90 Pathophysiology and Classification of Shock States
 Pathophysiology of Shock
Pathophysiology of Shock
Circulatory shock represents a final common pathway of cardiovascular failure. The mortality rate remains high, particularly for patients in cardiogenic and septic shock, for whom the overall mortality rate approximates 50%.1,2 From a physiologic perspective, circulatory shock can be defined as a syndrome in which tissue perfusion is reduced such that blood flow is inadequate to meet cellular metabolic requirements. Clinical manifestations of shock are those of organ hypoperfusion: altered mental status; cool, clammy extremities; decreased blood pressure; decreased pulses; and oliguria.
Hemodynamic Assessment
In patients with circulatory shock, blood pressure should be monitored using intravascular measurements. Vasoconstriction due to compensatory mechanisms to maintain arterial pressure and the use of pharmacologic agents limits the accuracy of noninvasive measurements. This is particularly true in hypodynamic forms of circulatory failure.3
For most vital organs, autoregulatory and neuronal mechanisms maintain blood flow independent of blood pressure at a mean arterial pressure of 60 to 130 mm Hg.4 At either higher or lower levels of pressure, blood flow becomes linearly dependent on blood pressure. Diseases such as hypertension can shift this relationship and increase the critical level of arterial pressure required for organ perfusion. Similarly, impaired autoregulatory mechanisms present in a variety of pathologic states expand the range of pressure-dependent blood flow.
The level of arterial pressure is not a reliable indicator of circulatory performance and tissue perfusion.5,6 In states of hypodynamic circulatory shock, hypotension is a late marker of critical hypoperfusion. As cardiac output falls, blood pressure is initially maintained by increases in peripheral vascular resistance largely mediated by the sympathoadrenal system, and it is only after these mechanisms have been exhausted that hypotension develops. In this setting, tissue hypoperfusion may be present despite normal levels of blood pressure as blood flow is redirected toward more vital organs.7,8 Conversely, hypotension may exist without evidence of organ hypoperfusion. In some vasodilated states, increases in cardiac output maintain vital organ blood flow despite decreased levels of arterial pressure.
Pulmonary artery wedge pressure and central venous pressure are indirect measures of ventricular preload. These measurements correlate poorly with blood volume, end-diastolic volumes, and fluid responsiveness.9,10 Filling pressures are determined by ventricular compliance, venous return, and systolic function. Factors such as ventricular interactions, positive airway pressure, and intrinsic cardiac disease may decrease ventricular compliance and lead to an overestimation of ventricular preload.9 Echocardiographic techniques can provide a more accurate assessment of ventricular loading conditions, while dynamic indicators such as pulse pressure variation or stroke volume variation may provide greater insight as to fluid responsiveness.11,12
 Classification of Shock
Classification of Shock
Hinshaw and Cox proposed a classification of circulatory shock involving four subsets: hypovolemic, cardiogenic, distributive, and obstructive shock.13 This classification can be simplified into two categories with typical hemodynamic profiles (Table 90-1). The first category is hypodynamic shock, which includes the hypovolemic, cardiogenic, and obstructive shock subsets. The second category, hyperdynamic shock, includes distributive shock.
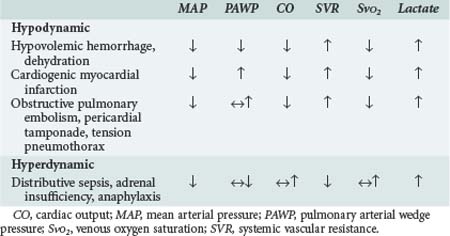
The central features of hypodynamic shock are a low cardiac index and a high-resistance vasoconstricted state. Increased oxygen extraction and lactic acidosis usually parallel the decrease in cardiac output. In cases of hypodynamic shock, the development of organ dysfunction is directly related to inadequate global blood flow. Common causes of hypovolemic shock are hemorrhage, dehydration, and massive capillary leak. Acute decreases in blood volume of 25% result in tachycardia and orthostatic hypotension, whereas decreases of 40% are associated with significant decreases in systolic blood pressure. Decreased filling pressures are the hallmark of hypovolemic shock, in contrast to cardiogenic shock where they are elevated. Acute myocardial infarction involving 40% or more of the ventricular mass is the most common cause of cardiogenic shock.14 Cardiomyopathies and severe valvular lesions are other important causes of cardiogenic shock. Finally, obstructive shock is most commonly due to pericardial tamponade, acute pulmonary embolism, and tension pneumothorax. Since filling pressures are usually increased in these settings (due to outflow obstruction, impaired ventricular filling, and decreased ventricular compliance), distinguishing between obstructive shock and cardiogenic shock can be difficult.
