fibres restores secure conduction. Neuropathol Appl Neurobiol. 1987;13:494.
37 Honmou O, Felts PA, Waxman SG, et al. Restoration of normal conduction properties in demyelinated spinal cord axons in the adult rat by transplantation of exogenous Schwann cells. J Neurosci. 1996;16:3199-3208.
38 Imaizumi T, Lankford KL, Kocsis JD. Transplantation of olfactory ensheathing cells or Schwann cells restores rapid and secure conduction across the transected spinal cord. Brain Res. 2000;854:70-78.
39 Colello RJ, Pott U. Signals that initiate myelination in the developing mammalian nervous system. Mol Neurobiol. 1997;15:83-100.
40 Stevens B, Fields RD. Response of Schwann cells to action potentials in development. Science. 2000;287:2267-2271.
41 Bennett V, Lambert S. Physiological roles of axonal ankyrins in survival of premyelinated axons and localization of voltagegated sodium channels. J Neurocytol. 1999;28:303-318.
42 Boiko T, Rasband MN, Levinson SR, et al. Compact myelin dictates the differential targeting of two sodium channel isoforms in the same axon. Neuron. 2001;30:91-104.
43 Kaplan MR, Cho MH, Ullian EM, et al. Differential control of clustering of the sodium channels Na(v)1.2 and Na(v)1.6 at developing CNS nodes of Ranvier. Neuron. 2001;30:105-119.
44 Kaplan MR, Meyer-Franke A, Lambert S, et al. Induction of sodium channel clustering by oligodendrocytes. Nature. 1997;386:724-728.
45 Rasband MN, Peles E, Trimmer JS, et al. Dependence of nodal sodium channel clustering on paranodal axoglial contact in the developing CNS. J Neurosci. 1999;19:7516-7528.
46 Rasband MN, Taylor CM, Bansal R. Paranodal transverse bands are required for maintenance but not initiation of Nav1.6 sodium channel clustering in CNS optic nerve axons. Glia. 2003;44:173-182.
47 Craner MJ, Lo AC, Black JA, et al. Abnormal sodium channel distribution in optic nerve axons in a model of inflammatory demyelination. Brain. 2003;126(Pt 7):1552-1561.
48 Craner MJ, Newcombe J, Black JA, et al. Molecular changes in neurons in multiple sclerosis: altered axonal expression of Nav1.2 and Nav1.6 sodium channels and Na+/Ca2+ exchanger. Proc Natl Acad Sci U S A. 2004;101:8168-8173.
49 Novakovic SD, Levinson SR, Schachner M, et al. Disruption and reorganization of sodium channels in experimental allergic neuritis. Muscle Nerve. 1998;21:1019-1032.
50 Rasband MN, Trimmer JS, Peles E, et al. K+ channel distribution and clustering in developing and hypomyelinated axons of the optic nerve. J Neurocytol. 1999;28:319-331.
51 Craner MJ, Hains BC, Lo AC, et al. Co-localization of sodium channel Nav1.6 and the sodium-calcium exchanger at sites of axonal injury in the spinal cord in EAE. Brain. 2004;127(Pt 2):294-303.
52 Black JA, Dib-Hajj S, Baker D, et al. Sensory neuron-specific sodium channel SNS is abnormally expressed in the brains of mice with experimental allergic encephalomyelitis and humans with multiple sclerosis. Proc Natl Acad Sci U S A. 2000;97:11598-11602.
53 Craner MJ, Kataoka Y, Lo AC, et al. Temporal course of upregulation of Na(v)1.8 in Purkinje neurons parallels the progression of clinical deficit in experimental allergic encephalomyelitis. J Neuropathol Exp Neurol. 2003;62:968-975.
54 Renganathan M, Gelderblom M, Black JA, et al. Expression of Nav1.8 sodium channels perturbs the firing patterns of cerebellar Purkinje cells. Brain Res. 2003;959:235-242.
55 Waxman SG. Cerebellar dysfunction in multiple sclerosis: evidence for an acquired channelopathy. Prog Brain Res. 2005;148:353-365.
