120e |
Carcinoma of Unknown Primary |
Carcinoma of unknown primary (CUP) is a biopsy-proven malignancy for which the anatomic site of origin remains unidentified after an intensive search. CUP is one of the 10 most frequently diagnosed cancers worldwide, accounting for 3–5% of all cancers. Most investigators limit CUP to epithelial cancers and do not include lymphomas, metastatic melanomas, and metastatic sarcomas because these cancers have specific histology- and stage-based treatments that guide management.
The emergence of sophisticated imaging, robust immunohistochemistry (IHC), and genomic and proteomic tools has challenged the “unknown” designation. Additionally, effective targeted therapies in several cancers have moved the paradigm from empiricism to considering a personalized approach to CUP management. The reasons cancers present as CUP remain unclear. One hypothesis is that the primary tumor either regresses after seeding the metastasis or remains so small that it is not detected. It is possible that CUP falls on the continuum of cancer presentation where the primary has been contained or eliminated by the natural body defenses. Alternatively, CUP may represent a specific malignant event that results in an increase in metastatic spread or survival relative to the primary. Whether the CUP metastases truly define a clone that is genetically and phenotypically unique to this diagnosis remains to be determined.
CUP BIOLOGY
Studies looking for unique signature abnormalities in CUP tumors have not been positive. Abnormalities in chromosomes 1 and 12 and other complex cytogenetic abnormalities have been reported. Aneuploidy has been described in 70% of CUP patients with metastatic adenocarcinoma or undifferentiated carcinoma. The overexpression of various genes, including Ras, bcl-2 (40%), her-2 (11%), and p53 (26–53%), has been studied in CUP samples, but they have no effect on response to therapy or survival. The extent of angiogenesis in CUP relative to that in metastases from known primaries has also been evaluated, but no consistent findings have emerged. Using the Sequenom (SQM) Massarray platform, a study in consecutive CUP patients showed that the overall mutational rate was surprisingly low (18%). No “new” low-frequency mutations were found using a panel of mutations involving the P13K/AKT pathway, MEK pathway, receptors, and downstream effectors. Nevertheless, it is possible that newer “deep sequencing” techniques in select patients may yield consistent abnormalities.
CLINICAL EVALUATION
Initial CUP evaluation has two goals: search for the primary tumor based on pathologic evaluation of the metastases and determine the extent of disease. Obtaining a thorough medical history from CUP patients is essential, including paying particular attention to previous surgeries, removed lesions, and family medical history to assess potential hereditary cancers. Adequate physical examination, including a digital rectal examination in men and breast and pelvic examinations in women, should be performed based on clinical presentation.
Role of Serum Tumor Markers and Cytogenetics Most tumor markers, including CEA, CA-125, CA 19-9, and CA 15-3, when elevated, are nonspecific and not helpful in determining the primary tumor site. Men who present with adenocarcinoma and osteoblastic metastasis should undergo a prostate-specific antigen (PSA) test. In patients with undifferentiated or poorly differentiated carcinoma (especially with a midline tumor), elevated β-human chorionic gonadotropin (β-hCG) and α fetoprotein (AFP) levels suggest the possibility of an extragonadal germ cell (testicular) tumor. With the availability of IHC, cytogenetic studies are rarely needed.
Role of Imaging Studies In the absence of contraindications, a baseline IV contrast computed tomography (CT) scan of the chest, abdomen, and pelvis is the standard of care. This helps to search for the primary tumor, evaluate the extent of disease, and select the most accessible biopsy site. Older studies suggested that the primary tumor site is detected in 20–35% of patients who undergo a CT scan of the abdomen and pelvis, although by current definition, these patients do not have CUP. These studies also suggest a latent primary tumor prevalence of 20%; with more sophisticated imaging, this has decreased to ≤5% today.
Mammography should be performed in all women who present with metastatic adenocarcinoma, especially in those with adenocarcinoma and isolated axillary lymphadenopathy. Magnetic resonance imaging (MRI) of the breast is a follow-up modality in patients with axillary adenopathy and suspected occult primary breast carcinoma following a negative mammography and ultrasound. The results of these imaging modalities can influence surgical management; a negative breast MRI result predicts a low tumor yield at mastectomy.
A conventional workup for a squamous cell carcinoma and cervical CUP (neck lymphadenopathy with no known primary tumor) includes a CT scan or MRI and invasive studies, including indirect and direct laryngoscopy, bronchoscopy, and upper endoscopy. Ipsilateral (or bilateral) staging tonsillectomy has been recommended for these patients. 18-Fluorodeoxyglucose positron emission tomography (18-FDG-PET) scans are useful in this patient population and may help guide the biopsy; determine the extent of disease; facilitate the appropriate treatment, including planning radiation fields; and help with disease surveillance. A smaller radiation field encompassing the primary (when found) and metastatic adenopathy decreases the risk of chronic xerostomia. Several studies have evaluated the utility of PET in patients with squamous cervical CUP, and head and neck primary tumors were identified in ~21–30%.
The diagnostic contribution of PET to the evaluation of other CUP (outside of the neck adenopathy indication) remains controversial and is not routinely recommended. PET-CT can be helpful for patients who are candidates for surgical intervention for solitary metastatic disease because the presence of disease outside the primary site may affect surgical planning.
Invasive studies, including upper endoscopy, colonoscopy, and bronchoscopy, should be limited to symptomatic patients or those with laboratory, imaging, or pathologic abnormalities that suggest that these techniques will result in a high yield in finding a primary cancer.
Role of Pathologic Studies A detailed pathologic examination of the most accessible biopsied tissue specimen is mandatory in CUP patients. Pathologic evaluation typically consists of hematoxylin and eosin stains and immunohistochemical tests.
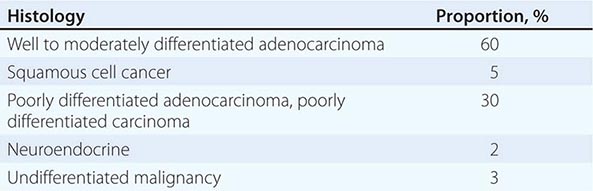
LIGHT MICROSCOPY EVALUATION Adequate tissue obtained preferably by excisional biopsy or core-needle biopsy (instead of only a fine-needle aspiration) is stained with hematoxylin and eosin and subjected to light microscopic examination. On light microscopy, 60–65% of CUP is adenocarcinoma, and 5% is squamous cell carcinoma. The remaining 30–35% is poorly differentiated adenocarcinoma, poorly differentiated carcinoma, or poorly differentiated neoplasm. A small percentage of lesions are diagnosed as neuroendocrine cancers (2%), mixed tumors (adenosquamous or sarcomatoid carcinomas), or undifferentiated neoplasms (Table 120e-1).
|
MAJOR HISTOLOGIES IN CARCINOMA OF UNKNOWN PRIMARY |
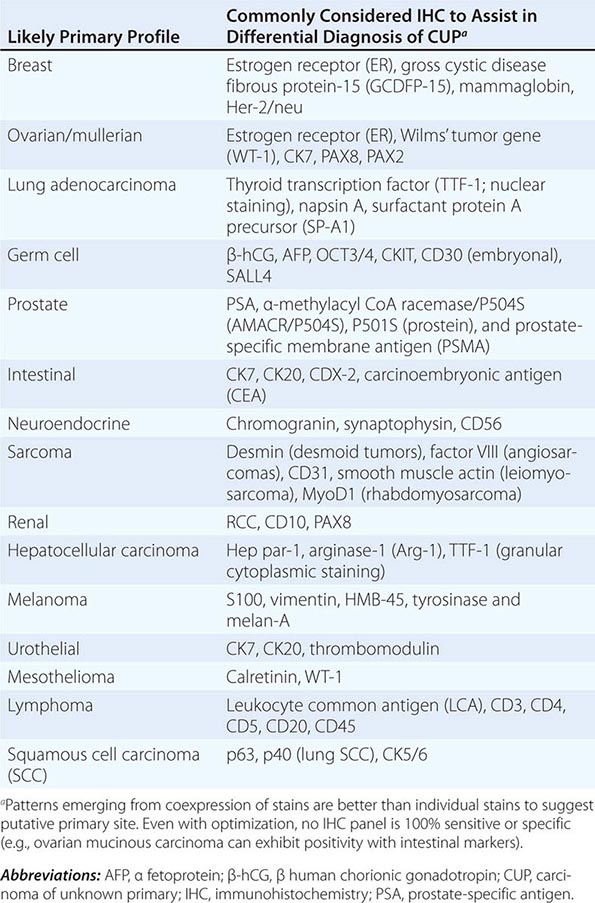
ROLE OF IMMUNOHISTOCHEMICAL ANALYSIS Immunohistochemical stains are peroxidase-labeled antibodies against specific tumor antigens that are used to define tumor lineage. The number of available immunohistochemical stains is ever-increasing. However, in CUP cases, more is not necessarily better, and immunohistochemical stains should be used in conjunction with the patient’s clinical presentation and imaging studies to select the best therapy. Communication between the clinician and pathologist is essential. No stain is 100% specific, and overinterpretation should be avoided. PSA and thyroglobulin tissue markers, which are positive in prostate and thyroid cancer, respectively, are the most specific of the current marker panel. However, these cancers rarely present as CUP, so the yield of these tests may be low. Fig. 120e-1 delineates a simple algorithm for immunohistochemical staining in CUP cases. Table 120e-2 lists additional tests that may be useful to further define the tumor lineage. A more comprehensive algorithm may improve the diagnostic accuracy but can make the process complex. With the use of immunohistochemical markers, electron microscopic analysis, which is time-consuming and expensive, is rarely needed.
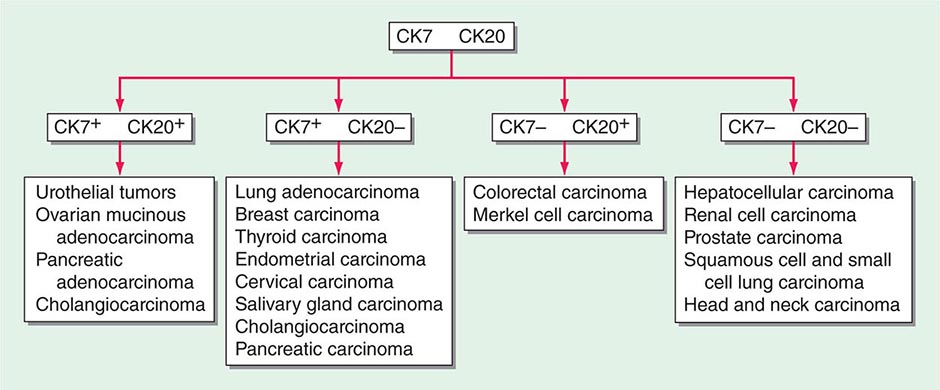
FIGURE 120e-1 Approach to cytokeratin (CK7 and CK20) markers used in adenocarcinoma of unknown primary.
|
SELECT IMMUNOHISTOCHEMICAL STAINS USEFUL IN THE DIAGNOSIS OF CARCINOMA OF UNKNOWN PRIMARY (CUP) |
There are >20 subtypes of cytokeratin (CK) intermediate filaments with different molecular weights and differential expression in various cell types and cancers. Monoclonal antibodies to specific CK subtypes have been used to help classify tumors according to their site of origin; commonly used CK stains in adenocarcinoma CUP are CK7 and CK20. CK7 is found in tumors of the lung, ovary, endometrium, breast, and upper gastrointestinal tract including pancreaticobiliary cancers, whereas CK20 is normally expressed in the gastrointestinal epithelium, urothelium, and Merkel cells. The nuclear CDX-2 transcription factor, which is the product of a homeobox gene necessary for intestinal organogenesis, is often used to aid in the diagnosis of gastrointestinal adenocarcinomas.
Thyroid transcription factor 1 (TTF-1) nuclear staining is typically positive in lung and thyroid cancers. Approximately 68% of adenocarcinomas and 25% of squamous cell lung cancers stain positive for TTF-1, which helps differentiate a lung primary tumor from metastatic adenocarcinoma in a pleural effusion, the mediastinum, or the lung parenchyma.
Gross cystic disease fibrous protein-15, a 15-kDa monomer protein, is a marker of apocrine differentiation that is detected in 62–72% of breast carcinomas. UROIII, high-molecular-weight cytokeratin, thrombomodulin, and CK20 are the markers used to diagnose lesions of urothelial origin.
IHC performs the best when used in groups that give rise to patterns that are strongly indicative of certain profiles. For example, the TTF-1/CK7+ and CK20+/CDX-2+/CK7– phenotypes have been reported as very suggestive of lung and lower gastrointestinal cancer profiles, respectively, although these patterns have not been validated prospectively in the absence of a primary cancer. IHC is not without its limitations; several factors affect tissue antigenicity (antigen retrieval, specimen processing, and fixation), interpretation of stains in tumor (nuclear, cytoplasmic, membrane) versus normal tissue, inter- and intraobserver variability, and tissue heterogeneity and inadequacy (given small biopsy sizes). Communication with the pathologist is critical to determine if additional tissue will be beneficial in the pathologic evaluation.
ROLE OF TISSUE OF ORIGIN MOLECULAR PROFILING In the absence of a known primary, developing therapeutic strategies for CUP is challenging. The current diagnostic yield with imaging and immunochemistry is ~20–30% for CUP patients. The use of gene expression studies holds the promise of substantially increasing this yield. Gene expression profiles are most commonly generated using quantitative reverse transcriptase polymerase chain reaction (RT-PCR) or DNA microarray.
Neural network programs have been used to develop predictive algorithms from the gene expression profiles. Typically, a training set of gene profiles from known cancers (preferably from metastatic sites) is used to train the software. The program can then be used to predict the putative origin of a test tumor and presumably of true CUP. Comprehensive gene expression databases that have become available for common malignancies may also be useful in CUP. These approaches have been effective in testing against known primary cancers and their metastases.
mRNA- or microRNA-based tissue of origin molecular profiling assays have been studied in prospective and retrospective CUP trials. Most of the CUP studies have evaluated assay performance, although the challenge with validating the accuracy of an assay for CUP is that, by definition, the primary cancer diagnosis cannot be verified. Thus, current estimates of tissue of origin test accuracy have relied on indirect metrics including comparison with IHC, clinical presentation, and appearance of latent primaries. Using these measures, the assays suggest a plausible primary in ~70% of patients studied. The only outcomes-based study is a single-arm study reporting a median survival of 12.5 months for patients who received assay-directed site-specific therapy. Firm conclusions of therapeutic impact cannot be drawn from this study given the nonrandomized design, statistical biases, confounding variables including use of subsequent lines of (empiric) therapy, and the heterogeneity of the CUP cancers. Additional studies are needed to better understand the clinical influence of tissue of origin profiling tools and how these assays complement IHC and help guide therapy.
TREATMENT CARCINOMA OF UNKNOWN PRIMARY
GENERAL CONSIDERATIONS
The treatment of CUP continues to evolve, albeit slowly. The median survival duration of most patients with disseminated CUP is ~6–10 months. Systemic chemotherapy is the primary treatment modality in most patients with disseminated disease, but the careful integration of surgery, radiation therapy, and even periods of observation is important in the overall management of this condition (Figs. 120e-2 and 120e-3). Prognostic factors include performance status, site and number of metastases, response to chemotherapy, and serum lactate dehydrogenase (LDH) levels. Culine and colleagues developed a prognostic model using performance status and serum LDH levels, which allowed the assignment of patients into two subgroups with divergent outcomes. Future prospective trials using this prognostic model are warranted. Clinically, some CUP diagnoses fall into a favorable prognostic subset. Others, including those with disseminated CUP, do not and have a more unfavorable prognosis.
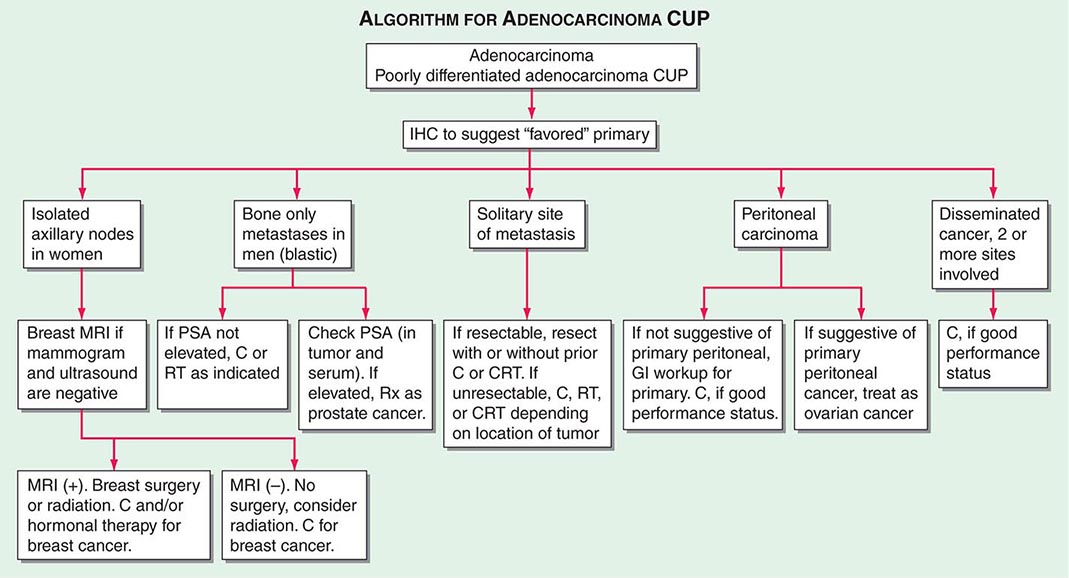
FIGURE 120e-2 Treatment algorithm for adenocarcinoma and poorly differentiated adenocarcinoma of unknown primary (CUP). C, chemotherapy; CRT, chemoradiation; GI, gastrointestinal; IHC, immunohistochemistry; MRI, magnetic resonance imaging; PSA, prostate-specific antigen; RT, radiation.
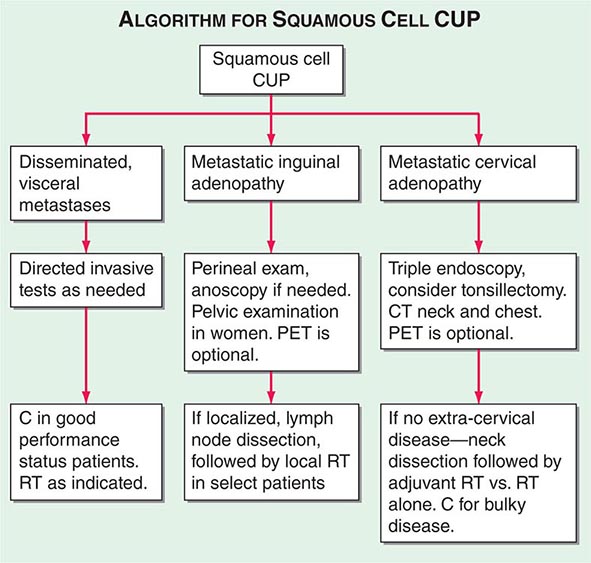
FIGURE 120e-3 Treatment algorithm for squamous cell carcinoma of unknown primary (CUP). C, chemotherapy; CT, computed tomography; PET, positron emission tomography; RT, radiation.
TREATMENT OF FAVORABLE CUP SUBSETS
Women with Isolated Axillary Adenopathy Women with isolated axillary adenopathy with adenocarcinoma or carcinoma are usually treated for stage II or III breast cancer based on pathologic findings. These patients should undergo a breast MRI if mammogram and ultrasound are negative. Radiation therapy to the ipsilateral breast is indicated if the breast MRI is positive. Chemotherapy and/or hormonal therapy are indicated based on patient’s age (premenopausal or postmenopausal), nodal disease bulk, and hormone receptor status (Chap. 108). It is important to verify that the pathology suggests a breast cancer profile (morphology, immunohistochemical breast markers including estrogen receptor, mammoglobin, GCDFP-15, HER-2 gene expression) before embarking on a breast cancer therapeutic program.
WOMEN WITH PERITONEAL CARCINOMATOSIS
The term primary peritoneal papillary serous carcinoma (PPSC) has been used to describe CUP with carcinomatosis with the pathologic and laboratory (elevated CA-125 antigen) characteristics of ovarian cancer but no ovarian primary tumor identified on transvaginal sonography or laparotomy. Studies suggest that ovarian cancer and PPSC, which are both of müllerian origin, have similar gene expression profiles. Similar to patients with ovarian cancer, patients with PPSC are candidates for cytoreductive surgery, followed by adjuvant taxane- and platinum-based chemotherapy. In one retrospective study of 258 women with peritoneal carcinomatosis who had undergone cytoreductive surgery and chemotherapy, 22% of patients had a complete response to chemotherapy; the median survival duration was 18 months (range 11–24 months). However, not all peritoneal carcinomatosis in women is PPSC. Careful pathologic evaluation can help diagnose a colon cancer profile (CDX-2+, CK-20+, CK7–) or a pancreaticobiliary cancer or even a mislabeled peritoneal mesothelioma (calretinin positive).
POORLY DIFFERENTIATED CARCINOMA WITH MIDLINE ADENOPATHY
Men with poorly differentiated or undifferentiated carcinoma that presents as a midline adenopathy should be evaluated for extragonadal germ cell malignancy. If diagnosed and treated as such, they often experience a good response to treatment with platinum-based combination chemotherapy. Response rates of >50% have been noted, and long-term survival rates of 10–15% long have been reported. Older patients (especially smokers) who present with mediastinal adenopathy are more likely to have a lung or head-and-neck cancer profile.
NEUROENDOCRINE CARCINOMA
Low-grade neuroendocrine carcinoma often has an indolent course, and treatment decisions are based on symptoms and tumor bulk. Urine 5-HIAA and serum chromogranin may be elevated and can be followed as markers. Often the patient is treated with somatostatin analogues alone for hormone-related symptoms (diarrhea, flushing, nausea). Specific local therapies or systemic therapy would only be indicated if the patient is symptomatic with local pain secondary to significant growth of the metastasis or the hormone-related symptoms are not controlled with endocrine therapy. Patients with high-grade neuroendocrine carcinoma are treated as having small-cell lung cancer and are responsive to chemotherapy; 20–25% show a complete response, and up to 10% patients survive more than 5 years.
SQUAMOUS CELL CARCINOMA PRESENTING AS NECK ADENOPATHY
Patients with early-stage squamous cell carcinoma involving the cervical lymph nodes are candidates for node dissection and radiation therapy, which can result in long-term survival. The role of chemotherapy in these patients is undefined, although chemoradiation therapy or induction chemotherapy is often used and is beneficial in bulky N2/N3 lymph node disease.
SOLITARY METASTATIC SITE
Patients with solitary metastases can also experience good treatment outcomes. Some patients who present with locoregional disease are candidates for aggressive trimodality management; both prolonged disease-free interval and occasionally cure are possible.
MEN WITH BLASTIC SKELETAL METASTASES AND ELEVATED PSA
Blastic bone-only metastasis is a rare presentation, and elevated serum PSA or tumor staining with PSA may provide confirmatory evidence of prostate cancer in these patients. Those with elevated levels are candidates for hormonal therapy for prostate cancer, although it is important to rule out other primary tumors (lung most common).
MANAGEMENT OF DISSEMINATED CUP
Patients who present with liver, brain, and adrenal metastatic disease usually have a poor prognosis. Patients with nonserous papillary primary peritoneal carcinomatosis can have a large differential diagnosis, which is mainly of gastrointestinal profile and includes gastric, appendiceal, colon, and pancreaticobiliary profiles.
Traditionally, platinum-based combination chemotherapy regimens have been used to treat CUP. Several broadly used regimens have been studied in the last two decades; these include paclitaxel-carboplatin, gemcitabine-cisplatin, gemcitabine-oxaliplatin, and irinotecan and fluoropyrimidine-based therapies. These chemotherapeutic agents used as empiric regimens have shown a response rate of 25–40%, and their use obtains median survival times of 6–13 months.
Outside of favorable subsets, there is a small group of patients with a “definitive” IHC. These patients usually have a single diagnosis based on their clinicopathologic presentation and are often treated for the putative primary tumor. This does not guarantee a response, although it increases the probability of response when select drugs are chosen from a class of drugs known to work in that cancer type. Patients who do not fall into those categories are candidates for broad-spectrum platinum-based regimens, clinical trials, and additional trial-based genomic and proteomic tests. Today, we do not have effective drugs for several CUP cancer profiles, and treatments overlap for some cancers. However, as novel therapies are developed for additional known cancers, tissue of origin and assessment of molecular features of the tumor will be important and might direct more selective treatment.
SUMMARY
Patients with CUP should undergo a directed diagnostic search for the primary tumor on the basis of clinical and pathologic data. Subsets of patients have prognostically favorable disease, as defined by clinical or histologic criteria, and may substantially benefit from aggressive treatment and expect prolonged survival. However, for most patients who present with advanced CUP, the prognosis remains poor with early resistance to available cytotoxic therapy. The current focus has shifted away from empirical chemotherapeutic trials to understanding the metastatic phenotype, tissue of origin profiling, and evaluating molecular targets in CUP patients.
121 |
Paraneoplastic Syndromes: Endocrinologic/Hematologic |
Neoplastic cells can produce a variety of products that can stimulate hormonal, hematologic, dermatologic, and neurologic responses. Paraneoplastic syndromes is the term used to refer to the disorders that accompany benign or malignant tumors but are not directly related to mass effects or invasion. Tumors of neuroendocrine origin, such as small-cell lung carcinoma (SCLC) and carcinoids, produce a wide array of peptide hormones and are common causes of paraneoplastic syndromes. However, almost every type of tumor has the potential to produce hormones or to induce cytokine and immunologic responses. Careful studies of the prevalence of paraneoplastic syndromes indicate that they are more common than is generally appreciated. The signs, symptoms, and metabolic alterations associated with paraneoplastic disorders may be overlooked in the context of a malignancy and its treatment. Consequently, atypical clinical manifestations in a patient with cancer should prompt consideration of a paraneoplastic syndrome. The most common endocrinologic and hematologic syndromes associated with underlying neoplasia will be discussed here.
ENDOCRINE PARANEOPLASTIC SYNDROMES
Etiology Hormones can be produced from eutopic or ectopic sources. Eutopic refers to the expression of a hormone from its normal tissue of origin, whereas ectopic refers to hormone production from an atypical tissue source. For example, adrenocorticotropic hormone (ACTH) is expressed eutopically by the corticotrope cells of the anterior pituitary, but it can be expressed ectopically in SCLC. Many hormones are produced at low levels from a wide array of tissues in addition to the classic endocrine source. Thus, ectopic expression is often a quantitative change rather than an absolute change in tissue expression. Nevertheless, the term ectopic expression is firmly entrenched and conveys the abnormal physiology associated with hormone production by neoplastic cells. In addition to high levels of hormones, ectopic expression typically is characterized by abnormal regulation of hormone production (e.g., defective feedback control) and peptide processing (resulting in large, unprocessed precursors).
A diverse array of molecular mechanisms has been suggested to cause ectopic hormone production. In rare instances, genetic rearrangements explain aberrant hormone expression. For example, translocation of the parathyroid hormone (PTH) gene can result in high levels of PTH expression in tissues other than the parathyroid gland because the genetic rearrangement brings the PTH gene under the control of atypical regulatory elements. A related phenomenon is well documented in many forms of leukemia and lymphoma, in which somatic genetic rearrangements confer a growth advantage and alter cellular differentiation and function (Chap. 134). Although genetic rearrangements cause selected cases of ectopic hormone production, this mechanism is rare, as many tumors are associated with excessive production of numerous peptides. Cellular dedifferentiation probably underlies most cases of ectopic hormone production. Many cancers are poorly differentiated, and certain tumor products, such as human chorionic gonadotropin (hCG), parathyroid hormone–related protein (PTHrP), and α fetoprotein, are characteristic of gene expression at earlier developmental stages. In contrast, the propensity of certain cancers to produce particular hormones (e.g., squamous cell carcinomas produce PTHrP) suggests that dedifferentiation is partial or that selective pathways are derepressed. These expression profiles probably reflect epigenetic modifications that alter transcriptional repression, microRNA expression, and other pathways that govern cell differentiation.
In SCLC, the pathway of differentiation has been relatively well defined. The neuroendocrine phenotype is dictated in part by the basic-helix-loop-helix (bHLH) transcription factor human achaete-scute homologue 1 (hASH-1), which is expressed at abnormally high levels in SCLC associated with ectopic ACTH. The activity of hASH-1 is inhibited by hairy enhancer of split 1 (HES-1) and by Notch proteins, which also are capable of inducing growth arrest. Thus, abnormal expression of these developmental transcription factors appears to provide a link between cell proliferation and differentiation.
Ectopic hormone production would be merely an epiphenomenon associated with cancer if it did not result in clinical manifestations. Excessive and unregulated production of hormones such as ACTH, PTHrP, and vasopressin can lead to substantial morbidity and complicate the cancer treatment plan. Moreover, the paraneoplastic endocrinopathies may be a presenting clinical feature of underlying malignancy and prompt the search for an unrecognized tumor.
