Chapter 36 Pathophysiologic Basis of Hibernating Myocardium
INTRODUCTION
Nuclear cardiology is increasingly used to identify viable chronically dysfunctional myocardium, and imaging is frequently the most significant factor in the decision to pursue coronary revascularization in patients with left ventricular dysfunction and ischemic cardiomyopathy. As summarized in Table 36-1, multiple pathophysiologies can account for viable dysfunctional myocardium in chronic coronary artery disease, and not all of these are the result of chronic repetitive ischemia distal to a coronary stenosis. Reversible dyssynergy has become lumped into a general category termed hibernating myocardium, but there appear to be important aspects of cellular remodeling, adaptation, and maladaptation that differ among entities responsible for viable dysfunctional myocardium. These can influence functional recovery, heart failure symptoms, and cardiovascular mortality, including sudden cardiac death. This chapter reviews our current understanding of the functional and metabolic consequences of ischemia. It also summarizes intrinsic mechanisms responsible for viable dysfunctional myocardium with an emphasis on how the physiologic changes impact the interpretation of viability imaging using single-photon emission computed tomography (SPECT), positron emission tomography (PET), and other imaging modalities.

IRREVERSIBLE ISCHEMIA AND THE EVOLUTION OF MYOCARDIAL INFARCTION
Evolution of Acute Myocardial Injury
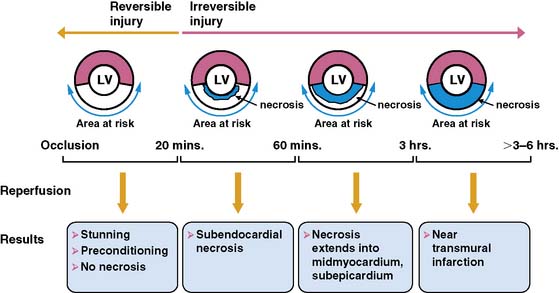
The temporal evolution and extent of irreversible myocardial injury after coronary occlusion is quite variable and strongly dependent on the magnitude of residual coronary flow, the hemodynamic determinants of oxygen consumption, and any endogenous protection afforded by preconditioning.1,2 Reimer and Jennings demonstrated that in the absence of significant collaterals, irreversible injury begins approximately 20 minutes after coronary occlusion3 and progresses in a wavefront from the subendocardium to the subepicardium (Fig. 36-1).4 This susceptibility of the subendocardium to injury reflects its higher oxygen consumption and the redistribution of collateral flow to the outer layers of the heart by the compressive determinants of flow at reduced coronary pressure.5 In experimental infarction, the entire subendocardium is irreversibly injured within 1 hour of occlusion, and the transmural progression of infarction is largely complete within 4 to 6 hours. Factors that increase myocardial oxygen consumption (e.g., tachycardia, hypertension) or reduce oxygen delivery (e.g., anemia, hypotension) accelerate the progression to irreversible injury. In contrast, repetitive reversible ischemia prior to occlusion can limit irreversible injury through a preconditioning effect.1
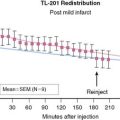
Residual Coronary Flow Limits Infarction
The magnitude of residual coronary flow, either due to subtotal coronary occlusion or the presence of intercoronary collaterals, is the most important determinant of the actual time course of irreversible injury. Ultimate infarct size as a percent of the area at risk of ischemia during a total occlusion is inversely related to collateral flow.6 Subendocardial flow of as little as 30% of resting values can prevent infarction during ischemic periods exceeding an hour. More moderate subendocardial ischemia (e.g., flow reduced by 50%) can persist for hours without resulting in significant irreversible injury.7 This phenomenon explains how the signs and symptoms of ischemia can be present for prolonged periods without producing significant myocardial necrosis. It also accounts for the observation that late coronary reperfusion can salvage myocardium beyond the 6-hour limit predicted from experimental models.
FUNCTIONAL CONSEQUENCES OF ACUTE REVERSIBLE ISCHEMIA
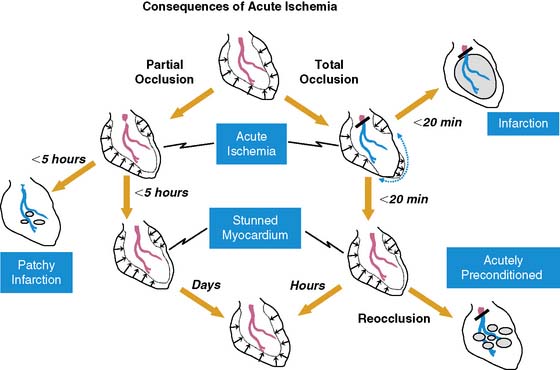
Fortunately, reversible ischemia is considerably more common than irreversible injury. The functional consequences of acute ischemia are summarized in Figure 36-2. Transient coronary occlusion as a result of coronary vasospasm or temporary thrombosis produces supply-mediated ischemia similar to that present at the onset of infarction (vide supra). Upon reperfusion, regional function can remain depressed as a result of myocardial stunning (vide infra). Demand-induced ischemia develops when regional perfusion is unable to sufficiently increase in response to increases in myocardial oxygen demand; this predominantly affects the subendocardium. Interestingly, these two mechanisms of acute ischemia have fundamentally different effects on myocardial diastolic relaxation, with supply-mediated ischemia increasing LV compliance and demand-induced ischemia reducing it. Because of the temporal delay in the development of angina, many episodes of acute myocardial ischemia associated with ST-segment depression are symptomatically silent. Very brief episodes of ischemia (as reflected by more sensitive indices such as regional dysfunction or elevations in end-diastolic pressure) can even be electrocardiographically silent.
Stunned Myocardium
There is a temporal delay in the recovery of function after single episodes of ischemia lasting more than 2 minutes. This regional dysfunction occurs despite the restoration of perfusion, with the time course of recovery determined by the duration and severity of the antecedent ischemia. Heyndrickx et al. were the first to demonstrate that regional dysfunction could persist for hours following a 15-minute occlusion, despite complete reperfusion (Fig. 36-3),8 a phenomenon that Kloner and Braunwald subsequently called stunned myocardium.9 The physiologic hallmark of stunned myocardium is a dissociation of the usual close relation between subendocardial flow and function, where regional dysfunction is present despite normal levels of resting perfusion. Stunned myocardium can also occur after demand-induced ischemia. For example, in the presence of a coronary stenosis, exercise can result in depressed regional function that persists for hours after perfusion is restored, and repetitive ischemia can lead to cumulative stunning.10 From a clinical perspective, the identification of stunned myocardium is important, since unlike other dysfunctional states, regional function will spontaneously normalize in the absence of recurrent ischemia. Furthermore, contractile reserve is invariably present with inotropic stimulation (e.g., dobutamine infusion), usually normalizing regional function (see Table 36-1). In contrast to animal models, acutely stunned myocardium in patients is not always a “pure entity” and frequently coexists with irreversibly injured and viable, chronically dysfunctional myocardium (i.e., chronically stunned and hibernating myocardium, vide infra).