56 Bostock H, Sears TA. Continuous conduction in demyelinated mammalian nerve fibers. Nature. 1976;263:786-787.
57 Deerinck TJ, Levinson SR, Bennett GV, et al. Clustering of voltage-sensitive sodium channels on axons is independent of direct Schwann cell contact in the dystrophic mouse. J Neurosci. 1997;17:5080-5088.
58 Novakovic SD, Eglen RM, Hunter JC. Regulation of Na+channel distribution in the nervous system. Trends Neurosci. 2001;24:473-478.
59 Bostock H, Sears TA. The internodal axon membrane: electrical excitability and continuous conduction in segmental demyelination. J Physiol (Lond). 1978;280:273-301.
60 Waxman SG. Demyelination in spinal cord injury. J Neurol Sci. 1989;91:1-14.
61 Shrager P, Rubinstein CT. Optical measurement of conduction in single demyelinated axons. J Gen Physiol. 1990;95:867-890.
62 Waxman SG, Brill MH. Conduction through demyelinated plaques in multiple sclerosis: computer simulations of facilitation by short internodes. J Neurol Neurosurg Psychiatry. 1978;41:408-416.
63 Ghatak NR, Hirano A, Lijtmaer H, et al. Asymptomatic demyelinated plaque in the spinal cord. Arch Neurol. 1974;30:484-486.
64 Namerow NS. The pathophysiology of multiple sclerosis. In: Wolfgram F, Ellison GW, Stevens JG, et al, editors. Multiple Sclerosis: Immunology, Virology and Ultrastructure. New York: Academic Press; 1972:143-172.
65 O’Riordan JI, Losseff NA, Phatouros C, et al. Asymptomatic spinal cord lesions in clinically isolated optic nerve, brain stem, and spinal cord syndromes suggestive of demyelination. J Neurol Neurosurg Psychiatry. 1998;64:353-357.
66 Phadke JG, Best PV. Atypical and clinically silent multiple sclerosis: a report of 12 cases discovered unexpectedly at necropsy. J Neurol Neurosurg Psychiatry. 1983;46:414-420.
67 Ulrich J, Groebke-Lorenz W. The optic nerve in multiple sclerosis: a morphological study with retrospective clinicopathological correlations. Neuroophthalmology. 1983;3:149-159.
68 Chalk JB, McCombe PA, Pender MP. Conduction abnormalities are restricted to the central nervous system in experimental autoimmune encephalomyelitis induced by inoculation with proteolipid protein but not with myelin basic protein. Brain. 1994;117:975-986.
69 Kaji R, Suzumura A, Sumner AJ. Physiological consequences of antiserum-mediated experimental demyelination in CNS. Brain. 1988;111:675-694.
70 Pender MP. The pathophysiology of acute experimental allergic encephalomyelitis induced by whole spinal cord in the Lewis rat. J Neurol Sci. 1988;84:209-222.
71 Smith KJ, Blakemore WF, McDonald WI. Central remyelination restores secure conduction. Nature. 1979;280:395-396.
72 Halliday AM, McDonald WI, Mushin J. Visual evoked response in diagnosis of multiple sclerosis. BMJ. 1973;4:661-664.
73 Frisen L, Hoyt WF, Bird AC, et al. Diagnostic uses of the Pulfrich phenomenon. Lancet. 1973;2:385-386.
74 Kerschensteiner M, Bareyre FM, Buddeberg BS, et al. Remodeling of axonal connections contributes to recovery in an animal model of multiple sclerosis. J Exp Med. 2004;200:1027-1038.
75 Pantano P, Iannetti GD, Caramia F, et al. Cortical motor reorganization after a single clinical attack of multiple sclerosis. Brain. 2002;125(Pt 7):1607-1615.
76 Reddy H, Narayanan S, Woolrich M, et al. Functional brain reorganization for hand movement in patients with multiple sclerosis: defining distinct effects of injury and disability. Brain. 2002;125(Pt 12):2646-2657.
77 Rocca MA, Mezzapesa DM, Falini A, et al. Evidence for axonal pathology and adaptive cortical reorganization in patients at presentation with clinically isolated syndromes suggestive of multiple sclerosis. Neuroimage. 2003;18:847-855.