A large number of paraneoplastic endocrine syndromes have been described, linking overproduction of particular hormones with specific types of tumors. However, certain recurring syndromes emerge from this group (Table 121-1). The most common paraneoplastic endocrine syndromes include hypercalcemia from overproduction of PTHrP and other factors, hyponatremia from excess vasopressin, and Cushing’s syndrome from ectopic ACTH.
|
PARANEOPLASTIC SYNDROMES CAUSED BY ECTOPIC HORMONE PRODUCTION |
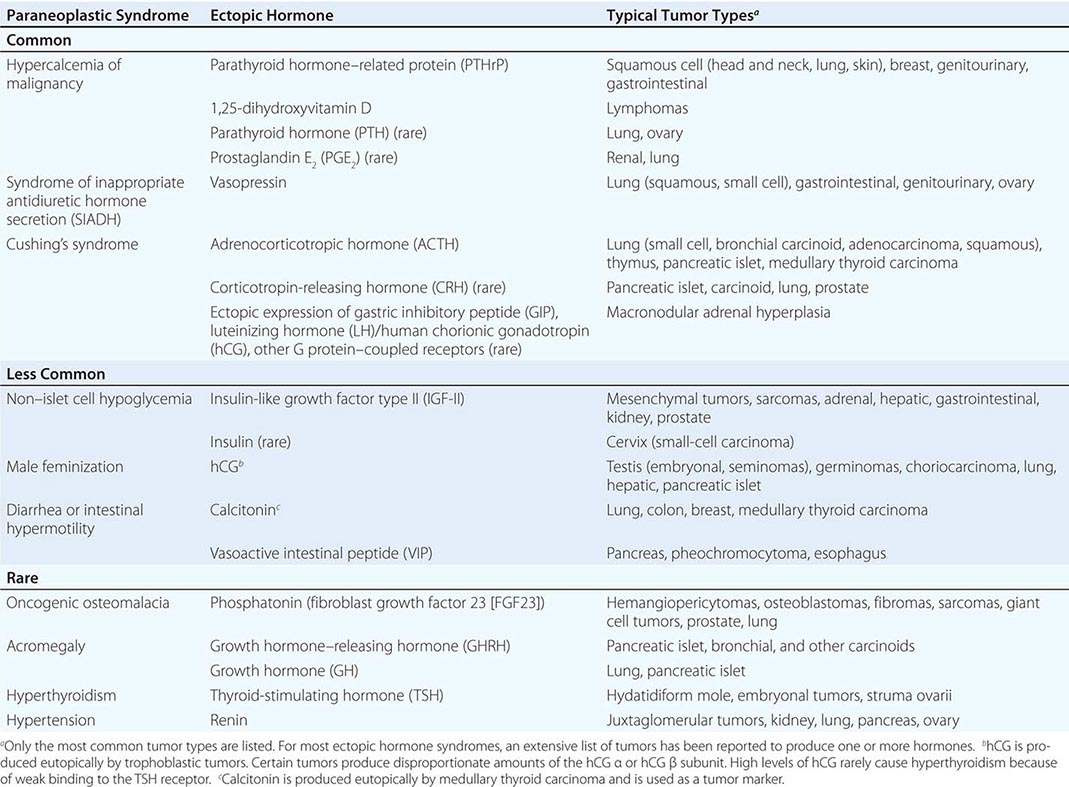
HYPERCALCEMIA CAUSED BY ECTOPIC PRODUCTION OF PTHrP
(See also Chap. 424)
Etiology Humoral hypercalcemia of malignancy (HHM) occurs in up to 20% of patients with cancer. HHM is most common in cancers of the lung, head and neck, skin, esophagus, breast, and genitourinary tract and in multiple myeloma and lymphomas. There are several distinct humoral causes of HHM, but it is caused most commonly by overproduction of PTHrP. In addition to acting as a circulating humoral factor, bone metastases (e.g., breast, multiple myeloma) may produce PTHrP, leading to local osteolysis and hypercalcemia. PTHrP may also affect the initiation and progression of tumors by acting through pro-survival and chemokine pathways.
PTHrP is structurally related to PTH and binds to the PTH receptor, explaining the similar biochemical features of HHM and hyperparathyroidism. PTHrP plays a key role in skeletal development and regulates cellular proliferation and differentiation in other tissues, including skin, bone marrow, breast, and hair follicles. The mechanism of PTHrP induction in malignancy is incompletely understood; however, tumor-bearing tissues commonly associated with HHM normally produce PTHrP during development or cell renewal. PTHrP expression is stimulated by hedgehog pathways and Gli transcription factors that are active in many malignancies. Transforming growth factor β (TGF-β), which is produced by many tumors, also stimulates PTHrP, in part by activating the Gli pathway. Mutations in certain oncogenes, such as Ras, also can activate PTHrP expression. In adult T cell lymphoma, the transactivating Tax protein produced by human T cell lymphotropic virus 1 (HTLV-1) stimulates PTHrP promoter activity. Metastatic lesions to bone are more likely to produce PTHrP than are metastases in other tissues, suggesting that bone produces factors (e.g., TGF-β) that enhance PTHrP production or that PTHrP-producing metastases have a selective growth advantage in bone. PTHrP activates the pro-survival AKT pathway and the chemokine receptor CXCR4. Thus, PTHrP production can be stimulated by mutations in oncogenes, altered expression of viral or cellular transcription factors, and local growth factors. In addition to its role in HHM, the PTHrP pathway may also provide a potential target for therapeutic intervention to impede cancer growth.
Another relatively common cause of HHM is excess production of 1,25-dihydroxyvitamin D. Like granulomatous disorders associated with hypercalcemia, lymphomas can produce an enzyme that converts 25-hydroxyvitamin D to the more active 1,25-dihydroxyvitamin D, leading to enhanced gastrointestinal calcium absorption. Other causes of HHM include tumor-mediated production of osteolytic cytokines and inflammatory mediators.
Clinical Manifestations The typical presentation of HHM is a patient with a known malignancy who is found to be hypercalcemic on routine laboratory tests. Less often, hypercalcemia is the initial presenting feature of malignancy. Particularly when calcium levels are markedly increased (>3.5 mmol/L [>14 mg/dL]), patients may experience fatigue, mental status changes, dehydration, or symptoms of nephrolithiasis.
Diagnosis Features that favor HHM, as opposed to primary hyperparathyroidism, include known malignancy, recent onset of hypercalcemia, and very high serum calcium levels. Like hyperparathyroidism, hypercalcemia caused by PTHrP is accompanied by hypercalciuria and hypophosphatemia. Patients with HHM typically have metabolic alkalosis rather than hyperchloremic acidosis, as is seen in hyperparathyroidism. Measurement of PTH is useful to exclude primary hyperparathyroidism; the PTH level should be suppressed in HHM. An elevated PTHrP level confirms the diagnosis, and it is increased in ~80% of hypercalcemic patients with cancer. 1,25-Dihydroxyvitamin D levels may be increased in patients with lymphoma.
ECTOPIC VASOPRESSIN: TUMOR-ASSOCIATED SIADH
(See also Chap. 63)
Etiology Vasopressin is an antidiuretic hormone normally produced by the posterior pituitary gland. Ectopic vasopressin production by tumors is a common cause of the syndrome of inappropriate antidiuretic hormone (SIADH), occurring in at least half of patients with SCLC. SIADH also can be caused by a number of nonneoplastic conditions, including central nervous system (CNS) trauma, infections, and medications (Chap. 404). Compensatory responses to SIADH, such as decreased thirst, may mitigate the development of hyponatremia. However, with prolonged production of excessive vasopressin, the osmostat controlling thirst and hypothalamic vasopressin secretion may become reset. In addition, intake of free water, orally or intravenously, can quickly worsen hyponatremia because of reduced renal diuresis.
Tumors with neuroendocrine features, such as SCLC and carcinoids, are the most common sources of ectopic vasopressin production, but it also occurs in other forms of lung cancer and with CNS lesions, head and neck cancer, and genitourinary, gastrointestinal, and ovarian cancers. The mechanism of activation of the vasopressin gene in these tumors is unknown but often involves concomitant expression of the adjacent oxytocin gene, suggesting derepression of this locus.
Clinical Manifestations Most patients with ectopic vasopressin secretion are asymptomatic and are identified because of the presence of hyponatremia on routine chemistry testing. Symptoms may include weakness, lethargy, nausea, confusion, depressed mental status, and seizures. The severity of symptoms reflects the rapidity of onset as well as the severity of hyponatremia. Hyponatremia usually develops slowly but may be exacerbated by the administration of IV fluids or the institution of new medications.
Diagnosis The diagnostic features of ectopic vasopressin production are the same as those of other causes of SIADH (Chaps. 63 and 404). Hyponatremia and reduced serum osmolality occur in the setting of an inappropriately normal or increased urine osmolality. Urine sodium excretion is normal or increased unless volume depletion is present. Other causes of hyponatremia should be excluded, including renal, adrenal, or thyroid insufficiency. Physiologic sources of vasopressin stimulation (CNS lesions, pulmonary disease, nausea), adaptive circulatory mechanisms (hypotension, heart failure, hepatic cirrhosis), and medications, including many chemotherapeutic agents, also should be considered as possible causes of hyponatremia. Vasopressin measurements are not usually necessary to make the diagnosis.
CUSHING’S SYNDROME CAUSED BY ECTOPIC ACTH PRODUCTION
(See also Chap. 406)
Etiology Ectopic ACTH production accounts for 10–20% of cases of Cushing’s syndrome. The syndrome is particularly common in neuroendocrine tumors. SCLC is the most common cause of ectopic ACTH, followed by bronchial and thymic carcinoids, islet cell tumors, other carcinoids, and pheochromocytomas. Ectopic ACTH production is caused by increased expression of the proopiomelanocortin (POMC) gene, which encodes ACTH, along with melanocyte-stimulating hormone (MSH), β lipotropin, and several other peptides. In many tumors, there is abundant but aberrant expression of the POMC gene from an internal promoter, proximal to the third exon, which encodes ACTH. However, because this product lacks the signal sequence necessary for protein processing, it is not secreted. Increased production of ACTH arises instead from less abundant, but unregulated, POMC expression from the same promoter site used in the pituitary. However, because the tumors lack many of the enzymes needed to process the POMC polypeptide, it is typically released as multiple large, biologically inactive fragments along with relatively small amounts of fully processed, active ACTH.
Rarely, corticotropin-releasing hormone (CRH) is produced by pancreatic islet cell tumors, SCLC, medullary thyroid cancer, carcinoids, or prostate cancer. When levels are high enough, CRH can cause pituitary corticotrope hyperplasia and Cushing’s syndrome. Tumors that produce CRH sometimes also produce ACTH, raising the possibility of a paracrine mechanism for ACTH production.
A distinct mechanism for ACTH-independent Cushing’s syndrome involves ectopic expression of various G protein–coupled receptors in the adrenal nodules. Ectopic expression of the gastric inhibitory peptide (GIP) receptor is the best-characterized example of this mechanism. In this case, meals induce GIP secretion, which inappropriately stimulates adrenal growth and glucocorticoid production.
Clinical Manifestations The clinical features of hypercortisolemia are detected in only a small fraction of patients with documented ectopic ACTH production. Patients with ectopic ACTH syndrome generally exhibit less marked weight gain and centripetal fat redistribution, probably because the exposure to excess glucocorticoids is relatively brief and because cachexia reduces the propensity for weight gain and fat deposition. The ectopic ACTH syndrome is associated with several clinical features that distinguish it from other causes of Cushing’s syndrome (e.g., pituitary adenomas, adrenal adenomas, iatrogenic glucocorticoid excess). The metabolic manifestations of ectopic ACTH syndrome are dominated by fluid retention and hypertension, hypokalemia, metabolic alkalosis, glucose intolerance, and occasionally steroid psychosis. The very high ACTH levels often cause increased pigmentation, and melanotrope-stimulating hormone (MSH) activity derived from the POMC precursor peptide is also increased. The extraordinarily high glucocorticoid levels in patients with ectopic sources of ACTH can lead to marked skin fragility and easy bruising. In addition, the high cortisol levels often overwhelm the renal 11β-hydroxysteroid dehydrogenase type II enzyme, which normally inactivates cortisol and prevents it from binding to renal mineralocorticoid receptors. Consequently, in addition to the excess mineralocorticoids produced by ACTH stimulation of the adrenal gland, high levels of cortisol exert activity through the mineralocorticoid receptor, leading to severe hypokalemia.
Diagnosis The diagnosis of ectopic ACTH syndrome is usually not difficult in the setting of a known malignancy. Urine free cortisol levels fluctuate but are typically greater than two to four times normal, and the plasma ACTH level is usually >22 pmol/L (>100 pg/mL). A suppressed ACTH level excludes this diagnosis and indicates an ACTH-independent cause of Cushing’s syndrome (e.g., adrenal or exogenous glucocorticoid). In contrast to pituitary sources of ACTH, most ectopic sources of ACTH do not respond to glucocorticoid suppression. Therefore, high-dose dexamethasone (8 mg PO) suppresses 8:00 A.M. serum cortisol (50% decrease from baseline) in ~80% of pituitary ACTH-producing adenomas but fails to suppress ectopic ACTH in ~90% of cases. Bronchial and other carcinoids are well-documented exceptions to these general guidelines, as these ectopic sources of ACTH may exhibit feedback regulation indistinguishable from pituitary adenomas, including suppression by high-dose dexamethasone, and ACTH responsiveness to adrenal blockade with metyrapone. If necessary, petrosal sinus catheterization can be used to evaluate a patient with ACTH-dependent Cushing’s syndrome when the source of ACTH is unclear. After CRH stimulation, a 3:1 petrosal sinus:peripheral ACTH ratio strongly suggests a pituitary ACTH source. Imaging studies (computed tomography or magnetic resonance imaging) are also useful in the evaluation of suspected carcinoid lesions, allowing biopsy and characterization of hormone production using special stains. If available, positron emission tomography or octreotide scanning may identify some sources of ACTH production.
TUMOR-INDUCED HYPOGLYCEMIA CAUSED BY EXCESS PRODUCTION OF IGF-II
(See also Chap. 420) Mesenchymal tumors, hemangiopericytomas, hepatocellular tumors, adrenal carcinomas, and a variety of other large tumors have been reported to produce excessive amounts of insulin-like growth factor type II (IGF-II) precursor, which binds weakly to insulin receptors and more strongly to IGF-I receptors, leading to insulin-like actions. The gene encoding IGF-II resides on a chromosome 11p15 locus that is normally imprinted (that is, expression is exclusively from a single parental allele). Biallelic expression of the IGF-II gene occurs in a subset of tumors, suggesting loss of methylation and loss of imprinting as a mechanism for gene induction. In addition to increased IGF-II production, IGF-II bioavailability is increased due to complex alterations in circulating binding proteins. Increased IGF-II suppresses growth hormone (GH) and insulin, resulting in reduced IGF binding protein 3 (IGFBP-3), IGF-I, and acid-labile subunit (ALS). The reduction in ALS and IGFBP-3, which normally sequester IGF-II, causes it to be displaced to a small circulating complex that has greater access to insulin target tissues. For this reason, circulating IGF-II levels may not be markedly increased despite causing hypoglycemia. In addition to IGF-II–mediated hypoglycemia, tumors may occupy enough of the liver to impair gluconeogenesis.
In most cases, a tumor causing hypoglycemia is clinically apparent (usually >10 cm in size) and hypoglycemia develops in association with fasting. The diagnosis is made by documenting low serum glucose and suppressed insulin levels in association with symptoms of hypoglycemia. Serum IGF-II levels may not be increased (IGF-II assays may not detect IGF-II precursors). Increased IGF-II mRNA expression is found in most of these tumors. Any medications associated with hypoglycemia should be eliminated. Treatment of the underlying malignancy, if possible, may reduce the predisposition to hypoglycemia. Frequent meals and IV glucose, especially during sleep or fasting, are often necessary to prevent hypoglycemia. Glucagon and glucocorticoids have also been used to enhance glucose production.
HUMAN CHORIONIC GONADOTROPIN
hCG is composed of α and β subunits and can be produced as intact hormone, which is biologically active, or as uncombined biologically inert subunits. Ectopic production of intact hCG occurs most often in association with testicular embryonal tumors, germ cell tumors, extragonadal germinomas, lung cancer, hepatoma, and pancreatic islet tumors. Eutopic production of hCG occurs with trophoblastic malignancies. hCG α subunit production is particularly common in lung cancer and pancreatic islet cancer. In men, high hCG levels stimulate steroidogenesis and aromatase activity in testicular Leydig cells, resulting in increased estrogen production and the development of gynecomastia. Precocious puberty in boys or gynecomastia in men should prompt measurement of hCG and consideration of a testicular tumor or another source of ectopic hCG production. Most women are asymptomatic. hCG is easily measured. Treatment should be directed at the underlying malignancy.
ONCOGENIC OSTEOMALACIA
Hypophosphatemic oncogenic osteomalacia, also called tumor-induced osteomalacia (TIO), is characterized by markedly reduced serum phosphorus and renal phosphate wasting, leading to muscle weakness, bone pain, and osteomalacia. Serum calcium and PTH levels are normal, and 1,25-dihydroxyvitamin D is low. Oncogenic osteomalacia is usually caused by benign mesenchymal tumors, such as hemangiopericytomas, fibromas, and giant cell tumors, often of the skeletal extremities or head. It has also been described in sarcomas and in patients with prostate and lung cancer. Resection of the tumor reverses the disorder, confirming its humoral basis. The circulating phosphaturic factor is called phosphatonin—a factor that inhibits renal tubular reabsorption of phosphate and renal conversion of 25-hydroxyvitamin D to 1,25-dihydroxyvitamin D. Phosphatonin has been identified as fibroblast growth factor 23 (FGF23). FGF23 levels are increased in some, but not all, patients with osteogenic osteomalacia. FGF23 forms a ternary complex with the klotho protein and renal FGF receptors to reduce renal phosphate reabsorption. Treatment involves removal of the tumor, if possible, and supplementation with phosphate and vitamin D. Octreotide treatment reduces phosphate wasting in some patients with tumors that express somatostatin receptor subtype 2. Octreotide scans may also be useful in detecting these tumors.
HEMATOLOGIC SYNDROMES
The elevation of granulocyte, platelet, and eosinophil counts in most patients with myeloproliferative disorders is caused by the proliferation of the myeloid elements due to the underlying disease rather than to a paraneoplastic syndrome. The paraneoplastic hematologic syndromes in patients with solid tumors are less well characterized than are the endocrine syndromes because the ectopic hormone(s) or cytokines responsible have not been identified in most of these tumors (Table 121-2). The extent of the paraneoplastic syndromes parallels the course of the cancer.
|
PARANEOPLASTIC HEMATOLOGIC SYNDROMES |
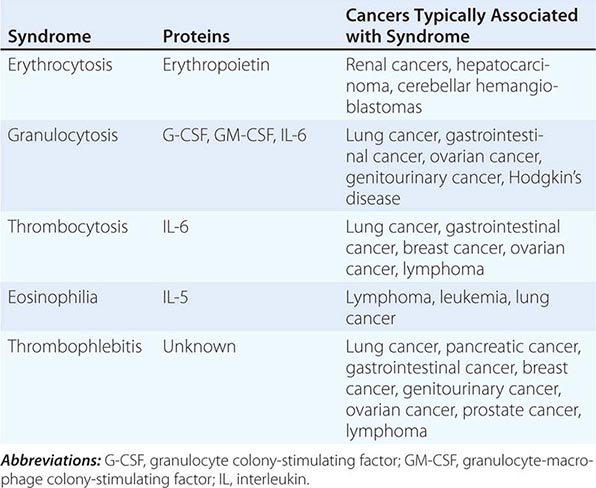
ERYTHROCYTOSIS
Ectopic production of erythropoietin by cancer cells causes most paraneoplastic erythrocytosis. The ectopically produced erythropoietin stimulates the production of red blood cells (RBCs) in the bone marrow and raises the hematocrit. Other lymphokines and hormones produced by cancer cells may stimulate erythropoietin release but have not been proved to cause erythrocytosis.
Most patients with erythrocytosis have an elevated hematocrit (>52% in men, >48% in women) that is detected on a routine blood count. Approximately 3% of patients with renal cell cancer, 10% of patients with hepatoma, and 15% of patients with cerebellar hemangioblastomas have erythrocytosis. In most cases, the erythrocytosis is asymptomatic.
Patients with erythrocytosis due to a renal cell cancer, hepatoma, or CNS cancer should have measurement of red cell mass. If the red cell mass is elevated, the serum erythropoietin level should be measured. Patients with an appropriate cancer, elevated erythropoietin levels, and no other explanation for erythrocytosis (e.g., hemoglobinopathy that causes increased O2 affinity; Chap. 77) have the paraneoplastic syndrome.
GRANULOCYTOSIS
Approximately 30% of patients with solid tumors have granulocytosis (granulocyte count >8000/μL). In about half of patients with granulocytosis and cancer, the granulocytosis has an identifiable nonparaneoplastic etiology (infection, tumor necrosis, glucocorticoid administration, etc.). The other patients have proteins in urine and serum that stimulate the growth of bone marrow cells. Tumors and tumor cell lines from patients with lung, ovarian, and bladder cancers have been documented to produce granulocyte colony-stimulating factor (G-CSF), granulocyte-macrophage colony-stimulating factor (GM-CSF), and/or interleukin 6 (IL-6). However, the etiology of granulocytosis has not been characterized in most patients.
Patients with granulocytosis are nearly all asymptomatic, and the differential white blood cell count does not have a shift to immature forms of neutrophils. Granulocytosis occurs in 40% of patients with lung and gastrointestinal cancers, 20% of patients with breast cancer, 30% of patients with brain tumors and ovarian cancers, 20% of patients with Hodgkin’s disease, and 10% of patients with renal cell carcinoma. Patients with advanced-stage disease are more likely to have granulocytosis than are those with early-stage disease.
Paraneoplastic granulocytosis does not require treatment. The granulocytosis resolves when the underlying cancer is treated.
THROMBOCYTOSIS
Some 35% of patients with thrombocytosis (platelet count >400,000/μL) have an underlying diagnosis of cancer. IL-6, a candidate molecule for the etiology of paraneoplastic thrombocytosis, stimulates the production of platelets in vitro and in vivo. Some patients with cancer and thrombocytosis have elevated levels of IL-6 in plasma. Another candidate molecule is thrombopoietin, a peptide hormone that stimulates megakaryocyte proliferation and platelet production. The etiology of thrombocytosis has not been established in most cases.
Patients with thrombocytosis are nearly all asymptomatic. Thrombocytosis is not clearly linked to thrombosis in patients with cancer. Thrombocytosis is present in 40% of patients with lung and gastrointestinal cancers; 20% of patients with breast, endometrial, and ovarian cancers; and 10% of patients with lymphoma. Patients with thrombocytosis are more likely to have advanced-stage disease and have a poorer prognosis than do patients without thrombocytosis. In ovarian cancer, IL-6 has been shown to directly promote tumor growth. Paraneoplastic thrombocytosis does not require treatment other than treatment of the underlying tumor.
EOSINOPHILIA
Eosinophilia is present in ~1% of patients with cancer. Tumors and tumor cell lines from patients with lymphomas or leukemia may produce IL-5, which stimulates eosinophil growth. Activation of IL-5 transcription in lymphomas and leukemias may involve translocation of the long arm of chromosome 5, to which the genes for IL-5 and other cytokines map.
Patients with eosinophilia are typically asymptomatic. Eosinophilia is present in 10% of patients with lymphoma, 3% of patients with lung cancer, and occasional patients with cervical, gastrointestinal, renal, and breast cancer. Patients with markedly elevated eosinophil counts (>5000/μL) can develop shortness of breath and wheezing. A chest radiograph may reveal diffuse pulmonary infiltrates from eosinophil infiltration and activation in the lungs.
THROMBOPHLEBITIS
Deep venous thrombosis and pulmonary embolism are the most common thrombotic conditions in patients with cancer. Migratory or recurrent thrombophlebitis may be the initial manifestation of cancer. Nearly 15% of patients who develop deep venous thrombosis or pulmonary embolism have a diagnosis of cancer (Chap. 142). The coexistence of peripheral venous thrombosis with visceral carcinoma, particularly pancreatic cancer, is called Trousseau’s syndrome.
Pathogenesis Patients with cancer are predisposed to thromboembolism because they are often at bed rest or immobilized, and tumors may obstruct or slow blood flow. Postoperative deep venous thrombosis is twice as common in cancer patients who undergo surgery. Chronic IV catheters also predispose to clotting. In addition, clotting may be promoted by release of procoagulants or cytokines from tumor cells or associated inflammatory cells or by platelet adhesion or aggregation. The specific molecules that promote thromboembolism have not been identified.
Chemotherapeutic agents, particularly those associated with endothelial damage, can induce venous thrombosis. The annual risk of venous thrombosis in patients with cancer receiving chemotherapy is about 11%, sixfold higher than the risk in the general population. Bleomycin, L-asparaginase, thalidomide analogues, cisplatin-based regimens, and high doses of busulfan and carmustine are all associated with an increased risk.
In addition to cancer and its treatment causing secondary thrombosis, primary thrombophilic diseases may be associated with cancer. For example, the antiphospholipid antibody syndrome is associated with a wide range of pathologic manifestations (Chap. 379). About 20% of patients with this syndrome have cancers. Among patients with cancer and antiphospholipid antibodies, 35–45% develop thrombosis.
Clinical Manifestations Patients with cancer who develop deep venous thrombosis usually develop swelling or pain in the leg, and physical examination reveals tenderness, warmth, and redness. Patients who present with pulmonary embolism develop dyspnea, chest pain, and syncope, and physical examination shows tachycardia, cyanosis, and hypotension. Some 5% of patients with no history of cancer who have a diagnosis of deep venous thrombosis or pulmonary embolism will have a diagnosis of cancer within 1 year. The most common cancers associated with thromboembolic episodes include lung, pancreatic, gastrointestinal, breast, ovarian, and genitourinary cancers; lymphomas; and brain tumors. Patients with cancer who undergo surgical procedures requiring general anesthesia have a 20–30% risk of deep venous thrombosis.
Diagnosis The diagnosis of deep venous thrombosis in patients with cancer is made by impedance plethysmography or bilateral compression ultrasonography of the leg veins. Patients with a noncompressible venous segment have deep venous thrombosis. If compression ultrasonography is normal and there is a high clinical suspicion for deep venous thrombosis, venography should be done to look for a luminal filling defect. Elevation of D-dimer is not as predictive of deep venous thrombosis in patients with cancer as it is in patients without cancer; elevations are seen in people over age 65 years without concomitant evidence of thrombosis, probably as a consequence of increased thrombin deposition and turnover in aging.
Patients with symptoms and signs suggesting a pulmonary embolism should be evaluated with a chest radiograph, electrocardiogram, arterial blood gas analysis, and ventilation-perfusion scan. Patients with mismatched segmental perfusion defects have a pulmonary embolus. Patients with equivocal ventilation-perfusion findings should be evaluated as described above for deep venous thrombosis in their legs. If deep venous thrombosis is detected, they should be anticoagulated. If deep venous thrombosis is not detected, they should be considered for a pulmonary angiogram.
Patients without a diagnosis of cancer who present with an initial episode of thrombophlebitis or pulmonary embolus need no additional tests for cancer other than a careful history and physical examination. In light of the many possible primary sites, diagnostic testing in asymptomatic patients is wasteful. However, if the clot is refractory to standard treatment or is in an unusual site or if the thrombophlebitis is migratory or recurrent, efforts to find an underlying cancer are indicated.
Cutaneous paraneoplastic syndromes are discussed in Chap. 72. Neurologic paraneoplastic syndromes are discussed in Chap. 122.
ACKNOWLEDGMENT
The authors acknowledge the contributions of Bruce E. Johnson to prior versions of this chapter.
