[level-membership-for-emergency-medicine-category]
Chapter 10 Pathophysiology of Heat-Related Illnesses
For online-only figures, please go to www.expertconsult.com 
Heat Stress and Thermoregulation
Four Avenues of Heat Exchange
Mammals and other homeotherms are capable of maintaining body temperature within a fairly narrow range (approximately 35° to 41° C [95° to 105.8° F]) despite large fluctuations in environmental temperature. Environmental variables that have the largest impact on heat exchange are temperature; humidity; radiation from the air, water, or land; and air or water motion.88 To maintain stable body temperature, organisms rely on four avenues of heat exchange: conduction, convection, radiation, and evaporation.
Dry heat exchange is achieved by conduction, convection, and radiation. The effectiveness of these mechanisms depends on differences between the skin and environmental temperatures. That is, dry heat loss occurs when skin temperature exceeds that of the environment, and dry heat gain occurs when environmental temperature exceeds that of the body. Conduction occurs when the body surface is in direct contact with a solid object and depends on the thermal conductivity of the object and the amount of surface area in contact with the object. Conduction is typically an ineffective mechanism of heat exchange because of behavioral adjustments that minimize contact with an object. For example, the wearing of shoes is an effective behavioral adjustment that minimizes conduction of heat from a hot surface (e.g., desert sand to the foot). Within the body, conductive heat transfer occurs between tissues that are in direct contact with one another, but is limited by poor conductivity of the tissues. For example, subcutaneous fat has approximately 60% lower conductivity than does the dermis and impedes conductive heat loss.332 Convection is a mechanism of dry heat transfer that occurs as air or water moves over the skin surface. The windchill index is an example of the convective cooling effect of wind velocity. The rate of convective heat transfer depends on the temperature gradient between the body and environment, thermal currents, bodily movements, and areas of the body surface that are exposed to the environment, which can vary dramatically with different clothing ensembles. Within the body, convective heat transfer occurs between blood vessels and tissues and is most efficient at the capillary beds, which are thin-walled and provide a large surface area for heat exchange. Radiative heat transfer is electromagnetic energy that is exchanged between the body and surrounding environmental objects and is independent of air velocity or temperature. It is effective even when air temperature is below that of the body. All objects within our environment absorb and emit thermal radiation, but clothing can reduce radiant heat that impinges on the skin from various environmental sources.
Evaporation represents a major avenue of heat loss when environmental temperatures are equal to or above skin temperature or when body temperature is increased by vigorous physical activity. In humans, evaporative cooling is achieved as sweat is vaporized and removes heat from the skin surface, with approximately 580 kcal of heat lost per each liter of evaporated sweat.94 The most important environmental variables affecting evaporative cooling are ambient humidity and wind velocity. Sweat is converted to water vapor and readily evaporates from the skin in dry air with wind, whereas the conversion of sweat to water vapor is limited in still or moist air. If sweat accumulates and fails to evaporate, sweat secretion is inhibited and the cooling benefit is negated. Small mammals, such as rodents, do not possess sweat glands but achieve evaporative cooling by grooming nonfurred and highly vascularized skin surfaces, such as the ears, paw pads, and tail, with saliva that evaporates in a manner similar to that of sweat in humans.104,294
Body Temperature Control
Regulation of a relatively constant internal temperature is critical for normal physiologic functioning of tissues and cells because membrane fluidity, electrical conductance, and enzyme functions are most efficient within a narrow temperature range. By convention, thermal physiologists describe body temperature control with a two-compartment model that consists of an internal core (i.e., viscera and brain) and an outer shell (i.e., subcutaneous fat and skin) (Figure 10-1).70
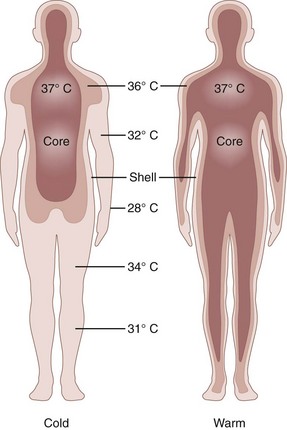
(From Elizondo RS: Human adaptation to hot environments. In Rhoades RA, Pflanger RG, editors: Human physiology, ed 3, Philadelphia, 1996, Saunders. Reprinted with permission of Brooks/Cole, a division of Thomson Learning. http://www.thomsonrights.com.)
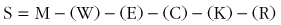
where M is metabolic rate, W is work, and E, C, K, and R are evaporative, convective, conductive, and radiant heat transfer, respectively.132 The impact of the four avenues of heat exchange on total body storage depends on a variety of organismal (e.g., age, gender, adiposity), environmental (e.g., humidity, wind velocity), and occupational (e.g., protective clothing, work intensity) variables. Under conditions in which heat production and/or heat gain exceeds heat loss, such as during exercise or heat exposure, positive heat storage occurs and body temperature increases. When heat loss exceeds heat production and/or heat gain, such as during prolonged cold exposure, negative heat storage occurs and body temperature decreases.88
Endothermic animals use both autonomic and behavioral thermoeffector mechanisms to regulate body temperature. Autonomic thermoeffector responses are often referred to as “involuntary” and include sweating, vasodilation, vasoconstriction, piloerection (furred mammals), and shivering and nonshivering thermogenesis (brown fat heat production). Behavioral thermoeffector mechanisms are considered “voluntary” and include clothing changes, use of heat or air conditioning systems, huddling or use of blankets, fan cooling, and seeking of shade or shelter. Rather than working independently of one another, autonomic and behavioral thermoeffector mechanisms typically function in concert to maintain temperature control. For example, evaporative cooling in rodents requires autonomic stimulation of salivation and behavioral spreading of saliva onto nonfurred surfaces.104,294 Many large species in the wild use natural water sources to facilitate cooling. Elephants spray water onto their skin surface, and hippopotamuses and other species are often observed near or in watering holes. Water is a more effective medium to facilitate convective heat transfer than air, because of its high heat-transfer coefficient (approximately 25 times greater than air),305 even if the water temperature is tepid. However, voluntary suppression of behavioral mechanisms of cooling in humans can increase the risk for thermal injury. This is illustrated by older adults who refuse to use air conditioning systems or leave their residences during heat waves, or highly motivated athletes and military personnel who voluntarily dehydrate and/or sustain a high rate of work in hot weather.
Regulation of body temperature is best conceptualized as a negative-feedback system consisting of sensors, integrators, and effectors. In vertebrates, neurons in the skin, spinal cord, and abdomen sense thermal stimuli and convert those signals to action potentials that are transmitted by afferent sensory neurons to the preoptic area of the anterior hypothalamus (POAH). The POAH is considered the main central nervous system (CNS) site for thermoregulatory control because it receives and integrates synaptic afferent inputs and evokes corrective autonomic and behavioral thermoeffector responses for body temperature regulation.28 A diagrammatic representation of this negative-feedback loop is shown in Figure 10-2.
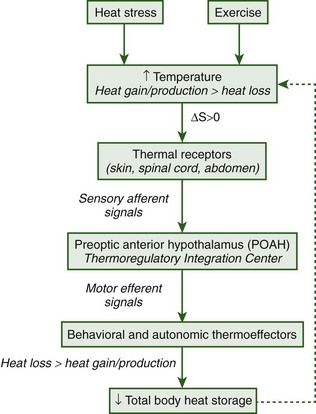
The concept of a temperature set point was developed as a theoretical framework to examine regulated and unregulated changes in body temperature.19 The temperature set point is analogous to a thermostat that controls a mechanical heating device; under homeostatic conditions, body temperature is approximately equal and oscillates around the temperature set point. Environmental perturbations, such as heat and exercise, cause body temperature to deviate from the set-point temperature as heat gain and/or production exceeds heat loss and the organism becomes hyperthermic (body temperature is greater than the set-point temperature). During prolonged cold exposure, heat loss exceeds heat gain and/or production and the organism becomes hypothermic (body temperature is less than the set-point temperature) (Figure 10-3).
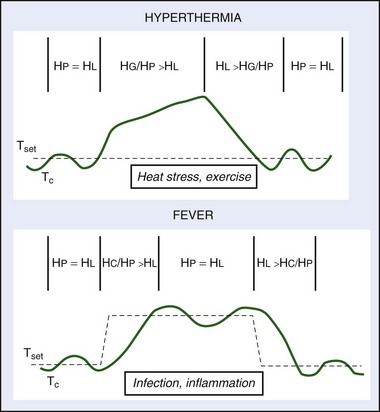
Regulated increases and decreases in the temperature set point are referred to as fever and regulated hypothermia (also called anapyrexia), respectively, and are protective immune responses to infection, inflammation, or trauma. Fever is defined as a regulated increase in the temperature set point and is actively established and defended by heat-producing (e.g., shivering and nonshivering thermogenesis) and heat-conserving (e.g., peripheral vasoconstriction, huddling to reduce exposed body surface area) thermoeffectors (see Figure 10-3).132 An individual is considered normothermic once fever is established and body temperature oscillates around the new elevated temperature set point (see Figure 10-3). The highly regulated nature of fever was first suggested by Liebermeister in the 1800s when it was observed that individuals actively reestablished an elevation in body temperature following experimental warming or cooling.180,286 Fever is a protective immune response used by invertebrates, fish, amphibians, reptiles, and mammals to survive infection or injury.* The protective effects of fever are mediated by increased mobility and activity of white blood cells,218,313 increased production of interferon (IFN; antiviral and antibacterial agent) antibodies,64 and reduced plasma iron concentrations, the effects of all of which inhibit the growth of pathogens.89,156 In mammals, inhibition of fever using antipyretic drugs (e.g., aspirin) increases mortality from bacterial and viral infections, which speaks to the importance of fever as an immune response.126,315
Many species also develop regulated hypothermia to survive severe environmental insults. Regulated hypothermia is elicited in response to a decrease in the temperature set point and is actively established and defended by behavioral and autonomic heat-loss mechanisms.132 The Q10 effect states that each 10° C (18° F) change in body temperature is associated with a twofold to threefold change in enzymatic reaction rates. Based on this relationship, a regulated decrease in body temperature is thought to protect against injury and inflammation by reducing production of harmful enzymatic end products that compromise tissue function under conditions of low oxygen supply. In bumblebees, infected worker bees spend significantly more time in cooler temperatures outside of the nest than healthy worker bees; this cold-seeking behavior is associated with increased survival from parasitic infection.118,213 Mice inoculated with influenza virus also show cold-seeking behavior and develop regulated hypothermia, which is associated with improved infection outcome.153 Other environmental insults that induce regulated hypothermia in small rodents include hypoglycemia,34,98 hypoxia,194,252 hemorrhage,119 dehydration,129 infection,153,176,264 and heatstroke.173,174
Mechanisms of Heat Dissipation During Thermal Stress
Cardiovascular mechanisms have evolved to shunt warm blood from the body core to the skin surface and increase heat loss during thermal stress. Arteriovenous anastomoses (AVAs) are collateral connections between adjacent blood vessels that increase the volume of blood that is delivered to a particular tissue. Mean skin blood flow can vary approximately 10-fold in humans depending on the thermal environment. The hands and feet are concentrated with AVAs that serve as effective areas for dry heat loss. The nonfurred surfaces of small rodents, such as the ears, tail, and paw pads, also have an abundance of AVAs and a large surface area to facilitate convective heat transfer.92,99 During exercise heat stress, increased blood flow to the skin surface is accompanied by sweat secretion. The density, secretion rate, and activation threshold of regional sweat glands determine the volume of sweat loss at a body site. In humans, the back and chest have the highest sweat rates for a given body temperature change, whereas only approximately 25% of total sweat is produced by the lower limbs.217 Additional factors affecting sweat rate include clothing characteristics, environmental conditions, and rate of metabolic heat production. Panting is an effective method of evaporative heat dissipation in large animals, such as birds, dogs, sheep, and rabbits, and occurs at a resonant ventilation frequency that requires minimal energy expenditure.107,257,329 Humans and rodents do not pant per se, but breathing frequency and minute volume increase during severe heat exposure to facilitate evaporative cooling from the respiratory surfaces. In humans, the contribution of respiratory evaporative cooling is small compared with that of skin evaporative cooling (Figure 10-4).
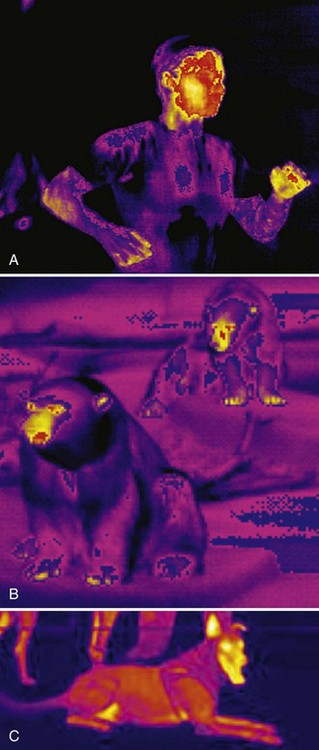
Dehydration and Electrolyte Imbalance
Water requirements during heat exposure are primarily determined by a person’s sweat losses. Water depletion dehydration develops when the rate of water replacement is not adequate, which can be a result of a mismatch between fluid intake and sweat loss, lack of water availability, or use of diuretic medications. Sweat rates may range from 0.3 to approximately 3 L/hr during athletic or occupational activities, depending on the environmental conditions and type, duration, and intensity of work.47,143 If high sweat rates are maintained without adequate replenishment of lost water, this can cause electrolyte imbalances that impede the efficiency of autonomic mechanisms of thermoregulatory control. For example, hyperosmolarity alters heat responsiveness of warm-sensitive neurons in the POAH and limits the effectiveness of evaporative heat loss.121,219,276 Severe hypernatremic dehydration is associated with brain edema, intracranial hemorrhage, hemorrhagic infarcts, and permanent brain damage (Figure 10-5, online).214
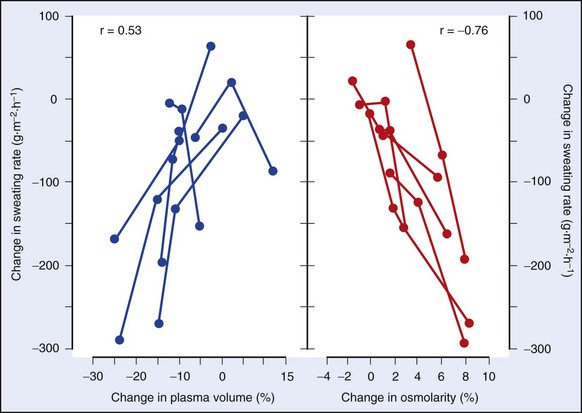
FIGURE 10-5 Effect of reduced plasma volume or increased osmolality on the sweat rates of six individuals.
(Modified from Sawka MN, Young AJ, Francesconi RP, et al: Thermoregulatory and blood responses during exercise at graded hypohydration levels, J Appl Physiol 59:1394, 1985.)
Severe reductions in electrolytes can have a profound impact on heatstroke outcome. Symptomatic hyponatremia (decreased serum sodium concentration) is a relatively rare condition, but it has been observed in marathon runners and military recruits during training exercises as a consequence of overconsumption of hypotonic fluids with inadequate replacement of sodium losses.210,230 Intracellular swelling is a severe consequence of hyponatremia that may cause CNS dysfunction. Hypokalemia (decreased serum potassium concentration) may be caused by overproduction of aldosterone, excessive sweating, or respiratory alkalosis. Any cause of overproduction of urine (polyuria) potentially causes urinary potassium loss.323 Potassium is a potent vasodilator of blood vessels to the skeletal and cardiac muscles, so excessive loss of this electrolyte can have detrimental effects, such as decreased sweat volume, cardiovascular instability, and reductions in muscle blood flow that predispose to skeletal muscle injury (i.e., rhabdomyolysis).158,279
Heat Illnesses
Heat illnesses are best viewed as existing along a continuum that transitions from the mild conditions of heat cramps and heat exhaustion to more serious conditions of heat injury and heatstroke (Table 10-1).
| Condition | Symptoms | Management |
|---|---|---|
| Heat cramps | Brief, painful skeletal muscle spasms | Rest; replacement of electrolytes; avoid salt tablets |
| Heat rash (miliaria rubra) | Blocked eccrine sweat glands | Cool, dry affected skin area; topical corticosteroids, aspirin |
| Heat exhaustion | Mild to moderate illness with inability to sustain cardiac output; moderate (>38.5° C [101.3° F]) to high (>40° C [104° F]) body temperature; often accompanied by dehydration | Move supine individual to cool, shaded environment, and elevate legs; loosen or remove clothing, and actively cool skin; administer oral fluids |
| Heatstroke | Profound CNS abnormalities (agitation, delirium, stupor, coma) with severe hyperthermia (>40° C [104° F]) | Ensure an open airway, and move to a cool environment. Immediately cool to <39° C (102.2° F) using ice packs, water bath, wetting with water and continuous fanning; IV fluid administration; reestablish normal CNS function; avoid antipyretics or drugs with liver toxicity |
CNS, Central nervous system; IV, intravenous.
Data from Bouchama A, Knochel JP: Heat stroke. N Engl J Med 346:1978, 2002; and Winkenwerder W, Sawka MN: Disorders due to heat and cold. In Goldman L, Ausiello DA, Arend W, et al, editors: Cecil textbook of medicine, ed 23, Philadelphia, 2007, Saunders, pp 763-767.
Heat exhaustion (also referred to as heat prostration or heat collapse) is a mild to moderate form of heat illness that is associated with moderate (>38.5° C [101.3° F]) to high (>40° C [104° F]) elevations in core temperature and an inability to sustain cardiac output.335 The signs and symptoms of heat exhaustion include fatigue, dizziness, headache, nausea, vomiting, malaise, hypotension, and tachycardia with potential for collapse. Heat exhaustion can occur with or without exercise in hot environments and may progress to a moderately severe condition without associated organ damage. Heat exhaustion is often observed in older adults as a result of medications (e.g., diuretics), inadequate water intake that leads to dehydration, or preexisting cardiovascular insufficiency that predisposes to collapse. Treatment should consist of placing the individual in a recumbent position in a cool environment to normalize blood pressure. Oral fluid ingestion with electrolytes is often adequate for recovery; intravenous (IV) fluid administration may be warranted in severely dehydrated individuals.
Heat injury is a moderate to severe condition characterized by tissue (e.g., skeletal muscle) or organ (e.g., gut, kidney, spleen, liver) damage and hyperthermia (core temperature usually, but not always >40° C [104° F]).335 Heat injury may progress to heatstroke if the patient is not rapidly cooled. Heatstroke is life threatening, with the patient presenting with profound CNS abnormalities, such as delirium, agitation, stupor, seizures, or coma and severe hyperthermia (core temperature typically, but not always >40° C [104° F]).335 Reliance on a specific core temperature value for clinical diagnosis of heatstroke is ill advised, because there is wide interindividual variability in documented cases. One of the main reasons for a lack of clinical treatments for heatstroke is the complicated nature of the syndrome, because there are different classifications based on etiology and pathophysiologic mechanisms of injury. Classic (also known as passive) heatstroke occurs at rest in vulnerable individuals, such as infants and older adults. Several intrinsic factors may predispose infants to heatstroke death. These include increased surface area–to–body mass ratio (accelerates heat gain), limited effective mechanisms of thermoregulation (e.g., suppressed behavioral adjustments), increased risk for dehydration (e.g., lack of water availability), and preexisting respiratory infections. Many older individuals have preexisting conditions, such as mental illness, prescription drug use (e.g., diuretics, anticholinergics), or infections that predispose to passive heatstroke (Box 10-1).6,56,312
BOX 10-1
Predisposing Risk Factors for Serious Heat Illness
Data from references 6, 58, 196, 318, and 340.
Exertional heatstroke (EHS) has a different etiology than classic heatstroke and affects young healthy populations that perform strenuous physical activity or work in temperate or hot weather. During exercise, approximately 80% of expended energy is released as heat that must be dissipated from the body to avoid hyperthermia. Military and athletic populations are composed of young, healthy individuals who are highly motivated to perform strenuous physical activity in hot weather, which increases the risk for EHS. A recent epidemiologic study identified a variety of factors that predispose to EHS, including gender (women greater than men), geographic region of origin (northern greater than southern states), preexisting illness, and race or ethnicity (whites greater than blacks).40 Unfortunately, exercise induces physiologic responses similar to those of heat stress, such that teasing apart the influence of these two factors in EHS is difficult.
Heatstroke Epidemiology and Risk Factors
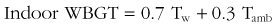
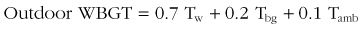
Heat waves are defined as three or more consecutive days during which the environmental temperature exceeds 32.2° C (90° F).41 In the summer of 2003, Europe experienced 22,000 to 45,000 heat-related deaths during a 2-week period in which the average temperature was 3.5° C (6.3° F) above normal.189,273 The European continent has experienced an increase in minimum daily temperatures over the last 30 years, and this trend will likely increase if average global temperatures continue to rise. A 1.4° to 5.8° C (2.5° to 10.4° F) increase in minimum daily temperatures in Europe is predicted over the next century.131,337 Most prediction models suggest that heat waves in the future will be more severe and longer in duration. Predictions based on climate variability data from the 1995 Chicago and 2003 Europe heat waves suggest that by 2090, heat waves in these cities will be 25% to 31% more frequent and last 3 to 4 days longer.203 Another prediction model suggests a 253% increase in annual heatstroke deaths in the United Kingdom by 2050.61
The impact of climate change is not equally distributed across the globe because of regional variability in thermal tolerance that influences the incidence of heatstroke mortality. A study of 11 U.S. cities showed that threshold temperatures for heatstroke mortality are higher in warmer southern cities than in cooler northern cities.53 A comparison of temperature–mortality relationships in southern Finland, southeastern New England, and North Carolina indicated that lower temperature thresholds in cooler climates are coupled with steeper temperature–mortality relationships.62 Similarly, the upper safety limits of environmental temperatures in the Netherlands, London, and Taiwan are 16.5°, 19°, and 29° C (61.7°, 66.2°, 84.2° F), respectively.192 A case study of 15 Marine recruits who collapsed from heatstroke during training exercises in South Carolina showed that 73% previously resided in northern states and that 60% of cases occurred during the second week of training during the hottest summer months.232 From 1980 to 2002, the highest EHS incidence in military recruits was in nonacclimatized individuals from northern, cold-climate states who were enlisted for less than 12 months.40 During July, many regions of the world have a WBGT index that is greater than 29° C (84.2° F), and military training often occurs in environments with a WBGT index that is greater than 35° C (95° F). During peacetime exercises, approximately 25% of fatal military EHS cases occur during the hottest summer months in recruits who have been in training camp less than approximately 2 weeks.192 Individuals from northern states are expected to be less acclimatized to hot, humid summer conditions than are those from southern states. Heat acclimatization improves thermotolerance but requires several days to weeks of exposure to similar heat stress and exercise conditions to be fully effective. This likely accounts for hot days early in the summer showing a greater impact on heatstroke morbidity and mortality than those cases occurring later in the training process, after the protective effects of heat acclimatization have been realized.105
Humanity’s impact on the landscape in conjunction with increased production of greenhouse gases may be creating the largest climate change. Urban heat islands are created in cities when vegetation is removed and blacktop roads and concrete buildings are erected. Temperatures may be 30° to 40° C (54° to 72° F) higher on asphalt roads and roof tops compared with those of the surrounding air.85 Since 1978 urban sprawl has accounted for an increase in city temperatures in southeastern Asia of approximately 0.05° C (0.09° F) per decade.343 Across the entire land mass of the United States, the surface temperature has increased approximately 0.27° C (0.49° F) per century because of changes in the land cover arising from agricultural and urban development.139 Concrete and asphalt surfaces cool slowly during the nighttime when air temperature decreases, and this increase in urban heat storage magnifies the intensity of heat exposure experienced by individuals living in concrete urban structures.49,165
Several social factors predispose older adults to heatstroke mortality, including living alone, inability or unwillingness to leave one’s home, residing on the top floor of buildings (heat rises), and annual income of less than $10,000.221 Most heat wave early warning systems emphasize use of air-conditioning systems, but availability and use of the units are limited by socioeconomic status because they are expensive to operate.56,221 A working air conditioner was the strongest protective factor against mortality during the 1999 heat wave in Chicago; fan cooling did not afford protection.221 High mortality rates were recorded in Chicago despite extensive programs to educate high-risk populations, such as advising older adults to seek cool shelters or use air-conditioning systems. Approximately 10,000 elders died during the France heat wave of 2003 primarily because of lack of air-conditioning units in residences and hospitals.63,312 In 2005, Hurricane Katrina ravaged the U.S. Gulf Coast, and electrical failures caused high heatstroke mortality of older adults confined to residences, retirement homes, and hospitals because local temperatures exceeded 43° C (109.4° F). Increases in the average human lifespan, global climate change, and use of medications that compromise cardiovascular adjustments to heat stress will necessitate increased reliance on artificial cooling systems and educational programs to prevent heatstroke deaths in vulnerable populations, such as older adults.