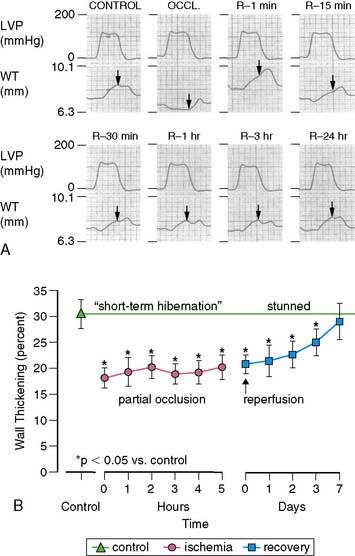
(A, Modified from Heyndrickx et al.84 and republished with permission from the American Physiological Society. B, From Matsuzaki M, Gallagher KP, Kemper WS, et al: Sustained regional dysfunction produced by prolonged coronary stenosis: Gradual recovery after reperfusion, Circulation 68:170–182, 1983. Reprinted with permission.)
Short-Term Hibernation During Prolonged Moderate Ischemia
When coronary pressure distal to a stenosis falls below the lower limit of autoregulation, flow reserve is exhausted, resulting in subendocardial ischemia during which reductions in subendocardial flow are closely coupled to reductions in regional contractile function.11 When moderate ischemia persists, the close matching between perfusion and contraction results in a new steady-state with reduced regional oxygen consumption and energy utilization, a phenomenon termed short-term hibernation.7,11 Despite persistent hypoperfusion, the balance between oxygen supply and demand is reestablished, as reflected by regeneration of creatine phosphate and ATP as well as the resolution of lactate production (Fig. 36-4).12 Importantly, this ability to restore an energetic balance allows myocyte viability to be maintained for a much longer period of time than when flow is absent. Reperfusion of short-term hibernation also leads to stunning, which may take up to a week to resolve (see Fig. 36-3).13
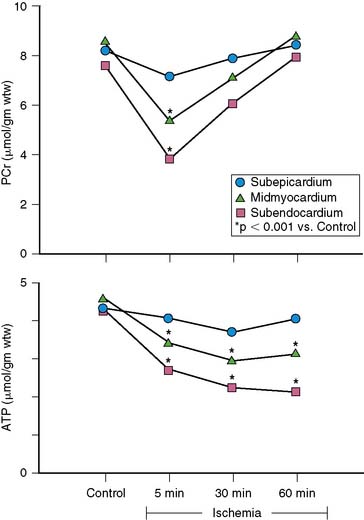
While short-term hibernation was originally hypothesized to be the mechanism of chronic hibernating myocardium, it is now clear that short-term hibernation is an extremely tenuous condition, with small increases in myocardial oxygen demand precipitating recurrent ischemia and a rapid deterioration in function and metabolism.6 Thus, the reversible nature of short-term hibernation is limited by the severity and duration of ischemia, with irreversible injury frequently occurring after more than 12 to 24 hours.14
PATHOPHYSIOLOGY OF CHRONIC REPETITIVE ISCHEMIA
Over 60% of patients with ischemic cardiomyopathy have regional dysfunction as a result of repetitive episodes of myocardial ischemia. This dysfunction is part of a pathophysiologic continuum from chronically stunned myocardium with normal resting perfusion to chronic hibernating myocardium with reduced resting perfusion, which appears to be related to the functional significance of the underlying coronary disease and therefore its propensity to cause ischemia (Fig. 36-5).15,16 The distinguishing physiologic features differentiating chronically stunned from hibernating myocardium are described in the following sections.
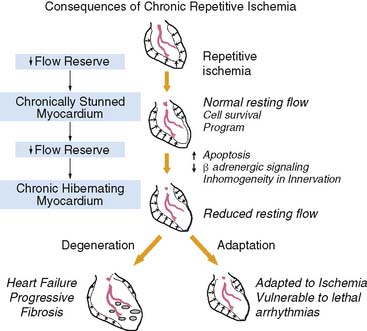
Figure 36-5 Functional consequences of repetitive ischemia. With the progression in severity of a stenosis, coronary flow reserve decreases, and ischemia becomes more frequent. Repetitive ischemia initially leads to chronic preconditioning against infarction and stunning (see Fig. 36-2), which maintains resting function and prevents the development of infarction. As stenosis severity and the frequency of spontaneous ischemia increase further, there is a gradual progression from contractile dysfunction with normal resting flow (chronically stunned myocardium) to contractile dysfunction with depressed resting flow (hibernating myocardium). This transition is related to the physiologic significance of a coronary stenosis and can occur in a time frame as short as 1 week. Alternatively, it can develop chronically and progress in the absence of symptomatic ischemia. The cellular responses during the progression to chronic hibernating myocardium are quite variable. While some patients exhibit little cell death and fibrosis (Adaptation), others develop a picture characterized by degenerative changes that include progressive fibrosis and myocyte death that is difficult to distinguish from subendocardial infarction (Degeneration). The factors that determine the variable progression have not been resolved.
(Modified from Canty JM Jr: Coronary blood flow and myocardial ischemia. In Libby P, Bonow RO, Mann DL, et al (eds): Braunwald’s Heart Disease, 8th ed. Philadelphia: Elsevier, 2007, pp 1167–1194.)
Chronically Stunned Myocardium
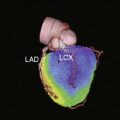
Basic studies have clearly shown that repetitive episodes of myocardial ischemia with17–19 and without a residual stenosis20,21 can result in chronic regional dysfunction with normal resting perfusion. In experimental animal models with a progressive stenosis, chronically stunned myocardium precedes the development of hibernating myocardium.18,22 Although resting flow is normal in chronically stunned myocardium, the regional uptake of the glucose analog, fluorine-18 2-fluorodeoxyglucose (FDG), is variably increased and appears to be related to the physiologic severity of a coronary stenosis. For example, pigs chronically instrumented with a fixed-diameter left anterior descending (LAD) coronary artery stenosis have regional dysfunction with normal resting perfusion at both 1 and 2 months after initial instrumentation (Fig. 36-6).18 Regional FDG uptake is initially normal, but it becomes increased as the reduction in flow reserve progresses, and this persists with the transition to hibernating myocardium.
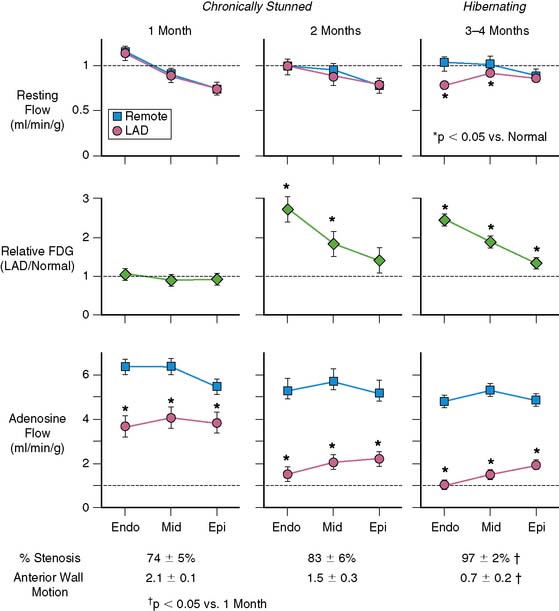
From a clinical perspective, it is important to recognize that viable, chronically dysfunctional myocardium with normal resting perfusion is much more common than hibernating myocardium,23,24 and it can develop independently of ischemia. For example, in patients with ischemic cardiomyopathy, regional dysfunction with normal resting perfusion may occur distant from a large dysfunctional region (viable or infarcted) as a result of pathologic remodeling. Dysfunction in this remodeled myocardium will not improve with regional revascularization, since it is not a direct result of ischemia. Thus, normal resting perfusion does not necessarily translate into reversible dyssynergy, and it is not surprising that noninfarcted but remodeled segments sometimes have persistent dysfunction after coronary revascularization.25
Chronic Hibernating Myocardium
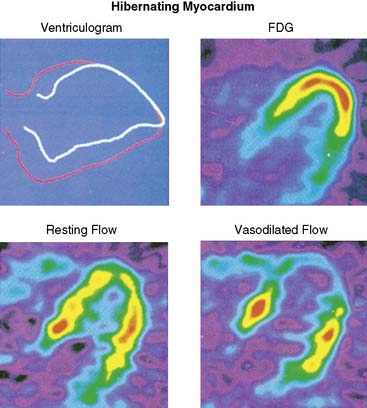
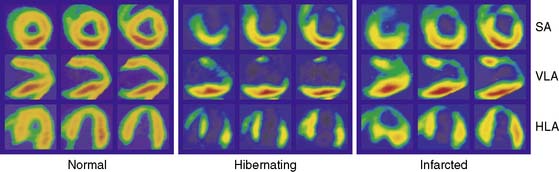
Hibernating myocardium is characterized by contractile dysfunction with reduced resting flow, in the absence of acute ischemia or significant necrosis. It is common in patients with ischemic cardiomyopathy,26 where it frequently coexists with nontransmural infarction and can also occur in the absence of heart failure or global LV dysfunction. The best clinical example of isolated hibernating myocardium is in patients with chronic coronary occlusions and collateral-dependent myocardium (Fig. 36-7).27,28 A similar constellation of findings has been reproduced in pigs in the absence of infarction or heart failure, which has facilitated the investigation of the underlying adaptations to ischemia that result in hibernating myocardium.22 Studies examining these key physiologic adaptations are summarized later.
