CHAPTER 101 PARANEOPLASTIC DISORDERS OF THE NERVOUS SYSTEM
Paraneoplastic neurological disorders (PNDs) are an extensive group of syndromes that can affect any part of the nervous system by mechanisms that are mostly immune mediated (Table 101-1).1 PNDs are more frequent than previously considered, with an incidence that varies with each type of tumor. The tumors most frequently involved are small cell lung cancer (SCLC) (∼3% of patients develop PND), thymoma (15%), and plasma cell dyscrasias associated with malignant monoclonal gammopathies (∼5% to 15%). With other solid tumors, the incidence of PND is less than 1%.2
TABLE 101-1 Classification of Immune-Mediated Paraneoplastic Neurological Disorders
| Area Involved | Classical Syndromes | Nonclassical Syndromes |
|---|---|---|
| Central nervous system | Encephalomyelitis | Brainstem encephalitis |
| Limbic encephalitis | Stiff-person syndrome | |
| Cerebellar degeneration | Necrotizing myelopathy | |
| Opsoclonus-myoclonus | Motor neuron disease | |
| Dorsal root ganglia or peripheral nerves | Subacute sensory neuronopathy | Acute sensorimotor neuropathy (Guillain-Barré syndrome, plexitis) |
| Gastrointestinal paresis or pseudoobstruction | Subacute and chronic sensorimotor neuropathies | |
| Neuropathy of plasma cell dyscrasias and lymphoma | ||
| Vasculitis of the nerve and muscle | ||
| Pure autonomic neuropathy | ||
| Muscle | Dermatomyositis | Acute necrotizing myopathy |
| Polymyositis | ||
| Neuromuscular junction | Lambert-Eaton myasthenic syndrome | Myasthenia gravis |
| Acquired neuromyotonia | ||
| Eye and retina | Cancer-associated retinopathy | Optic neuritis |
| Melanoma-associated retinopathy |
In 60% of patients with PND, symptoms develop before the presence of a tumor is known; the majority of these patients are seen by neurologists, who should be aware that prompt diagnosis and treatment of the tumor along with immunotherapy may stabilize or improve the PND. In 40% of patients, symptoms of PND develop after the tumor diagnosis or at tumor recurrence. In this group of patients, the differential diagnosis is extensive because PND may mimic many other neurological complications of cancer or its treatment. The diagnosis of PND has been facilitated by serological tests that are based on the detection of antineuronal antibodies in the patients’ serum or cerebrospinal fluid (CSF), but in at least 40% of patients, no antibodies are detected, and in some instances, the antibodies can be detected in patients who have cancer but not PND.3 Therefore, although testing for these antibodies is useful, it does not replace the importance of a comprehensive clinical assessment that should always rule out other complications of cancer. In addition to the immune-mediated PNDs, patients with cancer may develop neurological symptoms from a large and heterogeneous group of mechanisms unrelated to metastasis; these are not further discussed but are listed in Table 101-2.
TABLE 101-2 Nonmetastatic Neurological Complications of Cancer Different from Immune-Mediated Paraneoplastic Neurological Disorders
| Syndrome | Proposed Mechanism |
|---|---|
| Cerebrovascular disease | Coagulopathy |
| Wernicke-Korsakoff syndrome | Thiamine deficiency |
| Myelopathy, sensory neuropathy | Cobalamin deficiency |
| Pellagra-like syndrome | Niacin deficiency in carcinoid tumors |
| Diffuse metabolic encephalopathy | Hypoxia, organ failure, electrolyte imbalance, endocrine disorders |
| Opportunistic CNS infections | Cancer- or treatment-related immunodeficiency |
| POEMS neuropathy | Cytokines (IL-6, IL-1β, TNF-α), MMP, VEGF |
| Carcinoid myopathy | Increased serotonin secretion by carcinoid tumors |
| Cachectic myopathy | Proteolytic tumor-derived “toxohormone”-like peptides, cytokines (TNF-α, IFN γ, IL-6, IL-1β) |
CNS, central nervous system; IFN, interferon; IL, interleukin; MMP, matrix metalloproteinases; POEMS, syndrome of polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes; TNF, tumor necrosis factor; VEGF, vascular endothelial growth factor.
IMMUNOPATHOGENESIS OF PARANEOPLASTIC NEUROLOGICAL DISORDERS AND EFFECTS ON THE TUMOR
Paraneoplastic Neurological Disorders of the Central Nervous System
Many of these disorders occur in association with immune responses against intraneuronal antigens expressed by the underlying cancer (paraneoplastic or onconeuronal antigens). These immune responses are characterized by the presence of antibodies and cytotoxic T cell responses against the onconeuronal antigens. As far as the antibodies are concerned, there have been several attempts to reproduce PND by passive transfer of serum or immunoglobulin G (IgG) to animals or by immunization with recombinant antigens that prime B cell responses.4–6 These procedures did not result in neurological dysfunction, although immunization did result in high titers of serum antibodies.7 It has been argued that the antibodies cannot reach the intracellular antigens, but data from other immune-mediated disorders suggest that antibodies can enter cells and disrupt the function of the antigen.8 Prior studies may have failed in part because they did not reproduce the continuous intrathecal synthesis of antibodies that occurs in patients with PND.