The high death toll of older adults because of excessive heat per se may be small compared with that caused by aggravation of a preexisting illness. Heat stress refers to environmental and metabolic conditions that increase body temperature; heat strain refers to the physiologic consequences of heat stress. Heat strain imposes large cardiovascular demands on the body. Blood flow is shunted from the viscera to the skin surface to dissipate excess heat to the environment, making cardiovascular fitness a more important factor than age in determining an individual’s susceptibility to heatstroke. Austin and Berry10 examined 100 cases of heatstroke during three summer heat waves in St. Louis and found cardiovascular illness in 84% of patients. Levine181 found heatstroke deaths to be associated with arteriosclerotic heart disease (72%) and hypertension (12%). Cardiac deficiency impedes heat loss and compromises the ability to maintain cardiac output during prolonged heat exposure, leading to circulatory collapse and death. Older individuals may have impaired baroreceptor reflex modulation, lower sweat rates, longer time to onset of sweating, and diminished sympathetic nerve discharge, all of which increase the risk for heatstroke morbidity and mortality.130,144,291 Minson and colleagues210 demonstrated that during heat exposure, older men relied on a higher percentage of their cardiac chronotropic reserve compared with younger men.
Preexisting infection or inflammation may compromise an individual’s ability to appropriately respond to heat stress and can be a complicating factor, regardless of age. Fifty-seven percent of heatstroke patients more than 65 years old had evidence of infection upon clinical admission during a Chicago heat wave in 1995.56,157 In Singapore, a young EHS victim had been ill for 3 days before heatstroke collapse.43 It has been proposed that acute illness or inflammation can cause transient susceptibility to heatstroke in young, fit individuals who exercise in the heat. For example, idiosyncratic episodes of hyperthermia were associated with acute cellulitis and gastroenteritis in soldiers exercising in the heat.39,146 Four male Marine recruits presented with viral illness (mononucleosis, pneumonia) before collapse from exertional heat illness (EHI) during training exercises associated with “the Crucible” at Parris Island, South Carolina.289 Peripheral blood mononuclear cells (PBMCs) from these recruits expressed higher levels of IFN-inducible genes than did those from controls who participated in the training event but did not collapse.289 High plasma levels of IFN-α and IFN–γ mediate flulike symptoms during viral infection and are often associated with EHI/EHS.24,289 In rats, exposure to lipopolysaccharide (LPS), a cell wall component of gram-negative bacteria, exacerbated inflammation, coagulation, and multiorgan system dysfunction from heat exposure.182 Taken together, these findings suggest that a preexisting inflammatory state compromises an individual’s ability to respond to heat stress with appropriate thermoregulatory or immune responses to prevent collapse or multiorgan system failure and death.
The annual Muslim pilgrimage to Mecca (i.e., the Hajj) is associated with high heatstroke incidence each year and provides many lessons regarding etiologic factors that increase susceptibility. The Hajj takes place in the hot desert environment of Saudi Arabia during the extreme weather months of May to September, when temperatures range from 38° to 50° C (100.4° to 122° F).151 Hot weather combined with physical exertion (first day consists of a 3.5-km [2.2-mile] jog), heavy clothing that is traditional to the region (limits heat dissipation), and an older population (approximately 50 years is an advanced age for this region) predispose many individuals to heatstroke. Clothing has a significant impact on Muslim women because they are required to wear darker clothing that covers a larger surface area of the body than does clothing worn by men.114 Medical conditions, such as diabetes, cardiovascular abnormalities, or parasitic diseases, are common.151 Heatstroke is a major concern, but heat exhaustion with water or salt depletion is also prevalent. Overcrowding and congestion impose large demands on sanitation services, as exemplified in the 1980s, when approximately 2 million people participated in the Hajj. Advances in modern technologies, such as more rapid transport to the area, will likely introduce additional factors (e.g., lack of acclimatization, increased greenhouse gas production, increased congestion) to this already complex situation.
Protective clothing is a significant predisposing factor to EHS during athletic (heavy uniforms), military (chemical protective clothing), or occupational activities (e.g., pesticide application, firefighting, and race car driving). Protective clothing often consists of multiple layers that insulate anatomic sites from heat exchange, including the skin and head.251 The wearing of protective clothing during strenuous work can quickly result in a dangerous elevation in body temperature. Fifty-one cases of EHI were observed in military trainees in San Antonio, Texas, during participation in a 9.3-km (5.8-mile) march in full battle dress uniform and boots.285 Lack of acclimatization to athletic uniforms and high environmental temperatures results in the majority of cases of EHS in athletes occurring on the second or third day of exposure to hot weather before these individuals are acclimatized to the uniforms and environmental temperatures.97,260
Pathophysiology of Heatstroke
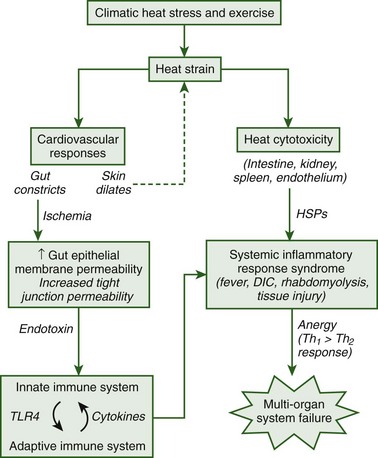
The pathophysiologic responses to heatstroke range from those conditions that are experienced immediately following collapse to long-term changes that persist for several weeks, months, or years following hospital treatment and release. Currently more is known about the immediate heatstroke responses because clinical records document symptoms during hospital treatment. However, clinical and experimental research has seen a shift within the past decade toward a focus on understanding the pathophysiologic responses that mediate long-term injury. It is now believed that the long-term pathophysiologic responses to heatstroke are caused by a systemic inflammatory response syndrome (SIRS) that ensues following heat-induced damage to the gut and other organs.24 Following damage to the epithelial membrane of the gut, endotoxin that is normally confined to the lumen of this organ is able to leak into the systemic circulation and elicit immune responses that cause tissue injury. The thermoregulatory, immune, coagulation, and tissue injury responses that ensue during the long-term progression of heatstroke closely resemble those observed during clinical sepsis and are likely mediated by similar cellular mechanisms. Clinical records have provided an extensive database of the immediate consequences of heatstroke, whereas the majority of knowledge regarding the pathophysiologic mechanisms of heat-induced SIRS has been obtained from experimental animal studies. Although there are several gaps in our knowledge of the specific factors that predispose to multiorgan system failure, this is an exciting area of research that is expected to progress at a rapid rate because of continued advancements in experimental and genetic technologies. Figure 10-6 provides an overview of the current understanding of the pathophysiologic responses that are thought to initiate and mediate heat-induced SIRS, which will be discussed in detail here.
Body Temperature Responses
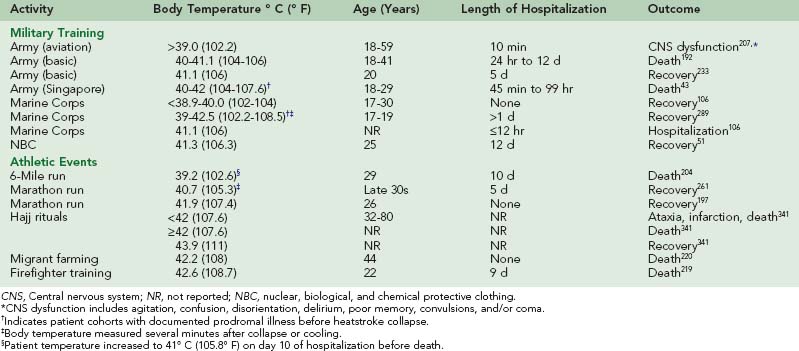
At the time of heatstroke collapse, the severity of hyperthermia varies widely between individuals, with reported core temperature values ranging from approximately 41° C (105.8° F) to 47° C (116.6° F).* During a summer heat wave in St. Louis in the 1950s, the core temperature of 100 heatstroke patients ranged from 38.5° to 44° C (101.3° to 111.2° F), with 10% of mortalities occurring below 41.1° C (106° F).10 In some instances, individuals may tolerate hyperthermia without adverse side effects. During a competitive marathon race in California, a 26-year-old man maintained a rectal temperature of 41.9° C (107.4° F) for approximately 45 minutes without clinical signs of heat illness.197 However, there are several reports of athletic, military, and occupational workers with core temperatures below 41.9° C (107.4° F) who were hospitalized, experienced permanent CNS impairment, or died from EHS (Table 10-2).
Hypothermia and fever are core temperature responses that are often observed in patients and experimental animal models during heatstroke recovery. Hypothermia is not a universal heatstroke recovery response in humans but has been anecdotally observed following aggressive cooling treatment. Hypothermia manifests as a rapid undershoot of body temperature below 37° C (98.6° F) and is thought to represent a loss of thermoregulatory control following heat-induced damage to the POAH. However, evidence in support of this hypothesis is lacking because autopsy reports and experimental animal studies have failed to detect histologic damage to the POAH despite extensive damage in other organs.174,192 Because hypothermia is not observed in all heatstroke patients, it continues to be regarded as a pathologic recovery response. In experimental animals, hypothermia is a natural heatstroke recovery response that is associated with behavioral and autonomic thermoeffector responses that support a decrease in core temperature. Mud puppies are ectothermic species that rely on behavioral adjustments, such as the selection of different microclimates, to control body temperature. Mud puppies heat shocked to approximately 34° C (93.2° F) behaviorally selected a cooler microclimate and maintained a significantly lower body temperature than did nonheated controls during 3 days of recovery.130 This study did not determine the impact of hypothermia on survival, but the association of decreased body temperature with the selection of cool microclimates indicated that this was a regulated response to a decrease in the temperature set point. Small rodents, such as mice, rats, and guinea pigs, showed reductions greater than 1.0° C (1.8° F) in body temperature that were associated with improved survival following passive heatstroke. In mice, hypothermia was associated with an approximately 35% decrease in metabolic heat production and the behavioral selection of microclimates that precisely regulated the depth and duration of this response.174 Exposure of mice to warm ambient temperatures that prevented heat-induced hypothermia caused increased intestinal damage and mortality.173,333 Hypothermia likely provides protection against heat-induced tissue injury in a manner similar to that shown for protection against other extreme environmental insults based on the temperature coefficient (Q10) effect.
A common heatstroke recovery response observed in patients and animal models is recurrent fever during the days and weeks of recovery.9,10,173,192,204 In mice, fever was observed within a day after passive heatstroke collapse and associated with an approximately 20% increase in metabolic heat production and increased plasma levels of the proinflammatory cytokine interleukin (IL)-6.154,172,174 IL-6 is an important regulator of fever during infection and inflammation and may regulate fever during heatstroke recovery.172 In patients, fever is reestablished following clinical cooling.192 This is reminiscent of Liebermeister’s experimental observations of the recurrence of fever following experimental cooling of the POAH of rats.180,192 In experimental animal models, the inability to recover from hypothermia and develop fever is associated with increased mortality, suggesting that fever may be important for the resolution of infection.173 However, in a case report of human heatstroke, fever was associated with poor outcome. An amateur long-distance runner was hospitalized for 10 days after collapsing from EHS during a 9.7-km (6-mile) footrace.204 Moderate fever (>38° C [100.4° F]) was evident during the first 4 days of hospitalization, but on the tenth day the patient experienced convulsions and a rapid increase of body temperature to 41° C (105.8° F). Rapid cooling and aspirin were ineffective in reducing body temperature, and the patient died.204 The inability of aspirin to inhibit the rapid rise in body temperature suggests that this was not a true fever response, but rather a pathologic response to increased metabolic heat production induced by the convulsions. It is important to recognize that there is an optimal temperature range above which the protective effects of fever are no longer realized because of the toxic effect of high body temperature on cell function.155
Immune Responses
During heat stress, blood flow to the skin is increased to facilitate heat loss to the environment and reduce the rate of total body heat storage. Increased skin blood flow is accompanied by a fall in splanchnic (i.e., visceral organ) blood flow as a compensatory mechanism to sustain blood pressure. Endotoxin is normally confined to the gut lumen by tight junctions of the epithelial membrane, but these junctions can become “leaky” following prolonged reductions in blood flow that cause ischemic stress.108,164 There are several lines of evidence that support the hypothesis that endotoxin leakage from the gut lumen into the systemic circulation is the initiating stimulus for heat-induced SIRS. First, systemic injection of LPS into experimental animals induces symptoms similar to those observed in heatstroke, including hyperthermia, hypothermia, fever, hypotension, cytokine production, coagulation, and tissue injury.175,268 Second, increased portal or systemic endotoxin levels are observed in heatstroke patients and animal models. In primates, circulating endotoxin was detected at rectal temperatures above 41.5° C (106.7° F) with a precipitous increase at approximately 43.0° C (109.4° F).90 Splanchnic blood flow shows an initial decrease at 40° C (104° F); the liver, which is an important clearance organ for endotoxin, shows damage at body temperatures of approximately 42° to 43° C (107.6° to 109.4° F).* In a young athlete with a body temperature of 40.6° C (105.1° F) on the second day of football practice, high circulating levels of endotoxin were associated with hemorrhagic necrosis of the liver.97 In heatstroke patients, endotoxin was detected at approximately 42.1° C (107.8° F) and remained elevated despite cooling.26 Third, rats rendered endotoxin tolerant following the systemic injection of LPS are protected from heatstroke mortality.66,67 The protective effect of endotoxin tolerance is related to enhanced stimulation of the liver reticuloendothelial system (RES), which is composed of monocytes, macrophages, and Kupffer cells that are important for endotoxin clearance.66,67 RES stimulation reduced and RES blockade increased mortality of heat-stressed rats.67 Fourth, antibiotic therapy protects against heatstroke in several species. In dogs, antibiotics reduced gut flora levels and improved 18-hour survival rates by more than threefold when provided before heat exposure.37 In rabbits with heatstroke, hyperthermia and endotoxemia were reduced following oral antibiotics.36 Anti-LPS hyperimmune serum reversed the heatstroke mortality rates of primates and returned plasma LPS levels to baseline, but it was ineffective at the highest body temperature of 43.8° C (110.8° F), indicating that hyperthermia can cause irreversible organ damage and death.91
Heat-induced SIRS is initiated by the innate and adaptive immune systems, which interact to sense the presence of endotoxin and orchestrate an immunologic response. The innate immune system comprises monocytes, macrophages, and neutrophils that use pattern recognition receptors (PRRs) on their cell surfaces to recognize pattern-associated molecular patterns (PAMPs) on the cell surface of endotoxin and other invading pathogens.137 Toll-like receptors (TLRs) are a class of PRRs that have been widely studied in the immune response to infection.208,314 Ten mammalian TLRs have been identified, and the specific pathogenic ligands that activate these PRRs are known (Table 10-3).
| Toll-Like Receptor | Ligand | Cell/Tissue Types |
|---|---|---|
| TLR1 | Triacyl lipopeptide | Monocytes, macrophages, DCs, polymorphonuclear leukocytes, B and T cells, NK cells |
| TLR2 | Lipopolysaccharide Peptidoglycan Lipoteichoic acid Measles virus Human cytomegalovirus Hepatitis C virus Zymosan Necrotic cells |
Monocytes, granulocytes Brain, heart, lung, spleen |
| TLR3 | Viral double-stranded RNA | DCs, T cells, NK cells, monocytes, granulocytes Placenta, pancreas |
| TLR4 | Lipopolysaccharide Fibrinogen Heat shock proteins High mobility group box 1 |
B cells, DCs, monocytes, macrophages, granulocytes, T cells Spleen |
| TLR5 | Flagellated bacteria | Monocytes Ovary, prostate |
| TLR6 | Diacyl lipopeptide | B cells, monocytes Thymus, spleen, lung |
| TLR7 | Single-stranded RNA | Monocytes, B cells, DCs Lung, placenta, spleen, lymph node, tonsil |
| TLR8 | Single-stranded RNA | Monocytes Lung, placenta, spleen, lymph node, bone marrow, PBLs |
| TLR9 | CpG DNA | B cells, DCs Spleen, lymph node, bone marrow, PBLs |
| TLR10 | Unknown | B cells Spleen, lymph node, thymus, tonsil |
CpG, Deoxycytidylate-phosphate-deoxyguanylate; DC, dendritic cell; DNA, deoxyribonucleic acid; NK, natural killer; PBL, peripheral blood leukocyte; RNA, ribonucleic acid.
Data from Medvedev AE, Sabroe I, Hasday JD, et al: Tolerance to microbial TLR ligands: Molecular mechanisms and relevance to disease, J Endotoxin Res 12:133, 2006; and Tsujimoto H, Ono S, Efron PA, et al: Role of Toll-like receptors in the development of sepsis, Shock 29:315, 2008.
TLR4 is the principal receptor for LPS that stimulates gene transcription factors, such as NF-κB, to increase the synthesis of a variety of immune modulators in response to endotoxin. Endotoxin infection (i.e., sepsis) is associated with increased expression of TLR4 on circulating human PBMCs, as well as on mouse liver and spleen macrophages.307,308 In the 1960s, a spontaneous mutation in the TLR4 gene was discovered in C3H/HeJ mice, which has been an important animal model to determine the role of TLR4 in endotoxin responsiveness. C3H/HeJ mice show a diminished response to bacterial infection, but increased mortality from SIRS, because of an inability to respond appropriately to endotoxin and induce the full complement of immune responses.103 Inability to respond to antigens is known as anergy and is a proposed mechanism that predisposes to increased risk and mortality from bacterial infection.117 Given that TLR4 mutations exist in humans, this may be one (of several) genetic factors that predispose to mortality associated with heat-induced SIRS, although the association of mortality with TLR4 polymorphisms remains controversial.5,75 C3H/HeJ mice have not been tested for their resistance to heatstroke morbidity/mortality but are a useful experimental model to determine the roles of TLR4 and anergy in this syndrome.
Specificity of immune responses is provided by B and T cells of the adaptive immune system. These cells respond to antigens by secreting cytokines, which are intercellular immune signals that elicit proinflammatory (Th1 type) and antiinflammatory (Th2 type) actions during the progression of SIRS. The actions of cytokines depend on the nature of the danger signal, the target cells with which they interact, and the cytokine “milieu” in which they function. Th1 and Th2 cytokines function in a negative feedback pathway to regulate each other’s production and maintain a delicate balance of inflammatory reactions. Anergy is thought to be a consequence of inadequate Th2 cytokine production late in SIRS. For example, increased patient mortality from peritonitis is associated with the inability to mount a Th2 cytokine response.117
Alarmins are endogenous PAMPs that are released from stressed or injured tissues and initiate restoration of homeostasis following an infectious or inflammatory insult.17 High mobility group box 1 (HMGB1) is a highly conserved nuclear protein that functions as an alarmin following release from necrotic (but not apoptotic) cells.272 Necrosis is the premature death of cells in a tissue or organ in response to external factors, such as pathogens and toxins. Because necrosis is detrimental to the host, it is associated with an inflammatory response. Apoptosis refers to genetically programmed cell death that does not elicit an inflammatory response, because apoptosis is beneficial to the host. Release of HMGB1 from necrotic cells stimulates Th1 cytokine production late in the sepsis syndrome and is a purported mediator of lethality; this shift in the balance of cytokines from a Th2 to Th1 phenotype is a potential mechanism of sepsis lethality. In human PBMCs, HMGB1 interacts with TLR2 and TLR4 to enhance Th1 cytokine production in synergy with LPS.124 Elevated serum HMGB1 levels are observed 8 to 32 hours following LPS injection in mice. Anti-HMGB1 antibodies did not protect against LPS-induced mortality unless the antibodies were provided 12 and 36 hours after LPS exposure.324 The delayed kinetics of HMGB1 and the association of elevated serum levels of this protein with poor outcome in sepsis patients suggest that HMGB1 detection late in SIRS may be a sensitive clinical marker of disease severity.115,297,324
Coagulation
Disseminated intravascular coagulation (DIC) is a common clinical symptom of heatstroke and manifests in two different forms (Figure 10-7).
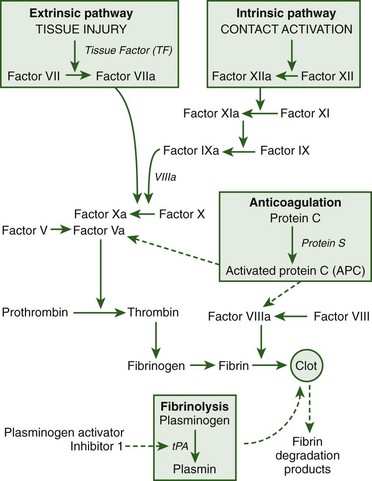
Microvascular thrombosis is a form of DIC characterized by fibrin deposition and/or platelet aggregation that occludes arterioles and capillaries and predisposes to multiorgan system dysfunction.177 Microvascular thrombosis is commonly observed in response to sepsis or trauma. DIC associated with consumptive coagulation is characterized by excessive blood loss when platelets and coagulation proteins are consumed faster than they are produced.8,12 Hemorrhagic complications in heatstroke victims include prolonged bleeding from venipuncture sites or other areas (e.g., gums), which can have a fatal outcome.148 The primary event that initiates coagulation in heatstroke patients is thermal injury to the vascular endothelium.21,24,215 In vitro studies have shown the ability of heat (43° to 44° C [109.4° to 111.2° F]) to directly activate platelet aggregation and cause irreversible hyperaggregation despite cooling.87,333 Cancer patients treated with whole-body hyperthermia (41.8° C [107.2° F] for 2 hours) showed decreased fibrinogen and plasminogen at body temperatures as low as 39° C (102.2° F), alterations in factor VII activity at 41.8° C (107.2° F), and decreased platelet concentrations from the time of maximum body temperature through 18 hours of recovery.295
Several proteins, including HMGB1, IL-1, tumor necrosis factor (TNF), and activated protein C (APC), affect the coagulation, anticoagulation, and fibrinolytic pathways. In rats, HMGB1 in combination with thrombin caused excess fibrin deposition in glomeruli, prolonged clotting times, and increased sepsis mortality compared with thrombin alone.133 The effect of HMGB1 protein was demonstrated in vitro to be caused by inhibition of the APC pathway and stimulation of tissue factor expression on monocytes.133 Cytokines stimulate microvascular thrombosis by interacting with neutrophils, macrophages, platelets, and endothelium to increase expression of intracellular adhesion molecules. Increased expression of cell adhesion molecules, neutrophil adhesion, and release of reactive oxygen species caused endothelial activation and injury.201,330 APC is an important component of the anticoagulation pathway that inactivates factors Va and VIIIa to inhibit fibrin clot formation. In septic patients, reduced APC production was associated with increased risk of mortality from systemic inflammation and DIC.78,179 In addition to its anticoagulation properties, APC possessed antiinflammatory and antiapoptotic properties that protected against experimental sepsis and heatstroke.44,302
Tissue Injury
Multiorgan system failure is the ultimate cause of heatstroke mortality and is a consequence of SIRS, which ensues following heat-induced damage to the gut and other tissues.24 A variety of noninfectious and infectious clinical conditions are associated with SIRS, and similar physiologic mechanisms are thought to mediate the pathogenesis of these conditions (Box 10-2).
