CHAPTER 71 PARKINSON’S DISEASE
Parkinson’s disease (PD) is a neurodegenerative disease with initial clinical features that are predominantly the result of loss of dopaminergic neurons in the substantia nigra pars compacta (SNc) of the midbrain. As the disease progresses, the involvement of additional brain areas in the degenerative process produces mainly nondopaminergic, nonmotor features. The discovery of dopamine deficiency in PD and the introduction of levodopa have provided patients with a significant improvement in both quality of life and life expectancy, but the treatment of nonmotor features and slowing of disease progression remain important unmet needs for patients.
HISTORICAL PERSPECTIVE
In 1960, Ehringer and Hornykiewicz identified the dopamine deficiency in PD striatum.1 Studies on the replacement of dopamine with DL-dopa produced equivocal results until used in sufficient quantity.2 This began the era of symptomatic treatment for PD, which has remained focused on the dopaminergic system for almost 40 years.
EPIDEMIOLOGY
Defining the epidemiology of PD is confounded by several variables that include the difficulty in diagnosis and the age dependence of the disease. Several studies have sought to define incidence. In the United States, the age-adjusted figure is 13.5 to 13.9 per 100,000 person-years.3,4 The age-adjusted prevalence is approximately 115 per 100,000 and is estimated as 1.3 per 100,000 under age 45 years and 1192.9 per 100,000 in those aged 75 to 85 years.3 A prevalence study in Holland found 3100 cases per 100,000 aged 75 to 85 years and 4300 per 100,000 for those over age 85 years.6 The geographical distribution of the disease appears similar across the United States and Japan, but failure to adjust population figures for age can lead to widely discrepant results, such as the prevalence of 10 per 100,000 in Nigeria.7
PATHOLOGY
Macroscopic examination of the sectioned PD brain shows depigmentation of the substantia nigra and locus ceruleus. The characteristic pathological change of PD is the loss of pigmented dopaminergic neurons, particularly in the ventral tier of the SNc with intracytoplasmic eosinophilic inclusions (Lewy bodies) in a proportion of the surviving neurons (Fig. 71-1). The SNc also contains activated microglia and extracellular neuromelanin. There is also cell loss in the locus ceruleus; the nucleus basalis of Meynert; the dorsal motor nucleus of the vagus; the Edinger-Westphal, raphe, and pedunculopontine nuclei; and the pons, midbrain, spinal cord, and peripheral sympathetic ganglia. Lewy bodies may be found in these areas and in the cortex. Thus, in addition to the dopaminergic system, the cholinergic, serotonergic, γ-amino butyric acid, and adrenergic transmitter systems are involved in PD.
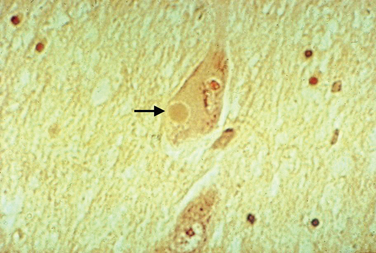
Lewy bodies have attracted considerable attention over the years, as they may hold important clues to pathogenesis of the disease. They are 5 to 30 μm in diameter with a hyaline eosinophilic core, which may be dense and composed of concentric lamellae; a pale halo may be seen around the core. Electron microscopy demonstrates 7-to 20-nm intermediate filaments. The Lewy body is composed of a number of different proteins, staining for ubiquitin, α-synuclein, and proteasomal components. It is not known whether these inclusions represent a protective response to aggregate abnormal or toxic proteins, or whether their formation is part of a toxic process that damages the cell.
Studies suggest that the earliest pathological changes are seen in the dorsal motor nucleus and in the olfactory bulbs and nucleus—Braak stages 1 and 28 (Fig. 71-2). In this context, it is noteworthy that loss of olfactory function can occur at a time prior to the onset of dopaminergic symptoms or signs and may serve to define an “at-risk” population.9 Lewy bodies then develop in the locus ceruleus and progress in the medulla and pons.10 The appearance of inclusions in the SNc defines the onset of Braak stage 3 with progression to stage 4. At this stage there is also degeneration in the pedunculopontine nucleus, the dorsal raphe nuclei, and the hypothalamus. Stages 5 and 6 involve progressive involvement of the cerebral cortex and neurodegeneration in those regions already affected.
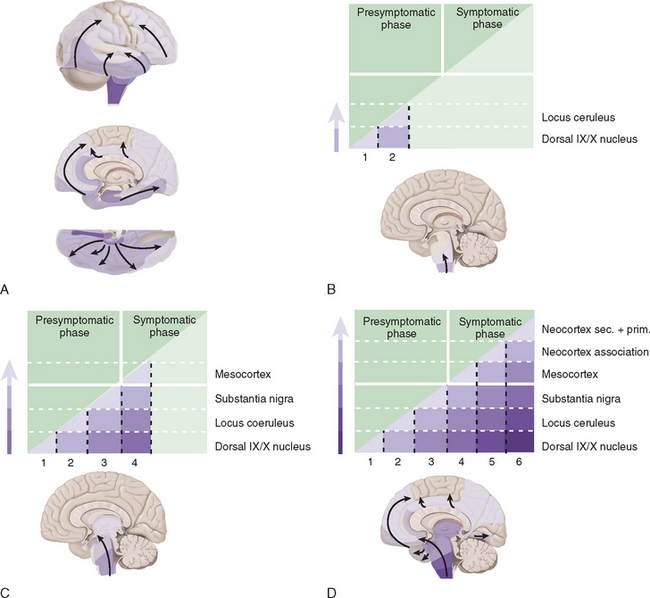
Figure 71-2 Braak staging of Parkinson’s disease.
(From Del Tredici K, Rub U, De Vos RA, et al: Where does Parkinson’s disease pathology begin in the brain? J Neuropathol Exp Neurol 2002; 61:413-426; and Braak H, Del Tredici K, Rub U, et al: Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 2003; 24:197-211.)
ETIOLOGY
Genetic Factors
Sir William Gowers recognized that there was an increased prevalence of PD among the relatives of sufferers and proposed a genetic cause.11 Epidemiological studies evaluating the genetic contribution of PD are complicated by ensuring an accurate clinical diagnosis, inability to identify presymptomatic cases and the need for an appropriate control population. Case-control studies have confirmed Gowers’ observation that PD is more common in relatives of PD cases compared with matched controls.12–16 The relative risk of developing PD in family members has varied widely between different studies, but in many, incomplete family information was obtained and only one study has been population based.16 Overall, the relative risk in first-degree relatives of PD cases is increased approximately two- to threefold.17
A large PD twin study showed no significant concordance for PD among monozygotic twins, suggesting that there was no significant genetic contribution to PD.18 However, there was significant concordance for those with onset before age 50 years, implying that young-onset PD is more likely genetically determined. Another smaller twin study using fluorodopa positron emission tomography (PET) to image dopaminergic function in both affected and unaffected monozygotic and dizygotic twin pairs demonstrated an increased concordance for PD among identical twins.19 At follow-up, the combined concordance levels for subclinical dopaminergic dysfunction and clinical PD were 75% in the 12 monozygotic twins and 22% in the 9 dizygotic twins evaluated twice.
Table 71-1 shows the current list of genes known to cause PD. PARK1 through PARK9 have been discovered using large single or combined pedigrees, whereas PARK10 and PARK11 have been identified using association techniques.
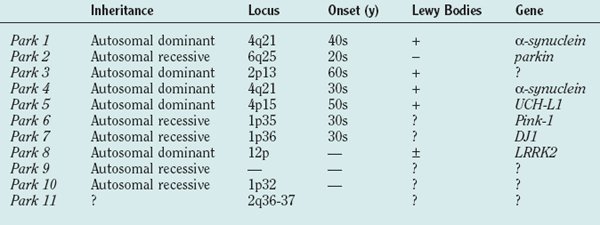
α-Synuclein (PARK1)
PARK1 involves mutations in the α-synuclein gene.20,21 The first families with an α-synuclein mutation originated from southern Italy and Greece, and with the description of several additional families with the same A53T mutation, it is believed that they have a common founder. Age at onset was young (mean, 46 years) with a low frequency of tremor and relatively rapid disease progression, although symptoms were levodopa responsive.22 Pathological analysis of the few brains available revealed the presence of Lewy bodies in the substantia nigra and locus ceruleus.23 Linkage to markers on chromosome 4q21-q23 was demonstrated and the locus designated PARK1.24 Further analyses identified a mutation in exon 4 of the gene encoding α-synuclein resulting in an alanine-to-threonine substitution at codon 53 (A53T), which was also found in affected members of three Greek families with earlyonset autosomal dominant PD.20 A second mutation (A30P) was later found in a small German family with PD,21 but extensive study in large groups of PD families and sporadic cases has not identified other patients or families with this mutation.25–28
α-Synuclein is a protein of 140 amino acids that is predominantly expressed in neurons and is one of the most common brain proteins. Its normal function remains unclear, although it plays a role in synaptic plasticity based on song-learning studies in the zebra finch29 and vesicular regulation of dopamine release from knockout mice.30 A key observation linking α-synuclein to PD was the demonstration that it is one of the principal components of Lewy bodies.31 Lewy bodies are intracytoplasmic aggregates comprising several proteins, including ubiquitin and α-synuclein, and have supported the notion that abnormal protein handling might be important in PD pathogenesis. Furthermore, mutant isoforms of α-synuclein more readily oligomerize and it has been suggested that its tendency to aggregate into misfolded structures may confer toxic properties to the protein. Indeed, overexpression of wild-type or mutant protein in transgenic mice or Drosophila reproduces many of the behavioral and pathological features of PD.32,33
Multiplications of the wild-type α-synuclein gene have been described in PD families. A triplication of the gene was identified in a large autosomal dominant kindred with PD and tremor,34 and duplication of the gene was found in one of 42 familial probands of early onset PD.35 A third α-synuclein point mutation (E46K) has been reported in an autosomal dominant family with parkinsonism and Lewy body dementia.36
Several models of abnormal α-synuclein expression have been developed. Knockout of the gene in mice resulted in no detectable abnormality other than an alteration of dopamine release in response to rapid stimulation, although this has no clear functional correlate.30 Overexpression of wild-type human α-synuclein in mice resulted in loss of dopaminergic terminals, intranuclear and cytoplasmic ubiquitin-rich nonfibrillar α-synuclein inclusions in the substantia nigra, hippocampus, cortex, and a rotor-rod motor deficit at 1 year.32 Overexpression of human wild-type and mutant α-synuclein in flies caused a loss of dopaminergic neurons, Lewy body–like inclusions with fibrillar α-synuclein, and a motor deficit with no significant difference between wild-type and mutant α-synuclein.33 Additional mouse models of α-synuclein expression have demonstrated inclusion formation and spinal cord pathology but no dopaminergic cell loss, and motor deficit at late stage.37 Virus-mediated overexpression of α-synuclein induces nigral degeneration in rodents.38
The α-synuclein mutations result in the protofibrillar form, which is considered the more toxic form of the protein. The A53T and A30P α-synuclein mutations promote protofibril formation and A30P inhibits conversion to fibrils.39 Catecholamines, including dopamine and levodopa, inhibit fibril formation in vitro, and this is reversed by antioxidants; that is, catechol oxidation promotes protofibril formation.40 This observation would support a protective role for Lewy bodies in PD. An important observation revealing a potential toxic mechanism for α-synuclein is that the mutant A30P form increases toxicity to dopamine, increasing cell death and free radical–mediated damage.41 The authors proposed that the mutation impaired vesicular uptake of dopamine, resulting in higher cytoplasmic or extravesicular synaptic concentrations of dopamine that would in turn cause free radical–mediated damage. Phosphorylation at the Ser129 residue is required to mediate the toxicity of α-synuclein and increased the formation of inclusions in SHSY-5Y cells.42 This phosphorylated form of α-synuclein is present in Lewy bodies.43 Prevention of this phosphorylation by substitution of an alanine residue reduced inclusion formation in the SHSY-5Y model, and in the Drosophila model this same mutation at 129 that prevents phosphorylation protected against dopaminergic neuronal loss.44
Parkin (PARK2)
PARK2 gene mutations were first identified in autosomal recessive juvenile-onset parkinsonism (ARJPD).45 ARJPD has been most commonly seen in the Japanese population and is characterized by onset before age 40 years, symptomatic improvement following sleep, mild dystonia, and a good response to levodopa.46 Resting tremor is seen less frequently than in idiopathic PD, and patients may have brisk tendon reflexes but no other pyramidal, cerebellar, or autonomic features. The disease is often symmetrical and dyskinesias develop early but progression is usually slow. Pathologically, there is dopaminergic cell loss in the SNc and locus ceruleus but no Lewy bodies are seen.47 The gene responsible for ARJPD was mapped to 6q25.2-q27,48 and in 1998 the gene was discovered and named parkin.45 Affected patients carry deletions or point mutations in various parts of the parkin gene.49,50 The absence of Lewy bodies in ARJPD may simply reflect the limited time over which the pathology has evolved. However, the relationship of parkin mutations to idiopathic PD has been highlighted by the identification of parkin mutations in apparently sporadic cases of PD and by the description of Lewy bodies in parkin positive patients with later-onset disease than ARJPD.51,52 Parkin mutations are a common cause of PD under age 25 years but rare over age 40 years.53,54 Parkin-related PD has been reported in multiple generations in families without consanguinity, suggesting a pseudo-autosomal dominant mode of inheritance for some mutations.55,56 Fluorodopa positron emission tomography (PET) in parkin patients demonstrates reduced uptake in the striatum, although there is some discordance regarding the symmetry and pattern of this reduction57,58 However, the rate of loss of fluorodopa PET signal was slower in the parkin patients than in sporadic PD.59 Asymptomatic heterozygous parkin mutation carriers had intermediate levels of striatal fluorodopa PET uptake compared with normal controls and homozygous symptomatic patients.58 This suggests an intermediate stage of nigrostriatal dysfunction that may interact with other genetic or environmental factors to induce PD. The frequency of heterozygous parkin mutation carriers is not known.
PARK2 encodes parkin, which functions as an E3 ligase, ubiquinating proteins for destruction by the proteosome.60,61 Several substrates for parkin have been identified, including a 22-kDa glycosylated form of α-synuclein, parkin-associated endothelin receptor-like receptor (Pael-R), and CDCrel-1. Overexpression of Pael-R causes it to become ubiquinated, insoluble, and unfolded and leads to endoplasmic reticulum stress and cell death.62 It has been demonstrated to accumulate in its insoluble form in the brains of patients with parkin mutations, suggesting a possible toxic mechanism. CDCrel-1 is a protein involved in cytokinesis and may influence synaptic vesicle function.61 Overexpression of parkin protected against dopaminergic loss in rodents coexpressing α-synuclein, suggesting a protective role for parkin.63
A parkin knockout mouse model has been described.64 This showed an increase in striatal extracellular dopamine, a reduction in synaptic excitability, and a mild nonprogressive motor deficit at 2 to 4 months. There was no loss of dopaminergic neurons and no inclusion formation. Dopamine receptor binding affinities and parkin E3 ligase substrate levels were normal. Interestingly, these mice had decreased striatal mitochondrial respiratory chain function and reductions in specific respiratory chain and antioxidant proteins.65 Parkin knockout flies developed muscle pathology, mitochondrial abnormalities and apoptotic cell death.66 Overexpression of parkin in PC12 cells indicated that it is associated with the mitochondrial outer membrane.67 Parkin-positive patients have decreased lymphocyte complex I activity.68 The ability of parkin to ubiquinate proteins may be impaired by S-nitrosylation, which in turn may be a consequence of excitotoxicity-mediated damage.69
UCH-L1 (PARK5)
A further mutation in the gene encoding ubiquitin carboxyhydrolase (UCH)-L1 again supported the relevance of the ubiquitin-proteosomal system (UPS) in PD pathogenesis.70 UCH-LI is an enzyme that hydrolyzes the C-terminus of ubiquitin to generate ubiquitin monomers that can be recycled to clear other proteins. A missense mutation was identified in two siblings with typical PD in a German family demonstrating apparent autosomal dominant inheritance.70 Age at onset was 49 years in one and 51 years in the other. The mutant form of UCH-L1 was shown to have reduced enzyme activity resulting in impaired protein clearance through the ubiquitin-proteasome pathway. However, no other mutations in this gene have been identified in other families, suggesting it is a rare cause of PD.71,72 Given that no further cases of PD have been described with mutations in this gene, some doubt has been cast on the relevance of UCH-L1 to PD.
PINK1 (PARK6)
The PARK6 locus (chromosome 1p3673) was first identified in a large consanguineous Italian family and subsequently in an additional three Italian families and others from Europe and elsewhere, including Asia.74–78 The mean age at onset ranges from 21 to 57 years. Progression is usually slow and patients exhibit a good response to levodopa. PARK6 mutations appear to be a rare cause of PD.
The PINK1 (PTEN-induced kinase 1) gene is ubiquitously transcribed and is believed to encode a mitochondrial kinase.74,79 It is believed that PINK1 may play a role in protecting cells against stress conditions that affect mitochondrial membrane potential, but the downstream targets through which PINK1 mediates its protection have not been identified. As 11 of the 14 reported mutations fall into the kinase domain of PINK1,74,76,77 altered phosphorylation of target proteins probably represents a key pathogenic mechanism, leading to abnormal stress response and neurodegeneration. The reversible phosphorylation of proteins is an important method of regulating cellular activities.80 Up to 30% of eukaryotic proteins are phosphorylated,81 and there are more than 500 human genes encoding protein kinases.82 The phosphorylation of mitochondrial proteins is considered pivotal to the regulation of respiratory activity in the cell and to signaling pathways leading to apoptosis, as well as for other vital mitochondrial processes. For instance the phosphorylation of α-synuclein is an important step in mediating its toxicity (see earlier), and Lewy bodies do contain the phosphorylated form of this protein.
DJ-1 (PARK7)
The PARK7 locus on chromosome 1p36, only about 25 cM from the PARK6 locus, was first identified in a small group of young-onset PD patients in a remote region of Holland.83 Average age at onset is 32 years, with a currently reported range of 25 to 40 years. Onset is asymmetrical, progress is slow, and there is a good response to levodopa. Tremor is infrequent, and psychiatric disturbances have been described in some. Fluorodopa PET scans demonstrate a symmetrical reduction in uptake. No pathological studies of PARK7 patients have been undertaken at the time of writing.
PARK7 encodes DJ-1; mutations are autosomal recessive and comprise both deletions and point mutations that result in a loss or inactivation of the protein. Its function is unknown, but it is widely distributed and conserved. It can protect against toxicity mediated by free radicals and transfers to the outer mitochondrial membrane under conditions of oxidative stress.84,85 Wild-type DJ-1 is also located in the mitochondrial matrix and intermembraneous space, and this distribution is not altered by mutations in the protein.86
LRRK2 (PARK8)
Mutations in the LRRK2 gene are the most common cause of either familial or “sporadic” PD identified to date. The LRRK2 G2019S mutation alone has been reported in 2.8% to 6.6% of autosomal dominant PD families87–89 and in 2% to 8% of sporadic cases.90–92 The G2019S mutation has variable penetrance, with 17% at 50 years and 85% at 70 years, a profile that mimics idiopathic, sporadic PD. Although other LRRK2 mutations are described, the G2019S mutation remains the most common cause of either sporadic or familial PD. This mutation has not been seen in Alzheimer’s disease or in parkinsonian syndromes other than idiopathic PD.93,94
Many of the reported cases of LRRK2 mutations have typical features of PD with asymmetrical onset of tremor, bradykinesia, and rigidity. As noted, the age at onset is variable with occasional very late onset cases89 and a report of one carrier male reaching 89 years with only subtle neurological changes.95 Patients have a good response to levodopa but develop motor complications including dyskinesias. Fluorodopa PET and imaging using ligands for the dopamine transporter with single-photon emission computed tomography (SPECT) demonstrate changes typical of those seen in idiopathic PD.96 Although all LRRK2 mutant brains examined to date demonstrate loss of dopaminergic neurons in the SNc, one of the morphological hallmarks of idiopathic PD, additional pathology may also be seen. Pure nigral neuronal degeneration was found in the first family linked to this locus97; neurofibrillary tangles, abnormal tau deposits, and widespread Lewy body synucleinopathy have been described in others, including one family with anterior horn cell loss.98,99 Three brains of “sporadic” PD with G2019S LRRK2 mutations have had pathological examination and all have demonstrated nigral neuronal loss and Lewy body formation typical of PD.90 All of these subjects had PD based on clinical criteria.
The LRRK2 gene encodes a 286-kDa cytoplasmic protein that is widely expressed in the brain.100 LRRK2 is a member of the ROCO protein family and appears to have multiple functions, at least by virtue of its predicted structure. These include a Ras/GTPase domain involved in cytoskeletal responses to external stimuli, vesicular trafficking and the stimulation of stress-activated kinase.101 The leucine-rich motif may have numerous functions, including protein-protein interactions and substrate binding for ubiquitination. The LRRK2 kinase domain belongs to the MAPKKK family of kinases with catalytic activity for both serine/threonine and tyrosine residues.
The G2019S mutation changes a highly conserved glycine at the start of the kinase activation segment, and it has been postulated that this has an activating effect causing a “gain of function” compatible with its autosomal dominant inheritance pattern.89 LRRK2 also has a WD40 domain, which again may be involved in cytoskeletal assembly and signal transduction.
PARK9, PARK10, and PARK11
The PARK9 locus on chromosome 1p36 was described in an autosomal recessive, juvenile-onset parkinsonian disorder with pyramidal features, ophthalmoplegia, and dementia.102,103 PARK10 on chromosome 1p32 was identified in the families of Icelandic PD patients with late-onset disease.104 PARK11 was obtained by association studies, and little information is available on phenotype.