122 |
Paraneoplastic Neurologic Syndromes and Autoimmune Encephalitis |
Paraneoplastic neurologic disorders (PNDs) are cancer-related syndromes that can affect any part of the nervous system (Table 122-1). They are caused by mechanisms other than metastasis or by any of the complications of cancer such as coagulopathy, stroke, metabolic and nutritional conditions, infections, and side effects of cancer therapy. In 60% of patients, the neurologic symptoms precede the cancer diagnosis. Clinically disabling PNDs occur in 0.5–1% of all cancer patients, but they affect 2–3% of patients with neuroblastoma or small-cell lung cancer (SCLC) and 30–50% of patients with thymoma or sclerotic myeloma.
|
PARANEOPLASTIC SYNDROMES OF THE NERVOUS SYSTEM |

PATHOGENESIS
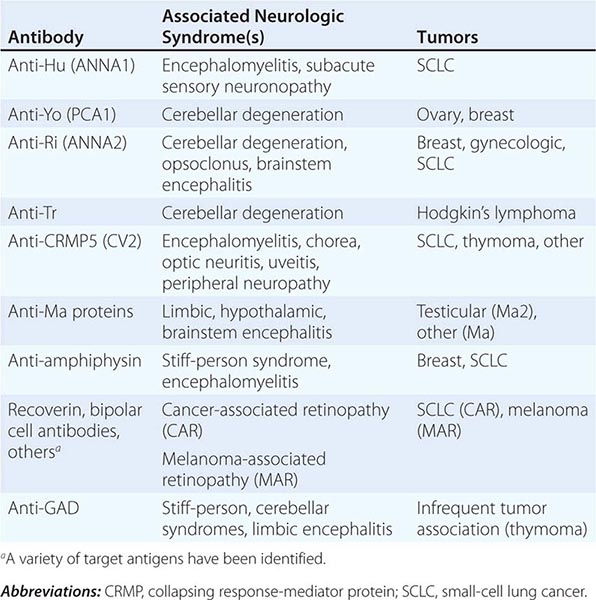
Most PNDs are mediated by immune responses triggered by neuronal proteins (onconeuronal antigens) expressed by tumors. In PNDs of the central nervous system (CNS), many antibody-associated immune responses have been identified (Table 122-2). These antibodies react with the patient’s tumor, and their detection in serum or cerebrospinal fluid (CSF) usually predicts the presence of cancer. When the antigens are intracellular, most syndromes are associated with extensive infiltrates of CD4+ and CD8+ T cells, microglial activation, gliosis, and variable neuronal loss. The infiltrating T cells are often in close contact with neurons undergoing degeneration, suggesting a primary pathogenic role. T cell–mediated cytotoxicity may contribute directly to cell death in these PNDs. Thus both humoral and cellular immune mechanisms participate in the pathogenesis of many PNDs. This complex immunopathogenesis may underlie the resistance of many of these conditions to therapy.
|
ANTIBODIES TO INTRACELLULAR ANTIGENS, SYNDROMES, AND ASSOCIATED CANCERS |
In contrast to the disorders associated with immune responses against intracellular antigens, those associated with antibodies to antigens expressed on the neuronal cell surface of the CNS or at the neuromuscular junction are more responsive to immunotherapy (Table 122-3, Fig. 122-1). These disorders occur with and without a cancer association and may affect children and young adults, and there is increasing evidence that they are mediated by the antibodies.
|
ANTIBODIES TO CELL SURFACE OR SYNAPTIC ANTIGENS, SYNDROMES, AND ASSOCIATED TUMORS |
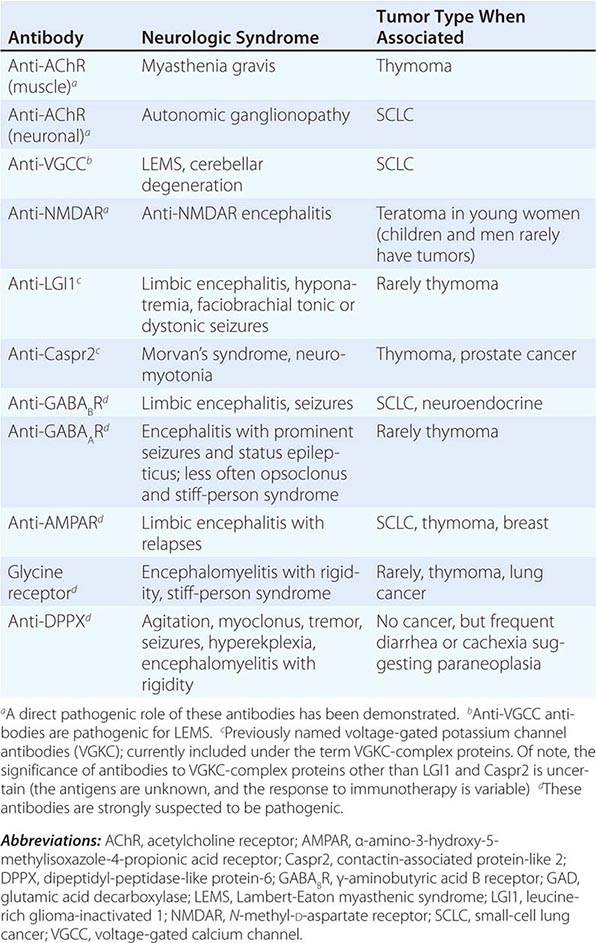
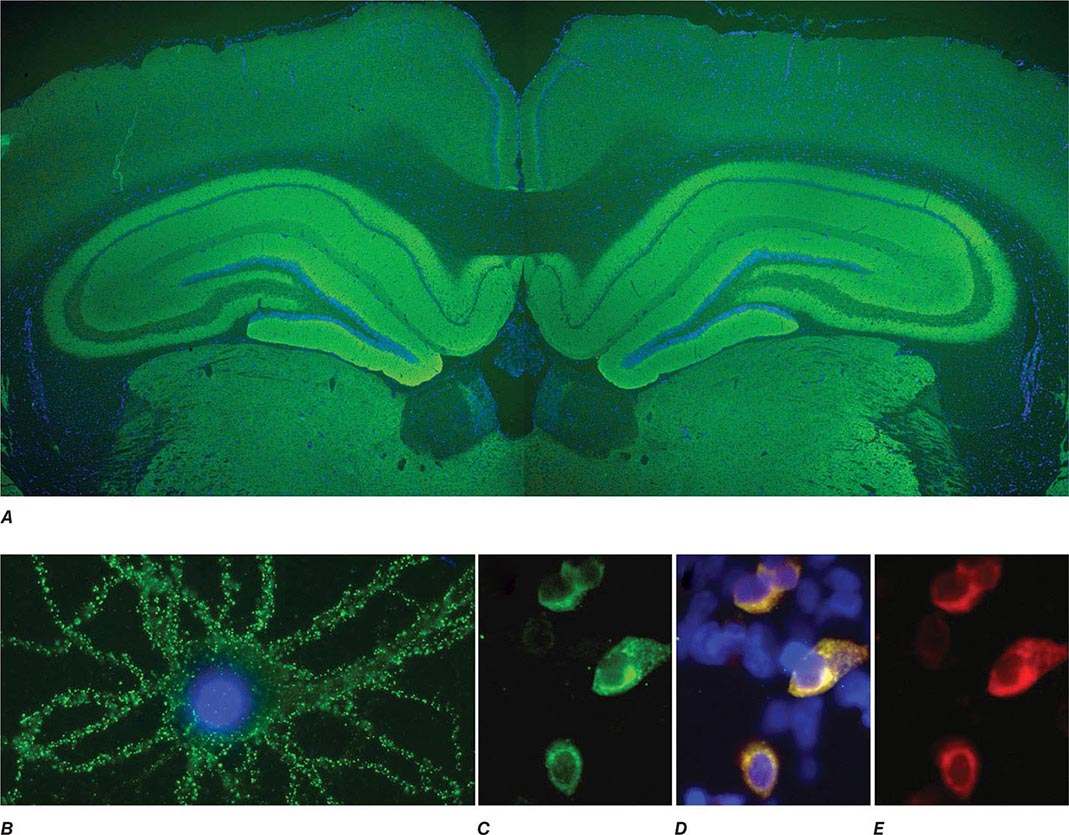
FIGURE 122-1 Antibodies to the GluN1 subunit of the N-methyl-D-aspartate (NMDA) receptor in a patient with anti-NMDA receptor encephalitis and ovarian teratoma. (A) Coronal section of rat brain immunolabeled (green fluorescence) with the patient’s antibodies. The reactivity predominates in the hippocampus, which is highly enriched in NMDA receptors. (B) This image shows the antibody reactivity with cultures of rat hippocampal neurons; the intense green immunolabeling is due to the antibodies against the GluN1 subunit of NMDA receptors. (C–E) Images of HEK cells (a human kidney cell line) transfected to express NMDA receptors, showing reactivity with patient’s antibodies (C) and with a commercial monoclonal antibody against NMDA receptors (E); the patient’s antibody reactivity co-labels only the cells that express NMDA receptors (D). (From J Dalmau et al: Lancet Neurol 7:1091, 2008; with permission.)
Other PNDs are likely immune-mediated, although their antigens are unknown. These include several syndromes of inflammatory neuropathies and myopathies. In addition, many patients with typical PND syndromes are antibody-negative.
For still other PNDs, the cause remains quite obscure. These include, among others, several neuropathies that occur in the terminal stages of cancer and a number of neuropathies associated with plasma cell dyscrasias or lymphoma without evidence of inflammatory infiltrates or deposits of immunoglobulin, cryoglobulin, or amyloid.
APPROACH TO THE PATIENT:
Paraneoplastic Neurologic Disorders
Three key concepts are important for the diagnosis and management of PNDs. First, it is common for symptoms to appear before the presence of a tumor is known; second, the neurologic syndrome usually develops rapidly, producing severe deficits in a short period of time; and third, there is evidence that prompt tumor control improves the neurologic outcome. Therefore, the major concern of the physician is to recognize a disorder promptly as paraneoplastic and to identify and treat the tumor.
PND OF THE CENTRAL NERVOUS SYSTEM AND DORSAL ROOT GANGLIA
When symptoms involve brain, spinal cord, or dorsal root ganglia, the suspicion of PND is usually based on a combination of clinical, radiologic, and CSF findings. Presence of antineuronal antibodies (Tables 122-2 and 122-3) may help in the diagnosis, but only 60–70% of PNDs of the CNS and less than 20% of those involving the peripheral nervous system have neuronal or neuromuscular junction antibodies that can be used as diagnostic tests.
Magnetic resonance imaging (MRI) and CSF studies are important to rule out neurologic complications due to the direct spread of cancer, particularly metastatic and leptomeningeal disease. In most PNDs, the MRI findings are nonspecific. Paraneoplastic limbic encephalitis is usually associated with characteristic MRI abnormalities in the mesial temporal lobes (see below), but similar findings can occur with other disorders (e.g., nonparaneoplastic autoimmune limbic encephalitis and human herpesvirus type 6 [HHV-6] encephalitis) (Fig. 122-2). The CSF profile of patients with PND of the CNS or dorsal root ganglia typically consists of mild to moderate pleocytosis (<200 mononuclear cells, predominantly lymphocytes), an increase in the protein concentration, and a variable presence of oligoclonal bands. There are no specific electrophysiologic tests that are diagnostic of PND. Moreover, a biopsy of the affected tissue is often difficult to obtain, and although useful to rule out other disorders (e.g., metastasis) the pathologic findings are not specific for PND.
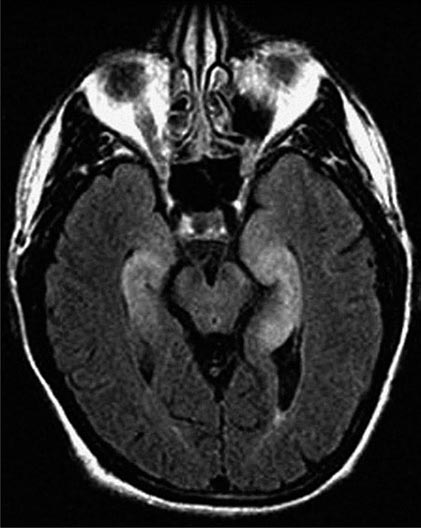
FIGURE 122-2 Fluid-attenuated inversion recovery sequence magnetic resonance imaging of a patient with limbic encephalitis and LGI1 antibodies. Note the abnormal hyperintensity involving the medial aspect of the temporal lobes.
PND OF NERVE AND MUSCLE
If symptoms involve peripheral nerve, neuromuscular junction, or muscle, the diagnosis of a specific PND is usually established on clinical, electrophysiologic, and pathologic grounds. The clinical history, accompanying symptoms (e.g., anorexia, weight loss), and type of syndrome dictate the studies and degree of effort needed to demonstrate a neoplasm. For example, the frequent association of Lambert-Eaton myasthenic syndrome (LEMS) with SCLC should lead to a chest and abdomen computed tomography (CT) or body positron emission tomography (PET) scan and, if negative, periodic tumor screening for at least 3 years after the neurologic diagnosis. In contrast, the weak association of polymyositis with cancer calls into question the need for repeated cancer screenings in this situation. Serum and urine immunofixation studies should be considered in patients with peripheral neuropathy of unknown cause; detection of a monoclonal gammopathy suggests the need for additional studies to uncover a B cell or plasma cell malignancy. In paraneoplastic neuropathies, diagnostically useful antineuronal antibodies are limited to anti-CV2/CRMP5 and anti-Hu.
For any type of PND, if antineuronal antibodies are negative, the diagnosis relies on the demonstration of cancer and the exclusion of other cancer-related or independent neurologic disorders. Combined CT and PET scans often uncover tumors undetected by other tests. For germ cell tumors of the testis and teratomas of the ovary, ultrasound and CT or MRI of the abdomen and pelvis may reveal tumors undetectable by PET.
SPECIFIC PARANEOPLASTIC NEUROLOGIC SYNDROMES
PARANEOPLASTIC ENCEPHALOMYELITIS AND FOCAL ENCEPHALITIS
The term encephalomyelitis describes an inflammatory process with multifocal involvement of the nervous system, including brain, brainstem, cerebellum, and spinal cord. It is often associated with dorsal root ganglia and autonomic dysfunction. For any given patient, the clinical manifestations are determined by the areas predominantly involved, but pathologic studies almost always reveal abnormalities beyond the symptomatic regions. Several clinicopathologic syndromes may occur alone or in combination: (1) cortical encephalitis, which may present as “epilepsia partialis continua”; (2) limbic encephalitis, characterized by confusion, depression, agitation, anxiety, severe short-term memory deficits, partial complex seizures, and sometimes dementia (the MRI usually shows unilateral or bilateral medial temporal lobe abnormalities, best seen with T2 and fluid-attenuated inversion recovery sequences); (3) brainstem encephalitis, resulting in eye movement disorders (nystagmus, opsoclonus, supranuclear or nuclear paresis), cranial nerve paresis, dysarthria, dysphagia, and central autonomic dysfunction; (4) cerebellar gait and limb ataxia; (5) myelitis, which may cause lower or upper motor neuron symptoms, myoclonus, muscle rigidity, and spasms; and (6) autonomic dysfunction as a result of involvement of the neuraxis at multiple levels, including hypothalamus, brainstem, and autonomic nerves (see Paraneoplastic Peripheral Neuropathies, below). Cardiac arrhythmias, postural hypotension, and central hypoventilation are frequent causes of death in patients with encephalomyelitis.
Paraneoplastic encephalomyelitis and focal encephalitis are usually associated with SCLC, but many other cancers have been implicated. Patients with SCLC and these syndromes usually have anti-Hu antibodies in serum and CSF. Anti-CRMP5 antibodies occur less frequently; some of these patients may develop chorea, uveitis, or optic neuritis. Antibodies to Ma proteins are associated with limbic, hypothalamic, and brainstem encephalitis and occasionally with cerebellar symptoms (Fig. 122-3); some patients develop hypersomnia, cataplexy, and severe hypokinesia. MRI abnormalities are frequent, including those described with limbic encephalitis and variable involvement of the hypothalamus, basal ganglia, or upper brainstem. The oncologic associations of these antibodies are shown in Table 122-2.
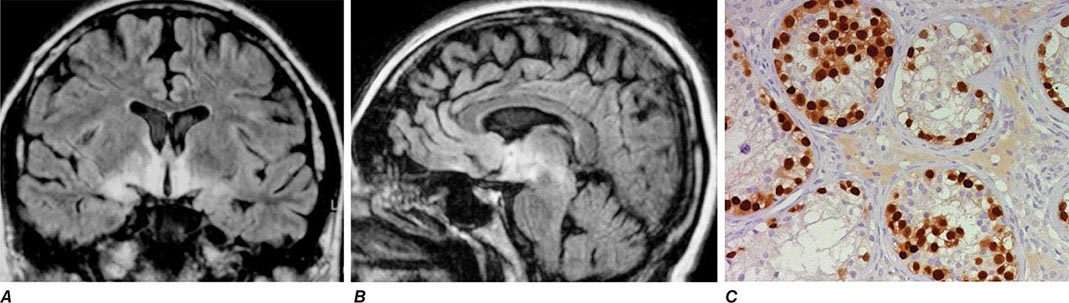
FIGURE 122-3 Magnetic resonance imaging (MRI) and tumor of a patient with anti-Ma2-associated encephalitis. (A and B) Fluid-attenuated inversion recovery MRI sequences showing abnormal hyperintensities in the medial temporal lobes, hypothalamus, and upper brainstem. (C) This image corresponds to a section of the patient’s orchiectomy incubated with a specific marker (Oct4) of germ cell tumors. The positive (brown) cells correspond to an intratubular germ cell neoplasm.
ENCEPHALITIDES WITH ANTIBODIES TO CELL-SURFACE OR SYNAPTIC PROTEINS (TABLE 122-3)
These disorders are important for three reasons: (1) they can occur with and without tumor association, (2) some syndromes predominate in young individuals and children, and (3) despite the severity of the symptoms patients usually respond to treatment of the tumor, if found, and immunotherapy (e.g., glucocorticoids, IVIg, plasma exchange, rituximab, or cyclophosphamide).
Encephalitis with N-methyl-D-aspartate (NMDA) receptor antibodies (Fig. 122-1) usually occurs in young women and children, but men and older patients of both sexes can be affected. The disorder has a characteristic pattern of symptom progression that includes a prodrome resembling a viral process, followed in a few days by the onset of severe psychiatric symptoms, memory loss, seizures, decreased level of consciousness, abnormal movements (orofacial, limb, and trunk dyskinesias, dystonic postures), autonomic instability, and frequent hypoventilation. Monosymptomatic episodes, such as pure psychosis, occur in 4% of the patients. Clinical relapses occur in 12–24% of patients (12% during the first 2 years after initial presentation). Most patients have intrathecal synthesis of antibodies, likely by infiltrating plasma cells in brain and meninges (Fig. 122-4A). The syndrome is often misdiagnosed as a viral or idiopathic encephalitis, neuroleptic malignant syndrome, or encephalitis lethargica, and many patients are initially evaluated by psychiatrists with the suspicion of acute psychosis. The detection of an associated teratoma is dependent on age and gender: 46% of female patients 12 years or older have uni- or bilateral ovarian teratomas, whereas less than 7% of girls younger than 12 have a teratoma (Fig. 122-4B). In male patients, the detection of a tumor is rare. Patients older than 45 years are more frequently male; about 20% of these patients have tumors (e.g., cancer of the breast, ovary, or lung).
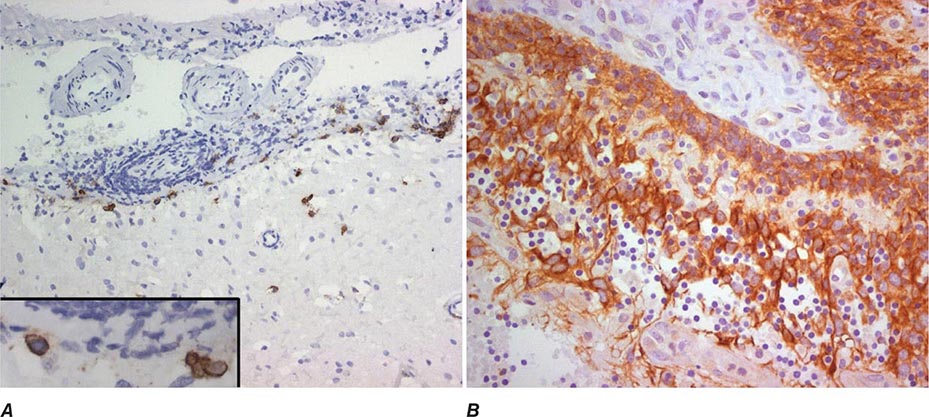
FIGURE 122-4 Pathologic findings in anti–N-methyl-D-aspartate (NMDA) receptor encephalitis. Infiltrates of plasma cells (brown cells; stained for CD138) in the meninges and brain of a patient (A); the inset is a magnification of some plasma cells. (B) Neurons and neuronal processes in the teratoma of a patient (brown cells; stained with MAP2); these neurons express NMDA receptors (not shown). (From E Martinez-Hernandez et al: Neurology 77:589, 2011, with permission.)
Encephalitis with leucine-rich glioma-inactivated 1 (LGI1) antibodies predominates in patients older than 50 years (65% male) and frequently presents with memory loss and seizures (limbic encephalopathy), along with hyponatremia and sleep dysfunction. In a small number of patients, the encephalitis is preceded by or occurs with myoclonic-like movements called faciobrachial dystonic or tonic seizures. Less than 10% of patients have thymoma.
Encephalitis with contactin-associated protein-like 2 (Caspr2) antibodies predominates in patients older than 50 years and is associated with Morvan’s syndrome (encephalitis, insomnia, confusion, hallucinations, autonomic dysfunction, and neuromyotonia) and, less frequently, with limbic encephalitis, neuromyotonia, and neuropathic pain. About 30–40% of patients have thymoma.
Encephalitis with γ-aminobutyric acid type B (GABAB) receptor antibodies is usually associated with limbic encephalitis and seizures. In rare instances, patients develop cerebellar symptoms and opsoclonus. Fifty percent of patients have SCLC or a neuroendocrine tumor of the lung. Patients may have additional antibodies to glutamic acid decarboxylase (GAD), which are of unclear significance. Other antibodies to nonneuronal proteins are often found in these patients as well as in patients with α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) receptor antibodies, indicating a general tendency to autoimmunity.
Encephalitis with GABAA receptor antibodies may affect children and adults. When antibodies are present at high titer in serum and CSF, the disorder associates with prominent seizures and status epilepticus, often requiring pharmacologically induced coma. Low titer antibodies in serum are often associated with other autoimmune conditions, and the spectrum of symptoms is wider, including encephalitis, seizures, opsoclonus, or stiff-person syndrome. Most patients do not have an underlying tumor, but some may have thymoma.
Encephalitis with AMPA receptor antibodies affects middle-aged women, who develop acute limbic dysfunction or, less frequently, prominent psychiatric symptoms; 70% of the patients have an underlying tumor in the lung, breast, or thymus. Neurologic relapses may occur; these also respond to immunotherapy and are not necessarily associated with tumor recurrence.
Encephalitis with glycine receptor (GlyR) antibodies has been described in adults with progressive encephalomyelitis with rigidity and myoclonus (PERM) and stiff-person spectrum of symptoms (with or without GAD antibodies). The disorder usually occurs without tumor association, although some patients have lung cancer, thymoma, or Hodgkin’s lymphoma.
Encephalitis with dipeptidyl-peptidase-like protein-6 (or DPPX) antibodies results in symptoms of CNS hyperexcitability including agitation, hallucinations, paranoid delusions, tremor, myoclonus, nystagmus, seizures, and sometimes hyperekplexia. Some patients develop progressive encephalomyelitis with rigidity and myoclonus. Diarrhea, other gastrointestinal symptoms, and substantial loss of weight often suggest the presence of an underlying tumor, but no tumor association has been identified. The disorder responds to immunotherapy.
PARANEOPLASTIC CEREBELLAR DEGENERATION
This disorder is often preceded by a prodrome that may include dizziness, oscillopsia, blurry or double vision, nausea, and vomiting. A few days or weeks later, patients develop dysarthria, gait and limb ataxia, and variable dysphagia. The examination usually shows downbeating nystagmus and, rarely, opsoclonus. Brainstem dysfunction, upgoing toes, or a mild neuropathy may occur. Early in the course, MRI studies are usually normal; later, the MRI reveals cerebellar atrophy. The disorder results from extensive degeneration of Purkinje cells, with variable involvement of other cerebellar cortical neurons, deep cerebellar nuclei, and spinocerebellar tracts. The tumors more frequently involved are SCLC, cancer of the breast and ovary, and Hodgkin’s lymphoma.
Anti-Yo antibodies in patients with breast and gynecologic cancers and anti-Tr antibodies in patients with Hodgkin’s lymphoma are the two immune responses typically associated with prominent or pure cerebellar degeneration. Antibodies to P/Q-type voltage-gated calcium channels (VGCC) occur in some patients with SCLC and cerebellar dysfunction; only some of these patients develop LEMS. A variable degree of cerebellar dysfunction can be associated with virtually any of the antibodies and PND of the CNS shown in Table 122-2.
A number of single case reports have described neurologic improvement after tumor removal, plasma exchange, IVIg, cyclophosphamide, rituximab, or glucocorticoids. However, most patients with paraneoplastic cerebellar degeneration do not improve with treatment.
PARANEOPLASTIC OPSOCLONUS-MYOCLONUS SYNDROME
Opsoclonus is a disorder of eye movement characterized by involuntary, chaotic saccades that occur in all directions of gaze; it is frequently associated with myoclonus and ataxia. Opsoclonus-myoclonus may be cancer-related or idiopathic. When the cause is paraneoplastic, the tumors involved are usually cancer of the lung and breast in adults, neuroblastoma in children, and ovarian teratoma in adolescents and young women. The pathologic substrate of opsoclonus-myoclonus is unclear, but studies suggest that disinhibition of the fastigial nucleus of the cerebellum is involved. Most patients do not have antineuronal antibodies. A small subset of patients with ataxia, opsoclonus, and other eye-movement disorders develop anti-Ri antibodies; in rare instances, muscle rigidity, laryngeal spasms, autonomic dysfunction, and dementia also occur. The tumors most frequently involved in anti-Ri-associated syndromes are breast and ovarian cancer. If the tumor is not successfully treated, the syndrome in adults often progresses to encephalopathy, coma, and death. In addition to treating the tumor, symptoms may respond to immunotherapy (glucocorticoids, plasma exchange, and/or IVIg).
At least 50% of children with opsoclonus-myoclonus have an underlying neuroblastoma. Hypotonia, ataxia, behavioral changes, and irritability are frequent accompanying symptoms. Neurologic symptoms often improve with treatment of the tumor and glucocorticoids, adrenocorticotropic hormone (ACTH), plasma exchange, IVIg, and rituximab. Many patients are left with psychomotor retardation and behavioral and sleep problems.
PARANEOPLASTIC SYNDROMES OF THE SPINAL CORD
The number of reports of paraneoplastic spinal cord syndromes, such as subacute motor neuronopathy and acute necrotizing myelopathy, has decreased in recent years. This may represent a true decrease in incidence, due to improved and prompt oncologic interventions, or the identification of nonparaneoplastic etiologies. Some patients with cancer develop upper or lower motor neuron dysfunction or both, resembling amyotrophic lateral sclerosis. It is unclear whether these disorders have a paraneoplastic etiology or simply coincide with the presence of cancer. There are isolated case reports of cancer patients with motor neuron dysfunction who had neurologic improvement after tumor treatment. A search for lymphoma should be undertaken in patients with a rapidly progressive motor neuron syndrome and a monoclonal protein in serum or CSF.
Paraneoplastic myelitis may present with upper or lower motor neuron symptoms, segmental myoclonus, and rigidity, and can be the first manifestation of encephalomyelitis. Neuromyelitis optica (NMO) with aquaporin 4 antibodies may occur in rare instances as a paraneoplastic manifestation of a cancer.
PARANEOPLASTIC STIFF-PERSON SYNDROME
This disorder is characterized by progressive muscle rigidity, stiffness, and painful spasms triggered by auditory, sensory, or emotional stimuli. Rigidity mainly involves the lower trunk and legs, but it can affect the upper extremities and neck. Sometimes, only one extremity is affected (stiff-limb syndrome). Symptoms improve with sleep and general anesthetics. Electrophysiologic studies demonstrate continuous motor unit activity. The associated antibodies target proteins (GAD, amphiphysin) involved in the function of inhibitory synapses using γ-aminobutyric acid (GABA) or glycine as neurotransmitters. The presence of amphiphysin antibodies usually indicates a paraneoplastic etiology related to SCLC and breast cancer. By contrast, GAD antibodies may occur in some cancer patients but are much more frequently present in the nonparaneoplastic disorder. GlyR antibodies may occur in some patients with stiff-person syndrome; these antibodies are also detectable in patients with PERM.
Optimal treatment of stiff-person syndrome requires therapy of the underlying tumor, glucocorticoids, and symptomatic use of drugs that enhance GABA-ergic transmission (diazepam, baclofen, sodium valproate, tiagabine, vigabatrin). IVIg and plasma exchange are transiently effective in some patients.
PARANEOPLASTIC SENSORY NEURONOPATHY OR DORSAL ROOT GANGLIONOPATHY
This syndrome is characterized by sensory deficits that may be symmetric or asymmetric, painful dysesthesias, radicular pain, and decreased or absent reflexes. All modalities of sensation and any part of the body including face and trunk can be involved. Specialized sensations such as taste and hearing can also be affected. Electrophysiologic studies show decreased or absent sensory nerve potentials with normal or near-normal motor conduction velocities. Symptoms result from an inflammatory, likely immune-mediated, process that targets the dorsal root ganglia, causing neuronal loss and secondary degeneration of the posterior columns of the spinal cord. The dorsal and, less frequently, the anterior nerve roots and peripheral nerves may also be involved. This disorder often precedes or is associated with encephalomyelitis and autonomic dysfunction and has the same immunologic and oncologic associations (Hu antibodies, SCLC).