Hyperdynamic circulatory shock is characterized by a high cardiac output and a low-resistance vasodilated state. Filling pressures can be increased or normal depending on volume status and myocardial competence. Common causes of hyperdynamic shock include sepsis, anaphylaxis, some drug intoxications, spinal shock, and adrenal insufficiency. The underlying hemodynamic defect is maldistribution of blood flow and/or blood volume such that effective nutrient blood flow is compromised. In contrast to hypodynamic shock, oxygen extraction may be normal or decreased despite evidence of hypoperfusion.15 Direct mediator-related effects coupled with tissue hypoperfusion produce cellular injury and organ dysfunction in patients with septic shock.
Considerable overlap may exist between these different syndromes. Early in septic and anaphylactic shock, prior to fluid infusion, a significant hypovolemic component usually exists.16 Hypovolemia may be present in a small group of patients presenting with shock due to acute myocardial infarction.17 In the presence of severe sepsis-related myocardial depression, patients with septic shock can develop a hypodynamic profile. Similarly, patients in cardiogenic shock after myocardial infarction and cardiac surgery may demonstrate significant vasodilation due to the activation of mediator cascades while on cardiopulmonary bypass.1,18
Progression of Shock
Critical reductions in tissue perfusion elicit a complex set of reflexes that are directed at maintaining cardiac output and arterial pressure.4 Activation of the sympathetic system increases heart rate and contractility. The release of catecholamines, angiotensin, vasopressin, and endothelins increases arteriolar and venous tone, thereby increasing arterial blood pressure and shifting blood volume from the capacitance vessels to the central circulation. In addition, blood flow is redirected from skeletal muscle, subcutaneous tissue, and the splanchnic circulation to the heart and brain. Vasopressin and activation of the renin-angiotensin system serve to enhance water and sodium retention, thereby protecting intravascular blood volume.
Progression of the shock state is marked by further declines in blood pressure that compromise coronary perfusion and cardiac performance. Increases in peripheral vascular resistance impede left ventricular ejection by increasing left ventricular afterload. Terminal phases of shock are marked by vasomotor dysfunction characterized by loss of arteriolar tone with paradoxical increased venular resistance. The resulting increase in capillary hydrostatic pressure coupled with increased microvascular permeability leads to a loss of intravascular volume and worsening of the shock state. Leukostasis and changes in erythrocyte rheology further impair microvascular blood flow. In animal models of hemorrhagic shock, a state of irreversible shock evolves from which the animals cannot be successfully resuscitated.19
This pathophysiology is altered in patients with hyperdynamic forms of circulatory failure such as septic shock, where inflammatory mediators play a prominent role.20 These patients are characterized by arterial and venous dilation and increased cardiac output. The influence of vasodilatory substances such as nitric oxide predominates over the effects of endogenous and exogenous vasopressor substances. In some forms of vasodilatory shock, inappropriately low levels of vasopressin and cortisol may contribute to vasodilation and refractoriness to catecholamines.21,22 Decreases in capillary cross-sectional area due to the interactions of activated leukocytes, platelets, endothelial cells, and the clotting cascade limit effective nutrient blood flow despite the increase in cardiac output.23,24 Progressive hypotension refractory to fluid infusion and vasopressors results in worsening tissue hypoperfusion, acidosis, and organ failure. A hypodynamic circulation develops as a terminal event.
Oxidative Metabolism in Shock
The primary metabolic defect in circulatory shock is impaired oxidative metabolism with resulting cellular and organ failure. This impairment is most commonly due to decreases in tissue oxygen supply caused by either global decreases in blood flow or maldistribution of blood flow on a regional or microcirculatory level. Systemic oxygen consumption may initially be increased yet inadequate to meet tissue metabolic requirements; however, the terminal phases of all forms of shock are characterized by decreases in oxygen consumption. In experimental studies, the risk of mortality is directly related to the total amount of accumulated oxygen debt.25
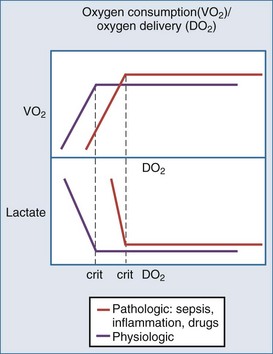