Genetic Associations
Only a minority of cases of PD are part of a clear familial pedigree. Some of the single-gene mutations described above may account for a proportion of the remaining patients. However, our current understanding is that such single-gene causes of PD will remain in the minority. Thus, the large proportion of PD patients may develop their disease as a result of environmental factors, polygenic influences, or a combination of the two. There have been several genetic association studies attempting to determine significant polymorphisms that may increase or decrease the risk for PD. Further evidence for the role of genes in PD comes from genome-wide screens.105,106 The first found 174 families with a minimum of two clinically affected individuals with PD per family and identified a marker from the intronic region of parkin in an earlyonset group and a region on 9q in a dopa-resistant group. In the total sample, areas of interest were found on chromosomes 5q, 8p, and 17q. Data from the second genome-wide linkage study used a sample of 113 affected sibling pairs with PD and identified suggestive linkage on chromosomes 1, 9, 10, and 16, with no evidence implicating the regions containing parkin, α-synuclein, or tau genes. However, additional studies have shown that α-synuclein promoter region variants can influence the risk for PD.107,108 Those alleles that increase α-synuclein expression lead to an increased risk for developing PD, an observation in line with the multiplications of the gene causing familial PD (see earlier). Similarly, variants that influence parkin expression can also modulate the risk for PD: in this case, those alleles that lower parkin expression enhance the risk for PD.109 Mutations in the gene for glucocerebrosidase have been associated with an increased risk for PD among Ashkenazi Jews.110 Glucocerebrosidase deficiency causes type 1 Gaucher’s disease. Thirty-one of 99 Ashkenazi PD patients had one or two mutant alleles of the glucocerebrosidase gene (GBA), and 95 of 1543 controls (Ashkenazi) were carriers. Those with GBA mutations had onset of their PD around age 60 years and clinical features typical of idiopathic disease.
Genetic Causes of Parkinsonism
Several disorders that include parkinsonism in their phenotype have been characterized at the genetic level (Table 71-2). The frontotemporal dementias are discussed in detail in Chapter 73, the dystonias in Chapter 35, Huntington’s disease in Chapter 67, Wilson’s disease in Chapter 108 and the inherited ataxias in Chapter 68, so these will be discussed only briefly here.
The frontotemporal dementias and parkinsonism linked to chromosome 17 (FTDP-17) usually have onset in the fifth decade, although the age range is wide (25 to 75 years). Symptoms are of gradual onset and include motor dysfunction in the forms of parkinsonism, behavioral, and personality disorders and cognitive decline.111 Patients may exhibit apathy, depression, aggression, disinhibition, obsessive-compulsive disorder, executive dysfunction, and nonfluent aphasia. Patients and families tend to fall into either the predominantly parkinsonian or dementia types. Pathological examination shows severe frontotemporal atrophy and degeneration that includes the substantia nigra and the basal ganglia. There are tau accumulations in the remaining neurons and glia. FTDP-17 is autosomal dominant and is due to mutations in the tau gene. Although the genotype-phenotype relationship is relatively loose, those with the parkinsonism predominant form more commonly have exon 10′ or 5′ mutations.
Certain of the spinocerebellar ataxias (SCAs) are associated with parkinsonism and indeed may even manifest with this feature. SCA2 dopa-responsive parkinsonism is most often observed in the Chinese Asian population.112,113 Patients may have asymmetrical disease; a resting tremor and the presence of ataxia and abnormal eye movements may make differentiation from other parkinsonian disorders difficult if genetic testing is not performed. Imaging with fluorodopa PET has produced variable results from changes typical of those seen in PD114 to severe involvement of the caudate.115 The demonstration of an abnormally expanded CAG repeat in the ataxin-2 gene confirms SCA2. SCA3 (Machado-Joseph disease) mutations in conjunction with parkinsonism have been found most often in Caribbean populations.
Fragile X mental retardation complex is a common cause of mental retardation. It is an X-linked disease caused by an abnormal CGG expansion in the FMRI gene, which results in reduced gene expression. Intermediate length repeats can be a cause of tremor-ataxia parkinsonism in men. About 60% of these patients have a postural tremor, ataxia, autonomic dysfunction, impaired cognition, and symmetrical parkinsonism.116,117
Dystonia in association with parkinsonism is seen in a number of genetic diseases. X-linked dystonia parkinsonism was first reported in men from an island in the Philippines who had early onset of action tremor, dystonia, blepharospasm, and parkinsonism in 40% (Lubag) with poor response to levodopa. This disease is referred to as DYT3.118 Rapid-onset dystonia parkinsonism (DYT12) is an autosomal dominant disorder associated with bulbar features including dysarthria and dysphagia, dystonia, postural instability, and bradykinesia. Symptoms progress rapidly over hours and may be precipitated by physical or emotional stress. There is usually a poor response to levodopa. Mutations in the ATP1A3 gene that encodes a subunit of the sodium-potassium channel have been described in this disorder.119
Mitochondria have their own DNA, and the mitochondrial genome encodes 13 proteins of the oxidative phosphorylation system in addition to 2 ribosomal and 22 transfer RNAs. The discovery of complex I deficiency in PD substantia nigra (see later) raised the possibility that the mutation of genes (nuclear or mitochondrial) encoding complex I subunits might be involved in determining the enzyme’s defective activity. As mitochondrial DNA (mtDNA) is inherited in a strictly maternal pattern, if there were full penetrance of such a mtDNA gene defect, mitochondrial inheritance should be identifiable in pedigrees with parkinsonism. Such maternal inheritance has been described in PD120 but appears rare. However, it is known that 40% of patients with proven mitochondrial diseases and mtDNA mutations present as sporadic cases. Thus maternal inheritance is not a sine qua non of mtDNA gene defects. However, molecular genetic investigations of mtDNA have so far been unable to identify any specific mutation that clearly co-segregates with PD.
Studies using age-matched controls found no increase in the 5-kilobase mitochondrial deletion mutation in PD substantia nigra.121 Several studies have sequenced mtDNA in PD, but these have all used unselected patients in terms of their complex I activity.122,123 Although some reports have suggested an increased frequency of certain mtDNA polymorphisms in PD, this has not been replicated in all studies.124–128 Two studies have demonstrated a relationship between mtDNA haplotypes and the risk for developing PD. The first showed a reduced risk for PD in individuals with haplotypes J and K,129 and the second, a 22% decrease in PD in those with the UKJT haplotype cluster.130 In contrast, a smaller study reported an increased risk for PD with haplotypes J and T.131
Mutations in the gene for mtDNA polymerase gamma (POLG) have been demonstrated in patients with progressive external ophthalmoplegia and parkinsonism. Autosomal dominant or recessive inheritance of progressive external ophthalmoplegia with age at onset ranging from 10 to 54 years was followed some years later (range, 6 to 40 years) by the development of an asymmetrical, levodopa-responsive bradykinetic rigid syndrome together with resting tremor in some patients. Additional features included variable limb, pharyngeal or facial weakness, cataracts, ataxia, peripheral neuropathy, and premature ovarian failure.132 Muscle biopsy demonstrated ragged red, cytochrome oxidase–negative fibers in all patients with multiple mtDNA deletions on Southern blotting. Symmetrically reduced striatal 18-fluorodopa PET was seen in two patients. Brain histology was available on an additional two patients; both showed severe loss of substantia nigral dopaminergic neurons but without the development of Lewy bodies or other synuclein aggregates. Four families had the same A2864G mutation inherited in autosomal fashion in three and with a founder effect in the fourth. Mutations in the exonuclease or polymerase portions of the gene were identified in the autosomal recessive families. Another patient with autosomal dominant progressive external ophthalmoplegia parkinsonism and an A2492G mutation has been reported.133
Environmental Factors
Several studies have sought to define the environmental contributions to the etiology of PD. A rural residency appears to increase the risk of the development of PD and, in particular, young-onset PD.134–136 However, this finding has not been confirmed in all studies.137 Rural living is associated with farming and pesticide use, and an association with the agricultural industry has been found with increased incidence in PD patients.138–140 In addition, another lifestyle study showed increased herbicide exposure in patients with PD.141 Organochloride pesticides were identified as risk factors in a German case-control study142 with the offending agent being identified as the organochloride dieldrin, which was found in 6 of 20 PD brains and none of 14 control brains.143 Another study identified dithiocarbamates as a risk factor for PD,137 a compound that has also been shown to enhance 1-methyl 4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) toxicity.144 Some studies have found that the significant association of PD with farming as an occupation cannot be accounted for by pesticide exposure alone.139 Another rural factor that has been linked to PD is the consumption of well water,145 although this may simply be further evidence in support of herbicides or pesticides as etiological factors for PD.
Carbon monoxide is a common environmental pollutant that may also play an important role in cell signaling. Acute carbon monoxide poisoning results in the complexing of carbon monoxide with the ferrous iron, protophorphyrin IX, and this prevents the carriage of oxygen. In addition, carbon monoxide is a potent inhibitor of cytochrome oxidase (complex IV) of the mitochondrial respiratory chain. Survivors of carbon monoxide poisoning have developed parkinsonism within a few days or weeks of exposure.146 The affected patients show necrosis of the globus pallidus on computed tomography scanning and magnetic resonance scanning.
Pyrethroid pesticides, when administered parenterally to rodents, cause a reduction in tyrosine hydroxylase–positive dopaminergic neurons in the nigrostriatum and an increase in dopamine transporter and brain-derived neurotrophic factor expression.147–150 These results indicate that commonly used pesticides can cross the blood-brain barrier and induce damage to the basal ganglia. Rotenone, a pesticide commonly used in the United States, when infused into rodents, can result in degeneration of nigrostriatal neurons and the formation of α-synuclein–rich Lewy-like inclusions.151 Paraquat, a widely used herbicide, has been shown to increase α-synuclein fibril formation in vitro in a dose-dependent fashion and to increase α-synuclein protein expression in mice with the reversible development of aggregates in substantia nigra dopaminergic neurons.152 Anonnacin, a component of sour-sop in the Caribbean, has been shown to produce a PD-like phenotype in humans and nigrostriatal loss in animals.153 MPTP, a meperidine analog designer drug, is known to produce parkinsonism in humans, other primates, and rodents through uptake and conversion mechanisms that target the nigrostriatal pathway,154 It is noteworthy that these agents result in inhibition of mitochondrial NADH CoQ reductase (complex 1) and are free radical generators, features of direct relevance to idiopathic PD.
Manganese is a constituent of several pesticides and herbicides as well as being an anti-knock additive to lead-free petrol. Manganese is neurotoxic to the basal ganglia and produces a parkinsonian syndrome with damage predominantly to the globus pallidum. There have been numerous reports of manganism developing among individuals exposed to manganese dioxide ore, usually by inhalation of manganese dust. Exposure is typically chronic over 6 months to 16 years, and the onset of manganism is slow, beginning with apathy, muscle weakness and cramps, and general irritability. Progression occurs and is characterized by dysarthria and psychosis followed by severe rigidity, anarthria, and dystonia.155 Manganese predominantly appears to affect the globus pallidus with sparing of the substantia nigra.156 There has been interest in the potential for manganese containing steel to induce parkinsonian features in welders,157 although this association is controversial. At present, it is not known if manganese-containing pesticides can induce PD.
Between 1917 and 1919, there was an epidemic of an influenza-like illness starting in Austria and France and spreading throughout Europe and North America. The illness was characterized by fever, headache, lethargy, and paralysis, particularly of the extraocular muscles. Following this, stupor, coma, sleep disturbance, and seizures could occur. Ocular gyric crises were seen in a high proportion of patients. Mortality was 30% to 40%, and parkinsonism developed in the majority of survivors over the next 10 years.158,159 The specific agent causing encephalitis lethargica was never identified. Although there has been no outbreak of encephalitis lethargica since the 1920s, infection as a cause for PD has still attracted some attention. There are numerous anecdotal reports of infections, particularly encephalitis, being associated with parkinsonism. These include a wide variety of viruses, bacteria (including Borrelia burgdorferii [Lyme disease]), and even fungi, such as Cryptococcus or Aspergillus. However, there is no evidence to suggest that any of these are relevant to the vast majority of patients with idiopathic PD. For instance, patients who develop PD before the age of 40 have no greater history of central nervous system infection than do patients who develop the disease over the age of 60. Intrauterine exposure to, for instance, the influenza viruses pandemic from 1890 to 1930 has not been supported by any association with year of birth.160 Searches for viral particles of antigens within the brains of patients with PD have not proved rewarding.161
Two environmental factors are recognized to lower the risk for PD: cigarette smoking162 and coffee drinking.163 The mechanisms through which they can reduce risk are not known. Coffee drinking appears more protective for men, so it is possible that there is an interaction with endocrine factors. There is evidence of active inflammatory change in the substantia nigra at the time of death in PD, with microglial activation, and expression of proinflammatory cytokines.164–166 The role of this inflammatory change is unknown but has been believed to be relevant to pathogenesis and neuronal damage. Similar changes have been seen in AD brain and prompted retrospective analyses that subsequently demonstrated the potential for antiinflammatory agents to reduce the risk for AD, although this effect remains controversial.167,168 A similar study in PD has also shown that use of a nonsteroidal antiinflammatory drug two or more times per week can produce a 45% lower risk for PD.169
PATHOGENESIS
Iron
High iron concentrations are found in control substantia nigra, globus pallidus and striatum. In PD, there is a 35% increase in substantia nigra iron levels.170,171 Other degenerative diseases involving cell loss in the basal ganglia also showed increased iron in these areas, such as progressive supranuclear palsy and multiple system atrophy. These studies suggested that increased iron concentrations were a reflection of neuronal cell loss rather than any specific pathogenetic factor. High concentrations of iron were also found in macrophages, astrocytes, and reactive microglia in the PD substantia nigra.172 One study, however, using x-ray microanalysis, found increased levels of iron in neuromelanin. In this respect, neuromelanin could again act as a toxic sink.173 In contrast, another study found no difference in iron concentrations between melanized and nonmelanized cells in controls but a significant increase in the cytoplasm of dopaminergic neurons.174 However, there was no apparent correlation between the high concentrations of iron and morphological alterations in the neurons that might suggest degeneration.
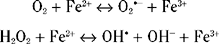
Thus, if iron is available in a free and reactive form, it has the potential for exacerbating oxidative stress and damage. Iron is normally bound to ferritin, which exists in two forms, H and L. Most brain ferritin is in the H form. Three studies have now been undertaken on ferritin concentrations in PD brain. One used a polyclonal antibody predominantly against L-ferritin and found a significant decrease in the concentration of this protein in PD substantia nigra and other areas.175 This decrease was not seen in other parkinsonian syndromes such as progressive supranuclear palsy or multiple system atrophy. Another study,176 again using an antibody against mainly L-ferritin, found an increase in the number of ferritin-positive microglia in substantia nigra. This latter work used immunohistochemistry and therefore is not directly quantifiable. In addition, this study incorporated parkinsonian syndromes as well as idiopathic PD into the disease group (M. Youdim, personal communication). A third and more comprehensive study involved monoclonal antibodies against both L and H ferritin together with a double capture technique incorporated into an enzyme-linked immunosorbent assay study together with Western blotting studies of PD substantia nigra protein.177 The results did not identify any significant difference in ferritin levels between control and PD substantia nigra. Thus, there are no hard data that ferritin levels are abnormal in PD. Indeed, ferritin has such a high iron binding capacity that the increase of iron noticed in PD brain may not require any increased buffering capacity from ferritin.
Oxidative Stress and Damage
There are several lines of evidence that suggest increased oxidative stress and oxidative damage to biomolecules in PD substantia nigra (Table 71-3).
The free radical gas nitric oxide (NO•) is present in many tissues, including the central nervous system. NO• is generated by the conversion of L-arginine to L-citrulline by nitric oxide synthase (NOS). At least three NOS isoforms are recognized and are all expressed within the brain. NO• acts as an atypical molecular messenger but at higher concentrations may have a toxic role and be implicated in the neurodegeneration that occurs in PD. As a free radical, NO• could potentially contribute to dopaminergic neuronal death by mechanisms such as increased lipid peroxidation, release of iron(II), and damage to DNA. It can also inhibit a number of enzymes such as cytochrome c oxidase and superoxide dismutase and affects mitochondrial function by inhibiting complexes II, III, and IV. Animal studies have implicated NO• in nigrostriatal neuronal loss. In addition to its possible neuroprotective effect with regard to 1-methyl-4 phenylpyridinium (MPP+) toxicity, 7-nitroindazole (7-NI) also protects in the methamphetamine animal model of PD.186 NOS activity is at its highest in the nigrostriatal system in nonhuman primates and humans. Attempts to demonstrate altered levels of NO• in the brains of PD patients have been inconclusive with both decreased and increased levels of cerebrospinal fluid nitrate, a marker of NOS activity, being seen.187–189
Mitochondrial Dysfunction
The first link between mitochondria and PD was made in 1989 when a defect in the activity of respiratory chain protein complex I was identified in substantia nigra from patients with PD.190,191 This study has been expanded over the years, and results to date show that there is a specific defect of approximately 35% complex I deficiency in PD nigra.177 This defect in complex I activity in PD brain does not affect any other part of the respiratory chain. In addition, no defect in mitochondrial activity has been identifiable in any other part of PD brain, including the caudate putamen, globus pallidus, tegmentum, cortex, cerebellum, and substantia innominata.192
Following the report of complex I deficiency in PD substantia nigra, respiratory chain abnormalities were described in skeletal muscle mitochondria from PD patients. This particular area has proved very contentious, with several groups either describing similar defects or no abnormality whatsoever (see Schapira193 for review). Two magnetic resonance spectroscopy studies on skeletal muscle mitochondrial function in PD have shown conflicting results.194,195 Finally, mitochondrial complex I deficiency was also identified in platelet mitochondria of PD patients.196–198 In contrast to skeletal muscle, there is a consensus among several laboratories that complex I deficiency does exist in PD platelet mitochondria. The majority of studies, however, suggest that this deficiency, as least based on a group-to-group analysis, is modest (about 20% to 25%) (see Schapira193 for review). The complex I deficiency in PD lacks the sensitivity to allow its use as a biomarker of PD.
CLINICAL FEATURES
Presymptomatic
Olfactory dysfunction is common in PD and eventually affects up to 90% of patients.199 It has been suggested that hyposmia may be a preclinical marker for PD,200,201 and olfactory deficits have been reported in asymptomatic relatives of patients with PD, some of whom subsequently developed PD.202,203 A prospective study involving 361 asymptomatic relatives of PD patients identified 40 with hyposmia.9 Within 2 years of follow-up, 10% of this subgroup had developed PD and another 12% had detectable presynaptic abnormalities on their dopamine transporter SPECT scan. No relative with normal smell had an abnormal SPECT scan or developed PD. It is tempting to relate these findings to Braak’s findings of Lewy body distribution in the olfactory nucleus in stage 1. Olfactory dysfunction has also been reported in diffuse Lewy body disease and multiple system atrophy. Vascular parkinsonism, cortocobasal degeneration, progressive supranuclear palsy, and parkin-associated PD usually have intact olfactory function.204–206
Rapid eye movement (REM) behavior disorder (RBD) is a common sleep disorder in PD. It is characterized by loss of the normal skeletal muscle atonia during REM sleep, thus enabling patients to physically enact their dreams, which are often vivid or unpleasant.207–210 Vocalizations (talking, shouting, vocal threats) and abnormal movements (arm/leg jerks, falling out of bed, violent assaults) are commonly reported by bed partners. In PD, up to one third of patients meet the diagnostic criteria of RBD.207,208 RBD appears to frequently precede the development of motor signs of PD and longitudinal data that suggest that RBD heralds the onset of motor symptoms in up to 40% of PD patients.211,212 In patients with isolated RBD, imaging studies have indicated a small but significant reduction in striatal dopaminergic uptake that may suggest preclinical PD.213,214 The anatomical basis of RBD is believed to involve the pontomedullary area resulting from degeneration of lower brainstem nuclei like the pedunculopontine and subceruleal nucleus; this area is consistent with Braak stages 1 and 2.
Constipation often precedes the diagnosis of PD.215–218 A prospective study of 7000 men for 24 years assessed inter alia for bowel habits found that those with constipation (less than one bowel movement per day) had a threefold risk of subsequently developing PD.219 The mean interval between the administration of the bowel questionnaire and the development of PD was 10 years. Colonic dopaminergic neurons degenerate with Lewy body formation in PD, although constipation does not respond well to dopaminergic treatment.220–222
Motor Features
Rigidity represents an increase in tone that is present throughout the range of movement and is independent of the speed at which the limb is moved. The tremor of PD may superimpose on rigidity to produce cog-wheeling, and this phenomenon may be absent if there is no tremor. Examination of the wrist with gentle flexion-extension movements is the best means to elicit cog-wheeling, and this can be repeated at the elbow. Rigidity affects the patient’s posture, producing a flexion at most joints including the spine, and this produces the simian posture typical of PD. An extreme form of this is known as camptocormia.223 Postural abnormalities also affect the distal limbs with extension of the fingers and flexion of the metacarpophalangeal joints or dorsiflexion of the great toe (striatal hand or toe).
The development of an asymmetrical intermittent resting tremor at 4 to 6 Hz is estimated to be a manifesting feature in 70% of PD patients. In addition, there is often a higher frequency (about 12 Hz) small-amplitude postural tremor.224 The resting hand tremor is referred to as “pill-rolling” in the style of the pharmacists of the nineteenth century, who would prepare their tablets by hand. A tremor may affect other parts including the foot or leg (in which it may first manifest), lips, jaw, and tongue.225 A head tremor, titubation, is more suggestive of essential tremor. The resting tremor is exacerbated by physical or emotional stress and can in the early stages be voluntarily inhibited for short periods. The tremor usually becomes bilateral after about 5 to 6 years, although the first affected side most often remains the more severe.226
Nonmotor Features
The widespread and progressive neurodegeneration in the PD brain leads to the emergence of a variety of features that are collectively grouped under the title of nonmotor symptoms. These are predominantly, but not exclusively, the consequence of loss of nondopaminergic pathways (Table 71-4). The nonmotor symptoms of PD range from cognitive problems such as apathy, depression, anxiety disorders, and hallucinations to fatigue, gait and balance disturbances, hypophonia, sleep disorders, sexual dysfunction, bowel problems, drenching sweats, sialorrhea, and pain. These symptoms are often the most troubling for patients and contribute significantly to morbidity and impaired quality of life.227 Diplopia is a frequent symptom even in early PD, although the neurological basis is not known.
| Motor | Postural instability |
| Gait disorders | |
| Speech problems | |
| Mental changes | Dementia |
| Depression | |
| Anxiety | |
| Apathy | |
| Autonomic nervous system dysfunction | Orthostatic hypotension |
| Constipation | |
| Sexual dysfunction | |
| Urinary problems | |
| Sweating | |
| Sensory phenomenon | Pain |
| Dysesthesias | |
| Sleep disturbances | Sleep fragmentation |
| Sleep apnea | |
| REM behavioral disorder |
Abnormalities of sleep are common in PD and are the result of a combination of the natural consequences of aging, the underlying disease pathology,228 motor and nonmotor complications,229,230 and drugs.231 Disordered sleep often results in excessive daytime sleepiness, and this in turn may be compounded by the sedative effect of dopaminergic drugs.232 Excessive daytime sleepiness and involuntary dozing affects up to 50% of PD patients and may be preclinical markers.233 In some, excessive daytime sleepiness has been linked to the development of sudden onset of sleep and a pattern reminiscent of narcolepsy with abnormal sleep latency period (<5 minutes) in 30% of PD patients. Polysomnographic studies have showed transition from wakefulness to sleep stage 2 within seconds without the sudden onset of REM sleep.234,235 Following reports of road traffic accidents caused by “sudden irresistible attacks of sleep” in eight PD patients, a large body of research focused on the possible effects of dopaminergic drugs and disease progression and the occurrence of sudden onset of sleep.232,236–238 The issue remains unclear, although is now regarded as part of the nonmotor complex of the disease progression in PD.239
Sexual and bladder dysfunction is common and occurs in both sexes. The dopaminergic treatment of PD may lead to increased sex drive, but the effects of the disease often result in impaired sexual performance.240 Bladder abnormalities particularly cause problems at night with nocturia, which when associated with bradykinesia during nocturnal “off” causes considerable discomfort.