As with anti-Hu-associated encephalomyelitis, the therapeutic approach focuses on prompt treatment of the tumor. Glucocorticoids occasionally produce clinical stabilization or improvement. The benefit of IVIg and plasma exchange is not proven.
PARANEOPLASTIC PERIPHERAL NEUROPATHIES
These disorders may develop any time during the course of the neoplastic disease. Neuropathies occurring at late stages of cancer or lymphoma usually cause mild to moderate sensorimotor deficits due to axonal degeneration of unclear etiology. These neuropathies are often masked by concurrent neurotoxicity from chemotherapy and other cancer therapies. In contrast, the neuropathies that develop in the early stages of cancer frequently show a rapid progression, sometimes with a relapsing and remitting course, and evidence of inflammatory infiltrates and axonal loss or demyelination. If demyelinating features predominate (Chaps. 459 and 460), IVIg, plasma exchange, or glucocorticoids may improve symptoms. Occasionally anti-CRMP5 antibodies are present; detection of anti-Hu suggests concurrent dorsal root ganglionitis.
Guillain-Barré syndrome and brachial plexitis have occasionally been reported in patients with lymphoma, but there is no clear evidence of a paraneoplastic association (Chap. 460).
Malignant monoclonal gammopathies include: (1) multiple myeloma and sclerotic myeloma associated with IgG or IgA monoclonal proteins; and (2) Waldenström’s macroglobulinemia, B cell lymphoma, and chronic B cell lymphocytic leukemia associated with IgM monoclonal proteins. These disorders may cause neuropathy by a variety of mechanisms, including compression of roots and plexuses by metastasis to vertebral bodies and pelvis, deposits of amyloid in peripheral nerves, and paraneoplastic mechanisms. The paraneoplastic variety has several distinctive features. Approximately half of patients with sclerotic myeloma develop a sensorimotor neuropathy with predominantly motor deficits, resembling a chronic inflammatory demyelinating neuropathy (Chap. 460); some patients develop elements of the POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, M protein, skin changes). Treatment of the plasmacytoma or sclerotic lesions usually improves the neuropathy. In contrast, the sensorimotor or sensory neuropathy associated with multiple myeloma is more refractory to treatment. Between 5 and 10% of patients with Waldenström’s macroglobulinemia develop a distal symmetric sensorimotor neuropathy with predominant involvement of large sensory fibers. These patients may have IgM antibodies in their serum against myelin-associated glycoprotein and various gangliosides (Chap. 460). In addition to treating the Waldenström’s macroglobulinemia, other therapies may improve the neuropathy, including plasma exchange, IVIg, chlorambucil, cyclophosphamide, fludarabine, or rituximab.
Vasculitis of the nerve and muscle causes a painful symmetric or asymmetric distal axonal sensorimotor neuropathy with variable proximal weakness. It predominantly affects elderly men and is associated with an elevated erythrocyte sedimentation rate and increased CSF protein concentration. SCLC and lymphoma are the primary tumors involved. Glucocorticoids and cyclophosphamide often result in neurologic improvement.
Peripheral nerve hyperexcitability (neuromyotonia, or Isaacs’ syndrome) is characterized by spontaneous and continuous muscle fiber activity of peripheral nerve origin. Clinical features include cramps, muscle twitching (fasciculations or myokymia), stiffness, delayed muscle relaxation (pseudomyotonia), and spontaneous or evoked carpal or pedal spasms. The involved muscles may be hypertrophic, and some patients develop paresthesias and hyperhidrosis. CNS dysfunction, including mood changes, sleep disorder, hallucinations, and autonomic symptoms may occur. The electromyogram (EMG) shows fibrillations; fasciculations; and doublet, triplet, or multiplet single-unit (myokymic) discharges that have a high intraburst frequency. Some patients have Caspr2 antibodies in the context of Morvan’s syndrome, but most cases of isolated neuromyotonia are antibody negative. The disorder often occurs without cancer; if paraneoplastic, benign and malignant thymomas and SCLC are the usual tumors. Phenytoin, carbamazepine, and plasma exchange improve symptoms.
Paraneoplastic autonomic neuropathy usually develops as a component of other disorders, such as LEMS and encephalomyelitis. It may rarely occur as a pure or predominantly autonomic neuropathy with cholinergic or adrenergic dysfunction at the pre- or postganglionic levels. Patients can develop several life-threatening complications, such as gastrointestinal paresis with pseudo-obstruction, cardiac dysrhythmias, and postural hypotension. Other clinical features include abnormal pupillary responses, dry mouth, anhidrosis, erectile dysfunction, and problems in sphincter control. The disorder occurs in association with several tumors, including SCLC, cancer of the pancreas or testis, carcinoid tumors, and lymphoma. Because autonomic symptoms can be the presenting feature of encephalomyelitis, serum anti-Hu and anti-CRMP5 antibodies should be sought. Antibodies to ganglionic (alpha3-type) neuronal acetylcholine receptors are the cause of autoimmune autonomic ganglionopathy, a disorder that frequently occurs without cancer association (Chap. 454).
LAMBERT-EATON MYASTHENIC SYNDROME
LEMS is discussed in Chap. 461.
MYASTHENIA GRAVIS
Myasthenia gravis is discussed in Chap. 461.
POLYMYOSITIS-DERMATOMYOSITIS
Polymyositis and dermatomyositis are discussed in detail in Chap. 388.
ACUTE NECROTIZING MYOPATHY
Patients with this syndrome develop myalgias and rapid progression of weakness involving the extremities and the pharyngeal and respiratory muscles, often resulting in death. Serum muscle enzymes are elevated, and muscle biopsy shows extensive necrosis with minimal or absent inflammation and sometimes deposits of complement. The disorder occurs as a paraneoplastic manifestation of a variety of cancers including SCLC and cancer of the gastrointestinal tract, breast, kidney, and prostate, among others. Glucocorticoids and treatment of the underlying tumor rarely control the disorder.
PARANEOPLASTIC VISUAL SYNDROMES
This group of disorders involves the retina and, less frequently, the uvea and optic nerves. The term cancer-associated retinopathy is used to describe paraneoplastic cone and rod dysfunction characterized by photosensitivity, progressive loss of vision and color perception, central or ring scotomas, night blindness, and attenuation of photopic and scotopic responses in the electroretinogram (ERG). The most commonly associated tumor is SCLC. Melanoma-associated retinopathy affects patients with metastatic cutaneous melanoma. Patients develop acute onset of night blindness and shimmering, flickering, or pulsating photopsias that often progress to visual loss. The ERG shows reduced b waves with normal dark adapted a waves. Paraneoplastic optic neuritis and uveitis are very uncommon and can develop in association with encephalomyelitis. Some patients with paraneoplastic uveitis and optic neuritis have anti-CRMP5 antibodies.
Some paraneoplastic retinopathies are associated with serum antibodies that specifically react with the subset of retinal cells undergoing degeneration, supporting an immune-mediated pathogenesis (Table 122-2). Paraneoplastic retinopathies usually fail to improve with treatment, although rare responses to glucocorticoids, plasma exchange, and IVIg have been reported.
123e |
Thymoma |
The thymus is derived from the third and fourth pharyngeal pouches and is located in the anterior mediastinum. It is composed of epithelial and stromal cells derived from the pharyngeal pouch and lymphoid precursors derived from mesodermal cells. It is the site to which bone marrow precursors that are committed to differentiate into T cells migrate to complete their differentiation. Like many organs, it is organized into functional regions, in this case the cortex and the medulla. The cortex of the thymus contains ~85% of the lymphoid cells, and the medulla contains ~15%. It appears that the primitive bone marrow progenitors enter the thymus at the corticomedullary junction and migrate first through the cortex toward the periphery of the gland and then toward the medulla as they mature. Medullary thymocytes have a phenotype that cannot be distinguished readily from that of mature peripheral blood and lymph node T cells.
Several things can go wrong with the thymus, but thymic abnormalities are very rare. If the thymus does not develop properly, serious deficiencies in T-cell development ensue and severe immunodeficiency is seen (e.g., DiGeorge syndrome, Chap. 374). If a lymphoid cell within the thymus becomes neoplastic, the disease that develops is a lymphoma. The majority of lymphoid tumors that develop in the thymus are derived from the precursor T cells, and the tumor is a precursor T-cell lymphoblastic lymphoma (Chap. 134). Rare B cells exist in the thymus, and when they become neoplastic, the tumor is a mediastinal (thymic) B cell lymphoma (Chap. 134). Hodgkin’s disease, particularly the nodular sclerosing subtype, often involves the anterior mediastinum. Extranodal marginal zone (mucosa-associated lymphoid tissue [MALT]) lymphomas have been reported to involve the thymus in the setting of Sjögren’s syndrome or other autoimmune disorders, and the lymphoma cells often express IgA instead of IgM on their surface. Castleman’s disease can involve the thymus. Germ cell tumors and carcinoid tumors occasionally may arise in the thymus. If the epithelial cells of the thymus become neoplastic, the tumor that develops is a thymoma.
CLINICAL PRESENTATION AND DIFFERENTIAL DIAGNOSIS
Thymoma, although rare (0.1–0.15 cases per 100,000 person-years), is the most common cause of an anterior mediastinal mass in adults, accounting for ~40% of all mediastinal masses. The other major causes of anterior mediastinal masses are lymphomas, germ cell tumors, and substernal thyroid tumors. Carcinoid tumors, lipomas, and thymic cysts also may produce radiographic masses. After combination chemotherapy for another malignancy, teenagers and young adults may develop a rebound thymic hyperplasia in the first few months after treatment. Granulomatous inflammatory diseases (tuberculosis, sarcoidosis) can produce thymic enlargement. Thymomas are most common in the fifth and sixth decades, are uncommon in children, and are distributed evenly between men and women.
About 40–50% of patients are asymptomatic; masses are detected incidentally on routine chest radiographs. When symptomatic, patients may have cough, chest pain, dyspnea, fever, wheezing, fatigue, weight loss, night sweats, or anorexia. Occasionally, thymomas may obstruct the superior vena cava. Pericardial effusion may be present. About 40% of patients with thymoma have another systemic autoimmune illness related to the thymoma. About 30% of patients with thymoma have myasthenia gravis, 5–8% have pure red cell aplasia, and ~5% have hypogammaglobulinemia. Thymoma with hypogammaglobulinemia also is called Good’s syndrome. Among patients with myasthenia gravis, ~10–15% have a thymoma. Thymoma more rarely may be associated with polymyositis, systemic lupus erythematosus, thyroiditis, Sjögren’s syndrome, ulcerative colitis, pernicious anemia, Addison’s disease, stiff person syndrome, scleroderma, and panhypopituitarism. In one series, 70% of patients with thymoma were found to have another systemic illness.
DIAGNOSIS AND STAGING
Once a mediastinal mass is detected, a surgical procedure is required for definitive diagnosis. An initial mediastinoscopy or limited thoracotomy can be undertaken to get sufficient tissue to make an accurate diagnosis. Fine-needle aspiration is poor at distinguishing between lymphomas and thymomas but is more reliable in diagnosing germ cell tumors and metastatic carcinoma. Thymomas and lymphomas require sufficient tissue to examine the tumor architecture to assure an accurate diagnosis and obtain prognostic information.
Once a diagnosis of thymoma is defined, subsequent staging generally occurs at surgery. However, chest computed tomography (CT) scans can assess local invasiveness in some instances. Magnetic resonance imaging (MRI) has a defined role in the staging of posterior mediastinal tumors, but it is not clear that it adds important information to the CT scan in anterior mediastinal tumors. Somatostatin receptor imaging with indium-labeled somatostatin analogues may be of value. If invasion is not distinguished by noninvasive testing, an effort to resect the entire tumor should be undertaken. If invasion is present, neoadjuvant chemotherapy may be warranted before surgery (see “Treatment” section below).
Some 90% of thymomas are in the anterior mediastinum, but some may be in other mediastinal sites or even the neck, based on aberrant migration of the developing thymic enlage.
The staging system for thymoma was developed by Masaoka and colleagues (Table 123e-1). It is an anatomic system in which the stage is increased on the basis of the degree of invasiveness. The 5-year survival of patients in the various stages is as follows: stage I, 96%; stage II, 86%; stage III, 69%; and stage IV, 50%. The French Study Group on Thymic Tumors (GETT) has proposed modifications to the Masaoka scheme based on the degree of surgical removal because the extent of surgery has been noted to be a prognostic indicator. In their system, stage I tumors are divided into A and B on the basis of whether the surgeon suspects adhesions to adjacent structures; stage III tumors are divided into A and B based on whether disease was subtotally resected or only biopsied. The concurrence between the two systems is high.
|
MASAOKA STAGING SYSTEM FOR THYMOMAS |
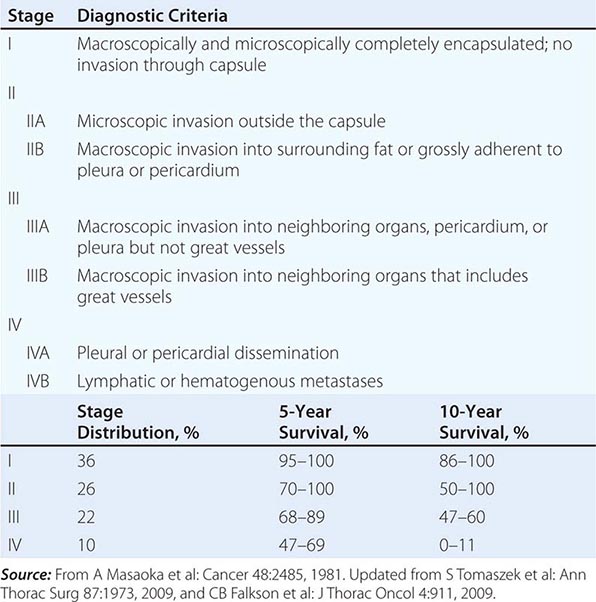
PATHOLOGY AND ETIOLOGY
Thymomas are epithelial tumors, and all of them have malignant potential. It is not worthwhile to try to divide them into benign and malignant forms; the key prognostic feature is whether they are noninvasive or invasive. About 65% of thymomas are encapsulated and noninvasive, and about 35% are invasive. They may have a variable percentage of lymphocytes within the tumor, but genetic studies suggest that the lymphocytes are benign polyclonal cells. The epithelial component of the tumor may consist primarily of round or oval cells derived mainly from the cortex or spindle-shaped cells derived mainly from the medulla or combinations of the two types (World Health Organization classification; Table 123e-2). Cytologic features are not reliable predictors of biologic behavior. In part, this unreliability may be related to the moderate reproducibility of the system. About 90% of A, AB, and B1 tumors are localized. A very small number of patients have aggressive histology features characteristic of carcinomas. Thymic carcinomas are invasive and have a poor prognosis.
|
WORLD HEALTH ORGANIZATION (WHO) HISTOLOGIC CLASSIFICATION OF THYMUS TUMORS |
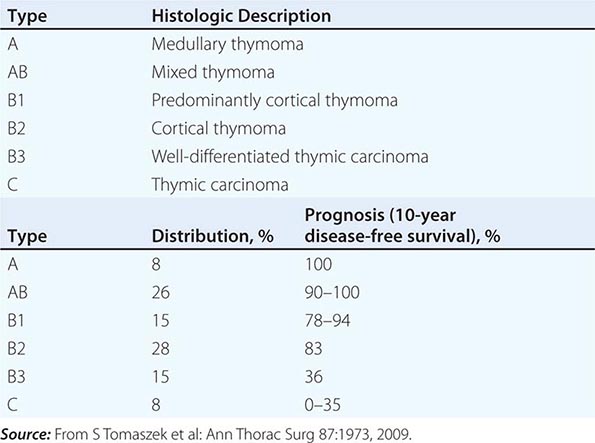
Genetic lesions are common in thymomas. The most common abnormalities affect chromosome 6p21.3 (the MHC locus) and 6p25.2-25.3 (usually loss of heterozygosity). Abnormalities affecting a number of other genes altered in other types of tumors are also seen, including p53, RB, FHIT, and APC. Thymic carcinomas may overexpress c-kit, HER2, or growth factor receptor genes (epidermal growth factor receptor and insulin-like growth factor receptor). Some data suggest that Epstein-Barr virus may be associated with thymomas. Some tumors overexpress the p21 ras gene product. However, molecular pathogenesis remains undefined. A thymoma susceptibility locus has been defined on rat chromosome 7, but the relationship between this gene locus, termed Tsr1, and human thymoma has not been examined.
INFLUENCE OF THYMECTOMY ON THE COURSE OF ACCOMPANYING DISEASES
Patients with myasthenia gravis have a high incidence of thymic abnormalities (~80%), but overt thymoma is present in only ~10–15% of patients with myasthenia gravis. It is thought that the thymus plays a role in breaking self-tolerance and generating T cells that recognize the acetylcholine receptor as a foreign antigen. Although patients with thymoma and myasthenia gravis are less likely to have a remission in the myasthenia as a consequence of thymectomy than are patients with thymic abnormalities other than thymoma, the course of myasthenia gravis is not significantly different in patients with or without thymoma. Thymectomy produces at least some symptomatic improvement in ~65% of patients with myasthenia gravis. In one large series, thymoma patients with myasthenia gravis had a better long-term survival from thymoma resection than did those without myasthenia gravis.
About 30–50% of patients with pure red cell aplasia have a thymoma. Thymectomy results in the resolution of pure red cell aplasia in ~30% of patients. About 10% of patients with hypogammaglobulinemia have a thymoma, but hypogammaglobulinemia rarely responds to thymectomy.
124e |
Neoplasia During Pregnancy |
Cancer complicates ~1 in every 1000 pregnancies. Of all the cancers that occur in women, less than 1% complicate pregnancies. The four cancers that most commonly complicate pregnancies are cervical cancer, breast cancer, melanoma, and lymphomas (particularly Hodgkin’s lymphoma); however, virtually every form of cancer has been reported in pregnant women (Table 124e-1). In addition to cancers developing in other organs of the mother, gestational trophoblastic tumors can arise from the placenta. The problem of cancer in a pregnant woman is complex. One must take into account (1) the possible influence of the pregnancy on the natural history of the cancer, (2) effects on the mother and fetus of complications from the malignancy (e.g., anorexia, nausea, vomiting, malnutrition), (3) potential effects of diagnostic and staging procedures, and (4) potential effects of cancer treatments on both the mother and the developing fetus. Generally, the management that optimizes maternal physiology is also best for the fetus. However, the dilemma occasionally arises that what is best for the mother may be harmful to the fetus, and what is best for the fetus may compromise the ultimate prognosis for the mother. The best way to approach management of a pregnant woman with cancer is to ask, “What would we do for this woman in this clinical situation if she was not pregnant? Now, which, if any, of those plans need to be modified because she is pregnant?”
|
INCIDENCE OF MALIGNANT TUMORS DURING GESTATION |
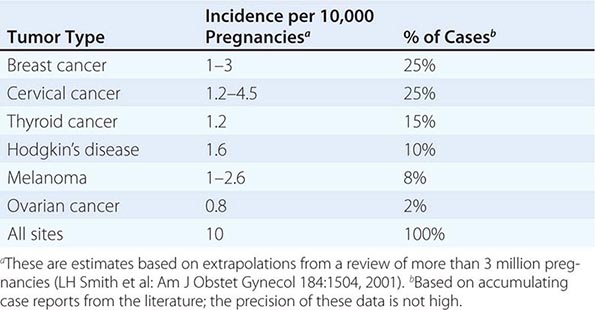
Pregnancy is associated with a number of physiologic changes that frequently result in symptoms that may make it difficult to recognize symptoms or physical findings suggestive of a neoplasm. Increased sensitivity of central chemoreceptors to PCO2 drives an increase in minute ventilation that many women perceive as dyspnea at rest or with minimal exertion. The combination of increased total body water, decreased colloid oncotic pressure, and some obstruction of venous return from the lower extremities causes demonstrable dependent edema in more than 50% of pregnant women. Decreased gastrointestinal motility due to high serum progesterone levels and mechanical compression from an enlarging uterus cause early satiety, gastroesophageal reflux, nausea, vomiting, and constipation. Hemorrhoids develop and often bleed. Breasts enlarge and increase in density and “lumpiness.” These changes may result in delayed recognition and more advanced disease at diagnosis.
Physiologic changes in the maternal immune system necessary to facilitate retention of the fetal semi-allograft raise concerns that the relationship of a cancer with its host may be altered to the detriment of the maternal host. One half of all the genes necessary to create a new individual by sexual reproduction come from each parent. This provides the opportunity for many antigenic differences between the conceptus and the mother. Mammalian placentation has been a very successful method of reproduction, but it has necessitated some combination of both fetal and maternal evolutionary immune adaptations. These mechanisms are incompletely understood and remain an area of active investigation. It does seem likely, however, that this has been accomplished without a general, nonspecific blunting of the maternal immune response, which would be maladaptive to the mother. The multiple mechanisms likely include some “masking” of fetal antigens from recognition by the maternal immune system, blunting the maternal inflammatory response locally at the placental–maternal interface and induction of fetal-specific maternal immune tolerance to avoid rejection. Attention has turned to a subset of CD4+ induced, peripherally produced regulatory T cells that express the × chromosome encoded transcription factor Foxp3 (so-called Tregs). When these Foxp3 cells develop centrally in the thymus, they are termed “Tregs.” When Foxp3-expressing cells develop peripherally, they are called “Pregs.” These regulatory cells suppress the immune response against “self” and foreign antigens. They seem to be capable of suppressing the maternal response to paternal antigens expressed by the fetus and creating memory cells that retain tolerance to the same paternal antigens in subsequent pregnancies. Unfortunately, in a mouse model, the interleukin (IL) 10 produced by these cells enhanced susceptibility to infection by Listeria and Salmonella, while ironically not proving essential for retaining the fetal graft. Undoubtedly much remains to be learned about this critical immune balance.
RADIATION IN PREGNANCY
Exposure of developing fetuses to ionizing radiation may cause adverse fetal effects; awareness among physicians of this potential toxicity has resulted in a disproportionate aversion to diagnostic imaging in pregnancy. First, it must be stated that there are very useful imaging modalities (i.e., ultrasound and magnetic resonance imaging [MRI]) that do not use any ionizing radiation and are not associated with any demonstrable adverse fetal effects. There are three potential adverse fetal effects of ionizing radiation: teratogenesis (induction of anatomic birth defects), mutagenesis, and carcinogenesis. The fetus is most sensitive to teratogenesis during organogenesis in the first trimester. The dose of ionizing radiation necessary to induce birth defects in human fetuses is derived from studies of the survivors of the atomic bomb explosions and by extrapolation from controlled experiments in nonhuman mammals. From these data sources, it is clear that a minimum of 5 rem and more likely greater than 10 rem exposure is needed to induce birth defects in the first trimester. The fetal doses of radiation associated with some common diagnostic radiologic procedures are displayed in Table 124e-2. The data in Table 124e-2 show that no single procedure or selective combination of diagnostic procedures will exceed the very conservative 5 rem teratogenic threshold. Teratogenic effects later in pregnancy are largely limited to microcephaly and require exposures exceeding 25 rem. The reason for the disproportionate concern about radiation exposure and birth defects is that 2.5% of all fetuses are affected with birth defects without radiation exposure and, therefore, 2.5% of women undergoing any diagnostic imaging procedure will deliver malformed fetuses. Spontaneous mutations occur relatively infrequently, and high doses of radiation (>150 rem) are required to cause a demonstrable increase in that rate. The magnitude of the risk of carcinogenesis in offspring exposed as fetuses to diagnostic doses of radiation has been very difficult to measure due to the relative rarity of cancer in children and the long duration of follow-up that might credibly be needed to see the effect. The inconsistent results and small effect sizes observed from diagnostic exposures make it likely that, if there is an effect, it is very small and, if there is not a significant effect, it will be impossible to prove that fact to everyone’s satisfaction. No imaging using ionizing radiation should be done without a compelling reason and due consideration to obtaining the necessary information by other imaging modalities. Exposure to diagnostic and therapeutic radionuclides, especially radioactive iodine, poses unique risks, but a full discussion of these is beyond the scope of this chapter. Radiation therapy uses radiation doses three orders of magnitude greater than diagnostic procedures, entails substantial risks if the fetus is in the radiation field, and is rarely appropriate in pregnancy. Finally, although difficult to prove, it is likely that more harm has come to pregnant women from failing to perform appropriate diagnostic procedures than has been done to their offspring from performing appropriate diagnostic procedures.
|
ESTIMATED FETAL EXPOSURE FROM SOME COMMON RADIOLOGIC PROCEDURES |
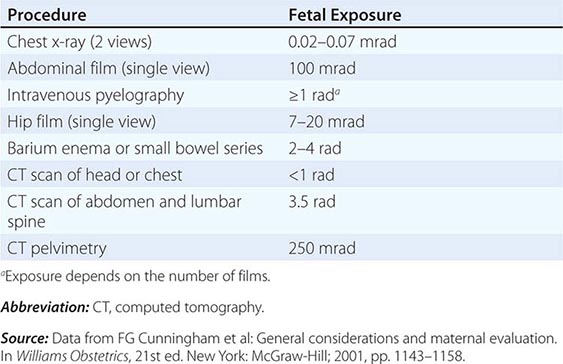
CHEMOTHERAPY IN PREGNANCY
There are a number of reasons why it is impossible to make many definitive statements regarding the safety and efficacy of chemotherapy in pregnancy. All of the available data in the literature are published as case reports or case series. The quality and completeness of the data are inconsistent and often poor. Reports may come from medical oncologists, obstetricians, pediatricians, or other treating physicians familiar with the information important to the report from their own perspective but missing information important for other specialty areas. Reports frequently lack critical details of drug administration, such as dose, duration, cumulative dose, and timing of exposure in gestation, and outcomes, including birth weight and gestational age at delivery, indication for or cause of premature delivery, and follow-up of offspring beyond the immediate neonatal period. There are a wide variety of agents available to treat cancer, and they are usually used in combinations. This results in the fact that every patient is almost unique (an experiment of one) in the combination of agents, doses, durations, and gestational ages of administration, making it very difficult to attribute what benefit or toxicity accrues to which agent. Fortunately, cancer in pregnant women is sufficiently rare that it takes quite a while to accumulate enough information for any one agent or combination of agents to be confident about what toxicities (including congenital malformations) are truly associated with which agents. There is such rapid progress in cancer chemotherapy that by the time there may seem to be enough information about the agents currently in use to use them intelligently and counsel patients meaningfully, the cancer community has moved on to newer, more efficacious, and hopefully less toxic agents for which there is little or no experience in pregnancy. Finally, for obvious reasons, there are no untreated controls for comparison. It may be very difficult to sort out the maternal consequences (nausea, vomiting, fever, weight loss, dehydration) that might result directly from the malignancy and cause adverse pregnancy outcomes from some of the toxicities of the chemotherapeutic agents used to treat the malignancy.
Generally, toxic chemotherapy should be avoided during pregnancy, if at all possible. It should virtually never be given in the first trimester. However, a variety of single agents and combinations have been given in the second and third trimesters, without a high frequency of toxic effects to the pregnancy or the fetus, but data on safety are sparse. Maternal factors that may influence the pharmacology of chemotherapeutic agents include the 50% increase in plasma volume, altered absorption and protein binding, increased glomerular filtration rate, increased hepatic mixed function oxidase activity, and third space created by amniotic fluid. The fetus is protected from some agents by placental expression of drug efflux pumps, but decreased fetal hepatic mixed function oxidase and glucuronidation activity may prolong the half-life of agents that do cross the placenta. A database on the risks associated with individual chemotherapy agents is available on the Internet (http://ntp.niehs.nih.gov/ntp/ohat/cancer_chemo_preg/chemopregnancy_monofinal_508.pdf).
Optimal management strategies have not been developed based on prospective clinical trials. Management of a malignancy complicating pregnancy will be critically determined by the gestational age when the malignancy is diagnosed and the anticipated natural history of the lesion, if left untreated. On one extreme, if the malignancy is slowly progressive, the patient is near her delivery date, and waiting until delivery to begin treatment would not be anticipated to compromise maternal prognosis, then treatment could be delayed until after delivery to avoid fetal exposure to chemotherapy. If there is a greater sense of urgency to begin definitive treatment to avoid compromising maternal prognosis, and the patient is beyond 24 weeks of gestation but remote from her delivery date, then treatment (surgical, medical, or both) might be initiated during pregnancy and plans made to deliver the fetus early to avoid exposure to more chemotherapy than absolutely necessary. Finally, if the patient is in her first trimester and toxic chemotherapy must be initiated promptly to avoid a very poor maternal outcome, then it may be necessary to consider therapeutic abortion to avoid maternal disaster and fetal survival with injury resulting in long-term morbid sequelae. No two cases are precisely alike, and inevitably, decision making must be individualized, preferably with consultation with a multidisciplinary team including medical oncology, surgical oncology if appropriate, maternal–fetal medicine, neonatology, and anesthesia. Pregnancy appears to have little or no impact on the natural history of malignancies, despite the hormonal influences. Spread of the mother’s cancer to the fetus (so-called vertical transmission) is exceedingly rare.