78 Rocca MA, Pagani E, Ghezzi A, et al. Functional cortical changes in patients with multiple sclerosis and nonspecific findings on conventional magnetic resonance imaging scans of the brain. Neuroimage. 2003;19:826-836.
79 Toosy AT, Hickman SJ, Miszkiel KA, et al. Adaptive cortical plasticity in higher visual areas after acute optic neuritis. Ann Neurol. 2005;57:622-633.
80 Toosy AT, Werring DJ, Bullmore ET, et al. Functional magnetic resonance imaging of the cortical response to photic stimulation in humans following optic neuritis recovery. Neurosci Lett. 2002;330:255-259.
81 Werring DJ, Bullmore ET, Toosy AT, et al. Recovery from optic neuritis is associated with a change in the distribution of cerebral response to visual stimulation: a functional magnetic resonance imaging study. J Neurol Neurosurg Psychiatry. 2000;68:441-449.
82 Burke D. Microneurography, impulse conduction, and paresthesias. Muscle Nerve. 1993;16:1025-1032.
83 Smith KJ, Felts PA, Kapoor R. Axonal hyperexcitability: mechanisms and role in symptom production in demyelinating diseases. Neuroscientist. 1997;3:237-246.
84 Baker M, Bostock H. Ectopic activity in demyelinated spinal root axons of the rat. J Physiol (Lond). 1992;451:539-552.
85 Smith KJ, McDonald WI. Spontaneous and evoked electrical discharges from a central demyelinating lesion. J Neurol Sci. 1982;55:39-47.
86 Baker MD. Axonal flip-flops and oscillators. Trends Neurosci. 2000;23:514-519.
87 Burchiel KJ. Ectopic impulse generation in demyelinated axons: effects of PaCO2, pH, and disodium edetate. Ann Neurol. 1981;9:378-383.
88 Davis FA, Becker FO, Michael JA, et al. Effect of intravenous sodium bicarbonate, disodium edetate (Na2EDTA), and hyperventilation on visual and oculomotor signs in multiple sclerosis. J Neurol Neurosurg Psychiatry. 1970;33:723-732.
89 Kapoor R, Li YG, Smith KJ. Slow sodium-dependent potential oscillations contribute to ectopic firing in mammalian demyelinated axons. Brain. 1997;120(Pt 4):647-652.
90 Kapoor R, Smith KJ, Felts PA, et al. Internodal potassium currents can generate ectopic impulses in mammalian myelinated axons. Brain Res. 1993;611:165-169.
91 Calvin WH, Devor M, Howe JF. Can neuralgias arise from minor demyelination? Spontaneous firing, mechanosensitivity, and afterdischarge from conducting axons. Exp Neurol. 1982;75:755-763.
92 Calvin WH, Loeser JD, Howe JF. A neurophysiological theory for the pain mechanism of tic douloureux. Pain. 1977;3:147-154.
93 Howe JF, Calvin WH, Loeser JD. Impulses reflected from dorsal root ganglia and from focal nerve injuries. Brain Res. 1976;116:139-144.
94 Burchiel KJ. Abnormal impulse generation in focally demyelinated trigeminal roots. J Neurosurg. 1980;53:674-683.
95 Huizar P, Kuno M, Miyata Y. Electrophysiological properties of spinal motoneurones of normal and dystrophic mice. J Physiol (Lond). 1975;248:231-246.
96 Chen Y, Devor M. Ectopic mechanosensitivity in injured sensory axons arises from the site of spontaneous electrogenesis. Eur J Pain. 1998;2:165-178.
97 Sedano MJ, Trejo JM, Macarron JL, et al. Continuous facial myokymia in multiple sclerosis: treatment with botulinum toxin. Eur Neurol. 2000;43:137-140.
98 Mogyoros I, Bostock H, Burke D. Mechanisms of paresthesias arising from healthy axons. Muscle Nerve. 2000;23:310-320.
99 Sorkin LS, Xiao WH, Wagner R, et al. Tumour necrosis factoralpha induces ectopic activity in nociceptive primary afferent fibres. Neuroscience. 1997;81:255-262.
100 Ahern GP, Hsu S-F, Klyachko VA, et al. Induction of persistent sodium current by exogenous and endogenous nitric oxide. J Biol Chem. 2000;275:28810-28815.