Oxygen delivery is determined by cardiac output, hemoglobin concentration, and the arterial oxygen saturation. Under normal circumstances, oxygen consumption is independent of oxygen delivery and cardiac output (Figure 90-1). Increases in cellular oxygen extraction from a normal level of 25% to a maximum of level of 80% maintain oxygen consumption as blood flow is reduced. When oxygen extraction is maximized, a critical level of oxygen delivery (DO2crit) is reached below which oxygen consumption decreases and anaerobic metabolism ensues. Alterations in vasomotor reflexes due to sepsis or drugs limit maximal oxygen extraction, resulting in critical tissue hypoxia and anaerobic metabolism at higher levels of oxygen delivery.26,27
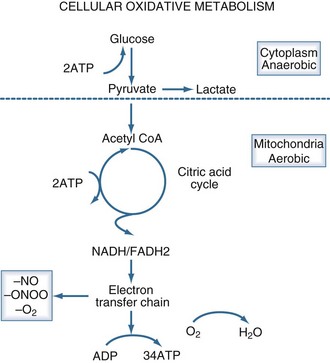
Aerobic adenosine triphosphate (ATP) generation is dependent on glycolysis occurring in the cytoplasm and oxidative phosphorylation occurring in the mitochondria (Figure 90-2). Under anaerobic conditions, ATP generation is limited to the two ATP generated in the cytoplasm, as compared to the 38 ATP generated aerobically. The decreased entry of pyruvate into the citric acid cycle results in the accumulation of lactic acid and the generation of additional hydrogen ions from the hydrolysis of ATP. Accordingly, the presence of lactic acidosis serves as an indicator of critical cellular deficits in high-energy phosphate metabolism. The normal level of lactate is 0.4 mEq/L to 1.2 mEq/L; levels greater than 2 mEq/L are associated with an increased mortality rate.28
Oxidative metabolism may also be impaired by mechanisms independent of tissue hypoperfusion. A number of inflammatory mediators including nitric oxide, endotoxin, oxygen radicals, calcium, and tumor necrosis factor impair mitochondrial function. Mitochondrial abnormalities have been observed in animal models of septic shock and in cases of reperfusion injury.29 Serum from patients with septic shock inhibits mitochondrial respiration and decreases cellular ATP concentration in vitro.30 A potential pathway of direct mitochondrial impairment involves nitric oxide and its metabolite, peroxynitrite. Both of these substances can directly impair mitochondrial electron chain complexes.31
Accumulation of tissue carbon dioxide (CO2) parallels the development of oxygen debt in circulatory shock.32 Clinically, increases in tissue CO2 levels are manifested by venous hypercapnia and decreases in venous pH. The result is a widening of the arterial-venous CO2 gradient proportional to the degree of circulatory failure. The normal gradient is less than 5 mm Hg, and it can increase to 40 mm Hg during cardiac arrest.33 Decreased clearance of CO2 generated by oxidative processes is responsible for the initial increase in tissue CO2 levels. With the onset of anaerobic metabolism, tissue CO2 excess is largely generated from titration of anaerobically derived acids by bicarbonate. The increase in tissue CO2 levels may have physiologic significance and has been associated with impaired myocardial performance in vitro.
Monitoring Perfusion Failure
Controversy exists over the optimal manner in which to monitor tissue perfusion in patients with circulatory shock. Commonly utilized parameters such as heart rate, arterial pressure, and cardiac output correlate poorly with survival in critically ill patients.5,6 This is particularly true in patients with septic shock and traumatic injury, in whom underlying deficits in tissue perfusion may exist despite initial resuscitative efforts.5,34 These observations have led to the use of indices of tissue oxygen metabolism as markers of tissue perfusion and the adequacy of resuscitative efforts.
Mixed venous oxygen saturation (SvO2) measured on blood taken from the pulmonary artery is used as an index of tissue oxygenation. Venous blood is in equilibrium with the tissues. Mixed venous blood, representing a weighted mean of all the venous effluents, reflects overall tissue oxygenation. Since increased oxygen extraction is the primary compensatory mechanism to maintain oxygen consumption, decreases in SvO2 are an early marker of compromised tissue perfusion. In cardiogenic shock, SvO2 tracks cardiac function and systemic perfusion.35 The same is not true in septic shock and other settings where the relationship between venous blood and tissue oxygenation is altered by maldistribution of blood flow.15 In these circumstances, the ability of the tissues to extract oxygen is limited by decreases in effective nutrient flow such that SvO2 may be increased or normal despite the presence of tissue hypoxia and anaerobic metabolism. Accordingly, while mixed venous desaturation is indicative of tissue hypoxia, normal levels do not preclude tissue hypoperfusion.