Pain is a frequent symptom in PD, and some patients present especially with shoulder pain. Pain, anxiety, akathisia, respiratory distress, depressive mood swings, and slowed and impaired thought are symptoms that may be experienced during “off” periods and that will respond, at least in part, to dopaminergic therapy.241–243
DIFFERENTIAL DIAGNOSIS
The diagnosis of PD is best predicted by the presence of an asymmetrical bradykinetic rigid syndrome with a resting tremor and a good response to levodopa.244 The diagnostic specificity of these criteria is estimated at 98.6%, and sensitivity, at 91.1%.245 However, many patients still present a diagnostic challenge, especially those who have no tremor or those with marked asymmetrical tremor but limited bradykinesia or rigidity246 (Tables 71-5 and 71-6). Imaging studies on early PD patients recruited into neuroprotection trials indicate that approximately 10% have normal PET or SPECT scans.247,248 The Parkinson-plus disorders, dementia with Lewy bodies, Wilson’s disease, and tremor are covered in Chapters 72, 70, 108, and 33, respectively, and some of the additional diseases that may mimic PD have been discussed earlier. The clinical features of the main disorders that require differentiation from PD are covered only briefly in the context of diagnosis (see Table 71-4).
TABLE 71-6 Clinical Features of Parkinsonian Disorders
| Multiple System Atrophy | Corticobasal Degeneration | Progressive Supranuclear Palsy |
|---|---|---|
Drug-Induced Parkinsonism
Any dopamine receptor blocker has the potential to induce parkinsonism. In practice, the most common causes are the major neuroleptics, but drugs such as metoclopramide can also induce symptoms that may be confused with PD. A drug history is essential when considering PD. Clinical clues for drug-induced parkinsonism include symmetry and the absence of a resting tremor, although their presence does not exclude a drug cause. The presence of akathisia and orofacial dyskinesias supports a drug cause.249 Withdrawal of the drug may result in remission, although this can take months or years, and in some patients, the parkinsonism and related movement disorders are permanent.
Essential Tremor
Typical essential tremor comprises a bilateral usually symmetrical, visible, and persistent upper limb postural or kinetic tremor.250 Bradykinesia, rigidity, and postural abnormalities are not present. The tremor of essential tremor is present at rest in only 10% of cases251 but, when present or when asymmetrical, can cause difficulty with a distinction from PD, although such patients usually evolve to PD.252 The presence of a head or voice tremor, a strong and usually autosomal dominant family history, and improvement with alcohol all favor a diagnosis of essential tremor. In contrast, clear asymmetry, the presence of bradykinesia or rigidity, and leg tremor support a diagnosis of PD.
Multiple System Atrophy
Multiple system atrophy is a multicentric neurodegenerative disease that includes degeneration of the SNc and hence clinical features that can mimic PD. However, multiple system atrophy patients exhibit additional symptoms that may be predominantly cerebellar or parkinsonian with autonomic failure.253 Onset is similar in age to PD. The diagnosis of multiple system atrophy is supported by the presence of early cerebellar signs, autonomic failure such as bladder dysfunction or postural hypotension, early falls, pyramidal signs, myoclonus, bulbar features, pronounced anterocollis, and a rapid course. The response to levodopa is usually limited in degree and time,254 although some patients can develop dyskinesias, particularly of the orofacial and cervical musculature; they are rare.255
Progressive Supranuclear Palsy
Progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome) typically manifests with gaze palsy, particularly of the vertical downward plane, a staring appearance, symmetrical parkinsonism, and pseudobulbar palsy.256 Axial greater than limb rigidity and falls within the first year are suggestive of progressive supranuclear palsy.257 Response to dopaminergic therapy is poor.
Corticobasal Degeneration
Corticobasal degeneration usually manifests as an asymmetrical disorder that may include abnormal limb posturing (alien limb), dystonic upper limb posturing with irregular jerks, cortical sensory deficits, primary progressive aphasia, apraxia, and parkinsonism. Tremor is uncommon; cognitive disturbance, particularly involving frontal lobe function, is often seen and frequently is the manifesting sign.258 The disease is rapidly progressive, and there is no or only a very poor response to levodopa.
Vascular Parkinsonism
This is a diagnosis that can only be made with confidence in the clinic when there is evidence of widespread vascular disease and usually a history of stepwise progression. The clinical features are usually bilateral if not symmetrical; tremor is rare and gait abnormality is common. The latter manifests as a type of apraxia with a wide-based small stepping gait and freezing, so-called lower body parkinsonism.259 There may be a mild to moderate response to levodopa.
Dementia With Lewy Bodies
Dementia will develop in approximately 30% to 40% of PD patients, although it occurs at least 12 months (and usually longer) after the appearance of the motor features.260,161 The clinical diagnosis of dementia with Lewy bodies will be based on the presence of a progressive dementia with fluctuating attentional and visuospatial components, hallucinations, and parkinsonism that usually develops within 1 year of the dementia.
INVESTIGATION
Imaging by computed tomography is normal in idiopathic PD. There may be superimposed changes of atrophy or cerebrovascular disease, although in the case of the latter this should not be so severe as to raise the diagnosis of vascular PD. CT is helpful in excluding normal pressure hydrocephalus or the rare case of tumor (usually benign) causing parkinsonian features. Magnetic resonance imaging in routine clinical practice has greater definition than CT but does not reveal any specific features for PD. However, magnetic resonance imaging may be useful in helping to differentiate PD from other parkinsonian syndromes. Patients with multiple system atrophy may demonstrate brainstem, cerebellar, and dentate atrophy with hyperintensity of the middle cerebellar peduncle, cerebellum, inferior olives, and pontine fibers producing the “hot cross bun” sign.262,263 There may be putaminal atrophy or lateral putaminal hyperintensity (slit sign). Magnetic resonance volumetry is the most sensitive in distinguishing PD, multiple system atrophy, and progressive supranuclear palsy but is not widely available.264
Imaging with SPECT for the dopamine transporter has become increasingly available and can be used to differentiate PD from essential tremors or drug-induced PD, even in early disease. The signal in PD shows an asymmetrical reduction in transporter binding, particularly affecting the putamen (Fig. 71-3). It cannot distinguish PD from multiple system atrophy or progressive supranuclear palsy unless performed using voxel-based statistical parametric mapping.265 SPECT can be used to follow progression in PD, and it has been calculated that there is an annual loss of 8% to 11% of transporter signal in the early stages of PD.266,267 However, as mentioned, approximately 10% of patients believed by neurologists to have PD have normal imaging on PET or SPECT. It remains unclear whether these patients could have been distinguished by more accurate clinical assessment. SPECT can also be used to image sympathetic cardiac innervation in PD with iodine-123–labeled meta-iodobenzylguanidine. This shows postganglionic sympathetic denervation in PD patients, in contrast to retained innervation in multiple system atrophy or progressive supranuclear palsy, with 90% specificity and sensitivity, although the meta-iodobenzylguanidine scan may be normal in early PD.268,269
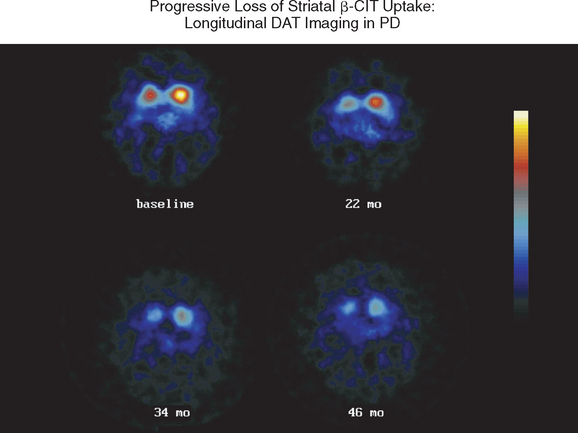
Figure 71-3 Sequential β-CIT uptake in Parkinson’s disease.
(From the Parkinson Study Group: Dopamine transporter brain imaging to assess the effects of pramipexole vs levodopa on Parkinson disease progression. JAMA 2002; 287:1653-1661. Copyright © 2002 American Medical Association. All rights reserved.)
Transcranial ultrasound has attracted increasing attention.270 This technique may demonstrate hyperechogenicity of the substantia nigra in over 90% of PD patients.271 Additional studies are required to assess its reproducibility in other centers and its application to diagnosis and the differentiation of PD from other parkinsonian diseases.
Routine neurophysiological studies are not helpful in the evaluation of PD patients. Autonomic function testing is useful in identifying patients with multiple system atrophy. For instance, 50% of multiple system atrophy patients have a 30 mm Hg systolic or greater than 15 mm Hg diastolic fall in blood pressure with head-up tilt compared with 20% of PD patients.246 Such drops in blood pressure occur earlier in the course of multiple system atrophy than in PD. The presence of sphincter denervation early in the course of disease favors multiple system atrophy, but none of the autonomic function studies accurately distinguishes PD from the parkinsonian syndromes, except progressive supranuclear palsy, in which autonomic function usually remains intact.272
A good response to dopaminergic therapy is considered an integral part of the confirmation of the diagnosis of PD. It has been suggested that a challenge with levodopa or apomorphine might be a useful aid when there is uncertainty regarding diagnosis.273,274 However, there are problems with interpretation, with 20% of parkinsonian multiple system atrophy patients having a positive response and positive responses may also be seen in progressive supranuclear palsy or vascular parkinsonism.275 In assessing a patient’s potential clinical response to levodopa, it is best to build up gradually to a dose of not less than 1000 mg for a duration of at least 2 months before an accurate view of responsiveness is obtained in the parkinsonian syndromes.
TREATMENT
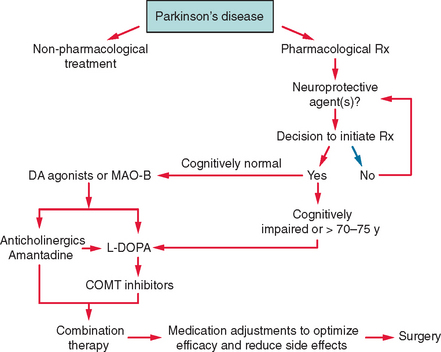
The treatment of PD comprises several stages determined by the natural progression of the disease and by the complications that can develop as a consequence of drug use. Dopaminergic agents are the drugs that are most effective in improving the motor deficits of PD and include levodopa, dopamine agonists, and the monoamine oxidase B (MAO-B) inhibitors. Several new drugs will shortly be released and reflect the rapid increase in treatment options for PD (Fig. 71-4). An algorithm for the use of drugs in PD is suggested in Figure 71-5.

Levodopa
Levodopa was the first drug to be used to replace the dopamine deficiency of PD and remains the “gold standard” against which the efficacy of others are judged. Only 1% of an oral dose of levodopa is absorbed into the blood because of extensive metabolism in the gut and so it is routinely combined with a dopadecarboxylase inhibitor to reduce peripheral metabolism that in turn both increases absorption to 10% and decreases side effects. Levodopa and other dopaminergic agents improve both the quality of life and life expectancy of PD patients.276–278 It provides rapid and effective relief of bradykinesia, rigidity, and associated pain and improves tremor in many patients. Levodopa improves symptoms in early PD patients by 12 or 13 Unified Parkinson’s Disease Rating Scale (UPDRS) points after 3 months.
Side effects are mainly gastrointestinal and consist of nausea, vomiting, and anorexia. These usually disappear over 2 to 3 weeks but may persist in some patients. They can be prevented or treated with domperidone 10 to 20 mg t.i.d., taken usually for a period of 2 to 4 weeks. Constipation, orthostatic hypotension, akathisia, hallucinosis, and daytime sleepiness are less common and are seen more often in the elderly population. Constipation, which can also be a consequence of PD itself, usually responds to standard treatments, including increased fluid, bowel training, timing of evacuation to the patient being “on,” and increased fiber intake. Symptomatic orthostatic hypotension may respond to simple advice regard ing postural change, maintaining hydration, the use of pressure stockings, antidiuretic hormone, midodrine, or fludrocortisone. Akathisia, hallucinosis, and daytime sleepiness can all occur as a consequence of PD itself and dopaminergic treatments in general. If due to the latter, they may respond to a reduction in dose. Hallucinations in particular are recognized as a consequence of PD pathology that develop in the mid to advanced stages of the disease.279 Some patients experience additional psychiatric effects from dopaminergic therapy, including obsessive traits, punding, and pathological gambling.280–282 These problems usually respond to a reduction in dopaminergic therapy but sometimes require the use of antipsychotic medication of the type less likely to exacerbate PD.
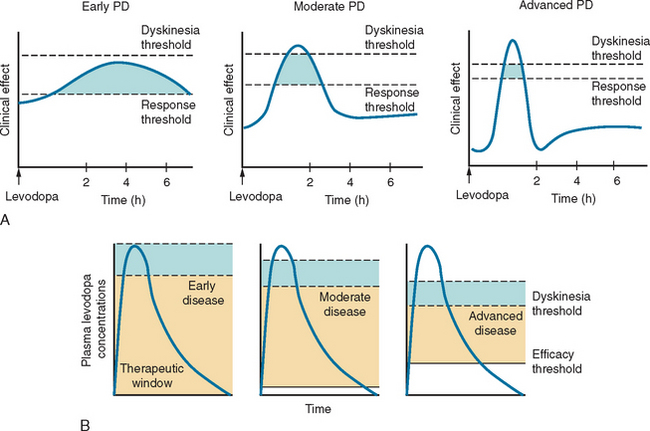
Levodopa has a long-duration response in early disease that enables adequate symptomatic control with dosage schedules of three times daily (Fig. 71-6). Disease progression, however, erodes the usefulness of levodopa as 70% of patients develop motor complications within 6 years of initiation of the drug.283,284 Wearing off effects frequently require modification of dosage and/or dose frequency or the introduction of additional or alternative therapies. Interestingly, so long as the plasma levodopa concentration is maintained, the clinical response will persist285,287 and “wearing off” does not occur if the drug is given by continuous infusion.288,289 A significant longterm complication of levodopa use is the development of dyskinesias. Dyskinesias develop at a rate of approximately 10% per annum, although this rate is much greater in young onset PD patients, of whom 70% will have dyskinesias within 3 years of levodopa initiation.290 The mechanisms by which these motor complications develop are not completely understood, but pulsatile stimulation of dopamine receptors by short-acting agents, including levodopa, and the degree of striatal denervation have been implicated.291 Dyskinesias may occur at the time of maximal clinical benefit and peak concentration of levodopa (peak dose dyskinesias) or appear at the onset and wearing off of the levodopa effect (diphasic dyskinesias). Motor complications can be an important source of disability for some patients who cycle between “on” periods, which are complicated by dyskinesias, and “off” periods, in which they suffer severe parkinsonism.
Catechol-O-methyl Transferase Inhibitors
The routine combination of levodopa with a dopa decarboxylase (DDC) inhibitor improves absorption but the majority of levodopa is still metabolized in the gut by catechol-O-methyl transferase (COMT), which produces 3-O-methyldopa. COMT inhibition therefore offers a strategy to increase levodopa absorption and improve kinetics (Fig. 71-7). Two selective COMT inhibitors are available for clinical use for the treatment of PD. These drugs exert profound influences on levodopa kinetics by increasing its bioavailability and elimination half-life. This allows more stable levodopa plasma levels to be obtained via the oral route and, conceivably, more sustained brain dopaminergic stimulation to be attained.
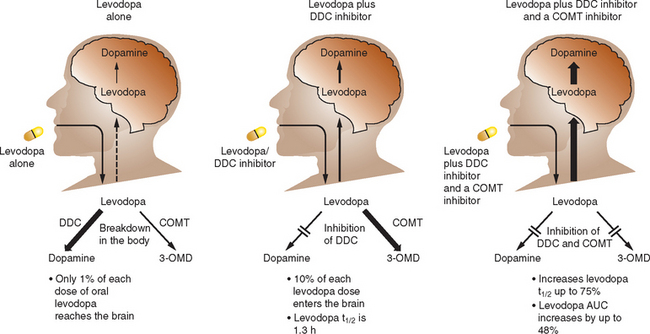
Entacapone is a selective, reversible inhibitor of COMT. It does not cross the blood-brain barrier and acts primarily in the gut. Entacapone essentially increases both the peripheral and central availability of levodopa. The plasma elimination of a 200-mg oral dose of entacapone is 1 to 2 hours. The pharmacokinetics of entacapone, particularly its elimination characteristics, are similar to those of levodopa, allowing coadministration of these agents. The recommended dose of entacapone is a 200-mg tablet administered with each dose of levodopa/carbidopa, up to a maximum of 10 times daily (in Europe) and 8 times daily (in the United States). It should be noted that only the dose of levodopa should be titrated; the dose of entacapone administered with each dose of levodopa should remain the same (200 mg). Entacapone is effective in patients with wearing-off–type motor fluctuations and can produce an increase in “on” time and a reduction in “off” time by an average of 60 minutes per day.292 The most common adverse effect seen with entacapone is dyskinesia, which reflects increased central dopaminergic activity. Reducing the daily levodopa dosage by about 25% may be necessary to minimize possible dopaminergic adverse effects. This reduction may be made at the time of entacapone introduction in those patients on more than 800 mg of levodopa daily or in those already with dyskinesias, but generally it is better to delay changing the levodopa dose until the patient’s response can be evaluated. Physicians should be aware that dopaminergic adverse events generally occur within 24 hours of initiating entacapone and may require an immediate adjustment of the levodopa dosage. Entacapone may be combined with both standard and controlled-release formulations of levodopa/carbidopa and may be administered with or without food.
Tolcapone (unlike entacapone) can cross the blood-brain barrier293 and may produce some central COMT inhibition, although its clinical effect is likely to be minimal. A study in newly diagnosed, levodopa-naïve patients with PD failed to show any clinical efficacy with the introduction of tolcapone either alone or with selegiline.294 Tolcapone has a similar half-life to entacapone; however, due to a greater bioavailability and smaller volume of distribution, tolcapone produces a greater inhibition of COMT and is required only on a three-times-daily regimen.295 Although tolcapone is available now in both Europe and North America, its use is restricted by its potential to cause severe hepatic toxicity.296 It should not be given to patients with impaired liver function, and those PD patients taking tolcapone require regular monitoring of hepatic enzymes. This effect on liver function is not seen with entacapone and probably reflects their differing potency in inducing mitochondrial permeabilization.297 The use of tolcapone is generally limited to those patients who have failed to derive significant benefit from entacapone. Both entacapone and tolcapone can induce diarrhea, which is more common and may be severe and explosive with the latter drug.298
Dopamine Agonists
Several dopamine agonists are available for use in PD and fall broadly into two groups: ergot and non-ergot. Ergot agonists include bromocriptine, cabergoline, lisuride, and pergolide, and non-ergot agonists include apomorphine, piribedil, ropinirole, and pramipexole. Bromocriptine, cabergoline, pergolide, pramipexole, and ropinirole have all been studied for monotherapy use in early PD,299–308 as well as for adjunctive treatment in more advanced PD.309–316 They have all demonstrated a significant beneficial effect on motor function and activities of daily living. Their side effect profile is similar to that of levodopa in terms of inducing dopaminergic related symptoms such as nausea, vomiting, and postural hypotension but are associated with a higher rate of peripheral edema, somnolence, and hallucinosis, particularly in the elderly. Somnolence with dopamine agonists is mainly seen during the early dose escalation phase, and patients should be warned of this and the rare but important side effect of sudden onset of sleep.317 In patients with early PD (mean age, 61 years), hallucinosis also occurred more frequently during dose escalation but, like sedation, settled to a rate no higher than levodopa during maintenance.
The use of dopamine agonists is rarely associated with the development of pleural, pericardial, or peritoneal fibrosis.318 A report has linked pergolide with fibrotic cardiac valvular disease319 in a pattern similar to that seen with other agents that also stimulate the 5-hydroxytryptamine2 receptor, including methysergide and fenfluramine. There are insufficient data at present to know whether this complication is associated with ergot agonists alone, all dopamine agonists, or all dopaminergic drugs and whether the effect is dose or time related or both. Until such time as additional information becomes available, vigilance is recommended and, when necessary, appropriate investigations (echocardiogram, chest radiography, and erythrocyte sedimentation rate) and referral to a cardiologist.
Dopamine agonist monotherapy can effectively control dopaminergic symptoms for a period of time. Longterm follow-up indicates that approximately 85%, 68%, 55%, 43%, and 34% of PD patients initiated on pramipexole or ropinirole are still controlled on monotherapy at 1, 2, 3, 4, and 5 years, respectively.317,320,321 However, this is dependent on the agonist being used at an appropriate dose. Nevertheless, patients will require levodopa supplementation at some point during their disease. If used correctly, agonists can produce symptom control comparable with levodopa. Although the two monotherapy studies quoted earlier showed superiority for levodopa in UPDRS scores by up to 5 points, patients in the agonist arms had comparable quality of life scores and could have taken supplemental levodopa if the physician or patient believed it was required. The explanation for this discrepancy might be because the UPDRS score does not capture all the benefit that a patient might derive from a dopamine agonist, including possible nonmotor effects such as an antidepressive action.