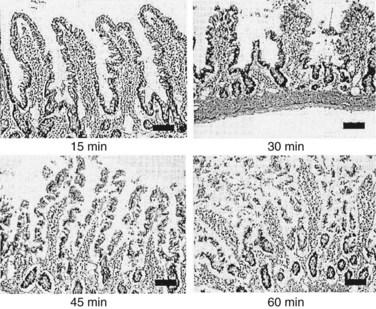
The severity of heatstroke is primarily related to the extent of damage to the brain, liver, and kidneys and is clinically identified by elevations in serum biomarkers, such as creatine kinase (CK), blood urea nitrogen (BUN), aspartate aminotransferase (AST), and alanine aminotransferase (ALT). CK is released from muscle and is a marker of skeletal muscle injury (also known as rhabdomyolysis), myocardial infarction, muscular dystrophy, and acute renal failure.1,274,322 BUN is a measure of the amount of nitrogen in the blood in the form of urea, which is secreted by the liver and removed from the blood by the kidneys. A high BUN concentration is typically regarded as an indication of impaired renal function, although BUN levels may be altered by conditions unrelated to heat illness, including malnutrition, high-protein diets, burns, fever, and pregnancy.1,267 AST is released by the liver and skeletal muscle and may be a clinical sign of congestive heart failure, viral hepatitis, mononucleosis, or muscle injury. ALT is released by the liver, red blood cells, cardiac muscle, skeletal muscle, kidneys, and brain tissue. AST and ALT are common clinical markers of liver function in heatstroke patients despite multiple tissue sources of these enzymes and occasional false-negative results that complicate interpretation. Unfortunately, all of these biomarkers are released by a variety of tissues and altered by heat-exhaustive exercise, so they do not always provide a precise measure of the extent of tissue injury.95,102,283 The extent and time course of organ injury vary widely between individuals. Tissue injury manifests as primary and/or secondary multiorgan dysfunction, depending on whether heat toxicity alone or in combination with SIRS causes cellular damage.4,192 Gut epithelial barrier disruption is an example of primary organ dysfunction that is evident at the time of heatstroke collapse. Hyperthermia degrades epithelial membrane integrity and causes microhemorrhages, dilation of the central lacteals of the microvilli, and blood clots within the stomach and small intestine (Figure 10-8).27,108,172
It is often difficult to determine if organ injury is caused by primary or secondary factors.* For example, protein clumping in kidney tubular epithelial cells may be a consequence of heat toxicity, elevated myoglobin levels, or DIC.† A conscious mouse model has shown that kidney damage is present within approximately 2 hours following heatstroke collapse and remains elevated through approximately 24 hours of recovery.172 In heatstroke patients, acute renal failure is a nearly universal finding that is accompanied by decrements in function within 24 hours of admission to the intensive care unit.244 In patients that survive more than 24 hours, severe hypotension, dehydration, BUN, and oliguria are associated with tubular necrosis or intertubular edema.192 Primary changes in the spleen are even less well understood, but cytoplasmic protein clumping is thought to be a consequence of this organ being “simply cooked and coagulated.”43,172
CNS dysfunction is a hallmark of heatstroke that is dominant early in the disorder. Patients are often confused, delirious, combatant, or comatose at the time of clinical presentation. Hyperthermia with exercise is also associated with reduced cerebral blood flow, which may account for these CNS abnormalities.228 Despite rapid treatment, approximately 30% of heatstroke survivors experience permanent decrements in neurologic function.6,24,56 CNS dysfunction is often associated with cerebral edema and microhemorrhages at autopsy in heatstroke patients.* The blood–brain barrier (BBB) is a semipermeable membrane that allows selective entry of substances (e.g., glucose) into the brain while blocking the entry of other substances (e.g., bacteria). Hyperthermia increases BBB permeability in experimental animal models, which permits leakage of proteins and pathogens from the systemic circulation into the brain. Computed tomography (CT) scans have been used to examine CNS changes in heatstroke patients. In the 1995 Chicago heat wave, atrophy, infarcts of the cerebellum, and edema were evident in older adult victims. CT scans also revealed severe loss of gray-white matter discrimination (GWMD), which was associated with headache, coma, absence of normal reflexive responses, and multiorgan dysfunction.299 Loss of GWMD is a consequence of increased brain water content, which is in line with occurrence of edema in heatstroke victims. If GWMD provides an early, sensitive measure of brain injury, it will be a powerful prognostic indicator of outcome for heatstroke patients.
EHS is often associated with rhabdomyolysis, which is a form of skeletal muscle injury caused by the leakage of muscle cell contents into the circulation or extracellular fluid. Myoglobin released from damaged muscle cells is filtered and metabolized by the kidneys. When severe muscle damage occurs, the renal threshold for filtration of myoglobin is exceeded, and this protein appears in the urine in a reddish brown color.86 Myoglobin is toxic to nephrons and causes overproduction of uric acid, which precipitates in the kidney tubules to cause acute renal failure, coagulopathy, and death if not rapidly detected and treated.11,86,185,242,323 Not all cases of rhabdomyolysis are associated with myoglobinuria; many patients can be asymptomatic. Clinical markers of rhabdomyolysis include elevated myoglobin, CK, aldolase, lactate dehydrogenase, ALT, and AST, which are influenced by a variety of factors (type, intensity, and duration of exercise; gender; temperature; altitude) and released by more than one organ or tissue.50,205,263 If a clinical diagnosis of rhabdomyolysis is confirmed, immediate medical attention is imperative because approximately 50% mortality rates from acute renal failure have been documented for this condition.
Liver failure is one of the most common causes of morbidity and mortality in patients during the later stages of recovery. The time course of liver damage differs from that of the other organs and often does not peak until approximately 24 to 48 hours after heat exposure. For example, liver damage consisting of centrilobular degeneration and necrosis with parenchymal damage was only evident in EHS patients that survived more than 30 hours.192 In addition, enhanced breakdown of fat or an inability of the mitochondria to use fat results in heatstroke-associated fatty liver changes.43 Disturbances in plasma glucose homeostasis are a sign of liver damage that may cause hyperglycemia or hypoglycemia as a result of dysfunction of phosphoenolpyruvate carboxykinase, which is a regulatory enzyme of the liver’s gluconeogenic pathway.21,171,172 Liver dysfunction may also contribute to increased circulating endotoxin levels because of the important bacterial-clearance function of this organ.30,225 Unfortunately, many heatstroke patients require liver transplantation. Use of antipyretic drugs, such as acetaminophen (Tylenol), has been associated with hepatic failure.93,113,114,269,319
Many patients are released from the hospital following several days or weeks of treatment and continue to experience organ dysfunction during the ensuing years of recovery. Following the 2003 heat wave in France, mortality rates increased from 58% at day 28 of hospitalization (mean hospital stay was 24 days) to 71% mortality by the second year of recovery.6 An epidemiologic study of military EHS patients showed an approximately twofold increased risk for death from cardiovascular, kidney, and liver disease within 30 years of hospitalization.321 Several of the clinical responses (hyperthermia, dehydration, kidney and liver damage) occurring during progression or shortly after heatstroke collapse are clinically recognized and treated. However, those occurring during the months and years following hospitalization are underreported. The mechanisms responsible for long-term decrements in organ function remain poorly understood.
Cytokines
Cytokines are a class of intercellular protein messengers released from macrophages, T and B cells, endothelial cells, astrocytes, and other cell types that mediate inflammatory reactions to disease and injury.46,141,195,280,309 Cytokine-inducing stimuli include bacterial and viral infection,65,243 psychological stress,191,234 heat stress,* whole-body hyperthermia,222 and exercise.38,209,224,298 The defining characteristics of cytokines include a lack of constitutive production, the ability to regulate each other’s production, and overlapping actions that depend on the target cell type and cytokine milieu in which they function. Cytokines act over short distances and time spans (half-life is generally less than 60 minutes) and are usually present at low concentrations in the circulation. Cytokines bind reversibly to high-affinity cell-surface receptors and stimulate intracellular signaling pathways (e.g., NF-κB) that alter the transcription of genes involved in immune responses.
There are several lines of evidence that link cytokines with symptoms of heat-induced SIRS. These include induction of heatstroke symptoms by cytokine injection in experimental animal models, association of increased circulating cytokine levels with heatstroke morbidity/mortality, and effectiveness of cytokine neutralization in altering heatstroke mortality in animal models. Peripheral injection of IL-1β, IL-2, IL-6, IL-10, TNF-α, and platelet-activating factor into experimental animals replicates the pathophysiologic responses observed in heatstroke, including hyperthermia, hypothermia, fever, increased vascular permeability, DIC, and death.* Simultaneous injection of multiple cytokines (e.g., IL-1 and TNF) is most effective in mimicking heatstroke symptoms and has shed light on cytokine interactions in vivo that orchestrate SIRS. Increased circulating levels of IL-1α, IL-1β, IL-1 receptor antagonist (IL-1ra, a naturally occurring antagonist of IL-1), IL-6, soluble IL-6 receptor (sIL-6R), IL-8, IL-10, IL-12, IFN-γ, TNF-α, and soluble TNF receptor (sTNFR) concentrations are commonly observed at the time of heatstroke collapse or shortly after cooling.† Sustained high IL-6 levels during cooling correlate with heatstroke severity, tissue injury, and death, whereas high circulating IL-8 levels are implicated in leukocyte activation and coagulation in EHS patients.23,125 The reciprocal regulation of IL-12 and IL-10 production suggests complex interactions in heat-induced SIRS, but the function of these cytokines has not been clearly delineated. As previously mentioned, high IFN-inducible gene expression and IFN-γ levels are clinical measures of viral or intracellular bacterial infection and are evident in EHS patients with preexisting infections.289
The failure of clinical and animal studies to correlate cytokine production with specific heatstroke responses is probably caused by the short half-lives of these proteins, local tissue concentrations exceeding those in the circulation, or the presence of soluble cytokine receptors that mask detection or alter cytokine action(s).‡ For example, the sTNFR inhibits the actions of TNF and is often higher in heatstroke survivors than nonsurvivors, suggesting that TNF may mediate lethality.109 On the other hand, the sIL-6R may potentiate endogenous IL-6 effects by increasing the concentration of available IL-6 signaling receptors on cell membranes (known as a trans-signaling effect) or reduce IL-6 signaling through competitive binding with IL-6 receptors that are already present on the cell membrane (Figure 10-9).
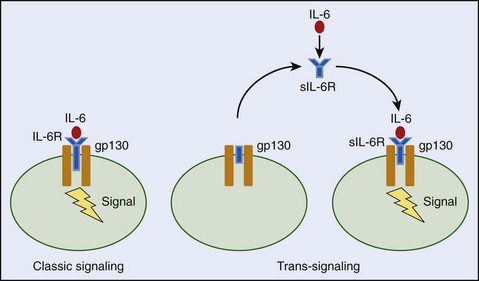
Although cytokines are known to interact with one another, their soluble receptors, and other endogenous stress hormones (e.g., glucocorticoids) during SIRS, it remains unknown how these interactions in vivo affect heatstroke outcome. Taken together, results from the few antagonism/neutralization studies that have been conducted to date indicate that high levels of cytokines may be detrimental for heatstroke recovery, but baseline (permissive) actions of some cytokines (e.g., IL-6, TNF, or proteins affected by their actions) appear to be essential for survival.170 Clearly, more research is required in this area to determine the multitude of cytokine actions in heat-induced SIRS and determine the protective versus detrimental effects of the proteins on multiorgan system function.
Heat Shock Proteins
Heat shock proteins (HSPs) are molecular chaperones that prevent misfolding and aggregation of cellular proteins during exposure to stressful stimuli.76,111,134,184 HSPs are found in organisms ranging from bacteria to humans. Their chaperoning activities protect against environmental (heavy metals, heat stress), physiologic (cell differentiation, protein translation), and pathologic (infections, ischemia/reperfusion) stimuli that cause cellular damage.134,160,184 HSPs were originally discovered in Drosophila melanogaster, when puffs associated with novel protein synthesis appeared on the giant chromosomes of the salivary glands in response to heat stress.259,303 It was later discovered that heat denaturation of mature proteins inside the cell was the cellular signal that increased protein synthesis in response to heat stress in Drosophila.120
HSPs are grouped into families according to their molecular mass, cellular localization, and function (Table 10-4).
| Family | Function | Attributes |
|---|---|---|
| HSP 27 (sHSP) | Antiapoptotic | Constitutively expressed |
| Cytoskeletal stabilization | Cytosolic and nuclear | |
| HSP 60 | Protein refolding | Mitochondria and cytosol |
| Prevents aggregation of denatured proteins | ||
| Immune responses | ||
| HSP 70 family | ||
| HSP 72 | Thermotolerance | Highly inducible |
| HSP 73 (HSC 70) | Molecular chaperone | Constitutively expressed |
| HSP 75 | Molecular chaperone | Mitochondrial |
| HSP 78 (GRP 79, Bip) | Cytoprotection | Endoplasmic reticulum |
| HSP 90 family: HSP 90 | ||
| GRP 96 | Glucocorticoid receptor functioning | Cytosolic and nuclear |
| Glucose regulation | Endoplasmic reticulum | |
| HSP 110/104 | Molecular chaperone | Cytosolic |
| Tumor antigen presentation |
Bip, Binding protein; GRP, glucose regulated protein-78; HSC, heat shock cognate; HSP, heat shock protein.
Data from Hartl FU, Hayer-Hartl M: Molecular chaperones in the cytosol: From nascent chain to folded protein, Science 295:1852, 2002; and Kregel KC: Heat shock proteins: Modifying factors in physiological stress responses and acquired thermotolerance, J Appl Physiol 92:2177, 2002.
HSP 27 (also referred to as sHSP) is a constitutively expressed cytosolic and nuclear protein with cytoskeletal stabilization and antiapoptotic functions.7,167,241 HSP 60 exists in the mitochondria and cytosol, is released from PBMCs on LPS stimulation, and functions as a “danger” signal for the innate immune system. HSPs interact with PAMPs, such as TLRs, to stimulate monocytes, macrophages, and dendritic cells to produce cytokines.35,226 The HSP 70 family has been extensively studied for protective function(s) against thermal stress,142,340 ischemia/reperfusion,196,248,293 tissue injury,33 glucose deprivation,334 and sepsis.166,317 HSP 70s function in concert with other molecular chaperones, such as HSP 90 and HSP 110, to facilitate LPS and antitumor responses.127
HSP gene expression is mediated primarily at the level of gene transcription by a family of heat shock transcription factors (HSFs) that interact with the heat shock regulatory element (HSE) in the promoter region of genes. HSF-1 is the major stress responsive element in mammalian cells that is activated by febrile-range temperatures.318,338 HSF-1 interacts with HSEs on cytokine genes to alter transcription and confer protection against endotoxin and other infectious/inflammatory stimuli. In gene-transfected human PBMCs, inhibition of TNF-α, IL-1β, IL-10, and IL-12 in response to LPS was specific to HSPs 70 overexpression.59 A lack of effect of HSP 70 on IL-6 gene transcription may be an indirect mechanism of protection, because IL-6 functions in a regulatory feedback loop to inhibit IL-1 and TNF production, which are Th1 cytokines with potent proinflammatory activities.59,71,73,325 In murine macrophages, HSP 70 inhibited IL-12 (Th1) and stimulated IL-10 (Th2) production in response to LPS.325 The shift from Th1 to Th2 cytokine production may be a mechanism by which HSP 70 protects against bacterial infection.
Heat strain is a consequence of the time and intensity of heat exposure. These factors interact in vivo to influence the magnitude and kinetics of HSP expression. In human PBMCs, maximal expression of intracellular HSPs 70 was observed between 4 and 6 hours after a brief heat shock (43° C [109.4° F] for 20 minutes).288 Increased expression of HSPs 10, 20, 40, 60, 70, 90, and 110 was observed in PBMCs from EHS patients or following exposure to hypoxia in vitro.287,289 Anatomic differences in the magnitude and kinetics of in vivo expression have also been observed, with HSP 70 expression occurring within 1 hour in the brain, lungs, and skin and delayed until 6 hours after heat exposure in the liver of rats.18 In mice, liver expression of HSP 70 showed a progressive increase from approximately 6 to 24 hours following collapse from passive heatstroke.171 In rats, a high rate of passive heating (0.175° C [0.315° F]/min) induced greater HSP 70 expression in the liver, small intestine, and kidneys than did a lower rate of heating (0.05° C [0.09° F]/min), despite attaining the same maximum body temperature (42° C [107.6° F]).80 Differences in tissue blood flow and metabolic activity likely account for regional differences in HSP expression during passive and exertional heat exposure.
Thermotolerance is the term used to describe the noninheritable, transient resistance to a lethal heat stress that is acquired following previous exposure to a nonlethal level of heat stress. Increased HSP 70 expression is a mechanism of thermotolerance that protects against heat-induced increases in epithelial permeability. A unique in vitro model system consisting of high-resistance Madin-Darby canine kidney (MDCK) epithelial cell monolayers was developed to examine the relationship between HSP 70 expression and changes in epithelial integrity with heat exposure. Following heat stress to 38.3° C (100.9° F), MDCK monolayers showed an increase in permeability that was reversible with cooling.212 If the monolayers were preexposed to a conditioning heat stress of 42° C (107.6° F) for 90 minutes, subsequent exposure to a higher temperature of 39.4° C (102.9° F) was required to increase monolayer permeability.21 The association of a thermotolerant state with increased HSP 70 expression suggests that HSPs shift the temperature threshold upward to prevent heat-induced disruptions in epithelial permeability.212 Follow-up studies showed that HSPs interact with proteins in the tight junctions of the epithelium to regulate permeability. Occludin is a plasma-membrane protein located at tight junctions that was increased, along with HSPs 27, 40, 70, and 90, in intestinal epithelial monolayer (Caco-2) cells exposed to 39° or 41° C (102.2° or 105.8° F).60 Treatment of Caco-2 cells with quercetin (an inhibitor of HSF-1) inhibited HSPs and occludin expression and reversed the thermotolerant state of these cells.60 These studies demonstrate a complex interaction between HSPs and tight-junction proteins for modulation of epithelial barrier function during thermal stress.
It is interesting to speculate that differences in HSP expression profiles may be a sensitive marker of heat stress susceptibility among different populations. During the life of an organism, there is an accumulation of protein damage caused by continual oxidant and free radical activity within cells. The lifespan of Drosophila was extended by heat shock treatment or the addition of HSP 70 gene copies, suggesting that increased protein chaperonin activity may protect against aging.148,301 In rats, aging was associated with a significant reduction in liver HSP expression following passive heat exposure, which was associated with greater liver damage compared with that observed in young rats.161,342 Older animals do not appear to have a global inability to express HSP, because exertional heat stress can induce expression profiles similar to those of mature rats.161 Rather, aging is associated with a reduction in the threshold for HSP stimulation.161 Similarly, Fargnoli and co-workers74 showed that global reduction in protein synthesis was not responsible for decreased HSP induction in aged lung fibroblasts. Alzheimer’s disease is thought to be a consequence of decreased HSP function that results in increased deposition of abnormally folded proteins.211 It is anticipated that screening for altered HSP titers will help to identify individuals with reduced thermotolerance caused by aging, infection, or other conditions that may predispose to heatstroke.150
Heatstroke Treatments (See Chapter 11)
Current heatstroke therapies fall into two categories: supportive therapies directed at the immediate clinical symptoms and therapies directed at the causative mechanisms of injury. The primary objectives of clinical heatstroke treatments are to reduce body temperature as rapidly as possible, reestablish normal CNS function, and stabilize peripheral multiorgan system function. Supportive therapies consist of rapid cooling and IV fluid administration for restoration of normal blood pressure and tissue perfusion, whereas advanced therapies are directed at the coagulation and inflammatory disturbances that cause organ failure. Despite these efforts, heatstroke morbidity and mortality rates remain quite high, and multiorgan system dysfunction continues to claim the lives of heatstroke victims during ensuing years of recovery.6,321 This section discusses conventional clinical treatments of heatstroke, as well as innovative treatment strategies targeted at SIRS to mitigate injury and death.
Cooling
Rapid cooling is considered the single most important treatment for protection against permanent CNS damage and death from heatstroke. To facilitate cooling, the individual should be placed into a supine position and as many clothes as possible removed to expose a large surface area of the body to facilitate heat transfer. If the individual is comatose, he or she should be placed onto his or her side to ensure an open airway. Conventional methods of cooling include cold or ice water immersion, packing ice around the body, sponging with (or without) fanning, or use of a hypothermia blanket. The goal of all cooling methods is to rapidly decrease and maintain body temperature below 39° C (102.2° F) and to prevent rebound hyperthermia. The use of an ice bath or ice packs on the skin surface has met with some resistance because it is thought that cooling of the skin will elicit peripheral vasoconstriction and shivering, which can increase heat production and counteract the cooling effect. This was shown in an experimental study in which young, fit test subjects were heat stressed to 40° C (104° F) and experienced shivering and cold sensations during immersion in cold water.339 Because the threshold for activation of shivering is increased in heatstroke patients, the risk for cold or ice water immersion eliciting such a response and compromising the benefits of cooling is unlikely. However, to minimize the possibility of peripheral vasoconstriction with cooling, the skin may be massaged to stimulate increases in cutaneous blood flow. Regardless of the treatment strategy, current medical doctrine dictates that heatstroke patients are cooled as rapidly as possible until normal CNS function is reestablished.
Dantrolene and nonsteroidal antiinflammatory drugs (NSAIDs) have been tested for their effects on body cooling, but neither class of drugs has shown efficacy for protection against heatstroke. Dantrolene is a calcium-lowering agent that protects against hyperthermia by lowering intracellular calcium concentrations in skeletal muscle to decrease muscle tone. Dantrolene is effective for the treatment of malignant hyperthermia (MH), which is a genetic mutation that predisposes to involuntary muscle contractions and rigidity following exposure to general anesthetics or muscle-depolarizing agents. Dantrolene has been considered as a treatment for heatstroke, but animal and human heatstroke studies have failed to validate its use for this condition. In a study of heatstroke patients, dantrolene decreased the required cooling time by approximately 20 minutes but had no effect on recovery. Similarly, a randomized, double-blind, placebo-controlled trial of Hajj heatstroke patients failed to show a cooling advantage of dantrolene over traditional cooling methods.22,43 MH is distinct from exertional or passive heatstroke, so it is not surprising that this drug does not protect equally against these diverse conditions. (MH is discussed in detail later in this chapter.)