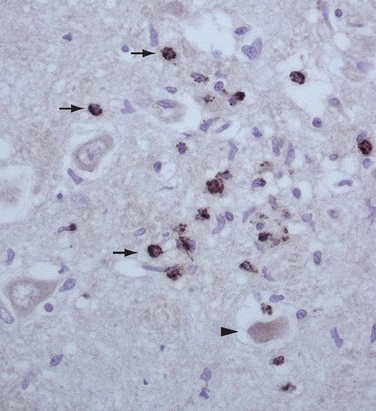
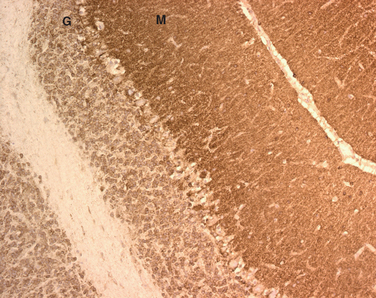
As far as the T cell response is concerned, a pathogenic role was initially suggested by the neuropathological findings in autopsy studies of patients with PND.9 These studies demonstrated prominent infiltrates of inflammatory cells, later characterized as CD4+ and CD8+ T cells that are usually accompanied by microglial activation and gliosis (Fig. 101-1).10,11 Analysis of the presence of antigen-specific T cells in the peripheral blood and brain infiltrates of patients and studies of antigen presentation by dendritic cells or fibroblasts modified to express the paraneoplastic antigens provide strong evidence of a role of cytotoxic T cells.12–15 An attempt to model the disease in animals by priming the T cell response resulted in minimal perivascular inflammatory infiltrates but did not reproduce the disease or result in interstitial T cell infiltrates with neuronophagia that occur in patients.16
Paraneoplastic Neurological Disorders of the Peripheral Nervous System
For many PNDs of the peripheral nerve and muscle, the evidence of immune-mediated mechanisms is also abundant, but the target antigens are less well defined than in PNDs of the CNS. Evidence of immune mechanisms is found mainly by the detection of inflammatory infiltrates composed of T cells.
Effects of Paraneoplastic Immunity on the Tumor
It has been suggested that paraneoplastic immunity is clinically effective in controlling tumor growth, accounting for the small size and limited metastatic burden of tumors associated with PNDs. However, despite the demonstration that some patients with PNDs develop cytotoxic T cell responses to tumor antigens, the oncological outcomes in studies of hundreds of patients with antibody-associated PND do not significantly differ from those of patients without PNDs.17–23 In addition, investigators who examined whether immunotherapy favored tumor growth did not identify a change in tumor behavior.24,25 By and large, these studies suggest either that the efficacy of the antitumor immune response is minimal or that the cancer usually overcomes the antitumor immune effect.1
GENERAL APPROACH TO THE DIAGNOSIS OF PARANEOPLASTIC NEUROLOGICAL DISORDERS
The diagnosis of PNDs is usually based on (1) the recognition of the neurological syndrome, (2) the demonstration of the associated cancer, and (3) the detection of serum and CSF paraneoplastic antibodies.3
Recognition of the Neurological Syndrome
An extensive group of disorders similar to PNDs may occur in the absence of cancer (Table 101-3). However, some syndromes are associated with cancer much more frequently than others, or the clinical features are characteristic enough that they readily suggest a paraneoplastic etiology. These syndromes are considered “classical PNDs” (see Table 101-1). Other syndromes may result from paraneoplastic mechanisms but occur more frequently in the absence of cancer. These syndromes are considered “nonclassical” and necessitate a more extensive differential diagnosis. For example, a brainstem syndrome, chorea, or the Guillain-Barré syndrome may be paraneoplastic manifestations of cancer but are mostly not cancer related, or their development in a patient with cancer may simply be coincidental.
TABLE 101-3 Differential Diagnosis of Paraneoplastic Neurological Disorders of the Central Nervous System
| Paraneoplastic Neurological Disorder | Differential Diagnosis | Additional Considerations in Patients Known to Have Cancer |
|---|---|---|
| Cerebellar degeneration | Alcohol-related degeneration | Cerebellar metastasis |
| Vitamin deficiency (B1, E) | Chemotherapy toxicity (5-FU, Ara-C) | |
| Toxins (anticonvulsants, other) | ||
| Infectious or postinfectious cerebellitis | ||
| Miller-Fisher syndrome | ||
| GAD-associated ataxia | ||
| Gliadin-associated ataxia | ||
| Idiopathic degeneration | ||
| Limbic encephalitis | Viral encephalitis (HSV) | Brain metastasis |
| Nonparaneoplastic (anti-VGKC) encephalitis | HHV 6 infection (particularly after bone marrow transplantation) | |
| Temporal lobe tumor | ||
| Systemic lupus erythematosus | ||
| Doxifluridine toxicity | ||
| Toxic-metabolic encephalopathy | ||
| Hashimoto’s encephalitis | ||
| Sjögren’s syndrome | ||
| Idiopathic encephalitis | ||
| Sensory neuronopathy | Sjögren’s syndrome | Chemotherapy (cisplatin, paclitaxel, docetaxel, vincristine) |
| Toxins (pyridoxine) | ||
| Idiopathic neuronopathy | ||
| Opsoclonus-myoclonus | Infectious, postinfectious encephalitis | Brain metastasis |
| Metabolic encephalopathy | ||
| Toxins | ||
| Idiopathic encephalitis |
Ara-C, cytosine arabinoside; 5-FU, 5-fluorouracil; GAD, glutamic-acid decarboxylase; HHV, human herpesvirus; HSV, herpes simplex virus; VGKC, voltage-gated potassium channel.