Several trials have now confirmed that bromocriptine, cabergoline, pergolide, pramipexole, and ropinirole are associated with a significantly reduced risk for the development of motor complications in comparison with levodopa303,307,320–322 (Fig. 71-8). In the pramipexole study, quality of life scores were also equivalent for the 4-year period.317 This implies that the patients were equally well controlled on agonist (with levodopa supplementation when required) or levodopa alone. Of course the levodopa group had more dyskinesias, but at 4 years these did not intrude significantly into patient quality of life or start to limit treatment options for motor control.
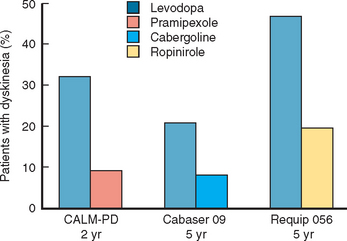
Figure 71-8 Dopamine agonists delay the development of dyskinesias.
(Results are from Rinne UK, Bracco F, Chouza C, et al: Early treatment of Parkinson’s disease with cabergoline delays the onset of motor complications. Results of a doubleblind levodopa controlled trial. The PKDS009. Study Group. Drugs 1998; 55[Suppl 1]:23-30; Rascol O, Brooks DJ, Korczyn AD, et al: A five-year study of the incidence of dyskinesia in patients with early Parkinson’s disease who were treated with ropinirole or levodopa. 056 Study Group. N Engl J Med 2000; 342:1484-1491; and Parkinson Study Group: Pramipexole vs levodopa as initial treatment for Parkinson disease. A randomized controlled trial. JAMA 2000; 284:1931-1938.)
Monoamine Oxidase-B inhibitors
Selegiline has been available for several years and showed benefit as adjunctive treatment for PD. The DATATOP study was a prospective doubleblind, placebo-controlled trial that investigated the effect of selegiline 5 mg twice daily or 2000 IU vitamin E, or both, as putative neuroprotective therapies.324 The time until PD patients required levodopa was used as the primary endpoint. No beneficial effect of vitamin E was detected at the dose given. In contrast, selegiline significantly delayed the need for levodopa compared with placebo, an effect consistent with slowing of disease progression (Fig. 71-9). However, selegiline was also found to exert a mild symptomatic effect that confounded interpretation of the study. In an attempt to avoid this confound, selegiline was compared with placebo using as the primary endpoint, the change in motor score between an untreated baseline visit and an untreated final visit performed after 12 months of treatment and 2 months of study drug withdrawal.325 In this study, PD patients treated with selegiline had less deterioration from baseline than those receiving placebo, again suggesting that selegiline might be neuroprotective. In a longterm follow-up study of the DATATOP cohort, levodopa patients who had been taking selegiline for 7 years, compared with those who were changed to placebo after 5 years, had a significantly slower decline and less wearing off, on-off, and freezing but more dyskinesias than in those on deprenyl.326 Although one study did suggest that selegiline use might be associated with excess mortality, a large meta-analysis indicates that no such effect is evident and confirms the clinical efficacy of this drug in PD with the total UPDRS score being improved by 2.7 points at 3 months.327 There is no evidence at present that MAO-B inhibition itself delays the development of motor fluctuations other than through the delay in introduction of levodopa and an ability to use a lower dose.
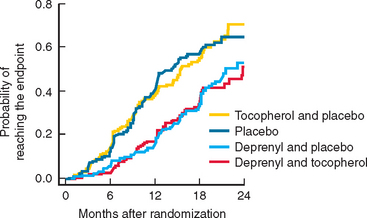
Figure 71-9 The results of the DATATOP study.
(From the Parkinson Study Group: Effects of tocopherol and deprenyl on the progression of disability in early Parkinson’s disease. N Engl J Med 1993; 328:176-183. Copyright 1993 Massachusetts Medical Society. All rights reserved.)
Rasagiline has been studied in patients with early PD328 (Fig. 71-10). Patients with early PD were randomized to placebo or rasagiline (1 or 2 mg/day). In the placebo and rasagiline 1-mg and 2-mg groups, 81%, 83%, and 80%, respectively, were still on “monotherapy” at 6 months, and there were no statistical differences in the rates for either levodopa supplementation or patient withdrawal. At the end of the 6-month period, the 1-mg rasagiline group had an improved UPDRS score compared with a placebo of 4.2 units, and this was 3.56 for the 2-mg group. The degree of motor improvement over the 6-month period was comparable with that seen for selegiline in the DATATOP study324 but not as great as that seen for dopamine agonists. There were no significant differences in the adverse event profile between the treatment arms and placebo. At 6 months, the two treatment arms were almost back to their respective baseline UPDRS scores. The initial 6-month period was extended by an additional 6 months.328 Patients were continued on their original dose of rasagiline or, if on placebo, were given rasagiline 2 mg/day. Patients requiring additional dopaminergic therapy were prescribed either levodopa or a dopamine agonist. For the entire 12-month period, deterioration from baseline scores was 3.01, 1.97, and 4.17 UPDRS units for the 1-mg, 2-mg, and delayed 2-mg cohorts, respectively. Those given rasagiline 1 mg/day for 12 months compared with those on the 2-mg dose for only the last 6 months maintained a total UPDRS improvement of 1.82 UPDRS units. The 12-month rasagiline 2-mg group had a 2.29-unit improvement over the 2-mg 6-month group. There was no significant excess of adverse events in the rasagiline arms compared with the placebo arm.
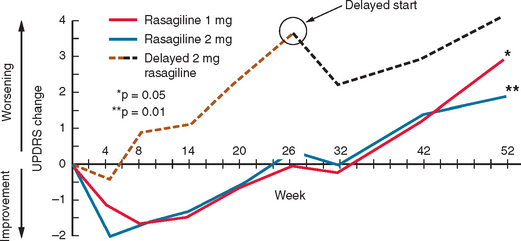
Figure 71-10 Results of the TEMPO study.
(From the Parkinson Study Group: A controlled, randomized, delayedstart study of rasagiline in early Parkinson disease. Arch Neurol 2004; 61:561-566. Copyright © 2004 American Medical Association. All rights reserved.)
Two studies have been published on the efficacy of rasagiline in PD patients already taking levodopa. The PRESTO trial investigated PD patients on stable levodopa with at least 2.5 hours of “off” (i.e., poor motor state).329 Placebo decreased “off” time by 0.9 hour (15% of “off” time) and rasagiline 1 mg/day by 1.9 hours, equating to a 29% reduction in “off” time. Benefits were seen within 6 weeks of randomization and maintained throughout the 26-week study period. The improvement in “off” time was accompanied by a corresponding increase in “on” time, but 32% of the extra “on” time in the 1-mg group was troublesome with dyskinesia although this did not lead to any early terminations. The 1-mg rasagiline dose also resulted in significant improvements in the UPDRS score. The LARGO study investigated the effect of 1 mg/day rasagiline compared with entacapone or placebo in PD patients on stable levodopa but with at least 1 hour of motor fluctuations per day.330 Placebo reduced “off” time by 0.4 hour, both rasagiline and entacapone decreased “off” time by 1.2 hours. There was a comparable and significant increase in “on” time without dyskinesias of 0.8 hour with both drugs. These two studies demonstrate that once-a-day rasagiline (1 mg) significantly improves PD control in patients optimized on levodopa with or without additional therapy. Its efficacy is comparable with entacapone but probably less than that of dopamine agonists, which induce a 1-to 2-hour improvement in PD control.314,315
Other Drugs
Anticholinergics were used to treat the symptoms of PD prior to the introduction of levodopa. Relatively little data are available on their potency and tolerance. Clinical trials have shown a modest benefit for anticholinergics in improving bradykinesia and rigidity,331–333 but this was at the expense of impaired cognitive function. Benztropine was equivalent to clozapine in producing a mild improvement in tremor.334
Amantadine produces mild and transitory improvements in PD symptoms, with benefits usually lasting 6 to 9 months.335 Although some have suggested that in pure PD patients the effects are more long lasting.336 It is generally considered unsuitable for monotherapy in PD and is mostly used as an adjunct. Improvements in bradykinesia and rigidity are generally of the same order of magnitude as anticholinergics, but their combination is additive.337,338 Amantadine use is also limited by its potential to induce cognitive defects.
Initiation of Treatment
Treatment for PD is always tailored to the specific needs and circumstances of the patient. Traditionally, drug treatment has been initiated only when the patient’s symptoms interfered significantly with their employment or social activities. The rationale for this was very reasonable: the treatments available were considered symptomatic only and incapable of modifying the course of the disease. Advances in our understanding of the pathophysiology and pharmacology of PD and the availability of new treatments for the disease have required reevaluation of this strategy.340
The clinical onset of PD motor features is directly associated with a series of functional changes in basal ganglia circuits and their target projections.341 Basal ganglia output becomes abnormal and clinical features appear, when dopamine levels fall to less than 7% in MPTP-treated nonhuman primates.342,343 The corresponding figure in humans is not known but may be around 20% to 30%. The estimated asymptomatic latent period of approximately 6 years344 in idiopathic PD (and longer in familial PD) indicates the remarkable capacity for the basal ganglia to cope with progressively lower levels of dopamine, the compensatory mechanisms maintaining apparently normal motor function over the intervening years to diagnosis. These compensatory mechanisms include increased striatal dopamine turnover and receptor sensitivity, upregulation of striatopallidal enkephalin levels, increased subthalamic excitation of the globus pallidum pars externa, and maintenance of cortical motor area activation.345,346 These observations, although neither completely defined nor understood, support the notion that declining dopamine levels during the early phase of PD put the basal ganglia level under stress. The onset of clinical symptoms denotes the point of failure to deal adequately with dopamine depletion. It might be that early correction of the basal ganglia functional abnormalities caused by dopaminergic cell loss and dopamine deficiency is a means to support the intrinsic physiological compensatory mechanisms and both limit and delay the circuitry changes that evolve as PD progresses. Review of the outcomes of the DATATOP, ELLDOPA (Fig. 71-11), and TEMPO studies may support such a proposition.340 In these studies, those patients who received effective symptomatic treatment earlier in the course of their PD fared significantly better clinically than those initiated on placebo even when, as in the case of TEMPO, they were switched to the active drug after only 6 months.
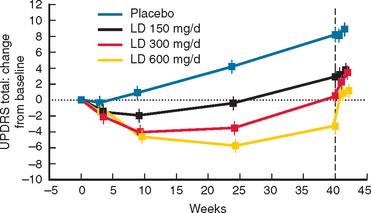
Maintenance of Treatment
Most PD patients respond well to the initiation of dopaminergic therapy in small doses. However, some require regular uptitration of their dose before an adequate control of motor function is reached. This is particularly true of the dopamine agonists that may need to be increased to, for example, 3 mg of pramipexole or 12 to 15 mg of ropinirole before there is a good response. Some patients will need these doses to be increased as their disease progresses. As indicated earlier, regardless of what drug is first used, PD patients will eventually require levodopa. This is most often introduced as a three-times-daily regimen, although the short half-life of the drug means that this falls well short of providing continuous dopaminergic stimulation. A higher frequency of administration would provide better symptom control and possibly less risk of the development of motor complications but needs to be balanced against a likely lower rate of compliance.
Motor Complications
The majority of PD patients will develop wearing off or dyskinesias at some point in the course of their disease. Although the use of dopamine agonists will delay the onset, once levodopa is introduced the risk for their appearance increases. It is possible that the initiation of levodopa with a COMT inhibitor might delay the onset of motor complications, but evidence for this is lacking at present. The development of dyskinesias is probably related to dose of levodopa347,348 as well as duration,349,350 so the continuation of the dopamine agonist or MAO-B inhibitor to limit the levodopa dose is beneficial.
Wearing off equates to the loss of effectiveness of a given dose of dopaminergic therapy and the emergence of the primary motor features of PD (i.e., bradykinesia and rigidity) that remain responsive to dopaminergic treatment. Patients recognize this as a return of symptoms prior to their next dose (“end-of-dose failure”), although some mistakenly associate it with the administration of the next dose given the medication’s latency of effect. As noted, wearing off is eliminated by continuous administration of either levodopa or a dopamine agonist.288,289,351 Although these methods are effective, they are not practical for the majority of patients. The simplest strategy is to increase the frequency of administration of levodopa, although this too may become difficult as dosage regimens increase to six or more times per day. Controlled-release formulations have proved disappointing with often little improvement in duration of response and problems with unpredictability of absorption and motor response. One open-label study demonstrated that the addition of cabergoline, a long-acting ergot dopamine agonist, to patients taking pramipexole or ropinirole, both non-ergots, resulted in improved motor control.352
The addition of a COMT or MAO-B inhibitor to levodopa significantly improves “off” time (see earlier) and is an easy and effective strategy for managing wearing off.329,330,353
The practical management of dyskinesias depends on the severity of the involuntary movements and their relationship to medication dosage schedule. They may be peak dose, biphasic, or random. Peak dose dyskinesias are related to high plasma concentrations of levodopa and can be managed by fractionating levodopa doses to avoid such peaks. This may or may not require an increase in the total daily dose. Alternative strategies include the introduction of a dopamine agonist if the patient is not already taking such an agent and if he or she remains a suitable candidate. Long-acting agonists are particularly useful in the management of dyskinesias, presumably due to their ability to provide more continuous dopaminergic stimulation while avoiding rapid fluctuations in receptor stimulation.298 Biphasic dyskinesias occur when plasma levodopa concentration is rising or falling and are associated with generally lower plasma levodopa concentrations. They tend to be more stereotypical and repetitive and to involve the lower extremities. They are more troublesome to manage but may respond to higher levodopa doses designed to keep the plasma concentrations above a critical level.354
Amantadine has demonstrated efficacy in improving peak dose dyskinesias.355–357 The effective dose is 200 to 400 mg/day in two divided doses. The severity of dyskinesias may be reduced by 24% to 56% and the effect sustained at 1 year.
Management of Nonmotor Complications
Depression and, to some extent, apathy (anhedonia) may respond to tricyclics such as amitriptyline or to selective serotonin reuptake inhibitors. Pramipexole may be useful as an antidepressant, separate from its action to improve the motor features of PD.358–360 Anxiety and panic attacks can be prominent in PD, and these may sometimes relate to “wearing off” and so respond to strategies outlined for this complication. However, additional anxiolytic therapy may be needed in some patients.
Hallucinations, if due to drugs, usually respond to a reduction in dose. However, in some patients, this is difficult due to re-emergence of motor features, and they may respond to clozapine or quetiapine.361–363 Hallucinations are, of course, an important symptom of diffuse Lewy body disease, and their emergence early in the course of PD is a risk factor for dementia. PD patients who demonstrated dementia after the 2 years diagnosis of PD showed a modest but significant improvement in cognitive function with rivastigmine, to a degree similar to that seen with this drug in Alzheimer’s disease.364
Several strategies are available to improve both nighttime sleep and daytime alertness in PD and include improving sleep hygiene, treating nocturnal motor problems, better managing nocturia, modifying medication,317 and using modafinil in patients with refractory daytime drowsiness.365
Viagra or apomorphine can, in select cases, usefully manage the sexual dysfunction associated with PD.366,367 Bladder abnormalities particularly cause problems at night but can be improved by a range of options that include nonpharmacological and pharmacological strategies. The latter include the use of oxybutinin, detrusitol, or amitriptyline in patients with concomitant depression. Sialorrhea and drooling are often the result of reduced frequency of swallowing and may be helped by simple things such as chewing gum or sucking sweets. Anticholinergic drugs may help but often cause unwanted side effects. Botulinum toxin can be used for refractory cases.368 Constipation may respond to dopaminergic drugs and bowel training. Aperients often need to be added.
Surgery
Surgical approaches to the management of PD have been practiced since the mid-twentieth century. The discovery of dopamine depletion and the subsequent introduction of oral levodopa made surgery less attractive. The recognition of motor complications and the development of severe dyskinesias in some patients led to a resurgence of interest in lesioning the brain to control these features. Advances in surgical technique in neurophysiology and in molecular cell biology have provided the stimulus for the generation of a wide range of nonmedical options for PD (Table 71-7).
Destructive Lesions
Thalamotomy may produce a reduction in tremor and bradykinesia; the best results have been achieved with lesion in the ventrointermediate nucleus.369 However, thalamotomy is not particularly helpful for bradykinesia or rigidity, and the procedure can be associated with significant morbidity related to the placement of the lesion.370 Thalamotomy has largely been replaced by medical therapies or deep brain stimulation. Posteroventral pallidotomy can provide long-lasting improvement in contralateral dyskinesia and some improvement in bradykinesia and rigidity in PD patients.371–374 Like thalamotomy, pallidotomy has become less common as deep brain stimulation has become more available. However, both destructive lesions may still be offered when symptoms significantly affect one side (bilateral destructive lesions cause increased complications including bulbar dysfunction) and when the opportunity for regular and expensive follow-up is limited.
Subthalamotomy has been shown to improve parkinsonian motor abnormalities including dyskinesias in animal models375–377 and in PD patients.378–380 However, dyskinesia and hemiballismus have also been reported, following subthalamotomy, which in a few cases have been permanent.381,382
Stimulation
Deep brain stimulation was first proposed as a treatment in PD by Benabid based on his experience with high-frequency stimulation as a means of confirming the target site for an ablative lesion.383 Deep brain stimulation can be used for bilateral procedures with relative safety. Also, the stimulator can be adjusted to maximize benefits and reduce side effects. Deep brain stimulation simulates the effect of a lesion but avoids the need to create a destructive brain lesion. The precise mechanism of action is unknown, but possibilities include depolarization blockade, release of inhibitory neurotransmitters, backfiring, and inhibition of aberrant neuronal signals.384
Deep brain stimulation of the ventral intermediate nucleus significantly improves contralateral tremor and is comparable in effect with destructive lesions but is superior in terms of side effects.385–387 Benefits are long-lasting and have been shown to persist for more than 10 years. However, only tremor is improved and there is no effect on bradykinesia, rigidity, or dyskinesias. Thus deep brain stimulation of the ventral intermediate nucleus is not as attractive as deep brain stimulation of other targets. Deep brain stimulation of the subthalamic nucleus388,389 or globus pallidum interna390–394 improves all of the cardinal features of PD as well as dyskinesias. Patients who could not be further improved with medical therapies (typically because of motor complications) experienced a substantial reduction in disability following deep brain stimulation of the subthalamic nucleus or globus pallidum interna. Longterm studies demonstrate that benefits of deep brain stimulation persist over more than 5 years of follow-up, although disability still progresses from year to year, reflecting degeneration in nondopaminergic sites.395
Adverse events with deep brain stimulation can be related to the intracranial procedure, the electrode system, and stimulation. The surgical procedure can be associated with hemorrhage, tissue damage, and infection. In a multicenter study, 7 of 143 patients experienced hemorrhage and neurological deficits persisted in 4.394 Problems can also occur in relation to the device including lead breaks, lead migration, infection, and skin erosion.397 These occur in about 2% to 3% of cases and occasionally require replacement of the electrode. Severe depression and suicidal ideations or riotous laughing have been observed with stimulation of the subthalamic nucleus,398 suggesting that basal ganglia circuits are involved with higher cortical and/or limbic as well as motor functions. The use of diathermy during surgical procedures should be avoided in patients with deep brain stimulation as excess heat might be conducted to the brain by the electrode wire and cause necrosis.
Deep brain stimulation, particularly of the subthalamic nucleus or globus pallidum interna, offers a significant benefit to those patients who have severe dyskinesias not controlled by standard means. Parkinsonian features are also improved but no more than can be achieved by dopaminergic medication. Deep brain stimulation is relatively safe if performed by an experienced surgeon but still carries some small risk of permanent neurological deficit (often quoted as less than 1%). Patients should be carefully selected; those with cognitive impairment are excluded because of the risk of exacerbating this with surgery. Continuous follow-up is required, and the procedure is expensive.
Fetal nigral transplantation has been evaluated in two doubleblind, placebo-controlled trials. The first randomized 40 patients to receive a transplantation or placebo procedure and followed them for 1 year.399 Modest benefits of transplantation were observed in UPDRS scores of activities of daily living and motor function in patients younger than 60 years. There was a significant increase in striatal fluorodopa uptake on PET, and there was modest survival of implanted cells at postmortem. The procedure was well tolerated, but approximately 15% of transplanted patients developed dyskinesias that persisted for days or weeks after levodopa was discontinued and this was a source of major disability in some patients.400 Quality of life, the primary endpoint, was not improved. The second trial was a 2-year doubleblind placebo-controlled study that used a slightly different implantation protocol.401 Transplanted patients were not significantly improved in comparison with placebo patients, despite having significant increases in striatal fluorodopa uptake on PET and survival of implanted dopaminergic neurons at postmortem. Over one half (56%) of the transplanted patients in this study developed dyskinesia during the practically defined off state when they had been held off levodopa for more than 12 hours (“off-medication dyskinesia”). This phenomenon was not observed in nontransplanted patients. The precise mechanism responsible for off-medication dyskinesia remains unknown.
Glial-derived neurotrophic factor has attracted attention as a potential treatment for PD because of its capacity to protect or rescue dopaminergic neurons in tissue culture402 and in MPTP-treated monkeys.403 Intraventricular administration of GDNF to PD patients did not produce benefit.404 One open-label study used an infusion pump to directly administer glial-derived neurotrophic factor into the striatum in five PD patients.405 There was an improvement in UPDRS motor scores during practically defined off as well as a small increase in striatal fluorodopa uptake around the catheter tip. However, a doubleblind trial comparing glial-derived neurotrophic factor with placebo was negative.406
NEUROPROTECTION
MAO-B Inhibitors
Selegiline can protect cultured dopaminergic neurons against the toxicity of MPP+ and, in animal models, can reduce dopaminergic cell loss in response to MPTP.407–410 Selegiline also protects against apoptotic cell death induced by serum and growth factor withdrawal,411,412 possibly via an increased production of Bcl2. Selegiline, by virtue of its MAO-B activity, will reduce the turnover of dopamine and so reduce free radical generation. The production of reactive oxygen species and free radical–mediated damage to lipids and proteins have been implicated in PD pathogenesis. Thus, this property, together with the ability for selegiline to protect against MPTP toxicity, led to the evaluation of this drug in the first clinical trial for neuroprotection in PD.