NSAIDs have been considered as therapeutics based on their potent antiinflammatory and antipyretic effects. The action(s) of classic NSAIDs, such as, aspirin, ibuprofen, and acetaminophen, are attributed primarily to blockade of the cyclooxygenase (COX) pathway of eicosanoid metabolism (Figure 10-10).
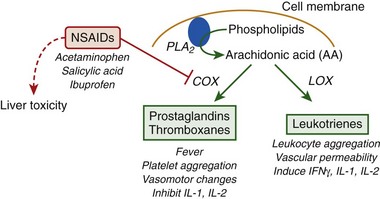
Prostaglandins are synthesized by the COX pathway in response to a variety of stimuli (e.g., bacterial infection, heat shock) and regulate a multitude of physiologic responses, including fever, inflammation, and cytokine production. During fever, prostaglandins are released in response to proinflammatory cytokines (e.g., IL-1, IL-6) and stimulate an increase in the temperature set point to induce fever.292 Inhibition of prostaglandin production by NSAIDs is the primary mechanism for the antipyretic (i.e., fever-reducing) actions of these drugs. However, hyperthermia in response to heat exposure is not caused by an increase in the temperature set point but by an unregulated increase in body temperature that occurs when heat gain exceeds heat loss in an absence of a change in the temperature set point. Rather than providing a beneficial effect, NSAIDs are contraindicated as a treatment for heatstroke because of the potential toxic effects of these drugs on liver function and a lack of clinical and/or experimental data to support their use. For example, pretreatment with sodium salicylate was without effect on skin temperature and pulse rate but significantly increased the rate of body temperature rise and potentiated hyperthermia in men walking in a hot environment.135 As previously mentioned, the use of antipyretic drugs, such as acetaminophen, has been anecdotally associated with the need for liver transplantation.93,113,114,269,319 Recurrent hyperthermia is a common heatstroke recovery response that is thought to be a true fever and a prostaglandin-mediated response to endotoxin leakage and cytokine stimulation. Whether the effects of NSAIDs on cytokine expression or body temperature will protect or exacerbate heatstroke sequelae in vivo is unknown but is an important area of investigation, given the high use of NSAIDs as over-the-counter medications for pain and/or fever relief in our society.
Fluid Resuscitation (See Chapter 70)
One of the first lines of defense against permanent tissue damage is treatment with resuscitation fluids. The objective of IV fluid administration is to restore intravascular volume and rehydrate the interstitium to stabilize cardiovascular functioning, improve tissue perfusion, and maintain immune function. The resuscitation fluid needs to be safe, efficacious, inexpensive, easy to transport for use in military or athletic settings, and have the capability to restore tissue oxygen perfusion and minimize cellular/tissue injury. Blood provides oxygen-carrying capacity and volume, but supply is limited and there is a risk for allergic or infectious reactions, difficulties with crossmatching, and potential for high hemoglobin levels to increase blood viscosity and reduce nutrient flow to the tissues.255,258 Balanced salt solutions, such as saline and lactated Ringer’s, have a long shelf life and are inexpensive and in unlimited supply with a minimal risk for disease transmission. However, they are able to freely cross semipermeable capillary membranes, which increases the risk for tissue edema and makes frequent transfusions necessary to maintain adequate plasma volume.110,145,304 Tissue edema increases the distance from blood vessels to tissue mitochondria and limits oxygen delivery to the tissues. There is a greater risk for edema in heatstroke patients because of increases in capillary permeability and lack of muscle movement that limits the flow of lymph following collapse.
To minimize the adverse consequences of balanced salt solutions, these fluids may be replaced with colloid solutions. Natural colloids, such as albumin, possess antioxidant properties that reduce tissue injury during times of oxidant stress but carry a risk for infection.84,328 Dextran is an artificial colloid that was in use after World War II until adverse hemostatic effects restricted its use to specific clinical conditions, such as deep vein thrombosis and pulmonary embolism.15,55 Hydroxyethyl starch (HES) is a blood plasma substitute that exerts high colloidal pressure to stimulate movement of fluid from the interstitial space into the blood vessel lumen for plasma volume expansion.239,336,344 Small-volume treatment with HES protected against heatstroke mortality in rats, but use of HES in other heatstroke animal models and humans has not been validated.187 Because of severe dehydration and acute renal failure with heatstroke, fluid shifts from the interstitial fluid into the vessel lumen may mean HES will not be well tolerated in severe heatstroke patients.
Anticoagulants
Anticoagulants (e.g., heparin, aspirin) have been examined for heatstroke protection, with mixed results. Heparin therapy has been associated with positive heatstroke outcome in patients, although it is difficult to dissociate the direct effects of this therapy from other clinical treatments.245,290 The mechanisms of heat-induced DIC may include prostaglandin synthesis, because aspirin has shown protection against platelet hyperaggregation in vitro and in animal models. In human volunteers, ingestion of aspirin 12 to15 hours before blood sampling or heat exposure of cells was effective in inhibiting platelet hyperaggregation.87 However, aspirin was ineffective if provided after heat exposure, even though complete inhibition of the arachidonic acid pathway was achieved.87 The ability of aspirin to protect guinea pigs from DIC induced by Staphylococcus aureus suggests that similar activities function in vivo to control platelet reactivity.227 However, there is currently insufficient evidence to support the use of aspirin as a preventive measure in heatstroke patients. Given the hormonal and metabolic alterations that accompany heatstroke, including dehydration, increased catecholamine levels, hypoxia, and others, the mechanisms responsible for DIC extend beyond those mediated by prostaglandins alone. Furthermore, aspirin and other antiinflammatory drugs can cause liver damage if consumed in large quantities, as previously mentioned.
Recombinant APC is an effective antiinflammatory drug for the treatment of sepsis and may also hold promise as a treatment for heatstroke patients. APC efficacy appears to depend on a variety of patient conditions, including age (most effective in patients over 50 years of age), extent of organ dysfunction (benefit not apparent if failure of only one organ), and the presence of shock at the time of infusion, which improves its efficacy.327 In rat heatstroke models, the efficacy of APC depends on the time of treatment. A single dose of recombinant human APC provided at the onset of heatstroke inhibited inflammation and coagulopathy, prevented organ failure, and improved survival; however, if treatment was delayed for 40 minutes following the onset of heatstroke, there was no beneficial effect on survival time.45 The efficacy of APC was less obvious in a baboon heatstroke model. Infusion for 12 hours following heatstroke onset attenuated plasma IL-6, thrombomodulin, and procoagulant components but had no effect on mortality.25 APC is the first biologic agent approved in the United States for the treatment of severe sepsis on account of two decades of research in this area,327 but there is insufficient evidence to justify the use of this treatment in heatstroke patients.
Anticytokine Therapies
As previously described, attenuations in splanchnic blood flow during heatstroke contribute to increased gut permeability and a rise in circulating endotoxin. This series of events is hypothesized to stimulate elevations in plasma cytokine levels that have been implicated in the adverse consequences of SIRS. Based on these findings, the question arises: Do anticytokine therapies represent an efficacious treatment strategy for heatstroke? There have been no controlled studies examining the efficacy of anticytokine therapies on patient outcome with heatstroke. However, clinical sepsis trials indicate that potential protective effects of anticytokine therapies need to be viewed with cautious optimism. Sepsis patients display high circulating IL-1 levels that correlate with morbidity and mortality, but IL-1ra treatment has been unsuccessful in reducing mortality.236,253 A comparison of 12 randomized, double-blind multicenter trials of more than 6200 sepsis patients showed no significant benefit of antiendotoxin antibodies, ibuprofen, plasminogen-activating factor receptor antagonist, anti-TNF monoclonal antibody, or IL-1ra on all-cause mortality.57 There are several explanations for negative results from anticytokine therapies, including the possibility that the mediator has no pathophysiologic role in the response, the agent failed to neutralize the protein (because of a lack of biologic activity, competition by other mediators, inadequate anatomic distribution), compensatory increase of other mediator(s) with similar activities, administration too early or late in the course of disease, too-short therapy duration, or the need for combination therapy.198 Kinetic studies have shown the half-life of IL-1ra to be approximately 20 minutes, and phase III clinical trials showed that circulating IL-1β levels at the time of clinical treatment were undetectable in 95% of the patient population.77,246 Exposure to anticytokine therapies before sepsis onset is thought to be desirable (but practically infeasible), although this may suppress shifts in Th1/Th2 immune responses that are important for the resolution of infection. On the other hand, anticytokine therapies given too late in sepsis progression may shift the Th1/Th2 milieu in an unpredictable manner or have no effect because of the overwhelming nature of the septic event.100 The transient nature of cytokine production and/or clearance and lack of correlation between serum levels and disease severity further complicate treatment scenarios. Given the short half-life of TNF-α (approximately 6 to 7 minutes)16 and biphasic clearance patterns of IL-1β and IL-6 (rapid disappearance in the first 3 minutes followed by an attenuated clearance over the next 1 to 4 hours),152 the narrow protective window in which anticytokine therapies may be effective is a difficult obstacle to overcome.
Interleukin-10 is a potent antiinflammatory cytokine that suppresses production of several proinflammatory cytokines, including IL-1β, IL-6, and TNF-α, and is an important negative feedback loop during infection/disease.101,311 Few, if any, studies have investigated the efficacy of IL-10 treatment for heatstroke recovery, although the cytokine has been commonly used in the treatment of multiple autoimmune diseases, including rheumatoid arthritis, Crohn’s disease, multiple sclerosis, and sepsis.31,229 As with other cytokines, the timing of IL-10 administration must be carefully considered. IL-10 decreases IFN-γ production by natural killer and Th1 cells, which may suppress the clearance of infectious organisms (via IFN-γ).254,320 Consistent with anticytokine therapies, beneficial effects of exogenous IL-10 administration depend on multiple factors, including time of administration, route, and the site to which the cytokine is targeted.229,265 These considerations, along with the immune suppressive and unpredictable nature of IL-10–based therapies, have resulted in diminished enthusiasm for anticytokine therapies for sepsis.
Heatstroke Prevention
Heatstroke is currently a more preventable than treatable disease. The most effective preventive measures include acclimatization to the heat, reductions in the duration and extent of physical activity, rescheduling of activities to cooler times of the day, increased consumption of nonalcoholic fluids, and removing vulnerable populations, such as those with preexisting viral or bacterial infections, from the heat stress environment. Fan cooling has not shown protection against heatstroke and is associated with increased thermal discomfort at temperatures higher than 38° C (100.4° F).149
Heat Acclimatization
Climatic heat stress and exercise interact synergistically to increase body temperature (core and skin) and cardiovascular strain and to decrease performance in the heat. Heat acclimatization is the within-lifetime changes in an organism (as opposed to evolutionary changes) that protect against these negative effects of heat strain. Heat acclimatization occurs following exposure to the natural environment, whereas heat acclimation develops following exposure to artificial conditions. However, these terms are used interchangeably because they induce similar physiologic adaptations.331 Heat acclimation occurs following repeated bouts of heat exposure that are of sufficient intensity, frequency, duration, and number to elevate core and skin temperature and induce profuse sweating. Heat acclimation may be achieved following exercise or rest in the heat, although the former method is more effective. It is important to note that heat acclimatization is specific to the climate and activity level; therefore, if individuals will be working in a hot, humid climate, heat acclimatization should be conducted under similar conditions (Box 10-3).
BOX 10-3 Heat Acclimation Strategies
Must Mimic Climate of Athletic Event or Occupational Setting and Include Adequate Heat Stress
Although heat acclimation does not require daily exposure to heat and exercise, the rapidity with which biological adaptations are achieved is slower with less frequent exposures. This was shown experimentally in human volunteers in whom heat acclimation was achieved following 10 days of daily heat exposure but required 27 days when the frequency of exposure was reduced to every third day of experimentation (conditions 47° C [116.6° F], 17% relative humidity).74 Continual 24-hour exposures are also not required, because daily 100-minute periods of exposure were adequate to produce heat acclimation in subjects exposed to dry heat.183 However, because of the transient nature of the biologic adaptations, continued heat exposures are required to maintain the acclimated state. Aerobically trained athletes retain heat acclimation benefits longer than do unfit individuals, because they are exposed to high body temperatures during training exercises.240
Improvements in thermal comfort and exercise performance are achieved in heat-acclimated individuals through a variety of physiologic mechanisms, including a lower threshold and higher rate of skin blood flow, reduction in metabolic rate, earlier onset and rate of sweating, and improvements in cardiovascular function and fluid balance.271 Figure 10-11, online illustrates the effect of 10 days of heat acclimation on heart rate, core temperature, and mean skin temperature responses of subjects walking on a treadmill in a desert type of environment.70
On the 10th day of acclimation, heart rate was lower by approximately 40 beats/min, and rectal and skin temperatures were reduced approximately 1° C (1.8° F) and 1.5° C (2.7° F), respectively (see Figure 10-11, online).69 Once heat acclimation is achieved, skin vasodilation and sweating are initiated at a lower core temperature threshold, and higher sweat rates can be sustained without the sweat glands becoming “fatigued.”52,81 Whereas an unacclimated individual will secrete sweat with a sodium concentration of approximately 60 mEq/L (or higher), the concentration of secreted sodium from the sweat glands of an acclimated individual is significantly lower at approximately 5 mEq/L.262 This effect of heat acclimation on salt conservation is thought to be caused by increased aldosterone secretion or responsiveness of the sweat glands to this steroid hormone, which is released by the adrenal cortex and increases the resorption of ions and water in the distal tubules and collecting ducts of the kidneys.82,151 Overall improvements in fluid balance with heat acclimation include reduced sweat sodium losses, a better matching of thirst to body water needs, and increased total body water and blood volume.190,270 Provided that fluids are not restricted during physical activities, heat-acclimated individuals will be better able to maintain hydration during exercise and showed a marked reduction in “voluntary” dehydration.13,69,271 Although there is controversy regarding the ability of heat acclimation to alter the maximum temperature that can be tolerated during exercise in the heat,223 individuals who live or train in hot environments may experience reduced incidence of syncope (Figure 10-12).13,96,247
An essential cellular adaptation of heat acclimation is altered expression or reprogramming of genes that encode constitutive and stress-inducible proteins.122 Heat acclimation is associated with down regulation of genes associated with energy metabolism, food intake, mitochondrial energy metabolism, and cellular maintenance processes and upregulation of genes that are linked with immune responsiveness.122 The HSPs are the most extensively studied heat-inducible proteins and show faster transcriptional response and elevated cellular reserves (HSP 72 specifically) in heat-acclimated versus nonacclimated individuals.122,123,193,200 That is, whereas nonacclimated individuals require de novo HSP 72 synthesis for cellular protection, individuals who reside in hot climates maintain elevated HSP 72 levels.190 The cellular mechanisms of heat acclimation are not fully understood but are thought to involve global and tissue-specific changes in genes involved in thermal responsiveness, DNA repair and synthesis, free radical scavenging, and apoptosis.122
Genetic Polymorphisms
Single Nucleotide Polymorphisms
Single nucleotide polymorphisms (SNPs) are variations in the nucleotide sequence of DNA that can affect physiologic responses to environmental stimuli. SNPs have been implicated in a variety of diseases, including sepsis, type 1 diabetes, arthritis, inflammatory bowel disease, and rheumatic fever.79,136,250,296 The identification of SNPs in the promoter region of genes suggests that disease susceptibility may be affected by altered transcription of immune determinants of clinical outcome. SNPs have been identified in IL-1, IL-2, TNF, IFNγ, IL-10, IL-1ra, TLR-2, and TLR-4. The risk for death from sepsis is significantly increased in patients with a genetic polymorphism in the TNF-α or TNF-β gene.83 Even for those cytokine polymorphisms located distal to a critical promoter region that do not directly affect gene transcription rates, coinheritance of multiple immune-responsive genes by a process known as linkage disequilibrium can alter immune function. Some TNF-α polymorphisms exist in linkage disequilibrium with HLA haplotypes that encode cell-surface antigens. Coinheritance of these genes may ultimately be responsible for poor sepsis outcome.138,296
Malignant Hyperthermia
MH is a genetic disorder that causes muscle rigidity, hyperthermia, tachycardia, and metabolic acidosis during exposure to volatile anesthetics or depolarizing skeletal muscle relaxants. Exercise, heat stress, and emotional stress also trigger reactions in approximately 5% to 10% of MH patients.58,326 MH reactions are a result of a massive release of calcium from the type 1 ryanodine receptor (RyR1) of the sarcoplasmic reticulum, which overwhelms cellular mechanisms of calcium homeostasis and activates actin–myosin filaments to cause muscle rigidity and hyperthermia.238 RyR1 is the most common mutation in skeletal muscle, but additional isoforms have been identified in B and T cells, thalamus, hippocampus, and heart.163,202,300 Activation of the RyR1 by a variety of pharmacologic compounds, including caffeine, halothane, and the muscle relaxant 4-chloro-m-cresol, has led to the development of an in vitro contracture test of skeletal muscle biopsies to identify MH individuals.266,345 Dantrolene is used to treat MH reactions by lowering intracellular calcium stores, decreasing muscle metabolic activity, and preventing hyperthermia. The use of dantrolene, in combination with improved monitoring standards and early detection using the in vitro contracture test, has helped to reduce mortality rates from greater than 70% to less than 5% in MH patients.186
MH has been identified in several animal species, including dogs,231 boars,278 cats,14 and horses,3 but the most common experimental animal is a porcine MH model that possesses a single mutation in the skeletal muscle RyR1 gene. These animals develop the MH syndrome in response to inhalational anesthetics, exercise, heat, and other stressors.314 Mild exercise exacerbates MH symptoms in response to anesthetics in MH pigs, suggesting that inflammatory mediators released by skeletal muscle may contribute to the MH syndrome.314 MH patients show an approximately fivefold higher expression of IL-1β when stimulated with caffeine and 4-chloro-m-cresol compared with control cells.94 Recent development of a transgenic mouse model that overexpresses the RyR1 receptor has proved useful to study the MH/EHS link and could shed light on the role played in this syndrome by inflammatory cytokine production from skeletal muscle or other organs.69 Association of RyR1 mutations with EHS incidence suggests that screening young, healthy individuals (e.g., athletes, military personnel) for the MH mutation could be a powerful tool to determine heatstroke susceptibility.
1 Adams KFJr, Uddin N, Patterson JH. Clinical predictors of in-hospital mortality in acutely decompensated heart failure—piecing together the outcome puzzle. Congest Heart Fail. 2008;14:127.
2 Aderka D, Engelmann H, Maor Y, et al. Stabilization of the bioactivity of tumor necrosis factor by its soluble receptors. J Exp Med. 1992;175:323.
3 Aleman M, Riehl J, Aldridge BM, et al. Association of a mutation in the ryanodine receptor 1 gene with equine malignant hyperthermia. Muscle Nerve. 2004;30:356.
4 American College of Chest Physicians (ACCP)/Society of Critical Care Medicine Consensus Conference. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Crit Care Med. 1992;20:864.
5 Arbour NC, Lorenz E, Schutte BC, et al. TLR4 mutations are associated with endotoxin hyporesponsiveness in humans. Nat Genet. 2000;25:187.
6 Argaud L, Ferry T, Le QH, et al. Short- and long-term outcomes of heatstroke following the 2003 heat wave in Lyon, France. Arch Intern Med. 2007;167:2177.
7 Arrigo AP, Suhan JP, Welch WJ. Dynamic changes in the structure and intracellular locale of the mammalian low-molecular-weight heat shock protein. Mol Cell Biol. 1988;8:5059.
8 Asakura H, Suga Y, Yoshida T, et al. Pathophysiology of disseminated intravascular coagulation (DIC) progresses at a different rate in tissue factor-induced and lipopolysaccharide-induced DIC models in rats. Blood Coagul Fibrinolysis. 2003;14:221.
9 Attia M, Khogali M, El-Khatib G, et al. Heat-stroke: An upward shift of temperature regulation set point at an elevated body temperature. Int Arch Occup Environ Health. 1983;53:9.
10 Austin MG, Berry JW. Observations on one hundred cases of heatstroke. JAMA. 1956;161:1525.
11 Bagley WH, Yang H, Shah KH. Rhabdomyolysis. Intern Emerg Med. 2007;2:210.
12 Barbui T, Falanga A. Disseminated intravascular coagulation in acute leukemia. Semin Thromb Hemost. 2001;27:593.
13 Bean WB, Eichna LW. Performance in relation to environmental temperature: Reactions of normal young men to simulated desert environment. Fed Proc. 1943;22:144.
14 Bellah JR, Robertson SA, Buergelt CD, et al. Suspected malignant hyperthermia after halothane anesthesia in a cat. Vet Surg. 1989;18:483.
15 Bergqvist D. Dextran and haemostasis. Acta Chir Scand. 1982;148:633.
16 Beutler BA, Milsark IW, Cerami A. Cachectin/tumor necrosis factor: Production, distribution, and metabolic fate in vivo. J Immunol. 1985;135:3972.
17 Bianchi ME. DAMPs, PAMPs and alarmins: All we need to know about danger. J Leukoc Biol. 2007;81:1.
18 Blake MJ, Gershon D, Fargnoli J, et al. Discordant expression of heat shock protein mRNAs in tissues of heat-stressed rats. J Biol Chem. 1990;265:15275.
19 Bligh J. Fever. In: Neuberger A, Tatum EL, editors. Temperature regulation in mammals and other vertebrates. Amsterdam: North-Holland Publishing, 1973.
20 Bouchama A, al-Sedairy S, Siddiqui S, et al. Elevated pyrogenic cytokines in heatstroke. Chest. 1993;104:1498.
21 Bouchama A, Bridey F, Hammami MM. Activation of coagulation and fibrinolysis in heatstroke. Thromb Haemost. 1996;76:909.
22 Bouchama A, Cafege A, Devol EB, et al. Ineffectiveness of dantrolene sodium in the treatment of heatstroke. Crit Care Med. 1991;19:176.
23 Bouchama A, Hammami MM, Al Shail E, et al. Differential effects of in vitro and in vivo hyperthermia on the production of interleukin-10. Intensive Care Med. 2000;26:1646.
24 Bouchama A, Knochel JP. Heat stroke. N Engl J Med. 2002;346:1978.
25 Bouchama A, Kunzelmann C, Dehbi M, et al. Recombinant activated protein C attenuates endothelial injury and inhibits procoagulant microparticles release in baboon heatstroke. Arterioscler Thromb Vasc Biol. 2008;28:1318.
26 Bouchama A, Parhar RS, el-Yazigi A, et al. Endotoxemia and release of tumor necrosis factor and interleukin 1 alpha in acute heatstroke. J Appl Physiol. 1991;70:2640.
27 Bouchama A, Roberts G, Al Mohanna F, et al. Inflammatory, hemostatic, and clinical changes in a baboon experimental model for heatstroke. J Appl Physiol. 2005;98:697.
28 Boulant JA. Hypothalamic neurons regulating body temperature. In: Fregly MJ, Blatteis CM, editors. Handbook of physiology, section 4: Environmental physiology. New York: Oxford University; 1996:105-125.
29 Bowers WD, Hubbard RW, Leav I. Alterations of rat liver subsequent to heat overload. Arch Pathol Med. 1978;102:154.
30 Bradfield JW. Control of spillover: The importance of Kupffer-cell function in clinical medicine. Lancet. 1974;2:883.
31 Brandtzaeg P, Osnes L, Ovstebø R, et al. Net inflammatory capacity of human septic shock plasma evaluated by a monocyte-based target cell assay: Identification of interleukin-10 as a major functional deactivator of human monocytes. J Exp Med. 1996;184:51.
