CHAPTER 72 PARKINSON PLUS DISORDERS
The term parkinson plus disorders has been coined to embrace a heterogeneous group of movement disorders with prominent signs of parkinsonism plus additional features that allow clinical separation of these entities from classic idiopathic Parkinson disease (IPD). A synonymous designation is “atypical parkinsonian disorders” where the prefix “atypical” again refers to features that either are not part of the clinical spectrum of IPD—such as cerebellar ataxia, pyramidal tract signs, myoclonus, supranuclear gaze palsy, or apraxia—or are more pronounced and/or occur earlier in the disease course compared with IPD—such as autonomic failure or dementia. Poor or absent responses to L-dopa is another criterion of “atypical” parkinsonian disorders (APDs).
MULTIPLE SYSTEM ATROPHY
MSA is a sporadic neurodegenerative disorder characterized clinically by any combination of parkinsonian, autonomic, cerebellar, or pyramidal symptoms and signs and pathologically by cell loss, gliosis, and glial cytoplasmic inclusions (GCIs) in several brain and spinal cord structures. The term multiple system atrophy was introduced in 19691; however, cases of MSA were previously reported under the rubrics of striatonigral degeneration (SND),2–4 olivopontocerebellar atrophy (OPCA),5,6 Shy-Drager syndrome (SDS),7 and idiopathic orthostatic hypotension (OH). In 1989, GCIs were first described in the brains of patients with MSA regardless of clinical presentation.8 GCIs were not present in a large series of patients with other neurodegenerative disorders. The abundant presence of GCIs in all clinical subtypes of MSA led to the recognition of SDS, SND, and sporadic OPCA as one disease entity characterized by neuronal multisystem degeneration with unique oligodendroglial inclusion pathology. In the late 1990s, α-synuclein immunostaining was recognized as a sensitive marker of inclusion pathology in MSA,9,10 and MSA is now classified among the “synucleinopathies” along with Parkinson disease and dementia with Lewy bodies (DLBs).
Epidemiology
There are only few descriptive epidemiological studies on MSA. Bower and colleagues reported the incidence of MSA over a 14-year period in Olmsted County, Minnesota. Nine incident cases of MSA were identified, none of which had an onset before the age of 50 years. The reported crude incidence rate was 0.6 case per 100,000 population per year; when the age band of greater than 50 years was examined, the estimate rose to 3 cases per 100,000 population. Estimates of the prevalence of MSA (per 100,000 in the population) in four studies ranged from 1.9 to 4.9.11–14 These prevalence figures are similar to those of other well-known neurodegenerative disorders such as Huntington disease and motor neuron disease. Although many studies report a possible role of environmental toxins in Parkinson disease, such a role is even more likely in MSA, as this is a sporadic disease. However, to date only three studies have addressed environmental risk factors in MSA.15–17 So far, no single environmental factor has been clearly established as conferring increased or reduced risks to develop MSA.
Clinical Presentation, Course, and Prognosis
The disease affects both men and women, it usually starts in the sixth decade, and it relentlessly progresses with a mean survival of 6 to 9 years.18–21 There is considerable variation of disease progression, with survival of longer than 15 years in some instances.
Clinically, cardinal features include autonomic failure, parkinsonism, cerebellar ataxia, and pyramidal signs in any combination. Previous studies suggest that 29% to 33% of patients with isolated late-onset cerebellar ataxia and 8% of patients presenting with parkinsonism eventually develop MSA.22–24 Two major motor presentations can be distinguished clinically. Parkinsonian features predominate in 80% of patients (MSA-P subtype), and cerebellar ataxia is the major motor feature in 20% of patients (MSA-C subtype).21,25 Importantly, both motor presentations of MSA are associated with similar survival times.20 However, MSA-P patients have a more rapid functional deterioration than MSA-C patients.18
MSA-P–associated parkinsonism is characterized by progressive akinesia and rigidity. Jerky postural tremor and, less commonly, tremor at rest may be superimposed. Frequently, patients exhibit orofacial or craniocervical dystonia (Fig. 72-1)26 associated with a characteristic quivering high-pitched dysarthria. Postural stability is compromised early on; however, recurrent falls at disease onset are unusual, in contrast to progressive supranuclear palsy (PSP). Differential diagnosis of MSA-P and Parkinson disease may be exceedingly difficult in the early stages due to a number of overlapping features such as rest tremor or asymmetrical akinesia and rigidity. Furthermore, L-dopa–induced improvement of parkinsonism may be seen in 30% of MSA-P patients. However, the benefit is transient in most of these subjects, leaving 90% of the MSA-P patients unresponsive to L-dopa in the long term. L-Dopa–induced dyskinesia affecting orofacial and neck muscles occurs in 50% of MSA-P patients, sometimes in the absence of motor benefit.27 In most instances, a fully developed clinical picture of MSA-P evolves within 5 years of disease onset, allowing a clinical diagnosis during follow-up.28
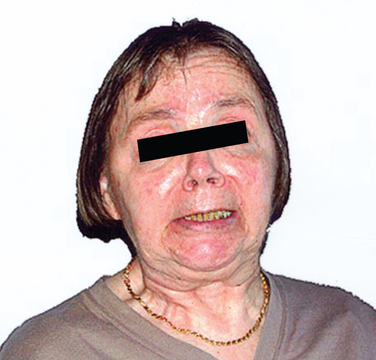
Figure 72-1 Orofacial dystonia in MSA.
(From Wenning GK, Geser F, Poewe W. The ‘risus sardonicus’ of multiple system atrophy. Mov Disord 2003; 18:1211.)
The cerebellar disorder of MSA-C comprises gait ataxia, limb kinetic ataxia, and scanning dysarthria as well as cerebellar oculomotor disturbances. Patients with MSA-C usually develop additional noncerebellar symptoms and signs but before doing so may be indistinguishable from other patients with idiopathic late-onset cerebellar ataxia, many of whom have a disease restricted clinically to cerebellar signs and pathologically to degeneration of the cerebellum and olives.22
Dysautonomia is characteristic of both MSA motor presentations, primarily comprising urogenital and orthostatic dysfunction. Early impotence (erectile dysfunction) is virtually universal in men with MSA, and urinary incontinence or retention, often early in the course or as presenting symptoms, is frequent.21 Disorders of micturition in MSA are due to changes in the complex peripheral and central innervation of the bladder29 and generally occur more commonly, earlier, and to a more severe degree than in Parkinson disease. In contrast, constipation occurs equally in Parkinson disease and MSA. Symptomatic OH is present in 68% of clinically diagnosed patients, but recurrent syncopes emerge in only 15%.21 Levodopa or dopamine agonists may provoke or worsen OH.
Diagnostic Criteria
Clinical diagnostic criteria for MSA were first proposed by Quinn,30,31 who classified cases as either SND or OPCA type MSA depending on the predominance of parkinsonism or cerebellar ataxia.
More recently, operationalized criteria have been proposed by an International Consensus Conference.25 The consensus criteria have since been widely established in the research community as well as movement disorders clinics. They define three diagnostic categories of increasing certainty: possible, probable, and definite. The diagnosis of possible and probable MSA is based on the presence of specific clinical features (Table 72-1). In addition, exclusion criteria have to be considered. A definite diagnosis requires a typical neuropathological lesion pattern with α-synuclein–positive GCIs.
| Domain | Criterion | Feature |
|---|---|---|
| Autonomic and urinary dysfunction | Orthostatic fall in blood pressure (by 30 mmHg systolic or 15 mmHg diastolic) | Orthostatic hypotension (by 20 mmHg systolic or 10 mmHg diastolic) |
| or | Urinary incontinence or incomplete bladder emptying | |
| persistent urinary incontinence with erectile dysfunction in men | ||
| or both | ||
| Parkinsonism | Bradykinesia plus rigidity | Bradykinesia (progressive reduction in speed and amplitude of voluntary movements during repetitive actions) |
| or | Rigidity | |
| postural instability | Postural instability (loss of primary postural reflexes) | |
| or | Tremor (postural, resting, or both) | |
| tremor | ||
| Cerebellar dysfunction | Gait ataxia plus ataxic dysarthria | Gait ataxia (wide-based stance with irregular steps) |
| or | Ataxic dysarthria | |
| limb ataxia | Limb ataxia | |
| or | Sustained gaze-evoked nystagmus | |
| sustained gaze-evoked nystagmus | ||
| Corticospinal tract dysfunction | No defining features | Extensor plantar responses with hyperreflexia |
Nomenclature of clinical domains, features (disease characteristics), and criteria (defining features or composite of features) used in the diagnosis of multiple system atrophy.
Modified from Gilman S, Sima AA, Junck L, et al: Spinocerebellar ataxia type 1 with multiple system degeneration and glial cytoplasmic inclusions. Ann Neurol 1996; 39: 241-255.
A recent retrospective evaluation of the Consensus criteria on pathologically proven cases showed excellent positive predictive values for both possible and probable MSA; however, sensitivity for probable MSA was poor.32 While such formal diagnostic criteria are important for certain types of clinical research, they add little to the problem of detecting early cases, and improved screening instruments are certainly needed.
Besides the poor response to levodopa, and the additional presence of pyramidal or cerebellar signs or autonomic failure as major diagnostic clues, certain other features (“red flags”) such as orofacial dystonia, stridor, or REM sleep behavior disorder (RBD) may raise suspicion of MSA.30,33 MSA patients may present with isolated RBD.34–36 RBD and other sleep disorders are more common in patients with MSA than in those with Parkinson disease matched for disease duration, reflecting both a profound striatal monoaminergic deficit37 and diffuse subcortical and brainstem disease in MSA.38
Genetics
MSA, as reflected in its current definition, is regarded as a sporadic disease,39 and no confirmed familial cases of MSA have been described; notwithstanding, it is conceivable that genetic factors may play a role in the etiology of the disease. This has, for example, been convincingly demonstrated for PSP,40 another disease that, in the vast majority of cases, is a sporadic disease. However, initial screening studies for candidate genes revealed no risk factors.41,42 Other studies have looked for polymorphisms or mutations in candidate genes, which may predispose an individual toward developing MSA. The apolipoprotein ε4 allele is not overrepresented in MSA when compared with controls, and there have been conflicting reports of the association of a cytochrome P450-2D6 polymorphism with MSA.43,44,45 Furthermore, there is no evidence to support an association between MSA and polymorphisms in the H5 pore region of the human homolog of the weaver mouse gene hiGIRK2, the insulin-like growth factor 1 receptor gene, or the ciliary neurotrophic factor gene.41 Genotyping of a functional polymorphism in the dopamine beta-hydroxylase (DBH) gene showed no association between the DBH–1021 C→T polymorphism and MSA.46 Increased expression of a brain specific protein called ZNF231 in cerebellar neurons has been reported to occur in patients with MSA.47 The gene is located on chromosome 3p21 and encodes a neuronal double zinc finger protein with a nuclear targeting sequence, suggesting that it might function as a transcription regulator. The importance of this finding is as yet uncertain, but it is possible that patients with MSA differ from unaffected individuals by sequence polymorphisms within, and flanking, the putative functional motifs of the ZNF231 gene.
Gilman and colleagues48 reported an MSA-like phenotype including GCIs in one SCA-1 (spinocerebellar ataxia type 1) family. Other SCA mutations, except for SCA-2,27 have not been reported to present with MSA-like features.49–54 Conversely, the majority of MSA-C patients do not appear to have expanded SCA1 and SCA3 alleles.41 Indeed, MSA-C appears to be a frequent form of sporadic cerebellar ataxia of late onset; 29% of sporadic adult-onset ataxia patients suffer from MSA.22 This finding corresponds well with data of a study of sporadic OPCA patients who were followed 3 months to 10 years.23 Within this period, 17 of 51 patients developed autonomic failure or parkinsonism indicating a diagnosis of MSA. There is significant overlap in clinical and radiological features between the fragile X–associated tremor/ataxia syndrome (FXTAS) and atypical parkinsonism, in particular, MSA-C. A considerable number of cases with FXTAS have already been described in male carriers of the fragile X premutation.55,56 Kamm and colleagues found an elevated frequency of fragile X mental retardation 1 gene (FMR1) “gray zone” alleles (40 to 54 repeats) in both male MSA and PSP patients compared with controls, suggesting that small repeat expansion in this gene may possibly act as a susceptibility factor for certain types of neurodegenerative diseases in apparently sporadic male patients, probably in combination with other genetic and environmental factors.57 In contrast, there was no significant difference in allele frequency for either FMR1 “gray zone” alleles for female patients or for FMR1 premutation alleles (55 to 200 repeats) in both male and female patients compared with healthy controls, indicating that FXTAS due to a premutation in the FMR1 gene represents only a rare cause of apparently sporadic atypical parkinsonism.57
Ancillary Investigations
The diagnosis of MSA still rests on the clinical history and neurological examination. According to the Consensus Conference on the diagnosis of MSA,25 additional investigations such as autonomic function tests, sphincter electromyography, and neuroimaging may be used to support the diagnosis or to exclude other conditions. The abnormalities reviewed below have been observed in patients with advanced rather than early disease. In the early stages the investigations may give equivocal results. Therefore, the Consensus Conference on MSA considered it premature to incorporate the results of laboratory investigations into the diagnostic guidelines that were established.