Most PNDs develop and progress rapidly until stabilization in a few weeks or months, causing severe disability. Patients often become wheelchair bound or bedridden over a short period of time. PNDs that affect the CNS, dorsal root ganglia, or proximal nerve roots are often accompanied by lymphocytic pleocytosis, an elevated IgG index, the presence of oligoclonal bands, or intrathecal synthesis of paraneoplastic antibodies. However, similar CSF abnormalities can be encountered in any inflammatory or immune-mediated disorder of the CNS, and some patients with PNDs may have normal findings on CSF studies. In most instances, CSF studies are necessary to rule out other cancer complications, such as leptomeningeal metastasis.26
All patients with PNDs of the CNS and some peripheral nerve syndromes (e.g., plexopathies) should undergo neuroimaging evaluation of the involved area. Magnetic resonance imaging (MRI) is the best technique for ruling out metastatic lesions or other complications that may suggest a PND. In most PNDs of the CNS, the function of the blood-brain barrier is preserved, and therefore the affected brain regions are rarely enhanced with contrast material. The abnormalities are usually demonstrated with T2-weighted imaging and fluid-attenuated inversion recovery (FLAIR) sequences. In syndromes such as limbic encephalitis with predominant hippocampal involvement (short-term memory loss, seizures), the MRI findings are often suggestive of the syndrome, although the etiology could be nonparaneoplastic.27 There is also increasing evidence that brain [F18] fluorodeoxyglucose positron emission tomography (FDG-PET) has diagnostic usefulness in PNDs.28 In the early stages of PNDs, FDG-PET may show hypermetabolism in the abnormal brain regions identified by MRI, but in some patients, the MRI findings are normal.29
Biopsy of an abnormal brain region identified by MRI or FDG-PET may be considered if a neoplastic process is suspected or if the clinical, CSF, and MRI findings are unusual. Abnormalities supporting but not specific to PND include infiltrates of mononuclear cells, neuronophagic nodules, neuronal degeneration, microglial proliferation, and gliosis.30
Demonstration of Associated Cancer
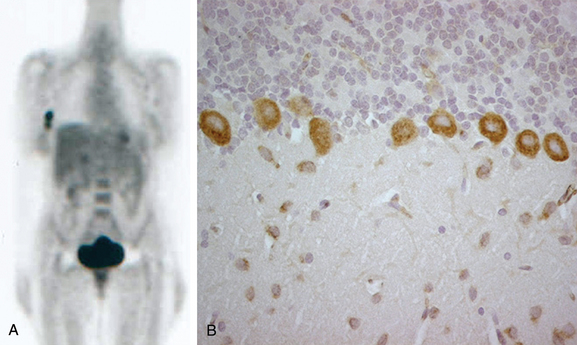
PNDs usually develop at early stages of cancer, and therefore the tumor (or tumor recurrence) may be difficult to demonstrate. Modern imaging techniques are able to demonstrate small tumors that were often missed by techniques used previously. In most instances, the tumor is revealed by computed tomography of the chest, abdomen, and pelvis. The type of syndrome and paraneoplastic antibody may suggest a specific underlying tumor and the need for additional tests, such as mammography or ultrasonography of the pelvis or testes. Whole-body FDG-PET is very useful in demonstrating occult neoplasms or small metastatic lesions that may be more accessible for biopsy than the primary tumor is (Fig. 101-2).31 In addition to imaging studies, serum cancer markers such as carcinoembryonic antigen, CA-125, CA-15.3, or prostate-specific antigen are helpful. All patients with a neuropathy of unclear etiology should be examined for the presence of a monoclonal gammopathy in the serum and urine and, if results are positive, undergo a skeletal survey and bone marrow biopsy; these studies may uncover a malignant plasma cell dyscrasia, amyloidosis, or B cell lymphoma.2
Detection of Paraneoplastic Antibodies
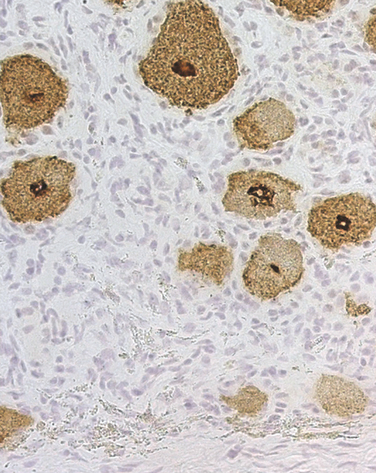
Paraneoplastic antibodies are antibodies whose presence serves as a marker of the paraneoplastic origin of a neurological syndrome. Several concepts are important in testing for paraneoplastic antibodies (Table 101-4). First, antibodies are present in approximately 60% of patients with PNDs of the CNS; therefore, the absence of antibodies does not preclude a paraneoplastic syndrome. Second, paraneoplastic antibodies may be identified (usually at low titers) in the serum of a variable proportion of patients with cancer but without PND (i.e., anti-Hu and anti-CV2/CRMP5 in 20% and 10% of patients with SCLC, respectively).32,33 Third, in PNDs of the CNS, the antibodies are found in serum and CSF; detection of CSF antibodies is a strong indicator that the associated neurological syndrome is paraneoplastic. Fourth, most PNDs of the peripheral nerve or muscle are not associated with paraneoplastic antibodies, except for anti-Hu (Fig. 101-3) and anti-CV2/CRMP5 antibodies. Fifth, not all paraneoplastic antibodies have the same sensitivity and specificity; on the basis of their clinical relevance, the paraneoplastic antibodies are classified in two categories: well-characterized paraneoplastic antibodies and partially characterized antibodies (see Table 101-4).3
| Antibody | Associated Syndrome | Most Frequent Cancers |
|---|---|---|
| Well-Characterized Paraneoplastic Antibodies | ||
| Hu (ANNA1) | Paraneoplastic encephalomyelitis, PSN, PCD, limbic encephalitis | Small cell lung cancer |
| Yo (PCA1) | PCD | Ovary, breast cancers |
| CV2/CRMP5 | Several | Small cell lung cancer |
| Ri (ANNA2) | Ataxia, opsoclonus-myoclonus, brainstem encephalitis | Breast, gynecological, small cell lung cancers |
| Ma2* | Limbic, diencephalic, brainstem encephalitis | Testicular, lung cancers |
| Amphiphysin | Stiff-person syndrome, paraneoplastic encephalomyelitis | Breast, small cell lung cancers |
| Partially Characterized Paraneoplastic Antibodies | ||
| Tr | PCD | Hodgkin’s disease |
| Zic4 | PCD | Small cell lung cancer |
| PCA2 | Several | Small cell lung cancer |
| ANNA3 | Several | Small cell lung cancer |
PCD, paraneoplastic cerebellar degeneration; PSN, paraneoplastic sensory neuronopathy.