32 Bronstein SM, Conner WE. Endotoxin-induced behavioural fever in the Madagascar cockroach, Gromphadorhina portentosa. J Insect Physiol. 1984;30:327.
33 Brown IR, Rush S, Ivy GO. Induction of a heat shock gene at the site of tissue injury in the rat brain. Neuron. 1989;2:1559.
34 Buchanan TA, Cane P, Eng CC, et al. Hypothermia is critical for survival during prolonged insulin-induced hypoglycemia in rats. Metabolism. 1991;40:330.
35 Bukau B, Horwich AL. The HSP70 and HSP60 chaperone machines. Cell. 1998;92:351.
36 Butkow N, Mitchell D, Laburn H, et al. Heat stroke and endotoxaemia in rabbits. In: Hales JRS, editor. Thermal physiology. New York: Raven Press; 1984:511-514.
37 Bynum G, Brown J, Dubose D. Increased survival in experimental dog heatstroke after reduction of gut flora. Aviat Space Environ Med. 1979;50:816.
38 Camus G, Poortmans J, Nys M, et al. Mild endotoxemia and the inflammatory response induced by a marathon race. Clin Sci (Lond). 1997;92:415.
39 Carter RIII, Cheuvront SN, Sawka MN. A case report of idiosyncratic hyperthermia and review of U.S. Army heat stroke hospitalizations. J Sport Rehabil. 2007;16:238.
40 Carter RIII, Cheuvront SN, Williams JO, et al. Epidemiology of hospitalizations and deaths from heat illness in soldiers from 1980 through 2002. Med Sci Sport Exerc. 2005;37:1328.
41 Centers for Disease Control and Prevention. Heat-related deaths: Chicago, Illinois, 1996-2001, and United States, 1979-1999. MMWR Morb Mortal Wkly Rep. 2003;52:610.
42 Chang DM. The role of cytokines in heat stroke. Immunol Invest. 1993;22:553.
43 Channa AB, Seraj MA, Saddique AA, et al. Is dantrolene effective in heat stroke patients? Crit Care Med. 1990;18:290.
44 Chao TC, Sinniah R, Pakiam JE. Acute heat stroke deaths. Pathology. 1981;13:145.
45 Chen CC, Chen ZC, Lin MT, et al. Activated protein C improves heatstroke outcomes through restoration of normal hypothalamic and thermoregulatory function. Am J Med Sci. 2009;338:382.
46 Chen CM, Hou CC, Cheng KC, et al. Activated protein C therapy in a rat heat stroke model. Crit Care Med. 2006;34:1960.
47 Chensue SW, Shmyr-Forsch C, Otterness IG, et al. The beta form is the dominant interleukin 1 released by murine peritoneal macrophages. Biochem Biophys Res Commun. 1989;160:404.
48 Cheuvront SN, Carter R, Sawka MN. Fluid balance and endurance exercise performance. Curr Sports Med Rep. 2003;2:202.
49 Chiu WT, Kao TY, Lin MT. Interleukin-1 receptor antagonist increases survival in rat heatstroke by reducing hypothalamic serotonin release. Neurosci Lett. 1995;202:33.
50 Clarke JF. Some effects of the urban structure on heat mortality. Environ Res. 1972;5:92.
51 Clarkson PM, Kearns AK, Rouzier P, et al. Serum creatine kinase levels and renal function measures in exertional muscle damage. Med Sci Sports Exerc. 2006;38:623.
52 Cole RD. Heat stroke during training with nuclear, biological, and chemical protective clothing: Case report. Mil Med. 1983;148:624.
53 Collins KJ, Crockford GW, Weiner JS. Sweat-gland training by drugs and thermal stress. Arch Environ Health. 1965;11:407.
54 Curriero FC, Heiner KS, Samet JM, et al. Temperature and mortality in 11 cities of the eastern United States. Am J Epidemiol. 2002;155:80.
55 D’Alecy LG, Kluger MJ. Avian febrile response. J Physiol (Lond). 1975;253:223.
56 Deitch EA. Animal models of sepsis and shock: A review and lessons learned. Shock. 1998;9:1.
57 de Jong E, Levi M. Effects of different plasma substitutes on blood coagulation: A comparative review. Crit Care Med. 2001;129:1261.
58 Dematte JE, O’Mara K, Suescher J, et al. Near-fatal heat stroke during the 1995 heat wave in Chicago. Ann Intern Med. 1998;129:173.
59 Denborough M. Malignant hyperthermia. Lancet. 1998;352:1131.
60 Ding XZ, Fernandez-Prada CM, Bhattacharjee AK, et al. Over-expression of HSP-70 inhibits bacterial lipopolysaccharide-induced production of cytokines in human monocyte-derived macrophages. Cytokine. 2001;16:210.
61 Dokladny K, Wharton W, Lobb R, et al. Induction of physiological thermotolerance in MDCK monolayers: Contribution of heat shock protein 70. Cell Stress Chaperones. 2006;11:268.
62 Donaldson G, Kovats RS, Keatinge WR, et al. Heat- and cold-related mortality and morbidity and climate change. In: Maynard RL, editor. Health effects of climate change in the UK. London: Department of Health; 2001:70-80.
63 Donaldson GC, Keatinge WR, Näyhä S. Changes in summer temperature and heat-related mortality since 1971 in North Carolina, South Finland, and Southeast England. Environ Res. 2003;91:1.
64 Dorozynski A. Chirac announces investigation into heat wave’s death toll. British Med J. 2003;327:465.
65 Downing JF, Taylor MW, Wei KM, et al. In vivo hyperthermia enhances plasma antiviral activity and stimulates peripheral lymphocytes for increased synthesis of interferon-gamma. J Interferon Res. 1987;7:185.
66 Drexler AM. Tumor necrosis factor: its role in HIV/AIDS. STEP Perspect. 1995;7:13.
67 Dubose DA, Basamania K, Maglione L, et al. Role of bacterial endotoxins of intestinal origin in rat heat stress mortality. J Appl Physiol. 1983;54:31.
68 DuBose DA, McCreary J, Sowders L, et al. Relationship between rat heat stress mortality and alterations in reticuloendothelial carbon clearance function. Aviat Space Environ Med. 1983;54:1090.
69 Durham WJ, Aracena-Parks P, Long C, et al. RyR1 S-nitrosylation underlies environmental heat stroke and sudden death in Y522S RyR1 knockin mice. Cell. 2008;133:53.
70 Eichna LW, Park CR, Nelson N, et al. Thermal regulation during acclimatization in a hot, dry (desert type) environment. Am J Physiol. 1950;163:585.
71 Elizondo RS. Regulation of body temperature. In: Rhoades RA, Tanner GA, editors. Medical physiology. Boston: Little, Brown; 1995:823-840.
72 Ensor JE, Crawford EK, Hasday JD. Warming macrophages to febrile range destabilizes tumor necrosis factor-α mRNA without inducing heat shock. Am J Physiol. 1995;269:C1140.
73 Ensor JE, Wiener SM, McCrea KA, et al. Differential effects of hyperthermia on macrophage interleukin-6 and tumor necrosis factor-α expression. Am J Physiol. 1994;266:C967.
74 Fargnoli J, Kunisada T, Fornace AJJr, et al. Decreased expression of heat shock protein 70 mRNA and protein after heat treatment in cells of aged rats. Proc Natl Acad Sci USA. 1990;87:846.
75 Fein JT, Haymes EM, Buskirk ER. Effects of daily and intermittent exposures on heat acclimation of women. Int J Biometeorol. 1975;19:41.
76 Feterowski C, Emmanuilidis K, Miethke T, et al. Effects of functional Toll-like receptor-4 mutations on the immune response to human and experimental sepsis. Immunology. 2003;109:426.
77 Fink AL. Chaperone-mediated protein folding. Physiol Rev. 1999;79:425.
78 Fischer E, Marano MA, Barber AE, et al. Comparison between effects of interleukin-1 alpha administration and sublethal endotoxemia in primates. Am J Physiol. 1991;261:R442.
79 Fisher CJJr, Yan SB. Protein C levels as a prognostic indicator of outcome in sepsis and related diseases. Crit Care Med. 2000;28:S49.
80 Fishman D, Faulds G, Jeffery R, et al. The effect of novel polymorphisms in the interleukin-6 (IL-6) gene on IL-6 transcription and plasma IL-6 levels, and an association with systemic-onset juvenile chronic arthritis. J Clin Invest. 1998;102:1369.
81 Flanagan SW, Ryan AJ, Gisolfi CV, et al. Tissue-specific HSP70 response in animals undergoing heat stress. Am J Physiol. 1995;268:R28.
82 Fox RH, Goldsmith R, Kidd DJ, et al. Acclimatization to heat in man by controlled elevation of body temperature. J Physiol (Lond). 1963;166:530.
83 Francesconi RP, Sawka MN, Pandolf KB. Hypohydration and heat acclimation: Plasma rennin and aldosterone during exercise. J Appl Physiol. 1983;55:1790.
84 Freeman BD, Buchman TG. Gene in a haystack: Tumor necrosis factor polymorphisms and outcome in sepsis. Crit Care Med. 2000;28:3090.
85 Frei B, Stocker R, Ames BN. Antioxidant defenses and lipid peroxidation in human blood plasma. Proc Natl Acad Sci USA. 1988;85:9748.
86 Frumkin H. Urban sprawl and public health. Public Health Rep. 2002;117:201.
87 Gabow PA, Kaehny WD, Kelleher SP. The spectrum of rhabdomyolysis. Medicine (Baltimore). 1982;61:141.
88 Gader AM, al-Mashhadani SA, al-Harthy SS, et al. Direct activation of platelets by heat is the possible trigger of the coagulopathy of heat stroke. Br J Haematol. 1990;74:86.
89 Gagge AP, Gonzalez RR. Mechanisms of heat exchange: Biophysics and physiology. In: Fregly MJ, Blatteis CM, editors. Handbook of physiology, section 4: Environmental physiology. New York: Oxford University; 1996:45-84.
90 Garibaldi JA. Influence of temperature on the biosynthesis of iron transport compounds by Salmonella typhimurium. J Bacteriol. 1972;110:262.
91 Gathiram P, Gaffin SL, Brock-Utne JG. Time course of endotoxemia and cardiovascular changes in heat-stressed primates. Aviat Space Environ Med. 1987;58:1071.
92 Gathiram P, Wells MT, Brock-Utne JG. Antilipopolysaccharide improves survival in primates subjected to heat stroke. Circ Shock. 1987;23:157.
93 Gemmell RT, Hales JR. Cutaneous arteriovenous anastomoses present in the tail but absent from the ear of the rat. J Anat. 1977;124:355.
94 Giercksky T, Boberg KM, Farstad IN, et al. Severe liver failure in exertional heat stroke. Scand J Gastroenterol. 1999;34:824.
95 Girard T, Cavagna D, Padovan E, et al. B-lymphocytes from malignant hyperthermia: Susceptible patients have an increased sensitivity to skeletal muscle ryanodine receptor activators. J Biol Chem. 2001;276:48077.
96 Gisolfi CV, Wenger CB. Temperature regulation during exercise: Old concepts new ideas. Exerc Sports Sci Rev. 1984;12:339.
97 Goddard CJ, Warnes TW. Raised liver enzymes in asymptomatic patients: Investigation and outcome. Dig Dis. 1992;10:218.
98 Gonzales-Alonso JC, Teller C, Anderson SL, et al. Influence of body temperature on the development of fatigue during prolonged exercise in the heat. J Appl Physiol. 1999;86:1032.
99 Graf R, Krishna S, Heller HC. Regulated nocturnal hypothermia induced in pigeons by food deprivation. Am J Physiol. 1989;256:R733.
100 Grahn D, Heller HC. The physiology of mammalian temperature homeostasis. TraumaCare. 2004;14:52.
101 Grant RT. Vasodilation and body warming in the rat. J Physiol. 1963;167:311.
102 Grau GE, Maennel DN. TNF inhibition and sepsis: Sounding a cautionary note. Nat Med. 1997;3:1193.
103 Greenberger MJ, Strieter RM, Kunkel SL, et al. Neutralization of IL-10 increases survival in a murine model of Klebsiella pneumoniae. J Immunol. 1995;155:722.
104 Haber MM, West AB, Haber AD, et al. Relationship of aminotransferases to liver histological status in chronic hepatitis C. Am J Gastroenterol. 1995;90:1250.
105 Hagberg L, Briles DE, Edén CS. Evidence for separate genetic defects in C3H/HeJ and C3HeB/FeJ mice that affect susceptibility to gram-negative infections. J Immunol. 1985;134:4118.
106 Hainsworth FR. Saliva spreading, activity and body temperature regulation in the rat. Am J Physiol. 1967;212:1288.
107 Hajat S, Kovats RS, Atkinson RW, et al. Impact of hot temperatures on death in London: A time series approach. J Epidemiol Community Health. 2002;56:367.
108 Hakre S, Gardner JW, Kark JA. Predictors of hospitalization in male Marine Corps recruits with exertional heat illness. Mil Med. 2004;169:169.
109 Hales JR, Brown GD. Net energetic and thermoregulatory efficiency during panting in the sheep. Comp Biochem Physiol. 1974;49:413.
110 Hall DM, Buettner GR, Oberley LW. Mechanisms of circulatory and intestinal barrier dysfunction during whole body hyperthermia. Am J Physiol. 2001;280:H509.
111 Hammami MM, Bouchama A, Al-Sedairy S, et al. Concentrations of soluble tumor necrosis factor and interleukin-6 receptors in heatstroke and heatstress. Crit Care Med. 1997;25:1314.
112 Handrigan MT, Bentley TB, Oliver JD, et al. Choice of fluid influences outcome in prolonged hypotensive resuscitation after hemorrhage in awake rats. Shock. 2005;23:337.
113 Hartl FU, Hayer-Hartl M. Molecular chaperones in the cytosol: From nascent chain to folded protein. Science. 2002;295:1852.
114 Hashim IA, Al-Zeer A, Al-Shohaib S, et al. Cytokine changes in patients with heatstroke during pilgrimage to Makkah. Mediators Inflamm. 1997;6:135.
115 Hassanein T, Perper JA, Tepperman L, et al. Liver failure occurring as a component of exertional heatstroke. Gastroenterology. 1991;100:1442.
116 Hassanein T, Razack A, Gavaler JS, et al. Heatstroke: Its clinical and pathological presentation, with particular attention to the liver. Am J Gastroenterol. 1992;87:1382.
117 Hatada T, Wada H, Nobori T, et al. Plasma concentrations and importance of high mobility group box protein in the prognosis of organ failure in patients with disseminated intravascular coagulation. Thromb Haemost. 2005;94:975.
118 Haveman J, Geerdink AG, Rodermond HM. Cytokine production after whole body and localized hyperthermia. Int J Hyperthermia. 1996;12:791.
119 Heidecke CD, Hensler T, Weighardt H, et al. Selective defects of T lymphocyte function in patients with lethal intraabdominal infection. Am J Surg. 1999;178:288.
120 Heinrich B, Heinrich MJE. Heterothermia in foraging workers and drones of the bumblebee Bombus terricola. Physiol Zool. 1983;56:563.
121 Henderson RA, Whitehurst ME, Morgan KR, et al. Reduced metabolic rate accompanies the hemorrhage-induced hypothermia in conscious rats. Resuscitation. 2000;44:129.
122 Hightower LE. Cultured animal cells exposed to amino acid analogues or puromycin rapidly synthesize several polypeptides. J Cell Physiol. 1980;102:407.
123 Horowitz M. Thermoregulation under conditions of impaired body fluid/osmotic balance in mammals. J Basic Clin Physiol Pharmacol. 1990;1:267.
124 Horowitz M. Heat acclimation and cross-tolerance against novel stressors: Genomic-physiological linkage. Prog Brain Res. 2007;162:373.
125 Hreggvidsdottir HS, Ostberg T, Wahamaa H, et al. The alarming HMGB1 acts in synergy with endogenous and exogenous danger signals to promote inflammation. J Leukoc Biol. 2009;86:655.
126 Hubbard RW, Matthew WT, Linduska JD, et al. The laboratory rat as a model for hyperthermic syndromes in humans. Am J Physiol. 1976;231:1119.
127 Huisse MG, Pease S, Hurtado-Nedelec M, et al. Leukocyte activation: The link between inflammation and coagulation during heatstroke: A study of patients during the 2003 heat wave in Paris. Crit Care Med. 2008;36:2288.
128 Husseini RH, Sweet C, Collie MH, et al. Elevation of nasal viral levels by suppression of fever in ferrets infected with influenza viruses of differing virulence. Infect Dis. 1982;145:520.
129 Hutchison KA, Dittmar KD, Czar MJ. Proof that HSP70 is required for assembly of the glucocorticoid receptor into a heterocomplex with HSP90. J Biol Chem. 1994;269:5043.
130 Hutchison VH, Murphy K. Behavioral thermoregulation in the salamander Necturus maculosus after heat shock. Comp Biochem Physiol. 1985;82A:391.
131 Ibuka N, Fukumura K. Unpredictable deprivation of water increases the probability of torpor in the Syrian hamster. Physiol Behav. 1997;62:551.
132 Inoue Y, Shibasaki M, Ueda H, et al. Mechanisms underlying the age-related decrement in the human sweating response. Eur J Appl Physiol. 1999;79:121.
133 Intergovernmental Panel on Climate Change. Climate change 2001: The scientific basis: Contribution of Working Group I to the third assessment report. Cambridge: Cambridge University Press; 2001. pp 1-944
134 International Union of Physiological Sciences. Glossary of terms for thermal physiology. Jap J Physiol. 2001;51:245.
135 Ito T, Kawahara K, Nakamura T, et al. High-mobility box 1 protein promotes development of microvascular thrombosis in rats. J Thromb Haemost. 2007;5:109.
136 Jaattela M. Heat shock proteins as cellular lifeguards. Ann Med. 1999;31:261.
137 Jacobson ED, Bass DE. Effects of sodium salicylate on physiological responses to work in heat. J Appl Physiol. 1964;19:33.
138 Jahromi MM, Millward BA, Demaine AG. A polymorphism in the promoter region of the gene for interleukin-6 is associated with susceptibility to type 1 diabetes mellitus. J Interferon Cytokine Res. 2000;20:885.
139 Janeway CAJr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197.
140 Jongeneel CV, Briant L, Udalova IA, et al. Extensive genetic polymorphism in the human tumor necrosis factor region and relation to extended HLA haplotypes. Proc Natl Acad Sci USA. 1991;88:9717.
141 Kalnay E, Cai M. Impact of urbanization and land-use change on climate. Nature. 2003;423:528.
142 Kao TY, Lin MT. Brain serotonin depletion attenuates heatstroke-induced cerebral ischemia and cell death in rats. Am J Physiol. 1996;80:680.
143 Kelker HC, Oppenheim JD, Stone-Wolff DS, et al. Characterization of human tumor necrosis factor produced by peripheral blood monocytes and its separation from lymphotoxin. Int J Cancer. 1985;36:69.
144 Kelty JD, Noseworthy PA, Feder ME, et al. Thermal preconditioning and heat-shock protein 72 preserve synaptic transmission during thermal stress. J Neurosci. 2002;22:1.
145 Kenefick RW, Cheuvront SN, Sawka MN. Thermoregulatory function during the marathon. Sports Med. 2007;37:312.
146 Kenney MJ, Fels RJ. Sympathetic nerve regulation to heating is altered in senescent rats. Am J Physiol. 2002;283:R513.
147 Kentner R, Safar P, Prueckner S, et al. Titrated hypertonic/hyperoncotic solution for hypotensive fluid resuscitation during uncontrolled hemorrhagic shock in rats. Resuscitation. 2005;65:87.
148 Keren G, Epstein Y, Magazanik A. Temporary heat intolerance in a heatstroke patient. Aviat Space Environ Med. 1981;52:116.
149 Kew M, Bersohn I, Seftel H. Liver damage in heat stroke. Am J Med. 1970;49:192.
150 Khazaeli AA, Tatar M, Pletcher SD, et al. Heat-induced longevity extension in Drosophila: I. Heat treatment, mortality, and thermotolerance. J Gerontol. 1997;52A:B48.
151 Khogali M. Epidemiology of heat illnesses during the Makkah pilgrimages in Saudi Arabia. Int J Epidemiol. 1983;12:267.
152 Kilbourne EM, Choi K, Jones TS, et al. Risk factors for heatstroke. A case-control study. JAMA. 1982;247:3332.
153 Kim H. Diagnostic significance of antibodies to heat shock proteins. Clin Chim Acta. 2003;337:1.
154 Kirby CR, Convertino VA. Plasma aldosterone and sweat sodium concentrations after exercise and heat acclimation. J Appl Physiol. 1986;61:967.
155 Klapproth J, Castell J, Geiger T, et al. Fate and biological action of human recombinant interleukin 1 beta in the rat in vivo. Eur J Immunol. 1989;19:1485.
156 Klein MS, Conn CA, Kluger MJ. Behavioral thermoregulation in mice inoculated with influenza virus. Physiol Behav. 1992;52:1133.
157 Kluger MJ. Fever: Role of pyrogens and cryogens. Physiol Rev. 1991;71:93.
158 Kluger MJ, Ringler DH, Anver MR. Fever and survival. Science. 1975;188:166.
159 Kluger MJ, Rothenburg BA. Fever and reduced iron: Their interaction as a host defense response to bacterial infection. Science. 1979;203:374.
160 Knochel JP. Heat and related disorders. Dis Mon. 1989;35:306.
161 Knochel JP, Dotin LN, Hamburger RJ. Pathophysiology of intense physical conditioning in a hot climate: I. Mechanisms of potassium depletion. J Clin Invest. 1972;51:242.
162 Knüpfer H, Preiss R. sIL-6R: More than an agonist? Immunol Cell Biol. 2008;86:87.
163 Kregel KC. Heat shock proteins: Modifying factors in physiological stress responses and acquired thermotolerance. J Appl Physiol. 2002;92:2177.
164 Kregel KC, Moseley PL. Differential effects of exercise and heat stress on liver HSP70 accumulation with aging. J Appl Physiol. 1996;80:547.
165 Kuwagata Y, Oda J, Irisawa T, et al. Effect of ibuprofen on interleukin-1beta-induced abnormalities in hemodynamics and oxygen metabolism in rabbits. Shock. 2003;20:558.
166 Lai FA, Erickson HP, Rousseau E, et al. Purification and reconstitution of the calcium release channel from skeletal muscle. Nature. 1988;331:315.
167 Lambert GP, Gisolfi CV, Berg DJ, et al. Selected contribution: Hyperthermia-induced intestinal permeability and the role of oxidative and nitrosative stress. J Appl Physiol. 2002;92:1750.
168 Landsberg HE. Climates and urban planning [W.M.O. tech. note No. 108. Urban climates. 1970, 129-136.
169 Lappas GD, Karl IE, Hotchkiss RS. Effect of ethanol and sodium arsenite on HSP-72 formation and on survival in a murine endotoxin model. Shock. 1994;2:34.
170 Lavoie JN, Gingras-Breton G, Tanguay RM, et al. Induction of Chinese hamster HSP27 gene expression in mouse cells confers resistance to heat shock: HSP27 stabilization of the microfilament organization. J Biol Chem. 1993;268:3420.