Cardiovascular Function
A history of postural faintness or other evidence of OH, such as neck ache on rising in the morning or posturally related changes of visual perception, should be sought in all patients in whom MSA is suspected. After taking a comprehensive history, testing of cardiovascular function should be performed. According the consensus statement of the American Autonomic Society and the American Academy of Neurology on the definition of OH, pure autonomic failure, and MSA, a drop in systolic blood pressure of 20 mm Hg or more, or in diastolic blood pressure of 10 mm Hg or more, compared with baseline is defined as OH and must lead to more specific assessment.58 This is based on continuous noninvasive measurement of blood pressure and heart rate during tilt-table testing.59 In MSA, cardiovascular dysregulation appears to be caused by central rather than peripheral autonomic failure. During supine rest, norepinephrine levels (representing postganglionic sympathetic efferent activity) are normal,60 and there is no denervation hypersensitivity.61 In contrast, mainly postganglionic sympathetic dysfunction is thought to account for autonomic failure associated with Parkinson disease. However, a study demonstrated that abnormal cardiovascular autonomic function tests failed to differentiate autonomic failure associated with Parkinson disease versus MSA.62 Although such abnormalities may be nonspecific, their presence within the first 3 to 5 years of disease onset make a diagnosis of MSA more likely than Parkinson disease.
Bladder Function
Assessment of bladder function is mandatory in MSA and usually provides evidence of involvement of the autonomic nervous system at an early stage of the disease (when bladder function is still normal in most Parkinson disease patients). Following a careful history regarding frequency of voiding, difficulties in initiating or suppressing voiding, and the presence of urinary incontinence, a standard urine analysis should exclude an infection. Postvoid residual volume needs to be determined sonographically or via catheterization to initiate intermittent self-catheterization in due course. In some patients, only cystometry can discriminate between hypocontractile detrusor function and a hyperreflexic sphincter-detrusor dyssynergy. The nature of bladder dysfunction is different in MSA and Parkinson disease. Although frequency and urgency are common in both disorders, marked urge or stress incontinence with continuous leakage is not a feature of Parkinson disease apart from very advanced cases. Urodynamic studies show a characteristic pattern of abnormality in MSA patients.63 In the early stages there is often detrusor hyperreflexia, often with bladder neck incompetence due to abnormal urethral sphincter function, which results in early frequency and urgency followed by urge incontinence. Later on, the ability to initiate a voluntary micturition reflex and the strength of the hyperreflexic detrusor contractions diminish, and the bladder may become atonic, accounting for increasing postmicturition residual urine volumes.
Sphincter Electromyography
An abnormal sphincter electromyogram (EMG) may be found in many patients with clinically definitive MSA, including those who, as yet, have no urological or anorectal problems. In at least 80% of patients with MSA, an EMG of the external anal sphincter reveals signs of neuronal degeneration in Onuf’s nucleus with spontaneous activity and increased polyphasia.29,64,65 The prevalence of abnormalities in early stages of MSA remains unclear. These findings do not reliably differentiate between MSA and other forms of APD such as PSP.66 Furthermore, neurogenic changes of external anal sphincter muscle have also been demonstrated in advanced stages of Parkinson disease.67 Also, chronic constipation, previous pelvic surgery, or vaginal deliveries can be confounding factors to induce nonspecific abnormalities.68 However, anal sphincter EMG abnormalities appear to distinguish MSA from Parkinson disease in the first 5 years after disease onset, and from pure autonomic failure, as well as from cerebellar ataxias, if other causes for sphincter denervation have been ruled out.69
Imaging
Magnetic Resonance Imaging
Routine 1.5-T magnetic resonance imaging (MRI) including diffusion weighted imaging (DWI) should be performed in all patients with suspected MSA because basal ganglia and/or brainstem abnormalities suggestive of MSA may be observed even during early disease stages. These changes include an OPCA-like atrophy pattern indistinguishable from autosomal dominant cerebellar ataxia.70 MRI measures of basal ganglia pathology in MSA are less well established, and naked eye assessments are often unreliable. In advanced cases, putaminal atrophy may be detectable and may correlate with severity of extrapyramidal symptoms (Fig. 72-2A, B).
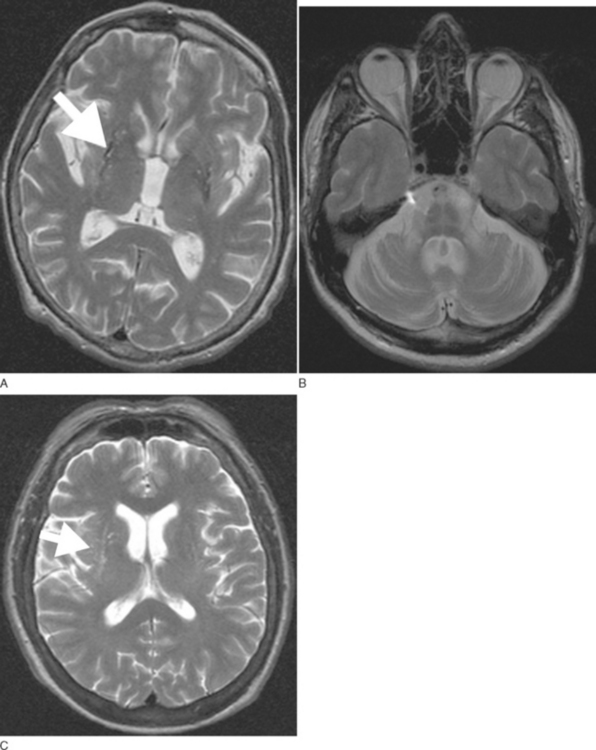
Abnormalities on MRI may include not only OPCA70 or putaminal atrophy71 but also signal abnormalities on T2-weighted images. Signal hyperintensities within the pons and middle cerebellar peduncles are thought to reflect degeneration of pontocerebellar fibers; these changes occasionally resemble a hot cross bun.71 Nonspecific putaminal hypointensities in patients with atypical parkinsonism including MSA were first reported in 1986 by two groups using a 1.5-T T2-weighted images.72,73 This change has subsequently been confirmed by others in cases with pathologically proven MSA.74–76 Similar MRI abnormalities may occur in patients with classic Parkinson disease.77 However, a study demonstrated that hypointense putaminal signal changes were more frequent in MSA than in Parkinson disease patients using T2*-weighted gradient echo (GE) instead of T2-weighted fast spin echo images, indicating that T2*-weighted GE sequences are of better diagnostic value for patients with parkinsonism.78 Increased putaminal hypointensities may be associated with a slit-like hyperintense band lateral to the putamen79,80 (Fig. 72-2C). The latter appears to be more specific for MSA than putaminal hypointensity71,79,80; however, further studies in larger cohorts of patients are needed to confirm this. The hyperintense slit-signal correlated with reactive microgliosis and astrogliosis in a case with pathologically proven MSA.76
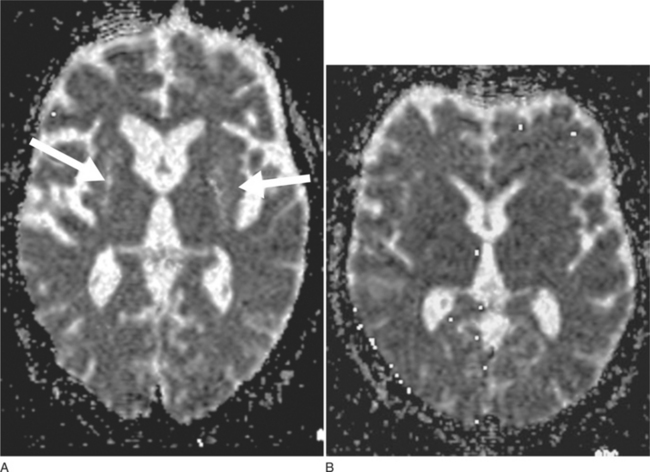
DWI may represent a useful diagnostic tool that can provide additional support for a diagnosis of MSA-P. DWI is able to discriminate MSA-P and both patients with Parkinson disease and healthy volunteers on the basis of putaminal rADC (regional apparent diffusion coefficient) values81 (Fig. 72-3A, B). The increased putaminal rADC values in MSA-P are likely to reflect ongoing striatal degeneration, whereas most neuropathological studies reveal intact striatum in Parkinson disease. But, since in PSP compared with Parkinson disease rADCs were also significantly increased in both putamen and globus pallidus,82 increased putaminal rADC values do not discriminate MSA-P from PSP.
Schulz et al.83 found significant reductions in mean striatal and brainstem volumes in patients with MSA-P, MSA-C, and PSP, whereas patients with MSA-C and MSA-P also showed a reduction in cerebellar volume. More recently, voxel-based morphometry confirmed previous region of interest (ROI)–based volumetric studies83 showing basal ganglia and infratentorial volume loss in MSA-P patients.81 These data also revealed prominent cortical volume loss in MSA-P mainly comprising the cortical targets of striatal projections such as the primary sensorimotor, lateral premotor cortices, and the prefrontal cortex. MR-based volumetry is a helpful tool to investigate the progression of cortical and subcortical atrophy patterns in MSA compared with other disorders; however, it cannot be applied for routine diagnostic workup of individual patients.
Functional Imaging (Single-Photon Emission Computed Tomography and Positron Emission Tomography)
Overall presynaptic and postsynaptic dopaminergic markers have yielded disappointing results regarding the differentiation between Parkinson disease, MSA, and PSP.85 PET studies using other ligands such as [11C]diprenorphine (nonselective opioid receptor antagonist)86 and 18F-fluorodeoxyglucose ([18F]FDG)87–89 have proved more consistent in detecting striatal degeneration and in distinguishing patients with MSA-P from those with Parkinson disease, particularly when combined with a dopamine D2 receptor scan.90 Widespread functional abnormalities in MSA-C have been demonstrated using [18F]FDG and PET.91 Reduced metabolism was most marked in the brainstem and cerebellum, but other areas such as the basal ganglia and cerebral cortex were also involved, supporting its nosological status as the cerebellar subtype of MSA.
SPECT evaluation of the dopamine transporter (DAT) using [123]β-CIT [2β-carboxymethoxy-3β-(4-iodophenyl)tropane] reflects the disruption of the nigrostriatal pathway, and therefore MSA and PSP cannot be separated from Parkinson disease with this method alone.92 However, a study using voxelwise SPM analysis of DAT-binding found significant differences between Parkinson disease and MSA patients in an area including the mesopontine junction, and this type of analysis may enhance the differential diagnostic potential DAT-SPECT in MSA versus Parkinson disease.93
SPECT studies using [123I]iodobenzamide ([123] IBZM) as D2 receptor ligand have revealed significant reductions of striatal IBZM binding in clinically probable MSA subjects compared with Parkinson disease patients or controls.94–96 However, striatal IBZM binding is also reduced in other APDs such as PSP95 limiting its predictive value for an early diagnosis of MSA.
Scintigraphic visualization of sympathetic cardiac neurons using scintigraphy with [123I]metaiodobenzylguanadine ([123I] MIBG) has been shown to reveal loss of binding in patients with Parkinson disease regardless of disease severity, reflecting postganglionic sympathetic denervation compared with preserved cardiac binding in MSA97,98 and PSP.95 Considering all reports published so far, MIBG scintigraphy was able to accurately discriminate a total of 246 Parkinson disease patients from 45 MSA patients with high sensitivity (90%) and specificity (95%).98 Similarly, 18F-dopa PET is able to demonstrate cardiac sympathetic denervation in pure autonomic failure and Parkinson disease in contrast with intact cardiac sympathetic innervation in MSA.99
Principles of Management
Symptomatic Therapy
There is virtually no effective treatment for the cerebellar features of the disease.100,101 Therefore, medical treatment is largely aimed at alleviating parkinsonism and dysautonomia.
Parkinsonism
The commonly held belief that patients with MSA are non–or poorly L-dopa–responsive is misleading. Clinical series have documented levodopa efficacy in up to 40% of patients with possible or probable MSA.21,102–104 Data obtained from series with pathological confirmation are more variable, with rates of beneficial L-dopa response ranging between 30% and 80%.105–108 L-Dopa responsiveness should be tested by administering escalating doses (with a peripheral decarboxylase inhibitor) over a 3-month period up to a least 1000 mg/day (if necessary and if tolerated).25 However, whatever response there is it usually declines after a few years of treatment.31 Dyskinesias emerge in half of the patients treated with L-dopa and they are often dystonic, predominantly affecting the orofacial district.27,105
Results with dopamine agonists are also variable, but these compounds are no more effective than levodopa and often poorly tolerated. Goetz and colleagues,100 using doses of 10 to 80 mg/day of bromocriptine, reported a benefit in five patients who had previously responded to L-dopa and one patient who had failed to respond to levodopa. In a controlled trial with lisuride in seven patients, only one, who had already responded to levodopa, showed improvement.109 No formal trials looking at the efficacy of pergolide, cabergoline, ropinirole, or pramipexole are available.
Despite anecdotal benefit in single cases, a short-term open trial with amantadine at high doses (400 to 600 mg/day) in five patients with MSA unresponsive to L-dopa was negative.110 Anticholinergics usually do not improve motor symptoms, but they may be helpful when sialorrhea is severe and disturbing.