* Some patients harbor Ma1 and Ma2 antibodies; the presence of Ma1 is usually associated with predominant brainstem and cerebellar involvement and with tumors other than testicular neoplasms. The prognosis in patients with tumors other than testicular neoplasms is poorer than that of patients with Ma2 antibodies and testicular neoplasms.
Several antibodies, including P/Q-type voltage-gated calcium channels, voltage-gated potassium channels, and nicotinic or ganglionic AChR antibodies can be detected in both the paraneoplastic and nonparaneoplastic forms of the associated disorder (Table 101-5). These antibodies may assist in the diagnosis of the neurological disorder but are not predictive of the presence of a tumor. The same P/Q-type voltage-gated calcium channel antibodies that are associated with the paraneoplastic and nonparaneoplastic forms of LEMS have also been encountered in a subset of patients with paraneoplastic cerebellar degeneration (PCD) and SCLC; therefore, detection of these antibodies in patients with a subacute cerebellar syndrome should prompt the search for a SCLC.34
TABLE 101-5 Antibodies Associated with the Paraneoplastic and Nonparaneoplastic Forms of the Neurological Disorder
| Antibody | Syndrome |
|---|---|
| AChR (nicotinic, neuromuscular junction) | Myasthenia gravis |
| AChR (ganglionic or neuronal) | Autonomic neuropathy |
| P/Q-type VGCC* | Lambert-Eaton myasthenic syndrome |
| VGKC | Neuromyotonia, limbic encephalitis |
AChR, acetylcholine receptor; VGCC, voltage-gated calcium channels; VGKC, voltage-gated potassium channels.
* In patients with cerebellar degeneration, with or without associated Lambert-Eaton myasthenic syndrome, detection of these antibodies should prompt the search of a small cell lung cancer.
Diagnostic Criteria of Paraneoplastic Neurological Disorders
Information obtained from the type of neurological syndrome, detection of the cancer, and presence or absence of paraneoplastic antibodies has been used to define general guidelines for the diagnosis of PNDs, with the caveat that both the diagnosis of the tumor and the development of the neurological syndrome should occur within 5 years (Table 101-6).3 Nowadays, the use of whole-body computed tomography and FDG-PET allows detection of the tumor at the time of neurological syndrome manifestation in 80% to 90% of the patients. The indicated guidelines are useful but not perfect, and patients with criteria of possible PND should be carefully considered for alternative diagnoses.
TABLE 101-6 Diagnostic Criteria for Paraneoplastic Neurological Disorders
SPECIFIC PARANEOPLASTIC SYNDROMES
Paraneoplastic Encephalomyelitis
Paraneoplastic encephalomyelitis refers to an immune-mediated inflammatory disorder that can affect any part of the CNS, dorsal root ganglia, and autonomic nerves. The main areas involved include the hippocampus (limbic encephalitis), the Purkinje cells of the cerebellum (cerebellar degeneration), the lower brainstem (brainstem encephalitis), the dorsal root ganglia (sensory neuronopathy), the spinal cord (myelitis) (Fig. 101-4), and the sympathetic or parasympathetic ganglia and nerves (orthostatic hypotension, gastrointestinal paresis or pseudoobstruction, cardiac arrhythmia, erectile dysfunction, and abnormal pupillary responses to light).17,19 Less frequently, patients may develop discrete focal cortical encephalitis, sometimes manifesting as epilepsia partialis continua. The diagnosis of paraneoplastic encephalomyelitis should be considered when the dominant symptoms result from involvement of two or more of the indicated areas.
Symptoms of paraneoplastic encephalomyelitis develop rapidly and progress over weeks or months until stabilization or death. The CSF is almost always abnormal, with mild to moderate lymphocytic pleocytosis, increased protein concentration, and oligoclonal bands or increased IgG index.18 Brain MRI findings are often abnormal; FLAIR or T2-weighted sequences reveal hyperintensities in involved areas and sometimes clinically silent regions; the abnormalities usually are not enhanced after administration of contrast material. For reasons that are unclear, contrast enhancement is more likely to occur in some forms of encephalitis (e.g., limbic-diencephalic encephalitis associated with anti-Ma2 antibodies) than in others (limbic encephalitis associated with anti-Hu antibodies).35,36
Several antibodies assist in the diagnosis of paraneoplastic encephalomyelitis and the underlying neoplasm (see Table 101-4). The management of paraneoplastic encephalomyelitis is based on prompt treatment of the tumor along with immunosuppression. Although the standard of care remains to be established, the use of corticosteroids and intravenous immunoglobulin (IVIg), combined with chemotherapy, may help stabilize or improve the neurological symptoms during the time that the tumor is treated. Afterward, if the neurological symptoms have stabilized or improved, patients should be considered for prolonged treatment with immunosuppressants that target not only the antibodies but also the T cell immunity (e.g., cyclophosphamide combined with corticosteroids, among other strategies; see Fig. 101-1). Although this approach has not been useful for patients with advanced disease,37 there is evidence that at early stages of PNDs, it may be effective.1,24 Patients with brainstem symptoms have a much poorer prognosis than do patients with involvement of other areas of the CNS.