Dopamine Agonists
Dopamine agonists have antioxidant activity as a result of their hydroxylated benzyl ring structure, and numerous laboratory studies have demonstrated neuronal protection against free radical–generating systems. These include attenuation of the effects of MPP+, dopamine, 6-hydroxydopamine, and nitric oxide and upregulation of protective scavenging enzymes such as catalase and superoxide dismutase.413–421 However, these benefits are predominantly seen at relatively high concentrations, which may not be relevant in clinical practice. Dopamine agonists have demonstrated antiapoptotic activity in laboratory studies. For instance, pramipexole reduces cell death, prevents the release of cytochrome c and caspase activation in dopaminergic cells treated with MPP+ or rotenone, and prevents a fall in mitochondrial membrane potential.422,423 Importantly, this dopamine agonist has also shown protective effects in the MPTP primate model of PD.424 Several studies suggest that dopamine agonists exert their protective effects not through stimulation of either D2 or D3 receptors, but rather via some alternative mechanism. The potential for dopamine agonists to protect nondopaminergic cells, if translated to the clinic, would have profound implications for disease modification and in particular for preventing the development of nonmotor features in PD.
Two studies have sought to determine whether the neuroprotective benefits of dopamine agonists seen in the laboratory can be transferred to patients to modify the course of PD (Fig. 71-12). The CALM-PD study used 2β-carboxymethoxy-3β(4-iodophenyl)tropane (β-CIT) SPECT to follow the rate of loss of dopamine transporter as a marker of dopaminergic nigrostriatal cell density.247 Patients with early PD were randomized to pramipexole or levodopa and followed for a total of 4 years; levodopa supplementation was allowed in both arms. At 2, 3, and 4 years, there was a significant reduction in the rate of transporter loss in the pramipexole group, averaging at approximately 40%, consistent with the drug having a relatively protective effect in comparison with levodopa. A similar result was seen in the REAL-PET ropinirole study that used a similar trial design but used PET to follow loss of nigrostriatal cell density with fluorodopa.248 This demonstrated an approximately 34% reduction over 2 years in the ropinirole group compared with those on levodopa. These studies have generated considerable interest and debate.425,426 Both studies demonstrate that dopamine agonists are associated with a significant delay in the rate of decline of a surrogate imaging marker of nigrostriatal function.
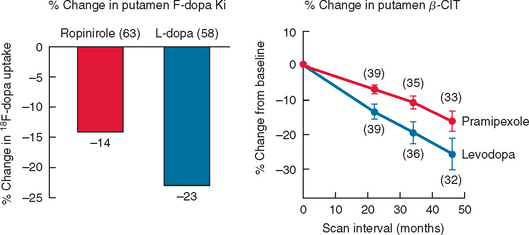
Figure 71-12 Results of SPECT and PET data from dopamine agonist neuroprotection studies.
(From the Parkinson Study Group: Dopamine transporter brain imaging to assess the effects of pramipexole vs levodopa on Parkinson disease progression. JAMA 2002; 287:1653-1661; and Whone AL, Watts RL, Stoessl AJ, et al: Slower progression of Parkinson’s disease with ropinirole versus levodopa: the REAL-PET study. Ann Neurol 2003; 54:93-101. Copyright © 2002 American Medical Association. All rights reserved.)
Another interpretation of these studies is that levodopa is toxic to nigral neurons. There is concern that levodopa might be toxic as it undergoes oxidative metabolism and has the potential to generate cytotoxic free radicals.427 Levodopa has been shown to be toxic to cultured dopamine neurons, but there is no convincing evidence that levodopa is toxic in in vivo models or in PD patients.428 The ELLDOPA trial investigated the possibility that levodopa may be toxic in PD patients but produced conflicting results. In this study, untreated PD patients were randomized to a total daily dose of 150, 300, or 600 mg of levodopa or placebo. β-CIT SPECT was used as an endpoint for integrity of the nigrostriatal system. Levodopa was associated with a significant increase in the rate of decline of imaging marker over 9 months compared with placebo, consistent with a toxic effect.429 Clinical evaluation, however, showed that those patients on levodopa had better UPDRS scores compared with placebo after 2 weeks of washout (see Fig. 71-9). This would, in contrast, be indicative of a protective effect of levodopa. However, intellectual parsimony would dictate that the simplest explanation for this clinical effect was that the washout period was too brief to eliminate the symptomatic benefits of levodopa.
Finally, it has been proposed that the differences between the effects of levodopa and dopamine agonists seen in the CALM-PD and REAL-PET studies are not related to any direct effect of the drugs on dopamine neuron survival or degeneration but rather to a pharmacologic difference in the capacity of these drugs to regulate the dopamine transporter or fluorodopa metabolism.426,430,431 However, a review of studies testing the effects of levodopa and dopamine agonists on transporter and fluorodopa metabolism reveals that the data are conflicting and that at present there is insufficient information for or against such an effect.425
Coenzyme Q10
Coenzyme Q10 has been evaluated in a pilot study of early PD patients to determine whether it might have disease-modifying capabilities.432 The rationale for the use of coenzyme Q10 in PD was based on the observation that mitochondrial complex I activity is decreased in the PD substantia nigra, PD patients have reduced levels of coenzyme Q10, and this compound protects against MPTP toxicity. Coenzyme Q10 is both an antioxidant and an integral component of oxidative phosphorylation that has been shown to enhance electron transport. It is presumed not to have any symptomatic effect. Patients were randomized to either a placebo arm or one of three doses of coenzyme Q10 (300, 600, or 1200 mg) and followed for 16 months. There was a significant benefit for coenzyme Q10 1200 mg in terms of change from baseline in total UPDRS compared with placebo at 16 months and a nonsignificant trend to benefit for lower doses (Fig. 71-13). This interesting and important result is sufficient to support further study of coenzyme Q10 but insufficient at present to advocate that PD patients should use this compound.
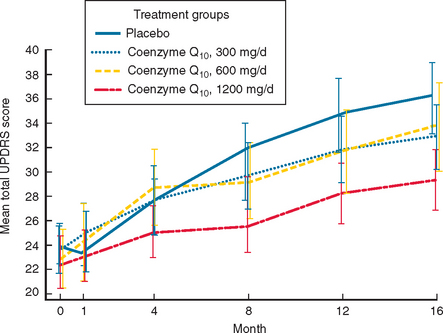
Figure 71-13 Results of the coenzyme Q10 study.
(From Shults CW, Oakes D, Kieburtz K, et al: Effects of coenzyme Q10 in early Parkinson disease: evidence of slowing of the functional decline. Arch Neurol 2002; 59:1541-1550. Copyright © 2002 American Medical Association. All rights reserved.)
Ebadi M, Pfeiffer RF, editors. Parkinson’s Disease. Boca Raton: CRC Press, 2005.
Nutt JG, Wooten GF. Clinical practice. Diagnosis and initial management of Parkinson’s disease. N Engl J Med. 2005;353:1021-1027.
Olanow CW, Schapira AHV, Agid Y. Neurodegeneration and prospects for neuroprotection and rescue in Parkinson’s disease. Ann Neurol. 2003;53(Suppl 3):S1-S170.
Samii A, Nutt JG, Ransom BR. Parkinson’s disease. Lancet. 2004;363:1783-1793.
Schapira AHV, Olanow CW, editors. Principles of Treatment in Parkinson’s Disease. Philadelphia: Butterworth Heinemann, 2005.
1 Ehringer H, Hornykiewicz O. Distribution of noradrenaline and dopamine (3-hydroxytyramine) in the human brain and their behavior in diseases of the extrapyramidal system. Klin Wochenschr. 1960;38:1236-1239.
2 Cotzias GC, Van Woert MH, Schiffer LM. Aromatic amino acids and modification of parkinsonism. N Engl J Med. 1967;276:374-379.
3 Mayeux R, Marder K, Cote LJ, et al. The frequency of idiopathic Parkinson’s disease by age, ethnic group, and sex in northern Manhattan, 1988–1993. Am J Epidemiol. 1995;142:820-827.
4 Bower JH, Maraganore DM, McDonnell SK, et al. Incidence and distribution of parkinsonism in Olmsted County, Minnesota, 1976–1990. Neurology. 1999;52:1214-1220.
5 Van Den Eeden SK, Tanner CM, Bernstein AL, et al. Incidence of Parkinson’s disease: variation by age, gender, and race/ethnicity. Am J Epidemiol. 2003;157:1015-1022.
6 de Rijk MC, Breteler MM, Graveland GA, et al. Prevalence of Parkinson’s disease in the elderly: the Rotterdam Study. Neurology. 1995;45:2143-2146.
7 Zhang ZX, Roman GC. Worldwide occurrence of Parkinson’s disease: an updated review. Neuroepidemiology. 1993;12:195-208.
8 Del Tredici K, Rub U, De Vos RA, et al. Where does parkinson disease pathology begin in the brain? J Neuropathol Exp Neurol. 2002;61:413-426.
9 Ponsen MM, Stoffers D, Booij J, et al. Idiopathic hyposmia as a preclinical sign of Parkinson’s disease. Ann Neurol. 2004;56:173-181.
10 Braak H, Del Tredici K, Rub U, et al. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197-211.
11 Gowers WR. A Manual of Diseases of the Nervous System. London: Churchill, 1888;996.
12 Payami H, Larsen K, Bernard S, et al. Increased risk of Parkinson’s disease in parents and siblings of patients. Ann Neurol. 1994;36:659-661.
13 Bonifati V, Fabrizio E, Vanacore N, et al. Familial Parkinson’s disease: a clinical genetic analysis. Can J Neurol Sci. 1995;22:272-279.
14 Vieregge P, Heberlein I. Increased risk of Parkinson’s disease in relatives of patients. Ann Neurol. 1995;37:685.
15 De Michele G, Filla A, Volpe G, et al. Environmental and genetic risk factors in Parkinson’s disease: a case-control study in southern Italy. Mov Disord. 1996;11:17-23.
16 Marder K, Tang MX, Mejia H, et al. Risk of Parkinson’s disease among first-degree relatives: A community-based study. Neurology. 1996;47:155-160.
17 Gasser T. Genetics of Parkinson’s disease. Ann Neurol. 1998;44(3 Suppl 1):S53-S57.
18 Tanner CM, Ottman R, Goldman SM, et al. Parkinson disease in twins: an etiologic study. JAMA. 1999;281:341-346.
19 Piccini P, Burn DJ, Ceravolo R, et al. The role of inheritance in sporadic Parkinson’s disease: evidence from a longitudinal study of dopaminergic function in twins. Ann Neurol. 1999;45:577-582.
20 Polymeropoulos MH, Lavedan C, Leroy E, et al. Mutation in the alphasynuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045-2047.
21 Kruger R, Kuhn W, Muller T, et al. Ala30Pro mutation in the gene encoding alphasynuclein in Parkinson’s disease. Nat Genet. 1998;18:106-108.
22 Golbe LI, Di Iorio G, Sanges G, et al. Clinical genetic analysis of Parkinson’s disease in the Contursi kindred. Ann Neurol. 1996;40:767-775.
23 Golbe LI, Di Iorio G, Bonavita V, et al. A large kindred with autosomal dominant Parkinson’s disease. Ann Neurol. 1990;27:276-282.
24 Polymeropoulos MH, Higgins JJ, Golbe LI, et al. Mapping of a gene for Parkinson’s disease to chromosome 4q21-q23. Science. 1996;274:1197-1199.
25 Chan P, Tanner CM, Jiang X, et al. Failure to find the alphasynuclein gene missense mutation (G209A) in 100 patients with younger onset Parkinson’s disease. Neurology. 1998;50:513-514.
26 Vaughan JR, Farrer MJ, Wszolek ZK, et al. Sequencing of the alphasynuclein gene in a large series of cases of familial Parkinson’s disease fails to reveal any further mutations. The European Consortium on Genetic Susceptibility in Parkinson’s Disease (GSPD). Hum Mol Genet. 1998;7:751-753.
27 Warner TT, Schapira AH. The role of the alphasynuclein gene mutation in patients with sporadic Parkinson’s disease in the United Kingdom. J Neurol Neurosurg Psychiatry. 1998;65:378-379.
28 Parsian A, Racette B, Zhang ZH, et al. Mutation, sequence analysis, and association studies of alphasynuclein in Parkinson’s disease. Neurology. 1998;51:1757-1759.
29 George JM, Jin H, Woods WS, et al. Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron. 1995;15:361-372.
30 Abeliovich A, Schmitz Y, Farinas I, et al. Mice lacking alphasynuclein display functional deficits in the nigrostriatal dopamine system. Neuron. 2000;25:239-252.
31 Spillantini MG, Schmidt ML, Lee VM, et al. Alphasynuclein in Lewy bodies. Nature. 1997;388:839-840.
32 Masliah E, Rockenstein E, Veinbergs I, et al. Dopaminergic loss and inclusion body formation in alphasynuclein mice: implications for neurodegenerative disorders. Science. 2000;287:1265-1269.
33 Feany MB, Bender WW. A Drosophila model of Parkinson’s disease. Nature. 2000;404:394-398.
34 Singleton AB, Farrer M, Johnson J, et al. Alphasynuclein locus triplication causes Parkinson’s disease. Science. 2003;302:841.
35 Farrer M, Kachergus J, Forno L, et al. Comparison of kindreds with parkinsonism and alphasynuclein genomic multiplications. Ann Neurol. 2004;55:174-179.
36 Zarranz JJ, Alegre J, Gomez-Esteban JC, et al. The new mutation, E46K, of alphasynuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55:164-173.
37 Giasson BI, Duda JE, Quinn SM, et al. Neuronal alphasynucleinopathy with severe movement disorder in mice expressing A53T human alphasynuclein. Neuron. 2002;34:521-533.
38 Kirik D, Rosenblad C, Burger C, et al. Parkinson-like neurodegeneration induced by targeted overexpression of alphasynuclein in the nigrostriatal system. J Neurosci. 2002;22:2780-2791.
39 Conway KA, Lee SJ, Rochet JC, et al. Acceleration of oligomerization, not fibrillization, is a shared property of both alphasynuclein mutations linked to earlyonset Parkinson’s disease: implications for pathogenesis and therapy. Proc Natl Acad Sci U S A. 2000;97:571-576.
40 Conway KA, Rochet JC, Bieganski RM, et al. Kinetic stabilization of the alphasynuclein protofibril by a dopaminealpha-synuclein adduct. Science. 2001;294:1346-1349.
41 Tabrizi SJ, Orth M, Wilkinson JM, et al. Expression of mutant alphasynuclein causes increased susceptibility to dopamine toxicity. Hum Mol Genet. 2000;9:2683-2689.
42 Smith WW, Margolis RL, Li X, et al. Alphasynuclein phosphorylation enhances eosinophilic cytoplasmic inclusion formation in SH-SY5Y cells. J Neurosci. 2005;25:5544-5552.
43 Hasegawa M, Fujiwara H, Nonaka T, et al. Phosphorylated alphasynuclein is ubiquitinated in alphasynucleinopathy lesions. J Biol Chem. 2002;277:49071-49076.
44 Chen L, Feany MB. Alphasynuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat Neurosci. 2005;8:657-663.
45 Kitada T, Asakawa S, Hattori N, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605-608.
46 Yamamura Y, Sobue I, Ando K, et al. Paralysis agitans of early onset with marked diurnal fluctuation of symptoms. Neurology. 1973;23:239-244.
47 Takahashi H, Ohama E, Suzuki S, et al. Familial juvenile parkinsonism: clinical and pathologic study in a family. Neurology. 1994;44(3 Pt 1):437-441.
48 Matsumine H, Saito M, Shimoda-Matsubayashi S, et al. Localization of a gene for an autosomal recessive form of juvenile Parkinsonism to chromosome 6q25.2–27. Am J Hum Genet. 1997;60:588-596.
49 Hattori N, Kitada T, Matsumine H, et al. Molecular genetic analysis of a novel parkin gene in Japanese families with autosomal recessive juvenile parkinsonism: evidence for variable homozygous deletions in the parkin gene in affected individuals. Ann Neurol. 1998;44:935-941.
50 Abbas N, Lucking CB, Ricard S, et al. A wide variety of mutations in the parkin gene are responsible for autosomal recessive parkinsonism in Europe. French Parkinson’s Disease Genetics Study Group and the European Consortium on Genetic Susceptibility in Parkinson’s Disease. Hum Mol Genet. 1999;8:567-574.
51 Farrer M, Chan P, Chen R, et al. Lewy bodies and parkinsonism in families with parkin mutations. Ann Neurol. 2001;50:293-300.
52 West A, Periquet M, Lincoln S, et al. Complex relationship between parkin mutations and Parkinson disease. Am J Med Genet. 2002;114:584-591.
53 Lucking CB, Durr A, Bonifati V, et al. Association between earlyonset Parkinson’s disease and mutations in the parkin gene. French Parkinson’s Disease Genetics Study Group. N Engl J Med. 2000;342:1560-1567.
54 Kann M, Jacobs H, Mohrmann K, et al. Role of parkin mutations in 111 community-based patients with earlyonset parkinsonism. Ann Neurol. 2002;51:621-625.
55 Klein C, Pramstaller PP, Kis B, et al. Parkin deletions in a family with adult-onset, tremor-dominant parkinsonism: expanding the phenotype. Ann Neurol. 2000;48:65-71.
56 Lucking CB, Bonifati V, Periquet M, et al. Pseudo-dominant inheritance and exon 2 triplication in a family with parkin gene mutations. Neurology. 2001;57:924-927.
57 Thobois S, Ribeiro MJ, Lohmann E, et al. Young-onset Parkinson disease with and without parkin gene mutations: a fluorodopa F 18 positron emission tomography study. Arch Neurol. 2003;60:713-718.
58 Hilker R, Klein C, Ghaemi M, et al. Positron emission tomographic analysis of the nigrostriatal dopaminergic system in familial parkinsonism associated with mutations in the parkin gene. Ann Neurol. 2001;49:367-376.
59 Khan NL, Brooks DJ, Pavese N, et al. Progression of nigrostriatal dysfunction in a parkin kindred: an [18F]dopa PET and clinical study. Brain. 2002;125:2248-2256.
60 Shimura H, Hattori N, Kubo S, et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet. 2000;25:302-305.
61 Zhang Y, Gao J, Chung KK, et al. Parkin functions as an E2-dependent ubiquitin-protein ligase and promotes the degradation of the synaptic vesicle-associated protein, CDCrel-1. Proc Natl Acad Sci U S A. 2000;97:13354-13359.
62 Imai Y, Soda M, Inoue H, et al. An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of parkin.. Cell. 2001;105:891-902.
63 Lo BC, Schneider BL, Bauer M, et al. Lentiviral vector delivery of parkin prevents dopaminergic degeneration in an alphasynuclein rat model of Parkinson’s disease. Proc Natl Acad Sci U S A. 2004;101:17510-17515.
64 Goldberg MS, Fleming SM, Palacino JJ, et al. Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J Biol Chem. 2003;278:43628-43635.
65 Palacino JJ, Sagi D, Goldberg MS, et al. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J Biol Chem. 2004;279:18614-18622.
66 Greene JC, Whitworth AJ, Kuo I, et al. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci U S A. 2003;100:4078-4083.
67 Darios F, Corti O, Lucking CB, et al. Parkin prevents mitochondrial swelling and cytochrome c release in mitochondria-dependent cell death. Hum Mol Genet. 2003;12:517-526.
68 Muftuoglu M, Elibol B, Dalmizrak O, et al. Mitochondrial complex I and IV activities in leukocytes from patients with parkin mutations. Mov Disord. 2004;19:544-548.
69 Chung KK, Thomas B, Li X, et al. S-nitrosylation of parkin regulates ubiquitination and compromises parkin’s protective function. Science. 2004;304:1328-1331.
70 Leroy E, Boyer R, Auburger G, et al. The ubiquitin pathway in Parkinson’s disease. Nature. 1998;395:451-452.
71 Harhangi BS, Farrer MJ, Lincoln S, et al. The Ile93Met mutation in the ubiquitin carboxy-terminal-hydrolase-L1 gene is not observed in European cases with familial Parkinson’s disease. Neurosci Lett. 1999;270:1-4.
72 Lincoln S, Vaughan J, Wood N, et al. Low frequency of pathogenic mutations in the ubiquitin carboxy-terminal hydrolase gene in familial Parkinson’s disease. Neuroreport. 1999;10:427-429.
73 Valente EM, Bentivoglio AR, Dixon PH, et al. Localization of a novel locus for autosomal recessive earlyonset parkinsonism, PARK6, on human chromosome 1p35-p36. Am J Hum Genet. 2001;68:895-900.
74 Valente EM, Abou-Sleiman PM, Caputo V, et al. Hereditary earlyonset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304:1158-1160.
75 Bentivoglio AR, Cortelli P, Valente EM, et al. Phenotypic characterisation of autosomal recessive PARK6-linked parkinsonism in three unrelated Italian families. Mov Disord. 2001;16:999-1006.
76 Rohe CF, Montagna P, Breedveld G, et al. Homozygous PINK1 C-terminus mutation causing earlyonset parkinsonism. Ann Neurol. 2004;56:427-431.
77 Hatano Y, Li Y, Sato K, et al. Novel PINK1 mutations in earlyonset parkinsonism. Ann Neurol. 2004;56:424-427.
78 Valente EM, Salvi S, Ialongo T, et al. PINK1 mutations are associated with sporadic earlyonset parkinsonism. Ann Neurol. 2004;56:336-341.
79 Unoki M, Nakamura Y. Growth-suppressive effects of BPOZ and EGR2, two genes involved in the PTEN signaling pathway. Oncogene. 2001;20:4457-4465.
80 Hunter T. Signaling: 2000 and beyond. Cell. 2000;100:113-127.
81 Cohen P. The origins of protein phosphorylation. Nat Cell Biol. 2002;4:E127-E130.
82 Manning G, Whyte DB, Martinez R, et al. The protein kinase complement of the human genome. Science. 2002;298:1912-1934.
83 van Duijn CM, Dekker MC, Bonifati V, et al. Park7: a novel locus for autosomal recessive earlyonset parkinsonism, on chromosome 1p36. Am J Hum Genet. 2001;69:629-634.