Ablative neurosurgical procedures such as medial pallidotomy fail to improve parkinsonian motor disturbance in MSA.111 However, beneficial short-term and long-term effects of bilateral subthalamic nucleus highfrequency stimulation have been reported in four patients with MSA-P.112 Further studies are needed to establish the scope of deep brain stimulation in MSA. Alternative therapeutic neurosurgical strategies such as trophic factors local delivery and neurotransplantation are currently being explored experimentally in animal models of MSA.113
Autonomic Dysfunction
Treatment of OH is often fraught with difficulties, but it is crucial to improve the quality of life in patients with MSA and autonomic dysfunction.114 The treating physician should not to be excessively concerned about a low standing blood pressure if the patient is asymptomatic. Patients with MSA can sometimes tolerate a decreased standing systolic blood pressure without symptoms, probably because their cerebral blood flow is kept at an adequate level thanks to a functioning autoregulation.115 The latter appears to be preserved down to a systolic blood pressure of 60 mm Hg, well below the 80 mm Hg at which autoregulation fails in normal subjects.116
When OH becomes disabling, it can often be alleviated by progressively avoiding aggravating factors, such as the effects of large meals, alcohol intake, drugs, straining during micturition and defecation, and exposure to a warm environment. Other nonpharmacological strategies that are also recommended include elastic stockings and head-up tilt of the bed at night, increasing salt intake, and, in selected cases, cardiac pacing.117
A variety of pharmacological agents with different mechanisms of action have been used to reduce OH; fludrocortisone and desmopressin act through plasma volume expansion and by reducing natriuresis; clonidine and yohimbine both induce release of norepinephrine. Midodrine (adrenergic agonist activating alpha 1-receptors on arterioles and veins) increases peripheral resistance, thereby significantly reducing OH, as shown by three randomized, controlled trials.118–120 DL–Threo-dihydroxyphenylserine increases endogenous production of norepinephrine,121 whereas ergot derivatives are venoconstrictor agents with direct action on β2-receptors. Most specialists now consider fludrocortisone and midodrine as the first-choice drugs for this condition, the option between the two being made according to the individual characteristics of the patient. Controlled trials comparing the different symptomatic drugs used for OH are not available, and therefore the ultimate choice among them should be made according to the experience and judgment of the treating physician. Supine hypertension (SH) may occasionally be associated with severe OH. SH does not require drug treatment if systolic blood pressure is below 200 mm Hg; if treatment is required, short-acting calcium antagonists given at nighttime are commonly used.
Urinary symptoms in MSA are due to a complex mixture of central and peripheral nervous problems, sometimes superimposed on local pathology such as prostatic hypertrophy and perineal laxity.122 Peripherally acting anticholinergic drugs may help incontinence, but often at the expense of inducing retention; the administration of desmopressin at night may reverse nocturia. Intermittent self-catheterization or even an indwelling catheter may be needed in the presence of incomplete bladder emptying.
Male impotence can be partially circumvented by the use of intracavernosal papaverine, prostaglandin E1 or penile implants. A double-blind, placebo-controlled study showed that sildenafil is efficacious in the treatment of erectile dysfunction in parkinsonism due to MSA, but it may unmask or exacerbate orthostatic hypotension.123 Therefore, measurement of lying and standing blood pressure before prescribing sildenafil to men with parkinsonism is recommended.
Inspiratory stridor develops in about 30% of patients, possibly due to progressive degeneration of the nucleus ambiguus124 and consequent bilateral laryngeal abductor weakness. Continuous positive airway pressure may be helpful in some of these patients but in about 4% a tracheostomy is needed, after having considered all the ethical issues related to this procedure.125,126 Sleep apnea may also occur and should be managed appropriately.
PROGRESSIVE SUPRANUCLEAR PALSY
Progressive supranuclear palsy (PSP) is a chronic progressive neurodegenerative disorder characterized by continuation of clinical features including akinetorigid parkinsonism, postural instability supranuclear vertical gaze palsy, axial dyskinesia, and frontolimbic dementia.127–129 It was originally delineated as a novel heterogeneous system disorder by Richardson and colleagues130 and later termed PSP by Steele, Richardson, and Olszewski in their seminal report of 1964.131 Its unique neuropathology involves neurodegeneration with neuronal and oligodendroglial deposition of abnormally phosphorylated tau protein forming abundant neurofibrillary tangles (NFTs) in distinct basal ganglia and brainstem regions but also in the frontal cortex and dentate nucleus of the cerebellum.131–133 Subcortical NFT formation with granulovacuolar neuronal degeneration and gliosis are especially marked in the globus pallidus, subthalamic nucleus, substantia nigra, as well as in the midbrain and pontine reticular formation including the midbrain oculomotor complex and superior colliculi, periaqueductal gray, and basis pontis.
Abnormal intraneuronal deposits of tau protein are the pathological hallmark of PSP and a group of neurodegenerative disease that may either present with dementia or a parkinsonian movement disorder or combinations thereof and have accordingly been collectively grouped as “tauopathies” (Table 72-2). At the ultrastructural and molecular levels, however, there are differences between the types of tau aggregates and tau composition in these various disorders.134,135
TABLE 72-2 Neurodegenerative Diseases with Intraneuronal Accumulation of Abnormal Tau Protein (“Tauopathies”)
| Presenting with parkinsonism or other movement disorder | Progressive supranuclear palsy (PSP) |
| Corticobasal degeneration (CBD) | |
| Postencephalitic parkinsonism | |
| Guadeloupean parkinsonism | |
| Presenting with dementia | Alzheimer disease |
| Dementia pugilistica | |
| Down syndrome | |
| Pick disease | |
| Parkinsonism dementia syndromes | FTDP-17 |
| Parkinsonism-dementia-complex of | |
| Guam | |
| Niemann-Pick disease type C |
Epidemiology
There are no data on incidence and prevalence of PSP from population-based studies. Some hospital-based studies have found low prevalence rates of 1 to 2 cases per 100,000136,137 and an age-dependent rise in the incidence rate from 1.7 cases per 100,000 at age 50 to 59 to 14.7/100,000 at age above 80.137 Using a hospital linkage system of general practitioners in an area of London serving a population of more than 120,000, Schrag and colleagues138 arrived at an age-adjusted prevalence rate of 6.4/100,000, which is similar to the value of 5.0 calculated from a nationwide ascertainment study performed in the United Kingdom.139 Given the considerable rate of misdiagnosis and underdiagnosis of PSP shown in a number of clinical and neuropathological studies,139,140 it appears likely that the true prevalence is above these estimates.
Neuropathological series from specialized centers suggest that PSP may be the second most common type of degenerative parkinsonism after idiopathic Parkinson disease, accounting for around 6% of cases diagnosed with a parkinsonian syndrome in life.141
Clinical Presentation, Course, and Prognosis
The classic clinical syndrome of PSP is characterized by symmetrical rigid-bradykinetic parkinsonism with prominent extensor rigidity and dystonia of neck muscles producing an erect posture with neck hyperextension. Facial immobility with markedly reduced blink rates and elevated eyebrows with frontalis muscle overactivity cause a characteristic staring and astonished facial expression, and further clinical hallmarks include pronounced postural instability with recurrent falls, typically backward. Rest tremor is distinctly uncommon in PSP and most patients lack a meaningful response of their parkinsonism to trials of L-dopa. Abnormalities of eye movement are considered pathognomic and include slowing of vertical and horizontal saccades, hypometric saccades, and the development of a highly distinctive supranuclear vertical gaze palsy with complete loss of vertical eye movements and fully preserved excursions of vestibulo-ocular reflex movements.
The mean age at disease onset is around 60 years and mean survival is 6 years.142 The disease is progressive despite any therapy. In the late stage, PSP patients are wheelchair or bed bound. Their speech shows a characteristic growling and groaning. Dressing and feeding themselves is impossible. Marked difficulty in swallowing bears the danger of aspiration and consequent pneumonia.
Goetz and colleagues have studied the progression of global disability in PSP by defining median latencies from disease onset to certain milestones of key disabilities. The median delay to any key motor disability was as short as 48 months in their study of 50 patients with PSP; loss of ambulation, inability to stand, and a wheelchair-bound state were reached after a median of 5 years; speech had become unintelligible after a median of 6 years; and tube feeding was required after a mean of 87 months.143
In some instances it can be difficult to distinguish PSP from Parkinson disease patients during the first 2 to 3 years from symptom onset if the former do not yet clearly exhibit postural instability or ophthalmoplegia, and when they may still show a response to levodopa. Lees and coworkers140 presented evidence that the syndrome of PSP is probably heterogeneous. In a careful clinicopathological study of 103 consecutive cases of pathologically confirmed PSP, they assessed early clinical features occurring within the first 2 years after disease onset. Supranuclear gaze palsy was present in less than half of all cases, and falls only occurred in some 60% (Table 72-3). Taken together, about 50% of cases were characterized by early postural instability and falls associated with supranuclear vertical gaze palsy and cognitive dysfunction, corresponding to the classic pattern captured in current diagnostic criteria (Table 72-4). A second group of almost one third of their cases, however, had a parkinsonian phenotype with asymmetrical onset, rest tremor, and a moderate therapeutic response to levodopa, therefore providing greater room for diagnostic error regarding the differentiation from Parkinson disease. These authors propose to subdivide PSP into at least two clinically distinct subtypes termed “Richardson’s syndrome”, corresponding to the classic syndrome and “PSP-parkinsonism” with greater similarity to Parkinson disease (Table 72-5).
TABLE 72-3 Early Clinical Features (First 2 Years) in Progressive Supranuclear Palsy
| Clinical Feature | % |
|---|---|
| Falls | 60 |
| Impaired postural reflexes | 62 |
| Bradykinesia | 75 |
| Rigidity | 42 |
| Cognitive decline | 29 |
| Speech disturbance | 39 |
| Supranuclear gaze palsy | 38 |
| Abnormal pursuit or saccades | 44 |
| Nonspecific visual symptoms | 21 |
| Asymmetrical onset | 28 |
N = 103.
From Williams DR, de Silva R, Paviour DC, et al: Charcteristics of two distinct clinical phenotypes in pathologically proven progressive supranuclear palsy: Richardson’s syndrome and PSP-parkinsonism. Brain 2005; 128:1247-1258.

TABLE 72-4 NINDS-SPSP Clinical Criteria for the Diagnosis of Progressive Supranuclear Palsy (PSP)
Rights were not granted to include this table in electronic media. Please refer to the printed book.
From Litvan I, Agid Y, Calne D, et al: Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology 1996; 47:1-9.
TABLE 72-5 Classification of Progressive Supranuclear Palsy (PSP)
| Richardson’s Syndrome | PSP-Parkinsonism |
|---|---|
| Falls, dementia | Asymmetrical onset tremor |
| Supranuclear gaze palsy | Mild/moderate L-dopa response |
| Poorer prognosis | Better prognosis |
| Overrepresentation of men | Equal sex distribution |
| 4R predominant tau tangles | 3R and 4R tau tangles |
From Williams DR, de Silva R, Paviour DC, et al: Characteristics of two distinct clinical phenotypes in pathologically proven progressive supranuclear palsy: Richardson’s syndrome and PSP-parkinsonism. Brain 2005; 128: 1247–1258.
Diagnostic Criteria
The NINDS diagnostic criteria for PSP have been established as the gold standard during the last 5 years144 (see Table 72-4). These criteria have been developed primarily as clinical research criteria and have been shown to have rather low sensitivity at first presentation in a recent clinicopathological study.145 PPPV at first visit, however, was 100% while sensitivity remained low at 34% even at the last visit before death. As Williams and colleagues have pointed out in their series of 103 pathologically confirmed cases of PSP, misdiagnosis as Parkinson disease is common and accounted for 23% of cases even in the hands of hospital specialists. As outlined earlier, this is probably due to the existence of at least one major clinical subtype of PSP that is not captured by the current NINDS criteria.
Genetics
In PSP (and CBD) pathological tau, composed of aggregated four-repeat (E10+) tau isoforms, accumulates in cells and glia in subcortical and cortical areas. Interestingly, recent genetic studies have indicated that a specific haplotype of the tau gene is overrepresented in PSP and CBD indicating a common genetic background of these tauopathies.146
Ancillary Investigations
The diagnosis of PSP is primarily clinical and relies on the NINDS criteria specified above. However, ancillary investiga tions have been explored in an attempt to improve the diagnostic accuracy. These include analysis of cerebrospinal fluid and protein biomarkers and structural and functional imaging as well as neurophysiological techniques.