CERVICAL CANCER DURING PREGNANCY
The incidence of cervical cancer in pregnant women is roughly comparable to that of age-matched controls who are not pregnant. Invasive cervical cancer complicates about 0.45 in 1000 live births, and carcinoma in situ is seen in 1 in 750 pregnancies. About 1% of women diagnosed with cervical cancer are pregnant at the time of diagnosis. Early signs of cervical cancer include vaginal spotting or discharge, pain, and postcoital bleeding, which are also common features of pregnancy. Early visual changes in the cervix related to invasive cancer can be mistaken for cervical decidualization or ectropion (columnar epithelium on the cervix) due to pregnancy. Women diagnosed with cervical cancer during pregnancy report having had symptoms for 4.5 months on average.
Approximately 95% of all cervical cancer is caused by human papillomavirus (HPV) infections, with types 16 and 18 accounting for about 70% of cervical cancer. The rate of carriage of these serotypes is highest among women in their early twenties and can be reduced with the use of vaccination before exposure. Women generally tend to clear the infection by age 30, with the risk of cervical cancer being highest among those who fail to clear the infection. Screening is recommended at the first prenatal visit and 6 weeks postpartum. The rate of cytologic abnormalities on Pap smear in pregnant women is about 5–8% and is not much different than the rate in nonpregnant women of the same age.
In 2012, several sets of recommendations were published for screening for cervical cancer: one by the American Cancer Society (ACS), the American Society for Colposcopy and Cervical Pathology (ASCCP), and the American Society for Clinical Pathology (ASCP); a second by the U.S. Preventive Services Task Force (USPSTF); and a third by the American College of Obstetricians and Gynecologists (ACOG). Although the details of the recommendations for screening and management of abnormal results differ slightly among the three sets of guidelines, there is general consensus that cytology screening should start at age 21 and continue every 3 years through age 29. After age 30, cytology screening frequency may be reduced to every 5 years if accompanied by co-testing for HPV. Recommendations for management of abnormal cytology findings are complex and determined by the degree of abnormality of the cytology finding (e.g., atypical squamous cells of undetermined significance; atypical squamous cells, cannot exclude high-grade squamous intraepithelial lesion; low-grade squamous intraepithelial lesion; or high-grade squamous intraepithelial lesion), the HPV status of the patient, the age of the patient, and whether this is the first abnormal finding or a persistent abnormality. A full discussion of all the treatment recommendations based on these factors is beyond the scope of this chapter. Some of the diagnostic procedures recommended for evaluation of nonpregnant women are contraindicated in pregnancy, and the indications for some procedures are modified in the setting of pregnancy. Suffice it to say that the evaluation of women with abnormal cervical cytology in pregnancy should be referred to knowledgeable and experienced gynecologists or gynecologic oncologists.
Cervical intraepithelial neoplasia is a slowly progressive lesion and has a low risk of progression to invasive cancer during pregnancy (~0.4%), and many low-grade lesions (36–70%) regress spontaneously. Accordingly, some physicians defer definitive diagnostic procedures in pregnant women until 6 weeks postpartum unless they are at high risk for invasive disease. If invasive disease is suspected and the pregnancy is between 16 and 20 weeks, a cone biopsy may be performed to make the diagnosis and may be curative for some lesions; however, the procedure may cause heavy bleeding due to the increased vasculature in the gravid cervix and increases the risk of premature rupture of membranes and preterm labor two- to threefold. Cone biopsy should not be done within 4 weeks of delivery. The only indication for therapy of cervical neoplasia in pregnant women is the documentation of invasive cancer.
Management of invasive disease is guided by the stage of disease, the gestational age of the fetus, and the desire of the mother to have the baby. If the disease is in early stage and the pregnancy is desired, it is safe to delay treatment regardless of gestational age until fetal maturity allows for safe delivery. Abortion followed by definitive therapy is recommended for women with advanced, but potentially curable, cancer in the first or second trimester (Chap. 117). If the disease is in an advanced stage in early pregnancy and the patient declines pregnancy termination to permit prompt definitive therapy, she must be informed of the fact that the maternal safety of delaying therapy is unproven. In women in the third trimester with advanced disease, the mother should be treated with betamethasone to accelerate fetal lung maturation and the baby should be delivered at the earliest possible gestational age followed immediately by stage-appropriate therapy. Most women with invasive cancer have early-stage disease. If the disease is microinvasive, vaginal delivery can take place and be followed by definitive treatment, usually conization. If a lesion is visible on the cervix, delivery is best done by caesarian section and followed by radical hysterectomy.
BREAST CANCER DURING PREGNANCY
Breast cancer complicates approximately 1 in 3000 to 10,000 live births. About 5% of all breast cancers occur in women age 40 years or younger. Among all premenopausal women with breast cancer, 25–30% were pregnant at the time of diagnosis. It has been recognized for some time that breast cancer associated with pregnancy generally seems to have a poorer prognosis for both overall survival and progression-free survival. The definition of pregnancy-associated breast cancer (PABC) has differed in various publications, but a generally accepted definition is breast cancer diagnosed during pregnancy or within 1 year of delivery. There are likely several reasons for the observation of the poorer prognosis. Breast cancers diagnosed during pregnancy are often diagnosed at a later stage of disease and so have a poorer outcome. The late diagnosis is often due to the fact that early physical signs of the disease are missed or attributed to the changes that occur in the breast normally as a function of pregnancy. However, a discreet breast mass in a pregnant woman should never be assumed to be normal. Another reason is the more aggressive behavior of the cancer possibly related to the hormonal milieu (estrogen increases 100-fold; progesterone increases 1000-fold) of the pregnancy. However, about 70% of the breast cancers found in pregnancy are estrogen receptor–negative. About 28–58% of the tumors express HER2, a biologically more aggressive breast cancer subset. Another factor is that aggressive, definitive chemotherapy and radiation therapy are often delayed due to concerns about the consequences of those treatments for the fetus. Younger women with breast cancer have a higher likelihood of having mutations in BRCA1 or BRCA2.
Differences in presentation between PABC and breast cancers diagnosed in nonpregnant women are shown in Table 124e-3. About 20% of breast cancers are detected in the first trimester, 45% in the second trimester, and 35% in the third trimester. Some argue that stage for stage, the outcome is the same for breast cancer diagnosed in pregnant and nonpregnant women. Primary tumors in pregnant women are 3.5 cm on average, compared to <2 cm in nonpregnant women. A dominant mass and a nipple discharge are the most common presenting signs, and they should prompt ultrasonography and breast MRI exam (if available) followed by lumpectomy if the mass is solid and aspiration if the mass is cystic. Mammography is less reliable in pregnancy due to the increased breast density. Needle aspirates of breast masses in pregnant women are often nondiagnostic or falsely positive. Even in pregnancy, most breast masses are benign (~80% are adenoma, lobular hyperplasia, milk retention cyst, fibrocystic disease, fibroadenoma, or other rarer entities).
|
DIFFERENCES IN BREAST CANCERS IN PREGNANT AND NONPREGNANT WOMEN |
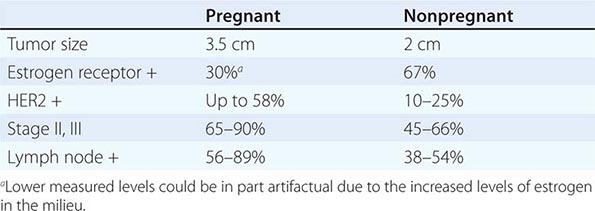
Many studies comparing outcomes among women with PABC to those of nonpregnant women have small sample sizes, and there is considerable heterogeneity among the study results, but a formal meta-analysis including multiple adjustments and sensitivity analyses confirms the clinical impression of poorer outcomes for women with PABC. The hazard ratios were 1.44 for poorer overall survival and 1.60 for poorer disease-free survival.
Although having had a pregnancy is a protective factor against breast cancer in women in general, it is questionable as to whether it retains its protective effect in carriers of BRCA1 and BRCA2 mutations. Cullinane et al. (Int J Cancer 117:988, 2005) found a statistically insignificant difference (odds ratio 0.94) in breast cancer risk among BRCA1 carriers who had ever been pregnant versus those who never had a pregnancy. Stratifying the risk of breast cancer according to the number of prior pregnancies versus no pregnancies, no statistically significant protective trend was observed. For BRCA2 carriers, there was a marginally statistically significant increased risk of breast cancer among women with prior pregnancies. In an international study with more than 65,000 person-years of observation (Andrieu J: Natl Cancer Inst 98:535, 2006), there was no significant effect in either direction of pregnancy on breast cancer risk for carriers of either mutation. Staging the axillary lymph nodes is currently somewhat controversial. Sentinel lymph node sampling is not straightforward in pregnant women. Blue dye has been carcinogenic in rats, and fetuses cannot be shielded from administered radionuclides. For this reason, many surgeons favor axillary node dissection to stage the nodes. Largely due to the typical delay in diagnosis, axillary nodes are more often positive in pregnant than in nonpregnant women.
As with other types of cancer in pregnant women, counseling following diagnosis in the first trimester should include a discussion of pregnancy termination to allow definitive therapeutic intervention at the earliest possible time without the potential for permanent injury to a surviving fetus. While definitive local surgery can safely be performed in the first trimester, radiation therapy and chemotherapy are considerably more risky. Delay in administration of systemic therapy can increase the risk of axillary spread. In the second and third trimesters, chemotherapy (particularly anthracycline-based combinations) is both safe and effective (Chap. 108). Lumpectomy followed by adjuvant chemotherapy is frequently used; fluorouracil and cyclophosphamide with either doxorubicin or epirubicin have been given without major risk to the fetus. Taxanes and gemcitabine are also beginning to be used; however, safety data are sparse. Methotrexate and other folate antagonists are to be avoided because of effects on the fetal nervous system. Myelotoxic therapy is generally not administered after 33 or 34 weeks of gestation to allow 3 weeks off therapy before delivery for recovery of blood counts. Endocrine therapy and trastuzumab are unsafe during pregnancy. Experience with lapatinib is anecdotal, but no fetal malformations have been reported. Antiemetics and colony-stimulating factors are also considered safe. Women being treated into the postpartum period should not breast-feed their babies because of excretion of cancer chemotherapy agents, particularly alkylating agents, in milk.
Subsequent pregnancies following gestational breast cancer do not appear to influence relapse rate or overall survival. A meta-analysis found that pregnancy in breast cancer survivors was associated with a reduced the risk of dying from breast cancer by as much as 42%. This finding, however, is heavily confounded by the “healthy survivor effect”; women with more extensive or advanced disease are more likely to avoid pregnancy.
MELANOMA DURING PREGNANCY
Speculation about melanoma occurring during pregnancy based largely on anecdotal evidence and small case series concluded that it occurred with increased frequency, was more aggressive in its natural history, and was caused in part by the hormonal changes that also produced hyperpigmentation (so-called melasma) during pregnancy. However, more complete epidemiologic data suggest that melanoma is no more frequent in pregnant women than in nonpregnant women in the same age group, that melanoma is not more aggressive during pregnancy, and that hormones seem to have little or nothing to do with the etiology. Pregnant and nonpregnant women do not differ in the location of primary tumor, depth of primary tumor, tumor ulceration, or vascular invasion.
Suspicious lesions should be looked for and managed definitively with excisional biopsy during pregnancy. Wide excision with sampling of regional lymph nodes is warranted. If lymph nodes are involved, the course of action is less clear. Several agents have demonstrated some activity in melanoma, but none have been used during pregnancy. Adjuvant interferon α is toxic, and its safety in pregnancy has not been documented. Agents active in advanced disease include dacarbazine, IL-2, ipilimumab (antibody to CTLA-4), and in those with BRAF mutation V600E, a BRAF kinase inhibitor. In the setting of metastatic disease, abortion may be indicated so that systemic therapy can be initiated as soon as possible (Chap. 105).
Melanoma is one of the very few cancers that are well documented to metastasize transplacentally to the fetus, where it seems to have a predilection for the head and neck. It has a very grave prognosis in the offspring. Fortunately, transplacental spread is rare.
Pregnancy subsequent to the diagnosis and treatment of melanoma is not associated with an increased risk of melanoma recurrence.
HODGKIN’S DISEASE AND NON-HODGKIN’S LYMPHOMA DURING PREGNANCY
(See Chap. 134) Hodgkin’s disease occurs mainly in the age range that coincides with child-bearing. However, Hodgkin’s disease is not more common in pregnant than nonpregnant women. Hodgkin’s disease is diagnosed in approximately 1 in 6000 pregnancies. It generally presents as a nontender lymph node swelling, most often in the left supraclavicular region. It may be accompanied by B symptoms (fever, night sweats, unexplained weight loss). Excisional biopsy is the preferred diagnostic procedure because fine-needle aspiration cannot reveal the architectural framework that is an essential component of Hodgkin’s disease diagnosis. The stage at presentation appears to be unaffected by pregnancy. Women diagnosed in the second and third trimester can be treated safely with combination chemotherapy, usually doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD). In general, the patient in the first trimester is asymptomatic, and a woman with a desired pregnancy can be followed until the second or third trimester when definitive multiagent chemotherapy can be safely given. Radiation therapy is not given during pregnancy and is not necessary for optimal management of the pregnant patient. If symptoms requiring treatment appear during the first trimester, anecdotal evidence suggests that Hodgkin’s disease symptoms can be controlled with weekly low-dose vinblastine. Such an approach has been safely used to avoid termination of pregnancy. Pregnancy does not have an adverse effect on treatment outcome.
Non-Hodgkin’s lymphomas are more unusual in pregnancy (approximately 0.8 per 100,000 pregnancies), but are usually tumors with an aggressive natural history, such as diffuse large B cell lymphoma, Burkitt’s lymphoma, or peripheral T cell lymphoma. Diagnosis relies on an excisional biopsy of a tumor mass, not fine-needle aspiration. Staging evaluation is generally limited to ultrasound or MRI examinations. Diagnosis in the first trimester should prompt termination of the pregnancy followed by definitive treatment with combination chemotherapy, because aggressive lymphomas are not likely to be held at bay with single-agent chemotherapy. Women diagnosed in the second or third trimesters can be treated with standard chemotherapy, such as with cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP). The experience with rituximab in this setting is anecdotal. However, infants born of mothers who have received rituximab may have transient delay in B cell development that typically normalizes by 6 months. The treatment outcome is similar in lymphomas diagnosed in pregnant and nonpregnant women of the same clinical stage.
THYROID CANCER DURING PREGNANCY
(See Chap. 405) Thyroid cancer, along with melanomas, brain tumors, and lymphomas, are cancers that are increasing in incidence in the general population. Thyroid cancers are rising faster among women in North America than the other increasing tumor types. The Endocrine Society has developed practice guidelines to inform the management of patients with thyroid disease during pregnancy (http://www.endocrine.org/~/media/endosociety/Files/Publications/Clinical%20Practice%20Guidelines/Thyroid-Exec-Summ.pdf). Thyroid nodules 1 cm or larger are approached by fine-needle aspiration. If a malignancy is diagnosed, surgery is generally recommended in the second and third trimesters. However, surgical complications appear to be twice as common when the patient is pregnant. Because the growth of thyroid tumors is often indolent, surgery can safely be postponed until after the first trimester. Patients with follicular cancer or early papillary cancer can be observed until the postpartum period. The fetal thyroid begins trapping iodine by 12 weeks of gestation and does so with very high avidity. Even small doses of radioactive iodine given during pregnancy can completely ablate the fetal thyroid with serious consequences for the fetus and should be avoided throughout pregnancy. Radioactive iodine can be safely administered after delivery. Patients with a history of thyroid cancer who become pregnant should be maintained on thyroid hormone replacement during pregnancy because of the adverse impact of maternal hypothyroidism on the fetus. Women who are breast-feeding should not be treated with radioactive iodine, and women treated with radioactive iodine should not become pregnant for 6–12 months after treatment.
The assessment of thyroid function during pregnancy is challenging because of the physiologic changes that occur during pregnancy. Women who have previously been treated for thyroid cancer are at risk of hypothyroidism. The demand for thyroid hormone increases during pregnancy, and doses to maintain normal function may increase by 30–50%. Total T4 levels are higher during pregnancy, but target therapeutic levels also increase (Table 124e-4). It is recommended that the upper and lower limits of the laboratory range be multiplied by 1.5 in the second and third trimester to establish a pregnancy-specific normal range. The target thyroid-stimulating hormone (TSH) level is <2.5 mIU/L.
|
THYROID FUNCTION TEST DURING PREGNANCY (MEAN LEVELS) |
GESTATIONAL TROPHOBLASTIC DISEASE
(See Chap. 117) Gestational trophoblastic disease encompasses hydatidiform mole, choriocarcinoma, placental site trophoblastic tumor, and assorted miscellaneous and unclassifiable trophoblastic tumors. Moles are the most common, occurring in 1 in 1500 pregnancies in the United States. The incidence is higher in Asia. In general, if the serum level of β-human chorionic gonadotropin (β-hCG) returns to normal after surgical removal (evacuation) of the mole, the illness is considered gestational trophoblastic disease. By contrast, if the β-hCG level remains elevated after mole evacuation, the patient is considered to have gestational trophoblastic neoplasia. Choriocarcinoma occurs in 1 in 25,000 pregnancies. Maternal age >45 years and prior history of molar pregnancy are risk factors. A previous molar pregnancy makes choriocarcinoma about 1000 times more likely to occur (incidence 1–2%).
Hydatidiform moles are characterized by clusters of villi with hydropic changes, trophoblastic hyperplasia, and absence of fetal blood vessels. Invasive moles are distinguished by invasion of the myometrium. Placental site trophoblastic tumors are composed mainly of cytotrophoblast cells arising at the site of placental implantation. Choriocarcinomas contain anaplastic trophoblastic tissue with both cytotrophoblast and syncytiotrophoblast features and no identifiable villi.
Moles can be partial, typically associated with fetal tissue, or complete, typically not associated with any fetal or embryonic tissue. Partial moles have a distinct molecular origin and usually are smaller tumors with less hydropic villi and considerably less potential for persistent or malignant disease. Partial moles result from fertilization of an egg by two sperm, resulting in diandric triploidy. Complete moles usually have a 46,XX genotype; 95% develop by a single male sperm fertilizing an empty egg and undergoing gene duplication (diandric diploidy); 5% develop from dispermic fertilization of an empty egg (diandric dispermy).
Women with molar gestations often present with first-trimester bleeding, disproportionately high serum β-hCG levels for menstrual age, unusually large uterine size for menstrual age, hyperemesis gravidarum, theca lutein cysts in the ovaries (due to β-hCG stimulation), and hyperthyroidism (due to cross-reactivity of β-hCG and TSH) and may develop preeclampsia before 20 weeks of menstrual age. Pelvic ultrasound imaging of complete moles shows absence of fetal parts, an enlarged echo-bright, hydropic placenta in an enlarged uterus, and enlarged multicystic ovaries. If the diagnosis is uncertain at the initial examination and the pregnancy is desired, then a serum β-hCG level should be obtained and the examination repeated in a week. If no embryo is seen within 7–10 days and the serum β-hCG is elevated, then this is a nonviable pregnancy that should be evacuated. Diagnosis of partial molar pregnancies can be more difficult because an embryo or fetus with visible heart motion is usually present, and the hydropic changes in the placenta, uterine enlargement, and elevations of β-hCG are not usually as dramatic. Although an embryo or fetus is present, it rarely grows normally with normal anatomy, and repeated ultrasound examinations usually make the diagnosis. Amniocentesis will also make the diagnosis by demonstration of triploidy.
Patients with molar pregnancies require prompt uterine evacuation with suction curettage, which may be complicated by very heavy bleeding. Following evacuation of complete moles, approximately 20% will result in persistent, invasive, or metastatic disease. Partial moles are considerably less likely (<5%) to result in persistent disease. Patients should be monitored with serial determinations of serum β-hCG until the values fall below the lower limit of the assay and remain low for at least 6 months. Patients should be advised not to become pregnant for at least 12 months.
A variety of criteria have been used to make the diagnosis of postmolar gestational trophoblastic disease, but current consensus guidelines as adopted by the International Federation of Gynecology and Obstetrics are listed below:
1. A β-hCG level plateau of four values plus or minus 10% recorded over a 3-week duration (days 1, 7, 14, and 21)
2. A β-hCG level increase of more than 10% in three values recorded over a 2-week duration (days 1, 7, and 14)
3. Persistence of detectable β-hCG for more than 6 months after molar evacuation
About half of choriocarcinomas develop after a molar pregnancy, and half develop after ectopic pregnancy or, rarely, after a normal full-term pregnancy. Disease is classified as stage I if it is confined to the uterus, stage II if disease is limited to genital structures (~30% have vaginal involvement), stage III if disease has spread to the lungs but no other organs, and stage IV if disease has spread to liver, brain, or other organs.
Patients without widely metastatic disease are generally managed with single-agent methotrexate (either 30 mg/m2 IM weekly until β-hCG normalizes or 1 mg/kg IM every other day for four doses followed by leucovorin 0.1 mg/kg IV 24 h after methotrexate), which cures >90% of patients. Patients with very high β-hCG levels, presenting >4 months after a pregnancy, with brain or liver metastases, or failing to be cured by single-agent methotrexate are treated with combination chemotherapy. Etoposide, methotrexate, and dactinomycin alternating with cyclophosphamide and vincristine (EMA-CO) is the most commonly used regimen, producing long-term survival in >80% of patients. Brain metastases can usually be controlled with brain radiation therapy. The vast majority of choriocarcinomas can be cured with chemotherapy alone. Hysterectomy is reserved for women who have completed their child-bearing, women with chemotherapy-resistant disease in the uterus, and women with rare placental site trophoblastic tumors confined to the uterus because these tumors are less reliably sensitive to chemotherapy. Women cured of trophoblastic disease who have not undergone hysterectomy do not appear to have increased risk of fetal abnormalities or maternal complications with subsequent pregnancies.
125 |
Late Consequences of Cancer and Its Treatment |
There are over 10 million American cancer survivors. The vast majority of these will bear some mark of their cancer and its treatment, and a large proportion will experience long-term consequences including medical problems, psychosocial dysfunction, economic hardship, sexual dysfunction, and discrimination regarding employment and insurance. Many of these problems are directly related to cancer treatment. As patients survive longer from more types of malignancies, we are increasingly recognizing the biologic toll our very imperfect therapies take in terms of morbidity and mortality. The human face of these consequences of therapy confronts the cancer specialist who treats them every day. Although long-term survivors of childhood leukemias, Hodgkin’s lymphoma, and testicular cancer, as examples, have taught us much about the consequences of cancer treatment, we keep learning more as patients survive longer with newer therapies. Newer “targeted” chemotherapy drugs have their own, often unique, long-term toxicities about which we remain in a learning process. Cancer “survivorship” clinics are increasing to expressly follow patients for long-term toxicities of cancer treatment.
The pace of developing therapies that mitigate treatment-related consequences has been slow, partly due to an understandable aversion to alter regimens that work and partly due to a lack of new, effective, less toxic therapeutic agents with less “collateral damage” to replace known agents with known toxicities. The types of damage from cancer treatment vary. Often, a final common pathway is irreparable damage to DNA. Surgery can create dysfunction, including blind gut loops with absorption problems and loss of function of removed body parts. Radiation may damage end-organ function, for example, loss of potency in prostate cancer patients, pulmonary fibrosis, and neurocognitive impairment, and may act as a direct carcinogen. Cancer chemotherapy can be a direct carcinogen and has a kaleidoscope of other toxicities discussed in this chapter. Table 125-1 lists the late effects of cancer treatment.
|
LATE EFFECTS OF CANCER THERAPY |
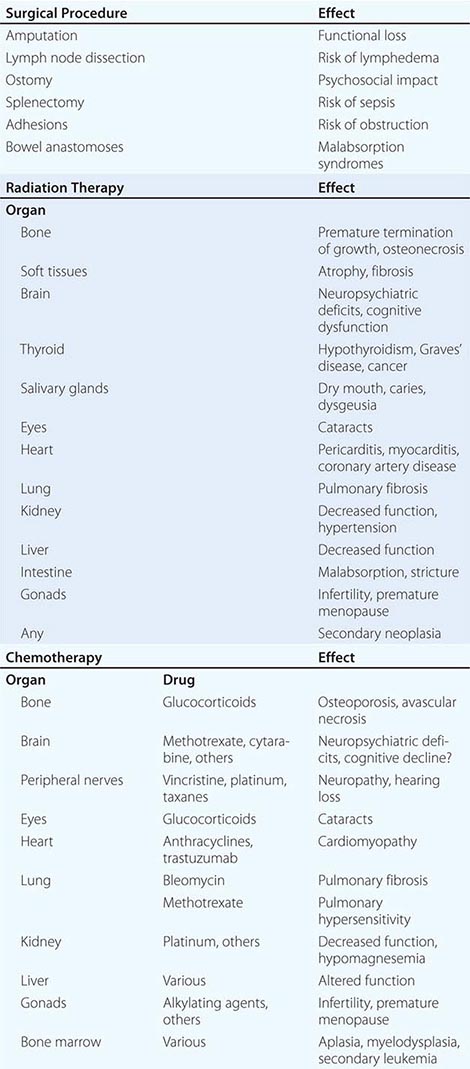
The first goal of therapy is to eradicate or control the malignancy. Late treatment consequences are, indeed, testimony to the increasing success of such treatment. Their occurrence sharply underlines the necessity to develop more effective therapies with less long-term morbidity and mortality. At the same time, a sense of perspective and relative risk is necessary; fear of long-term complications should not prevent the application of effective, particularly curative, cancer treatment.
CARDIOVASCULAR DYSFUNCTION
CHEMOTHERAPEUTIC AGENTS
Cardiovascular toxicity of cancer chemotherapeutic agents includes dysrhythmias, cardiac ischemia, cardiomyopathic congestive heart failure (CHF), pericardial disease, and peripheral vascular disease. Because these cardiac toxicities are difficult to distinguish from disease that is not associated with cancer treatment, clear etiologic implication of cancer chemotherapeutic agents may be difficult. Cardiovascular complications occurring in an unexpected clinical setting in patients who have undergone cancer therapy are often important in raising suspicion. Dose-dependent myocardial toxicity of anthracyclines with characteristic myofibrillar dropout is pathologically pathognomonic on endomyocardial biopsy. Anthracycline cardiotoxicity occurs through a root mechanism of chemical free radical damage. Fe3+-doxorubicin complexes damage DNA, nuclear and cytoplasmic membranes, and mitochondria. About 5% of patients receiving >450–550 mg/m2 of doxorubicin will develop CHF. Cardiotoxicity in relation to the dose of anthracycline is clearly not a step function, but rather a continuous function, and occasional patients are seen with CHF at substantially lower doses. Advanced age, other concomitant cardiac disease, hypertension, diabetes, and thoracic radiation therapy are all important cofactors in promoting anthracycline-associated CHF. The risk of cardiac failure appears to be substantially lower when doxorubicin is administered by continuous infusion. Anthracycline-related CHF is difficult to reverse and has a mortality rate as high as 50%, making prevention crucial. Some anthracyclines such as mitoxantrone are associated with less cardiotoxicity, and continuous-infusion regimens and liposomally encapsulated doxorubicin are associated with less cardiotoxicity. Dexrazoxane, an intracellular iron chelator, may limit anthracycline toxicity, but the concern of limiting chemotherapeutic efficacy has somewhat limited its use. Monitoring patients for cardiac toxicity typically involves periodic gated nuclear cardiac blood pool ejection fraction testing (multigated acquisition scan [MUGA]) or cardiac ultrasonography. More recently, cardiac magnetic resonance imaging (MRI) has been used, but MRI is not standard or widespread. Testing is performed more frequently at higher cumulative doses, with additional risk factors, and certainly for any newly developing CHF or other symptoms of cardiac dysfunction.
After anthracyclines, trastuzumab is the next most frequent cardiotoxic drug currently in use. Trastuzumab is frequently used as adjuvant breast cancer therapy, sometimes in conjunction with anthracyclines, which is believed to result in additive or possibly synergistic toxicity. In contrast to anthracyclines, cardiotoxicity is not dose-related, is usually reversible, is not associated with pathologic changes of anthracyclines on cardiac myofibrils, and has a different biochemical mechanism inhibiting intrinsic cardiac repair mechanisms. Toxicity is typically routinely monitored every three to four doses using functional cardiac testing as mentioned earlier for anthracyclines.