Limbic Encephalitis
The dominant symptom of limbic encephalitis is short-term memory loss in association with confusion, confabulation, seizures, affective or mood disorders, and hypersomnia or insomnia.27 Patients may present with acute behavioral change, delusional thoughts, poor judgment, or status epilepticus, which lead to an initial diagnosis of psychosis, drug abuse, or malingering. FLAIR or T2-weighted MRI demonstrates abnormalities in the medial aspect of the temporal lobes, but sometimes the involvement is more extensive or affects areas outside the limbic system. FDG-PET may reveal hyperactivity in regions that are normal in MRI (Fig. 101-5).35 Electroencephalography usually demonstrates unilateral or bilateral temporal lobe epileptic discharges or slow background activity. Electroencephalographic monitoring is important in patients who appear confused or have a low level of consciousness; many of them are in nonconvulsive status epilepticus.
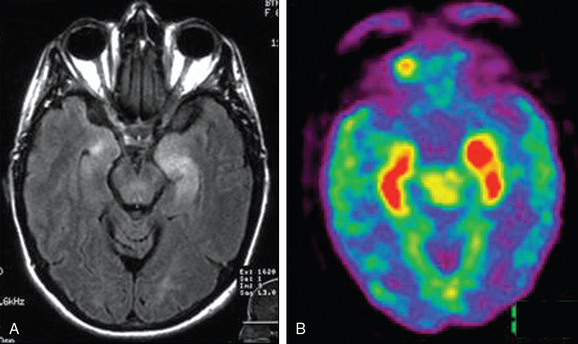
The antibodies more frequently associated with paraneoplastic limbic encephalitis are anti-Hu, anti-CV2/CRMP5, and anti-Ma2.35,36,38 Patients with anti-Hu antibodies usually have SCLC, and those with anti-CV2/CRMP5 antibodies often have SCLC or thymoma. Anti-Ma2 antibodies in young men is usually associated with testicular neoplasms; in elderly men or women, the leading neoplasm is non–small cell lung cancer.
The response of paraneoplastic limbic encephalitis to treatment is variable; some patients exhibit dramatic improvement if the tumor is treated promptly or sometimes with corticosteroids and IVIg, whereas others with similar symptoms and antibodies do not respond to therapy. However, improvement is unlikely if the tumor is not treated. There is some evidence that patients without paraneoplastic antibodies or young patients with anti-Ma2 antibodies are more likely to experience improvement than are patients with anti-Hu or CV2/CRMP5 antibodies.35,36
A subset of patients with limbic encephalitis harbors antibodies to voltage-gated potassium channels; most of these patients do not have cancer, but in a few instances a SCLC has been found.39 Although this type of limbic encephalitis usually responds to IVIg, plasma exchange, or corticosteroids, some patients develop life-threatening complications (e.g., status epilepticus and hyponatremia) that are difficult to control.
Paraneoplastic Cerebellar Degeneration
Patients with PCD usually present with dizziness, vertigo, oscillopsia, or gait unsteadiness that rapidly evolves to frank gait and limb ataxia. Other symptoms include dysarthria, dysphagia, diplopia (often without obvious oculoparesis), and predominant downbeat nystagmus. MRI of the brain usually yields normal findings at symptom presentation and reveals cerebellar atrophy as the disease evolves.40
Cerebellar symptoms may occur in association with any of the paraneoplastic antibodies listed in Table 101-4. The tumors more frequently associated with PCD are lung, ovary, and breast cancers and lymphoma. A few antibodies (anti-Yo, anti-Tr) are associated with dominant cerebellar symptoms without significant involvement of other areas of the nervous system (see Fig. 101-2B).41,42 Other antibodies are usually associated with additional neurological symptoms. Patients with PCD and SCLC should be evaluated for motor weakness, because some of these patients may have LEMS.40
PCD results from degeneration of the Purkinje cells of the cerebellum along with inflammatory infiltrates in the deep cerebellar nuclei and lower brainstem. As occurs with other PNDs, 30% to 40% of patients with PCD do not harbor antineuronal antibodies. In these patients, the differential diagnosis is extensive (see Table 101-3). Although PCD is refractory to most treatments, isolated case reports of response to plasma exchange, IVIg, and T cell immunosuppression (e.g., cyclophosphamide) have been reported. Prompt diagnosis and treatment of the tumor, along with immunosuppression, may stabilize the disorder or prevent involvement of other areas of the CNS.
Opsoclonus-Myoclonus
Opsoclonus is a disorder of eye motility with involuntary, chaotic, conjugate saccades. It is almost always associated with myoclonus of the trunk and limbs and sometimes with truncal or limb ataxia. Some patients develop symptoms of diffuse encephalopathy that may lead to stupor, coma, and death. In adults, the tumors more frequently involved are SCLC and gynecological and breast cancers.43 In children, opsoclonus-myoclonus is usually accompanied by hypotonia, irritability, behavioral changes, and psychomotor retardation, and the underlying tumor is a neuroblastoma.44
Most patients with paraneoplastic opsoclonus-myoclonus do not have detectable paraneoplastic antibodies. An exception is the anti-Ri antibody that identifies a subgroup of patients with opsoclonus-myoclonus, ataxia, breast or gynecological cancers, and, less frequently, SCLC.45 Other antibodies (e.g., anti-Yo, anti-Ma2, anti-Hu) have been reported in a few patients with opsoclonus associated with cerebellar or brainstem encephalitis.