Cerebrospinal Fluid Studies
Several investigators have attempted to identify biomarkers in the cerebrospinal fluid to achieve early and accurate diagnosis of PSP. Cerebrospinal fluid concentrations of tau appear to be higher in corticobasal degeneration than in PSP, with sensitivity and specificity of 100% and 87.5%.147 High cerebrospinal fluid concentrations of neurofilament have been observed in atypical parkinsonian disorders; however, PSP could not be differentiated from other atypical disorders such as MSA. Furthermore, sensitivity was suboptimal due to overlapping ranges of neurofilament concentrations.148 It has been suggested that the concomitant use of a levodopa test and the neurofilament protein assay could improve diagnostic accuracy for atypical parkinsonism to 90%.149
Magnetic Resonance Imaging
A number of findings suggestive of PSP, such as midbrain atrophy with enlargement of the third ventricle and tegmental atrophy, signal increase in the midbrain and in the inferior olives, as well frontal cortex and temporal lobe atrophy, have been described.71 Indirect parameters of midbrain atrophy comprising reduced anteroposterior midbrain diameter and abnormal superior profile of the midbrain (flat or concave versus convex aspect in PSP patients) may assist in the differential diagnosis of PSP (Fig. 72-4).150 Visual assessment of atrophy of the superior peduncle has been shown to differentiate PSP patients from controls and patients with other parkin sonian disorders including Parkinson disease and MSA, with a sensitivity of 74% and a specificity of 94%.151
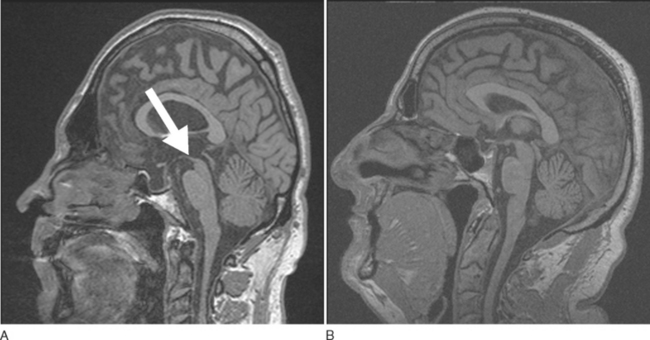
Using MRI-based volumetry (MRV) with semiautomatic segmentation techniques, patients with PSP showed significant reductions in whole brain, striatal, brainstem (especially midbrain), and frontal volumes compared with Parkinson disease patients.152,153
Groschel and colleagues154 used a mathematical model derived from a discriminant analysis using only postmortem confirmed cases of PSP and CBD as well as controls. The volumes of midbrain, parietal white matter, temporal gray matter, brainstem, frontal white matter, and pons were identified to separate best between groups, predicting correctly the diagnosis in 95% of controls as well as in 76% of all PSP and 83% of all CBD patients.
Voxel-based morphometry (VBM) has been used to study neurodegenerative parkinsonian disorders including PSP, MSA, and Parkinson disease. VBM permits operator-independent and semiautomated detection of significant differences in different tissue types of the whole brain, avoiding a priori range-of-interest (ROI) selection. Two studies reported the use of VBM in PSP patients.155,156 Brenneis and colleagues155 showed gray matter loss in frontotemporal, insular, supplementary motor, and mediotemporal areas compared with healthy controls. Further, white matter loss was observed in the midbrain and frontotemporal areas. The second study confirmed these findings comparing PSP patients not only with healthy controls but also Parkinson disease patients.156 Interestingly, this study tested the clinical utility of the VBM results as a guide for the differential diagnosis of PSP from Parkinson disease and healthy controls. On neuroradiological review of the T1-weighted MR images, the study participants were allocated to either PSP or non-PSP based on the presence or absence of the midbrain tissue loss in the PSP group highlighted using VBM. With these regional differences on VBM as a guide, neuroradiological diagnosis achieved a sensitivity of 83% and a specificity of 79%.
Using magnetization transfer imaging (MTI), abnormalities of the basal ganglia and substantia nigra have been reported in patients with parkinsonism.157 Although this technique may be useful to separate Parkinson disease from atypical parkinsonian disorders, discrimination of PSP and MSA is suboptimal.
More recently, DWI has been applied to patients with various parkinsonian disorders including PSP.
Pathological DWI findings in Parkinson disease are very rare. In contrast, abnormal putaminal diffusivity appears to be common in clinically established atypical parkinsonian disorders.81,82 Therefore, DWI seems useful to discriminate Parkinson disease from atypical parkinsonism; however, DWI fails to separate MSA from PSP on the basis of elevated putaminal diffusivity.
Proton magnetic resonance spectroscopy can provide an indirect measure of neuronal loss in vivo. Several investigators have reported spectral changes in the lentiform nucleus of PSP patients.158,159 The discriminatory value of this technique on an individual basis remains questionable.160
Functional Imaging
Several studies of blood flow and oxygen or glucose metabolism on PET in patients with PSP have demonstrated frontal lobe hypometabolism.161 Investigation of the nigrostriatal dopaminergic system with cocaine analogs and PET or SPECT imaging can reliably show presynaptic dopaminergic degeneration in PSP and also progression of this degeneration.162 However, the pattern of abnormality is nonspecific and cannot differentiate PSP from other parkinsonian syndromes, even when used in combination with a dopamine D2 receptor ligand.163 Ligands for the cholinergic system may offer more hope in improving diagnostic accuracy for PSP. Use of carbon-11–labeled N-methyl-4-piperidyl acetate and PET to measure acetylcholinesterase activity showed a preferential loss of cholinergic innervation to the thalamus in PSP compared with Parkinson disease and controls.164
More recently, imaging of activated microglia with carbon-11–labeled PK11195 and PET in four PSP patients revealed increased binding in the lentiform nucleus and pons in particular, although dorsolateral prefrontal cortex, caudate, substantia nigra, and thalamus were also involved.165 These findings raise the possibility that disease activity could be monitored.
Neurophysiology
Various neurophysiological techniques have been used to study PSP, aiming to improve diagnostic accuracy and to understand the underlying pathophysiological disease process.166 Electro-oculography has shown a distinct and progressive profile of deficits characterized by decreased saccadic velocity, frequent square-wave jerks, and increased error rates on antisaccade tasks.167 Autonomic function testing is usually normal in PSP compared with MSA.168 Sphincter EMG can differentiate Parkinson disease from atypical parkinsonism but fails to reliably discriminate PSP from MSA.69 The auditory startle response is delayed or absent in PSP in contrast to frequent hyperexcitability in MSA.169 The clinical usefulness and pathophysiological basis of evoked potential abnormalities in PSP remain to be elucidated in further studies.
Principles of Management
Parkinsonism
To date, there have been no randomized double-blind controlled trials of dopaminergic replacement therapies (L-dopa and dopamine agonists) in PSP patients except for one short-term trial of pergolide limited by small sample size.170 The literature evidence is based on anecdotal case reports, small-scale open-label trials or retrospective chart reviews using clinical, at times not validated, criteria for diagnosing PSP. Taken together, the results suggest that dopaminergic agents are usually ineffective in PSP, reflecting striatal dopamine receptor loss as well as lesions in nondopaminergic neurotransmitter systems including cholinergic brainstem and basal forebrain nuclei. Overall, only around 20% to 40% of the patients show transient mild benefit to L-dopa.171–173 L-Dopa–induced motor or psychiatric complications appear to be rare. In a review of 82 consecutive patients treated with open-label L-dopa (mean maximum daily dose, 1.015 mg) only three had mild dyskinesias, and one had an acute psychotic reaction when amitriptyline was added to the L-dopa regimen.171 In theory, postsynaptic dopamine receptor agonists should be more effective in ameliorating PSP symptoms than L-dopa, as the former agents do not require metabolic conversion by degenerating dopaminergic neurons in order to become pharmacologically effective. In practice, however, dopamine agonists have proved no less ineffective. Williams and colleagues174 failed to demonstrate any significant improvement in most of nine PSP patients treated with bromocriptine in a double-blind placebo-controlled trial. However, transient antiparkinsonian efficacy has been observed in a retrospective chart review of 12 autopsy-proved PSP patients treated with bromocriptine.172 Modest 20% improvement of motor disability has also been reported in a double-blind controlled trial of pergolide conducted in 3 PSP patients.170 In contrast, lack of antiparkinsonian efficacy was reported for lisuride175 and pramipexole176 in two small-scale open-label studies. Nevertheless, in the absence of more effective antiparkinsonian treatment these therapies should be tried when parkinsonism is present in PSP because of the possibility of a sometimes moderate, but useful effect, and because the lack of a sustained or marked benefit from L-dopa effectively rules out Parkinson disease, and may therefore support the diagnosis of PSP or other atypical parkinsonian disorders.
A mild (rarely dramatic) symptomatic improvement of parkinsonism may also be seen with tricyclic antidepressants in a minority of patients.177–179 Amitriptyline and desipramine were both shown to ameliorate parkinsonian features in a small-scale double-blind crossover trial of four PSP patients.177 In addition, gaze palsy was improved in one patient and apraxia of eyelid opening in two patients. Side effects were mild and included reversible urinary retention and a dry mouth. A recent open-label trial in two patients with PSP reported antiparkinsonian efficacy and improved tolerability of low-dose amitriptyline.178 Nortriptiline may also be used in patients with PSP; however, future multicenter controlled trials are necessary to definitively establish the role of antidepressants.
Idazoxan is a potent and selective alpha-2 presynaptic inhibitor drug whose overall effect is to increase norepinephrine neurotransmission. A double-blind crossover trial in nine PSP patients revealed significant improvement of mobility, balance, gait, and finger dexterity.180 In contrast, efaroxan, a more potent enhancer of norepinephrine neurotransmission compared with idaxozan, failed to confer any benefit to 14 PSP patients in a double-blind crossover trial.181 Methysergide, a serotonin antagonist, has been reported to improve swallowing, speech, parkinsonian features, and oculomotor disturbances in an open-label study of 12 patients,182 but this observation could not be confirmed by others.183 Serotonin antagonists such as methysergide may cause severe side effects such as retroperitoneal fibrosis or pleuropulmonary fibrosis. Aniracetam, a metabolic activator with acetylcholine-like properties, was reported to improve motor and cognitive function in two PSP patients; however, there is no further evidence to substantiate these findings.184 Other drugs with minimal or absent efficacy in PSP include amantadine, baclofen, bupropion, fluoxetine, selegeline, and valproate.185,186,171 Electroconvulsive therapy was reported to ameliorate motor dysfunction in some PSP patients; however, hospitalization was prolonged and patients experienced treatment-induced confusion, thus limiting the usefulness of this technique.187,188 Implantation of adrenal medullary tissue into the caudate nucleus has been performed in a few PSP patients; however, the procedure proved to be ineffective as well as hazardous with substantial perioperative morbidity and mortality.189,190
Oculomotor and Related Disturbances
Zolpidem, a short-acting hypnotic drug and selective agonist of the benzodiazepine receptor subtype BZ1, was shown to improve saccadic eye movements and parkinsonism in a double-blind crossover study of 10 patients with probable PSP; however, the benefit was limited by drowsiness, particularly at dosages higher than 5 mg/day.191 Involuntary eye closure may be treated with botulinum toxin injections.192–195 Visual prisms are rarely of help; patients may instead resort to books on tape. Artificial tears are useful to avoid exposure keratitis secondary to decreased eye blink rate.
Cognitive Disturbance
Administration of cholinergic agents is not beneficial, indeed, mental status and gait of patients may worsen with these drugs.196–198 To date, only three cholinergic agents have been used to treat PSP. Foster et al.196 evaluated the effects of RS-86, an M1-M2 muscarinic agonist, on motor and cognitive function in 10 PSP patients during a 9-week double-blind randomized controlled trial. No effects were found with regard to either cognitive tasks or motor functions. Litvan and colleagues reported lack of therapeutic efficacy for physostigmine and scopalamine in a double-blind placebo-controlled study of nine PSP patients.197 Fabbrini and colleagues198 demonstrated lack of efficacy for donezepil, a centrally acting cholinesterase inhibitor, in an open-label trial of six PSP patients.
Other Features
Because gait instability shows only minimal or no response to drug therapy, weighted walkers should be considered. Swallowing disturbances should be regularly evaluated by speech therapists to avoid aspiration pneumonia. Dysphagia can be managed by the use of straws, food thickeners, or soft processed food. The question of whether a nasogastric tube or percutaneous endoscopic gastrostomy could reduce the chances of aspiration pneumonia needs further assessment. Patients can also be helped by a variety of communication aids. Although drooling can be managed with anticholinergics, these drugs should be used cautiously because they can worsen patient symptomatology. Emotional incontinence has been reported to improve with amitriptyline.199
CORTICOBASAL DEGENERATION
Epidemiology
The incidence and prevalence of CBD are largely unknown. Schrag and coworkers138 failed to identify a single case in a community-based prevalence study of parkinsonism covering more than 120,000 persons, and the series by Hughes and colleagues covering 143 cases of postmortem confirmed parkinsonian syndromes in a highly specialized movement disorder unit only included four CBD cases.200 This would suggest that CBD might count for only some 3% of patients with degenerative parkinsonism.