Central venous oxygen saturation (ScvO2) measured on samples taken from the superior vena cava and right atrium serves as an alternative to SvO2.36,37 In critically ill patients, the ScvO2 is generally 5% higher than SvO2; however, their correlation is inconsistent, depending in part on the location of the tip of the central venous catheter. In one study, patients with septic shock demonstrated improved survival when therapy was titrated to ScvO2 ≥ 70%.36
Lactate concentration is a useful marker of critical hypoperfusion. Increases in lactate levels indicate the presence of anaerobic metabolism and tissue energy deficits.28 Although the initial blood level of lactate has prognostic significance, the inability to clear lactate over time is more discriminating.38,39 In patients with septic shock, factors other than hypoperfusion may contribute to lactate accumulation. These factors include increased muscle ATPase activity, increased hepatic flux of alanine from skeletal muscle, decreased pyruvate dehydrogenase activity, decreased hepatic clearance of lactate, and dysfunctional mitochondrial respiration. Despite these concerns, increases in lactate concentration are associated with decreases in the intracellular redox potential in patients with septic shock, suggesting that it is a useful marker of cellular energy metabolism.40 When titration of therapy to ScvO2 above 70% was compared to achieving a lactate clearance of 10% over 6 hours in patients with septic shock, the outcome was similar.41
Oxygen consumption and oxygen delivery are global markers of systemic oxygen metabolism. Oxygen consumption, a measure of overall metabolic requirements, is calculated from cardiac index, hemoglobin, and arterial and venous oxygen saturation. It can also be measured directly from expired gases. Oxygen delivery is calculated from cardiac output, hemoglobin, and arterial saturation and is a measure of the total amount of oxygen being delivered to the tissues. Although increased values of oxygen consumption and oxygen delivery have been observed in survivors compared with non-survivors, considerable overlap exists between the two groups. Efforts to titrate therapy to values associated with survival—“optimal goals”—have produced mixed results.42,43 These differences may in part reflect the varying metabolic requirements of individual patients.
The decrease in CO2 clearance in circulatory shock is the basis for end-tidal CO2 and tissue CO2 measurements. End-tidal CO2 measurements are useful in monitoring perfusion during cardiopulmonary resuscitation.44 Cardiac arrest results in marked decreases in pulmonary blood flow and accompanying decreases in CO2 excretion. Consequently, end-tidal CO2 values move toward zero during arrest and increase with successful resuscitation.
There have been multiple attempts to use measures of local or regional perfusion as indices of overall systemic perfusion. Toe temperature, subcutaneous oxygen tensions, transcutaneous oxygen tension, and laser Doppler are some examples of regional measures previously studied. Gastric tonometry, and more recently sublingual tonometry, have been used to assess those vascular beds for CO2 excess as a marker of systemic hypoperfusion. Current attention has focused on two measures of microvascular blood flow. One approach is the use of near-infrared spectroscopy (NIRS) to assess the level of oxygenated hemoglobin in thenar skeletal muscle. Both the actual value and the response of tissue hemoglobin saturation to reactive hyperemia have been reported to predict survival.45,46 The other techniques, orthogonal polarization spectral (OPS) imaging and sidestream dark-field (SDF) imaging, have been used to directly visualize microcirculatory flow. Decreases in capillary blood flow have been observed in patients with septic shock and cardiogenic shock which correlated with survival.24,47 Evidence of persistent hypoperfusion using these measurements has been reported in patients with septic shock, despite improvement in systemic indices of perfusion.48 Whether titration of therapy to these measures of local perfusion will impact on outcome remains to be determined.
Organ Failure
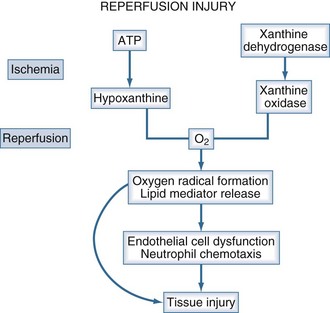
The primary causes of organ dysfunction in circulatory shock are ischemic injury, mediator-related organ dysfunction, and reperfusion injury. Ischemic injury occurs when anaerobic metabolism ensues and high-energy phosphate production falls below the level required for cellular function and membrane integrity. It is the major factor contributing to organ failure in patients with cardiogenic and hypovolemic shock. The direct effect of inflammatory mediators, coupled with an ischemic injury, plays a major role in organ dysfunction in septic shock. Tumor necrosis factor, nitric oxide, and superoxide radicals are examples of mediators directly affecting cellular and organ function. Reperfusion injury occurs upon restoration of tissue perfusion following an absence of blood flow (Figure 90-3). Activated neutrophils, oxygen radicals, endothelial cell dysfunction and apoptosis play important roles in this process.49 Reperfusion injury may be important in hemorrhagic and traumatic shock; its role in cardiogenic shock and septic shock is less clear.
In cases of acute myocardial infarction shock, cardiac dysfunction is related to ischemia and myocardial necrosis. Reperfusion injury may also play a role in patients following acute coronary revascularization. Cardiac dysfunction is frequently observed in patients in shock. Myocardial depressant substances cause myocardial depression in patients in septic shock and may also play a role in cases of hemorrhagic shock.50 Down-regulation of β-receptor density and affinity contribute to myocardial failure in sepsis and other syndromes. Increases in pulmonary vascular resistance are the cause of acute right ventricular failure in patients with pulmonary embolism and may also be important in septic shock, particularly when it is complicated by the acute respiratory distress syndrome.