Other cardiotoxic drugs include lapatinib, phosphoramide mustards (cyclophosphamide), ifosfamide, interleukin 2, ponatinib, imatinib, and sunitinib.
RADIATION THERAPY
Radiation therapy that includes the heart can cause interstitial myocardial fibrosis, acute and chronic pericarditis, valvular disease, and accelerated premature atherosclerotic coronary artery disease. Repeated or high (>6000 cGy) radiation doses are associated with greater risk, as is concomitant or distant cardiotoxic cancer chemotherapy exposure. Symptoms of acute pericarditis, which peaks about 9 months after treatment, include dyspnea, chest pain, and fever. Chronic constrictive pericarditis may develop 5–10 years following radiation therapy. Cardiac valvular disease includes aortic insufficiency from fibrosis or papillary muscle dysfunction resulting in mitral regurgitation. A threefold increased risk of fatal myocardial infarction is associated with mantle field radiation with accelerated coronary artery disease. Carotid radiation similarly increases the risk of embolic stroke.
PULMONARY DYSFUNCTION
CHEMOTHERAPEUTIC AGENTS
Bleomycin generates activated free radical oxygen species and causes pneumonitis associated with a radiographic or interstitial ground-glass appearance diffusely throughout both lungs, often worse in the lower lobes. A nonproductive cough with or without fever may be an early sign. This toxicity is dose-related and dose-limiting. The diffusion capacity of the lungs for carbon dioxide (DLCO) is a sensitive measure of toxicity and recovery, and a baseline value is generally obtained for future comparison prior to bleomycin therapy. Additive or synergistic risk factors include age, prior lung disease, and concomitant use of other chemotherapy, lung irradiation, and high concentrations of inspired oxygen. Other chemotherapeutic agents notable for pulmonary toxicity include mitomycin, nitrosoureas, doxorubicin with radiation, gemcitabine combined with weekly docetaxel, methotrexate, and fludarabine. High-dose alkylating agents, cyclophosphamide, ifosfamide, and melphalan are frequently used in the hematopoietic stem cell transplant setting, often with whole-body radiation. This therapy may result in severe pulmonary fibrosis and/or pulmonary venoocclusive disease.
RADIATION THERAPY
Risk factors for radiation pneumonitis include advanced age, poor performance status, preexisting compromised pulmonary function, and radiation volume and dose. The dose “threshold” is thought to be in the range of 5 to 20 Gy. Hypoxemia and dyspnea on exertion are characteristic. Fine, high-pitched “Velcro rales” may be an accompanying physical finding, and fever, cough, and pleuritic chest pain are common symptoms. The DLCO is the most sensitive measure of pulmonary functional impairment, and ground-glass infiltrates often correspond with relatively sharp edges to the irradiated volume, although the pneumonitis may progress beyond the field and even occasionally involve the contralateral unirradiated lung.
NEUROLOGIC DYSFUNCTION
CHEMOTHERAPEUTIC AGENTS
Chemotherapy- and radiation-induced neurologic dysfunction is unfortunately increasing in both incidence and severity as a result of improved supportive care leading to more aggressive regimens and longer cancer survivorship allowing the development of late toxicity. Direct effects on myelin, glial cells, and neurons have all been implicated, with alterations in cellular cytoskeleton, axonal transport, and cellular metabolism as mechanisms.
Vinca alkaloids produce a characteristic “stocking-glove” neuropathy with numbness and tingling advancing to loss of motor function, which is highly dose related. Distal sensorimotor polyneuropathy prominently involves loss of deep tendon reflexes with initially loss of pain and temperature sensation, followed by proprioceptive and vibratory loss. This requires careful patient history and physical examination by experienced oncologists to decide when the drug must be stopped due to toxicity. Milder toxicity often slowly completely resolves. Vinca alkaloids may sometimes be associated with jaw claudication, autonomic neuropathy, ileus, cranial nerve palsies, and, in severe cases, encephalopathy, seizures, and coma.
Cisplatin is associated with sensorimotor neuropathy and hearing loss, especially at doses >400 mg/m2, requiring audiometry in patients with preexisting hearing compromise. Carboplatin is often substituted in such cases given its lesser effect on hearing.
Many of the agents that target kinase enzymes in tumor cells and 5-fluorouracil congeners produce dysesthesias and painful hands and feet known as hand-foot syndrome or palmar-plantar erythrodysesthesia. Symptoms usually abate when the agent is stopped.
Neurocognitive dysfunction has been well described in childhood survivors of acute lymphoblastic leukemia (ALL) treatment, including intrathecal methotrexate or cytosine arabinoside in conjunction with prophylactic cranial irradiation. Methotrexate alone may cause acute leukoencephalopathy characterized by somnolence and confusion that is often reversible. Acute toxicity is dose related, especially at doses >3 g/m2, with younger patients being at greater risk. Subacute methotrexate toxicity occurs weeks after therapy and is often ameliorated with glucocorticoid therapy. Chronic methotrexate toxicity (leukoencephalopathy) develops months or years after treatment and is characterized clinically as progressive loss of cognitive function and focal neurologic signs, which are irreversible, promoted by synchronous or metachronous radiation therapy, and more pronounced at a younger age.
Neurocognitive decline following chemotherapy alone occurs notably in breast cancer patients receiving adjuvant chemotherapy; this has been referred to as “chemo brain.” It is clinically associated with impaired memory, learning, attention, and speed of information processing. There is no clear mechanistic explanation for its cause and no clearly effective therapy. This entity is justifiably attracting more attention and clearly needs to be studied to develop effective therapy or prophylaxis.
Many cancer patients experience intrusive or debilitating concerns about cancer recurrence following successful therapy. In addition, these patients may experience job, insurance, stress, relationship, financial, and sexual difficulties. Oncologists need to ask about and address these issues explicitly with patients and provide appropriate counseling or support systems. Suicidal ideation and suicide have an increased incidence in cancer patients and survivors.
RADIATION THERAPY
Acute radiation central nervous system (CNS) toxicity occurs within weeks; is characterized by nausea, drowsiness, hypersomnia, and ataxia; and is most often associated with recovery. Early delayed toxicity occurring weeks to 3 months following therapy is associated with similar symptoms as acute toxicity and is pathologically associated with reversible demyelination. Chronic, late radiation injury occurs 9 months to up to 10 years following therapy. Focal necrosis is a common pathologic finding, and glucocorticoid therapy may be helpful. Diffuse radiation injury is associated with global CNS neurologic dysfunction and diffuse white matter changes on computed tomography (CT) or MRI. Pathologically, small vessel changes are prominent. Glucocorticoids may be symptomatically useful but do not alter the course. Necrotizing encephalopathy is the most severe form of radiation injury and almost always is associated with chemotherapy, notably methotrexate.
Cranial radiation may also be associated with an array of endocrine abnormalities with disruption of normal pituitary/hypothalamic axis function, and a high index of suspicion needs to be maintained to identify and treat this toxicity.
Radiation-associated spinal cord injury (myelopathy) is highly dose-dependent and rarely occurs with modern radiation therapy. An early, self-limited form involving electric sensations down the spine on neck flexion (Lhermitte’s sign) is seen 6–12 weeks after treatment and generally resolves over weeks. Peripheral nerve toxicity is quite rare owing to relative radiation resistance.
HEPATIC DYSFUNCTION
CHEMOTHERAPEUTIC AGENTS
Long-term hepatic damage from standard chemotherapy regimens is rare. Long-term methotrexate or high-dose chemotherapy alone or with radiation therapy, for example, in preparative regimens for bone marrow transplantation, may result in venoocclusive disease of the liver. This potentially lethal complication classically presents with anicteric ascites, elevated alkaline phosphatase, and hepatosplenomegaly. Pathologically, there is venous congestion, epithelial cell proliferation, and hepatocyte atrophy progressing to frank fibrosis. Frequent monitoring of liver function tests during any chemotherapy is necessary to avoid both idiosyncratic and expected toxicities.
Certain nucleoside drugs have been associated with hepatic dysfunction; however, this complication is rare in oncology.
RADIATION THERAPY
Hepatic radiation damage depends on dose, volume, fractionation, preexisting liver disease, and synchronous or metachronous chemotherapy. In general, radiation doses to the liver >1500 cGy can produce hepatic dysfunction with a steep dose-injury curve. Radiation-induced liver disease closely mimics hepatic venoocclusive disease.
RENAL/BLADDER DYSFUNCTION
Cisplatin produces reversible decrements in renal function, but may also produce severe irreversible toxicity in the presence of renal disease and may predispose to accentuated damage with subsequent renal insults. Cyclophosphamide and ifosfamide, as prodrugs primarily activated in the liver, have cleavage products (acrolein) that can produce hemorrhagic cystitis. This can be prevented with the free radical scavenger MESNA (mercaptoethane sulfonate), which is required for ifosfamide administration. Hemorrhagic cystitis caused by these agents may predispose to bladder cancer.
REPRODUCTIVE AND ENDOCRINE DYSFUNCTION
CHEMOTHERAPEUTIC AGENTS
Alkylating agents are associated with the highest rates of male and female infertility, which is directly dependent on age, dose, and duration of treatment. The age at treatment is an important determinant of fertility outcome, with prepubertal patients having the highest tolerance. Ovarian failure is age related, and females who resume menses after treatment are still at increased risk for premature menopause. Males generally have reversible azoospermia during lower intensity alkylator chemotherapy, and long-term infertility is associated with doses of cyclophosphamide >9 g/m2 and with high-intensity therapy, such as that used in hematopoietic stem cell transplantation. Males undergoing potentially sterilizing chemotherapy should be offered sperm banking. Gonadotropin-releasing hormone (GnRH) analogs remain experimental to preserve ovarian function. Assisted reproductive technologies can be helpful to couples with chemotherapy-induced infertility.
RADIATION THERAPY
Testicles and ovaries in prepubertal patients are less sensitive to radiation damage; spermatogenesis is affected by low doses of radiation, and complete azoospermia occurs at 600–700 cGy. Leydig cell dysfunction, in contrast, occurs at <2000 cGy, and hence, endocrine function is lost at much higher radiation doses than spermatogenesis. Erectile dysfunction occurs in up to 80% of men treated with external-beam radiation therapy for prostate cancer. Sildenafil may be useful in reversing erectile dysfunction. Ovarian function damage with radiation is age related and occurs at doses of 150–500 cGy. Premature induction of menopause can have serious medical and psychological sequelae. Hormone replacement therapy is often contraindicated (as in estrogen receptor–positive breast cancer). Attention must be paid to maintenance of bone mass with calcium and vitamin D supplements and oral bisphosphonates, and bone mass should be monitored using bone density determinations. Paroxetine, clonidine, pregabalin, and other drugs may be useful in symptomatically controlling hot flashes.
Long-term survivors of childhood cancer (e.g., ALL) who have received cranial radiation may have altered leptin biology and growth hormone deficiency, leading to obesity and reduced strength, exercise tolerance, and bone density.
Radiation therapy to the neck (e.g., in Hodgkin’s lymphoma) may lead to hypothyroidism, Graves’ disease, thyroiditis, and thyroid malignancies. Thyroid-stimulating hormone (TSH) is followed routinely in such patients to prevent hypothyroidism, and to suppress persistently elevated levels of TSH which may cause or drive thyroid cancer.
OCULAR COMPLICATIONS
Cataracts may be caused by glucocorticoids, depending on duration and dose; radiation therapy; and uncommonly tamoxifen. Orbital radiation therapy may cause blindness.
ORAL COMPLICATIONS
Radiation therapy can produce xerostomia (dry mouth), with an attendant increase in caries and poor dentition. Taste and appetite may be suppressed. Bisphosphonate use may result in osteonecrosis of the jaw.
RAYNAUD’S PHENOMENON
Up to 40% of patients treated with bleomycin may develop Raynaud’s phenomenon as a result of an unknown mechanism.
SECOND MALIGNANCIES
Second malignancies in patients cured of cancer are a major cause of death, and treated cancer patients must be monitored for their occurrence. The induction of second malignancies is governed by the complex interplay of a number of factors including age, gender, environmental exposures, genetic susceptibility, and cancer treatment itself. In a number of settings, the events leading to the primary cancer themselves increase the risk of second malignancies. Patients with lung cancer are at increased risk of esophageal and head and neck cancers, and vice versa, due to shared risk factors including alcohol and tobacco abuse. Indeed, the risk of developing a second primary head and neck, esophageal, or lung cancer is also increased in these patients. Patients with breast cancer are at increased risk of breast cancer in the opposite breast. Patients with Hodgkin’s lymphoma are at risk for non- Hodgkin’s lymphomas. Genetic cancer syndromes (e.g., multiple endocrine neoplasia or Li-Fraumeni, Lynch’s, Cowden’s, and Gardner’s syndromes) are examples of genetically based second malignancies of specific types. Cancer treatment itself does not appear to be responsible for the risk of these secondary malignancies. Deficient DNA repair can greatly increase the risk of cancers from DNA-damaging agents, as in ataxia-telangiectasia. Importantly, the risk of treatment-related second malignancies is at least additive and often synergistic with combined chemotherapy and radiation therapy, and hence for such combined-therapy treatment approaches, it is important to establish the necessity of each in the treatment program. All of these patients require special surveillance or, in some cases, prophylactic surgery as part of appropriate treatment and follow-up.
CHEMOTHERAPEUTIC AGENTS
Chemotherapy is significantly associated with two fatal second malignancies, acute leukemia and myelodysplastic syndromes. Two types of leukemia have been described; in patients treated with alkylating agents, acute myeloid leukemia is associated with deletions in chromosome 5 or 7. The lifetime risk is about 1–5%, is increased by radiation therapy, and increases with age. The incidence of these leukemias peaks at 4–6 years, with risk returning close to baseline at 10 years. The other type of acute myeloid leukemia is related to therapy with topoisomerase inhibitors, is associated with chromosome 10q23 translocations, has an incidence of <1%, and generally occurs 1.5–3 years after treatment. Both of these acute myeloid leukemias are refractory to treatment and have a high mortality. The development of myelodysplastic syndromes is increased following chemotherapy, and these are often associated with leukemic progression and a dismal prognosis.
RADIATION THERAPY
Patients receiving radiation have an increasing and lifelong risk of second malignancies that is 1–2% in the second decade following treatment but increases to >25% after 25 years. These malignancies include cancers of the thyroid and breast, sarcomas, and CNS cancers, which often tend to be aggressive and have a poor prognosis. An example of organ-, age-, and sex-dependent radiation-induced secondary malignancy is breast cancer, in which the risk is small with radiation in women under age 30 but increases about 20-fold over baseline in women over 30. A 25-year-old woman treated with mantle radiation for Hodgkin’s lymphoma has a 29% actuarial risk of developing breast cancer by age 55.
HORMONAL THERAPY
Treatment of breast cancer with tamoxifen for 5 years or longer is associated with a 1–2% risk of endometrial cancer. Surveillance is generally effective at finding these cancers at an early stage. The risk of mortality from tamoxifen-induced endometrial cancer is low compared to the benefit of tamoxifen as adjuvant therapy for breast cancer.
IMMUNOSUPPRESSIVE THERAPY
Immunosuppressive therapy, as used in allogeneic bone marrow transplantation, particularly with T cell depletion using antithymocyte globulin or other means, increases the risk of Epstein Barr virus–associated B cell lymphoproliferative disorder. The incidence at 10 years after T cell depletion is 9–12%. Discontinuing immunosuppressive therapy, if possible, is often associated with complete disease regression.
RECOMMENDATIONS FOR FOLLOW-UP
All former cancer patients should be followed indefinitely. This is most often done by oncologists, but demographic changes suggest that more primary care physicians will need to be trained in the follow-up of treated cancer patients in remission. Cancer patients need to be educated about signs and symptoms of recurrence and potentially adverse effects related to therapy. Localized pain or palpable abnormality in a previously radiated field should prompt radiographic evaluation. Screening tests, when available and validated, should be used on a routine and regular basis (e.g., mammography and Pap smear), particularly in patients receiving radiation to specific organs. Annual mammography should start no later than 10 years after breast radiation. Patients receiving radiation fields encompassing thyroid tissue should have regular thyroid exams and TSH testing. Patients treated with alkylating agents or topoisomerase inhibitors should have a complete blood count every 6–12 months, and cytopenias, abnormal cells on peripheral smear, or macrocytosis should be evaluated with bone marrow biopsy and aspirate, to include cytogenetics, flow cytometry, or fluorescence in situ hybridization (FISH) studies as appropriate.
As the population of cancer survivors lives longer and grows, cancer survivorship has become an increasingly recognized subject, and the Institute of Medicine and National Research Council have published a monograph entitled From Cancer Patient to Cancer Survivor: Lost in Transition. The monograph proposes a plan that would inform clinicians caring for cancer survivors in complete detail of their previous treatments, complications thereof, signs and symptoms of late effects, and recommended screening and follow-up procedures. Table 125-2 lists long-term treatment effects by cancer type.
|
LONG-TERM TREATMENT EFFECTS BY CANCER TYPE |
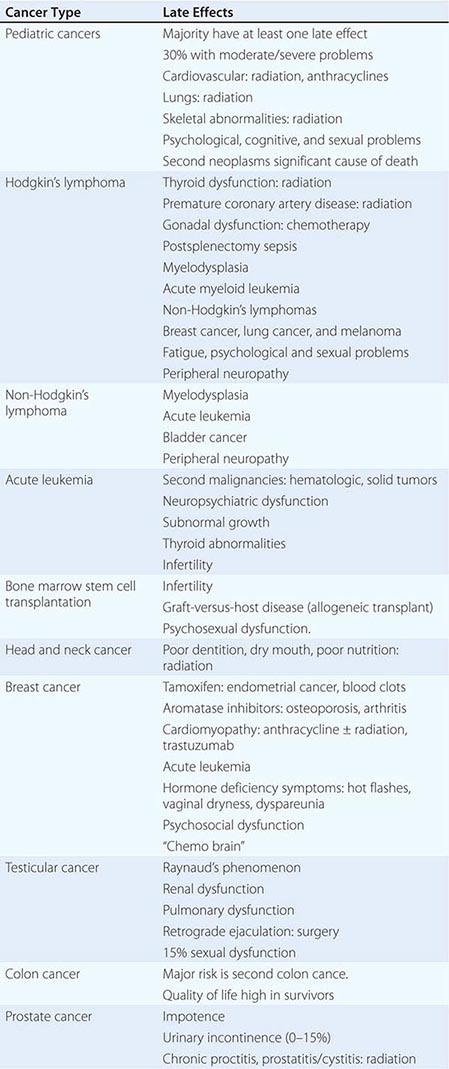
OUTLOOK
Clearly, the challenge for the future is to combine chemotherapy, targeted agents, biologic therapies, radiation, and surgery to produce better outcomes with less toxicity, including late effects of therapy. This is easily said but less easily accomplished. As treatment becomes more effective in new patient populations (ovarian, bladder, anal, and laryngeal cancers, for example), we will expect to discover new populations at risk for late effects. These populations will need to be followed carefully, so that such effects are recognized and treated. Cancer survivors represent an underused resource for prevention studies. Childhood cancer survivors, especially, suffer multiple chronic health impairments. The incidence of these late treatment consequences appears to have no plateau with age, throwing in stark relief the necessity of close monitoring and therapies with fewer late consequences of treatment.
SECTION 2 HEMATOPOIETIC DISORDERS
126 |
Iron Deficiency and Other Hypoproliferative Anemias |
Anemias associated with normocytic and normochromic red cells and an inappropriately low reticulocyte response (reticulocyte index <2–2.5) are hypoproliferative anemias. This category includes early iron deficiency (before hypochromic microcytic red cells develop), acute and chronic inflammation (including many malignancies), renal disease, hypometabolic states such as protein malnutrition and endocrine deficiencies, and anemias from marrow damage. Marrow damage states are discussed in Chap. 130.
Hypoproliferative anemias are the most common anemias, and in the clinic, iron deficiency anemia is the most common of these followed by the anemia of inflammation. The anemia of inflammation, similar to iron deficiency, is related in part to abnormal iron metabolism. The anemias associated with renal disease, inflammation, cancer, and hypometabolic states are characterized by a suboptimal erythropoietin response to the anemia.
IRON METABOLISM
Iron is a critical element in the function of all cells, although the amount of iron required by individual tissues varies during development. At the same time, the body must protect itself from free iron, which is highly toxic in that it participates in chemical reactions that generate free radicals such as singlet O2 or OH–. Consequently, elaborate mechanisms have evolved that allow iron to be made available for physiologic functions while at the same time conserving this element and handling it in such a way that toxicity is avoided.
The major role of iron in mammals is to carry O2 as part of hemoglobin. O2 is also bound by myoglobin in muscle. Iron is a critical element in iron-containing enzymes, including the cytochrome system in mitochondria. Iron distribution in the body is shown in Table 126-1. Without iron, cells lose their capacity for electron transport and energy metabolism. In erythroid cells, hemoglobin synthesis is impaired, resulting in anemia and reduced O2 delivery to tissue.
|
BODY IRON DISTRIBUTION |
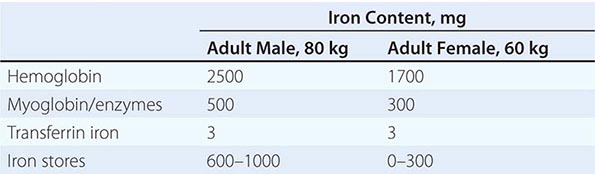
THE IRON CYCLE IN HUMANS
Figure 126-1 outlines the major pathways of internal iron exchange in humans. Iron absorbed from the diet or released from stores circulates in the plasma bound to transferrin, the iron transport protein. Transferrin is a bilobed glycoprotein with two iron binding sites. Transferrin that carries iron exists in two forms—monoferric (one iron atom) or diferric (two iron atoms). The turnover (half-clearance time) of transferrin-bound iron is very rapid—typically 60–90 min. Because almost all of the iron transported by transferrin is delivered to the erythroid marrow, the clearance time of transferrin-bound iron from the circulation is affected most by the plasma iron level and the erythroid marrow activity. When erythropoiesis is markedly stimulated, the pool of erythroid cells requiring iron increases and the clearance time of iron from the circulation decreases. The half-clearance time of iron in the presence of iron deficiency is as short as 10–15 min. With suppression of erythropoiesis, the plasma iron level typically increases and the half-clearance time may be prolonged to several hours. Normally, the iron bound to transferrin turns over 6–8 times per day. Assuming a normal plasma iron level of 80–100 μg/dL, the amount of iron passing through the transferrin pool is 20–24 mg/d.
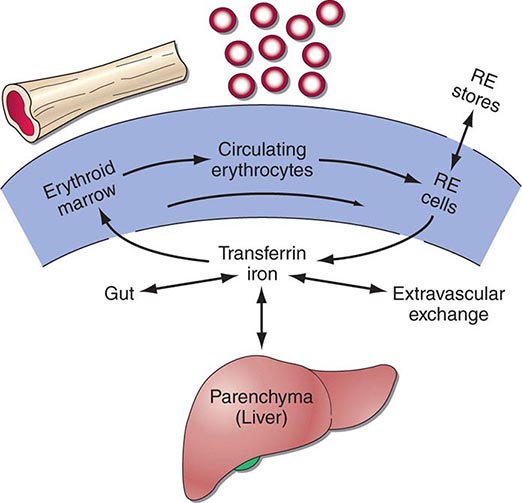
FIGURE 126-1 Internal iron exchange. Normally 80% of iron passing through the plasma transferrin pool is recycled from senescent red cells. Absorption of approximately 1 mg/d is required from the diet in men, and 1.4 mg/d in women to maintain homeostasis. As long as transferrin saturation is maintained between 20 and 60% and erythropoiesis is not increased, use of iron stores is not required. However, in the event of blood loss, dietary iron deficiency, or inadequate iron absorption, up to 40 mg/d of iron can be mobilized from stores. RE, reticuloendothelial.
The iron-transferrin complex circulates in the plasma until it interacts with specific transferrin receptors on the surface of marrow erythroid cells. Diferric transferrin has the highest affinity for transferrin receptors; apotransferrin (not carrying iron) has very little affinity. Although transferrin receptors are found on cells in many tissues within the body—and all cells at some time during development will display transferrin receptors—the cell having the greatest number of receptors (300,000–400,000/cell) is the developing erythroblast.
Once the iron-bearing transferrin interacts with its receptor, the complex is internalized via clathrin-coated pits and transported to an acidic endosome, where the iron is released at the low pH. The iron is then made available for heme synthesis while the transferrin-receptor complex is recycled to the surface of the cell, where the bulk of the transferrin is released back into circulation and the transferrin receptor reanchors into the cell membrane. At this point a certain amount of the transferrin receptor protein may be released into circulation and can be measured as soluble transferrin receptor protein. Within the erythroid cell, iron in excess of the amount needed for hemoglobin synthesis binds to a storage protein, apoferritin, forming ferritin. This mechanism of iron exchange also takes place in other cells of the body expressing transferrin receptors, especially liver parenchymal cells where the iron can be incorporated into heme-containing enzymes or stored. The iron incorporated into hemoglobin subsequently enters the circulation as new red cells are released from the bone marrow. The iron is then part of the red cell mass and will not become available for reutilization until the red cell dies.
In a normal individual, the average red cell life span is 120 days. Thus, 0.8–1% of red cells are replaced each day. At the end of its life span, the red cell is recognized as senescent by the cells of the reticuloendothelial (RE) system, and the red cell undergoes phagocytosis. Once within the RE cell, the ingested hemoglobin is broken down, the globin and other proteins are returned to the amino acid pool, and the iron is shuttled back to the surface of the RE cell, where it is presented to circulating transferrin. It is the efficient and highly conserved recycling of iron from senescent red cells that supports steady-state (and even mildly accelerated) erythropoiesis.
Because each milliliter of red cells contains 1 mg of elemental iron, the amount of iron needed to replace those red cells lost through senescence amounts to 20 mg/d (assuming an adult with a red cell mass of 2 L). Any additional iron required for daily red cell production comes from the diet. Normally, an adult male will need to absorb at least 1 mg of elemental iron daily to meet needs, while females in the childbearing years will need to absorb an average of 1.4 mg/d. However, to achieve a maximum proliferative erythroid marrow response to anemia, additional iron must be available. With markedly stimulated erythropoiesis, demands for iron are increased by as much as six- to eightfold. With extravascular hemolytic anemia, the rate of red cell destruction is increased, but the iron recovered from the red cells is efficiently reutilized for hemoglobin synthesis. In contrast, with intravascular hemolysis or blood loss anemia, the rate of red cell production is limited by the amount of iron that can be mobilized from stores. Typically, the rate of mobilization under these circumstances will not support red cell production more than 2.5 times normal. If the delivery of iron to the stimulated marrow is suboptimal, the marrow’s proliferative response is blunted, and hemoglobin synthesis is impaired. The result is a hypoproliferative marrow accompanied by microcytic, hypochromic anemia.
Whereas blood loss or hemolysis places a demand on the iron supply, inflammatory conditions interfere with iron release from stores and can result in a rapid decrease in the serum iron (see below).
NUTRITIONAL IRON BALANCE
The balance of iron in humans is tightly controlled and designed to conserve iron for reutilization. There is no regulated excretory pathway for iron, and the only mechanisms by which iron is lost are blood loss (via gastrointestinal bleeding, menses, or other forms of bleeding) and the loss of epithelial cells from the skin, gut, and genitourinary tract. Normally, the only route by which iron comes into the body is via absorption from food or from medicinal iron taken orally. Iron may also enter the body through red cell transfusions or injection of iron complexes. The margin between the amount of iron available for absorption and the requirement for iron in growing infants and the adult female is narrow; this accounts for the great prevalence of iron deficiency worldwide—currently estimated at one-half billion people.
The amount of iron required from the diet to replace losses averages approximately 10% of body iron content a year in men and 15% in women of childbearing age. Dietary iron content is closely related to total caloric intake (approximately 6 mg of elemental iron per 1000 calories). Iron bioavailability is affected by the nature of the foodstuff, with heme iron (e.g., red meat) being most readily absorbed. In the United States, the average iron intake in an adult male is 15 mg/d with 6% absorption; for the average female, the daily intake is 11 mg/d with 12% absorption. An individual with iron deficiency can increase iron absorption to approximately 20% of the iron present in a meat-containing diet but only 5–10% of the iron in a vegetarian diet. As a result, one-third of the female population in the United States has virtually no iron stores. Vegetarians are at an additional disadvantage because certain foodstuffs that include phytates and phosphates reduce iron absorption by approximately 50%. When ionizable iron salts are given together with food, the amount of iron absorbed is reduced. When the percentage of iron absorbed from individual food items is compared with the percentage for an equivalent amount of ferrous salt, iron in vegetables is only about one-twentieth as available, egg iron one-eighth, liver iron one-half, and heme iron one-half to two-thirds.
Infants, children, and adolescents may be unable to maintain normal iron balance because of the demands of body growth and lower dietary intake of iron. During the last two trimesters of pregnancy, daily iron requirements increase to 5–6 mg, and iron supplements are strongly recommended for pregnant women in developed countries.