101 Hammarstrom AKM, Gage PW. Nitric oxide increases persistent sodium current in rat hippocampal neurons. J Physiol (Lond). 1999;520:451-461.
102 Hartmann M, Rottach KG, Wohlgemuth WA, et al. Trigeminal neuralgia triggered by auditory stimuli in multiple sclerosis. Arch Neurol. 1999;56:731-733.
103 Matthews B. Symptoms and signs of multiple sclerosis. In: Compston A, Ebers G, Lassmann H, et al, editors. McAlpine’s Multiple Sclerosis. London: Churchill Livingstone; 1998:145-190.
104 Matthews WB. Paroxysmal symptoms in multiple sclerosis. J Neurol Neurosurg Psychiatry. 1975;38:619-623.
105 Rasminsky M. Ephaptic transmission between single nerve fibres in the spinal nerve roots of dystrophic mice. J Physiol (Lond). 1980;305:151-169.
106 Rasminsky M. Ectopic generation of impulses and crosstalk in spinal nerve roots of “dystrophic” mice. Ann Neurol. 1978;3:351-357.
107 Smith KJ, McDonald WI. The pathophysiology of multiple sclerosis: the mechanisms underlying the production of symptoms and the natural history of the disease. Phil Trans R Soc Lond B. 1999;354:1649-1673.
108 Smith KJ, McDonald WI. Spontaneous and mechanically evoked activity due to central demyelinating lesion. Nature. 1980;286:154-155.
109 Kanchandani R, Howe JG. Lhermitte’s sign in multiple sclerosis: a clinical survey and review of the literature. J Neurol Neurosurg Psychiatry. 1982;45:308-312.
110 Lhermitte J, Bollack J, Nicholas M. Les douleurs à type de décharge électrique consécutives à la flexion céphalique dans la sclérose en plaques. Rev Neurol. 1924;2:56-62.
111 Nordin M, Nystrom B, Wallin U, et al. Ectopic sensory discharges and paresthesiae in patients with disorders of peripheral nerves, dorsal roots and dorsal columns. Pain. 1984;20:231-245.
112 Bitsch A, Wegener C, Da Costa C, et al. Lesion development in Marburg’s type of acute multiple sclerosis: from inflammation to demyelination. Mult Scler. 1999;5:138-146.
113 Youl BD, Turano G, Miller DH, et al. The pathophysiology of acute optic neuritis. An association of gadolinium leakage with clinical and electrophysiological deficits. Brain. 1991;114:2437-2450.
114 Moreau T, Coles A, Wing M, et al. Transient increase in symptoms associated with cytokine release in patients with multiple sclerosis. Brain. 1996;119(Pt 1):225-237.
115 Coles AJ, Wing MG, Molyneux P, et al. Monoclonal antibody treatment exposes three mechanisms underlying the clinical course of multiple sclerosis. Ann Neurol. 1999;46:296-304.
116 Hua LL, Liu JSH, Brosnan CF, et al. Selective inhibition of human glial inducible nitric oxide synthase by interferon-beta: implications for multiple sclerosis. Ann Neurol. 1998;43:384-387.
117 Giovannoni G, Miller RF, Heales SJR, et al. Elevated cerebrospinal fluid and serum nitrate and nitrite levels in patients with central nervous system complications of HIV-1 infection: a correlation with blood-brain-barrier dysfunction. J Neurol Sci. 1998;156:53-58.
118 Smith KJ, Lassmann H. The role of nitric oxide in multiple sclerosis. Lancet Neurol. 2002;1:232-241.
119 Redford EJ, Kapoor R, Smith KJ. Nitric oxide donors reversibly block axonal conduction: demyelinated axons are especially susceptible. Brain. 1997;120(Pt 12):2149-2157.
120 Shrager P, Custer AW, Kazarinova K, et al. Nerve conduction block by nitric oxide that is mediated by the axonal environment. J Neurophysiol. 1998;79:529-536.
121 Ahern GP, Hsu S-F, Jackson MB. Direct actions of nitric oxide on rat neurohypophysial K+ channels. J Physiol (Lond). 1999;520:165-176.