171 Leon LR. Cytokine regulation of fever: Studies using gene knockout mice. J Appl Physiol. 2002;92:2648.
172 Leon LR. Hypothermia in systemic inflammation: Role of cytokines. Frontiers Biosci. 2004;9:1877.
173 Leon LR. The thermoregulatory consequences of heat stroke: Are cytokines involved? J Therm Biol. 2006;31:67.
174 Leon LR. Heat stroke and cytokines. Prog Brain Res. 2007;162:481.
175 Leon LR, Blaha MD, DuBose DA. Time course of cytokine, corticosterone and tissue injury responses in mice during heat strain recovery. J Appl Physiol. 2006;100:1400.
176 Leon LR, DuBose DA, Mason CD. Heat stress induces a biphasic thermoregulatory response in mice. Am J Physiol. 2005;288:R197.
177 Leon LR, Gordon CJ, Helwig BG, et al. Thermoregulatory, behavioral, and metabolic responses to heat stroke in a conscious mouse model. Am J Physiol. 2010;299:R241.
178 Leon LR, Kozak W, Rudolph K, et al. An antipyretic role for interleukin-10 in LPS fever in mice. Am J Physiol. 1999;276:R81.
179 Leon LR, White AA, Kluger MJ. Role of IL-6 and TNF in thermoregulation and survival during sepsis in mice. Am J Physiol. 1998;275:R269.
180 Levi M, ten Cate H. Disseminated intravascular coagulation. N Engl J Med. 1999;341:586.
181 Levine JA. Heat stroke in the aged. Am J Med. 1969;47:251.
182 Liaw PC, Esmon CT, Kahnamoui K, et al. Patients with severe sepsis vary markedly in their ability to generate activated protein C. Blood. 2004;104:3958.
183 Liebermeister C. Vorlesungen uber specielle pathologie und therapie. Leipzig: Verlag von FCW Vogel; 1887.
184 Lin MT, Kao TY, Su CF, et al. Interleukin-1 beta production during the onset of heat stroke in rabbits. Neurosci Lett. 1994;174:17.
185 Lin XJ, Li YJ, Li ZL, et al. Pre-existing lipopolysaccharide may increase the risk of heatstroke in rats. Am J Med Sci. 2009;337:265.
186 Lind AR, Bass DE. Optimal exposure time for development of acclimatization to heat. Fed Proc. 1963;22:704.
187 Lindquist S, Craig EA. The heat shock proteins. Annu Rev Genet. 1988;22:631.
188 Line RL, Rust GS. Acute exertional rhabdomyolysis. Am Fam Physician. 1995;52:502.
189 Litman RS, Rosenberg H. Malignant hyperthermia: Update on susceptibility testing. JAMA. 2005;293:2918.
190 Liu CC, Cheng BC, Lin MT, et al. Small volume resuscitation in a rat model of heatstroke. Am J Med Sci. 2009;337:79.
191 Lu K-C, Wang J-Y, Lin S-H, et al. Role of circulating cytokines and chemokines in exertional heatstroke. Crit Care Med. 2004;32:399.
192 Luterbacher J, Dietrich D, Xoplaki E, et al. European seasonal and annual temperature variability, trends, and extremes since 1500. Science. 2004;303:1499.
193 Lyashko VN, Vikulova VK, Chernicov VG, et al. Comparison of the heat shock response in ethnically and ecologically different human populations. Proc Natl Acad Sci. 1994;91:12492.
194 Mack GW, Nadel ER. Body fluid balance during heat stress in humans. In: Fregly MJ, Blatteis CM, editors. Handbook of physiology, section 4: Environmental physiology. New York: Oxford University; 1996:187-214.
195 Maes M, Christophe A, Bosman E, et al. In humans, serum polyunsaturated fatty acid levels predict the response of proinflammatory cytokines to psychological stress. Biol Psychiatry. 2000;47:910.
196 Malamud N, Haymaker W, Custer RP. Heat stroke. Mil Surg. 1946;99:397.
197 Maloyan A, Palmon A, Horowitz M. Heat acclimation increases the basal HSP72 level and alters its production dynamics during heat stress. Am J Physiol. 1999;276:R1506.
198 Malvin GM, Wood SC. Behavioral hypothermia and survival of hypoxic protozoans Paramecium caudatum. Science. 1992;255:1423.
199 Malyak M, Smith MFJr, Abel AA, et al. Peripheral blood neutrophil production of interleukin-1 receptor antagonist and interleukin-1β. J Clin Immunol. 1994;14:20.
200 Marber MS, Mestril R, Chi SH, et al. Overexpression of the rat inducible 70-kD heat stress protein in a transgenic mouse increases the resistance of the heart to ischemic injury. J Clin Invest. 1995;95:1446.
201 Maron MB, Wagner JA, Horvath SM. Thermoregulatory responses during competitive marathon running. J Appl Physiol. 1977;42:909.
202 Marshall JC. The failure of clinical trials in sepsis: Challenges of clinical trial design. In: Redl H, Schlag G, editors. Cytokines in severe sepsis and septic shock. Basel/Boston/Berlin: Birkhauser; 1999:349-362.
203 McClung JP, Hasday JD, He JR, et al. Exercise-heat acclimation in humans alters baseline levels and ex vivo heat inducibility of HSP72 and HSP90 in peripheral blood mononuclear cells. Am J Physiol Regul Integr Comp Physiol. 2008;294:R185.
204 McGill SN, Ahmed NA, Christou NV. Endothelial cells: Role in infection and inflammation. World J Surg. 1998;22:171.
205 McPherson PS, Campbell KP. The ryanodine receptor/Ca2+ release channel. J Biol Chem. 1993;268:13765.
206 Medvedev AE, Sabroe I, Hasday JD, et al. Tolerance to microbial TLR ligands: Molecular mechanisms and relevance to disease. J Endotoxin Res. 2006;12:133.
207 Meehl GA, Tebaldi C. More intense, more frequent, and longer lasting heat waves in the 21st century. Science. 2004;305:994.
208 Mendeloff AI, Smith DE. Extreme physical effort in summer heat followed by collapse, stupor, purpura, jaundice and azotemia. Am J Med. 1955;18:659.
209 Mikkelsen TS, Toft P. Prognostic value, kinetics and effect of CVVHDF on serum of the myoglobin and creatine kinase in critically ill patients with rhabdomyolysis. Acta Anaesthesiol Scand. 2005;49:859.
210 Minson CT, Wladkowski SL, Cardell AF, et al. Age alters the cardiovascular response to direct passive heating. J Appl Physiol. 1998;84:1323.
211 Mitchell GW. Rapid onset of severe heat illness: A case report. Aviat Space Environ Med. 1991;62:779.
212 Modlin RL, Brightbill HD, Godowski PJ. The toll of innate immunity on microbial pathogens. N Engl J Med. 1999;340:1834.
213 Moldoveanu AI, Shephard RJ, Shek PN. The cytokine response to physical activity and training. Sports Med. 2001;31:115.
214 Montain SJ, Sawka MN, Wenger CB. Hyponatremia associated with exercise: Risk factors and pathogenesis. Exerc Sport Sci Rev. 2001;29:113.
215 Morrison-Bogorad M, Zimmerman AL, Pardue S. Heat-shock 70 messenger RNA levels in human brain: Correlation with agonal fever. J Neurochem. 1995;64:235.
216 Moseley PL, Gapen C, Wallen ES, et al. Thermal stress induced epithelial permeability. Am J Physiol. 1994;267:C425.
217 Muller CB, Schmid-Hempel P. Exploitation of cold temperature as defence against parasitoids in bumblebees. Nature. 1993;363:65.
218 Musapasaoglu H, Agildere AM, Teksam M, et al. Hypernatraemic dehydration in a neonate: Brain MRI findings. Br J Radiol. 2008;81:e57.
219 Mustafa KY, Omer O, Khogali M, et al. Blood coagulation and fibrinolysis in heat stroke. Br J Haematol. 1985;61:517.
220 Myhre KM, Cabanac M, Myhre G. Fever and behavioural temperature regulation in the frog Rana esculenta. Acta Physiol Scand. 1977;101:219.
221 Nadel ER, Mitchell JW, Saltin B, et al. Peripheral modifications to the central drive for sweating. J Appl Physiol. 1971;31:828.
222 Nahas GG, Tannieres ML, Lennon JF. Direct measurement of leukocyte motility: Effects of pH and temperature. Proc Soc Exp Biol Med. 1971;138:350.
223 Nakashima T, Hori T, Kiyohara T, et al. Osmosensitivity of preoptic thermosensitive neurons in hypothalamic slices in vitro. Pflugers Arch. 1985;405:112.
224 National Institute of Occupational Health and Safety: Recruit fire fighter suffers heat stroke during physical fitness training and dies nine days later, U.S. Department of Health and Human Services, Fatality assessment and control evaluation investigative report No. F2005-26, Cincinnati, Ohio, 2006.
225 National Institute of Occupational Health and Safety: Migrant farm worker dies from heat stroke while working on a tobacco farm—North Carolina, U.S. Department of Health and Human Services, Fatality assessment and control evaluation investigative report No. 2006-04, 2007.
226 Naughton MP, Henderson A, Mirabelli MC, et al. Heat-related mortality during a 1999 heat wave in Chicago. Am J Prev Med. 2002;22:221.
227 Neville AJ, Sauder DN. Whole body hyperthermia (41-42° C) induces interleukin-1 in vivo. Lymphokine Res. 1988;7:201.
228 Nielsen B, Hales JRS, Strange S, et al. Human circulatory and thermoregulatory adaptations with heat acclimation and exercise in a hot, dry environment. J Physiol. 1993;460:467.
229 Nieman DC, Henson DA, Smith LL, et al. Cytokine changes after a marathon race. J Appl Physiol. 2001;91:109.
230 Nolan JP. Endotoxin, reticuloendothelial function, and liver injury. Hepatology. 1981;1:458.
231 Nomoto K, Yoshikai Y. Heat-shock proteins and immunopathology: Regulatory role of heat-shock protein-specific T cells. Springer Semin Immunopathol. 1991;13:63.
232 Nussbaum E, Tuazon CU, Kessler CM. Aspirin (ASA) prevents the disseminated intravascular coagulation (DIC) induced by S. aureus and its purified peptidoglycan (PEP). Blood. 1988;72:305a.
233 Nybo L. Exercise and heat stress: Cerebral challenges and consequences. Prog Brain Res. 2007;162:29.
234 Oberholzer A, Oberholzer C, Moldawer LL. Interleukin-10: A complex role in the pathogenesis of sepsis syndromes and its potential as an anti-inflammatory drug. Crit Care Med. 2002;30:S58.
235 O’Brien KK, Montain SJ, Corr WP, et al. Hyponatremia associated with overhydration in U.S. Army trainees. Mil Med. 2001;166:405.
236 O’Brien PJ, Cribb PH, White RJ, et al. Canine malignant hyperthermia: Diagnosis of susceptibility in a breeding colony. Can Vet J. 1983;24:172.
237 O’Donnell TFJr. Acute heat stroke: Epidemiologic, biochemical, renal, and coagulation studies. JAMA. 1975;234:824.
238 Oh RCH, Oh RCH, Henning JS. Exertional heatstroke in an infantry soldier taking ephedra-containing dietary supplements. Mil Med. 2003;168:429.
239 Oka T, Oka K, Hori T. Mechanisms and mediators of psychological stress-induced rise in core temperature. Psychosomatic Med. 2001;63:476.
240 Okusawa S, Gelfand JA, Ikejima T, et al. Interleukin 1 induces a shock-like state in rabbits: Synergism with tumor necrosis factor and the effect of cyclooxygenase inhibition. J Clin Invest. 1988;81:1162.
241 Opal SM, Fisher CJJr, Dhainaut JF, et al. Confirmatory interleukin-1 receptor antagonist trial in severe sepsis: A phase III, randomized, double-blind, placebo-controlled, multicenter trial: The Interleukin-1 Receptor Antagonist Sepsis Investigator Group. Crit Care Med. 1997;25:1115.
242 Oshima T, Laroux FS, Coe LL, et al. Interferon-γ and interleukin-10 reciprocally regulate endothelial junction integrity and barrier function. Microvasc Res. 2001;61:130.
243 O’Sullivan GH, McIntosh JM, Heffron JJ. Abnormal uptake and release of Ca2+ ions from human malignant hyperthermia-susceptible sarcoplasmic reticulum. Biochem Pharmacol. 2001;61:1479.
244 Oz MC, Zikria BA, McLeod PF, et al. Hydroxyethyl starch macromolecule and superoxide dismutase effects on myocardial reperfusion injury. Am J Surg. 1991;162:59.
245 Pandolf KB, Burse RL, Goldman RF. Role of physical fitness in heat acclimatisation, decay and reinduction. Ergonomics. 1977;20:399.
246 Parcellier A, Gurbuxani S, Schmitt E, et al. Heat shock proteins, cellular chaperones that modulate mitochondrial cell death pathways. Biochem Biophys Res Commun. 2003;304:505.
247 Patel DR, Gyamfi R, Torres A. Exertional rhabdomyolysis and acute kidney injury. Phys Sportsmed. 2009;37:71.
248 Patel RT, Deen KI, Youngs D, et al. Interleukin 6 is a prognostic indicator of outcome in severe intra-abdominal sepsis. Br J Surg. 1994;81:1306.
249 Pease S, Bouadma L, Kermarrec N, et al. Early organ dysfunction course, cooling time and outcome in classic heatstroke. Intensive Care Med. 2009;35:1454.
250 Perchick JS, Winkelstein A, Shadduck RK. Disseminated intravascular coagulation in heat stroke: Response to heparin therapy. JAMA. 1975;231:480.
251 Pruitt JH, Welborn MB, Edwards PD, et al. Increased soluble interleukin-1 type II receptor concentrations in postoperative patients and in patients with sepsis syndrome. Blood. 1996;7:3282.
252 Pugh LGCE, Corbett JL, Johnson RH. Rectal temperatures, weight losses, and sweat rates in marathon running. J Appl Physiol. 1967;23:347.
253 Rajdev S, Hara K, Kokubo Y, et al. Mice overexpressing rat heat shock protein 70 are protected against cerebral infarction. Ann Neurol. 2000;47:782.
254 Raju SF, Robinson GH, Bower JO. The pathogenesis of acute renal failure in heat stroke. South Med J. 1973;66:330.
255 Ramasawmy R, Fae KC, Spina G, et al. Association of polymorphisms within the promoter region of the tumor necrosis factor-alpha with clinical outcomes of rheumatic fever. Mol Immunol. 2007;44:1873.
256 Rasch W, Samson P, Cote J, et al. Heat loss from the human head during exercise. J Appl Physiol. 1991;71:590.
257 Rausch RN, Crawshaw LI, Wallace HL. Effects of hypoxia, anoxia, and endogenous ethanol on thermoregulation in goldfish, Carassius auratus. Am J Physiol. 2000;278:R545.
258 Remick DG. Cytokine therapeutics for the treatment of sepsis: Why has nothing worked? Curr Pharm Des. 2003;9:75.
259 Rennick DM, Fort MM, Davidson NJ. Studies with IL-10-/- mice: An overview. J Leukoc Biol. 1997;61:389.
260 Replogle RL, Merrill W. Experimental polycythemia and hemodilution: Physiologic and rheologic effects. J Thorac Cardiovasc Surg. 1970;60:582.
261 Reynolds WW, Casterlin ME, Covert JB. Behavioural fever in teleost fishes. Nature Lond. 1976;259:41.
262 Richards SA. The biology and comparative physiology of thermal panting. Biol Rev Camb Philos Soc. 1970;45:223.
263 Richardson TQ, Guyton A. Effects of polycythemia and anemia on cardiac output and other circulatory factors. Am J Physiol. 1959;197:1167.
264 Ritossa F. A new puffing pattern induced by temperature shock and DNP in Drosophila melanogaster: Relation to chromosome puffs. Experientia. 1962;18:571.
265 Roberts WO. Death in the heat: Can football heat stroke be prevented? Current Sports Med Rep. 2004;3:1.
266 Roberts WO. Exertional heat stroke during a cool weather marathon: A case study. Med Sci Sports Exerc. 2006;38:1197.
267 Robinson S, Robinson AH. Chemical composition of sweat. Physiol Rev. 1954;34:202.
268 Rogers MA, Stull GA, Apple FS. Creatine kinase isoenzyme activities in men and women following a marathon race. Med Sci Sports Exerc. 1985;17:679.
269 Romanovsky AA, Shido O, Sakurada S, et al. Endotoxin shock-associated hypothermia: How and why does it occur? Ann NY Acad Sci. 1997;813:733.
270 Roncarolo MG, Battaglia M, Gregori S. The role of interleukin 10 in the control of autoimmunity. J Autoimmun. 2003;20:269.
271 Rosenberg H, Antognini JF, Muldoon S. Testing for malignant hyperthermia. Anesthesiology. 2002;96:232.
272 Rosner MH. Urinary biomarkers for the detection of renal injury. Adv Clin Chem. 2009;49:73.
273 Ryan AJ, Matthes RD, Mitros FA, et al. Heat stress does not sensitize rats to the toxic effects of bacterial lipopolysaccharide. Med Sci Sports Exerc. 1994;26:687.
274 Saissy JM. Liver transplantation in a case of fulminant liver failure after exertion. Intensive Care Med. 1996;22:831.
275 Sawka MN, Coyle EF. Influence of body water and blood volume on thermoregulation and exercise performance in the heat. In: Hollozsy JO, editor. Exercise and sport sciences reviews. Baltimore: Williams & Wilkins; 1999:167-218.
276 Sawka MN, Wenger CB, Pandolf KB. Thermoregulatory responses to acute exercise heat stress and heat acclimation. In: Fregly MJ, Blatteis CM, editors. Handbook of physiology, Section 4: Environmental physiology. New York: Oxford University; 1996:157-185.
277 Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191.
278 Schär C, Vidale PL, Lüthi D, et al. The role of increasing temperature variability in European summer heat waves. Nature. 2004;427:332.
279 Schlattner U, Tokarska-Schlattner M, Wallimann T. Mitochondrial creatine kinase in human health and disease. Biochim Biophys Acta. 2006;1762:164.
280 Schobitz B, Pezeshki G, Pohl T, et al. Soluble interleukin-6 (IL-6) receptor augments central effects of IL-6 in vivo. FASEB J. 1995;9:659.
281 Schwimmer H, Eli-Berchoer L, Horowitz M. Acclimatory-phase specificity of gene expression during the course of heat acclimation and superimposed hypohydration in the rat hypothalamus. J Appl Physiol. 2006;100:1992.
282 Sculier JP, Bron D, Verboven N, et al. Multiple organ failure during interleukin-2 administration and LAK cells infusion. Intensive Care Med. 1988;14:666.
283 Seeler DC, McDonell WN, Basrur PK. Halothane and halothane/succinylcholine induced malignant hyperthermia (porcine stress syndrome) in a population of Ontario boars. Can J Comp Med. 1983;47:284.
284 Senay LCJr. Relationship of evaporative rates to serum [Na+], [K+], and osmolarity in acute heat stress. J Appl Physiol. 1968;25:149.
285 Sharif SF, Hariri RJ, Chang VA, et al. Human astrocyte production of tumour necrosis factor-α, interleukin-1β and interleukin-6 following exposure to lipopolysaccharide endotoxin. Neurol Res. 1993;15:109.
286 Sharma HS, Dey PK. Probable involvement of 5-hydroxytryptamine in increased permeability of blood-brain barrier under heat stress in young rats. Neuropharmacology. 1986;25:161.
287 Sharma HS, Dey PK. Influence of long-term acute heat exposure on regional blood-brain barrier permeability, cerebral blood flow and 5-HT level in conscious normotensive young rats. Brain Res. 1987;424:153.
288 Sherman KE. Alanine aminotransferase in clinical practice. Arch Intern Med. 1991;151:260.
289 Shibolet S, Lancaster MC, Danon Y. Heat stroke: A review. Aviat Space Environ Med. 1976;47:280.
290 Smalley B, Janke RM, Cole D. Exertional heat illness in Air Force basic military trainees. Mil Med. 2003;168:298.
291 Snell ES, Atkins E. The mechanisms of fever. In: Bittar EE, Bittar N, editors. The biological basis of medicine. New York: Academic Press; 1968:397-419.
292 Sonna LA, Cullivan ML, Sheldon HK, et al. Effect of hypoxia on gene expression by human hepatocytes (HepG2). Physiol Genomics. 2003;12:195.
293 Sonna LA, Gaffin SL, Pratt RE, et al. Effect of acute heat shock on gene expression by human peripheral blood mononuclear cells. J Appl Physiol. 2002;92:2208.
294 Sonna LA, Wenger CB, Flinn S, et al. Exertional heat injury and gene expression changes: A DNA microarray analysis study. J Appl Physiol. 2004;96:1943.
295 Srichaikul T, Prayoonwiwat W, Sanguanwong S, et al. Clinical manifestations and therapy of heat stroke: Consumptive coagulopathy successfully treated by exchange transfusion and heparin. Southeast Asian J Trop Med Public Health. 1989;20:479.
296 Stauss HM, Morgan DA, Anderson KE, et al. Modulation of baroreflex sensitivity and spectral power of blood pressure by heat stress and aging. Am J Physiol. 1997;272:H776.
297 Stitt JT. Prostaglandin E as the neural mediator of the febrile response. Yale J Biol Med. 1986;59:137.
298 Stojadinovic A, Kiang J, Smallridge R, et al. Induction of heat-shock protein 72 protects against ischemia/reperfusion in rat small intestine. Gastroenterology. 1995;109:505.
299 Stricker EM, Hainsworth FR. Evaporative cooling in the rat: Interaction with heat loss from the tail. Q J Exp Physiol Cogn Med Sci. 1971;56:231.
300 Strother SV, Bull JM, Branham SA. Activation of coagulation during therapeutic whole body hyperthermia. Thromb Res. 1986;43:353.
301 Stuber F, Petersen M, Bokelmann F, et al. A genomic polymorphism within the tumor necrosis factor locus influences plasma tumor necrosis factor-alpha concentrations and outcome of patients with severe sepsis. Crit Care Med. 1996;24:381.
302 Sundén-Cullberg J, Norrby-Teglund A, Rouhiainen A, et al. Persistent elevation of high mobility group box-1 protein (HMGB1) in patients with severe sepsis and septic shock. Crit Care Med. 2005;33:564.
303 Suzuki K, Nakaji S, Yamada M, et al. Impact of a competitive marathon race on systemic cytokine and neutrophil response. Med Sci Sports Exerc. 2003;35:348.
304 Szold O, Reider-Groswasser II, Ben Abraham R, et al. Gray-white matter discrimination: A possible marker for brain damage in heat stroke? Eur J Radiol. 2002;43:1.
305 Takeshima H, Nishimura S, Matsumoto T, et al. Primary structure and expression from complementary DNA of skeletal muscle ryanodine receptor. Nature. 1989;339:439.
306 Tatar M, Khazaeli AA, Curtsinger JW. Chaperoning extended life. Nature. 1997;390:30.
307 Taylor FBJr, Chang A, Esmon CT, et al. Protein C prevents the coagulopathic and lethal effects of Escherichia coli infusion in the baboon. J Clin Invest. 1987;79:918.
308 Tissières A, Mitchell HK, Tracy VM. Protein synthesis in salivary glands of Drosophila melanogaster: Relation to chromosome puffs. J Mol Biol. 1974;84:389.
309 Tollofsrud S, Bjerkelund CE, Kongsgaard U, et al. Cold and warm infusion of Ringer’s acetate in healthy volunteers: The effects on haemodynamic parameters, transcapillary fluid balance, diuresis and atrial peptides. Acta Anaesthesiol Scand. 1993;37:768.