The unpredictable progression of clinical coronary disease precludes defining the temporal evolution of hibernating myocardium in patients, and it was originally thought that hibernating myocardium arose from a primary reduction in flow in a fashion similar to experimental models of prolonged moderate ischemia and short-term hibernation.7 While this is a plausible mechanism for the development of hibernating myocardium in association with an acute coronary syndrome, more recent studies have highlighted the role of the physiologic severity of a coronary stenosis and repetitive ischemia as the major determinants of the progression from stunned to hibernating myocardium. This paradigm developed from animal studies with a slowly progressive stenosis that clearly demonstrated that regional dysfunction with normal resting flow (chronically stunned myocardium) preceded the development of hibernating myocardium, with the down-regulation in resting flow associated with a critical impairment in subendocardial flow reserve (see Fig. 36-6).15,18,22,29 This progression is not merely a function of time, since the transition from chronically stunned to hibernating myocardium can be experimentally produced in as little as 1 week after placement of a critical stenosis which exhausts coronary flow reserve.19 A common conclusion of all serial studies is that reduced resting flow in hibernating myocardium is a result rather than a cause of the contractile dysfunction. As will be discussed, the down-regulation of resting flow in hibernating myocardium appears to be a physiologic marker reflecting numerous intrinsic metabolic adaptations of the heart to repetitive episodes of ischemia.
Metabolic and Energetic Adaptations to Chronic Ischemia
Myocardial Glucose Uptake
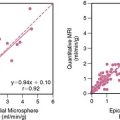
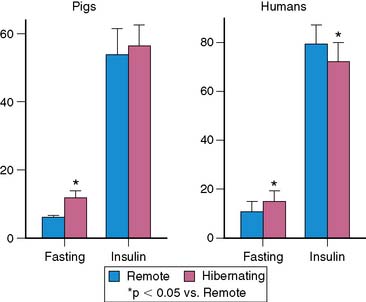
Acute myocardial ischemia is obviously associated with acute reductions in oxidative metabolism and an increased dependence on anaerobic glycolysis. This is facilitated by the mobilization of glycogen stores, as well as an increase in myocardial glucose uptake secondary to the translocation of the glucose transporter GLUT4 from intracellular vesicles. This underlies the basis of “hot spot” imaging with the glucose analog FDG for exercise-induced ischemia and acute coronary syndromes30 and is likely responsible for the enhanced FDG uptake in viable dysfunctional myocardium following dobutamine infusion.31 Chronically dysfunctional myocardium also demonstrates enhanced basal glucose utilization,32 and this adaptation can occur in chronically stunned myocardium.21 Although the mechanism by which glucose utilization is increased in viable dysfunctional myocardium is still under investigation, studies in chronically instrumented pigs have shown an increase in membrane-bound GLUT4, likely due to an up-regulation in p38 mitogen-activated protein kinase.33 Preliminary studies had suggested a role for up-regulation of glucose transporters in chronic ischemia34; however, maximal FDG uptake during insulin stimulation was not altered in pigs with hibernating myocardium (Fig. 36-8).35 A small decrease in maximal insulin-stimulated FDG uptake has been reported in humans.36
Oxygen Consumption
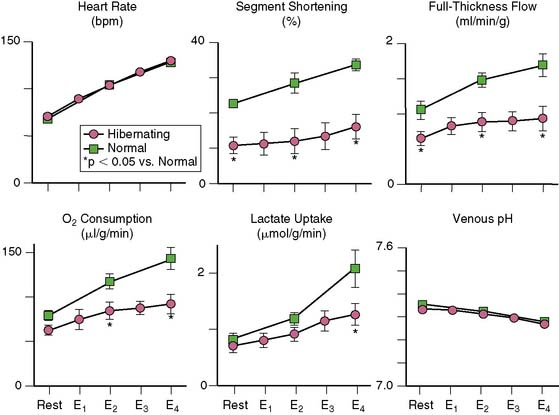
Since myocardial oxygen extraction is near maximal at rest, resting oxygen consumption must also be reduced in hibernating myocardium, and this has been clearly documented using invasive techniques in animals (Fig. 36-9).37 Other investigators have noninvasively documented regionally reduced oxygen consumption in viable, chronically dysfunctional myocardium using PET and carbon-11 acetate.27 Most but not all32,38 studies have shown an energetic balance despite hypoperfusion and some ability to increase metabolism during submaximal stress. For example, in pigs with hibernating myocardium, there was net lactate uptake and normal venous pH during submaximal increases in heart rate, despite the fact that flow and function were lower than in normal myocardium.37 In humans, coronary venous lactate levels39 and myocardial tissue lactate concentrations in biopsy specimens were normal.40 Both basic and clinical studies have also demonstrated preserved creatine phosphate/ATP ratios41,42 and ATP/adenosine diphosphate (ADP) ratios in hibernating myocardium.40,42 Absolute ATP levels are mildly reduced in hibernating myocardium,41 but these alterations are also present in the remote, normally perfused myocardium,42,43 similar to those occurring in normally perfused periinfarct regions of pigs with myocardial infarction.
Fatty Acid Metabolism
Coincident with the increase in glucose utilization during acute ischemia is a down-regulation in fatty acid utilization, which can be imaged with PET using carbon-11 palmitic acid44 and other tracers.45 This down-regulation persists in acute and chronically stunned myocardium,21 but the limited data currently available suggest that fatty acid metabolism can normalize in chronically dysfunctional myocardium.46
Pathologic Remodeling in Hibernating Myocardium
Myocardial biopsy specimens obtained from patients at the time of coronary bypass surgery have demonstrated a broad array of cellular changes that could contribute to the phenotype of hibernating myocardium. The heterogeneity of these findings has led to controversy over whether the myocyte pathologic alterations represent an adaptive or maladaptive response to chronic repetitive ischemia. Many studies have supported adaptation, with a reversion to a fetal cellular phenotype and a protein expression pattern that would serve to protect hibernating cardiac myocytes from ischemic stress.47,48 In contrast, other investigators have found a degenerative phenotype characterized by progressive cell death and marked fibrosis.49,50 The range of findings reported in human biopsies is illustrated in Figure 36-10. The factors that determine a path of progressive structural degeneration, as opposed to successful adaptation to chronic repetitive ischemia, are currently unknown. Nevertheless, degenerative changes appear to be more common when global LV dysfunction and heart failure are present, and adaptation is more common in stable patients with only regional cardiac dysfunction. This suggests that adaptive changes may be importantly modified by neurohormonal activation. The subsequent discussion of potential mechanisms will primarily focus on observations from animals and humans where hibernating myocardium occurs regionally in the absence of heart failure.