Minute ventilation and respiratory rate increase in patients with shock. Overt respiratory failure may result from pulmonary edema or acute lung injury and leads to additional increases in the work of breathing. Decreased respiratory muscle perfusion coupled with hypoxia contributes to respiratory muscle failure. In patients with septic shock, inflammatory mediators may also directly impair respiratory muscle activity.51
Renal dysfunction in shock is related to ischemic and reperfusion injury. Initially, as cardiac output decreases, glomerular filtration is maintained by increases in efferent arteriolar tone. Release of atrial natriuretic peptide due to increased atrial pressures may help protect renal blood flow in patients with cardiogenic shock. However, as shock progresses, the increases in afferent arteriolar tone result in renal ischemia and acute tubular necrosis. Activation of neutrophils, dendritic cells, and lymphocytes during reperfusion all play an important role in renal injury associated with shock.52 In septic shock, alterations in intrarenal blood flow may also impair effective glomerular filtration.
A characteristic pattern that involves centrilobar necrosis and marked transaminase elevation is observed in patients with ischemic hepatic injury associated with hypodynamic circulatory states.53 Activation of Kupffer cells and the release of inflammatory mediators exacerbate ischemic injury in patients in septic shock and traumatic shock. In septic shock, canalicular cell function is impaired, resulting in intrahepatic cholestasis. Hepatic metabolic failure and impaired amino acid clearance are also features of septic shock.
Splanchnic mucosal blood flow is compromised early in shock. Intestinal injury may result from hypoperfusion and/or the release of oxygen radicals and activation of neutrophils during reperfusion. Loss of the integrity of the intestinal barrier can lead to translocation of bacteria and toxins, which in turn contributes to organ failure.54 Splanchnic hypoperfusion related either to shock or to the use of vasopressors also contributes to the development of stress ulceration, acalculous cholecystitis, intestinal necrosis, and pancreatitis. Pancreatic hypoperfusion may also predispose to the release of myocardial depressant factors.
Disorientation and delirium are common in patients in shock. Hypotension, metabolic abnormalities, and hypoxia all contribute to neurologic dysfunction. Alterations in cerebral vascular reactivity and direct toxic effects of inflammatory mediators may also play a role in cerebral injury.55 Severe hypotension, mean arterial pressure well below 60 mm Hg, can result in ischemic injury of the arterial border zones in the cortex and spinal cord.
Microvascular blood flow is impaired in all forms of circulatory failure.24,47,56 The microcirculation is characterized by heterogeneous blood flow and decreased capillary perfusion. Rheologic abnormalities of neutrophils and erythrocytes impede microvascular blood flow. Increased expression of the neutrophil integrins, platelet P-selectin, and the endothelial cell adhesion molecules result in cellular aggregation and microvascular obstruction. Platelet-fibrin interactions mediated through platelet expression of glycoprotein IIB/IIIA receptors accentuate this process.23 Decreased endothelial cell nitric oxide synthetase activity impairs normal vasodilatory reflexes and decreases the microvascular response to hypoxia. Increased microvascular permeability and tissue edema may also impede the diffusion of oxygen from the capillaries into the cells.
Shock is associated with down-regulation of immunologic function. Immunosuppressive substances including interleukin (IL)-10, prostaglandin E2, and adenosine are released and decrease cellular and humoral immunity. Dendritic cell– and monocyte-mediated antigen processing is impaired, as is neutrophil function. Apoptosis of lymphocytes, dendritic cells, and monocytes is increased. An immunologic profile of decreased monocyte HLA-DR expression and impaired monocyte responsiveness to inflammatory stimuli has been associated with an increased risk of secondary infection and mortality.57,58
 Clinical Aspects of Shock
Clinical Aspects of Shock
Initial Approach to Circulatory Shock
The approach to patients with circulatory shock involves a rapid assessment of the underlying disease process and restoration of cardiopulmonary stability. The patient should be assessed for the cause of the shock syndrome and for evidence of end-organ hypoperfusion. A complete blood cell count, coagulation studies, blood gases, and electrolytes measurement should be performed. Blood lactate measurement is helpful to confirm the severity of perfusion failure. An electrocardiogram and chest radiograph should also be obtained. The need for additional studies such as cultures, cardiac enzymes, and other tests depends on the suspected cause of the shock state. Efforts to achieve cardiopulmonary stability should occur simultaneously. The VIP approach can be used to prioritize these efforts by focusing on ventilation, infusion, and pump activity.59 A systematic approach that incorporates physiologic endpoints for resuscitation with monitoring indices of systemic perfusion and an algorithm for therapeutic interventions based on the pathophysiology of the underlying shock state results in the best outcomes.60
Critical hypovolemia is present in the majority of patients presenting with circulatory shock in the medical-surgical setting and a significant portion of patients presenting with shock and acute myocardial infarction. Fluids should be infused in boluses and titrated to specific endpoints of heart rate, blood pressure, urine output, central venous or mixed venous saturation, and clearance of blood lactate. Determining the adequacy of fluid infusion based on intracavitary measurements such as central venous pressure may be difficult and should be guided by dynamic measurements when possible.10–12 Attention should be given to the hemoglobin level, which will decrease with significant asanguineous fluid resuscitation. Although many patients tolerate a hemoglobin level of 7 g/dL to 10 g/dL, higher levels may be required in patients with cardiac dysfunction.