310 Toner MM, McArdle WD. Human thermoregulatory responses to acute cold stress with special reference to water immersion. In: Fregly MJ, Blatteis CM, editors. Handbook of physiology, section 4: Environmental physiology. New York: Oxford University; 1996:379-418.
311 Tracey KJ, Lowry SF, Cerami A. Physiological responses to cachectin. Ciba Found Symp. 1987;131:88.
312 Tsujimoto H, Ono S, Efron PA, et al. Role of Toll-like receptors in the development of sepsis. Shock. 2008;29:315.
313 Tsujimoto H, Ono S, Majima T, et al. Neutrophil elastase, MIP-2, and TLR-4 expression during human and experimental sepsis. Shock. 2005;23:39.
314 van den HC, Lister D, Muylle E, et al. Malignant hyperthermia in Belgian Landrace pigs rested or exercised before exposure to halothane. Br J Anaesth. 1976;48:821.
315 Vandentorren S, Suzan F, Medina S, et al. Mortality in 13 French cities during the August 2003 heat wave. Am J Public Health. 2004;94:1518.
316 van der PT, Jansen J, Levi M, et al. Regulation of interleukin 10 release by tumor necrosis factor in humans and chimpanzees. J Exp Med. 1994;180:1985.
317 van Oss CJ, Absolom DR, Moore LL, et al. Effect of temperature on the chemotaxis, phagocytic engulfment, digestion and O2 consumption of human polymorphonuclear leukocytes. Reticuloendothel Soc. 1980;27:561.
318 Varghese GM, John G, Thomas K, et al. Predictors of multi-organ dysfunction in heatstroke. Emerg Med J. 2005;22:185.
319 Vasselon T, Detmers PA. Toll receptors: A central element in innate immune responses. Infect Immun. 2002;70:1033.
320 Vaughn LK, Bernheim HA, Kluger MJ. Fever in the lizard Dipsosaurus dorsalis. Nature Lond. 1974;252:473.
321 Vaughn LK, Veale WL, Cooper KE. Antipyresis: Its effect on mortality rate of bacterially infected rabbits. Brain Res Bull. 1980;5:69.
322 Villar J, Ribeiro SP, Mullen JBM, et al. Induction of the heat shock response reduces mortality rate and organ damage in a sepsis-induced acute lung injury model. Crit Care Med. 1994;22:914.
323 Voellmy R. Transduction of the stress signal and mechanisms of transcriptional regulation of heat shock/stress protein gene expression in higher eukaryotes. Crit Rev Eukaryot Gene Expr. 1994;4:357.
324 Wagner M, Kaufmann P, Fickert P, et al. Successful conservative management of acute hepatic failure following exertional heatstroke. Eur J Gastroenterol Hepatol. 2003;15:1135.
325 Wagner RD, Maroushek NM, Brown JF, et al. Treatment with anti-interleukin-10 monoclonal antibody enhances early resistance to but impairs complete clearance of Listeria monocytogenes infection in mice. Infect Immun. 1994;62:2345.
326 Wallace RF, Kriebel D, Punnett L, et al. Prior heat illness hospitalization and risk of early death. Environ Res. 2007;104:290.
327 Wallimann T, Hemmer W. Creatine kinase in non-muscle tissues and cells. Mol Cell Biochem. 1994;133-134:193.
328 Wang AY, Li PK, Lui SF, et al. Renal failure and heatstroke. Ren Fail. 1995;17:171.
329 Wang H, Bloom O, Zhang M, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248.
330 Wang X, Zou Y, Wang Y, et al. Differential regulation of interleukin-12 and interleukin-10 by heat shock response in murine peritoneal macrophages. Biochem Biophys Res Commun. 2001;287:1041.
331 Wappler F, Fiege M, Steinfath M, et al. Evidence for susceptibility to malignant hyperthermia in patients with exercise-induced rhabdomyolysis. Anesthesiology. 2001;94:95.
332 Warren HS, Suffredini AF, Eichacker PQ, et al. Risks and benefits of activated protein C treatment for severe sepsis. N Engl J Med. 2002;347:1027.
333 Wayner DD, Burton GW, Ingold KU, et al. The relative contributions of vitamin E, urate, ascorbate and proteins to the total peroxyl radical-trapping antioxidant activity of human blood plasma. Biochim Biophys Acta. 1987;924:408.
334 Weiss SJ. Tissue destruction by neutrophils. N Eng J Med. 1989;320:365.
335 Wenger CB. Human heat acclimation. In: Pandolf KB, Sawka MN, Gonzalez RR, editors. Human performance physiology and environmental medicine at terrestrial extremes. Indianapolis: Benchmark; 1988:153-197.
336 Wenger CB. The regulation of body temperature. In: Rhoades RA, Tanner GA, editors. Medical physiology. Boston: Little, Brown; 1995:590.
337 White JG. Effects of heat on platelet structure and function. Blood. 1968;32:324.
338 Wilkinson DA, Burholt DR, Shrivastava PN. Hypothermia following whole-body heating of mice: Effect of heating time and temperature. Int J Hyperthermia. 1998;4:171.
339 Williams RS, Thomas JA, Fina M, et al. Human heat shock protein 70 (HSP70) protects murine cells from injury during metabolic stress. J Clin Invest. 1993;92:503.
340 Winkenwerder W, Sawka MN. Disorders due to heat and cold. In: Goldman L, Ausiello DA, Arend W, et al, editors. Cecil textbook of medicine. ed 23. Philadelphia: Saunders; 2007:763-767.
341 Wisselink W, Patetsios P, Panetta TF, et al. Medium molecular weight pentastarch reduces reperfusion injury by decreasing capillary leak in an animal model of spinal cord ischemia. J Vasc Surg. 1998;27:109.
342 World Health Organization (WHO) Regional Committee for Europe. Heatwaves: Impacts and responses: Briefing note for the fifty-third session of the WHO regional committee for Europe, Vienna. Geneva: WHO; 2003.
343 Wu C. Heat shock transcription factors: Structure and regulation. Annu Rev Cell Dev Biol. 1995;11:441.
344 Wyndham CH, Strydom NB, Cooke HM, et al. Methods of cooling subjects with hyperpyrexia. J Appl Physiol. 1959;14:771.
345 Yang YL, Lu KT, Tsay HJ, et al. Heat shock protein expression protects against death following exposure to heatstroke in rats. Neurosci Lett. 1998;252:9.
* Approved for public release: Distribution is unlimited.
* References 32, 54, 154, 216, 256, 316.
* References 26, 42, 109, 112, 188, 289.
* References 29, 30, 43, 108, 147, 225.
* References 21, 56, 97, 171, 172, 192.
† Reference 27, 43, 97, 147, 172, 188, 249, 323.
* References 48, 140, 192, 281, 282, 284, 299.
* References 20, 27, 36, 116, 172, 181.
* References 154, 162, 168, 169, 235, 237, 277, 306.
[/level-membership-for-emergency-medicine-category][not-level-membership-for-emergency-medicine-category]
Chapter 10 Pathophysiology of Heat-Related Illnesses
For online-only figures, please go to www.expertconsult.com
Heat Stress and Thermoregulation
Four Avenues of Heat Exchange
Mammals and other homeotherms are capable of maintaining body temperature within a fairly narrow range (approximately 35° to 41° C [95° to 105.8° F]) despite large fluctuations in environmental temperature. Environmental variables that have the largest impact on heat exchange are temperature; humidity; radiation from the air, water, or land; and air or water motion.88 To maintain stable body temperature, organisms rely on four avenues of heat exchange: conduction, convection, radiation, and evaporation.
Dry heat exchange is achieved by conduction, convection, and radiation. The effectiveness of these mechanisms depends on differences between the skin and environmental temperatures. That is, dry heat loss occurs when skin temperature exceeds that of the environment, and dry heat gain occurs when environmental temperature exceeds that of the body. Conduction occurs when the body surface is in direct contact with a solid object and depends on the thermal conductivity of the object and the amount of surface area in contact with the object. Conduction is typically an ineffective mechanism of heat exchange because of behavioral adjustments that minimize contact with an object. For example, the wearing of shoes is an effective behavioral adjustment that minimizes conduction of heat from a hot surface (e.g., desert sand to the foot). Within the body, conductive heat transfer occurs between tissues that are in direct contact with one another, but is limited by poor conductivity of the tissues. For example, subcutaneous fat has approximately 60% lower conductivity than does the dermis and impedes conductive heat loss.332 Convection is a mechanism of dry heat transfer that occurs as air or water moves over the skin surface. The windchill index is an example of the convective cooling effect of wind velocity. The rate of convective heat transfer depends on the temperature gradient between the body and environment, thermal currents, bodily movements, and areas of the body surface that are exposed to the environment, which can vary dramatically with different clothing ensembles. Within the body, convective heat transfer occurs between blood vessels and tissues and is most efficient at the capillary beds, which are thin-walled and provide a large surface area for heat exchange. Radiative heat transfer is electromagnetic energy that is exchanged between the body and surrounding environmental objects and is independent of air velocity or temperature. It is effective even when air temperature is below that of the body. All objects within our environment absorb and emit thermal radiation, but clothing can reduce radiant heat that impinges on the skin from various environmental sources.
Evaporation represents a major avenue of heat loss when environmental temperatures are equal to or above skin temperature or when body temperature is increased by vigorous physical activity. In humans, evaporative cooling is achieved as sweat is vaporized and removes heat from the skin surface, with approximately 580 kcal of heat lost per each liter of evaporated sweat.94 The most important environmental variables affecting evaporative cooling are ambient humidity and wind velocity. Sweat is converted to water vapor and readily evaporates from the skin in dry air with wind, whereas the conversion of sweat to water vapor is limited in still or moist air. If sweat accumulates and fails to evaporate, sweat secretion is inhibited and the cooling benefit is negated. Small mammals, such as rodents, do not possess sweat glands but achieve evaporative cooling by grooming nonfurred and highly vascularized skin surfaces, such as the ears, paw pads, and tail, with saliva that evaporates in a manner similar to that of sweat in humans.104,294
Body Temperature Control
Regulation of a relatively constant internal temperature is critical for normal physiologic functioning of tissues and cells because membrane fluidity, electrical conductance, and enzyme functions are most efficient within a narrow temperature range. By convention, thermal physiologists describe body temperature control with a two-compartment model that consists of an internal core (i.e., viscera and brain) and an outer shell (i.e., subcutaneous fat and skin) (Figure 10-1).70
(From Elizondo RS: Human adaptation to hot environments. In Rhoades RA, Pflanger RG, editors: Human physiology, ed 3, Philadelphia, 1996, Saunders. Reprinted with permission of Brooks/Cole, a division of Thomson Learning. http://www.thomsonrights.com.)
where M is metabolic rate, W is work, and E, C, K, and R are evaporative, convective, conductive, and radiant heat transfer, respectively.132 The impact of the four avenues of heat exchange on total body storage depends on a variety of organismal (e.g., age, gender, adiposity), environmental (e.g., humidity, wind velocity), and occupational (e.g., protective clothing, work intensity) variables. Under conditions in which heat production and/or heat gain exceeds heat loss, such as during exercise or heat exposure, positive heat storage occurs and body temperature increases. When heat loss exceeds heat production and/or heat gain, such as during prolonged cold exposure, negative heat storage occurs and body temperature decreases.88
Endothermic animals use both autonomic and behavioral thermoeffector mechanisms to regulate body temperature. Autonomic thermoeffector responses are often referred to as “involuntary” and include sweating, vasodilation, vasoconstriction, piloerection (furred mammals), and shivering and nonshivering thermogenesis (brown fat heat production). Behavioral thermoeffector mechanisms are considered “voluntary” and include clothing changes, use of heat or air conditioning systems, huddling or use of blankets, fan cooling, and seeking of shade or shelter. Rather than working independently of one another, autonomic and behavioral thermoeffector mechanisms typically function in concert to maintain temperature control. For example, evaporative cooling in rodents requires autonomic stimulation of salivation and behavioral spreading of saliva onto nonfurred surfaces.104,294 Many large species in the wild use natural water sources to facilitate cooling. Elephants spray water onto their skin surface, and hippopotamuses and other species are often observed near or in watering holes. Water is a more effective medium to facilitate convective heat transfer than air, because of its high heat-transfer coefficient (approximately 25 times greater than air),305 even if the water temperature is tepid. However, voluntary suppression of behavioral mechanisms of cooling in humans can increase the risk for thermal injury. This is illustrated by older adults who refuse to use air conditioning systems or leave their residences during heat waves, or highly motivated athletes and military personnel who voluntarily dehydrate and/or sustain a high rate of work in hot weather.
Regulation of body temperature is best conceptualized as a negative-feedback system consisting of sensors, integrators, and effectors. In vertebrates, neurons in the skin, spinal cord, and abdomen sense thermal stimuli and convert those signals to action potentials that are transmitted by afferent sensory neurons to the preoptic area of the anterior hypothalamus (POAH). The POAH is considered the main central nervous system (CNS) site for thermoregulatory control because it receives and integrates synaptic afferent inputs and evokes corrective autonomic and behavioral thermoeffector responses for body temperature regulation.28 A diagrammatic representation of this negative-feedback loop is shown in Figure 10-2.
The concept of a temperature set point was developed as a theoretical framework to examine regulated and unregulated changes in body temperature.19 The temperature set point is analogous to a thermostat that controls a mechanical heating device; under homeostatic conditions, body temperature is approximately equal and oscillates around the temperature set point. Environmental perturbations, such as heat and exercise, cause body temperature to deviate from the set-point temperature as heat gain and/or production exceeds heat loss and the organism becomes hyperthermic (body temperature is greater than the set-point temperature). During prolonged cold exposure, heat loss exceeds heat gain and/or production and the organism becomes hypothermic (body temperature is less than the set-point temperature) (Figure 10-3).
Regulated increases and decreases in the temperature set point are referred to as fever and regulated hypothermia (also called anapyrexia), respectively, and are protective immune responses to infection, inflammation, or trauma. Fever is defined as a regulated increase in the temperature set point and is actively established and defended by heat-producing (e.g., shivering and nonshivering thermogenesis) and heat-conserving (e.g., peripheral vasoconstriction, huddling to reduce exposed body surface area) thermoeffectors (see Figure 10-3).132 An individual is considered normothermic once fever is established and body temperature oscillates around the new elevated temperature set point (see Figure 10-3). The highly regulated nature of fever was first suggested by Liebermeister in the 1800s when it was observed that individuals actively reestablished an elevation in body temperature following experimental warming or cooling.180,286 Fever is a protective immune response used by invertebrates, fish, amphibians, reptiles, and mammals to survive infection or injury.* The protective effects of fever are mediated by increased mobility and activity of white blood cells,218,313 increased production of interferon (IFN; antiviral and antibacterial agent) antibodies,64 and reduced plasma iron concentrations, the effects of all of which inhibit the growth of pathogens.89,156 In mammals, inhibition of fever using antipyretic drugs (e.g., aspirin) increases mortality from bacterial and viral infections, which speaks to the importance of fever as an immune response.126,315
Many species also develop regulated hypothermia to survive severe environmental insults. Regulated hypothermia is elicited in response to a decrease in the temperature set point and is actively established and defended by behavioral and autonomic heat-loss mechanisms.132 The Q10 effect states that each 10° C (18° F) change in body temperature is associated with a twofold to threefold change in enzymatic reaction rates. Based on this relationship, a regulated decrease in body temperature is thought to protect against injury and inflammation by reducing production of harmful enzymatic end products that compromise tissue function under conditions of low oxygen supply. In bumblebees, infected worker bees spend significantly more time in cooler temperatures outside of the nest than healthy worker bees; this cold-seeking behavior is associated with increased survival from parasitic infection.118,213 Mice inoculated with influenza virus also show cold-seeking behavior and develop regulated hypothermia, which is associated with improved infection outcome.153 Other environmental insults that induce regulated hypothermia in small rodents include hypoglycemia,34,98 hypoxia,194,252 hemorrhage,119 dehydration,129 infection,153,176,264 and heatstroke.173,174
Mechanisms of Heat Dissipation During Thermal Stress
Cardiovascular mechanisms have evolved to shunt warm blood from the body core to the skin surface and increase heat loss during thermal stress. Arteriovenous anastomoses (AVAs) are collateral connections between adjacent blood vessels that increase the volume of blood that is delivered to a particular tissue. Mean skin blood flow can vary approximately 10-fold in humans depending on the thermal environment. The hands and feet are concentrated with AVAs that serve as effective areas for dry heat loss. The nonfurred surfaces of small rodents, such as the ears, tail, and paw pads, also have an abundance of AVAs and a large surface area to facilitate convective heat transfer.92,99 During exercise heat stress, increased blood flow to the skin surface is accompanied by sweat secretion. The density, secretion rate, and activation threshold of regional sweat glands determine the volume of sweat loss at a body site. In humans, the back and chest have the highest sweat rates for a given body temperature change, whereas only approximately 25% of total sweat is produced by the lower limbs.217 Additional factors affecting sweat rate include clothing characteristics, environmental conditions, and rate of metabolic heat production. Panting is an effective method of evaporative heat dissipation in large animals, such as birds, dogs, sheep, and rabbits, and occurs at a resonant ventilation frequency that requires minimal energy expenditure.107,257,329 Humans and rodents do not pant per se, but breathing frequency and minute volume increase during severe heat exposure to facilitate evaporative cooling from the respiratory surfaces. In humans, the contribution of respiratory evaporative cooling is small compared with that of skin evaporative cooling (Figure 10-4).
Dehydration and Electrolyte Imbalance
Water requirements during heat exposure are primarily determined by a person’s sweat losses. Water depletion dehydration develops when the rate of water replacement is not adequate, which can be a result of a mismatch between fluid intake and sweat loss, lack of water availability, or use of diuretic medications. Sweat rates may range from 0.3 to approximately 3 L/hr during athletic or occupational activities, depending on the environmental conditions and type, duration, and intensity of work.47,143 If high sweat rates are maintained without adequate replenishment of lost water, this can cause electrolyte imbalances that impede the efficiency of autonomic mechanisms of thermoregulatory control. For example, hyperosmolarity alters heat responsiveness of warm-sensitive neurons in the POAH and limits the effectiveness of evaporative heat loss.121,219,276 Severe hypernatremic dehydration is associated with brain edema, intracranial hemorrhage, hemorrhagic infarcts, and permanent brain damage (Figure 10-5, online).214
FIGURE 10-5 Effect of reduced plasma volume or increased osmolality on the sweat rates of six individuals.
(Modified from Sawka MN, Young AJ, Francesconi RP, et al: Thermoregulatory and blood responses during exercise at graded hypohydration levels, J Appl Physiol 59:1394, 1985.)
Severe reductions in electrolytes can have a profound impact on heatstroke outcome. Symptomatic hyponatremia (decreased serum sodium concentration) is a relatively rare condition, but it has been observed in marathon runners and military recruits during training exercises as a consequence of overconsumption of hypotonic fluids with inadequate replacement of sodium losses.210,230 Intracellular swelling is a severe consequence of hyponatremia that may cause CNS dysfunction. Hypokalemia (decreased serum potassium concentration) may be caused by overproduction of aldosterone, excessive sweating, or respiratory alkalosis. Any cause of overproduction of urine (polyuria) potentially causes urinary potassium loss.323 Potassium is a potent vasodilator of blood vessels to the skeletal and cardiac muscles, so excessive loss of this electrolyte can have detrimental effects, such as decreased sweat volume, cardiovascular instability, and reductions in muscle blood flow that predispose to skeletal muscle injury (i.e., rhabdomyolysis).158,279
Heat Illnesses
Heat illnesses are best viewed as existing along a continuum that transitions from the mild conditions of heat cramps and heat exhaustion to more serious conditions of heat injury and heatstroke (Table 10-1).
| Condition | Symptoms | Management |
|---|---|---|
| Heat cramps | Brief, painful skeletal muscle spasms | Rest; replacement of electrolytes; avoid salt tablets |
| Heat rash (miliaria rubra) | Blocked eccrine sweat glands | Cool, dry affected skin area; topical corticosteroids, aspirin |
| Heat exhaustion | Mild to moderate illness with inability to sustain cardiac output; moderate (>38.5° C [101.3° F]) to high (>40° C [104° F]) body temperature; often accompanied by dehydration | Move supine individual to cool, shaded environment, and elevate legs; loosen or remove clothing, and actively cool skin; administer oral fluids |
| Heatstroke | Profound CNS abnormalities (agitation, delirium, stupor, coma) with severe hyperthermia (>40° C [104° F]) | Ensure an open airway, and move to a cool environment. Immediately cool to <39° C (102.2° F) using ice packs, water bath, wetting with water and continuous fanning; IV fluid administration; reestablish normal CNS function; avoid antipyretics or drugs with liver toxicity |
CNS, Central nervous system; IV, intravenous.
Data from Bouchama A, Knochel JP: Heat stroke. N Engl J Med 346:1978, 2002; and Winkenwerder W, Sawka MN: Disorders due to heat and cold. In Goldman L, Ausiello DA, Arend W, et al, editors: Cecil textbook of medicine, ed 23, Philadelphia, 2007, Saunders, pp 763-767.
Heat exhaustion (also referred to as heat prostration or heat collapse) is a mild to moderate form of heat illness that is associated with moderate (>38.5° C [101.3° F]) to high (>40° C [104° F]) elevations in core temperature and an inability to sustain cardiac output.335 The signs and symptoms of heat exhaustion include fatigue, dizziness, headache, nausea, vomiting, malaise, hypotension, and tachycardia with potential for collapse. Heat exhaustion can occur with or without exercise in hot environments and may progress to a moderately severe condition without associated organ damage. Heat exhaustion is often observed in older adults as a result of medications (e.g., diuretics), inadequate water intake that leads to dehydration, or preexisting cardiovascular insufficiency that predisposes to collapse. Treatment should consist of placing the individual in a recumbent position in a cool environment to normalize blood pressure. Oral fluid ingestion with electrolytes is often adequate for recovery; intravenous (IV) fluid administration may be warranted in severely dehydrated individuals.
Heat injury is a moderate to severe condition characterized by tissue (e.g., skeletal muscle) or organ (e.g., gut, kidney, spleen, liver) damage and hyperthermia (core temperature usually, but not always >40° C [104° F]).335 Heat injury may progress to heatstroke if the patient is not rapidly cooled. Heatstroke is life threatening, with the patient presenting with profound CNS abnormalities, such as delirium, agitation, stupor, seizures, or coma and severe hyperthermia (core temperature typically, but not always >40° C [104° F]).335 Reliance on a specific core temperature value for clinical diagnosis of heatstroke is ill advised, because there is wide interindividual variability in documented cases. One of the main reasons for a lack of clinical treatments for heatstroke is the complicated nature of the syndrome, because there are different classifications based on etiology and pathophysiologic mechanisms of injury. Classic (also known as passive) heatstroke occurs at rest in vulnerable individuals, such as infants and older adults. Several intrinsic factors may predispose infants to heatstroke death. These include increased surface area–to–body mass ratio (accelerates heat gain), limited effective mechanisms of thermoregulation (e.g., suppressed behavioral adjustments), increased risk for dehydration (e.g., lack of water availability), and preexisting respiratory infections. Many older individuals have preexisting conditions, such as mental illness, prescription drug use (e.g., diuretics, anticholinergics), or infections that predispose to passive heatstroke (Box 10-1).6,56,312
BOX 10-1
Predisposing Risk Factors for Serious Heat Illness
Data from references 6, 58, 196, 318, and 340.