84 Bonifati V, Rizzu P, van Baren MJ, et al. Mutations in the DJ-1 gene associated with autosomal recessive earlyonset parkinsonism. Science. 2003;299:256-259.
85 Canet-Aviles RM, Wilson MA, Miller DW, et al. The Parkinson’s disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. Proc Natl Acad Sci U S A. 2004;101:9103-9108.
86 Zhang L, Shimoji M, Thomas B, et al. Mitochondrial localization of the Parkinson’s disease related protein DJ-1: implications for pathogenesis. Hum Mol Genet. 2005;14:2063-2073.
87 Di Fonzo A, Rohe CF, Ferreira J, et al. A frequent LRRK2 gene mutation associated with autosomal dominant Parkinson’s disease. Lancet. 2005;365:412-415.
88 Nichols WC, Pankratz N, Hernandez D, et al. Genetic screening for a single common LRRK2 mutation in familial Parkinson’s disease. Lancet. 2005;365:410-412.
89 Kachergus J, Mata IF, Hulihan M, et al. Identification of a novel LRRK2 mutation linked to autosomal dominant parkinsonism: evidence of a common founder across European populations. Am J Hum Genet. 2005;76:672-680.
90 Gilks WP, Abou-Sleiman PM, Gandhi S, et al. A common LRRK2 mutation in idiopathic Parkinson’s disease. Lancet. 2005;365:415-416.
91 Deng H, Le W, Guo Y, et al. Genetic and clinical identification of Parkinson’s disease patients with LRRK2 G2019S mutation. Ann Neurol. 2005;57:933-934.
92 Zabetian CP, Samii A, Mosley AD, et al. A clinic-based study of the LRRK2 gene in Parkinson disease yields new mutations. Neurology. 2005;65:741-744.
93 Hernandez D, Paisan RC, Crawley A, et al. The dardarin G 2019 S mutation is a common cause of Parkinson’s disease but not other neurodegenerative diseases. Neurosci Lett. 2005;389:137-139.
94 Toft M, Sando SB, Melquist S, et al. LRRK2 mutations are not common in Alzheimer’s disease. Mech Ageing Dev. 2005;126:1201-1205.
95 Kay DM, Kramer P, Higgins D, et al. Escaping Parkinson’s disease: a neurologically healthy octogenarian with the LRRK2 G2019S mutation. Mov Disord. 2005;20:1077-1078.
96 Adams JR, van Netten H, Schulzer M, et al. PET in LRRK2 mutations: comparison to sporadic Parkinson’s disease and evidence for presymptomatic compensation. Brain. 2005;128:2777-2785.
97 Funayama M, Hasegawa K, Kowa H, et al. A new locus for Parkinson’s disease (PARK8) maps to chromosome 12p11.2-q13.1. Ann Neurol. 2002;51:296-301.
98 Wszolek ZK, Pfeiffer RF, Tsuboi Y, et al. Autosomal dominant parkinsonism associated with variable synuclein and tau pathology. Neurology. 2004;62:1619-1622.
99 Zimprich A, Biskup S, Leitner P, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601-607.
100 Ross OA, Farrer MJ. Pathophysiology, pleiotrophy and paradigm shifts: genetic lessons from Parkinson’s disease. Biochem Soc Trans. 2005;33:586-590.
101 Ridley AJ. Rho family proteins: coordinating cell responses. Trends Cell Biol. 2001;11:471-477.
102 Hampshire DJ, Roberts E, Crow Y, et al. Kufor-Rakeb syndrome, pallido-pyramidal degeneration with supranuclear upgaze paresis and dementia, maps to 1p36. J Med Genet. 2001;38:680-682.
103 Najim al-Din AS, Wriekat A, Mubaidin A, et al. Pallido-pyramidal degeneration, supranuclear upgaze paresis and dementia: Kufor-Rakeb syndrome. Acta Neurol Scand. 1994;89:347-352.
104 Hicks AA, Petursson H, Jonsson T, et al. A susceptibility gene for late-onset idiopathic Parkinson’s disease. Ann Neurol. 2002;52:549-555.
105 Scott WK, Nance MA, Watts RL, et al. Complete genomic screen in Parkinson disease: evidence for multiple genes. JAMA. 2001;286:2239-2244.
106 DeStefano AL, Golbe LI, Mark MH, et al. Genome-wide scan for Parkinson’s disease: the GenePD Study. Neurology. 2001;57:1124-1126.
107 Farrer M, Maraganore DM, Lockhart P, et al. alpha-Synuclein gene haplotypes are associated with Parkinson’s disease. Hum Mol Genet. 2001;10:1847-1851.
108 Tan EK, Tan C, Shen H, et al. Alpha synuclein promoter and risk of Parkinson’s disease: microsatellite and allelic size variability. Neurosci Lett. 2003;336:70-72.
109 West AB, Maraganore D, Crook J, et al. Functional association of the parkin gene promoter with idiopathic Parkinson’s disease. Hum Mol Genet. 2002;11:2787-2792.
110 Aharon-Peretz J, Rosenbaum H, Gershoni-Baruch R. Mutations in the glucocerebrosidase gene and Parkinson’s disease in Ashkenazi Jews. N Engl J Med. 2004;351:1972-1977.
111 Foster NL, Wilhelmsen K, Sima AA, et al. Frontotemporal dementia and parkinsonism linked to chromosome 17: a consensus conference. Conference Participants. Ann Neurol. 1997;41:706-715.
112 Hulette CM, Pericak-Vance MA, Roses AD, et al. Neuropathological features of frontotemporal dementia and parkinsonism linked to chromosome 17q21–22 (FTDP-17): Duke Family 1684. J Neuropathol Exp Neurol. 1999;58:859-866.
113 Gwinn-Hardy K, Chen JY, Liu HC, et al. Spinocerebellar ataxia type 2 with parkinsonism in ethnic Chinese. Neurology. 2000;55:800-805.
114 Lu CS, Wu Chou YH, Yen TC, et al. Dopa-responsive parkinsonism phenotype of spinocerebellar ataxia type 2. Mov Disord. 2002;17:1046-1051.
115 Shan DE, Soong BW, Sun CM, et al. Spinocerebellar ataxia type 2 presenting as familial levodopa-responsive parkinsonism. Ann Neurol. 2001;50:812-815.
116 Hagerman RJ, Leehey M, Heinrichs W, et al. Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology. 2001;57:127-130.
117 Jacquemont S, Hagerman RJ, Leehey M, et al. Fragile X premutation tremor/ataxia syndrome: molecular, clinical, and neuroimaging correlates. Am J Hum Genet. 2003;72:869-878.
118 Haberhausen G, Schmitt I, Kohler A, et al. Assignment of the dystonia-parkinsonism syndrome locus, DYT3, to a small region within a 1.8-Mb YAC contig of Xq13.1. Am J Hum Genet. 1995;57:644-650.
119 de Carvalho AP, Sweadner KJ, Penniston JT, et al. Mutations in the Na+/K+-ATPase alpha3 gene ATP1A3 are associated with rapid-onset dystonia parkinsonism. Neuron. 2004;43:169-175.
120 Wooten GF, Currie LJ, Bennett JP, et al. Maternal inheritance in Parkinson’s disease. Ann Neurol. 1997;41:265-268.
121 Schapira AH, Holt IJ, Sweeney M, et al. Mitochondrial DNA analysis in Parkinson’s disease. Mov Disord. 1990;5:294-297.
122 Ozawa T, Tanaka M, Ino H, et al. Distinct clustering of point mutations in mitochondrial DNA among patients with mitochondrial encephalomyopathies and with Parkinson’s disease. Biochem Biophys Res Commun. 1991;176:938-946.
123 Ikebe S, Tanaka M, Ozawa T. Point mutations of mitochondrial genome in Parkinson’s disease. Brain Res Mol Brain Res. 1995;28:281-295.
124 Shoffner JM, Brown MD, Torroni A, et al. Mitochondrial DNA variants observed in Alzheimer disease and Parkinson disease patients. Genomics. 1993;17:171-184.
125 Lucking CB, Kosel S, Mehraein P, et al. Absence of the mitochondrial A7237T mutation in Parkinson’s disease. Biochem Biophys Res Commun. 1995;211:700-704.
126 Kosel S, Grasbon-Frodl EM, Hagenah JM, et al. Parkinson disease: analysis of mitochondrial DNA in monozygotic twins. Neurogenetics. 2000;2:227-230.
127 Richter G, Sonnenschein A, Grunewald T, et al. Novel mitochondrial DNA mutations in Parkinson’s disease. J Neural Transm. 2002;109:721-729.
128 Vives-Bauza C, Andreu AL, Manfredi G, et al. Sequence analysis of the entire mitochondrial genome in Parkinson’s disease. Biochem Biophys Res Commun. 2002;290:1593-1601.
129 van der Walt JM, Nicodemus KK, Martin ER, et al. Mitochondrial polymorphisms significantly reduce the risk of Parkinson disease. Am J Hum Genet. 2003;72:804-811.
130 Pyle A, Foltynie T, Tiangyou W, et al. Mitochondrial DNA haplogroup cluster UKJT reduces the risk of PD. Ann Neurol. 2005;57:564-567.
131 Ross OA, McCormack R, Maxwell LD, et al. mt4216C variant in linkage with the mtDNA TJ cluster may confer a susceptibility to mitochondrial dysfunction resulting in an increased risk of Parkinson’s disease in the Irish. Exp Gerontol. 2003;38:397-405.
132 Luoma P, Melberg A, Rinne JO, et al. Parkinsonism, premature menopause, and mitochondrial DNA polymerase gamma mutations: clinical and molecular genetic study. Lancet. 2004;364:875-882.
133 Mancuso M, Filosto M, Oh SJ, et al. A novel polymerase gamma mutation in a family with ophthalmoplegia, neuropathy, and parkinsonism. Arch Neurol. 2004;61:1777-1779.
134 Rajput AH, Uitti RJ, Stern W, et al. Early onset Parkinson’s disease in Saskatchewan-environmental considerations for etiology. Can J Neurol Sci. 1986;13:312-316.
135 Rajput AH, Uitti RJ, Rajput AH. Neurological disorders and services in Saskatchewan: a report based on provincial health care records. Neuroepidemiology. 1988;7:145-151.
136 Behari M, Srivastava AK, Das RR, et al. Risk factors of Parkinson’s disease in Indian patients. J Neurol Sci. 2001;190:49-55.
137 Semchuk KM, Love EJ, Lee RG. Parkinson’s disease and exposure to rural environmental factors: a population based case-control study. Can J Neurol Sci. 1991;18:279-286.
138 Barbeau A, Roy M, Bernier G, et al. Ecogenetics of Parkinson’s disease: prevalence and environmental aspects in rural areas. Can J Neurol Sci. 1987;14:36-41.
139 Gorrell JM, DiMonte D, Graham D. The role of the environment in Parkinson’s disease. Environ Health Perspect. 1996;104:652-654.
140 Fall PA, Fredrikson M, Axelson O, et al. Nutritional and occupational factors influencing the risk of Parkinson’s disease: a case-control study in southeastern Sweden. Mov Disord. 1999;14:28-37.
141 Semchuk KM, Love EJ, Lee RG. Parkinson’s disease and exposure to agricultural work and pesticide chemicals. Neurology. 1992;42:1328-1335.
142 Seidler A, Hellenbrand W, Robra BP, et al. Possible environmental, occupational, and other etiologic factors for Parkinson’s disease: a case-control study in Germany. Neurology. 1996;46:1275-1284.
143 Fleming L, Mann JB, Bean J, et al. Parkinson’s disease and brain levels of organochlorine pesticides. Ann Neurol. 1994;36:100-103.
144 Corsini GU, Pintus S, Chiueh CC, et al. 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) neurotoxicity in mice is enhanced by pretreatment with diethyldithiocarbamate. Eur J Pharmacol. 1985;119:127-128.
145 Tanner CM, Goldman SM. Epidemiology of Parkinson’s disease. Neurol Clin. 1996;14:317-335.
146 Klawans HL, Stein RW, Tanner CM, et al. A pure parkinsonian syndrome following acute carbon monoxide intoxication. Arch Neurol. 1982;39:302-304.
147 Elwan MA, Richardson JR, Guillot TS, et al. Pyrethroid pesticide-induced alterations in dopamine transporter function. Toxicol Appl Pharmacol. 2005.
148 Pittman JT, Dodd CA, Klein BG. Immunohistochemical changes in the mouse striatum induced by the pyrethroid insecticide permethrin. Int J Toxicol. 2003;22:359-370.
149 Bloomquist JR, Barlow RL, Gillette JS, et al. Selective effects of insecticides on nigrostriatal dopaminergic nerve pathways. Neurotoxicology. 2002;23(4–5):537-544.
150 Imamura L, Yasuda M, Kuramitsu K, et al. Deltamethrin, a pyrethroid insecticide, is a potent inducer for the activity-dependent gene expression of brain-derived neurotrophic factor in neurons. J Pharmacol Exp Ther. 2006;316:136-143.
151 Betarbet R, Sherer TB, MacKenzie G, et al. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat Neurosci. 2000;3:1301-1306.
152 Manning-Bog AB, McCormack AL, Li J, et al. The herbicide paraquat causes upregulation and aggregation of alphasynuclein in mice: paraquat and alphasynuclein. J Biol Chem. 2002;277:1641-1644.
153 Champy P, Hoglinger GU, Feger J, et al. Annonacin, a lipophilic inhibitor of mitochondrial complex I, induces nigral and striatal neurodegeneration in rats: possible relevance for atypical parkinsonism in Guadeloupe. J Neurochem. 2004;88:63-69.
154 Tipton KF, Singer TP. Advances in our understanding of the mechanisms of the neurotoxicity of MPTP and related compounds. J Neurochem. 1993;61:1191-1206.
155 Hochberg F, Lilienfeld D, Olanow W, et al. Progression after chronic manganese exposure. Neurology. 1993;43:1479-1483.
156 Mena I. Manganese poisoning. In: Vinken PJ, Bruyn GW, editors. Handbook of Clinical Neurology. Amsterdam: North Holland Publishing Company; 1979:217-237.
157 Jankovic J. Searching for a relationship between manganese and welding and Parkinson’s disease. Neurology. 2005;64:2021-2028.
158 Ziegler L. Follow-up studies in persons who have had epidemic encephalitis. JAMA. 1928;21:138-141.
159 Von Economo C. The Sequelae of Encephalitis Lethargica. New York: Oxford University Press, 1931;105-106.
160 Ebmeier KP, Mutch WJ, Calder SA, et al. Does idiopathic parkinsonism in Aberdeen follow intrauterine influenza? J Neurol Neurosurg Psychiatry. 1989;52:911-913.
161 Schwartz J, Elizan TS. Search for viral particles and virusspecific products in idiopathic Parkinson disease brain material. Ann Neurol. 1979;6:261-263.
162 Baron JA. Cigarette smoking and Parkinson’s disease. Neurology. 1986;36:1490-1496.
163 Ascherio A, Zhang SM, Hernan MA, et al. Prospective study of caffeine consumption and risk of Parkinson’s disease in men and women. Ann Neurol. 2001;50:56-63.
164 McGeer PL, Itagaki S, Boyes BE, et al. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology. 1988;38:1285-1291.
165 Boka G, Anglade P, Wallach D, et al. Immunocytochemical analysis of tumor necrosis factor and its receptors in Parkinson’s disease. Neurosci Lett. 1994;172:151-154.
166 Hunot S, Dugas N, Faucheux B, et al. FcepsilonRII/CD23 is expressed in Parkinson’s disease and induces, in vitro, production of nitric oxide and tumor necrosis factor-alpha in glial cells. J Neurosci. 1999;19:3440-3447.
167 McGeer PL, Schulzer M, McGeer EG. Arthritis and antiinflammatory agents as possible protective factors for Alzheimer’s disease: a review of 17 epidemiologic studies. Neurology. 1996;47:425-432.
168 In t’ Veld BA, Ruitenberg A, Hofman A, et al. Nonsteroidal antiinflammatory drugs and the risk of Alzheimer’s disease. N Engl J Med. 2001;345:1515-1521.
169 Chen H, Zhang SM, Hernan MA, et al. Nonsteroidal antiinflammatory drugs and the risk of Parkinson disease. Arch Neurol. 2003;60:1059-1064.
170 Dexter DT, Wells FR, Agid F, et al. Increased nigral iron content in postmortem parkinsonian brain. Lancet. 1987;2:1219-1220.
171 Sofic E, Paulus W, Jellinger K, et al. Selective increase of iron in substantia nigra zona compacta of parkinsonian brains. J Neurochem. 1991;56:978-982.
172 Jellinger K, Paulus W, Grundke-Iqbal I, et al. Brain iron and ferritin in Parkinson’s and Alzheimer’s diseases. J Neural Transm Park Dis Dement Sect. 1990;2:327-340.
173 Jellinger K, Kienzl E, Rumpelmair G, et al. Iron-melanin complex in substantia nigra of parkinsonian brains: an x-ray microanalysis. J Neurochem. 1992;59:1168-1171.
174 Hirsch EC, Brandel JP, Galle P, et al. Iron and aluminum increase in the substantia nigra of patients with Parkinson’s disease: an X-ray microanalysis. J Neurochem. 1991;56:446-451.
175 Dexter DT, Carayon A, Vidailhet M, et al. Decreased ferritin levels in brain in Parkinson’s disease. J Neurochem. 1990;55:16-20.
176 Riederer P, Sofic E, Rausch WD, et al. Transition metals, ferritin, glutathione, and ascorbic acid in parkinsonian brains. J Neurochem. 1989;52:515-520.
177 Mann VM, Cooper JM, Daniel SE, et al. Complex I, iron, and ferritin in Parkinson’s disease substantia nigra. Ann Neurol. Dec. 1994;36:876-881.
178 Perry TL, Godin DV, Hansen S. Parkinson’s disease: a disorder due to nigral glutathione deficiency? Neurosci Lett. 1982;33:305-310.
179 Sian J, Dexter DT, Lees AJ, et al. Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia. Ann Neurol. 1994;36:348-355.
180 Marttila RJ, Lorentz H, Rinne UK. Oxygen toxicity protecting enzymes in Parkinson’s disease. Increase of superoxide dismutase-like activity in the substantia nigra and basal nucleus. J Neurol Sci. 1988;86:321-331.
181 Saggu H, Cooksey J, Dexter D, et al. A selective increase in particulate superoxide dismutase activity in parkinsonian substantia nigra. J Neurochem. 1989;53:692-697.
182 Hirsch EC, Graybiel AM, Agid Y. Selective vulnerability of pigmented dopaminergic neurons in Parkinson’s disease. Acta Neurol Scand Suppl. 1989;126:19-22.
183 Ceballos I, Lafon M, Javoy-Agid F, et al. Superoxide dismutase and Parkinson’s disease. Lancet. 1990;335:1035-1036.
184 Dexter D, Carter C, Agid F, et al. Lipid peroxidation as cause of nigral cell death in Parkinson’s disease. Lancet. 1986;2:639-640.
185 Sanchez-Ramos J, Ocervik E, Ames BN. A marker of oxyradical-mediated DNA damage (8-hydroxy-2′-deoxyguanosine) is increased in nigrostriatum of Parkinson’s disease brain. Neurodegeneration. 1994;3:197-204.
186 Itzhak Y, Ali SF. The neuronal nitric oxide synthase inhibitor, 7-nitroindazole, protects against methamphetamine-induced neurotoxicity in vivo. J Neurochem. 1996;67:1770-1773.
187 Kuiper MA, Visser JJ, Bergmans PL, et al. Decreased cerebrospinal fluid nitrate levels in Parkinson’s disease, Alzheimer’s disease and multiple system atrophy patients. J Neurol Sci. 1994;121:46-49.
188 Molina JA, Jimenez-Jimenez FJ, Navarro JA, et al. Cerebrospinal fluid nitrate levels in patients with Parkinson’s disease. Acta Neurol Scand. 1996;93(2–3):123-126.
189 Qureshi GA, Baig S, Bednar I, et al. Increased cerebrospinal fluid concentration of nitrite in Parkinson’s disease. Neuroreport. 1995;6:1642-1644.
190 Schapira AH, Cooper JM, Dexter D. Mitochondrial complex 1 deficienty in Parkinson’s disease. Annals of neurology. 1989;26:122-123.
191 Schapira AH, Cooper JM, Dexter D, et al. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet. 1989;1:1269.
192 Schapira AH, Mann VM, Cooper JM, et al. Anatomic and disease specificity of NADH CoQ1 reductase (complex I) deficiency in Parkinson’s disease. J Neurochem. 1990;55:2142-2145.
193 Schapira AH. Evidence for mitochondrial dysfunction in Parkinson’s disease—a critical appraisal. Mov Disord. 1994;9:125-138.
194 Taylor DJ, Krige D, Barnes PR, et al. A 31P magnetic resonance spectroscopy study of mitochondrial function in skeletal muscle of patients with Parkinson’s disease. J Neurol Sci. 1994;125:77-81.
195 Penn AM, Roberts T, Hodder J, et al. Generalized mitochondrial dysfunction in Parkinson’s disease detected by magnetic resonance spectroscopy of muscle. Neurology. 1995;45:2097-2099.
196 Parker WDJr, Boyson SJ, Parks JK. Abnormalities of the electron transport chain in idiopathic Parkinson’s disease. Ann Neurol. 1989;26:719-723.
197 Krige D, Carroll MT, Cooper JM, et al. Platelet mitochondrial function in Parkinson’s disease. The Royal Kings and Queens Parkinson Disease Research Group. Ann Neurol. 1992;32:782-788.
198 Haas RH, Nasirian F, Nakano K, et al. Low platelet mitochondrial complex I and complex II/III activity in early untreated Parkinson’s disease. Ann Neurol. 1995;37:714-722.
199 Ansari KA, Johnson A. Olfactory function in patients with Parkinson’s disease. J Chronic Dis. 1975;28:493-497.
200 Doty RL, Deems DA, Stellar S. Olfactory dysfunction in parkinsonism: a general deficit unrelated to neurologic signs, disease stage, or disease duration. Neurology. 1988;38:1237-1244.