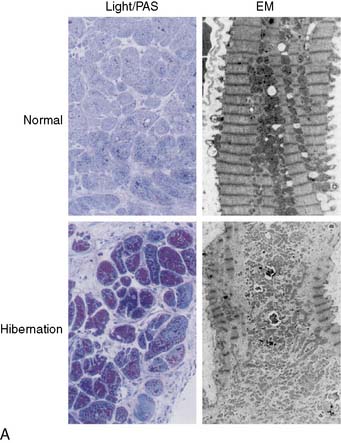
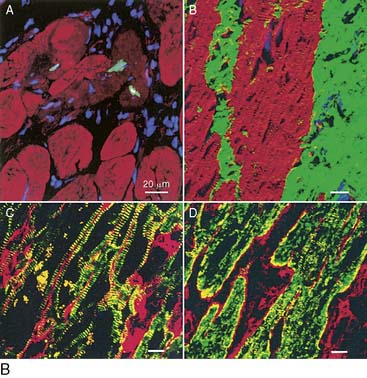
Figure 36-10 Diversity of pathologic findings from transmural biopsies of humans with hibernating myocardium. A, Adapted myocyte phenotype. In patients selected so as to avoid the confounding effects of superimposed infarction, Borgers et al. have reported ultrastructural characteristics that suggest reversion to a fetal phenotype.48 The major electron microscopic features (EM; right photomicrographs) are myofibrillar loss and mini-mitochondria. At the light microscopy level, there is increased glycogen content and minimal interstitial fibrosis (Light/PAS; left photomicrographs). Necrosis, apoptosis, and autophagy are absent. B, Degenerative myocyte phenotype. In consecutive unselected patients with viable dysfunctional myocardium and heart failure, the amount of fibrosis associated with reversible dyssynergy is substantially greater. Myocardial biopsies reveal apoptotic (panel A) and autophagic myocyte cell death, as well as interstitial remodeling with increased collagen (panel B) and fibronectin (panels C and D) that averages as much as 30% of the biopsy. The physiologic and clinical variables determining the progression to each of the two phenotypes is currently unclear.
(A, Adapted from Vanoverschelde J-L, Wijns W, Borgers M, et al: Chronic myocardial hibernation in humans. From bedside to bench, Circulation 95:1961–1971, 1997. B, Panel A adapted from Elsasser A, Vogt AM, Nef H, et al: Human hibernating myocardium is jeopardized by apoptotic and autophagic cell death, J Am Coll Cardiol 43:2191–2199, 2004. Panels B-D adapted from Elsasser A, Schlepper M, Klovekorn WP, et al: Hibernating myocardium: An incomplete adaptation to ischemia, Circulation 96:2920–2931, 1997.)
Myofibrillar Loss and Glycogen Accumulation
Biopsies from patients with hibernating myocardium reveal several prototypical features that include myofibrillar loss or myolysis, increased glycogen content, and small mitochondria.51 While highly affected areas can be identified, less than 40% of the myocytes in a hibernating region exhibit these morphologic changes, so normal myocytes still predominate. In addition, when the myofibrillar volume fraction was quantified, there was less than 10% loss of myofibrils (Fig. 36-11).19 Many of the cellular changes reported are consistent with a reversion to a fetal myocyte phenotype.47 The extent that this reflects dedifferentiated adult myocytes versus regenerating myocytes from resident cardiac stem cells is unknown. Nevertheless, these cellular changes can develop rapidly and be recapitulated in animal models in a period of as little as 2 weeks.19,52,53 They are also seen in disease states such as atrial fibrillation, dilated cardiomyopathy, and in the border zone between normal and infarcted segments of the heart, reviewed elsewhere.16
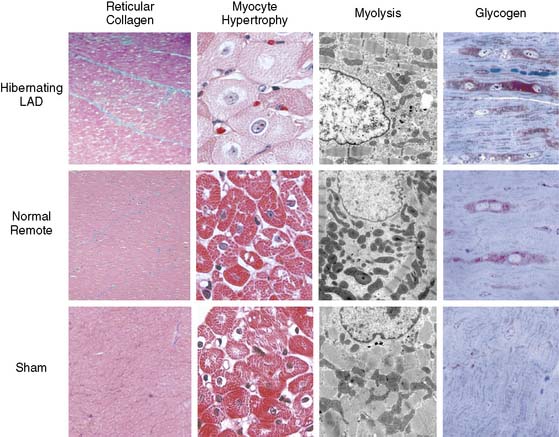
Figure 36-11 Myocyte cellular changes in swine with hibernating myocardium in the absence of heart failure. Swine with hibernating myocardium from a chronic LAD occlusion and collateral-dependent myocardium without infarction or heart failure develop an adapted phenotype. Like the results described by Borgers et al., there is increased reticular connective tissue that is only about 2% greater than values in the remote normally perfused region (left column). Hearts appear grossly normal, but there is cellular hypertrophy in the hibernating region that reflects the consequences of apoptosis-induced myocyte loss (second column). The electron microscopic characteristics of hibernating myocardium are similar to humans with an adapted phenotype and demonstrate myofibrillar loss (Myolysis), numerous small mitochondria, and increased glycogen content (right column). Interestingly, while these are markedly different from normal hearts, biopsies of remote nonischemic segments show similar morphologic changes. Global ultrastructural changes are also seen for other structural proteins.52 These data indicate that the “cellular hibernating phenotype” is not directly related to ischemia, nor is it the cause of regional contractile dysfunction. This accounts for its presence in disease states not associated with myocardial ischemia. LAD, left anterior descending coronary artery.
(Adapted from Canty JM Jr, Fallavollita JA: Chronic hibernation and chronic stunning: A continuum, J Nucl Cardiol 7:509–527, 2000.)