Iron absorption takes place largely in the proximal small intestine and is a carefully regulated process. For absorption, iron must be taken up by the luminal cell. That process is facilitated by the acidic contents of the stomach, which maintains the iron in solution. At the brush border of the absorptive cell, the ferric iron is converted to the ferrous form by a ferrireductase. Transport across the membrane is accomplished by divalent metal transporter type 1 (DMT-1, also known as natural resistance macrophage-associated protein type 2 [Nramp 2] or DCT-1). DMT-1 is a general cation transporter. Once inside the gut cell, iron may be stored as ferritin or transported through the cell to be released at the basolateral surface to plasma transferrin through the membrane-embedded iron exporter, ferroportin. The function of ferroportin is negatively regulated by hepcidin, the principal iron regulatory hormone. In the process of release, iron interacts with another ferroxidase, hephaestin, which oxidizes the iron to the ferric form for transferrin binding. Hephaestin is similar to ceruloplasmin, the copper-carrying protein.
Iron absorption is influenced by a number of physiologic states. Erythroid hyperplasia stimulates iron absorption even in the face of normal or increased iron stores, and hepcidin levels are inappropriately low. Thus, patients with anemias associated with high levels of ineffective erythropoiesis absorb excess amounts of dietary iron. The molecular mechanism underlying this relationship is not known. Over time, this may lead to iron overload and tissue damage. In iron deficiency, hepcidin levels are also low and iron is much more efficiently absorbed; the contrary is true in states of secondary iron overload. The normal individual can reduce iron absorption in situations of excessive intake or medicinal iron intake; however, while the percentage of iron absorbed goes down, the absolute amount goes up. This accounts for the acute iron toxicity occasionally seen when children ingest large numbers of iron tablets. Under these circumstances, the amount of iron absorbed exceeds the transferrin binding capacity of the plasma, resulting in free iron that affects critical organs such as cardiac muscle cells.
IRON-DEFICIENCY ANEMIA
![]() Iron deficiency is one of the most prevalent forms of malnutrition. Globally, 50% of anemia is attributable to iron deficiency and accounts for approximately 841,000 deaths annually worldwide. Africa and parts of Asia bear 71% of the global mortality burden; North America represents only 1.4% of the total morbidity and mortality associated with iron deficiency.
Iron deficiency is one of the most prevalent forms of malnutrition. Globally, 50% of anemia is attributable to iron deficiency and accounts for approximately 841,000 deaths annually worldwide. Africa and parts of Asia bear 71% of the global mortality burden; North America represents only 1.4% of the total morbidity and mortality associated with iron deficiency.
STAGES OF IRON DEFICIENCY
The progression to iron deficiency can be divided into three stages (Fig. 126-2). The first stage is negative iron balance, in which the demands for (or losses of) iron exceed the body’s ability to absorb iron from the diet. This stage results from a number of physiologic mechanisms, including blood loss, pregnancy (in which the demands for red cell production by the fetus outstrip the mother’s ability to provide iron), rapid growth spurts in the adolescent, or inadequate dietary iron intake. Blood loss in excess of 10–20 mL of red cells per day is greater than the amount of iron that the gut can absorb from a normal diet. Under these circumstances, the iron deficit must be made up by mobilization of iron from RE storage sites. During this period, iron stores—reflected by the serum ferritin level or the appearance of stainable iron on bone marrow aspirations—decrease. As long as iron stores are present and can be mobilized, the serum iron, total iron-binding capacity (TIBC), and red cell protoporphyrin levels remain within normal limits. At this stage, red cell morphology and indices are normal.
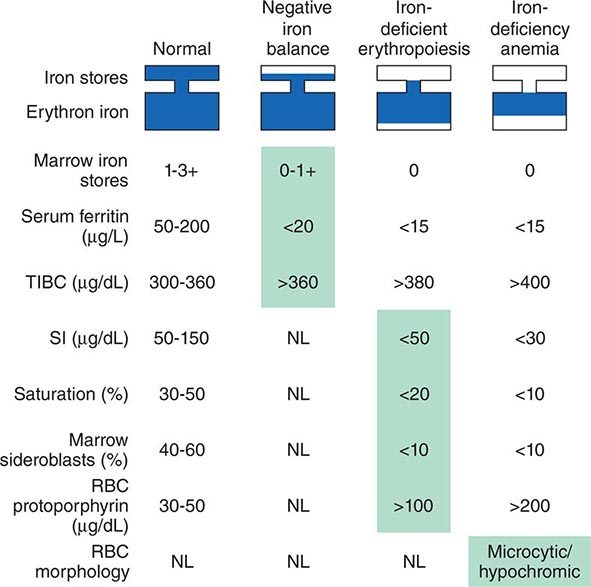
FIGURE 126-2 Laboratory studies in the evolution of iron deficiency. Measurements of marrow iron stores, serum ferritin, and total iron-binding capacity (TIBC) are sensitive to early iron-store depletion. Iron-deficient erythropoiesis is recognized from additional abnormalities in the serum iron (SI), percent transferrin saturation, the pattern of marrow sideroblasts, and the red blood cell (RBC) protoporphyrin level. Patients with iron-deficiency anemia demonstrate all the same abnormalities plus hypochromic microcytic anemia. (From RS Hillman, CA Finch: The Red Cell Manual, 7th ed. Philadelphia, F.A. Davis and Co., 1996, with permission.)
When iron stores become depleted, the serum iron begins to fall. Gradually, the TIBC increases, as do red cell protoporphyrin levels. By definition, marrow iron stores are absent when the serum ferritin level is <15 μg/L. As long as the serum iron remains within the normal range, hemoglobin synthesis is unaffected despite the dwindling iron stores. Once the transferrin saturation falls to 15–20%, hemoglobin synthesis becomes impaired. This is a period of iron-deficient erythropoiesis. Careful evaluation of the peripheral blood smear reveals the first appearance of microcytic cells, and if the laboratory technology is available, one finds hypochromic reticulocytes in circulation. Gradually, the hemoglobin and hematocrit begin to fall, reflecting iron-deficiency anemia. The transferrin saturation at this point is 10–15%.
When moderate anemia is present (hemoglobin 10–13 g/dL), the bone marrow remains hypoproliferative. With more severe anemia (hemoglobin 7–8 g/dL), hypochromia and microcytosis become more prominent, target cells and misshapen red cells (poikilocytes) appear on the blood smear as cigar- or pencil-shaped forms, and the erythroid marrow becomes increasingly ineffective. Consequently, with severe prolonged iron-deficiency anemia, erythroid hyperplasia of the marrow develops, rather than hypoproliferation.
CAUSES OF IRON DEFICIENCY
Conditions that increase demand for iron, increase iron loss, or decrease iron intake or absorption can produce iron deficiency (Table 126-2).
|
CAUSES OF IRON DEFICIENCY |
CLINICAL PRESENTATION OF IRON DEFICIENCY
Certain clinical conditions carry an increased likelihood of iron deficiency. Pregnancy, adolescence, periods of rapid growth, and an intermittent history of blood loss of any kind should alert the clinician to possible iron deficiency. A cardinal rule is that the appearance of iron deficiency in an adult male means gastrointestinal blood loss until proven otherwise. Signs related to iron deficiency depend on the severity and chronicity of the anemia in addition to the usual signs of anemia—fatigue, pallor, and reduced exercise capacity. Cheilosis (fissures at the corners of the mouth) and koilonychia (spooning of the fingernails) are signs of advanced tissue iron deficiency. The diagnosis of iron deficiency is typically based on laboratory results.
LABORATORY IRON STUDIES
Serum Iron and Total Iron-Binding Capacity The serum iron level represents the amount of circulating iron bound to transferrin. The TIBC is an indirect measure of the circulating transferrin. The normal range for the serum iron is 50–150 μg/dL; the normal range for TIBC is 300–360 μg/dL. Transferrin saturation, which is normally 25–50%, is obtained by the following formula: serum iron × 100 ÷ TIBC. Iron-deficiency states are associated with saturation levels below 20%. There is a diurnal variation in the serum iron. A transferrin saturation % >50% indicates that a disproportionate amount of the iron bound to transferrin is being delivered to nonerythroid tissues. If this persists for an extended time, tissue iron overload may occur.
Serum Ferritin Free iron is toxic to cells, and the body has established an elaborate set of protective mechanisms to bind iron in various tissue compartments. Within cells, iron is stored complexed to protein as ferritin or hemosiderin. Apoferritin binds to free ferrous iron and stores it in the ferric state. As ferritin accumulates within cells of the RE system, protein aggregates are formed as hemosiderin. Iron in ferritin or hemosiderin can be extracted for release by the RE cells, although hemosiderin is less readily available. Under steady-state conditions, the serum ferritin level correlates with total body iron stores; thus, the serum ferritin level is the most convenient laboratory test to estimate iron stores. The normal value for ferritin varies according to the age and gender of the individual (Fig. 126-3). Adult males have serum ferritin values averaging 100 μg/L, while adult females have levels averaging 30 μg/L. As iron stores are depleted, the serum ferritin falls to <15 μg/L. Such levels are diagnostic of absent body iron stores.
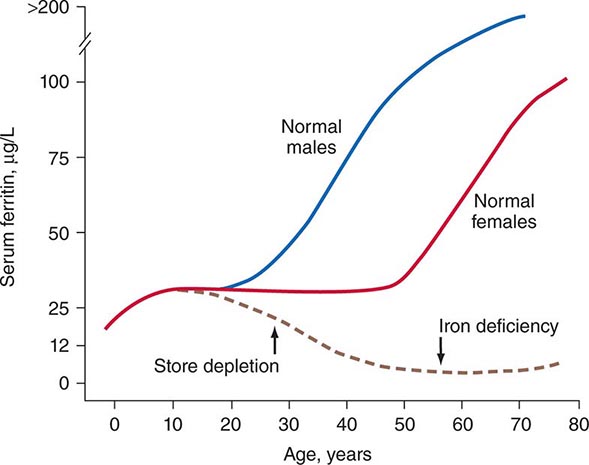
FIGURE 126-3 Serum ferritin levels as a function of sex and age. Iron store depletion and iron deficiency are accompanied by a decrease in serum ferritin level below 20 μg/L. (From RS Hillman et al: Hematology in Clinical Practice, 5th ed. New York, McGraw-Hill, 2011, with permission.)
Evaluation of Bone Marrow Iron Stores Although RE iron stores can be estimated from the iron stain of a bone marrow aspirate or biopsy, the measurement of serum ferritin has largely supplanted these procedures for determination of storage iron (Table 126-3). The serum ferritin level is a better indicator of iron overload than the marrow iron stain. However, in addition to storage iron, the marrow iron stain provides information about the effective delivery of iron to developing erythroblasts. Normally, when the marrow smear is stained for iron, 20–40% of developing erythroblasts—called sideroblasts—will have visible ferritin granules in their cytoplasm. This represents iron in excess of that needed for hemoglobin synthesis. In states in which release of iron from storage sites is blocked, RE iron will be detectable, and there will be few or no sideroblasts. In the myelodysplastic syndromes, mitochondrial dysfunction can occur, and accumulation of iron in mitochondria appears in a necklace fashion around the nucleus of the erythroblast. Such cells are referred to as ringed sideroblasts.
|
IRON STORE MEASUREMENTS |
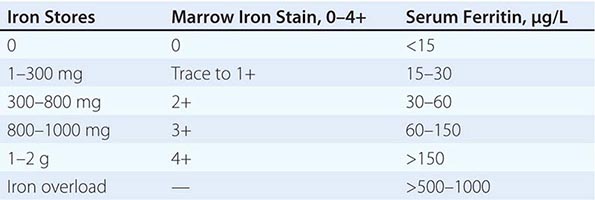
Red Cell Protoporphyrin Levels Protoporphyrin is an intermediate in the pathway to heme synthesis. Under conditions in which heme synthesis is impaired, protoporphyrin accumulates within the red cell. This reflects an inadequate iron supply to erythroid precursors to support hemoglobin synthesis. Normal values are <30 μg/dL of red cells. In iron deficiency, values in excess of 100 μg/dL are seen. The most common causes of increased red cell protoporphyrin levels are absolute or relative iron deficiency and lead poisoning.
Serum Levels of Transferrin Receptor Protein Because erythroid cells have the highest numbers of transferrin receptors of any cell in the body, and because transferrin receptor protein (TRP) is released by cells into the circulation, serum levels of TRP reflect the total erythroid marrow mass. Another condition in which TRP levels are elevated is absolute iron deficiency. Normal values are 4–9 μg/L determined by immunoassay. This laboratory test is becoming increasingly available and, along with the serum ferritin, has been proposed to distinguish between iron deficiency and the anemia of inflammation (see below).
DIFFERENTIAL DIAGNOSIS
Other than iron deficiency, only three conditions need to be considered in the differential diagnosis of a hypochromic microcytic anemia (Table 126-4). The first is an inherited defect in globin chain synthesis: the thalassemias. These are differentiated from iron deficiency most readily by serum iron values; normal or increased serum iron levels and transferrin saturation are characteristic of the thalassemias. In addition, the red blood cell distribution width (RDW) index is generally normal in thalassemia and elevated in iron deficiency.
|
DIAGNOSIS OF MICROCYTIC ANEMIA |
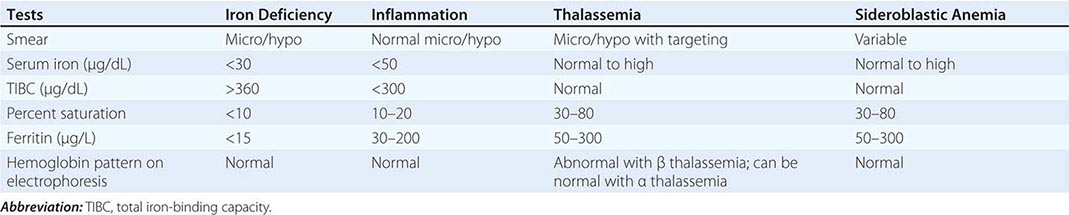
The second condition is the anemia of inflammation (AI; also referred to as the anemia of chronic disease) with inadequate iron supply to the erythroid marrow. The distinction between true iron-deficiency anemia and AI is among the most common diagnostic problems encountered by clinicians (see below). Usually, AI is normocytic and normochromic. The iron values usually make the differential diagnosis clear, as the ferritin level is normal or increased and the percent transferrin saturation and TIBC are typically below normal.
Finally, the myelodysplastic syndromes represent the third and least common condition. Occasionally, patients with myelodysplasia have impaired hemoglobin synthesis with mitochondrial dysfunction, resulting in impaired iron incorporation into heme. The iron values again reveal normal stores and more than an adequate supply to the marrow, despite the microcytosis and hypochromia.
TREATMENT IRON-DEFICIENCY ANEMIA
The severity and cause of iron-deficiency anemia will determine the appropriate approach to treatment. As an example, symptomatic elderly patients with severe iron-deficiency anemia and cardiovascular instability may require red cell transfusions. Younger individuals who have compensated for their anemia can be treated more conservatively with iron replacement. The foremost issue for the latter patient is the precise identification of the cause of the iron deficiency.
For the majority of cases of iron deficiency (pregnant women, growing children and adolescents, patients with infrequent episodes of bleeding, and those with inadequate dietary intake of iron), oral iron therapy will suffice. For patients with unusual blood loss or malabsorption, specific diagnostic tests and appropriate therapy take priority. Once the diagnosis of iron-deficiency anemia and its cause is made, there are three major therapeutic approaches.
RED CELL TRANSFUSION
Transfusion therapy is reserved for individuals who have symptoms of anemia, cardiovascular instability, and continued and excessive blood loss from whatever source and who require immediate intervention. The management of these patients is less related to the iron deficiency than it is to the consequences of the severe anemia. Not only do transfusions correct the anemia acutely, but the transfused red cells provide a source of iron for reutilization, assuming they are not lost through continued bleeding. Transfusion therapy will stabilize the patient while other options are reviewed.
ORAL IRON THERAPY
In the asymptomatic patient with established iron-deficiency anemia, treatment with oral iron is usually adequate. Multiple preparations are available, ranging from simple iron salts to complex iron compounds designed for sustained release throughout the small intestine (Table 126-5). Although the various preparations contain different amounts of iron, they are generally all absorbed well and are effective in treatment. Some come with other compounds designed to enhance iron absorption, such as ascorbic acid. It is not clear whether the benefits of such compounds justify their costs. Typically, for iron replacement therapy, up to 200 mg of elemental iron per day is given, usually as three or four iron tablets (each containing 50–65 mg elemental iron) given over the course of the day. Ideally, oral iron preparations should be taken on an empty stomach, since food may inhibit iron absorption. Some patients with gastric disease or prior gastric surgery require special treatment with iron solutions, because the retention capacity of the stomach may be reduced. The retention capacity is necessary for dissolving the shell of the iron tablet before the release of iron. A dose of 200 mg of elemental iron per day should result in the absorption of iron up to 50 mg/d. This supports a red cell production level of two to three times normal in an individual with a normally functioning marrow and appropriate erythropoietin stimulus. However, as the hemoglobin level rises, erythropoietin stimulation decreases, and the amount of iron absorbed is reduced. The goal of therapy in individuals with iron-deficiency anemia is not only to repair the anemia, but also to provide stores of at least 0.5–1 g of iron. Sustained treatment for a period of 6–12 months after correction of the anemia will be necessary to achieve this.
|
ORAL IRON PREPARATIONS |
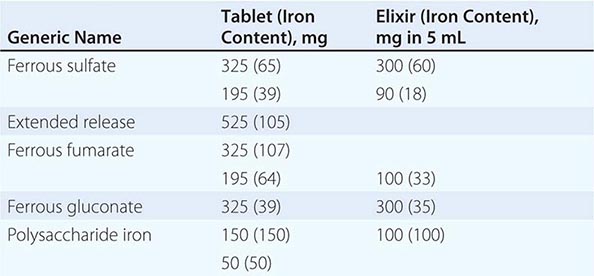
Of the complications of oral iron therapy, gastrointestinal distress is the most prominent and is seen in 15–20% of patients. Abdominal pain, nausea, vomiting, or constipation may lead to noncompliance. Although small doses of iron or iron preparations with delayed release may help somewhat, the gastrointestinal side effects are a major impediment to the effective treatment of a number of patients.
The response to iron therapy varies, depending on the erythropoietin stimulus and the rate of absorption. Typically, the reticulocyte count should begin to increase within 4–7 days after initiation of therapy and peak at 1–1½ weeks. The absence of a response may be due to poor absorption, noncompliance (which is common), or a confounding diagnosis. A useful test in the clinic to determine the patient’s ability to absorb iron is the iron tolerance test. Two iron tablets are given to the patient on an empty stomach, and the serum iron is measured serially over the subsequent 2 h. Normal absorption will result in an increase in the serum iron of at least 100 μg/dL. If iron deficiency persists despite adequate treatment, it may be necessary to switch to parenteral iron therapy.
PARENTERAL IRON THERAPY
Intravenous iron can be given to patients who are unable to tolerate oral iron; whose needs are relatively acute; or who need iron on an ongoing basis, usually due to persistent gastrointestinal blood loss. Parenteral iron use has been increasing rapidly in the last several years with the recognition that recombinant erythropoietin (EPO) therapy induces a large demand for iron—a demand that frequently cannot be met through the physiologic release of iron from RE sources or oral iron absorption. The safety of parenteral iron—particularly iron dextran—has been a concern. The serious adverse reaction rate to intravenous high-molecular-weight iron dextran is 0.7%. Fortunately, newer iron complexes are available in the United States, such as ferumoxytol (Feraheme), sodium ferric gluconate (Ferrlecit), iron sucrose (Venofer), and ferric carboxymaltose (Injectafer), that have much lower rates of adverse effects. Ferumoxytol delivers 510 mg of iron per injection; ferric gluconate 125 mg per injection, ferric carboxymaltose 750 mg per injection, and iron sucrose 200 mg per injection.
Parenteral iron is used in two ways: one is to administer the total dose of iron required to correct the hemoglobin deficit and provide the patient with at least 500 mg of iron stores; the second is to give repeated small doses of parenteral iron over a protracted period. The latter approach is common in dialysis centers, where it is not unusual for 100 mg of elemental iron to be given weekly for 10 weeks to augment the response to recombinant EPO therapy. The amount of iron needed by an individual patient is calculated by the following formula:
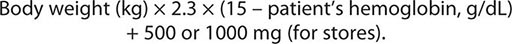
In administering intravenous iron dextran, anaphylaxis is a concern. Anaphylaxis is much rarer with the newer preparations. The factors that have correlated with an anaphylactic-like reaction include a history of multiple allergies or a prior allergic reaction to dextran (in the case of iron dextran). Generalized symptoms appearing several days after the infusion of a large dose of iron can include arthralgias, skin rash, and low-grade fever. These may be dose-related, but they do not preclude the further use of parenteral iron in the patient. To date, patients with sensitivity to iron dextran have been safely treated with other parenteral iron preparations. If a large dose of iron dextran is to be given (>100 mg), the iron preparation should be diluted in 5% dextrose in water or 0.9% NaCl solution. The iron solution can then be infused over a 60- to 90-min period (for larger doses) or at a rate convenient for the attending nurse or physician. Although a test dose (25 mg) of parenteral iron dextran is recommended, in reality a slow infusion of a larger dose of parenteral iron solution will afford the same kind of early warning as a separately injected test dose. Early in the infusion of iron, if chest pain, wheezing, a fall in blood pressure, or other systemic symptoms occur, the infusion of iron should be stopped immediately.
OTHER HYPOPROLIFERATIVE ANEMIAS
In addition to mild to moderate iron-deficiency anemia, the hypoproliferative anemias can be divided into four categories: (1) chronic inflammation, (2) renal disease, (3) endocrine and nutritional deficiencies (hypometabolic states), and (4) marrow damage (Chap. 130). With chronic inflammation, renal disease, or hypometabolism, endogenous EPO production is inadequate for the degree of anemia observed. For the anemia of chronic inflammation, the erythroid marrow also responds inadequately to stimulation, due in part to defective iron reutilization. As a result of the lack of adequate EPO stimulation, an examination of the peripheral blood smear will disclose only an occasional polychromatophilic (“shift”) reticulocyte. In cases of iron deficiency or marrow damage, appropriate elevations in endogenous EPO levels are typically found, and shift reticulocytes will be present on the blood smear.
ANEMIA OF ACUTE AND CHRONIC INFLAMMATION/INFECTION (AI)
AI—which encompasses inflammation, infection, tissue injury, and conditions (such as cancer) associated with the release of proinflammatory cytokines—is one of the most common forms of anemia seen clinically. It is the most important anemia in the differential diagnosis of iron deficiency, because many of the features of the anemia are brought about by inadequate iron delivery to the marrow, despite the presence of normal or increased iron stores. This is reflected by a low serum iron, increased red cell protoporphyrin, a hypoproliferative marrow, transferrin saturation in the range of 15–20%, and a normal or increased serum ferritin. The serum ferritin values are often the most distinguishing features between true iron-deficiency anemia and the iron-restricted erythropoiesis associated with inflammation. Typically, serum ferritin values increase threefold over basal levels in the face of inflammation. These changes are due to the effects of inflammatory cytokines and hepcidin, the key iron regulatory hormone, acting at several levels of erythropoiesis (Fig. 126-4).
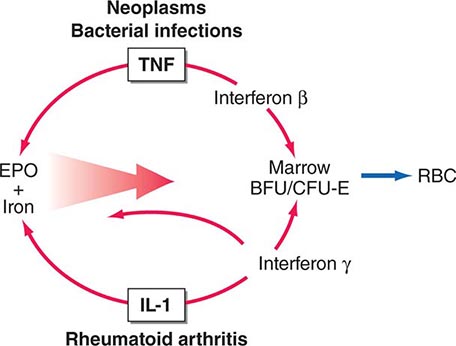
FIGURE 126-4 Suppression of erythropoiesis by inflammatory cytokines. Through the release of tumor necrosis factor (TNF) and interferon γ (IFN-γ), neoplasms and bacterial infections suppress erythropoietin (EPO) production and the proliferation of erythroid progenitors (erythroid burst-forming units and erythroid colony-forming units [BFU/CFU-E]). The mediators in patients with vasculitis and rheumatoid arthritis include interleukin 1 (IL-1) and IFN-γ. The red arrows indicate sites of inflammatory cytokine inhibitory effects. RBC, red blood cell.
Interleukin 1 (IL-1) directly decreases EPO production in response to anemia. IL-1, acting through accessory cell release of interferon γ (IFN-γ), suppresses the response of the erythroid marrow to EPO—an effect that can be overcome by EPO administration in vitro and in vivo. In addition, tumor necrosis factor (TNF), acting through the release of IFN-γ by marrow stromal cells, also suppresses the response to EPO. Hepcidin, made by the liver, is increased in inflammation via an IL-6 mediated pathway, and acts to suppress iron absorption and iron release from storage sites. The overall result is a chronic hypoproliferative anemia with classic changes in iron metabolism. The anemia is further compounded by a mild to moderate shortening in red cell survival.
With chronic inflammation, the primary disease will determine the severity and characteristics of the anemia. For example, many patients with cancer also have anemia that is typically normocytic and normochromic. In contrast, patients with long-standing active rheumatoid arthritis or chronic infections such as tuberculosis will have a microcytic, hypochromic anemia. In both cases, the bone marrow is hypoproliferative, but the differences in red cell indices reflect differences in the availability of iron for hemoglobin synthesis. Occasionally, conditions associated with chronic inflammation are also associated with chronic blood loss. Under these circumstances, a bone marrow aspirate stained for iron may be necessary to rule out absolute iron deficiency. However, the administration of iron in this case will correct the iron deficiency component of the anemia and leave the inflammatory component unaffected.
The anemia associated with acute infection or inflammation is typically mild but becomes more pronounced over time. Acute infection can produce a decrease in hemoglobin levels of 2–3 g/dL within 1 or 2 days; this is largely related to the hemolysis of red cells near the end of their natural life span. The fever and cytokines released exert a selective pressure against cells with more limited capacity to maintain the red cell membrane. In most individuals, the mild anemia is reasonably well tolerated, and symptoms, if present, are associated with the underlying disease. Occasionally, in patients with preexisting cardiac disease, moderate anemia (hemoglobin 10–11 g/dL) may be associated with angina, exercise intolerance, and shortness of breath. The erythropoietic profile that distinguishes the anemia of inflammation from the other causes of hypoproliferative anemias is shown in Table 126-6.
|
DIAGNOSIS OF HYPOPROLIFERATIVE ANEMIAS |
ANEMIA OF CHRONIC KIDNEY DISEASE (CKD)
Progressive CKD is usually associated with a moderate to severe hypoproliferative anemia; the level of the anemia correlates with the stage of CKD. Red cells are typically normocytic and normochromic, and reticulocytes are decreased. The anemia is primarily due to a failure of EPO production by the diseased kidney and a reduction in red cell survival. In certain forms of acute renal failure, the correlation between the anemia and renal function is weaker. Patients with the hemolytic-uremic syndrome increase erythropoiesis in response to the hemolysis, despite renal failure requiring dialysis. Polycystic kidney disease also shows a smaller degree of EPO deficiency for a given level of renal failure. By contrast, patients with diabetes or myeloma have more severe EPO deficiency for a given level of renal failure.
Assessment of iron status provides information to distinguish the anemia of CKD from the other forms of hypoproliferative anemia (Table 126-6) and to guide management. Patients with the anemia of CKD usually present with normal serum iron, TIBC, and ferritin levels. However, those maintained on chronic hemodialysis may develop iron deficiency from blood loss through the dialysis procedure. Iron must be replenished in these patients to ensure an adequate response to EPO therapy (see below).
ANEMIA IN HYPOMETABOLIC STATES
Patients who are starving, particularly for protein, and those with a variety of endocrine disorders that produce lower metabolic rates, may develop a mild to moderate hypoproliferative anemia. The release of EPO from the kidney is sensitive to the need for O2, not just O2 levels. Thus, EPO production is triggered at lower levels of blood O2 content in disease states (such as hypothyroidism and starvation) where metabolic activity, and thus O2 demand, is decreased.
Endocrine Deficiency States The difference in the levels of hemoglobin between men and women is related to the effects of androgen and estrogen on erythropoiesis. Testosterone and anabolic steroids augment erythropoiesis; castration and estrogen administration to males decrease erythropoiesis. Patients who are hypothyroid or have deficits in pituitary hormones also may develop a mild anemia. Pathogenesis may be complicated by other nutritional deficiencies because iron and folic acid absorption can be affected by these disorders. Usually, correction of the hormone deficiency reverses the anemia.
Anemia may be more severe in Addison’s disease, depending on the level of thyroid and androgen hormone dysfunction; however, anemia may be masked by decreases in plasma volume. Once such patients are given cortisol and volume replacement, the hemoglobin level may fall rapidly. Mild anemia complicating hyperparathyroidism may be due to decreased EPO production as a consequence of the renal effects of hypercalcemia or to impaired proliferation of erythroid progenitors.