201 Doty RL, Stern MB, Pfeiffer C, et al. Bilateral olfactory dysfunction in early stage treated and untreated idiopathic Parkinson’s disease. J Neurol Neurosurg Psychiatry. 1992;55:138-142.
202 Montgomery EBJr, Baker KB, Lyons K, et al. Abnormal performance on the PD test battery by asymptomatic first-degree relatives. Neurology. 1999;52:757-762.
203 Markopoulou K, Larsen KW, Wszolek EK, et al. Olfactory dysfunction in familial parkinsonism. Neurology. 1997;49:1262-1267.
204 Hawkes C. Olfaction in neurodegenerative disorder. Mov Disord. 2003;18:364-372.
205 Boeve BF, Silber MH, Ferman TJ, et al. Association of REM sleep behavior disorder and neurodegenerative disease may reflect an underlying synucleinopathy. Mov Disord. 2001;16:622-630.
206 Khan NL, Katzenschlager R, Watt H, et al. Olfaction differentiates parkin disease from earlyonset parkinsonism and Parkinson disease. Neurology. 2004;62:1224-1226.
207 Gagnon JF, Bedard MA, Fantini ML, et al. REM sleep behavior disorder and REM sleep without atonia in Parkinson’s disease. Neurology. 2002;59:585-589.
208 Comella CL, Tanner CM, Ristanovic RK. Polysomnographic sleep measures in Parkinson’s disease patients with treatment-induced hallucinations. Ann Neurol. 1993;34:710-714.
209 Ohayon MM, Caulet M, Priest RG. Violent behavior during sleep. J Clin Psychiatry. 1997;58:369-376.
210 Fantini ML, Ferini-Strambi L, Montplaisir J. Idiopathic REM sleep behavior disorder: toward a better nosologic definition. Neurology. 2005;64:780-786.
211 Schenck CH, Bundlie SR, Mahowald MW. Delayed emergence of a parkinsonian disorder in 38% of 29 older men initially diagnosed with idiopathic rapid eye movement sleep behaviour disorder. Neurology. 1996;46:388-393.
212 Olson EJ, Boeve BF, Silber MH. Rapid eye movement sleep behaviour disorder: demographic, clinical and laboratory findings in 93 cases. Brain. 2000;123:331-339.
213 Albin RL, Koeppe RA, Chervin RD, et al. Decreased striatal dopaminergic innervation in REM sleep behavior disorder. Neurology. 2000;55:1410-1412.
214 Eisensehr I, Linke R, Noachtar S, et al. Reduced striatal dopamine transporters in idiopathic rapid eye movement sleep behaviour disorder. Comparison with Parkinson’s disease and controls. Brain. 2000;123:1155-1160.
215 Byrne KG, Pfeiffer R, Quigley EM. Gastrointestinal dysfunction in Parkinson’s disease. A report of clinical experience at a single center. J Clin Gastroenterol. 1994;19:11-16.
216 Korczyn AD. Autonomic nervous system screening in patients with early Parkinson’s disease. In: Przuntek H, Riederer P, editors. Early Diagnosis and Preventive Therapy in Parkinson’s Disease. Vienna: Springer-Velag; 1989:41-48.
217 Ashraf W, Pfeiffer RF, Park F, et al. Constipation in Parkinson’s disease: objective assessment and response to psyllium. Mov Disord. 1997;12:946-951.
218 Astarloa R, Mena MA, Sanchez V, et al. Clinical and pharmacokinetic effects of a diet rich in insoluble fiber on Parkinson disease. Clin Neuropharmacol. 1992;15:375-380.
219 Abbott RD, Petrovitch H, White LR, et al. Frequency of bowel movements and the future risk of Parkinson’s disease. Neurology. 2001;57:456-462.
220 Singaram C, Ashraf W, Gaumnitz EA, et al. Dopaminergic defect of enteric nervous system in Parkinson’s disease patients with chronic constipation. Lancet. 1995;346:861-864.
221 Mathers SE, Kempster PA, Swash M, et al. Constipation and paradoxical puborectalis contraction in anismus and Parkinson’s disease: a dystonic phenomenon? J Neurol Neurosurg Psychiatry. 1988;51:1503-1507.
222 Edwards LL, Quigley EM, Harned RK, et al. Defecatory function in Parkinson’s disease: response to apomorphine. Ann Neurol. 1993;33:490-493.
223 Djaldetti R, Mosberg-Galili R, Sroka H, et al. Camptocormia (bent spine) in patients with Parkinson’s disease—characterization and possible pathogenesis of an unusual phenomenon. Mov Disord. 1999;14:443-447.
224 Hoehn MM, Yahr MD. Parkinsonism: onset, progression and mortality. Neurology. 1967;17:427-442.
225 Hunker CJ, Abbs JH. Uniform frequency of parkinsonian resting tremor in the lips, jaw, tongue, and index finger. Mov Disord. 1990;5:71-77.
226 Scott RM, Brody JA, Schwab RS, et al. Progression of unilateral tremor and rigidity in Parkinson’s disease. Neurology. 1970;20:710-714.
227 Hely MA, Morris JG, Reid WG, et al. Sydney Multicenter Study of Parkinson’s disease: non-L-dopa-responsive problems dominate at 15 years. Mov Disord. 2004;20:190-199.
228 Jellinger KA. Pathology of Parkinson’s disease. Changes other than the nigrostriatal pathway. Mol Chem Neuropathol. 1991;14:153-197.
229 Lees AJ, Blackburn NA, Campbell VL. The nighttime problems of Parkinson’s disease. Clin Neuropharmacol. 1988;11:512-519.
230 van Hilten B, Hoff JI, Middelkoop HA, et al. Sleep disruption in Parkinson’s disease. Assessment by continuous activity monitoring. Arch Neurol. 1994;51:922-928.
231 Paus S, Brecht HM, Koster J, et al. Sleep attacks, daytime sleepiness, and dopamine agonists in Parkinson’s disease. Mov Disord. 2003;18:659-667.
232 Hobson DE, Lang AE, Martin WR, et al. Excessive daytime sleepiness and sudden-onset sleep in Parkinson disease: a survey by the Canadian Movement Disorders Group. JAMA. 2002;287:455-463.
233 Garcia-Borreguero D, Larosa O, Bravo M. Parkinson’s disease and sleep. Sleep Med Rev. 2003;7:115-129.
234 Tracik F, Ebersbach G. Sudden daytime sleep onset in Parkinson’s disease: polysomnographic recordings. Mov Disord. 2001;16:500-506.
235 Ulivelli M, Rossi S, Lombardi C, et al. Polysomnographic characterization of pergolide-induced sleep attacks in idiopathic PD. Neurology. 2002;58:462-465.
236 Frucht S, Rogers JD, Greene PE, et al. Falling asleep at the wheel: motor vehicle mishaps in persons taking pramipexole and ropinirole. Neurology. 1999;52:1908-1910.
237 Schapira AH. Sleep attacks (sleep episodes) with pergolide. Lancet. 2000;355:1332-1333.
238 Olanow CW, Schapira AH, Roth T. Waking up to sleep episodes in Parkinson’s disease. Mov Disord. 2000;15:212-215.
239 Rye DB, Bliwise DL, Dihenia B, et al. FAST TRACK: daytime sleepiness in Parkinson’s disease. J Sleep Res. 2000;9:63-69.
240 Basson R. Sex and idiopathic Parkinson’s disease. Adv Neurol. 2001;86:295-300.
241 Marsden CD. Problems with longterm levodopa therapy for Parkinson’s disease. Clin Neuropharmacol. 1994;17(Suppl 2):S32-S44.
242 Hillen ME, Sage JI. Nonmotor fluctuations in patients with Parkinson’s disease. Neurology. 1996;47:1180-1183.
243 Goetz CG, Tanner CM, Levy M, et al. Pain in Parkinson’s disease. Mov Disord. 1986;1:45-49.
244 Hughes AJ, Daniel SE, Kilford L, et al. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry. 1992;55:181-184.
245 Hughes AJ, Daniel SE, Ben Shlomo Y, et al. The accuracy of diagnosis of parkinsonian syndromes in a specialist movement disorder service. Brain. 2002;125(Pt 4):861-870.
246 Tolosa E, Wenning G, Poewe W. The diagnosis of Parkinson’s disease. Lancet Neurol. 2006;5:75-86.
247 Dopamine transporter brain imaging to assess the effects of pramipexole vs levodopa on Parkinson disease progression. JAMA. 2002;287:1653-1661.
248 Whone AL, Watts RL, Stoessl AJ, et al. Slower progression of Parkinson’s disease with ropinirole versus levodopa: the REAL-PET study. Ann Neurol. 2003;54:93-101.
249 Hardie RJ, Lees AJ. Neuroleptic-induced Parkinson’s syndrome: clinical features and results of treatment with levodopa. J Neurol Neurosurg Psychiatry. 1988;51:850-854.
250 Deuschl G, Bain P, Brin M. Consensus statement of the Movement Disorder Society on Tremor. Ad Hoc Scientific Committee. Mov Disord. 1998;13((Suppl) 3):2-23.
251 Cohen O, Pullman S, Jurewicz E, et al. Rest tremor in patients with essential tremor: prevalence, clinical correlates, and electrophysiologic characteristics. Arch Neurol. 2003;60:405-410.
252 Chaudhuri KR, Buxton-Thomas M, Dhawan V, et al. Long duration asymmetrical postural tremor is likely to predict development of Parkinson’s disease and not essential tremor: clinical follow up study of 13 cases. J Neurol Neurosurg Psychiatry. 2005;76:115-117.
253 Wenning GK, Ben Shlomo Y, Magalhaes M, et al. Clinical features and natural history of multiple system atrophy. An analysis of 100 cases. Brain. 1994;117:835-845.
254 Colosimo C, Albanese A, Hughes AJ, et al. Some specific clinical features differentiate multiple system atrophy (striatonigral variety) from Parkinson’s disease. Arch Neurol. 1995;52:294-298.
255 Hughes AJ, Colosimo C, Kleedorfer B, et al. The dopaminergic response in multiple system atrophy. J Neurol Neurosurg Psychiatry. 1992;55:1009-1013.
256 Litvan I, Agid Y, Calne D, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology. 1996;47:1-9.
257 Collins SJ, Ahlskog JE, Parisi JE, et al. Progressive supranuclear palsy: neuropathologically based diagnostic clinical criteria. J Neurol Neurosurg Psychiatry. 1995;58:167-173.
258 Grimes DA, Lang AE, Bergeron CB. Dementia as the most common presentation of cortical-basal ganglionic degeneration. Neurology. 1999;53:1969-1974.
259 Winikates J, Jankovic J. Clinical correlates of vascular parkinsonism. Arch Neurol. 1999;56:98-102.
260 Marder K, Tang MX, Cote L, et al. The frequency and associated risk factors for dementia in patients with Parkinson’s disease. Arch Neurol. 1995;52:695-701.
261 McKeith I, Mintzer J, Aarsland D, et al. Dementia with Lewy bodies. Lancet Neurol. 2004;3:19-28.
262 Schrag A, Good CD, Miszkiel K, et al. Differentiation of atypical parkinsonian syndromes with routine MRI. Neurology. 2000;54:697-702.
263 Kraft E, Trenkwalder C, Auer DP. T2*-weighted MRI differentiates multiple system atrophy from Parkinson’s disease. Neurology. 2002;59:1265-1267.
264 Schulz JB, Skalej M, Wedekind D, et al. Magnetic resonance imaging-based volumetry differentiates idiopathic Parkinson’s syndrome from multiple system atrophy and progressive supranuclear palsy. Ann Neurol. 1999;45:65-74.
265 Scherfler C, Seppi K, Donnemiller E, et al. Voxel-wise analysis of [123I]beta-CIT SPECT differentiates the Parkinson variant of multiple system atrophy from idiopathic Parkinson’s disease. Brain. 2005;128:1605-1612.
266 Marek KL, Seibyl JP, Zoghbi SS, et al. [123I] beta-CIT/SPECT imaging demonstrates bilateral loss of dopamine transporters in hemi-Parkinson’s disease. Neurology. 1996;46:231-237.
267 Poewe W, Scherfler C. Role of dopamine transporter imaging in investigation of parkinsonian syndromes in routine clinical practice. Mov Disord. 2003;18(Suppl 7):S16-S21.
268 Braune S, Reinhardt M, Schnitzer R, et al. Cardiac uptake of [123I]MIBG separates Parkinson’s disease from multiple system atrophy. Neurology. 1999;53:1020-1025.
269 Courbon F, Brefel-Courbon C, Thalamas C, et al. Cardiac MIBG scintigraphy is a sensitive tool for detecting cardiac sympathetic denervation in Parkinson’s disease. Mov Disord. 2003;18:890-897.
270 Becker G, Seufert J, Bogdahn U, et al. Degeneration of substantia nigra in chronic Parkinson’s disease visualized by transcranial color-coded real-time sonography. Neurology. 1995;45:182-184.
271 Berg D, Siefker C, Becker G. Echogenicity of the substantia nigra in Parkinson’s disease and its relation to clinical findings. J Neurol. 2001;248:684-689.
272 Kimber J, Mathias CJ, Lees AJ, et al. Physiological, pharmacological and neurohormonal assessment of autonomic function in progressive supranuclear palsy. Brain. 2000;123:1422-1430.
273 Barker R, Duncan J, Lees A. Subcutaneous apomorphine as a diagnostic test for dopaminergic responsiveness in parkinsonian syndromes. Lancet. 1989;1:675.
274 Merello M, Nouzeilles MI, Arce GP, et al. Accuracy of acute levodopa challenge for clinical prediction of sustained longterm levodopa response as a major criterion for idiopathic Parkinson’s disease diagnosis. Mov Disord. 2002;17:795-798.
275 Clarke CE, Davies P. Systematic review of acute levodopa and apomorphine challenge tests in the diagnosis of idiopathic Parkinson’s disease. J Neurol Neurosurg Psychiatry. 2000;69:590-594.
276 Rajput AH. Levodopa prolongs life expectancy and is nontoxic to substantia nigra. Parkinsonism Relat Disord. 2001;8:95-100.
277 Karlsen KH, Tandberg E, Arsland D, et al. Health related quality of life in Parkinson’s disease: a prospective longitudinal study. J Neurol Neurosurg Psychiatry. 2000;69:584-589.
278 Clarke CE, Zobkiw RM, Gullaksen E. Quality of life and care in Parkinson’s disease. Br J Clin Pract. 1995;49:288-293.
279 Williams DR, Lees AJ. Visual hallucinations in the diagnosis of idiopathic Parkinson’s disease: a retrospective autopsy study. Lancet Neurol. 2005;4:605-610.
280 Evans AH, Katzenschlager R, Paviour D, et al. Punding in Parkinson’s disease: its relation to the dopamine dysregulation syndrome. Mov Disord. 2004;19:397-405.
281 Molina JA, Sainz-Artiga MJ, Fraile A, et al. Pathologic gambling in Parkinson’s disease: a behavioral manifestation of pharmacologic treatment? Mov Disord. 2000;15:869-872.
282 Dodd ML, Klos KJ, Bower JH, et al. Pathological gambling caused by drugs used to treat Parkinson disease. Arch Neurol. 2005;62:1377-1381.
283 Fahn S. Adverse effects of levodopa. In: Olanow CW, editor. The Scientific Basis for the Treatment of Parkinson’s Disease. Carnforth: Parthenon Publishing Group; 1992:89-112.
284 Contin M, Riva R, Martinelli P, et al. A levodopa kineticdynamic study of the rate of progression in Parkinson’s disease. Neurology. 1998;51:1075-1080.
285 Shoulson I, Glaubiger GA, Chase TN. On-off response. Clinical and biochemical correlations during oral and intravenous levodopa administration in parkinsonian patients. Neurology. 1975;25:1144-1148.
286 Hardie RJ, Lees AJ, Stern GM. On-off fluctuations in Parkinson’s disease. A clinical and neuropharmacological study. Brain. 1984;107:487-506.
287 Mouradian MM, Heuser IJ, Baronti F, et al. Modification of central dopaminergic mechanisms by continuous levodopa therapy for advanced Parkinson’s disease. Ann Neurol. 1990;27:18-23.
288 Nutt JG. On-off phenomenon: relation to levodopa pharmacokinetics and pharmacodynamics. Ann Neurol. 1987;22:535-540.
289 Kurth MC, Tetrud JW, Tanner CM, et al. Doubleblind, placebo-controlled, crossover study of duodenal infusion of levodopa/carbidopa in Parkinson’s disease patients with “on-off” fluctuations. Neurology. 1993;43:1698-1703.
290 Kostic V, Przedborski S, Flaster E, et al. Early development of levodopa-induced dyskinesias and response fluctuations in young-onset Parkinson’s disease. Neurology. 1991;41:202-205.
291 Obeso JA, Rodriguez-Oroz C, Chana P, et al. The evolution and origin of motor complications in Parkinson’s disease. Neuroogy. 2000;55(Suppl 4):S13-S20.
292 Parkinson Study Group. Entacapone improves motor fluctuations in levodopa-treated Parkinson’s disease patients. Ann Neurol. 1997;42:747-755.
293 Russ H, Muller T, Woitalla D, Rahbar A, Hahn J, Kuhn W. Detection of tolcapone in the cerebrospinal fluid of parkinsonian subjects. Naunyn Schmiedebergs Arch Pharmacol. 1999;360:719-720.
294 Hauser RA, Molho E, Shale H, et al. A pilot evaluation of the tolerability, safety, and efficacy of tolcapone alone and in combination with oral selegiline in untreated Parkinson’s disease patients. Tolcapone De Novo Study Group. Mov Disord. 1998;13:643-647.
295 Jorga KM. COMT inhibitors: pharmacokinetic and pharma-codynamic comparisons. Clin Neuropharmacol. 1998;21:S9-S16.
296 Assal F, Spahr L, Hadengue A, et al. Tolcapone and fulminant hepatitis. Lancet. 1998;352:958.
297 Korlipara LV, Cooper JM, Schapira AH. Differences in toxicity of the catechol-O-methyl transferase inhibitors, tolcapone and entacapone to cultured human neuroblastoma cells. Neuropharmacology. 2004;46:562-569.
298 Marco AD, Appiah-Kubi LS, Chaudhuri KR. Use of the dopamine agonist cabergoline in the treatment of movement disorders. Exp Opin Pharmacother. 2002;3:1481-1487.
299 Olanow CW. Single blind double observer-controlled study of carbidopa/levodopa vs bromocriptine in untreated Parkinson patients. Arch Neurol. 1988;45:205.
300 Staal-Schreinemachers AL, Wesseling H, Kamphuis DJ, et al. Low-dose bromocriptine therapy in Parkinson’s disease: doubleblind, placebo-controlled study. Neurology. 1986;36:291-293.
301 Rinne UK, Bracco F, Chouza C, et al. Cabergoline in the treatment of early Parkinson’s disease: results of the first year of treatment in a doubleblind comparison of cabergoline and levodopa. The PKDS009 Collaborative Study Group. Neurology. 1997;48:363-368.
302 Rinne UK, Bracco F, Chouza C, et al. Early treatment of Parkinson’s disease with cabergoline delays the onset of motor complications. Results of a doubleblind levodopa controlled trial. The PKDS009 Study Group. Drugs. 1998;55(Suppl 1):23-30.
303 Barone P, Bravi D, Bermejo-Pareja F, et al. Pergolide monotherapy in the treatment of early PD: a randomized, controlled study. Pergolide Monotherapy Study Group. Neurology. 1999;53:573-579.
304 Parkinson Study Group. Safety and efficacy of pramipexole in early Parkinson disease. A randomized dose-ranging study. JAMA. 1997;278:125-130.
305 Shannon KM, Bennett JPJr, Friedman JH. Efficacy of pramipexole, a novel dopamine agonist, as monotherapy in mild to moderate Parkinson’s disease. The Pramipexole Study Group. Neurology. 1997;49:724-728.
306 Korczyn AD, Brunt ER, Larsen JP, et al. A 3-year randomized trial of ropinirole and bromocriptine in early Parkinson’s disease. The 053 Study Group. Neurology. 1999;53:364-370.
307 Adler CH, Sethi KD, Hauser RA, et al. Ropinirole for the treatment of early Parkinson’s disease. The Ropinirole Study Group. Neurology. 1997;49:393-399.
308 Hoehn MM, Elton RL. Low dosages of bromocriptine added to levodopa in Parkinson’s disease. Neurology. 1985;35:199-206.
309 Toyokura Y, Mizuno Y, Kase M, et al. Effects of bromocriptine on parkinsonism. A nation-wide collaborative doubleblind study. Acta Neurol Scand. 1985;72:157-170.
310 Inzelberg R, Nisipeanu P, Rabey JM, et al. Doubleblind comparison of cabergoline and bromocriptine in Parkinson’s disease patients with motor fluctuations. Neurology. 1996;47:785-788.
311 Olanow CW, Fahn S, Muenter M, et al. A multicenter doubleblind placebo-controlled trial of pergolide as an adjunct to Sinemet in Parkinson’s disease. Mov Disord. 1994;9:40-47.
312 Diamond SG, Markham CH, Treciokas LJ. Doubleblind trial of pergolide for Parkinson’s disease. Neurology. 1985;35:291-295.
313 Guttman M. Doubleblind comparison of pramipexole and bromocriptine treatment with placebo in advanced Parkinson’s disease. International Pramipexole-Bromocriptine Study Group. Neurology. 1997;49:1060-1065.
314 Lieberman A, Ranhosky A, Korts D. Clinical evaluation of pramipexole in advanced Parkinson’s disease: results of a doubleblind, placebo-controlled, parallel-group study. Neurology. 1997;49:162-168.
315 Lieberman A, Olanow CW, Sethi K, et al. A multicenter trial of ropinirole as adjunct treatment for Parkinson’s disease. Ropinirole Study Group. Neurology. 1998;51:1057-1062.
316 Schapira AH. Excessive daytime sleepiness in Parkinson’s disease. Neurology. 2004;63(Suppl 3):S24-S27.
317 Holloway RG, Shoulson I, Fahn S, et al. Pramipexole vs levodopa as initial treatment for Parkinson disease: a 4-year randomized controlled trial. Arch Neurol. 2004;61:1044-1053.
318 Muller T, Fritze J. Fibrosis associated with dopamine agonist therapy in Parkinson’s disease. Clin Neuropharmacol. 2003;26:109-111.