Paraneoplastic opsoclonus-myoclonus may respond to IgG-depleting strategies (IVIg or plasma exchange and corticosteroids) along with treatment of the tumor; improvement is rare if the tumor is not treated. There is an idiopathic form of opsoclonus-myoclonus that tends to occur in younger patients and responds better to immunotherapy.43
Stiff-Person Syndrome
In about 80% of patients with stiff-person syndrome, the disorder develops as a nonparaneoplastic phenomenon in association with diabetes, polyendocrinopathy, and antibodies to glutamic acid decarboxylase. Neurological symptoms include fluctuating rigidity of the axial musculature with superimposed spasms precipitated by emotional upset and auditory or somesthetic stimuli. Muscle stiffness affects primarily the lower trunk and legs, but it can extend to the arms. Electrophysiological studies show continuous activity of motor units in the stiffened muscles, which improves after treatment with diazepam. The rigidity lessens during sleep or general anesthesia.46
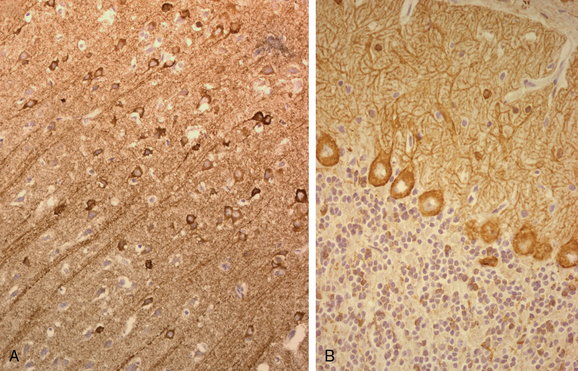
A similar syndrome, although with more frequent involvement of the arms, may occur as a paraneoplastic manifestation of cancers of the breast and lung and, less frequently, Hodgkin’s lymphoma. Paraneoplastic stiff-person syndrome is characterized by the presence of antibodies to amphiphysin (Fig. 101-6) and, in rare cases, with antibodies to glutamic acid decarboxylase.47 Muscle rigidity may also occur with less frequent syndromes: paraneoplastic encephalomyelitis with rigidity (usually with brainstem involvement) or spinal myoclonus with rigidity. Some of these patients harbor anti-Ri antibodies and have acute episodes of flushing and piloerection, which is suggestive of autonomic dysfunction.
Paraneoplastic Sensory Neuronopathy
Paraneoplastic sensory neuronopathy (PSN) is characterized by progressive numbness and often painful dysesthesias involving the limbs, trunk, and, less frequently, the cranial nerves, causing face numbness or sensorineural hearing loss. The symptom manifestation is frequently asymmetrical and is accompanied by decreased or abolished reflexes and relative preservation of strength. All types of sensation can be affected, but loss of proprioception is often predominant. As a result, patients develop sensory ataxia and pseudoathetoid movements of the extremities (predominantly the hands), demonstrated when the patient closes the eyes or is distracted during the examination.48 PSN results from inflammatory involvement of the dorsal root ganglia, usually accompanied by dorsal nerve root inflammation. For this reason, symptoms are often asymmetrical and the CSF exhibits inflammatory abnormalities. PSN is frequently associated with paraneoplastic encephalomyelitis, particularly in patients with SCLC. These patients almost always harbor anti-Hu antibodies (see Fig. 101-3).17
Electrophysiological studies demonstrate that patients with PSN have small-amplitude or no sensory nerve action potentials with relative preservation of motor conduction velocities. Because PSN often overlaps with paraneoplastic encephalomyelitis that may affect lower motor neurons and is sometimes associated with peripheral neuropathy, the electrophysiological studies may show motor abnormalities.49,50 Autopsy studies indicate that when anti-Hu antibodies are detected, the sensory symptoms result from involvement of the dorsal root ganglia and dorsal nerve roots.17 Some of these patients harbor additional antibodies to CV2/CRMP5 (Fig. 101-7).51
Prompt treatment of patients with corticosteroids and IVIg (along with treatment of the tumor) may result in stabilization or mild improvement of the dorsal root ganglia dysfunction, sometimes confirmed with improvements in results of electrophysiological studies.52
Paraneoplastic Neuropathies
Subacute paraneoplastic neuropathies that occur at early stages of cancer or tumor recurrence include PSN (that, as discussed previously, result from dorsal root ganglia involvement rather than peripheral nerve involvement) and a group of debilitating sensorimotor neuropathies whose appearance may precede the diagnosis of the tumor. The course of these neuropathies is usually rapid and progressive, rarely with relapsing and remitting episodes.53 The dominant symptoms are sensorimotor deficits, sometimes with pain as a major complaint (i.e., vasculitis of the nerve and muscle). In rare instances, the sympathetic or parasympathetic nerves are the main target of the paraneoplastic disorder, which results in acute pandysautonomia. Life-threatening symptoms include gastrointestinal dysmotility with intestinal pseudoobstruction,54,55 orthostatic hypotension,56,57 and cardiac dysrhythmias.17,58,59 Other symptoms may include dry mouth, erectile dysfunction, anhidrosis, and sphincter dysfunction.60
At advanced stages of cancer, the paraneoplastic neuropathies are more frequent but milder than those that precede the cancer diagnosis. An extensive number of chemotherapeutic agents can cause a neuropathy, and these should be considered in the differential diagnosis.61
Several malignancies involving plasma cells and B lymphocytes, such as myeloma or B cell lymphoma, are associated with sensorimotor neuropathies that often resemble chronic inflammatory demyelinating neuropathy. Patients with Waldenström’s macroglobulinemia develop distal symmetrical sensorimotor polyneuropathy with predominant involvement of large sensory fibers (vibration sense is particularly affected).62
In patients with paraneoplastic neuropathies, antibody markers are often not found except for anti-Hu antibodies, which usually indicate dorsal root ganglia involvement, and anti-CV2/CRMP5 antibodies, which may be associated with neuropathies with mixed axonal and demyelinating features. Antibodies against myelin-associated glycoprotein are often found in patients with Waldenström’s macroglobulinemia, but similar antibodies can be identified in patients with immunoglobulin M monoclonal gammopathies without an underlying malignancy. Several studies indicate that antiganglioside antibodies are absent in most paraneoplastic neuropathies,63 although a few isolated cases have been reported.64 Patients with autonomic neuropathy may harbor antibodies to the ganglionic AChR; similar antibodies are found in patients without cancer.65
In general, paraneoplastic neuropathies with predominant demyelinating features respond to immunotherapy (corticosteroids, IVIg, or plasma exchange), whereas those with axonal features are more difficult to treat.53 The neuropathy associated with vasculitis of the nerve and muscle may respond dramatically to corticosteroids and cyclophosphamide.66 Other treatment-responsive neuropathies include those associated with Waldenström’s macroglobulinemia (treatment of the malignancy, IVIg, plasma exchange, corticosteroids, or rituximab, among others); sclerotic myeloma (resection of the sclerotic lesion, focal radiation, or chemotherapy); and autonomic neuropathy with ganglionic AChR antibodies (IgG-depleting strategies).65 The neuropathy of multiple myeloma is refractory to most treatments (for a detailed review of paraneoplastic neuropathies, see Rudnicki and Dalmau, 20002).