Exertional heatstroke (EHS) has a different etiology than classic heatstroke and affects young healthy populations that perform strenuous physical activity or work in temperate or hot weather. During exercise, approximately 80% of expended energy is released as heat that must be dissipated from the body to avoid hyperthermia. Military and athletic populations are composed of young, healthy individuals who are highly motivated to perform strenuous physical activity in hot weather, which increases the risk for EHS. A recent epidemiologic study identified a variety of factors that predispose to EHS, including gender (women greater than men), geographic region of origin (northern greater than southern states), preexisting illness, and race or ethnicity (whites greater than blacks).40 Unfortunately, exercise induces physiologic responses similar to those of heat stress, such that teasing apart the influence of these two factors in EHS is difficult.
Heatstroke Epidemiology and Risk Factors
Heat waves are defined as three or more consecutive days during which the environmental temperature exceeds 32.2° C (90° F).41 In the summer of 2003, Europe experienced 22,000 to 45,000 heat-related deaths during a 2-week period in which the average temperature was 3.5° C (6.3° F) above normal.189,273 The European continent has experienced an increase in minimum daily temperatures over the last 30 years, and this trend will likely increase if average global temperatures continue to rise. A 1.4° to 5.8° C (2.5° to 10.4° F) increase in minimum daily temperatures in Europe is predicted over the next century.131,337 Most prediction models suggest that heat waves in the future will be more severe and longer in duration. Predictions based on climate variability data from the 1995 Chicago and 2003 Europe heat waves suggest that by 2090, heat waves in these cities will be 25% to 31% more frequent and last 3 to 4 days longer.203 Another prediction model suggests a 253% increase in annual heatstroke deaths in the United Kingdom by 2050.61
The impact of climate change is not equally distributed across the globe because of regional variability in thermal tolerance that influences the incidence of heatstroke mortality. A study of 11 U.S. cities showed that threshold temperatures for heatstroke mortality are higher in warmer southern cities than in cooler northern cities.53 A comparison of temperature–mortality relationships in southern Finland, southeastern New England, and North Carolina indicated that lower temperature thresholds in cooler climates are coupled with steeper temperature–mortality relationships.62 Similarly, the upper safety limits of environmental temperatures in the Netherlands, London, and Taiwan are 16.5°, 19°, and 29° C (61.7°, 66.2°, 84.2° F), respectively.192 A case study of 15 Marine recruits who collapsed from heatstroke during training exercises in South Carolina showed that 73% previously resided in northern states and that 60% of cases occurred during the second week of training during the hottest summer months.232 From 1980 to 2002, the highest EHS incidence in military recruits was in nonacclimatized individuals from northern, cold-climate states who were enlisted for less than 12 months.40 During July, many regions of the world have a WBGT index that is greater than 29° C (84.2° F), and military training often occurs in environments with a WBGT index that is greater than 35° C (95° F). During peacetime exercises, approximately 25% of fatal military EHS cases occur during the hottest summer months in recruits who have been in training camp less than approximately 2 weeks.192 Individuals from northern states are expected to be less acclimatized to hot, humid summer conditions than are those from southern states. Heat acclimatization improves thermotolerance but requires several days to weeks of exposure to similar heat stress and exercise conditions to be fully effective. This likely accounts for hot days early in the summer showing a greater impact on heatstroke morbidity and mortality than those cases occurring later in the training process, after the protective effects of heat acclimatization have been realized.105
Humanity’s impact on the landscape in conjunction with increased production of greenhouse gases may be creating the largest climate change. Urban heat islands are created in cities when vegetation is removed and blacktop roads and concrete buildings are erected. Temperatures may be 30° to 40° C (54° to 72° F) higher on asphalt roads and roof tops compared with those of the surrounding air.85 Since 1978 urban sprawl has accounted for an increase in city temperatures in southeastern Asia of approximately 0.05° C (0.09° F) per decade.343 Across the entire land mass of the United States, the surface temperature has increased approximately 0.27° C (0.49° F) per century because of changes in the land cover arising from agricultural and urban development.139 Concrete and asphalt surfaces cool slowly during the nighttime when air temperature decreases, and this increase in urban heat storage magnifies the intensity of heat exposure experienced by individuals living in concrete urban structures.49,165
Several social factors predispose older adults to heatstroke mortality, including living alone, inability or unwillingness to leave one’s home, residing on the top floor of buildings (heat rises), and annual income of less than $10,000.221 Most heat wave early warning systems emphasize use of air-conditioning systems, but availability and use of the units are limited by socioeconomic status because they are expensive to operate.56,221 A working air conditioner was the strongest protective factor against mortality during the 1999 heat wave in Chicago; fan cooling did not afford protection.221 High mortality rates were recorded in Chicago despite extensive programs to educate high-risk populations, such as advising older adults to seek cool shelters or use air-conditioning systems. Approximately 10,000 elders died during the France heat wave of 2003 primarily because of lack of air-conditioning units in residences and hospitals.63,312 In 2005, Hurricane Katrina ravaged the U.S. Gulf Coast, and electrical failures caused high heatstroke mortality of older adults confined to residences, retirement homes, and hospitals because local temperatures exceeded 43° C (109.4° F). Increases in the average human lifespan, global climate change, and use of medications that compromise cardiovascular adjustments to heat stress will necessitate increased reliance on artificial cooling systems and educational programs to prevent heatstroke deaths in vulnerable populations, such as older adults.
The high death toll of older adults because of excessive heat per se may be small compared with that caused by aggravation of a preexisting illness. Heat stress refers to environmental and metabolic conditions that increase body temperature; heat strain refers to the physiologic consequences of heat stress. Heat strain imposes large cardiovascular demands on the body. Blood flow is shunted from the viscera to the skin surface to dissipate excess heat to the environment, making cardiovascular fitness a more important factor than age in determining an individual’s susceptibility to heatstroke. Austin and Berry10 examined 100 cases of heatstroke during three summer heat waves in St. Louis and found cardiovascular illness in 84% of patients. Levine181 found heatstroke deaths to be associated with arteriosclerotic heart disease (72%) and hypertension (12%). Cardiac deficiency impedes heat loss and compromises the ability to maintain cardiac output during prolonged heat exposure, leading to circulatory collapse and death. Older individuals may have impaired baroreceptor reflex modulation, lower sweat rates, longer time to onset of sweating, and diminished sympathetic nerve discharge, all of which increase the risk for heatstroke morbidity and mortality.130,144,291 Minson and colleagues210 demonstrated that during heat exposure, older men relied on a higher percentage of their cardiac chronotropic reserve compared with younger men.
Preexisting infection or inflammation may compromise an individual’s ability to appropriately respond to heat stress and can be a complicating factor, regardless of age. Fifty-seven percent of heatstroke patients more than 65 years old had evidence of infection upon clinical admission during a Chicago heat wave in 1995.56,157 In Singapore, a young EHS victim had been ill for 3 days before heatstroke collapse.43 It has been proposed that acute illness or inflammation can cause transient susceptibility to heatstroke in young, fit individuals who exercise in the heat. For example, idiosyncratic episodes of hyperthermia were associated with acute cellulitis and gastroenteritis in soldiers exercising in the heat.39,146 Four male Marine recruits presented with viral illness (mononucleosis, pneumonia) before collapse from exertional heat illness (EHI) during training exercises associated with “the Crucible” at Parris Island, South Carolina.289 Peripheral blood mononuclear cells (PBMCs) from these recruits expressed higher levels of IFN-inducible genes than did those from controls who participated in the training event but did not collapse.289 High plasma levels of IFN-α and IFN–γ mediate flulike symptoms during viral infection and are often associated with EHI/EHS.24,289 In rats, exposure to lipopolysaccharide (LPS), a cell wall component of gram-negative bacteria, exacerbated inflammation, coagulation, and multiorgan system dysfunction from heat exposure.182 Taken together, these findings suggest that a preexisting inflammatory state compromises an individual’s ability to respond to heat stress with appropriate thermoregulatory or immune responses to prevent collapse or multiorgan system failure and death.
The annual Muslim pilgrimage to Mecca (i.e., the Hajj) is associated with high heatstroke incidence each year and provides many lessons regarding etiologic factors that increase susceptibility. The Hajj takes place in the hot desert environment of Saudi Arabia during the extreme weather months of May to September, when temperatures range from 38° to 50° C (100.4° to 122° F).151 Hot weather combined with physical exertion (first day consists of a 3.5-km [2.2-mile] jog), heavy clothing that is traditional to the region (limits heat dissipation), and an older population (approximately 50 years is an advanced age for this region) predispose many individuals to heatstroke. Clothing has a significant impact on Muslim women because they are required to wear darker clothing that covers a larger surface area of the body than does clothing worn by men.114 Medical conditions, such as diabetes, cardiovascular abnormalities, or parasitic diseases, are common.151 Heatstroke is a major concern, but heat exhaustion with water or salt depletion is also prevalent. Overcrowding and congestion impose large demands on sanitation services, as exemplified in the 1980s, when approximately 2 million people participated in the Hajj. Advances in modern technologies, such as more rapid transport to the area, will likely introduce additional factors (e.g., lack of acclimatization, increased greenhouse gas production, increased congestion) to this already complex situation.
Protective clothing is a significant predisposing factor to EHS during athletic (heavy uniforms), military (chemical protective clothing), or occupational activities (e.g., pesticide application, firefighting, and race car driving). Protective clothing often consists of multiple layers that insulate anatomic sites from heat exchange, including the skin and head.251 The wearing of protective clothing during strenuous work can quickly result in a dangerous elevation in body temperature. Fifty-one cases of EHI were observed in military trainees in San Antonio, Texas, during participation in a 9.3-km (5.8-mile) march in full battle dress uniform and boots.285 Lack of acclimatization to athletic uniforms and high environmental temperatures results in the majority of cases of EHS in athletes occurring on the second or third day of exposure to hot weather before these individuals are acclimatized to the uniforms and environmental temperatures.97,260
Pathophysiology of Heatstroke
The pathophysiologic responses to heatstroke range from those conditions that are experienced immediately following collapse to long-term changes that persist for several weeks, months, or years following hospital treatment and release. Currently more is known about the immediate heatstroke responses because clinical records document symptoms during hospital treatment. However, clinical and experimental research has seen a shift within the past decade toward a focus on understanding the pathophysiologic responses that mediate long-term injury. It is now believed that the long-term pathophysiologic responses to heatstroke are caused by a systemic inflammatory response syndrome (SIRS) that ensues following heat-induced damage to the gut and other organs.24 Following damage to the epithelial membrane of the gut, endotoxin that is normally confined to the lumen of this organ is able to leak into the systemic circulation and elicit immune responses that cause tissue injury. The thermoregulatory, immune, coagulation, and tissue injury responses that ensue during the long-term progression of heatstroke closely resemble those observed during clinical sepsis and are likely mediated by similar cellular mechanisms. Clinical records have provided an extensive database of the immediate consequences of heatstroke, whereas the majority of knowledge regarding the pathophysiologic mechanisms of heat-induced SIRS has been obtained from experimental animal studies. Although there are several gaps in our knowledge of the specific factors that predispose to multiorgan system failure, this is an exciting area of research that is expected to progress at a rapid rate because of continued advancements in experimental and genetic technologies. Figure 10-6 provides an overview of the current understanding of the pathophysiologic responses that are thought to initiate and mediate heat-induced SIRS, which will be discussed in detail here.
Body Temperature Responses
At the time of heatstroke collapse, the severity of hyperthermia varies widely between individuals, with reported core temperature values ranging from approximately 41° C (105.8° F) to 47° C (116.6° F).* During a summer heat wave in St. Louis in the 1950s, the core temperature of 100 heatstroke patients ranged from 38.5° to 44° C (101.3° to 111.2° F), with 10% of mortalities occurring below 41.1° C (106° F).10 In some instances, individuals may tolerate hyperthermia without adverse side effects. During a competitive marathon race in California, a 26-year-old man maintained a rectal temperature of 41.9° C (107.4° F) for approximately 45 minutes without clinical signs of heat illness.197 However, there are several reports of athletic, military, and occupational workers with core temperatures below 41.9° C (107.4° F) who were hospitalized, experienced permanent CNS impairment, or died from EHS (Table 10-2).
Hypothermia and fever are core temperature responses that are often observed in patients and experimental animal models during heatstroke recovery. Hypothermia is not a universal heatstroke recovery response in humans but has been anecdotally observed following aggressive cooling treatment. Hypothermia manifests as a rapid undershoot of body temperature below 37° C (98.6° F) and is thought to represent a loss of thermoregulatory control following heat-induced damage to the POAH. However, evidence in support of this hypothesis is lacking because autopsy reports and experimental animal studies have failed to detect histologic damage to the POAH despite extensive damage in other organs.174,192 Because hypothermia is not observed in all heatstroke patients, it continues to be regarded as a pathologic recovery response. In experimental animals, hypothermia is a natural heatstroke recovery response that is associated with behavioral and autonomic thermoeffector responses that support a decrease in core temperature. Mud puppies are ectothermic species that rely on behavioral adjustments, such as the selection of different microclimates, to control body temperature. Mud puppies heat shocked to approximately 34° C (93.2° F) behaviorally selected a cooler microclimate and maintained a significantly lower body temperature than did nonheated controls during 3 days of recovery.130 This study did not determine the impact of hypothermia on survival, but the association of decreased body temperature with the selection of cool microclimates indicated that this was a regulated response to a decrease in the temperature set point. Small rodents, such as mice, rats, and guinea pigs, showed reductions greater than 1.0° C (1.8° F) in body temperature that were associated with improved survival following passive heatstroke. In mice, hypothermia was associated with an approximately 35% decrease in metabolic heat production and the behavioral selection of microclimates that precisely regulated the depth and duration of this response.174 Exposure of mice to warm ambient temperatures that prevented heat-induced hypothermia caused increased intestinal damage and mortality.173,333 Hypothermia likely provides protection against heat-induced tissue injury in a manner similar to that shown for protection against other extreme environmental insults based on the temperature coefficient (Q10) effect.
A common heatstroke recovery response observed in patients and animal models is recurrent fever during the days and weeks of recovery.9,10,173,192,204 In mice, fever was observed within a day after passive heatstroke collapse and associated with an approximately 20% increase in metabolic heat production and increased plasma levels of the proinflammatory cytokine interleukin (IL)-6.154,172,174 IL-6 is an important regulator of fever during infection and inflammation and may regulate fever during heatstroke recovery.172 In patients, fever is reestablished following clinical cooling.192 This is reminiscent of Liebermeister’s experimental observations of the recurrence of fever following experimental cooling of the POAH of rats.180,192 In experimental animal models, the inability to recover from hypothermia and develop fever is associated with increased mortality, suggesting that fever may be important for the resolution of infection.173 However, in a case report of human heatstroke, fever was associated with poor outcome. An amateur long-distance runner was hospitalized for 10 days after collapsing from EHS during a 9.7-km (6-mile) footrace.204 Moderate fever (>38° C [100.4° F]) was evident during the first 4 days of hospitalization, but on the tenth day the patient experienced convulsions and a rapid increase of body temperature to 41° C (105.8° F). Rapid cooling and aspirin were ineffective in reducing body temperature, and the patient died.204 The inability of aspirin to inhibit the rapid rise in body temperature suggests that this was not a true fever response, but rather a pathologic response to increased metabolic heat production induced by the convulsions. It is important to recognize that there is an optimal temperature range above which the protective effects of fever are no longer realized because of the toxic effect of high body temperature on cell function.155
Immune Responses
During heat stress, blood flow to the skin is increased to facilitate heat loss to the environment and reduce the rate of total body heat storage. Increased skin blood flow is accompanied by a fall in splanchnic (i.e., visceral organ) blood flow as a compensatory mechanism to sustain blood pressure. Endotoxin is normally confined to the gut lumen by tight junctions of the epithelial membrane, but these junctions can become “leaky” following prolonged reductions in blood flow that cause ischemic stress.108,164 There are several lines of evidence that support the hypothesis that endotoxin leakage from the gut lumen into the systemic circulation is the initiating stimulus for heat-induced SIRS. First, systemic injection of LPS into experimental animals induces symptoms similar to those observed in heatstroke, including hyperthermia, hypothermia, fever, hypotension, cytokine production, coagulation, and tissue injury.175,268 Second, increased portal or systemic endotoxin levels are observed in heatstroke patients and animal models. In primates, circulating endotoxin was detected at rectal temperatures above 41.5° C (106.7° F) with a precipitous increase at approximately 43.0° C (109.4° F).90 Splanchnic blood flow shows an initial decrease at 40° C (104° F); the liver, which is an important clearance organ for endotoxin, shows damage at body temperatures of approximately 42° to 43° C (107.6° to 109.4° F).* In a young athlete with a body temperature of 40.6° C (105.1° F) on the second day of football practice, high circulating levels of endotoxin were associated with hemorrhagic necrosis of the liver.97 In heatstroke patients, endotoxin was detected at approximately 42.1° C (107.8° F) and remained elevated despite cooling.26 Third, rats rendered endotoxin tolerant following the systemic injection of LPS are protected from heatstroke mortality.66,67 The protective effect of endotoxin tolerance is related to enhanced stimulation of the liver reticuloendothelial system (RES), which is composed of monocytes, macrophages, and Kupffer cells that are important for endotoxin clearance.66,67 RES stimulation reduced and RES blockade increased mortality of heat-stressed rats.67 Fourth, antibiotic therapy protects against heatstroke in several species. In dogs, antibiotics reduced gut flora levels and improved 18-hour survival rates by more than threefold when provided before heat exposure.37 In rabbits with heatstroke, hyperthermia and endotoxemia were reduced following oral antibiotics.36 Anti-LPS hyperimmune serum reversed the heatstroke mortality rates of primates and returned plasma LPS levels to baseline, but it was ineffective at the highest body temperature of 43.8° C (110.8° F), indicating that hyperthermia can cause irreversible organ damage and death.91
Heat-induced SIRS is initiated by the innate and adaptive immune systems, which interact to sense the presence of endotoxin and orchestrate an immunologic response. The innate immune system comprises monocytes, macrophages, and neutrophils that use pattern recognition receptors (PRRs) on their cell surfaces to recognize pattern-associated molecular patterns (PAMPs) on the cell surface of endotoxin and other invading pathogens.137 Toll-like receptors (TLRs) are a class of PRRs that have been widely studied in the immune response to infection.208,314 Ten mammalian TLRs have been identified, and the specific pathogenic ligands that activate these PRRs are known (Table 10-3).
| Toll-Like Receptor | Ligand | Cell/Tissue Types |
|---|---|---|
| TLR1 | Triacyl lipopeptide | Monocytes, macrophages, DCs, polymorphonuclear leukocytes, B and T cells, NK cells |
| TLR2 | Lipopolysaccharide Peptidoglycan Lipoteichoic acid Measles virus Human cytomegalovirus Hepatitis C virus Zymosan Necrotic cells |
Monocytes, granulocytes Brain, heart, lung, spleen |
| TLR3 | Viral double-stranded RNA | DCs, T cells, NK cells, monocytes, granulocytes Placenta, pancreas |
| TLR4 | Lipopolysaccharide Fibrinogen Heat shock proteins High mobility group box 1 |
B cells, DCs, monocytes, macrophages, granulocytes, T cells Spleen |
| TLR5 | Flagellated bacteria | Monocytes Ovary, prostate |
| TLR6 | Diacyl lipopeptide | B cells, monocytes Thymus, spleen, lung |
| TLR7 | Single-stranded RNA | Monocytes, B cells, DCs Lung, placenta, spleen, lymph node, tonsil |
| TLR8 | Single-stranded RNA | Monocytes Lung, placenta, spleen, lymph node, bone marrow, PBLs |
| TLR9 | CpG DNA | B cells, DCs Spleen, lymph node, bone marrow, PBLs |
| TLR10 | Unknown | B cells Spleen, lymph node, thymus, tonsil |
CpG, Deoxycytidylate-phosphate-deoxyguanylate; DC, dendritic cell; DNA, deoxyribonucleic acid; NK, natural killer; PBL, peripheral blood leukocyte; RNA, ribonucleic acid.
Data from Medvedev AE, Sabroe I, Hasday JD, et al: Tolerance to microbial TLR ligands: Molecular mechanisms and relevance to disease, J Endotoxin Res 12:133, 2006; and Tsujimoto H, Ono S, Efron PA, et al: Role of Toll-like receptors in the development of sepsis, Shock 29:315, 2008.
TLR4 is the principal receptor for LPS that stimulates gene transcription factors, such as NF-κB, to increase the synthesis of a variety of immune modulators in response to endotoxin. Endotoxin infection (i.e., sepsis) is associated with increased expression of TLR4 on circulating human PBMCs, as well as on mouse liver and spleen macrophages.307,308 In the 1960s, a spontaneous mutation in the TLR4 gene was discovered in C3H/HeJ mice, which has been an important animal model to determine the role of TLR4 in endotoxin responsiveness. C3H/HeJ mice show a diminished response to bacterial infection, but increased mortality from SIRS, because of an inability to respond appropriately to endotoxin and induce the full complement of immune responses.103 Inability to respond to antigens is known as anergy and is a proposed mechanism that predisposes to increased risk and mortality from bacterial infection.117 Given that TLR4 mutations exist in humans, this may be one (of several) genetic factors that predispose to mortality associated with heat-induced SIRS, although the association of mortality with TLR4 polymorphisms remains controversial.5,75 C3H/HeJ mice have not been tested for their resistance to heatstroke morbidity/mortality but are a useful experimental model to determine the roles of TLR4 and anergy in this syndrome.
Specificity of immune responses is provided by B and T cells of the adaptive immune system. These cells respond to antigens by secreting cytokines, which are intercellular immune signals that elicit proinflammatory (Th1 type) and antiinflammatory (Th2 type) actions during the progression of SIRS. The actions of cytokines depend on the nature of the danger signal, the target cells with which they interact, and the cytokine “milieu” in which they function. Th1 and Th2 cytokines function in a negative feedback pathway to regulate each other’s production and maintain a delicate balance of inflammatory reactions. Anergy is thought to be a consequence of inadequate Th2 cytokine production late in SIRS. For example, increased patient mortality from peritonitis is associated with the inability to mount a Th2 cytokine response.117
Alarmins are endogenous PAMPs that are released from stressed or injured tissues and initiate restoration of homeostasis following an infectious or inflammatory insult.17 High mobility group box 1 (HMGB1) is a highly conserved nuclear protein that functions as an alarmin following release from necrotic (but not apoptotic) cells.272 Necrosis is the premature death of cells in a tissue or organ in response to external factors, such as pathogens and toxins. Because necrosis is detrimental to the host, it is associated with an inflammatory response. Apoptosis refers to genetically programmed cell death that does not elicit an inflammatory response, because apoptosis is beneficial to the host. Release of HMGB1 from necrotic cells stimulates Th1 cytokine production late in the sepsis syndrome and is a purported mediator of lethality; this shift in the balance of cytokines from a Th2 to Th1 phenotype is a potential mechanism of sepsis lethality. In human PBMCs, HMGB1 interacts with TLR2 and TLR4 to enhance Th1 cytokine production in synergy with LPS.124 Elevated serum HMGB1 levels are observed 8 to 32 hours following LPS injection in mice. Anti-HMGB1 antibodies did not protect against LPS-induced mortality unless the antibodies were provided 12 and 36 hours after LPS exposure.324
[/not-level-membership-for-emergency-medicine-category]