Clinical Presentation, Course, and Prognosis
Similar to the other conditions discussed in this chapter, CBD is a multisystem disorder affecting the nigrostriatal motor system plus a variety of other subcortical structures, including variable cell loss in the thalamus, subthalamic nucleus, pallidum, red nucleus, and dentate nucleus, and scattered changes in other brainstem nuclei. In addition, there is prominent and usually asymmetrical cortical degeneration involving frontoparietal areas. The resulting clinical picture is one of a strikingly asymmetrical akinetorigid parkinsonian syndrome associated with other movement disorders—most often dystonia and myoclonus—in combination with cortical signs including apraxia and “alien limb” phenomena, cortical sensory loss, and variable degrees of dysphasia. Between 30% and 50% of patients eventually show signs of depression and frontal-lobe type behavioral changes including apathy or disinhibition, impulsiveness, and irritability,201–203 and some series suggest that 25% may become demented.172
The clinical picture of CBD in its full expression including limb apraxia, alien limb behavior, and strictly asymmetrical parkinsonism, and jerky dystonia of the limbs is so characteristic that there is little room for clinical diagnostic error. On the other hand, early CBD with unilateral limb rigidity and clumsiness can be confused with idiopathic Parkinson disease; patients with significant postural instability and falls may be confused with PSP patients when there is some additional limited vertical gaze or frontal executive dysfunction. However, the supranuclear gaze palsy in CBD usually affects horizontal and vertical gaze equally, whereas vertical gaze is more severely affected in PSP. CBD presenting with cognitive impairment is also difficult to diagnose, and there is some controversy that frontotemporal dementia and CBD may represent different ends of a single disease spectrum.204 Overall, diagnostic sensitivity to CBD is suboptimal even among expert neurologists who only detected 30% of postmortem confirmed CBD cases on the basis of clinical presentation at first visit.205 Table 72-6 summarizes the most common presenting symptoms of CBD as described in two clinicopathological series.
| Presenting Symptom | % |
|---|---|
| Limb, clumsiness (arm) | ≈50 |
| Gait disorder | ≈30 |
| Tremor | ≈20 |
| Speech disturbance | Rare |
| Behavioral disturbance | Rare |
Data from Rinne JO, Lee MS, Thompson PD, et al: Corticobasal degeneration: a clinical study of 36 cases. Brain 1994;117:1183-1196; and Wenning GK, Litvan I, Jankovic J, et al: Natural history and survival of 14 patients with corticobasal degeneration confirmed at postmortem examination. J Neurol Neurosurg Psychiatry 1998; 64:184-189.
Clinical Course
Although publications on CBD have multiplied since the late 1980s, there are still no available data on its incidence and prevalence. There appears to be no sex predominance.201 Classically, patients with CBD present in the sixth or seventh decade of life with a unilateral jerky tremulous akinetorigid and apraxic extremity held in a fixed posture and displaying the alien limb syndrome.202 However, presentations can vary widely; they may relate to difficulty in walking, speech, or, less commonly, limb sensation. Symptoms usually remain clearly asymmetrical, eventually spreading from the affected arm to the ipsilateral leg, and progress steadily until death which usually occurs 4 to 8 years after disease onset. Bilateral parkinsonian features at first neurological evaluation predict shorter survival, particularly in the presence of frontal lobe dysfunction.201 The etiology and pathogenesis of CBD remain to be resolved. There is abnormal aggregation of tau affecting both basal ganglia and motor cortex. Recent evidence suggests that there may be a genetic predisposition toward tau accumulation that is shared by PSP patients.146
Diagnostic Criteria
Several schemes for diagnostic criteria for CBD have been proposed and most focus on the presence of a markedly asymmetrical parkinsonian syndrome with relentless progression and various combinations of movement disorders and cortical dysfunction. Table 72-7 summarizes the key components of clinical diagnostic criteria proposed by Kumar and colleagues.206
Adapted from Kumar R, Bergeron C, Lang AE. Corticobasal degeneration. In Jankovic J, Tolosa E, eds: Parkinson’s Disease and Movement Disorders, 4th ed. Philadelphia: Lippincott Williams & Wilkins, 2002, pp 185-198.
Genetics
CBD is usually a sporadic disorder but there have been rare reports of familial occurrence of a CBD like syndrome, with autopsy-proved CBD in one family member.207 Although there have been no documented tau mutations in cases of CBD, some studies found higher prevalences of the H1/H1-genotype in CBD, indicating that tau gene polymorphisms may play a role in this condition.146,208
Ancillary Investigations
While routine laboratory studies or cerebrospinal fluid studies are normal in patients with CBD, classic patients usually show asymmetrical frontoparietal cortical atrophy, which was present in 88% of cases on MR imaging in one series.209 Functional imaging using SPECT and dopamine transporter ligands show nigrostriatal terminal dopaminergic dysfunction which is similar to Parkinson disease or other degenerative forms of parkinsonism including PSP but may be helpful in differentiating CBD from Pick disease. PET studies have shown asymmetrical reductions in glucose metabolism and blood flow predominantly in the frontoparietal area but also subcortical nuclei like the thalamus or striatum,210 and PET studies using PK11195 have shown asymmetrical basal ganglia and cortical microglial activation in CBD.211 Neurophysiological studies are of limited usefulness in CBD, but electroencephalograms may show asymmetrical cortical slowing over the hemisphere contralateral to the most effected extremities in full-blown cases.
Principles of Management
Overall, CBD is often considered an untreatable condition due to its relentless progression and the at-best modest response to various symptomatic interventions. Nonetheless, at least temporary improvement can be achieved for several of the clinical problems highlighted in up to two thirds of patients depending on the target symptom (Table 72-8).172 It has to be pointed out, however, that due to the rarity of CBD (even tertiary referral centers usually follow less than 20 patients),172 to date there has been no single controlled or even uncontrolled prospective clinical trial of any intervention, and all recommendations given below are based on retrospective uncontrolled case series.
| Medication | Exposed, n (%) | Clinical Improvement, n (%) |
|---|---|---|
| Dopaminergic agents | 135 (92) | 33 (24) |
| Levodopa/carbidopa | 128 (87) | 33 (26) |
| Agonist | 33 (25) | 2 (6) |
| Selegiline | 30 (20) | 3 (10) |
| Amantadine | 24 (16) | 3 (13) |
| Benzodiazepines | 47 (32) | 19 (40) |
| Anticholinergics | 38 (27) | 8 (21) |
| Baclofen | 28 (19) | 2 (7) |
| Antidepressants | 16 (11) | 1 (6) |
| Anticonvulsants | 13 (9) | 3 (23) |
| Propanolol | 11 (8) | 2 (18) |
| Neuroleptics | 6 (4) | 4 (67) |
| Botulinum toxin | 9 (6) | 6 (67) |
Values are number of patients of a total possible of 147.
From Kompoliti K, Goetz CG, Litvan I, et al: Pharmacological therapy in progressive supranuclear palsy. Arch Neurol 1998; 55:1099-1102.
Treatment of Parkinsonism in Corticobasal Degeneration
Parkinsonism in CBD is dominated by rigidity and bradykinesia while tremor is present in only 30% to 50% of cases and is often irregular and jerky.172,201 In addition parkinsonism contributes to the gait disorder of CBD, which becomes a major source of disability in the course of disease with marked postural instability and falls. Due to the impact of cerebellar dysfunction and apraxia, antiparkinsonian medications have a variable effect on the gait problems of CBD patients.
The drugs that have been used to improve parkinsonian features in CBD include levodopa, dopamine agonists, selegiline, amantadine, and anticholinergics. The largest case series with uncontrolled retrospective data on drug responses was published by Kompoliti and colleagues (1998)211a who had included 147 patients followed at eight movement disorder centers.
L-Dopa
In the series by Kompoliti and coworkers211a, 87% of cases had been exposed to therapeutic trials of L-dopa with a mean daily dose of 300 mg (range, 100 to 2000 mg). Some clinical improvement of parkinsonism was noted in 26% of patients, and bradykinesia and rigidity responded the most, but there are no data on the magnitude or duration of this response. One patient each of a total of 33 responding to L-dopa was noted to improve regarding dystonic or alien limb features while 5% had some degree of worsening either of parkinsonism or gait dysfunction or dystonia and myoclonus.
L-Dopa–induced dyskinesias were not observed in this series even with high-dose treatment, and there are also no other reports in the literature noting the occurrence of dyskinesias in response to L-dopa in CBD.212 Gastrointestinal complaints were present in 15%, followed by confusion, dizziness and somnolence (4% each), and hallucinosis (2%).
Treatment of Dystonia in Corticobasal Degeneration
Dystonia is one of the cardinal motor features of CBD, affecting between 50% and 80% of patients,213 most often as asymmetrical limb dystonia producing jerky or fixed postural deformities that may the render the affected extremity functionally useless and may also be painful. It is therefore an area of great therapeutic need, but again there are no controlled prospective or controlled trials of antidystonic interventions on which to base treatment decisions. Retrospective uncontrolled observations point to the possible efficacy of drugs like anticholinergics, benzodiazepines, baclofen and—most strongly—of local botulinum toxin injections.
Systemic Drug Therapy
Anticholinergics as well as baclofen were reported to have improved dystonia in individual cases of the series of Goetz and coworkers214 and Kompoliti and colleagues212 and these authors as well as Vanek and Jankovic213 remark on the efficacy of clonazepam to improve myoclonic dystonia. Details on doses employed, effect size, or side effects are absent from all these reports.
Botulinum Toxin Injections
Because asymmetrical focal limb dystonia is a typical presentation of dystonia in CBD,201,202,213 localized injections of botulinum toxin have been tried in this situation—in analogy to their successful use in adult-onset focal idiopathic dystonia. Again no systematic or controlled trials of this form of treatment for dystonia in CBD are available. Of nine patients in the large series reported by Kompoliti and coworkers (1998), who received local botulinum toxin injections to treat focal limb dystonia, six (67%) reportedly showed some improvement. All 6 patients in the series of 66 cases reported by Jankovic and colleagues213 had some response of their focal dystonic symptoms to botulinum toxin injections—two of these experienced marked degrees of improvement of both dystonia and pain with this form of therapy. Unfortunately, none of these reports gives any detailed dose of botulinum toxin or muscle selection, so that it is difficult to derive practical treatment recommendations from them. Müller and colleagues195 recently reported on two patients with CBD who received successful treatment with botulinum toxin injections for focal limb dystonia. Dosages used were 40 to 120 units Dysport for finger flexor muscles, 80 to 120 units Dysport for wrist flexor and extensor muscles, and 160 to 240 units Dysport for elbow flexor muscles.
Treatment of Myoclonus in Corticobasal Degeneration
The use of benzodiazepines—most often clonazepam—as well as of valproate, mysoline, and piracetam to treat myoclonic jerking in CBD has been reported in a number of studies in anecdotal fashion.172,205,213 Results are inconsistent and the drug most often reported as beneficial is clonazepam ameliorating myoclonic jerking in 23% of patients in the series of Kompoliti and coworkers in 1998. Sedation is the most common side effect, affecting 26% of patients exposed to clonazepam in that report.
Practical Management
Mahapatra RK, Edwards MJ, Schott JM, et al. Corticobasal degeneration. Lancet Neurol. 2004;3:736-743.
Steele JC, Richardson JC, Olszewski J. Progressive supranuclear palsy. A heterogeneous degeneration involving the brainstem, basal ganglia and cerebellum with vertical gaze and pseudobulbar palsy, nuchal dystonia and dementia. Arch Neurol. 1964;10:333-359.
Wenning GK, Ben Shloma Y, Magalhaes M, et al. Clinical features and natural history of multiple system atrophy. An analysis of 100 cases. Brain. 1994;117:835-845.
Wenning GK, Colosimo C, Geser F, et al. Multiple system atrophy. Lancet Neurol. 2004;3:93-103.
Williams DR, de Silva R, Paviour DC, et al. Charcteristics of two distinct clinical phenotypes in pathologically proven progressive supranuclear palsy. Richardson’s syndrome and PSP-parkinsonism. Brain. 2005;128:1247-1258.
1 Graham J, Oppenheimer DR. Orthostatic-hypotension and nicotine sensitivity in a case of multiple system atrophy. J Neurol Neurosurg Psychiatry. 1969;32:28-34.
2 van der Eecken H, Adams RD, van Bogaert L. Striopallidal-nigral degeneration. A hitherto undescribed lesion in paralysis agitans. J Neuropathol Exp Neurol. 1960;19:159-161.
3 Adams R, Van Bogaert L, Van der Eecken H. Degenerescenes nigro-striees et cerebello-nigro-striees. Psychiat Neurol (Basel). 1961;142:219-259.
4 Adams R, Van Bogaert L, Van der Eecken H. Striatonigral degeneration. J Neuropathol Exp Neurosci. 1964;23:584-698.
5 Dejerine J, Thomas A. L’atrophie olivo-ponto-cerebelleuse. Nouvelle iconographie de la Salpetriere: Clinique des Malacies du Systeme Nerveux. 1900;13:330-370.
6 Stauffenberg: Zur Kenntnis des extrapyramidalen motorischen Systems und Mitteilung eines Falles von sog. “Atrophie olivo-pontocérébelleuse.”. Z Gesamte Neurol Psychiatrie. 1918;39:1-55.
7 Shy G, Drager GA. A neurological syndrome associated with orthostatic hypotension. A clinicopathological study. Arch Neurol. 1960;2:511-527.
8 Papp M, Kahn JE, Lantos PL. Glial cytoplasmic inclusions in the CNS of patients with multiple system atrophy (striatonigral degeneration, olivopontocerebellar atrophy and Shy-Drager syndrome). J Neurol Sci. 1989;94:79-100.
9 Wakabayashi K, Yoshimoto M, Tsuji S, et al. Alpha-synuclein immunoreactivity in glial cytoplasmic inclusions in multiple system atrophy. Neurosci Lett. 1998;249:180-182.
10 Spillantini MG, Crowther RA, Jakes R, et al. Filamentous alpha-synuclein inclusions link multiple system atrophy with Parkinson disease and dementia with Lewy bodies. Neurosci Lett. 1998;251:205-208.