319 Van Camp G, Flamez A, Cosyns B, et al. Treatment of Parkinson’s disease with pergolide and relation to restrictive valvular heart disease. Lancet. 2004;363:1179-1183.
320 Rascol O, Brooks DJ, Korczyn AD, et al. A five-year study of the incidence of dyskinesia in patients with early Parkinson’s disease who were treated with ropinirole or levodopa. 056 Study Group. N Engl J Med. 2000;342:1484-1491.
321 Parkinson Study Group. Pramipexole vs levodopa as initial treatment for Parkinson disease: A randomized controlled trial. JAMA. 2000;284:1931-1938.
322 Oertel WH. Pergolide versus levodopa (PELMOPET). Mov Disord. 2000;15(Suppl 3):4.
323 Schapira AH. Disease modification in Parkinson’s disease. Lancet Neurol. 2004;3:362-368.
324 Parkinson Study Group. Effects of tocopherol and deprenyl on the progression of disability in early Parkinson’s disease. N Engl J Med. 1993;328:176-183.
325 Olanow CW, Hauser RA, Gauger L, et al. The effect of deprenyl and levodopa on the progression of Parkinson’s disease. Ann Neurol. 1995;38:771-777.
326 Shoulson I, Oakes D, Fahn S, et al. Impact of sustained deprenyl (selegiline) in levodopa-treated Parkinson’s disease: a randomized placebo-controlled extension of the deprenyl and tocopherol antioxidative therapy of parkinsonism trial. Ann Neurol. 2002;51:604-612.
327 Ives NJ, Stowe RL, Marro J, et al. Monoamine oxidase type B inhibitors in early Parkinson’s disease: meta-analysis of 17 randomised trials involving 3525. patients. BMJ. 2004;11:329.
328 Parkinson Study Group. A controlled, randomized, delayedstart study of rasagiline in early Parkinson disease. Arch Neurol. 2004;61:561-566.
329 Parkinson Study Group. A randomized placebo-controlled trial of rasagiline in levodopa-treated patients with Parkinson disease and motor fluctuations: the PRESTO study. Arch Neurol. 2005;62:241-248.
330 Rascol O, Brooks DJ, Melamed E, et al. Rasagiline as an adjunct to levodopa in patients with Parkinson’s disease and motor fluctuations (LARGO, Lasting effect in Adjunct therapy with Rasagiline Given Once daily, study): a randomised, doubleblind, parallel-group trial. Lancet. 2005;365:947-954.
331 Cantello R, Riccio A, Gilli M, et al. Bornaprine vs placebo in Parkinson disease: doubleblind controlled crossover trial in 30. patients. Ital J Neurol Sci. 1986;7:139-143.
332 Martin WE, Loewenson RB, Resch JA, et al. A controlled study comparing trihexyphenidyl hydrochloride plus levodopa with placebo plus levodopa in patients with Parkinson’s disease. Neurology. 1974;24:912-919.
333 Cooper JA, Sagar HJ, Doherty SM, et al. Different effects of dopaminergic and anticholinergic therapies on cognitive and motor function in Parkinson’s disease. A follow-up study of untreated patients. Brain. 1992;115:1701-1725.
334 Friedman JH, Koller WC, Lannon MC, et al. Benztropine versus clozapine for the treatment of tremor in Parkinson’s disease. Neurology. 1997;48:1077-1081.
335 Schwab RS, England ACJr, Poskanzer DC, Young RR. Amantadine in the treatment of Parkinson’s disease. JAMA. 1969;208:1168-1170.
336 Factor SA, Molho ES. Transient benefit of amantadine in Parkinson’s disease: the facts about the myth. Mov Disord. 1999;14:515-517.
337 Parkes JD, Baxter RC, Marsden CD, Rees JE. Comparative trial of benzhexol, amantadine, and levodopa in the treatment of Parkinson’s disease. J Neurol Neurosurg Psychiatry. 1974;37:422-426.
338 Walker JE, Albers JW, Tourtellotte WW, et al. A qualitative and quantitative evaluation of amantadine in the treatment of Parkinson’s disease. J Chronic Dis. 1972;25:149-182.
339 Shulman LM, Minagar A, Rabinstein A, et al. The use of dopamine agonists in very elderly patients with Parkinson’s disease. Mov Disord. 2000;15:664-668.
340 Schapira AH, Obeso JA. Timing of treatment initiation in Parkinson’s disease: a need for re-appraisal? Ann Neurol. 2006;59:559-562.
341 Obeso JA, Rodriguez-Oroz MC, Rodriguez M, et al. Pathophysiology of the basal ganglia in Parkinson’s disease. Trends Neurosci. 2000;23(Suppl):S8-S19.
342 Pifl C, Schingnitz G, Hornykiewicz O. Striatal and nonstriatal neurotransmitter changes in MPTP-parkinsonism in rhesus monkey: the symptomatic versus the asymptomatic condition. Neurochem Int. 1992;20(Suppl):295S-297S.
343 Sossi V, Fuente-Fernandez R, Holden JE, et al. Changes of dopamine turnover in the progression of Parkinson’s disease as measured by positron emission tomography: their relation to disease-compensatory mechanisms. J Cereb Blood Flow Metab. 2004;24:869-876.
344 Hilker R, Schweitzer K, Coburger S, et al. Nonlinear progression of Parkinson disease as determined by serial positron emission tomographic imaging of striatal fluorodopa F 18 activity. Arch Neurol. 2005;62:378-382.
345 Bezard E, Boraud T, Bioulac B, et al. Involvement of the subthalamic nucleus in glutamatergic compensatory mechanisms. Eur J Neurosci. 1999;11:2167-2170.
346 Bezard E, Dovero S, Prunier C, et al. Relationship between the appearance of symptoms and the level of nigrostriatal degeneration in a progressive 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned macaque model of Parkinson’s disease. J Neurosci. 2001;21:6853-6861.
347 Fabbrini G, Mouradian MM, Juncos JL, et al. Motor fluctuations in Parkinson’s disease: central pathophysiological mechanisms, Part I. Ann Neurol. 1988;24:366-371.
348 Horstink MW, Zijlmans JC, Pasman JW, et al. Which risk factors predict the levodopa response in fluctuating Parkinson’s disease? Ann Neurol. 1990;27:537-543.
349 Roos RA, Vredevoogd CB, van der Velde EA. Response fluctuations in Parkinson’s disease. Neurology. 1990;40:1344-1346.
350 Koller WC, Hutton JT, Tolosa E, et al. Immediate-release and controlled-release carbidopa/levodopa in PD: a 5-year randomized multicenter study. Carbidopa/Levodopa Study Group. Neurology. 1999;53:1012-1019.
351 Stocchi F, Ruggieri S, Vacca L, et al. Prospective randomized trial of lisuride infusion versus oral levodopa in patients with Parkinson’s disease. Brain. 2002;125(Pt 9):2058-2066.
352 Stocchi F, Vacca L, Berardelli A, et al. Dual dopamine agonist treatment in Parkinson’s disease. J Neurol. 2003;250:822-826.
353 Rinne UK, Larsen JP, Siden A, et al. Entacapone enhances the response to levodopa in parkinsonian patients with motor fluctuations. Nomecomt Study Group. Neurology. 1998;51:1309-1314.
354 Lhermitte F, Agid Y, Signoret JL. Onset and end-of-dose levodopa-induced dyskinesias. Possible treatment by increasing the daily doses of levodopa. Arch Neurol. 1978;35:261-263.
355 Verhagen ML, Del Dotto P, van den MP, et al. Amantadine as treatment for dyskinesias and motor fluctuations in Parkinson’s disease. Neurology. 1998;50:1323-1326.
356 Metman LV, Del Dotto P, LePoole K, et al. Amantadine for levodopa-induced dyskinesias: a 1-year follow-up study. Arch Neurol. 1999;56:1383-1386.
357 Snow BJ, Macdonald L, Mcauley D, et al. The effect of amantadine on levodopa-induced dyskinesias in Parkinson’s disease: a doubleblind, placebo-controlled study. Clin Neuropharmacol. 2000;23:82-85.
358 Corrigan MH, Denahan AQ, Wright CE, et al. Comparison of pramipexole, fluoxetine, and placebo in patients with major depression. Depress Anxiety. 2000;11:58-65.
359 Lemke MR, Brecht HM, Koester J, et al. Anhedonia, depression, and motor functioning in Parkinson’s disease during treatment with pramipexole. J Neuropsychiatry Clin Neurosci. 2005;17:214-220.
360 Rektorova I, Rektor I, Bares M, et al. Pramipexole and pergolide in the treatment of depression in Parkinson’s disease: a national multicentre prospective randomized study. Eur J Neurol. 2003;10:399-406.
361 Pollak P, Tison F, Rascol O, et al. Clozapine in drug induced psychosis in Parkinson’s disease: a randomised, placebo controlled study with open follow up. J Neurol Neurosurg Psychiatry. 2004;75:689-695.
362 Fernandez HH, Trieschmann ME, Burke MA, et al. Longterm outcome of quetiapine use for psychosis among Parkinsonian patients. Mov Disord. 2003;18:510-514.
363 Mancini F, Tassorelli C, Martignoni E, et al. Longterm evaluation of the effect of quetiapine on hallucinations, delusions and motor function in advanced Parkinson disease. Clin Neuropharmacol. 2004;27:33-37.
364 Emre M, Aarsland D, Albanese A, et al. Rivastigmine for dementia associated with Parkinson’s disease. N Engl J Med. 2004;351:2509-2518.
365 Happe S, Pirker W, Sauter C, et al. Successful treatment of excessive daytime sleepiness in Parkinson’s disease with modafinil. J Neurol. 2001;248:632-634.
366 Raffaele R, Vecchio I, Giammusso B, et al. Efficacy and safety of fixed-dose oral sildenafil in the treatment of sexual dysfunction in depressed patients with idiopathic Parkinson’s disease. Eur Urol. 2002;41:382-386.
367 O’Sullivan JD. Apomorphine as an alternative to sildenafil in Parkinson’s disease. J Neurol Neurosurg Psychiatry. 2002;72:681.
368 Mancini F, Zangaglia R, Cristina S, et al. Doubleblind, placebo-controlled study to evaluate the efficacy and safety of botulinum toxin type A in the treatment of drooling in parkinsonism. Mov Disord. 2003;18:685-688.
369 Narabayashi H. Stereotaxic Vim thalamotomy for treatment of tremor. Eur Neurol. 1989;29(Suppl 1):29-32.
370 Tasker RR. Thalamotomy. Neurosurg Clin N Am. 1990;1:841-864.
371 Dogali M, Fazzini E, Kolodny E, et al. Stereotactic ventral pallidotomy for Parkinson’s disease. Neurology. 1995;45:753-761.
372 Baron MS, Vitek JL, Bakay RA, et al. Treatment of advanced Parkinson’s disease by posterior GPi pallidotomy: 1-year results of a pilot study. Ann Neurol. 1996;40:355-366.
373 Lang AE, Lozano AM, Montgomery E, et al. Posteroventral medial pallidotomy in advanced Parkinson’s disease. N Engl J Med. 1997;337:1036-1042.
374 Fine J, Duff J, Chen R, et al. Longterm follow-up of unilateral pallidotomy in advanced Parkinson’s disease. N Engl J Med. 2000;342:1708-1714.
375 Bergman H, Wichmann T, DeLong MR. Reversal of experimental parkinsonism by lesions of the subthalamic nucleus. Science. 1990;249:1436-1438.
376 Aziz TZ, Peggs D, Sambrook MA, et al. Lesion of the subthalamic nucleus for the alleviation of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced parkinsonism in the primate. Mov Disord. 1991;6:288-292.
377 Guridi J, Herrero MT, Luquin MR, et al. Subthalamotomy in parkinsonian monkeys. Behavioural and biochemical analysis. Brain. 1996;119:1717-1727.
378 Alvarez L, Macias R, Guridi J, et al. Dorsal subthalamotomy for Parkinson’s disease. Mov Disord. 2001;16:72-78.
379 Su PC, Tseng HM, Liu HM, et al. Treatment of advanced Parkinson’s disease by subthalamotomy: one-year results. Mov Disord. 2003;18:531-538.
380 Alvarez L, Macias R, Lopez G, et al. Bilateral subthalamotomy in Parkinson’s disease: initial and longterm response. Brain. 2005;128:570-583.
381 Chen CC, Lee ST, Wu T, et al. Hemiballism after subthalamotomy in patients with Parkinson’s disease: report of 2 cases. Mov Disord. 2002;17:1367-1371.
382 Tseng HM, Su PC, Liu HM. Persistent hemiballism after subthalamotomy: the size of the lesion matters more than the location. Mov Disord. 2003;18:1209-1211.
383 Benabid AL, Pollak P, Louveau A, et al. Combined (thalamotomy and stimulation) stereotactic surgery of the VIM thalamic nucleus for bilateral Parkinson disease. Appl Neurophysiol. 1987;50:344-346.
384 Benazzouz A, Hallett M. Mechanism of action of deep brain stimulation. Neurology. 2000;55(Suppl 6):S13-S16.
385 Blond S, Caparros-Lefebvre D, Parker F, et al. Control of tremor and involuntary movement disorders by chronic stereotactic stimulation of the ventral intermediate thalamic nucleus. J Neurosurg. 1992;77:62-68.
386 Benabid AL, Pollak P, Gao D, et al. Chronic electrical stimulation of the ventralis intermedius nucleus of the thalamus as a treatment of movement disorders. J Neurosurg. 1996;84:203-214.
387 Koller W, Pahwa R, Busenbark K, et al. High-frequency unilateral thalamic stimulation in the treatment of essential and parkinsonian tremor. Ann Neurol. 1997;42:292-299.
388 Limousin P, Pollak P, Benazzouz A, et al. Effect of parkinsonian signs and symptoms of bilateral subthalamic nucleus stimulation. Lancet. 1995;345:91-95.
389 Limousin P, Krack P, Pollak P, et al. Electrical stimulation of the subthalamic nucleus in advanced Parkinson’s disease. N Engl J Med. 1998;339:1105-1111.
390 Kumar R, Lozano AM, Kim YJ, et al. Doubleblind evaluation of subthalamic nucleus deep brain stimulation in advanced Parkinson’s disease. Neurology. 1998;51:850-855.
391 Bejjani B, Damier P, Arnulf I, et al. Pallidal stimulation for Parkinson’s disease. Two targets? Neurology. 1997;49:1564-1569.
392 Pahwa R, Wilkinson S, Smith D, et al. High-frequency stimulation of the globus pallidus for the treatment of Parkinson’s disease. Neurology. 1997;49:249-253.
393 Siegfried J, Lippitz B. Bilateral chronic electrostimulation of ventroposterolateral pallidum: a new therapeutic approach for alleviating all parkinsonian symptoms. Neurosurgery. 1994;35:1126-1129.
394 Volkmann J, Sturm V, Weiss P, et al. Bilateral high-frequency stimulation of the internal globus pallidus in advanced Parkinson’s disease. Ann Neurol. 1998;44:953-961.
395 Krack P, Batir A, Van Blercom N, et al. Five-year follow-up of bilateral stimulation of the subthalamic nucleus in advanced Parkinson’s disease. N Engl J Med. 2003;349:1925-1934.
396 The Deep-Brain Stimulation for Parkinson’s Disease Study Group. Deep-brain stimulation of the subthalamic nucleus or the pars interna of the globus pallidus in Parkinson’s disease. N Engl J Med. 2001;345:956-963.
397 Oh MY, Abosch A, Kim SH, et al. Longterm hardware-related complications of deep brain stimulation. Neurosurgery. 2002;50:1268-1274.
398 Krack P, Pollak P, Limousin P, et al. Opposite motor effects of pallidal stimulation in Parkinson’s disease. Ann Neurol. 1998;43:180-192.
399 Freed CR, Greene PE, Breeze RE, et al. Transplantation of embryonic dopamine neurons for severe Parkinson’s disease. N Eng J Med. 2001;344:710-719.
400 Greene PE, Fahn S, Tsai WY, et al. Severe spontaneous dyskinesias: a disabling complication of embryonic dopaminergic tissue implants in a subset of transplanted patients with advanced parkinson’s disease. Mov Disor. 1999;14:904.
401 Olanow CW, Goetz CG, Kordower JH, et al. A double blind controlled trial of bilateral fetal nigral transplantation in Parkinson’s disease. Ann Neurol. 2003;54:403-414.
402 Bjorklund LM, Sanchez-Pernaute R, Chung S, et al. Embryonic stem cells develop into functional dopaminergic neurons after transplantation in a Parkinson rat model. Proc Natl Acad Sci U S A. 2002;99:2344-2349.
403 Gash DM, Zhang Z, Cass WA, et al. Morphological and functional effects of intranigrally administered GDNF in normal rhesus monkeys. J Comp Neurol. 1995;363:345-358.
404 Nutt JG, Burchiel KJ, Comella CL, et al. Randomized, doubleblind trial of glial cell line-derived neurotrophic factor (GDNF) in PD. Neurology. 2003;60:69-73.
405 Gill SS, Patel NK, Hotton GR, et al. Direct brain infusion of glial cell line-derived neurotrophic factor in Parkinson disease. Nat Med. 2003;9:589-595.
406 Lang AE, Gill S, Patel NK, et al. Randomized control trial of intraputaminal glial cell line-derived neurotrophic factor infusion in Parkinson’s disease. Ann Neurol. 2006;59:459-466.
407 Mytilineou C, Cohen G. Deprenyl protects dopamine neurons from the neurotoxic effect of 1-methyl-4-phenylpyridinium ion. J Neurochem. 1985;45:1951-1953.
408 Tatton WG, Greenwood CE. Rescue of dying neurons: a new action for deprenyl in MPTP parkinsonism. J Neurosci Res. 1991;30:666-672.
409 Wu RM, Chiueh CC, Pert A, et al. Apparent antioxidant effect of l-deprenyl on hydroxyl radical formation and nigral injury elicited by MPP+ in vivo. Eur J Pharmacol. 1993;243:241-247.
410 Wu RM, Chen RC, Chiueh CC. Effect of MAO-B inhibitors on MPP+ toxicity in Vivo. Ann N Y Acad Sci. 2000;899:255-261.
411 Tatton WG, Ju WY, Holland DP, et al. (-)-Deprenyl reduces PC12 cell apoptosis by inducing new protein synthesis. J Neurochem. 1994;63:1572-1575.
412 Wadia JS, Chalmers-Redman RM, Ju WJ, et al. Mitochondrial membrane potential and nuclear changes in apoptosis caused by serum and nerve growth factor withdrawal: time course and modification by (-)-deprenyl. J Neurosci. 1998;18:932-947.
413 Ogawa N, Tanaka K, Asanuma M, et al. Bromocriptine protects mice against 6-hydroxydopamine and scavenges hydroxyl free radicals in vitro. Brain Res. 1994;657(1–2):207-213.
414 Muralikrishnan D, Mohanakumar KP. Neuroprotection by bromocriptine against 1-methyl-4-phenyl-1,2,3,6-tetrahy-dropyridine-induced neurotoxicity in mice. FASEB J. 1998;12:905-912.
415 Kondo T, Ito T, Sugita Y. Bromocriptine scavenges metham-phetamine-induced hydroxyl radicals and attenuates dopamine depletion in mouse striatum. Ann N Y Acad Sci. 1994;738:222-229.
416 Finotti N, Castagna L, Moretti A, et al. Reduction of lipid peroxidation in different rat brain areas after cabergoline treatment. Pharmacol Res. 2000;42:287-291.
417 Yoshioka M, Tanaka K, Miyazaki I, et al. The dopamine agonist cabergoline provides neuroprotection by activation of the glutathione system and scavenging free radicals. Neurosci Res. 2002;43:259-267.
418 Nishibayashi S, Asanuma M, Kohno M, et al. Scavenging effects of dopamine agonists on nitric oxide radicals. J Neurochem. 1996;67:2208-2211.
419 Gomez-Vargas M, Nishibayashi-Asanuma S, Asanuma M, et al. Pergolide scavenges both hydroxyl and nitric oxide free radicals in vitro and inhibits lipid peroxidation in different regions of the rat brain. Brain Res. 1998;790:202-208.
420 Iida M, Miyazaki I, Tanaka K, et al. Dopamine D2. receptor-mediated antioxidant and neuroprotective effects of ropinirole, a dopamine agonist. Brain Res. 1999;838:51-59.
421 Clow A, Freestone C, Lewis E, et al. The effect of pergolide and MDL 72974 on rat brain CuZn superoxide dismutase. Neurosci Lett. 1993;164:41-43.
422 Kakimura J, Kitamura Y, Takata K, et al. Release and aggregation of cytochrome c and alphasynuclein are inhibited by the antiparkinsonian drugs, talipexole and pramipexole. Eur J Pharmacol. 2001;417:59-67.
423 Gu M, Iravani MM, Cooper JM, King D, et al. Pramipexole protects against apoptotic cell death by nondopaminergic mechanisms. J Neurochem. 2004;91:1075-1081.
424 Iravani MM, Haddon CO, Cooper JM, et al. Pramipexole protects against MPTP toxicity in nonhuman primates. J Neurochem. 2006;96:1315-1321.
425 Schapira AH, Olanow CW. Neuroprotection in Parkinson disease: mysteries, myths, and misconceptions. JAMA. 2004;291:358-364.
426 Ahlskog JE. Slowing Parkinson’s disease progression: recent dopamine agonist trials. Neurology. 2003;60:381-389.
427 Olanow CW. A radical hypothesis for neurodegeneration. Trends Neurosci. 1993;16:439-444.
428 Agid Y, Olanow CW, Mizuno Y. Levodopa: why the controversy? Lancet. 2002;360:575.
429 Fahn S, Oakes D, Shoulson I, et al. Levodopa and the progression of Parkinson’s disease. N Engl J Med. 2004;351:2498-2508.
430 Albin RL, Frey KA. Initial agonist treatment of Parkinson disease: a critique. Neurology. 2003;60:390-394.
431 Wooten GF. Agonists vs levodopa in PD: the thrilla of whitha. Neurology. 2003;60:360-362.
432 Shults CW, Oakes D, Kieburtz K, et al. Effects of coenzyme Q10 in early Parkinson disease: evidence of slowing of the functional decline. Arch Neurol. 2002;59:1541-1550.