Lambert-Eaton Myasthenic Syndrome
LEMS results from an antibody-mediated attack against the P/Q-type voltage-gated calcium channels located at the presynaptic level of the neuromuscular junction. This interferes with the release of acetylcholine and results in muscle weakness and fatigability. LEMS should be suspected in patients with proximal weakness, dry mouth, and decreased or absent reflexes, particularly if the patient is known to have SCLC or a history of smoking.67 In general, the legs are more involved than the arms. Mild muscle aches and distal paresthesias are common.68 Cranial nerve involvement is frequent but mild and transient and is almost always accompanied by motor weakness in the extremities. The symptom presentation of isolated ocular weakness virtually excludes the diagnosis of LEMS.69 In addition to dry mouth, patients may have other signs of autonomic dysfunction (orthostatic hypotension, erectile dysfunction, blurred vision). Electrophysiological studies show small-amplitude compound muscle action potentials. At slow rates of repetitive nerve stimulation (2 to 5 Hz), there is a decremental response, whereas at fast rates (20 Hz or more) or after maximal voluntary muscle contraction, facilitation occurs with an incremental response of at least 100%.
Approximately 60% of patients with LEMS have an underlying neoplasm, usually SCLC or, in rare cases, other tumors such as lymphoma.67 Paraneoplastic LEMS may be associated with PCD or paraneoplastic encephalomyelitis.40 The nonparaneoplastic cases often have a slower symptom presentation and are associated with other autoimmune conditions such as thyroiditis or insulin-dependant diabetes mellitus.67 Treatment of the tumor and medications that enhance acetylcholine release (3,4-diaminopyridine or a combination of pyridostigmine and guanidine) are usually effective.70 IVIg and plasma exchange ameliorate symptoms within 2 to 4 weeks, but the benefit is transient. Long-term immunotherapy with prednisone or azathioprine is an alternative for patients who do not experience improvement with 3,4-diaminopyridine.71
Myasthenia Gravis
Myasthenia gravis is a disorder of the postsynaptic neuromuscular junction, usually caused by anti-AChR antibodies. Patients without these antibodies may harbor antibodies against muscle-specific kinase. In contrast to LEMS, in which the motor weakness tends to progress in a caudocranial direction with decreased or absent reflexes, myasthenia gravis tends to progress in the opposite direction with early and prominent ocular paresis and preservation of the reflexes.69 Approximately 10% of patients with myasthenia gravis have thymoma. Thymoma-related myasthenia gravis is almost invariably accompanied by AChR antibodies but not by anti–muscle-specific kinase antibodies. After resection of the thymoma, the treatment is similar to that in patients without thymoma and may include plasma exchange, IVIg, anticholinesterase inhibitors, and long-term immunosuppression.71
Paraneoplastic Dermatomyositis and Polymyositis
These two disorders have similar clinical features, including proximal muscle weakness, myalgia, and muscle tenderness. Pharyngeal, esophageal, and neck flexor muscles are often involved. Reflexes and sensation are unaffected. Dermatomyositis is accompanied by purplish discoloration of the eyelids (heliotrope rash) and erythematous scaly lesions over the knuckles. Associated conditions may include arthralgias and contractures, myocarditis, and interstitial lung disease.72
The diagnoses of both inflammatory myopathies are based on clinical and electrophysiological findings, detection of elevated serum muscle enzyme levels (which can be normal in some patients), and demonstration of inflammatory infiltrates in the muscle biopsy. Dermatomyositis is probably caused by an immune-mediated intramuscular angiopathy, leading to ischemia, muscle fiber necrosis, and perifascicular atrophy. The inflammatory infiltrates are composed of macrophages, B cells, and CD4+ T cells.73,74 In contrast, polymyositis is mediated by a CD8+ T cell response.