11 Tison F, Yekhlef F, Chrysostome V, et al. Prevalence of multiple system atrophy. Lancet. 2000;355:495-496.
12 Schrag A, Ben-Shlomo Y, Quinn NP. Prevalence of progressive supranuclear palsy and multiple system atrophy: a crosssectional study. Lancet. 1999;354:1771-1775.
13 Wermuth L, Joensen P, Bunger N, et al. High prevalence of Parkinson disease in the Faroe Islands. Neurology. 1997;49:426-432.
14 Chio A, Magnani C, Schiffer D. Prevalence of Parkinson disease in Northwestern Italy: comparison of tracer methodology and clinical ascertainment of cases. Mov Disord. 1998;13:400-405.
15 Nee LE, Gomez MR, Dambrosia J, et al. Environmental-occupational risk factors and familial associations in multiple system atrophy: a preliminary investigation. Clin Auton Res. 1991;1:9-13.
16 Vanacore N, Bonifati V, Fabbrini G, et al. Smoking habits in multiple system atrophy and progressive supranuclear palsy. Neurology. 2000;54:114-119.
17 Hanna P, Jankovic J, Kirkpatrick JB. Multiple system atrophy. The putative causative role of environmental toxins. Arch Neurol. 1999;56:90-94.
18 Watanabe H, Saito Y, Terao S, et al. Progression and prognosis in multiple system atrophy: an analysis of 230 Japanese patients. Brain. 2002;125:1070-1083.
19 Testa D, Monza D, Ferrarini M, et al. Comparison of natural histories of progressive supranuclear palsy and multiple system atrophy. Neurol Sci. 2001;22:247-251.
20 Ben-Shlomo Y, Wenning G, Tison F, et al. Survival of patients with pathologically proven multiple system atrophy: a meta-analysis. Neurology. 1997;48:384-393.
21 Wenning GK, Ben Shlomo Y, Magalhaes M, et al. Clinical features and natural history of multiple system atrophy. An analysis of 100 cases. Brain. 1994;117:835-845.
22 Abele M, Burk K, Schols L, et al. The aetiology of sporadic adult-onset ataxia. Brain. 2002;125:961-998.
23 Gilman S, Little R, Johanns J, et al. Evolution of sporadic olivopontocerebellar atrophy into multiple system atrophy. Neurology. 2000;55:527-532.
24 Schwarz J, Tatsch K, Gasser T, et al. 123I-IBZM binding compared with long-term clinical follow up in patients with de novo parkinsonism. Mov Disord. 1998;13:16-19.
25 Gilman S, Low P, Quinn N, et al. Consensus statement on the diagnosis of multiple system atrophy. Clin Auton Res. 1998;8:359-362.
26 Wenning GK, Geser F, Poewe W. The ‘risus sardonicus’ of multiple system atrophy. Mov Disord. 2003;18:1211.
27 Boesch SM, Wenning GK, Ransmayr G, et al. Dystonia in multiple system atrophy. J Neurol Neurosurg Psychiatry. 2002;72:300-303.
28 Wenning GK, Ben Shlomo Y, Hughes A, et al. What clinical features are most useful to distinguish definite multiple system atrophy from Parkinson disease? J Neurol Neurosurg. Psychiatry. 2000;68:434-440.
29 Beck R, Betts C, Fowler C. Genitourinary dysfunction in multiple system atrophy: clinical features and treatment in 62 cases. J Urol. 1994;151:1336-1341.
30 Quinn N. Multiple system atrophy—the nature of the beast. J Neurol Neurosurg Psychiatry. 1989;52(suppl):78-89.
31 Quinn N. Multiple system atrophy. In: Marsden CD, Fahn S, editors. Movement Disorders 3. London: Butterworth-Heinemann; 1994:262-281.
32 Osaki Y, Wenning GK, Daniel SE, et al. Do published criteria improve clinical diagnostic accuracy in multiple system atrophy? Neurology. 2002;59:1486-1491.
33 Gouider-Khouja N, Vidailhet M, Bonnet AM, et al. “Pure” striatonigral degeneration and Parkinson disease: a comparative clinical study. Mov Disord. 1995;10:288-294.
34 Tison F, Wenning GK, Quinn NP, et al. REM sleep behaviour disorder as the presenting symptom of multiple system atrophy. J Neurol Neurosurg Psychiatry. 1995;58:379-389.
35 Plazzi G, Corsini R, Provini F. REM sleep behavior disorders in multiple system atrophy. Neurology. 1997;48:1094-1097.
36 Olson EJ, Boeve BF, Silber MH. Rapid eye movement sleep behaviour disorder: demographic, clinical and laboratory findings in 93 cases. Brain. 2000;123:331-339.
37 Gilman S, Koeppe RA, Chervin RD, et al. REM sleep behavior disorder is related to striatal monoaminergic deficit in MSA. Neurology. 2003;61:29-34.
38 Ghorayeb I, Yekhlef F, Chrysostome V, et al. Sleep disorders and their determinants in multiple system atrophy. J Neurol Neurosurg Psychiatry. 2002;72:798-800.
39 Wenning GK, Wagner S, Daniel S, et al. Multiple system atrophy: sporadic or familial? Lancet. 1993;342:681.
40 Baker M, Litvan I, Houlden H, et al. Association of an extended haplotype in the tau gene with progressive supranuclear palsy. Hum Mol Genet. 1999;8:711-715.
41 Bandmann O, Sweeney MG, Daniel SE, et al. Multiple-system atrophy is genetically distinct from identified inherited causes of spinocerebellar degeneration. Neurology. 1997;49:1598-1604.
42 Nicholl DJ, Bennett P, Hiller L, et al. A study of five candidate genes in Parkinson disease and related neurodegenerative disorders. European Study Group on atypical parkinsonism. Neurology. 1999;53:1415-1421.
43 Iwahashi K, Miyatake R, Tsuneoka Y, et al. A novel cytochrome P-450IID6 (CYPIID6) mutant gene associated with multiple system atrophy. J Neurol Neurosurg Psychiatry. 1995;58:263-264.
44 Bandmann O, Wenning GK, Quinn NP, et al. Arg296 to Cys296 polymorphism in exon 6 of cytochrome P-450–2D6 (CYP2D6) is not associated with multiple system atrophy. J Neurol Neurosurg Psychiatry. 1995;59:557.
45 Cairns NJ, Atkinson PF, Kovacs T, et al. Apolipoprotein E e4 allele frequency in patients with multiple system atrophy. Neurosci Lett. 1997;221:161-164.
46 Healy DG, Abou-Sleiman PM, Jain S, et al. Assessment of a DJ-1 (PARK7) polymorphism in Finnish Parkinson disease. Neurology. 2003;61:1000-1002.
47 Hashida H, Goto J, Zhao N, et al. Cloning and mapping of ZNF231, a novel brain-specific gene encoding neuronal double zinc finger protein whose expression is enhanced in a neurodegenerative disorder, multiple system atrophy (MSA). Genomics. 1998;54:50-58.
48 Gilman S, Sima AA, Junck L, et al. Spinocerebellar ataxia type 1 with multiple system degeneration and glial cytoplasmic inclusions. Ann Neurol. 1996;39:241-255.
49 Schols L, Peters S, Szymanski S, et al. Extrapyramidal motor signs in degenerative ataxias. Arch Neurol. 2000;57:1495-1500.
50 Ranum LP, Lundgren JK, Schut LJ, et al. Spinocerebellar ataxia type 1 and Machado-Joseph disease: incidence of CAG expansions among adult-onset ataxia patients from 311 families with dominant, recessive, or sporadic ataxia. Am J Hum Genet. 1995;57:603-608.
51 Silveira I, Lopes-Cendes I, Kish S, et al. Frequency of spinocerebellar ataxia type 1, dentatorubropallidoluysian atrophy, and Machado-Joseph disease mutations in a large group of spinocerebellar ataxia patients. Neurology. 1996;46:4-8.
52 Leggo J, Dalton A, Morrison PJ, et al. Analysis of spinocerebellar ataxia types 1, 2, 3, and 6, dentatorubral-pallidoluysian atrophy, and Friedreich’s ataxia genes in spinocerebellar ataxia patients in the UK. J Med Genet. 1997;34:982-985.
53 Futamura N, Matsumura R, Fujimoto Y, et al. CAG repeat expansions in patients with sporadic cerebellar ataxia. Acta Neurol Scand. 1998;98:55-59.
54 Moseley ML, Benzow KA, Schut LJ, et al. Incidence of dominant spinocerebellar and Friedreich triplet repeats among 361 ataxia families. Neurology. 1998;51:1666-1671.
55 Hagermann RJ, Leehey M, Heinrichs W, et al. Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology. 2002;58:987.
56 Jacquemont S, Hagerman RJ, Leehey M, et al. Fragile X premutation tremor/ataxia syndrome: molecular, clinical, and neuroimaging correlates. Am J Hum Genet. 2003;72:869-878.
57 Kamm C, Leung J, Joseph S, et al. Refinded linkage to the RDP/DYT12 locus on 19q13.2 and evaluation of GRIK5 as a candidate gene. Mov Disord. 2004;19:845-847.
58 Consensus statement on the definition of orthostatic hypotension, pure autonomic failure, and multiple system atrophy. J Neurol Sci. 1996;144:218-219.
59 Bannister R, Mathias C. Investigation of autonomic disorders. In: Mathias C, Bannister R, editors. Autonomic Failure: A Text-book of Clinical Disorders of the Autonomic Nervous System. Oxford: Oxford University Press; 1999:169-195.
60 Ziegler M, Lake C, Kopin I. The sympathetic-nervous-system defect in primary orthostatic hypotension. N Engl J Med. 1977;296:293-297.
61 Polinsky R, Kopin I, Ebert M, et al. Pharmacologic distinction of different orthostatic hypotension syndromes. Neurology. 1981;31:1-7.
62 Riley DE, Chelimsky TC. Autonomic nervous system testing may not distinguish multiple system atrophy from Parkinson disease. J Neurol Neurosurg Psychiatry. 2003;74:56-60.
63 Kirby R, Fowler CJ, Gosling J, et al. Urethro-vesical dysfunction in progressive autonomic failure with multiple system atrophy. J Neurol Neurosurg Psychiatry. 1986;49:554-562.
64 Pramstaller PP, Wenning GK, Smith SJ, et al. Nerve conduction studies, skeletal muscle EMG, and sphincter EMG in multiple system atrophy. J Neurol Neurosurg Psychiatry. 1995;58:618-621.
65 Palace J, Chandiramani VA, Fowler CJ. Value of sphincter electromyography in the diagnosis of multiple system atrophy. Muscle Nerve. 1997;20:1396-1403.
66 Valldeoriola F, Valls-Sole J, Tolosa ES, et al. Striated anal sphincter denervation in patients with progressive supranuclear palsy. Mov Disord. 1995;10:550-555.
67 Giladi N, Simon ES, Korczyn Alzheimer disease, et al. Anal sphincter EMG does not distinguish between multiple system atrophy and Parkinson disease. Muscle Nerve. 2000;23:731-734.
68 Colosimo C, Inghilleri M, Chaudhuri KR. Parkinson disease misdiagnosed as multiple system atrophy by sphincter electromyography. J Neurol. 2000;247:559-561.
69 Vodusek B. Sphincter EMG and differential diagnosis of multiple system atrophy. Mov Disord. 2001;16:600-607.
70 Wüllner U, Klockgether T, Petersen D, et al. Magnetic resonance imaging in hereditary and idiopathic ataxia. Neurology. 1993;43:318-325.
71 Schrag A, Good CD, Miszkiel K, et al. Differentiation of atypical parkinsonian syndromes with routine MRI. Neurology. 2000;54:697-702.
72 Drayer B, Olanow W, Burger P, et al. Parkinson plus syndrome: diagnosis using high field MR imaging of brain iron. Radiology. 1986;159:493-498.
73 Pastakia B, Polinsky R, Di Chiro G, et al. Multiple system atrophy (Shy-Drager syndrome): MR imaging. Radiology. 1986;159:499-502.
74 O’Brien C, Sung JH, McGeachie RE, et al. Striatonigral degeneration: clinical, MRI, and pathologic correlation. Neurology. 1990;40:710-711.
75 Lang A, Curran T, Provias J, et al. Striatonigral degeneration: iron deposition in putamen correlated with the slit-like void signal of magnetic resonance imaging. Can J Neurol Sci. 1994;21:311-318.
76 Schwarz J, Weis S, Kraft E. Signal changes on MRI and increases in reactive microgliosis, astrogliosis, and iron in the putamen of two patients with multiple system atrophy. J Neurol Neurosurg Psychiatry. 1996;60:98-101.
77 Schrag A, Kingsley D, Phatouros C, et al. Clinical usefulness of magnetic resonance imaging in multiple system atrophy. J Neurol Neurosurg Psychiatry. 1998;65:65-71.
78 Kraft E, Trenkwalder C, Auer DP. T2*-weighted MRI differentiates multiple system atrophy from Parkinson disease. Neurology. 2002;59:1265-1267.
79 Konagaya M, Konagaya Y, Iida M. Clinical and magnetic resonance imaging study of extrapyramidal symptoms in multiple system atrophy. J Neurol Neurosurg Psychiatry. 1994;57:1528-1531.