122 Li Z, Chapleau MW, Bates JN, et al. Nitric oxide as an autocrine regulator of sodium currents in baroreceptor neurons. Neuron. 1998;20:1039-1049.
123 Renganathan M, Cummins TR, Hormuzdiar WN, et al. Nitric oxide is an autocrine regulator of Na+ currents in axotomized C-type DRG neurons. J Neurophysiol. 2000;83:2431-2442.
124 Bolanos JP, Almeida A, Stewart V, et al. Nitric oxide–mediated mitochondrial damage in the brain: mechanisms and implications for neurodegenerative diseases. J Neurochem. 1997;68:2227-2240.
125 Brown GC, Bolanos JP, Heales SJ, et al. Nitric oxide produced by activated astrocytes rapidly and reversibly inhibits cellular respiration. Neurosci Lett. 1995;193:201-204.
126 Garthwaite G, Goodwin DA, Garthwaite J. Nitric oxide stimulates cGMP formation in rat optic nerve axons, providing a specific marker of axon viability. Eur J Neurosci. 1999;11:4367-4372.
127 Willenborg DO, Staykova MA, Cowden WB. Our shifting understanding of the role of nitric oxide in autoimmune encephalomyelitis: a review. J Neuroimmunol. 1999;100:21-35.
128 Acarin N, Rio J, Fernandez AL, et al. Different antiganglioside antibody pattern between relapsing-remitting and progressive multiple sclerosis. Acta Neurol Scand. 1996;93:99-103.
129 Sadatipour BT, Greer JM, Pender MP. Increased circulating antiganglioside antibodies in primary and secondary progressive multiple sclerosis. Ann Neurol. 1998;44:980-983.
130 Willison HJ, Yuki N. Peripheral neuropathies and antiglycolipid antibodies. Brain. 2002;125(Pt 12):2591-2625.
131 Davie CA, Barker GJ, Webb S, et al. Persistent functional deficit in multiple sclerosis and autosomal dominant cerebellar ataxia is associated with axon loss. Brain. 1995;118(Pt 6):1583-1592.
132 De Stefano N, Matthews PM, Fu L, et al. Axonal damage correlates with disability in patients with relapsing-remitting multiple sclerosis. Results of a longitudinal magnetic resonance spectroscopy study. Brain. 1998;121:1469-1477.
133 Losseff NA, Webb SL, O’Riordan JI, et al. Spinal cord atrophy and disability in multiple sclerosis. A new reproducible and sensitive MRI method with potential to monitor disease progression. Brain. 1996;119(Pt 3):701-708.
134 Matthews PM, De Stefano N, Narayanan S, et al. Putting magnetic resonance spectroscopy studies in context: axonal damage and disability in multiple sclerosis. Semin Neurol. 1998;18:327-336.
135 Stevenson VL, Leary SM, Losseff NA, et al. Spinal cord atrophy and disability in MS: a longitudinal study. Neurology. 1998;51:234-238.
136 Truyen L, Van Waesberghe JHTM, Van Walderveen MAA, et al. Accumulation of hypointense lesions (“black holes”) on T1 spin-echo MRI correlates with disease progression in multiple sclerosis. Neurology. 1996;47:1469-1476.
137 Bjartmar C, Trapp BD. Axonal and neuronal degeneration in multiple sclerosis: mechanisms and functional consequences. Curr Opin Neurol. 2001;14:271-278.
138 Ferguson B, Matyszak MK, Esiri MM, et al. Axonal damage in acute multiple sclerosis lesions. Brain. 1997;120(Pt 3):393-399.
139 Bjartmar C, Kidd G, Mork S, et al. Neurological disability correlates with spinal cord axonal loss and reduced N-acetyl aspartate in chronic multiple sclerosis patients. Ann Neurol. 2000;48:893-901.
140 Smith KJ, Kapoor R, Hall SM, et al. Electrically active axons degenerate when exposed to nitric oxide. Ann Neurol. 2001;49:470-476.
141 Lovas G, Szilagyi N, Majtenyi K, et al. Axonal changes in chronic demyelinated cervical spinal cord plaques. Brain. 2000;123:308-317.