The treatment of lactic acidosis with alkali solutions is controversial. Sodium bicarbonate solutions increase serum osmolality and potentially worsen intracellular acidosis as bicarbonate is titrated to CO2 and water. Prospective randomized trials have not demonstrated any benefit in either oxygen metabolism or circulatory function after alkali infusion for severe lactic acidosis.61
Definitive therapy depends on the cause of the shock state and may require additional diagnostic and therapeutic interventions. These efforts should be pursued in a timely manner. Endoscopic or surgical interventions may be required for patients in hemorrhagic and traumatic shock. Circulatory assist devices coupled with prompt efforts at revascularization enhance outcome in patients with cardiogenic shock.1 Antibiotics and drainage procedures are required for septic shock. Steroids and activated protein C may also benefit patients with septic shock.22,62 Acute pulmonary embolism and shock can be treated with thrombolysis, catheter embolectomy, or, in more extreme circumstances, surgical embolectomy.
Newer Therapies
There is continuing interest in modulating the activity of inflammatory mediators in septic shock, hemorrhagic shock, and even cardiogenic shock.1 Similarly there is an ongoing focus on attenuating reperfusion injury through interventions directed at neutrophils, reactive oxygen species, and reactive nitrogen species in multiple settings.49 Efforts to avoid the adverse sequelae of resuscitation including the use of excessive fluids and the use of fluids that may exacerbate the inflammatory response are being examined. Newer fluids are being developed which, in addition to their volume-expanding capacity, have antiinflammatory activity. The role of the cholinergic antiinflammatory pathway and its manipulation is being elucidated. Hydrogen sulfide is being studied for its antiinflammatory and metabolic effects. Mitochondrial targeted therapies are being investigated in an effort to enhance mitochondrial function and recovery in shock. The role of apoptosis in the development of immune dysfunction and organ failure is being examined, with possible interventions directed at altering this process. Finally, the genetic underpinning of the immune response and its role in circulatory shock is another area of active interest.63 Progress in this important area will ultimately allow for the development of more focused interventions that have the greatest likelihood of benefiting individual patients.
Key Points
Brealey D, Brand M, Hargreaves I, et al. Association between mitochondrial dysfunction and severity and outcome in septic shock. Lancet. 2002;360:219-223.
Hinshaw LB, Cox BG. The fundamental mechanisms of shock. New York: Plenum Press; 1972.
Rivers E, Nguyen B, Havstad S, et al. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med. 2001;345:1368-1377.
Weil MH, Afifi AA. Experimental and clinical studies in lactate and pyruvate as indicators of the severity of acute circulatory failure (shock). Circulation. 1970;41:989-1000.
Weil MH, Rackow EC, Trevino R, et al. Differences in acid-base state between venous and arterial blood during cardiopulmonary resuscitation. N Engl J Med. 1986;315:153-156.
1 Reynolds H, Hochman J. Cardiogenic shock: Current concepts and improving outcomes. Circ. 2008;117:686-697.
2 Kumar A, Ellis P, Arabi Y, et al. Initiation of inappropriate antimicrobial therapy results in a fivefold reduction of survival in human septic shock. Chest. 2009;136:1237-1248.
3 Cohn JN. Blood pressure measurements in shock: Mechanism of inaccuracy in auscultatory and palpatory methods. JAMA. 1967;199:118-122.
4 Guyton A, Hall J. Textbook of Medical Physiology. Philadelphia, Pa: Elsevier Saunders; 2006.
5 Shoemaker W, Czer L. Evaluation of the biologic importance of various hemodynamic and oxygen transport variables. Crit Care Med. 1979;7:424-429.
6 Cady L, Weil MH, Afifi A, et al. Quantification of critical illness with special reference to blood lactate. Crit Care Med. 1973;1:75-80.
7 Edouard A, Degremont A, Duranteau J, et al. Heterogeneous regional vascular responses to simulated transient hypovolemia in man. Intensive Care Med. 1994;20:414-420.