80 Kraft E, Schwarz J, Trenkwalder C, et al. The combination of hypointense and hyperintense signal changes on T2-weighted magnetic resonance imaging sequences: a specific marker of multiple system atrophy? Arch. Neurol. 1999;56:225-228.
81 Schocke M, Seppi K, Esterhammer R, et al. Diffusion-weighted MRI differentiates the Parinson variant of multiple system atrophy from Parkinson disease. Neurology. 2002;58:575-580.
82 Seppi K, Schocke MF, Esterhammer R, et al. Diffusion-weighted imaging discriminates progressive supranuclear palsy from Parkinson disease, but not from the parkinson variant of multiple system atrophy. Neurology. 2003;60:922-927.
83 Schulz J, Skalej M, Wedekind D, et al. Magnetic resonance imaging-based volumetry differentiates idiopathic Parkinson syndrome from multiple system atrophy and progressive supranuclear palsy. Ann Neurol. 1999;45:65-74.
84 Brenneis C, Seppi K, Schocke M, et al. Voxel-based morphometry detects cortical atrophy in the Parkinson variant of multiple system atrophy. Mov Disord. 2003;18:1132-1138.
85 Brooks D, Ibanez V, Sawle GV, et al. Differing patterns of striatal 18F-dopa uptake in Parkinson disease, multiple system atrophy, and progressive supranuclear palsy. Ann Neurol. 1990;28:547-555.
86 Burn DJ, Rinne JO, Quinn NP, et al. Striatal opioid receptor binding in Parkinson disease, striatonigral degeneration and Steele-Richardson-Olszewski syndrome, A [11C]diprenor-phine PET study. Brain. 1995;118:951-958.
87 De Volder A, Francart J, Laterre C, et al. Decreased glucose utilization in the striatum and frontal lobe in probable striatonigral degeneration. Ann Neurol. 1989;26:239-247.
88 Perani D, Bressi S, Testa D, et al. Clinical/metabolic correlations in multiple system atrophy. A fludeoxyglucose F 18 positron emission tomographic study. Arch Neurol. 1995;52:179-185.
89 Antonini A, Kazamuta K, Feigin A, et al. Differential diagnosis of parkinsonism with 18F-fluorodeoxyglucose and PET. Mov Disord. 1998;13:268-274.
90 Ghaemi M, Hilker R, Rudolf J, et al. Differentiating multiple system atrophy from Parkinson disease: contribution of striatal and midbrain MRI volumetry and multi-tracer PET imaging. J Neurol Neurosurg Psychiatry. 2002;73:517-523.
91 Gilman S, Koeppe RA, Junck L, et al. Patterns of cerebral glucose metabolism detected with positron emission tomography differ in multiple system atrophy and olivopontocerebellar atrophy. Ann Neurol. 1994;36:166-175.
92 Brücke T, Djamshidian S, Bencsits G, et al. SPECT and PET imaging of the dopaminergic system in Parkinson disease. J Neurol. 2000;247(Suppl 4):2-7.
93 Scherfler C, Seppi K, Donnemiller E, et al. Voxelwise analysis of [123I]beta-CIT SPECT differentiates the Parkinson variant of multiple system atrophy from idiopathic Parkinson disease. Brain. 2005;128:1605-1612.
94 Schulz J, Klockgether T, Petersen D, et al. Multiple system atrophy: natural history, MRI morphology, and dopamine receptor imaging with 123IBZM-SPECT. J Neurol Neurosurg Psychiatry. 1994;57:1047-1056.
95 van Royen E, Verhoeff NFLG, Speelman JD, et al. Multiple system atrophy and progressive supranuclear palsy: diminished striatal D2 dopamine receptor activity demonstrated by 123I-IBZM sigle photon emission computed tomography. Arch Neurol. 1993;50:13-16.
96 Brücke T, Wenger S, Asenbaum S. Dopamine D2 receptor imaging and measurement with SPECT. Adv Neurol. 1993;60:494-500.
97 Braune S, Reinhardt M, Schnitzer R, et al. Cardiac uptake of (123I)MIBG separates Parkinson disease from multiple system atrophy. Neurology. 1999;53:1020-1025.
98 Braune S. The role of cardiac metaiodobenzylguanidine uptake in the differential diagnosis of parkinsonian syndromes. Clin Auton Res. 2001;11:351-355.
99 Goldstein DS, Holmes C, Cannon ROIII, et al. Sympathetic cardioneuropathy in dysautonomias. N Engl J Med. 1997;336:696-702.
100 Goetz CG, Tanner CM, Klawans HL. The pharmacology of olivopontocerebellar atrophy. Adv Neurol. 1984;41:143-148.
101 Botez MI, Botez-Marquard T, Elie R, et al. Amantadine hydrochloride treatment in heredodegenerative ataxias: a double blind study. J Neurol Neurosurg Psychiatry. 1996;61:259-264.
102 Albanese A, Colosimo C, Bentivoglio A, et al. Multiple system astrophy presenting as parkinsonism: clinical features and diagnostic criteria. J Neurol Neurosurg Psychiatry. 1995;59:144-151.
103 Parati E, Fetoni V, Geminiani C, et al. Response to L-DOPA in multiple system atrophy. Clin Neuropharmacol. 1993;16:139-144.
104 Lees A. The treatment of the motor disorder of multiple system atrophy. In: Mathias C, Bannister R, editors. Autonomic Failure. Oxford: Oxford University Press; 1999:357-363.
105 Hughes A, Colosimo C, Kleedorfer B, et al. The dopaminergic response in multiple system atrophy. J Neurol Neurosurg Psychiatry. 1992;55:1009-1013.
106 Colosimo C, Albanese A, Hughes A. Some specific clinical features differentiate multiple system atrophy (striatonigral variety) from Parkinson disease. Arch Neurol. 1995;52:294-298.
107 Wenning GK, Ben Shlomo Y, Magalhaes M, et al. Clinicopathological study of 35 cases of multiple system atrophy. J Neurol Neurosurg Psychiatry. 1995;58:160-166.
108 Rajput AH, Rozdilsky B, Rajput A, et al. Levodopa efficacy and pathological basis of Parkinson syndrome. Clin Neuropharmacol. 1990;13:553-558.
109 Lees A, Bannister R. The use of lisuride in the treatment of multiple system atrophy with autonomic failure (Shy Drager syndrome). J Neurol Neurosurg Psychiatry. 1981;44:347-351.
110 Colosimo C, Merello M, Pontieri FE. Amantadine in parkinsonian patients unresponsive to levodopa: a pilot study. J Neurol. 1996;243:422-425.
111 Lang AE, Lozano A, Duff J, et al. Medial pallidotomy in latestage Parkinson disease and striatonigral degeneration. Adv Neurol. 1997;74:199-211.
112 Visser-Vandewalle V, Temel Y, Colle H, et al. Bilateral highfrequency stimulation of the subthalamic nucleus in patients with multiple system atrophy–parkinsonism. Report of four cases. J Neurosurg. 2003;98:882-887.
113 Wenning G, Tison F, Scherfler C, et al. Towards neurotransplantation in multiple system atrophy: clinical rationale, pathophysiological basis, and preliminary experimental evidence. Cell Transpl. 2000;9:279-288.
114 Mathias CJ. Autonomic disorders and their recognition. N Engl J Med. 1997;336:721-724.
115 Mathias C, Mallipeddi R, Bleasdale-Barr KM. Symptoms associated with orthostatic hypotension in pure autonomic failure and multiple system atrophy. J Neurol. 1999;246:893-898.
116 Thomas DJ, Bannister R. Preservation of autoregulation of cerebral blood flow in autonomic failure. J Neurol Sci. 1980;44:205-212.
117 Chaudhuri K, Hu M. Central autonomic dysfunction. In: Appenzeller O, editor. Handbook of Clinical Neurology. Part II. Amsterdam: Elsevier Science; 2000:161-202.
118 Low PA, Gilden JL, Freeman R, et al. Efficacy of midodrine vs placebo in neurogenic orthostatic hypotension. A randomized, double-blind multicenter study. Midodrine Study Group. JAMA. 1997;277:1046-1051.
119 Wright RA, Kaufmann HC, Perera R, et al. A double-blind, dose-response study of midodrine in neurogenic orthostatic hypotension. Neurology. 1998;51:120-124.
120 Jankovic J, Gilden JL, Hiner BC, et al. Neurogenic orthostatic hypotension: a double-blind, placebo-controlled study with midodrine. Am J Med. 1993;95:38-48.
121 Freeman R, Landsberg L, Young J. The treatment of neurogenic orthostatic hypotension with 3,4-DL-threo-dihydrox-yphenylserine: a randomized, placebo-controlled, crossover trial. Neurology. 1999;53:2151-2157.
122 Bonnet AM, Pichon J, Vidailhet M, et al. Urinary disturbances in striatonigral degeneration and Parkinson disease: clinical and urodynamic aspects. Mov Disord. 1997;12:509-513.
123 Hussain IF, Brady CM, Swinn MJ, et al. Treatment of erectile dysfunction with sildenafil citrate (Viagra) in parkinsonism due to Parkinson disease or multiple system atrophy with observations on orthostatic hypotension. J Neurol Neurosurg Psychiatry. 2001;71:371-374.
124 Hayashi M, Isozaki E, Oda M, et al. Loss of large myelinated nerve fibers of the recurrent laryngeal nerve in patients with multiple system atrophy and vocal cord palsy. J Neurol Neurosurg Psychiatry. 1997;62:234-238.
125 Silber M, Levine S. Stridor and death in multiple system atrophy. Mov Disord. 2000;15:699-704.
126 Iranzo A, Santamaria J, Tolosa E, Barcelona Multiple System Atrophy Study Group. Continuous positive air pressure eliminates nocturnal stridor in multiple system atrophy. Lancet. 2000;356:1329-1330.
127 Maher ER, Lees AJ. The clinical features and natural history of the Steele-Richardson-Olszewski syndrome (progressive supranuclear palsy). Neurology. 1986;36:1005-1008.
128 Litvan I. Diagnosis and management of progressive supranuclear palsy. Semin Neurol. 2001;21:41-48.
129 Nath U, Ben-Shlomo Y, Thomson RG, et al. Clinical features and natural history of progressive supranuclear palsy: a clinical cohort study. Neurology. 2003;60:910-916.
130 Richardson JC, Steele J, Olszewski J. Supranuclear opthalmoplegia, pseudobulbar palsy, nuchal dystonia and dementia. A clinical report on eight cases of “heterogeneous system degeneration.”. Trans Am Neurol Assoc. 1963;88:25-29.
131 Steele JC, Richardson JC, Olszewski J. Progressive supranuclear palsy. A heterogeneous degeneration involving the brainstem, basal ganglia and cerebellum with vertical gaze and pseudobulbar palsy, nuchal dystonia and dementia. Arch Neurol. 1964;10:333-359.
132 Hauw JJ, Daniel SE, Dickson D, et al. Preliminary NINDS neuropathologic criteria for Steele-Richardson-Olszewski syndrome (progressive supranuclear palsy). Review Neurology. 1994;44:2015-2019.
133 Morris HR, Gibb G, Katzenschlager R. Pathological, clinical and genetic heterogeneity in progressive supranuclear palsy. Brain. 2002;125:969-975.
134 Spillantini MG, Goedert M. Tau protein pathology in neurodegenerative diseases. Trends Neurosci. 1998;21:428-433.
135 Arvanitakis Z, Wszolek ZK. Recent advances in the understanding of tau protein and movement disorders. Curr Opin Neurol. 2001;14:491-497.
136 Golbe LI, Davis PH, Schoenberg BS, et al. Prevalence and natural history of progressive supranuclear palsy. Neurology. 1988;38:1031-1034.
137 Bower JH, Maraganore DM, McDonnell SK, et al. Incidence of progressive supranuclear palsy and multiple system atrophy in Olmsted County, Minnesota, 1976 to 1990. Neurology. 1997;49:1284-1288.
138 Schrag A, Ben-Shlomo Y, Quinn NP. Prevalence of progressive supranuclear palsy and multiple system atrophy: a crosssectional study. Lancet. 1999;354:1771-1775.
139 Nath U, Ben-Shlomo Y, Thomson RG, et al. The prevalence of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome) in the UK. Brain. 2001;124:1438-1449.
140 Williams DR, de Silva R, Paviour DC, et al. Charcteristics of two distinct clinical phenotypes in pathologically proven progressive supranuclear palsy: Richardson’s syndrome and PSP-parkinsonism. Brain. 2005;128:1247-1258.
141 Hughes AJ, Ben-Shlomo Y, Daniel SE, et al. What features improve the accuracy of linical diagnosis in Parkinson disease: a clinicopathologic study 1992. Neurology. 2001;57(Suppl 3):34-38.
142 Litvan I, Agid Y, Jankovic J, et al. Accuracy of clinical criteria for the diagnosis of progressive supranuclear palsy. (Steele-Richardson-Olszewski syndrome). Neurology. 1996;46:922-930.
143 Goetz CG, Leurgans S, Lang AE, et al. Progression of gait, speech and swallowing deficits in progressive supranuclear palsy. Neurology. 2003;60:917-922.
144 Litvan I, Agid Y, Calne D, et al. Clinical research criteria for the diagnosos of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP internation workshop. Neurology. 1996;47:1-9.