Myocardial Fibrosis, Cell Death, and Myocyte Hypertrophy
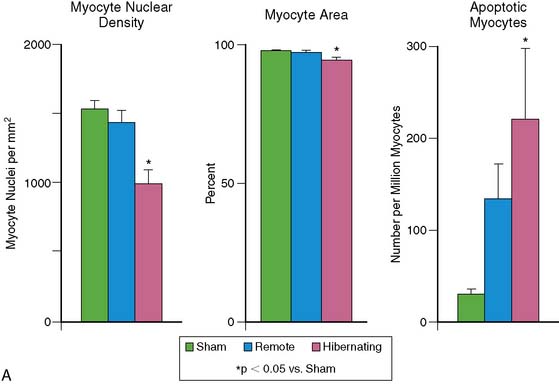
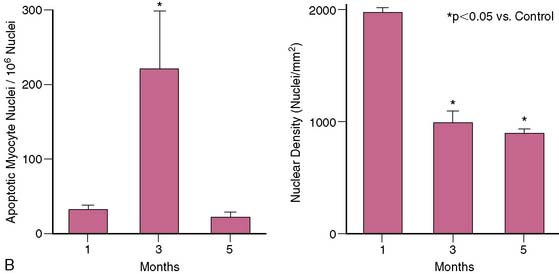
In patients with single-vessel disease and hibernating myocardium, there is no necrosis, and interstitial fibrosis averages less than 15% of the tissue volume.51 The limited number of myocytes in a clinical biopsy precludes accurate quantification of cell death, but porcine studies have identified apoptotic nuclei in about 1 to 2 per 10,000 myocyte nuclei. Although hibernating myocardium appears grossly normal, the chronic nature of apoptosis results in an approximately 30% regional loss of myocytes, with compensatory cellular hypertrophy occurring to maintain normal wall thickness.54 In contrast to some clinical studies that have suggested that cell death and fibrosis inexorably progress over time,55,56 pigs with hibernating myocardium demonstrate stable function and connective-tissue staining for at least 2 months.57,58 Some clinical studies have also demonstrated stability rather than deterioration in function and viability.59 The intrinsic molecular adaptations discussed later eventually serve to abrogate myocyte apoptosis (Fig. 36-12). Nevertheless, myocyte loss and/or remodeling, even in the absence of fibrosis, may ultimately limit functional recovery after revascularization.60,61
When biopsies are obtained from patients with ischemic cardiomyopathy and heart failure, reversible dyssynergy can be demonstrated, with fibrosis comprising as much as 30% of the sample.49 In this setting, myocyte death is more evident, with autophagy and even areas of patchy necrosis.50 The fibrosis may be exacerbated by the elevation of pro-inflammatory cytokines associated with active interstitial remodeling.62–64 Owing to the heterogeneous nature of the histologic changes in ischemic cardiomyopathy and the frequent association with patchy infarction, it is difficult to determine whether there is myocyte adaptation superimposed on nontransmural infarction or an incomplete adaptation to chronic ischemia in hibernating myocardium. In addition, interstitial fibrosis following repetitive ischemia and stunning may actually be reversible after reperfusion in some experimental models,65 similar to the reduction in scar volume after myocardial infarction.66
Global Versus Regional Changes
The cellular alterations described earlier were originally felt to be pathognomonic of hibernating myocardium, but it is now clear that identical changes occur in a variety of disease states associated with elevated preload, such as nonischemic cardiomyopathy and atrial fibrillation.16 Indeed, when remote normally perfused regions from patients as well as pigs with hibernating myocardium were evaluated, there were identical pathologic findings in both regions (see Fig. 36-11).19,52,40 Thus, it appears that the majority of histologic changes reflect myocyte stretch rather than the consequences of ischemia. It is also clear that there is a continuum of changes as the heart progresses from chronically stunned to hibernating myocardium, and although cellular dedifferentiation had been emphasized as a mechanism of adaptation, the ultrastructural changes are probably not causally related to the regional responses to ischemia in hibernating myocardium.16,14
Molecular Remodeling in Hibernating Myocardium
Mitochondrial Proteins and Metabolism
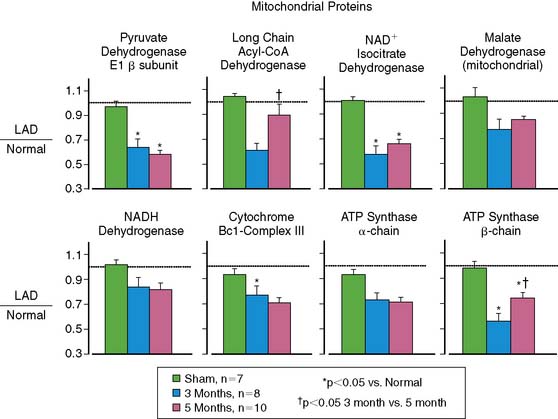
A key target of the intrinsic adaptations of the myocyte to repetitive ischemia appears to be mitochondrial function. Proteomic analysis demonstrates numerous down-regulated proteins, including the entry points to oxidative metabolism and members of the mitochondrial electron-transport chain (Fig. 36-13).67 Collectively, these changes reduce mitochondrial respiration and regional oxygen consumption at any external workload37 and protect myocytes from oxidative injury following simulated ischemia in vitro. One potential mechanism is through the up-regulation of uncoupling protein 2.68 The mitochondrial adaptations eventually prevent progressive apoptosis-induced myocyte loss at the expense of a reduced contractile function and metabolism.
Contractile Proteins and Cellular Calcium Handling
In swine models, there are variable reductions in the sarcoplasmic reticulum (SR) calcium uptake proteins that are similar to global alterations in cardiomyopathy. In hibernating myocardium, these changes are regional and occur in the absence of heart failure.69 Like other adaptations, they vary with the physiologic significance of the stenosis and are initially absent in chronically stunned myocardium.70 Isolated myocytes from swine with viable dysfunctional myocardium demonstrate that changes in SR calcium proteins are functionally significant,71 and human studies also support an important role of the SR in the depressed contractile response.72
Cell Survival Program and Antiapoptotic Program in Response to Repetitive Ischemia
Cell survival pathways are also induced by repetitive ischemia and appear to be particularly prominent during the transition from stunned to hibernating myocardium. Although the specific pathways vary depending upon model and physiologic state, they include up-regulation of heat shock proteins69 and down-regulation of glycogen synthase kinase-3β.21 There is variability in antiapoptotic protein expression in degenerating tissue, with proapoptotic proteins such as Bax increased when a profile of progressive cell death and fibrosis are present in human biopsies.50 It is likely that the variability of findings among studies reflect differences in the frequency and severity of ischemia, as well as the complex temporal expression of myocardial responses to chronic repetitive ischemia.