8 Higgins CB, Vatner SF, Franklin D, et al. Pattern of differential vasoconstriction in response to acute and chronic low-output states in the conscious dog. Cardiovasc Res. 1974;8:92-98.
9 Raper R, Sibbald W. Misled by the wedge? Chest. 1986;89:427-434.
10 Marik P, Baram M, Vahid B. Does central venous pressure predict fluid responsiveness? Chest. 2008;134:172-178.
11 Michard F, Teboul JL. Predicting fluid responsiveness in ICU patients. Chest. 2002;121:2000-2008.
12 Monnet X, Rienzo M, Osman D, et al. Passive leg raising predicts fluid responsiveness in the critically ill. Crit Care Med. 2006;34:1402-1407.
13 Hinshaw LB, Cox BG. The Fundamental Mechanisms of Shock. New York: Plenum Press; 1972.
14 Page DL, Caulfield JB, Kastor JA, et al. Myocardial changes associated with cardiogenic shock. N Engl J Med. 1971;285:133-137.
15 Astiz ME, Rackow EC, Kaufman BS, et al. Relationship of oxygen delivery and mixed venous oxygenation to lactic acidosis in patients with sepsis and acute myocardial infarction. Crit Care Med. 1988;16:655-658.
16 Rackow EC, Kaufman BS, Falk JL, et al. Hemodynamic response to fluid repletion in patients with septic shock: Evidence for early depression of cardiac performance. Circ Shock. 1987;22:11-22.
17 Forrester JS, Diamond C, Swan JH. Correlative classification of clinical and hemodynamic function after acute myocardial infarction. Am J Cardiol. 1977;31:137-145.
18 Kristof A, Magder S. Low systemic vascular resistance state in patients undergoing cardiopulmonary bypass. Crit Care Med. 1999;27:1121-1127.
19 Wiggers CJ. The present status of the shock problem. Physiol Rev. 1942;22:74-123.
20 Cinel I, Opal S. Molecular biology of inflammation and sepsis: A primer. Crit Care Med. 2009;37:291-304.
21 Landry D, Oliver J. The pathogenesis of the vasodilatory shock. N Engl J Med. 2001;345:588-595.
22 Annane D, Bellissant E, Bollaert PE, et al. Corticosteroids in the treatment of severe sepsis and septic shock. JAMA. 2009;301:2362-2375.
23 Kirschenbaum L, McKevitt D, Rullan M, et al. The importance of platelets and fibrinogen in neutrophil-endothelial cell interactions in septic shock. Crit Care Med. 2004;32:1904-1909.
24 De Backer D, Creteur J, Preiser JC, et al. Microvascular blood flow is altered in sepsis. Am J Crit Care Med. 2002;166:98-104.
25 Crowell JW, Smith EE. Oxygen deficit and irreversible hemorrhagic shock. Am J Physiol. 1964;206:313-316.
26 Samsel R, Nelson D, Sandes W, et al. Effect of endotoxin on systemic and skeletal muscle O2 extraction. J Appl Physiol. 1988;65:1377-1382.
27 Van Der Linen P, Gilbart E, Engelman E, et al. Effects of anesthetic agents on systemic critical O2 delivery. J Appl Physiol. 1991;71:83-93.
28 Weil MH, Afifi AA. Experimental and clinical studies in lactate and pyruvate as indicators of the severity of acute circulatory failure (shock). Circulation. 1970;41:989-1000.
29 Brealey D, Brand M, Hargreaves I, et al. Association between mitochondrial dysfunction and severity and outcome in septic shock. Lancet. 2002;360:219-223.
30 Boulos M, Astiz M, Barua R, Osman M. Impaired mitochondrial function induced by septic serum from septic shock patients is attenuated by inhibition of nitric oxide synthase and poly (ADP-ribose) synthase. Crit Care Med. 2003;31:151-154.
31 Pacher P, Szabo C. The role of peroxynitrite-poly (ADP-ribose) polymerase pathway in human disease. Am J Pathol. 2008;173:2-13.
32 Sato Y, Weil MH, Tang W. Tissue hypercarbic acidosis as a marker of acute circulatory failure. Chest. 1998;114:263-274.
33 Tang W, Weil M, Gazmuri R, et al. Reversible impairment of myocardial contractility due to hypercarbic acidosis in the isolated perfused rat heart. Crit Care Med. 1991;19:218-224.
34 Wu C, Shoemaker W, Appel P, et al. Unreliability of blood pressure and heart rate to evaluate cardiac output in emergency resuscitation and critical illness. Crit Care Med. 1993;21:218-223.
35 Creamer J, Edwards J, Nightingale P. Hemodynamic and oxygen transport variables in cardiogenic shock secondary to acute myocardial infarction, and response to treatment. Am J Cardiol. 1990;65:1297-1300.