145 Osaki Y, Ben-Shlomo Y, Lees AJ, et al. Accuracy of clinical diagnosis of progressive supranuclear palsy. Mov Disord. 2004;19:181-189.
146 Di Maria E, Tabaton M, Vigo T, et al. Corticobasal degeneration shares a common genetic background with progressive supranuclear palsy. Ann Neurol. 2000;47:374-377.
147 Urakami K, Wada K, Arai H, et al. Diagnostic significance of Tau protein in cerebrospinal fluid from patients with corticobasal degeneration or progressive supranuclear palsy. J Neurol Sci. 2001;183:95-98.
148 Holmberg B, Rosengren L, Karlsson JE, et al. Increased cerebrospinal fluid levels of neurofilament protein in progressive supranuclear palsy and multiple-system atrophy compared with Parkinson disease. Mov Disord. 1998;13:70-77.
149 Holmberg B, Johnels B, Ingvarsson P, et al. cerebrospinal fluid-neurofilament and levodopa tests combined with discriminant analysis may contribute to the differential diagnosis of Parkinsonian syndromes. Parkinsonism Relat Disord. 2001;8:23-31.
150 Warmuth-Metz M, Neumann M, Csoti I, et al. Measurement of the midbrain diameter on routine magnetic resonance imaging: a simple and accurate method of differentiating between Parkinson disease and progressive supranuclear palsy. Arch Neurol. 2001;58:1076-1079.
151 Paviour DC, Price SL, Stevens JM, et al. Quantitative MRI measurement of superior cerebellar peduncle in progressive supranuclear palsy. Neurology. 2005;64:675-679.
152 Schulz JB, Skalej M, Wedekind D, et al. Magnetic resonance imaging–based volumetry differentiates idiopathic Parkinson syndrome from multiple system atrophy and progressive supranuclear palsy. Ann Neurol. 1999;45:65-74.
153 Cordato NJ, Pantelis C, Halliday GM, et al. Frontal atrophy correlates with behavioural changes in progressive supranuclear palsy. Brain. 2002;125:789-800.
154 Groschel K, Hauser TK, Luft A, et al. Magnetic resonance imaging–based volumetry differentiates progressive supranuclear palsy from corticobasal degeneration. Neuroimage. 2004;21:714-724.
155 Brenneis C, Seppi K, Schocke M, et al. Voxel based morphometry reveals a distinct pattern of frontal atrophy in progressive supranuclear palsy. J Neurol Neurosurg Psychiatry. 2004;75:246-249.
156 Price S, Paviour D, Scahill R, et al. Voxel-based morphometry detects patterns of atrophy that help differentiate progressive supranuclear palsy and Parkinson disease. Neuroimage. 2004;23:663-669.
157 Eckert T, Sailer M, Kaufmann J, et al. Differntiation of idiopathic Parkinson disease, multiple system atrophy, progressive supranuclear palsy, and healthy controls using magnetization transfer imaging. Neuroimage. 2004;21:229-235.
158 Davie CA, Barker GJ, Machado C, et al. Proton magnetic resonance spectroscopy in Steele-Richardson-Olszewski syndrome. Mov Disord. 1997;12:767-771.
159 Tedeschi G, Litvan I, Bonavita S, et al. Proton magnetic resonance spectroscopic imaging in progressive supranuclear palsy, Parkinson disease and corticobasal degeneration. Brain. 1997;120:1541-1552.
160 Clarke CE, Lowry M. Systematic review of proton magnetic resonance spectroscopy of the striatum in parkinsonian syndromes. Eur J Neurol. 2001;8:573.
161 Brooks DJ. Functional imaging in relation to parkinsonian syndromes. J Neurol Sci. 1993;115:1-17.
162 Pirker W, Djamshidian S, Assenbaum S, et al. Progression of dopaminergic degeneration in Parkinson disease and atypical parkinsonism: a longitudinal beta-CIT SPECT study. Mov Disord. 2002;17:45-53.
163 Kim YJ, Ichise M, Ballinger JR, et al. Combination of dopamine transporter and D2 receptor SPECT in the diagnostic evaluation of Parkinson disease, MSA, and PSP. Mov Disord. 2002;17:303-312.
164 Shinotoh H, Namba H, Yamaguchi M, et al. Positron emission tomographic measurement of acetylcholinesterase activity reeals differential loss of ascending cholinergic systems in Parkinson disease and progressive supranuclear palsy. Ann Neurol. 1999;46:62-69.
165 Gerhard A, Trender-Gerhard I, Turkheimer F, et al. In vivo imaging of miroglial activation with [C]®-PK11195 PET in progressive supranuclear palsy. Mov Disord. 2006;21:89-93.
166 Valls-Sole J. Neurophysiological characterization of parkinsonian syndromes. Neurophysiol Clin. 2000;30:352-367.
167 Rivaud-Pechoux S, Vidailhet M, Gallouedec G, et al. Longitudinal ocular motor study in corticobasal degeneration and progressive supranuclear palsy. Neurology. 2000;54:1029-1032.
168 Kimber J, Mathias CJ, Lees AJ, et al. Physiological, pharmacological and neurohormonal assessment of autonomic function in progressive supranuclear palsy. Brain. 2000;123:1422-1430.
169 Vodusek DB. Sphincter EMG and differential diagnosis of multiple system atrophy. Mov Disord. 2001;16:600-607.
170 Kofler M, Wenning GK, Poewe W, et al. Cortical and brainstem hyperexcitability in a pathologically confirmed case of multiple system atrophy. Mov Disord. 2000;15:362-363.
171 Nieforth KA, Golbe LI. Retrospective study of drug response in 87 patients with progressive supranuclear palsy. Clin Neuropharmacol. 1993;16:338-346.
172 Kompoliti K, Goetz CG, Litvan I, et al. Pharmacological therapy in progressive supranuclear palsy. Arch Neurol. 1998;55:1099-1102.
173 Litvan I, Agid Y, Calne D, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology. 1996;47:1-9.
174 Williams AC, Nutt J, Lakes CR, et al. Actions of bromocriptine in the Shy-Drager and Steele-Richardson-Olszewski syndrome. In: Fixe K, Calne DB, editors. Dopaminergic Ergots and Motor Control. Oxford: Pergamon Press; 1979:271-283.
175 Neophytides A, Lieberman AN, Goldstein M, et al. The use of lisuride, a potent dopamine and serotonin agonist, in the treatment of progressive supranuclear palsy. J Neurol Neurosurg Psychiatry. 1982;45:261-263.
176 Weiner WJ, Minagar A, Shulman LM. Pramipexole in progressive supranuclear palsy. Neurology. 1999;52:873-874.
177 Newman GC. Treatment of progressive supranuclear palsy with tricyclic antidepressants. Neurology. 1985;35:1189-1193.
178 Engel PA. Treatment of progressive supranuclear palsy with amitriptyline: therapeutic and toxic effects. J Am Geriatr Soc. 1996;44:1072-1074.
179 Tamai S, Almeida OP. Nortriptyline for the treatment of depression in progressive supranuclear palsy. J Am Geriatr Soc. 1997;45:1033-1034.
180 Ghika J, Tennis M, Hoffman E, et al. Idazoxan treatment in progressive supranuclear palsy. Neurology. 1991;41:986-991.
181 Rascol O, Sieradzan K, Peyro-Saint-Paul H, et al. Efaroxan, an alpha-2 antagonist, in the treatment of progressive supranuclear palsy. Mov Disord. 1998;13:673-676.
182 Rafal RD, Grimm RJ. Progressive supranuclear palsy: functional analysis of the response to methysergide and antiparkinsonian agents. Neurology. 1981;31:1507-1518.
183 Paulson GW, Lowery HW, Taylor GC. Progressive supranuclear palsy: pneumoencephalography, electronystagmography and treatment with methysergide. Eur Neurol. 1981;20:13-16.
184 Nagasaka T, Togashi S, Amino A, et al. Aniracetam for treatment of patients with progressive supranuclear palsy. Eur Neurol. 1997;37:195-198.
185 Colosimo C, Merello M, Pontieri FE. Amantadine in parkinsonian patients unresponsive to levodopa: a pilot study. J Neurol. 1996;243:422-425.
186 Golbe LI, Langston JW, Shoulson I. Selegiline and Parkinson disease. Protective and symptomatic considerations. Drugs. 1990;39:646-651.
187 Barclay CL, Duff J, Sandor P, et al. Limited usefulness of electroconvulsive therapy in progressive supranuclear palsy. Neurology. 1996;46:1284-1286.
188 Hauser RA, Trehan R. Initial experience with electroconvulsive therapy for progressive supranuclear palsy. Mov Disord. 1994;9:467-469.
189 Koller WC, Morantz R, Vetere-Overfield B, et al. Autologous adrenal medullary transplant in progressive supranuclear palsy. Neurology. 1989;39:1066-1068.
190 Waxman MJ, Morantz RA, Koller WC, et al. High incidence of cardiopulmonary complications associated with implantation of adrenal medullary tissue into the caudate nucleus in patients with advanced neurologic disease. Crit Care Med. 1991;19:181-186.
191 Daniele A, Moro E, Bentivoglio AR. Zolpidem in progressive supranuclear palsy. N Engl J Med. 1999;341:543-544.
192 Polo KB, Jabbari B. Botulinum toxin-A improves the rigidity of progressive supranuclear palsy. Ann Neurol. 1994;35:237-239.
193 Piccione F, Mancini E, Tonin P, et al. Botulinum toxin treatment of apraxia of eyelid opening in progressive supranuclear palsy: report of two cases. Arch Phys Med Rehabil. 1997;78:525-529.
194 Barclay CL, Lang AE. Dystonia in progressive supranuclear palsy. J Neurol Neurosurg. Psychiat. 1997;62:352-356.
195 Müller J, Wenning GK, Wissel J, et al. Botulinum toxin treatment in atypical parkinsonian disorders associated with disabling focal dystonia. J Neurol. 2002;249:300-304.
196 Foster NL, Aldrich MS, Bluemlein L, et al. Failure of cholinergic agonist RS-86 to improve cognition and movement in PSP despite effects on sleep. Neurology. 1989;39:257-261.
197 Litvan I, Blesa R, Clark K, et al. Pharmacological evaluation of the cholinergic system in progressive supranuclear palsy. Ann Neurol. 1994;36:55-61.
198 Fabbrini G, Barbanti P, Bonifati V, et al. Donepezil in the treatment of progressive supranuclear palsy. Acta Neurol Scand. 2001;103:123-125.
199 Newman GC. Treatment of progressive supranuclear palsy with tricyclic antidepressants. Neurology. 1985;35:1189-1193.
200 Hughes AJ, Daniel SE, Ben Shlomo Y, et al. The accuracy of diagnosis of parkinsonian syndromes in a specialist movement disorder service. Brain. 2002;125:861-870.
201 Wenning GK, Litvan I, Jankovic J, et al. Natural history and survival of 14 patients with corticobasal degeneration confirmed at postmortem examination. J Neurol Neurosurg Psychiatry. 1998;64:184-189.
202 Rinne JO, Lee MS, Thompson PD, Marsden CD. Corticobasal degeneration: a clinical study of 36 cases. Brain. 1994;117:1183-1196.
203 Cummings JL, Litvan I. Neuropsychiatric aspects of corticobasal degeneration. Adv Neurol. 2000;82:147-152.
204 Kertesz A, Martinez-Lage P, Davidson W, et al. The corticobasal degeneration syndrome overlaps progressive aphasia and frontotemporal dementia. Neurology. 2000;55:1368-1375.
205 Litvan I. Progressive supranuclear palsy and corticobasal degeneration. Bailliere’s Clin Neurol. 1997;6:167-185.
206 Kumar R, Bergeron C, Lang AE. Corticobasal degeneration. In: Jankovic J, Tolosa E, editors. Parkinson Disease and Movement Disorders. 4th ed. Philadelphia: Lippincott, Williams & Wilkins; 2002:185-198.
207 Brown J, Lantos PL, Roques P, et al. Familial dementia with swollen achromatic neurons and corticobasal inclusion bodies: a clinical and pathological study. J Neurol Sci. 1996;135:21-30.
208 Houlden H, Baker M, Morris HR, et al. Corticobasal degeneration and progressive supranuclear palsy share a common tau haplotype. Neurology. 2001;56:1702-1706.
209 Soliveri P, Monza P, Paridi D, et al. Cognitive and magnetic resonance imaging aspects of corticobasal degeneration and progressive supranuclear palsy. Neurology. 2000;54:1878.
210 Brooks DJ. Functional imaging studies in corticobasal degeneration. Adv Neurol. 2000;82:209-215.
211 Gerhard A, Watts J, Trender-Gerhard I, et al. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in corticobasal degeneration. Mov Disord. 2004;19:1221-1226.
211a Kompoliti K, Goetz CG, Boeve BF, Maraganore DM, et al. Clinical presentation and pharmacological therapy in corticobasal degeneration. Arch Neurol. 1998;55(7):957-961.
212 Kompoliti K, Goetz CG. Therapeutic approaches. Adv Neurol. 2000;82:217-221.
213 Vanek ZF, Jankovic J. Dystonia in corticobasal degeneration. Adv Neurol. 2000;82:61-67.