Inhomogeneity in Sympathetic Innervation and β-Adrenergic Signaling
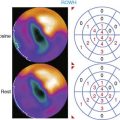
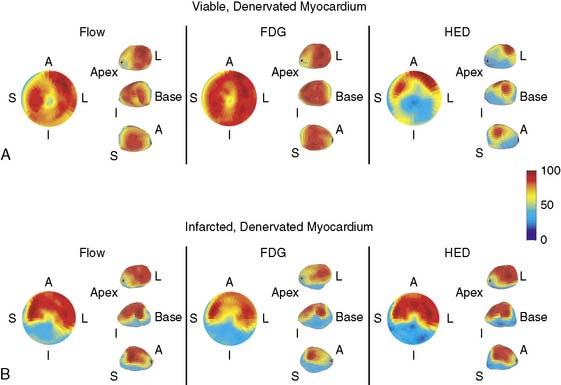
Although contractile reserve to β-adrenergic stimulation is present in hibernating myocardium, compared to normal, the response is attenuated,37 which likely accounts for the underestimation of viability with inotropic stimulation as compared to nuclear imaging.73 Despite a critical limitation in subendocardial flow reserve, increases in regional function can occur in the absence of acute metabolic ischemia during steady-state submaximal stimulation.37 At a cellular and molecular level, there are profound regional alterations in myocardial sympathetic innervation, with attenuation of both pre- and post synaptic adrenergic function. Previous studies in isolated vesicular preparations have demonstrated a reduction in cyclic adenosine monophosphate (cAMP) production, as well as a shift from a high- to low-affinity β-receptor subtype.74 In addition, there is partial sympathetic denervation75 associated with profound reductions in presynaptic sympathetic norepinephrine uptake, as assessed using iodine-131 metaiodobenzylguanidine (MIBG)76 or carbon-11 metahydroxyephedrine (HED) with PET imaging (Fig. 36-14).77 The magnitude of these changes is similar to those reported in infarcted myocardium and lead to profound inhomogeneity in myocardial sympathetic innervation. They are functionally significant and attenuate in vivo responses to sympathetic nerve stimulation as well as exogenous β-adrenergic stimulation.75 The temporal development of these changes and their potential reversibility are currently under study. As will be discussed, inhomogeneity in sympathetic nerve function in hibernating myocardium may identify a myocardial substrate at risk of lethal ventricular arrhythmias, even in the absence of infarction.78
TRANSLATION OF MECHANISTIC STUDIES TO THE CLINICAL ASSESSMENT OF VIABILITY
Blunted Contractile Reserve in Viable Myocardium
It is clear that contractile reserve during β-adrenergic stimulation has a high positive predictive value for functional recovery. However, the molecular alterations in hibernating myocardium that are responsible for the adaptation to chronic ischemia attenuate inotropic responsiveness. For example, the blunted contractile response in pigs with hibernating myocardium37,79,80 reflects a reduction in cAMP production due to a shift in the β-adrenergic receptor to a low-affinity subtype.74 In addition, reductions in the SR calcium-handling proteins reduce calcium uptake and release for any level of inotropic stimulation.19,69 These intrinsic responses in hibernating myocardium may underlie the clinical observation that contractile reserve generally underestimates the frequency of viable dysfunctional myocardium.73
Roles of Myocyte Loss and Cellular Remodeling in Limiting Functional Recovery
Even when myocardial scarring is carefully excluded with gadolinium magnetic resonance imaging (MRI),25 up to 25% of noninfarcted segments fail to improve after revascularization. The reasons for persistent dysfunction in the absence of recurrent ischemia or scar are not entirely clear but likely reflect the importance of irreversible myocyte remodeling. In support of this, pigs with hibernating myocardium and apoptosis-induced myocyte loss exhibit persistent dysfunction following revascularization, in the absence of any infarction. Similar myocardial changes can result in irreversible LV dysfunction after surgical intervention for advanced valvular regurgitation. This cellular remodeling is likely a major contributor to the LV dysfunction in advanced ischemic cardiomyopathy, where roughly 80% of the left ventricle is viable and less than 20% irreversibly scarred. Thus, developing approaches to identify myocyte remodeling in the absence of fibrosis may be useful in identifying the likelihood of reversible dyssynergy.
The Impact of Hibernating Myocardium on Prognosis (See Chapters 37 and 38)
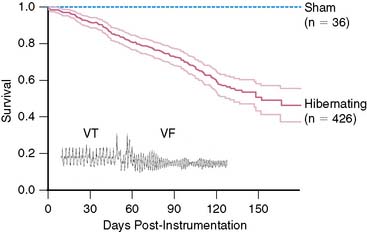
While nonrandomized clinical studies have demonstrated the profound negative effect of hibernating myocardium on survival and the amelioration of this risk with revascularization,26 cause-specific mortality remains undefined. Studies have primarily focused on the potential reversibility of myocardial dysfunction and heart failure; however, some data suggest that the major impact of hibernating myocardium may relate to an increased risk of sudden death.81 Basic studies support this contention78 and have demonstrated inhomogeneity in cellular myocyte remodeling and regional inhomogeneity in presynaptic and postsynaptic sympathetic nerve function, which can lead to a substrate that is electrically unstable. Indeed, sudden death from ventricular tachycardia/ventricular fibrillation (VT/VF) develops in pigs with chronic collateral-dependent myocardium in the absence of heart failure (Fig. 36-15). This has given rise to the notion that although adapted to ischemia, the myocyte cellular alterations associated with hibernating myocardium lead to vulnerability to lethal arrhythmias. Since asymptomatic myocardial ischemia is so common, it is plausible that similar neural and myocyte remodeling underlie the progression of coronary artery disease and development of sudden death.82 Thus, while the amount of hibernating myocardium can predict the likelihood of improvement from the standpoint of myocardial dysfunction and heart failure, even small amounts may increase the risk of sudden death. This hypothesis is the basis of an ongoing prospective study to determine whether the presence of hibernating myocardium and/or regional inhomogeneity in sympathetic innervation can predict an increased risk of sudden death in patients who are candidates to receive an implantable cardiac defibrillator (Fig. 36-16).83
1. Downey J.M., Cohen M.V. Reducing infarct size in the setting of acute myocardial infarction. Prog Cardiovasc Dis. 2006;48:363-371.
2. Cokkinos D.V., Pantos C. Myocardial protection in man–from research concept to clinical practice. Heart Fail Rev. 2007;12:345-362.
3. Reimer K.A., Jennings R.B. The “wavefront phenomenon” of myocardial ischemic cell death. II. Transmural progression of necrosis within the framework of ischemic bed size (myocardium at risk) and collateral flow. Lab Invest. 1979;40:633-644.
4. Kloner R.A., Jennings R.B. Consequences of brief ischemia: stunning, preconditioning, and their clinical implications: part 1. Circulation. 2001;104:2981-2989.
5. Canty J.M.Jr. Coronary blood flow and myocardial ischemia. In: Libby P., Bonow R.O., Mann D.L., et al, editors. Braunwald’s heart disease. ed 8. Philadelphia: Elsevier; 2007:1167-1194.
6. Schulz R., Rose J., Martin C., et al. Development of short-term myocardial hibernation: Its limitation by the severity of ischemia and inotropic stimulation. Circulation. 1993;88:684-695.
7. Heusch G. Hibernating myocardium. Physiol Rev. 1998;78:1055-1085.
8. Heyndrickx G.R., Millard R.W., McRitchie R.J., et al. Regional myocardial functional and electrophysiological alterations after brief coronary artery occlusion in conscious dogs. J Clin Invest. 1975;56:978-985.
9. Kloner R.A., Jennings R.B. Consequences of brief ischemia: stunning, preconditioning, and their clinical implications: part 2. Circulation. 2001;104:3158-3167.
10. Homans D.C., Laxson D.D., Sublett E., et al. Cumulative deterioration of myocardial function after repeated episodes of exercise-induced ischemia. Am J Physiol. 1989;256:H1462-H1471.
11. Ross J.Jr. Myocardial perfusion-contraction matching: Implications for coronary heart disease and hibernation. Circulation. 1991;83:1076-1083.
12. Pantely G.A., Malone S.A., Rhen W.S., et al. Regeneration of myocardial phosphocreatine in pigs despite continued moderate ischemia. Circ Res. 1990;67:1481-1493.
13. Matsuzaki M., Gallagher K.P., Kemper W.S., et al. Sustained regional dysfunction produced by prolonged coronary stenosis: Gradual recovery after reperfusion. Circulation. 1983;68:170-182.
14. Heusch G., Schulz R., Rahimtoola S.H. Myocardial hibernation: a delicate balance. Am J Physiol Heart Circ Physiol. 2005;288:H984-H999.
15. Canty J.M.Jr., Fallavollita J.A. Chronic hibernation and chronic stunning: a continuum. J Nucl Cardiol. 2000;7:509-527.
16. Canty J.M., Fallavollita J.A. Hibernating myocardium. J Nucl Cardiol. 2005;12:104-119.
17. Shen Y.-T., Vatner S.F. Mechanism of impaired myocardial function during progressive coronary stenosis in conscious pigs: Hibernation versus stunning? Circ Res. 1995;76:479-488.
18. Fallavollita J.A., Canty J.M.Jr. Differential 18F-2-deoxyglucose uptake in viable dysfunctional myocardium with normal resting perfusion: Evidence for chronic stunning in pigs. Circulation. 1999;99:2798-2805.