36 Rivers E, Nguyen B, Havstad S, et al. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med. 2001;345:1368-1377.
37 Scalea T, Hartnett R, Duncan A, et al. Central venous oxygen saturation: A useful tool in trauma patients. J Trauma. 1990;30:1539-1543.
38 Abrahamson D, Scalea T, Hitchcock R, et al. Lactate clearance and survival following trauma. J Trauma. 1993;35:584-589.
39 Bakker J, Coffernils M, Leon M, et al. Blood lactate levels are superior to oxygen derived variables in predicting outcome in human septic shock. Chest. 1991;99:956-962.
40 Levy B, Sadoune LO, Gelot AM, et al. Evolution of lactate pyruvate and arterial ketone body ratios in the early course of catecholamine-treated septic shock. Crit Care Med. 2000;28:114-119.
41 Jones A, Shapiro N, Trzeciak S, et al. Lactate clearance versus central; venous saturation as endpoints of early sepsis therapy: A randomized trial. Crit Care Med. 2009;379(suppl 12):A24.
42 Shoemaker W, Appel P, Kram H, et al. Prospective trial of supranormal values of survivors as therapeutic goals in high risk surgical patients. Chest. 1988;94:1176-1186.
43 Heyland D, Cook D, King D, et al. Maximizing oxygen delivery in critically ill patients: A methodologic appraisal of the evidence. Crit Care Med. 1996;24:517-524.
44 Weil MH, Rackow EC, Trevino R, et al. Differences in acid-base state between venous and arterial blood during cardiopulmonary resuscitation. N Engl J Med. 1986;315:153-156.
45 Cohn S, Nathens A, Moore F, et al. Tissue oxygen saturation predicts the development of organ dysfunction during traumatic shock resuscitation. J Trauma. 2007;82:44-55.
46 Creuter J, Carollo T, Soldati G, et al. The prognostic value of StO2 in septic patients. Intensive Care Med. 2007;33:1549-1556.
47 De Backer D, Creteur J, Dubois M, et al. Microvascular alterations in patients with acute severe heart failure and cardiogenic shock. Am Heart J. 2004;147:91-99.
48 Sakr Y, Dubois MJ, DeBacker D, et al. Persistent microcirculatory alterations are associated with organ failure and death in patients with septic shock. Crit Care Med. 2004;32:1825-1831.
49 Rushing G, Britt L. Reperfusion injury after hemorrhage. Ann Surg. 2008;247:929-937.
50 Rudiger A, Singer M. Mechanisms of sepsis-induced cardiac dysfunction. Crit Care Med. 2007;35:1599-1608.
51 Lanone S, Taille C, Boczkowski J, et al. Diaphragmatic fatigue during sepsis and septic shock. Intensive Care Med. 2005;31:1611-1617.
52 Kinsey G, Okusa M. Inflammation in acute kidney injury. Nephron Exp Nephrol. 2008;109:102-107.
53 Birgens H, Henrickson J, Matzen P, et al. The shock liver: Clinical and biochemical finding in patients with centrilobular liver necrosis following cardiogenic shock. Acta Med Scand. 1978;204:417-423.
54 Dietch EA, Xu D, Kaise X. Role of the gut in the development of injury and shock induced SIRS and MODS: the gut-lymph hypothesis, a review. Front Biosci. 2006;11:520-528.
55 Sharshar T, Gray F, Porn F, et al. Multifocal necrotizing leukoencephalopathy in septic shock. Crit Care Med. 2002;30:2371-2375.
56 Klijn E, Uil D, Bakker J, Ince C. The heterogeneity of the microcirculation in critical illness. Clin Chest Med. 2008;29:643-654.
57 Munoz C, Carlet J, Fitting C, et al. Dysregulation of in vitro cytokine production by monocytes during sepsis. J Clin Invest. 1991;88:1747-1754.
58 Tschoeke SK, Ertel W. Immunoparalysis after multiple trauma. Injury. 2007;38:1346-1357.
59 Weil MH, Shubin H. “The VIP” approach to the bedside management of shock. JAMA. 1969;207:337-341.
60 Jones A, Brown M, Trzeciak S, et al. The effect of a quantitative resuscitation strategy on mortality in patients with sepsis: a meta analysis. Crit Care Med. 2008;36:2734-2739.
61 Forsythe S, Schmidt G. Sodium bicarbonate for the treatment of lactic acidosis. Chest. 2000;117:260-267.
62 Bernard G, Vincent JL, Laterre P, et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001;344:699-709.
63 Namath A, Paterson AJ. Genetic polymorphisms in sepsis. Crit Care Clin. 2009;25:835-856.