142 McGavern DB, Murray PD, Rivera-Quinones C, et al. Axonal loss results in spinal cord atrophy, electrophysiological abnormalities and neurological deficits following demyelination in a chronic inflammatory model of multiple sclerosis. Brain. 2000;123:519-531.
143 Bechtold DA, Smith KJ. Sodium-mediated axonal degeneration in inflammatory demyelinating disease. J Neurol Sci. 2005;233:27-35.
144 Stys PK. White matter injury mechanisms. Curr Mol Med. 2004;4:113-130.
145 Kornek B, Storch MK, Bauer J, et al. Distribution of a calcium channel subunit in dystrophic axons in multiple sclerosis and experimental autoimmune encephalomyelitis. Brain. 2001;124(Pt 6):1114-1124.
145a Dutta R, McDonough J, Yin X, et al. Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Ann Neurol. 2006;59:478-489.
146 Lo AC, Black JA, Waxman SG. Neuroprotection of axons with phenytoin in experimental allergic encephalomyelitis. Neuroreport. 2002;13:1909-1912.
147 Bechtold DA, Kapoor R, Smith KJ. Axonal protection using flecainide in experimental autoimmune encephalomyelitis. Ann Neurol. 2004;55:607-616.
148 Bechtold DA, Yue X, Evans RM, et al. Axonal protection in experimental autoimmune neuritis by the sodium channel blocking agent flecainide. Brain. 2005;128(Pt 1):18-28.
149 Kapoor R, Davies M, Blaker PA, et al. Blockers of sodium and calcium entry protect axons from nitric oxide–mediated degeneration. Ann Neurol. 2003;53:174-180.
150 Smith KJ, Blaker PA, Kapoor R, et al. Protection of axons from degeneration resulting from exposure to nitric oxide. J Neurol Neurosurg Psychiatry. 2001;70:282.
151 Piani D, Frei K, Do KQ, et al. Murine brain macrophages induce NMDA receptor mediated neurotoxicity in vitro by secreting glutamate. Neurosci Lett. 1991;133:159-162.
152 Pitt D, Werner P, Raine CS. Glutamate excitotoxicity in a model of multiple sclerosis. Nat Med. 2000;6:67-70.
153 Smith T, Groom A, Zhu B, et al. Autoimmune encephalomyelitis ameliorated by AMPA antagonists. Nat Med. 2000;6:62-66.
154 Jung S, Zielasek J, Kollner G, et al. Preventive but not therapeutic application of rolipram ameliorates experimental autoimmune encephalomyelitis in Lewis rats. J Neuroimmunol. 1996;68:1-11.
155 Martinez I, Puerta C, Redondo C, et al. Type IV phosphodiesterase inhibition in experimental allergic encephalomyelitis of Lewis rats: sequential gene expression analysis of cytokines, adhesion molecules and the inducible nitric oxide synthase. J Neurol Sci. 1999;164:13-23.
156 Baker D, Pryce G. The therapeutic potential of cannabis in multiple sclerosis. Expert Opin Investig Drugs. 2003;12:561-567.
157 Kidd D, Barkhof F, McConnell R, et al. Cortical lesions in multiple sclerosis. Brain. 1999;122:17-26.
158 Peterson JW, Bo L, Mork S, et al. Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Ann Neurol. 2001;50:389-400.
159 Cifelli A, Matthews PM. Cerebral plasticity in multiple sclerosis: insights from fMRI. Mult Scler. 2002;8:193-199.
160 Dalton CM, Chard DT, Davies GR, et al. Early development of multiple sclerosis is associated with progressive grey matter atrophy in patients presenting with clinically isolated syndromes. Brain. 2004;127:1101-1107.
161 Chard DT, Griffin CM, McLean MA, et al. Brain metabolite changes in cortical grey and normal-appearing white matter in clinically early relapsing-remitting multiple sclerosis. Brain. 2002;125:2342-2352.
162 Chard DT, Griffin CM, Parker GJ, et al. Brain atrophy in clinically early relapsing-remitting multiple sclerosis. Brain. 2002;125:327-337.
163 De Stefano N, Matthews PM, Filippi M, et al. Evidence of early cortical atrophy in MS: relevance to white matter changes and disability. Neurol. 2003;60:1157-1162.