19. Thomas S.A., Fallavollita J.A., Borgers M., et al. Dissociation of regional adaptations to ischemia and global myolysis in an accelerated swine model of chronic hibernating myocardium. Circ Res. 2002;91:970-977.
20. Bolli R. Myocardial ‘stunning’ in man. Circulation. 1992;86:1671-1691.
21. Kim S.J., Peppas A., Hong S.K., et al. Persistent stunning induces myocardial hibernation and protection: flow/function and metabolic mechanisms. Cir Res. 2003;92:1233-1239.
22. Fallavollita J.A., Perry B.J., Canty J.M.Jr. 18F-2-deoxyglucose deposition and regional flow in pigs with chronically dysfunctional myocardium: Evidence for transmural variations in chronic hibernating myocardium. Circulation. 1997;95:1900-1909.
23. Melon P.G., de Landsheere C.M., Degueldre C., et al. Relation between contractile reserve and positron emission tomographic patterns of perfusion and glucose utilization in chronic ischemic left ventricular dysfunction: implications for identification of myocardial viability. J Am Coll Cardiol. 1997;30:1651-1659.
24. Sawada S., Elsner G., Segar D.S., et al. Evaluation of patterns of perfusion and metabolism in dobutamine-responsive myocardium. J Am Coll Cardiol. 1997;29:55-61.
25. Kim R.J., Wu E., Rafael A., et al. The use of contrast-enhanced magnetic resonance imaging to identify reversible myocardial dysfunction. New Engl J Med. 2000;343:1445-1453.
26. Allman K.C., Shaw L.J., Hachamovitch R., et al. Myocardial viability testing and impact of revascularization on prognosis in patients with coronary artery disease and left ventricular dysfunction: a meta-analysis. J Am Coll Cardiol. 2002;39:1151-1158.
27. Vanoverschelde J-LJ, Wijns W., Depre C., et al. Mechanisms of chronic regional postischemic dysfunction in humans: New insights from the study of noninfarcted collateral-dependent myocardium. Circulation. 1993;87:1513-1523.
28. Arani D.T., Greene D.G., Bunnell I.L., et al. Reductions in coronary flow under resting conditions in collateral-dependent myocardium of patients with complete occlusion of the left anterior descending coronary artery. J Am Coll Cardiol. 1984;3:668-674.
29. Canty J.M.Jr., Klocke F.J. Reductions in regional myocardial function at rest in conscious dogs with chronically reduced regional coronary artery pressure. Circ Res. 1987;61(Suppl II):II-107–II-116.
30. He Z.X., Shi R.F., Wu Y.J., et al. Direct imaging of exercise-induced myocardial ischemia with fluorine-18-labeled deoxyglucose and Tc-99m-sestamibi in coronary artery disease. Circulation. 2003;108:1208-1213.
31. McFalls E.O., Murad B., Haspel H.C., et al. Myocardial glucose uptake after dobutamine stress in chronic hibernating swine myocardium. J Nucl Cardiol. 2003;10:385-394.
32. Vogt A.M., Elsasser A., Nef H., et al. Increased glycolysis as protective adaptation of energy depleted, degenerating human hibernating myocardium. Mol Cell Biochem. 2003;242:101-107.
33. McFalls E.O., Hou M., Bache R.J., et al. Activation of p38 MAPK and increased glucose transport in chronic hibernating swine myocardium. Am J Physiol Heart Circ Physiol. 2004;287:H1328-H1334.
34. Brosius F.C., Nguyen N., Egert S., et al. Increased sarcolemmal glucose transporter abundance in myocardial ischemia. Am J Cardiol. 1997;80:77A-84A.
35. Fallavollita J.A. Spatial heterogeneity in fasting and insulin-stimulated 18F-2-deoxyglucose uptake in pigs with hibernating myocardium. Circulation. 2000;102:908-914.
36. Maki M., Luotolahti M., Nuutila P., et al. Glucose uptake in the chronically dysfunctional but viable myocardium. Circulation. 1996;93:1658-1666.
37. Fallavollita J.A., Malm B.J., Canty J.M.Jr. Hibernating myocardium retains metabolic and contractile reserve despite regional reductions in flow, function, and oxygen consumption at rest. Circ Res. 2003;92:48-55.
38. Elsasser A., Muller K.D., Skwara W., et al. Severe energy deprivation of human hibernating myocardium as possible common pathomechanism of contractile dysfunction, structural degeneration and cell death. J Am Coll Cardiol. 2002;39:1189-1198.
39. Indolfi C., Piscione F., Perrone-Filardi P., et al. Inotropic stimulation by dobutamine increases left ventricular regional function at the expense of metabolism in hibernating myocardium. Am Heart J. 1996;132:542-549.
40. Wiggers H., Noreng M., Paulsen P.K., et al. Energy stores and metabolites in chronic reversibly and irreversibly dysfunctional myocardium in humans. J Am Coll Cardiol. 2001;37:100-108.
41. McFalls E.O., Baldwin D., Palmer B., et al. Regional glucose uptake within hypoperfused swine myocardium as measured by positron emission tomography. Am J Physiol. 1997;272:H343-H349.
42. Hu Q., Suzuki G., Young R.F, et al. Reductions in mitochondrial O2 consumption and preservation of high energy phosphate levels after simulated ischemia in chronic hibernating myocardium. Am J Physiol Heart Circ Physiol. 2009;297:H223-H232.
43. McFalls E.O., Kelly R.F., Hu Q., et al. The energetic state within hibernating myocardium is normal during dobutamine despite inhibition of ATP-dependent potassium channel opening with glibenclamide. Am J Physiol Heart Circ Physiol. 2007;293:H2945-H2951.
44. Schwaiger M., Schelbert H.R., Keen R., et al. Retention and clearance of C-11 palmitic acid in ischemic and reperfused canine myocardium. J Am Coll Cardiol. 1985;6:311-320.
45. Shoup T.M., Elmaleh D.R., Bonab A.A., et al. Evaluation of trans-9–18F-fluoro-3,4-methyleneheptadecanoic acid as a PET tracer for myocardial fatty acid imaging. J Nucl Med. 2005;46:297-304.
46. Maki M.T., Haaparanta M.T., Luotolahti M.S., et al. Fatty acid uptake is preserved in chronically dysfunctional but viable myocardium. Am J Physiol. 1997;273:H2473-H2480.
47. Ausma J., Schaart G., Thon F., et al. Chronic ischemic viable myocardium in man: Aspects of dedifferentiation. Cardiovasc Pathol. 1995;4:29-37.
48. Vanoverschelde J.-L., Wijns W., Borgers M., et al. Chronic myocardial hibernation in humans. From bedside to bench. Circulation. 1997;95:1961-1971.
49. Elsasser A., Schlepper M., Klovekorn W.P., et al. Hibernating myocardium: an incomplete adaptation to ischemia. Circulation. 1997;96:2920-2931.
50. Elsasser A., Vogt A.M., Nef H., et al. Human hibernating myocardium is jeopardized by apoptotic and autophagic cell death. J Am Coll Cardiol. 2004;43:2191-2199.
51. Maes A., Flameng W., Nuyts J., et al. Histological alterations in chronically hypoperfused myocardium: Correlation with PET findings. Circulation. 1994;90:735-745.
52. Thijssen V.L., Borgers M., Lenders M.-H., et al. Temporal and spatial variations in structural protein expression during the progression from stunned to hibernating myocardium. Circulation. 2004;110:3313-3321.