Protein Starvation Decreased dietary intake of protein may lead to mild to moderate hypoproliferative anemia; this form of anemia may be prevalent in the elderly. The anemia can be more severe in patients with a greater degree of starvation. In marasmus, where patients are both protein and calorie deficient, the release of EPO is impaired in proportion to the reduction in metabolic rate; however, the degree of anemia may be masked by volume depletion and becomes apparent after refeeding. Deficiencies in other nutrients (iron, folate) may also complicate the clinical picture but may not be apparent at diagnosis. Changes in the erythrocyte indices on refeeding should prompt evaluation of iron, folate, and B12 status.
Anemia in Liver Disease A mild hypoproliferative anemia may develop in patients with chronic liver disease from nearly any cause. The peripheral blood smear may show spur cells and stomatocytes from the accumulation of excess cholesterol in the membrane from a deficiency of lecithin-cholesterol acyltransferase. Red cell survival is shortened, and the production of EPO is inadequate to compensate. In alcoholic liver disease, nutritional deficiencies are common and complicate the management. Folate deficiency from inadequate intake, as well as iron deficiency from blood loss and inadequate intake, can alter the red cell indices.
127 |
Disorders of Hemoglobin |
Hemoglobin is critical for normal oxygen delivery to tissues; it is also present in erythrocytes in such high concentrations that it can alter red cell shape, deformability, and viscosity. Hemoglobinopathies are disorders affecting the structure, function, or production of hemoglobin. These conditions are usually inherited and range in severity from asymptomatic laboratory abnormalities to death in utero. Different forms may present as hemolytic anemia, erythrocytosis, cyanosis, or vasoocclusive stigmata.
PROPERTIES OF THE HUMAN HEMOGLOBINS
HEMOGLOBIN STRUCTURE
Different hemoglobins are produced during embryonic, fetal, and adult life (Fig. 127-1). Each consists of a tetramer of globin polypeptide chains: a pair of α-like chains 141 amino acids long and a pair of β-like chains 146 amino acids long. The major adult hemoglobin, HbA, has the structure α2β2. HbF (α2γ2) predominates during most of gestation, and HbA2 (α2δ2) is minor adult hemoglobin. Embryonic hemoglobins need not be considered here.
FIGURE 127-1 The globin genes. The α-like genes (α, ζ) are encoded on chromosome 16; the β-like genes (β, γ, δ, ε) are encoded on chromosome 11. The ζ and ε genes encode embryonic globins.
Each globin chain enfolds a single heme moiety, consisting of a protoporphyrin IX ring complexed with a single iron atom in the ferrous state (Fe2+). Each heme moiety can bind a single oxygen molecule; a molecule of hemoglobin can transport up to four oxygen molecules.
The amino acid sequences of the various globins are highly homologous to one another. Each has a highly helical secondary structure. Their globular tertiary structures cause the exterior surfaces to be rich in polar (hydrophilic) amino acids that enhance solubility, and the interior to be lined with nonpolar groups, forming a hydrophobic pocket into which heme is inserted. The tetrameric quaternary structure of HbA contains two αβ dimers. Numerous tight interactions (i.e., α1β1 contacts) hold the α and β chains together. The complete tetramer is held together by interfaces (i.e., α1β2 contacts) between the α-like chain of one dimer and the non-α chain of the other dimer.
The hemoglobin tetramer is highly soluble, but individual globin chains are insoluble. Unpaired globin precipitates, forming inclusions that damage the cell and can trigger apoptosis. Normal globin chain synthesis is balanced so that each newly synthesized α or non-α globin chain will have an available partner with which to pair.
Solubility and reversible oxygen binding are the key properties deranged in hemoglobinopathies. Both depend most on the hydrophilic surface amino acids, the hydrophobic amino acids lining the heme pocket, a key histidine in the F helix, and the amino acids forming the α1β1 and α1β2 contact points. Mutations in these strategic regions tend to be the ones that alter oxygen affinity or solubility.
FUNCTION OF HEMOGLOBIN
To support oxygen transport, hemoglobin must bind O2 efficiently at the partial pressure of oxygen (Po2) of the alveolus, retain it in the circulation, and release it to tissues at the Po2 of tissue capillary beds. Oxygen acquisition and delivery over a relatively narrow range of oxygen tensions depend on a property inherent in the tetrameric arrangement of heme and globin subunits within the hemoglobin molecule called cooperativity or heme-heme interaction.
At low oxygen tensions, the hemoglobin tetramer is fully deoxygenated (Fig. 127-2). Oxygen binding begins slowly as O2 tension rises. However, as soon as some oxygen has been bound by the tetramer, an abrupt increase occurs in the slope of the curve. Thus, hemoglobin molecules that have bound some oxygen develop a higher oxygen affinity, greatly accelerating their ability to combine with more oxygen. This S-shaped oxygen equilibrium curve (Fig. 127-2), along which substantial amounts of oxygen loading and unloading can occur over a narrow range of oxygen tensions, is physiologically more useful than the high-affinity hyperbolic curve of individual monomers.
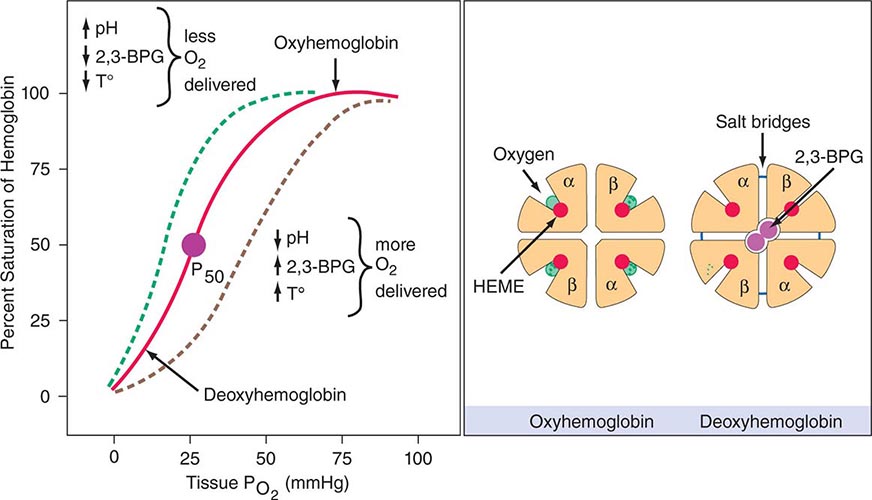
FIGURE 127-2 Hemoglobin-oxygen dissociation curve. The hemoglobin tetramer can bind up to four molecules of oxygen in the iron- containing sites of the heme molecules. As oxygen is bound, 2,3-bisphosphoglycerate (2,3-BPG) and carbon dioxide (CO2) are expelled. Salt bridges are broken, and each of the globin molecules changes its conformation to facilitate oxygen binding. Oxygen release to the tissues is the reverse process, with salt bridges being formed and 2,3-BPG and CO2 bound. Deoxyhemoglobin does not bind oxygen efficiently until the cell returns to conditions of higher pH, the most important modulator of O2 affinity (Bohr effect). When acid is produced in the tissues, the dissociation curve shifts to the right, facilitating oxygen release and CO2 binding. Alkalosis has the opposite effect, reducing oxygen delivery.
Oxygen affinity is modulated by several factors. The Bohr effect is the ability of hemoglobin to deliver more oxygen to tissues at low pH. It arises from the stabilizing action of protons on deoxyhemoglobin, which binds protons more readily than oxyhemoglobin because the latter is a weaker acid (Fig. 127-2). Thus, hemoglobin has a lower oxygen affinity at low pH. The major small molecule that alters oxygen affinity in humans is 2,3-bisphosphoglycerate (2,3-BPG; formerly 2,3-DPG), which lowers oxygen affinity when bound to hemoglobin. HbA has a reasonably high affinity for 2,3-BPG. HbF does not bind 2,3-BPG, so it tends to have a higher oxygen affinity in vivo. Hemoglobin also binds nitric oxide reversibly; this interaction influences vascular tone, but its clinical relevance remains incompletely understood.
Proper oxygen transport depends on the tetrameric structure of the proteins, the proper arrangement of hydrophilic and hydrophobic amino acids, and interaction with protons or 2,3-BPG.
DEVELOPMENTAL BIOLOGY OF HUMAN HEMOGLOBINS
Red cells first appearing at about 6 weeks after conception contain the embryonic hemoglobins Hb Portland (ζ2γ2), Hb Gower I (ζ2ε2), and Hb Gower II (α2ε2). At 10–11 weeks, fetal hemoglobin (HbF; α2γ2) becomes predominant. The switch to nearly exclusive synthesis of adult hemoglobin (HbA; α2β2) occurs at about 38 weeks (Fig. 127-1). Fetuses and newborns therefore require α-globin but not β-globin for normal gestation. A major advance in understanding the HbF to HbA transition has been the demonstration that the transcription factor Bcl11a plays a pivotal role in its regulation. Small amounts of HbF are produced during postnatal life. A few red cell clones called F cells are progeny of a small pool of immature committed erythroid precursors (BFU-e) that retain the ability to produce HbF. Profound erythroid stresses, such as severe hemolytic anemias, bone marrow transplantation, or cancer chemotherapy, cause more of the F-potent BFU-e to be recruited. HbF levels thus tend to rise in some patients with sickle cell anemia or thalassemia. This phenomenon probably explains the ability of hydroxyurea to increase levels of HbF in adults. Agents such as butyrate and histone deacetylase inhibitors can also activate fetal globin genes partially after birth.
GENETICS AND BIOSYNTHESIS OF HUMAN HEMOGLOBIN
![]() The human hemoglobins are encoded in two tightly linked gene clusters; the α-like globin genes are clustered on chromosome 16 and the β-like genes on chromosome 11 (Fig. 127-1). The α-like cluster consists of two α-globin genes and a single copy of the ζ gene. The non-α gene cluster consists of a single ε gene, the Gγ and Aγ fetal globin genes, and the adult δ and β genes.
The human hemoglobins are encoded in two tightly linked gene clusters; the α-like globin genes are clustered on chromosome 16 and the β-like genes on chromosome 11 (Fig. 127-1). The α-like cluster consists of two α-globin genes and a single copy of the ζ gene. The non-α gene cluster consists of a single ε gene, the Gγ and Aγ fetal globin genes, and the adult δ and β genes.
Important regulatory sequences flank each gene. Immediately upstream are typical promoter elements needed for the assembly of the transcription initiation complex. Sequences in the 5′ flanking region of the γ and the β genes appear to be crucial for the correct developmental regulation of these genes, whereas elements that function like classic enhancers and silencers are in the 3′ flanking regions. The locus control region (LCR) elements located far upstream appear to control the overall level of expression of each cluster. These elements achieve their regulatory effects by interacting with trans-acting transcription factors. Some of these factors are ubiquitous (e.g., Sp1 and YY1), while others are more or less limited to erythroid cells or hematopoietic cells (e.g., GATA-1, NFE-2, and EKLF). The LCR controlling the α-globin gene cluster is modulated by a SWI/SNF-like protein called ATRX; this protein appears to influence chromatin remodeling and DNA methylation. The association of α thalassemia with mental retardation and myelodysplasia in some families appears to be related to mutations in the ATRX pathway. This pathway also modulates genes specifically expressed during erythropoiesis, such as those that encode the enzymes for heme biosynthesis. Normal red blood cell (RBC) differentiation requires the coordinated expression of the globin genes with the genes responsible for heme and iron metabolism. RBC precursors contain a protein, α-hemoglobin-stabilizing protein (AHSP), that enhances the folding and solubility of α globin, which is otherwise easily denatured, leading to insoluble precipitates. These precipitates play an important role in the thalassemia syndromes and certain unstable hemoglobin disorders. Polymorphic variation in the amounts and/or functional capacity of AHSP might explain some of the clinical variability seen in patients inheriting identical thalassemia mutations.
CLASSIFICATION OF HEMOGLOBINOPATHIES
There are five major classes of hemoglobinopathies (Table 127-1). Structural hemoglobinopathies occur when mutations alter the amino acid sequence of a globin chain, altering the physiologic properties of the variant hemoglobins and producing the characteristic clinical abnormalities. The most clinically relevant variant hemoglobins polymerize abnormally, as in sickle cell anemia, or exhibit altered solubility or oxygen-binding affinity. Thalassemia syndromes arise from mutations that impair production or translation of globin mRNA, leading to deficient globin chain biosynthesis. Clinical abnormalities are attributable to the inadequate supply of hemoglobin and the imbalances in the production of individual globin chains, leading to premature destruction of erythroblasts and RBC. Thalassemic hemoglobin variants combine features of thalassemia (e.g., abnormal globin biosynthesis) and of structural hemoglobinopathies (e.g., an abnormal amino acid sequence). Hereditary persistence of fetal hemoglobin (HPFH) is characterized by synthesis of high levels of fetal hemoglobin in adult life. Acquired hemoglobinopathies include modifications of the hemoglobin molecule by toxins (e.g., acquired methemoglobinemia) and clonal abnormalities of hemoglobin synthesis (e.g., high levels of HbF production in preleukemia and α thalassemia in myeloproliferative disorders).
|
CLASSIFICATION OF HEMOGLOBINOPATHIES |
EPIDEMIOLOGY
![]() Hemoglobinopathies are especially common in areas in which malaria is endemic. This clustering of hemoglobinopathies is assumed to reflect a selective survival advantage for the abnormal RBC, which presumably provides a less hospitable environment during the obligate RBC stages of the parasitic life cycle. Very young children with α thalassemia are more susceptible to infection with the nonlethal Plasmodium vivax. Thalassemia might then favor a natural protection against infection with the more lethal Plasmodium falciparum.
Hemoglobinopathies are especially common in areas in which malaria is endemic. This clustering of hemoglobinopathies is assumed to reflect a selective survival advantage for the abnormal RBC, which presumably provides a less hospitable environment during the obligate RBC stages of the parasitic life cycle. Very young children with α thalassemia are more susceptible to infection with the nonlethal Plasmodium vivax. Thalassemia might then favor a natural protection against infection with the more lethal Plasmodium falciparum.
Thalassemias are the most common genetic disorders in the world, affecting nearly 200 million people worldwide. About 15% of African Americans are silent carriers for α thalassemia; α thalassemia trait (minor) occurs in 3% of African American and in 1–15% of persons of Mediterranean origin. β Thalassemia has a 10–15% incidence in individuals from the Mediterranean and Southeast Asia and 0.8% in African Americans. The number of severe cases of thalassemia in the United States is about 1000. Sickle cell disease is the most common structural hemoglobinopathy, occurring in heterozygous form in ~8% of African Americans and in homozygous form in 1 in 400. Between 2 and 3% of African Americans carry a hemoglobin C allele.
INHERITANCE AND ONTOGENY
Hemoglobinopathies are autosomal codominant traits—thus, compound heterozygotes who inherit a different abnormal mutant allele from each parent exhibit composite features of each. For example, patients inheriting sickle β thalassemia exhibit features of β thalassemia and sickle cell anemia. The α chain is present in HbA, HbA2, and HbF; α-chain mutations thus cause abnormalities in all three. The α-globin hemoglobinopathies are symptomatic in utero and after birth because normal function of the α-globin gene is required throughout gestation and adult life. In contrast, infants with β-globin hemoglobinopathies tend to be asymptomatic until 3–9 months of age, when HbA has largely replaced HbF. Prevention or partial reversion of the switch should thus be an effective therapeutic strategy for β-chain hemoglobinopathies.
DETECTION AND CHARACTERIZATION OF HEMOGLOBINOPATHIES–GENERAL METHODS
Electrophoretic techniques are still widely used for hemoglobin analysis. Electrophoresis at pH 8.6 on cellulose acetate membranes is especially simple, inexpensive, and reliable for initial screening. Agar gel electrophoresis at pH 6.1 in citrate buffer is often used as a complementary method because each method detects different variants. Some important variants are electrophoretically silent. These mutant hemoglobins can usually be characterized by more specialized techniques such as mass spectroscopy, which is rapidly replacing electrophoresis for initial analysis.
Quantitation of the hemoglobin profile is often desirable. HbA2 is frequently elevated in β thalassemia trait and depressed in iron deficiency. HbF is elevated in HPFH, some β thalassemia syndromes, and occasional periods of erythroid stress or marrow dysplasia. For characterization of sickle cell trait, sickle thalassemia syndromes, or HbSC disease, and for monitoring the progress of exchange transfusion therapy to lower the percentage of circulating HbS, quantitation of individual hemoglobins is also required. In most laboratories, quantitation is performed only if the test is specifically ordered. Complete characterization, including amino acid sequencing or gene cloning and sequencing, is readily available from several reference laboratories.
Because some variants can comigrate with HbA or HbS (sickle hemoglobin), electrophoretic assessment should always be regarded as incomplete unless functional assays for hemoglobin sickling, solubility, or oxygen affinity are also performed, as dictated by the clinical presentation. The best sickling assays involve measurement of the degree to which the hemoglobin sample becomes insoluble, or gelated, as it is deoxygenated (i.e., sickle solubility test). Unstable hemoglobins are detected by their precipitation in isopropanol or after heating to 50°C. High-O2 affinity and low-O2 affinity variants are detected by quantitating the P50, the partial pressure of oxygen at which the hemoglobin sample becomes 50% saturated with oxygen. Direct tests for the percent carboxyhemoglobin and methemoglobin, using spectrophotometric techniques, can readily be obtained from most clinical laboratories on an urgent basis.
Laboratory evaluation remains an adjunct, rather than the sole diagnostic aid. Diagnosis is best established by recognition of a characteristic history, physical findings, peripheral blood smear morphology, and abnormalities of the complete blood cell count (e.g., profound microcytosis with minimal anemia in thalassemia trait).
STRUCTURALLY ABNORMAL HEMOGLOBINS
SICKLE CELL SYNDROMES
The sickle cell syndromes are caused by a mutation in the β-globin gene that changes the sixth amino acid from glutamic acid to valine. HbS (α2β26 Glu→Val) polymerizes reversibly when deoxygenated to form a gelatinous network of fibrous polymers that stiffen the RBC membrane, increase viscosity, and cause dehydration due to potassium leakage and calcium influx (Fig. 127-3). These changes also produce the sickle shape. Sickled cells lose the pliability needed to traverse small capillaries. They possess altered “sticky” membranes that are abnormally adherent to the endothelium of small venules. These abnormalities provoke unpredictable episodes of microvascular vasoocclusion and premature RBC destruction (hemolytic anemia). Hemolysis occurs because the spleen destroys the abnormal RBC. The rigid adherent cells clog small capillaries and venules, causing tissue ischemia, acute pain, and gradual end-organ damage. This venoocclusive component usually dominates the clinical course. Prominent manifestations include episodes of ischemic pain (i.e., painful crises) and ischemic malfunction or frank infarction in the spleen, central nervous system, bones, joints, liver, kidneys, and lungs (Fig. 127-3).
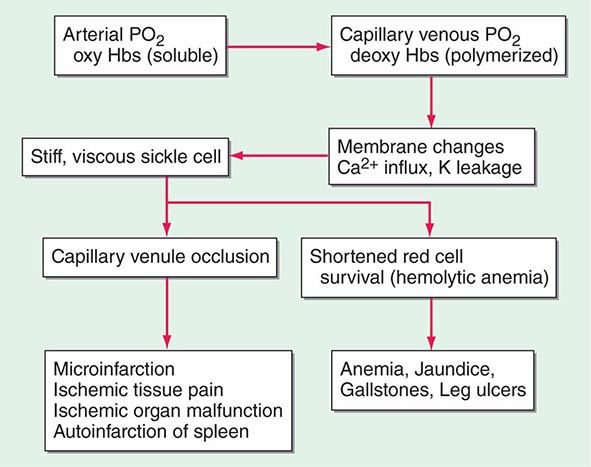
FIGURE 127-3 Pathophysiology of sickle cell crisis.
Several sickle syndromes occur as the result of inheritance of HbS from one parent and another hemoglobinopathy, such as β thalassemia or HbC (α2β26 Glu→Lys), from the other parent. The prototype disease, sickle cell anemia, is the homozygous state for HbS (Table 127-2).
|
CLINICAL FEATURES OF SICKLE HEMOGLOBINOPATHIES |
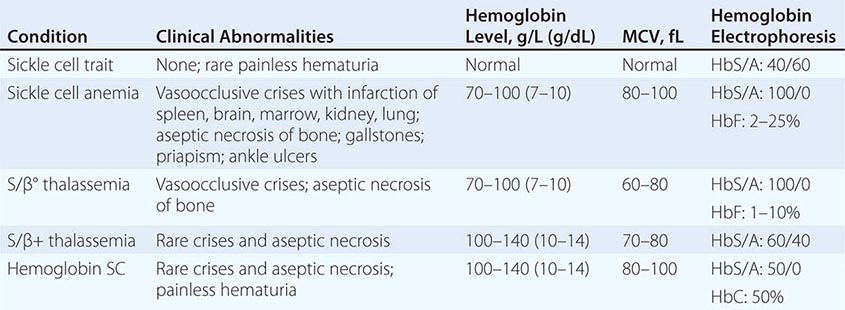
Clinical Manifestations of Sickle Cell Anemia Most patients with sickling syndromes suffer from hemolytic anemia, with hematocrits from 15 to 30%, and significant reticulocytosis. Anemia was once thought to exert protective effects against vasoocclusion by reducing blood viscosity. However, natural history and drug therapy trials suggest that an increase in the hematocrit and feedback inhibition of reticulocytosis might be beneficial, even at the expense of increased blood viscosity. The role of adhesive reticulocytes in vasoocclusion might account for these paradoxical effects.
Granulocytosis is common. The white count can fluctuate substantially and unpredictably during and between painful crises, infectious episodes, and other intercurrent illnesses.
Vasoocclusion causes protean manifestations. Intermittent episodes of vasoocclusion in connective and musculoskeletal structures produce ischemia manifested by acute pain and tenderness, fever, tachycardia, and anxiety. These recurrent episodes, called painful crises, are the most common clinical manifestation. Their frequency and severity vary greatly. Pain can develop almost anywhere in the body and may last from a few hours to 2 weeks. Repeated crises requiring hospitalization (>3 episodes per year) correlate with reduced survival in adult life, suggesting that these episodes are associated with accumulation of chronic end-organ damage. Provocative factors include infection, fever, excessive exercise, anxiety, abrupt changes in temperature, hypoxia, or hypertonic dyes.
Repeated microinfarction can destroy tissues having microvascular beds prone to sickling. Thus, splenic function is frequently lost within the first 18–36 months of life, causing susceptibility to infection, particularly by pneumococci. Acute venous obstruction of the spleen (splenic sequestration crisis), a rare occurrence in early childhood, may require emergency transfusion and/or splenectomy to prevent trapping of the entire arterial output in the obstructed spleen. Occlusion of retinal vessels can produce hemorrhage, neovascularization, and eventual detachments. Renal papillary necrosis invariably produces isosthenuria. More widespread renal necrosis leads to renal failure in adults, a common late cause of death. Bone and joint ischemia can lead to aseptic necrosis, especially of the femoral or humeral heads; chronic arthropathy; and unusual susceptibility to osteomyelitis, which may be caused by organisms, such as Salmonella, rarely encountered in other settings. The hand-foot syndrome is caused by painful infarcts of the digits and dactylitis. Stroke is especially common in children; a small subset tends to suffer repeated episodes. Stroke is less common in adults and is often hemorrhagic. A particularly painful complication in males is priapism, due to infarction of the penile venous outflow tracts; permanent impotence is a frequent consequence. Chronic lower leg ulcers probably arise from ischemia and superinfection in the distal circulation.
Acute chest syndrome is a distinctive manifestation characterized by chest pain, tachypnea, fever, cough, and arterial oxygen desaturation. It can mimic pneumonia, pulmonary emboli, bone marrow infarction and embolism, myocardial ischemia, or in situ lung infarction. Acute chest syndrome is thought to reflect in situ sickling within the lung, producing pain and temporary pulmonary dysfunction. Often it is difficult or impossible to distinguish among other possibilities. Pulmonary infarction and pneumonia are the most frequent underlying or concomitant conditions in patients with this syndrome. Repeated episodes of acute chest pain correlate with reduced survival. Acutely, reduction in arterial oxygen saturation is especially ominous because it promotes sickling on a massive scale. Chronic acute or subacute pulmonary crises lead to pulmonary hypertension and cor pulmonale, an increasingly common cause of death as patients survive longer. Considerable controversy exists about the possible role played by free plasma HbS in scavenging nitrogen dioxide (NO2), thus raising pulmonary vascular tone. Trials of sildenafil to restore NO2 levels were terminated because of adverse effects.
Chronic subacute central nervous system damage in the absence of an overt stroke is a distressingly common phenomenon beginning in early childhood. Modern functional imaging techniques have pinpointed circulatory dysfunction due to a likely CNS sickle vasculopathy; these changes correlate with an array of cognitive and behavioral abnormalities in children and young adults. It is important to be aware of these often subtle changes because they can complicate clinical management or be misinterpreted as “difficult patient” behaviors.
Sickle cell syndromes are remarkable for their clinical heterogeneity. Some patients remain virtually asymptomatic into or even through adult life, while others suffer repeated crises requiring hospitalization from early childhood. Patients with sickle thalassemia and sickle-HbE tend to have similar, slightly milder symptoms, perhaps because of the ameliorating effects of production of other hemoglobins within the RBC. Hemoglobin SC disease, one of the more common variants of sickle cell anemia, is frequently marked by lesser degrees of hemolytic anemia and a greater propensity for the development of retinopathy and aseptic necrosis of bones. In most respects, however, the clinical manifestations resemble sickle cell anemia. Some rare hemoglobin variants actually aggravate the sickling phenomenon.
The clinical variability in different patients inheriting the same disease-causing mutation (sickle hemoglobin) has made sickle cell disease the focus of efforts to identify modifying genetic polymorphisms in other genes that might account for the heterogeneity. The complexity of the data obtained thus far has dampened the expectation that genome-wide analysis will yield individualized profiles that predict a patient’s clinical course. Nevertheless, a number of interesting patterns have emerged from these modifying gene analyses. For example, genes affecting the inflammatory response or cytokine expression appear to be modifying candidates. Genes that affect transcriptional regulation of lymphocytes may also be involved.
Clinical Manifestations of Sickle Cell Trait Sickle cell trait is often asymptomatic. Anemia and painful crises are rare. An uncommon but highly distinctive symptom is painless hematuria often occurring in adolescent males, probably due to papillary necrosis. Isosthenuria is a more common manifestation of the same process. Sloughing of papillae with urethral obstruction has been reported, as have isolated cases of massive sickling or sudden death due to exposure to high altitudes or extremes of exercise and dehydration. Avoidance of dehydration or extreme physical stress should be advised.
Diagnosis Sickle cell syndromes are suspected on the basis of hemolytic anemia, RBC morphology (Fig. 127-4), and intermittent episodes of ischemic pain. Diagnosis is confirmed by hemoglobin electrophoresis, mass spectroscopy, and the sickling tests already discussed. Thorough characterization of the exact hemoglobin profile of the patient is important, because sickle thalassemia and hemoglobin SC disease have distinct prognoses or clinical features. Diagnosis is usually established in childhood, but occasional patients, often with compound heterozygous states, do not develop symptoms until the onset of puberty, pregnancy, or early adult life. Genotyping of family members and potential parental partners is critical for genetic counseling. Details of the childhood history establish prognosis and need for aggressive or experimental therapies. Factors associated with increased morbidity and reduced survival include more than three crises requiring hospitalization per year, chronic neutrophilia, a history of splenic sequestration or hand-foot syndrome, and second episodes of acute chest syndrome. Patients with a history of cerebrovascular accidents are at higher risk for repeated episodes and require partial exchange transfusion and especially close monitoring using Doppler carotid flow measurements. Patients with severe or repeated episodes of acute chest syndrome may need lifelong transfusion support, using partial exchange transfusion, if possible.
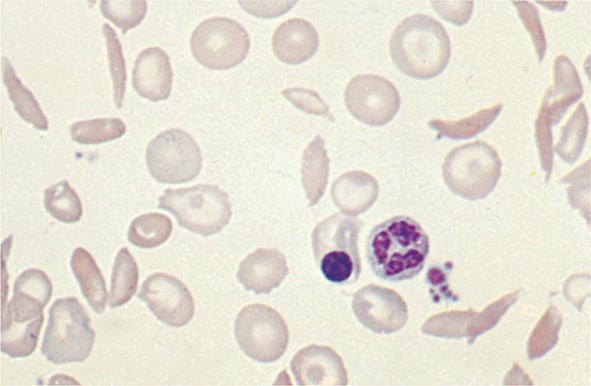
FIGURE 127-4 Sickle cell anemia. The elongated and crescent- shaped red blood cells seen on this smear represent circulating irreversibly sickled cells. Target cells and a nucleated red blood cell are also seen.