53. Chen C., Liu J., Hua D., et al. Impact of delayed reperfusion of myocardial hibernation on myocardial ultrastructure and function and their recoveries after reperfusion in a pig model of myocardial hibernation. Cardiovasc Pathol. 2000;9:67-84.
54. Lim H., Fallavollita J.A., Hard R., et al. Profound apoptosis-mediated regional myocyte loss and compensatory hypertrophy in pigs with hibernating myocardium. Circulation. 1999;100:2380-2386.
55. Schwarz E.R., Schaper J., vom D.J., et al. Myocyte degeneration and cell death in hibernating human myocardium. J Am Coll Cardiol. 1996;27:1577-1585.
56. Elsasser A., Decker E., Kostin S., et al. A self-perpetuating vicious cycle of tissue damage in human hibernating myocardium. Mol Cell Biochem. 2000;213:17-28.
57. Fallavollita J.A., Logue M., Canty J.M.Jr. Stability of hibernating myocardium in pigs with a chronic left anterior descending coronary artery stenosis: Absence of progressive fibrosis in the setting of stable reductions in flow, function and coronary flow reserve. J Am Coll Cardiol. 2001;37:1989-1995.
58. Suzuki G., Lee T.C., Fallavollita J.A., et al. Adenoviral gene transfer of FGF-5 to hibernating myocardium improves function and stimulates myocytes to hypertrophy and reenter the cell cycle. Circ Res. 2005;96:767-775.
59. Wiggers H., Nielsen S.S., Holdgaard P., et al. Adaptation of nonrevascularized human hibernating and chronically stunned myocardium to long-term chronic myocardial ischemia. Am J Cardiol. 2006;98:1574-1580.
60. Angelini A., Maiolino G., La Canna G., et al. Relevance of apoptosis in influencing recovery of hibernating myocardium. Eur J Heart Fail. 2007;9:377-383.
61. Banas M.D., Page B., Young R.F., et al. Residual dysfunction after revascularization of hibernating myocardium is independent of fibrosis and secondary to myocyte loss and persistent regional reduction in mitochondrial oxidative enzymes. Circulation. 2006;114(Suppl II):II-66.
62. Frangogiannis N.G., Shimoni S., Chang S.M., et al. Active interstitial remodeling: an important process in the hibernating human myocardium. J Am Coll Cardiol. 2002;39:1468-1474.
63. Frangogiannis N.G., Shimoni S., Chang S.M., et al. Evidence for an active inflammatory process in the hibernating human myocardium. Am J Pathol. 2002;160:1425-1433.
64. Frangogiannis N.G. The pathological basis of myocardial hibernation. Histol Histopathol. 2003;18:647-655.
65. Dewald O., Frangogiannis N.G., Zoerlein M., et al. Development of murine ischemic cardiomyopathy is associated with a transient inflammatory reaction and depends on reactive oxygen species. Pro Natl Acad Sci U S A. 2003;100:2700-2705.
66. Fieno D.S., Hillenbrand H.B., Rehwald W.G., et al. Infarct resorption, compensatory hypertrophy, and differing patterns of ventricular remodeling following myocardial infarctions of varying size. J Am Coll Cardiol. 2004;43:2124-2131.
67. Page B., Young R., Iyer V., et al. Persistent regional downregulation in mitochondrial enzymes and upregulation of stress proteins in swine with chronic hibernating myocardium. Circ Res. 2008;102:103-112.
68. McFalls E.O., Sluiter W., Schoonderwoerd K., et al. Mitochondrial adaptations within chronically ischemic swine myocardium. J Mol Cell Cardiol. 2006;41:980-988.
69. Fallavollita J.A., Jacob S.C., Young R.F., et al. Regional alterations in SR Ca2+-ATPase, phospholamban, and HSP-70 expression in chronic hibernating myocardium. Am J Physiol. 1999;277:H1418-H1428.
70. Fallavollita J.A., Lim H., Canty J.M.Jr. Myocyte apoptosis and reduced SR gene expression precede the transition from chronically stunned to hibernating myocardium. J Mol Cell Cardiol. 2001;33:1937-1944.
71. Bito V., van der Velden J., Claus P., et al. Reduced force generating capacity in myocytes from chronically ischemic, hibernating myocardium. Circ Res. 2007;100:229-237.
72. Nef H.M., Mollmann H., Skwara W., et al. Reduced sarcoplasmic reticulum Ca2+-ATPase activity and dephosphorylated phospholamban contribute to contractile dysfunction in human hibernating myocardium. Mol Cell Biochem. 2006;282:53-63.
73. Underwood S.R., Bax J.J., vom Dahl J., et al. Imaging techniques for the assessment of myocardial hibernation. Report of a Study Group of the European Society of Cardiology. Eur Heart J. 2004;25:815-836.
74. Iyer V., Canty J.M.Jr. Regional desensitization of β-adrenergic receptor signaling in swine with chronic hibernating myocardium. Circ Res. 2005;97:789-795.
75. Ovchinnikov V., Canty J.M.Jr, Fallavollita J.A. Hibernating myocardium leads to an upregulation in nerve growth factor and partial subendocardial sympathetic denervation. J Am Coll Cardiol. 2007;49(Suppl A):34A.
76. Luisi A.J.Jr, Fallavollita J.A., Suzuki G., et al. Spatial inhomogeneity of sympathetic nerve function in hibernating myocardium. Circulation. 2002;106:779-781.
77. Luisi A.J.Jr., Suzuki G., deKemp R., et al. Regional 11C-hydroxyephedrine retention in hibernating myocardium: Chronic inhomogeneity of sympathetic innervation in the absence of infarction. J Nucl Med. 2005;46:1368-1374.
78. Canty J.M.Jr, Suzuki G., Banas M.D., et al. Hibernating myocardium: Chronically adapted to ischemia but vulnerable to sudden death. Circ Res. 2004;94:1142-1149.
79. Malm B.J., Suzuki G., Canty J.M.Jr, et al. Variability of contractile reserve in hibernating myocardium: Dependence on the method of stimulation. Cardiovasc Res. 2002;56:422-433.
80. Ovchinnikov V., Suzuki G., Canty J.M.Jr, et al. Blunted functional responses to pre- and postjunctional sympathetic stimulation in hibernating myocardium. Am J Physiol Heart Circ Physiol. 2005;289:H1719-H1728.
81. Di Carli M.F., Maddahi J., Rokhsar S., et al. Long-term survival of patients with coronary artery disease and left ventricular dysfunction: implications for the role of myocardial viability assessment in management decisions. J Thorac Cardiovasc Surg. 1998;116:997-1004.
82. Burke A.P., Kolodgie F.D., Farb A., et al. Healed plaque ruptures and sudden coronary death: evidence that subclinical rupture has a role in plaque progression. Circulation. 2001;103:934-940.
83. Fallavollita J.A., Luisi A.J., Michalek S.M., et al. Prediction of Arrhythmic Events with Positron Emission Tomography: PAREPET study design and methods. Contemp Clin Trials. 2006;27:374-388.
84. Heyndrickx G.R., Baig H., Nellens P., et al. Depression of regional blood flow and wall thickening after brief coronary occlusions. Am J Physiol. 1978;234:H653-H659.
85. Fallavollita J.A., Lim H., Canty J.M.Jr. Apoptosis and reduced SR gene expression precede the transition from chronically stunned to hibernating myocardium. J Mol and Cell Cardiol. 2001;33:1937-1944.