Although patients with polymyositis may have cancer, studies suggest that in most instances, the presence of both is coincidental.75 In contrast, many patients with dermatomyositis (particularly those older than 50 years) have cancer; the tumors more involved are ovarian and breast cancers in women and lung and gastrointestinal cancers in men. There are no serological tests indicative of paraneoplasia. Besides treatment of the tumor, the management of paraneoplastic dermatomyositis and polymyositis is similar to that of the nonparaneoplastic form of these disorders and includes corticosteroids, azathioprine, and IVIg.76,77
Acute Necrotizing Myopathy
This disorder is a rare paraneoplastic manifestation of cancer that manifests with acute and severe muscle weakness, markedly increased serum muscle enzyme levels, and extensive muscle necrosis with mild or no inflammatory infiltrates. Patients often die as a result of the extensive muscle involvement; a few patients respond to treatment of the tumor and corticosteroids.78 The disorder has been reported in association with other syndromes, including myasthenia gravis with thymoma and rhabdomyolysis.79,80
Paraneoplastic Visual Syndromes
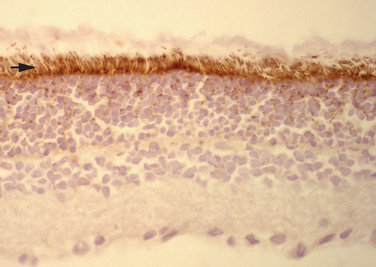
Paraneoplastic retinopathy is characterized by progressive loss of photoreceptor function that usually precedes the diagnosis of cancer.81 Symptoms include painless loss of visual acuity and color vision, light-induced glare, photosensitivity, and peripheral and ringlike scotomas. Funduscopic examination findings are normal or demonstrate arteriolar narrowing. The electroretinogram shows flat photopic responses. Paraneoplastic retinopathy associated with antirecoverin antibodies is known as cancer-associated retinopathy and is usually associated with SCLC (Fig. 101-8).82 There is evidence that antirecoverin antibodies can gain access to the aqueous humor and may cause apoptotic death of photoreceptor cells.83,84
Patients with metastatic melanoma may develop a syndrome known as melanoma-associated retinopathy. Symptoms include night blindness, relative preservation of visual acuity, shimmering or flickering sensations, midperipheral field defect, and prolonged dark adaptation. Retinal vessel attenuation and vitreous infiltrates are found in 30% of patients. The electroretinogram shows no or decreased adapted B wave response. Some patients with melanoma-associated retinopathy have antibodies against unknown antigens expressed by bipolar cells of the retina.85,86
The treatment of paraneoplastic retinopathies is based on controlling the tumor; there are a few reports of visual improvement after corticosteroids, IVIg, or plasma exchange, but the majority of patients do not experience improvement with immunotherapy.82,86
Optic neuritis is a very rare paraneoplastic manifestation of cancer that almost always occurs in association with encephalomyelitis and SCLC. Symptoms include painless bilateral visual loss, swollen optic discs, and field defects with tunnel vision. Several associated antibodies have been identified, including anti-Hu, anti-Tr, anti-Yo, and, more frequently, anti-CV2/CRMP5.87 Patients may have concurrent retinitis, vitreal infiltrates, and abnormal electroretinographic patterns. The visual outcome is usually poor.
Optic neuritis and myelitis resembling Devic’s disease has been reported in a patient with myasthenia gravis, thymoma, and necrotizing myopathy. An antibody against unknown antigens expressed by the CNS and thymoma was identified; all neurological symptoms except visual acuity were ameliorated by immunotherapy.79
Bataller L, Dalmau JO. Paraneoplastic disorders of the central nervous system: update on diagnostic criteria and treatment. Semin Neurol. 2004;24:461-471.
Darnell RB, Posner JB. Paraneoplastic syndromes involving the nervous system. N Engl J Med. 2003;349:1543-1554.
Graus F, Delattre JY, Antoine JC, et al. Recommended diagnostic criteria for paraneoplastic neurological syndromes. J Neurol Neurosurg Psychiatry. 2004;75:1135-1140.
Rosenfeld MR, Dalmau J. Current therapies for paraneoplastic neurologic syndromes. Curr Treat Options Neurol. 2003;5:69-77.
Rudnicki SA, Dalmau J. Paraneoplastic syndromes of the spinal cord, nerve, and muscle. Muscle Nerve. 2000;23:1800-1818.
1 Bataller L, Dalmau JO. Paraneoplastic disorders of the central nervous system: update on diagnostic criteria and treatment. Semin Neurol. 2004;24:461-471.
2 Rudnicki SA, Dalmau J. Paraneoplastic syndromes of the spinal cord, nerve, and muscle. Muscle Nerve. 2000;23:1800-1818.
3 Graus F, Delattre JY, Antoine JC, et al. Recommended diagnostic criteria for paraneoplastic neurological syndromes. J Neurol Neurosurg Psychiatry. 2004;75:1135-1140.
4 Tanaka K, Tanaka M, Onodera O, et al. Passive transfer and active immunization with the recombinant leucine-zipper (Yo) protein as an attempt to establish an animal model of paraneoplastic cerebellar degeneration. J Neurol Sci. 1994;127:153-158.
5 Tanaka K, Tanaka M, Igarashi S, et al. Trial to establish an animal model of paraneoplastic cerebellar degeneration with anti-Yo antibody. 2. Passive transfer of murine mononuclear cells activated with recombinant Yo protein to paraneoplastic cerebellar degeneration lymphocytes in severe combined immunodeficiency mice. Clin Neurol Neurosurg. 1995;97:101-105.
6 Graus F, Illa I, Agusti M, et al. Effect of intraventricular injection of an anti–Purkinje cell antibody (anti-Yo) in a guinea pig model. J Neurol Sci. 1991;106:82-87.
7 Sillevis Smitt PA, Manley GT, et al. Immunization with the paraneoplastic encephalomyelitis antigen HuD does not cause neurologic disease in mice. Neurology. 1995;45:1873-1878.
8 Levin MC, Lee SM, Kalume F, et al. Autoimmunity due to molecular mimicry as a cause of neurological disease. Nat Med. 2002;8:509-513.
9 Henson RA, Hoffman HL, Urich H. Encephalomyelitis with carcinoma. Brain. 1965;88:449-464.
10 Graus F, Ribalta T, Campo E, et al. Immunohistochemical analysis of the immune reaction in the nervous system in paraneoplastic encephalomyelitis. Neurology. 1990;40:219-222.
11 Jean WC, Dalmau J, Ho A, et al. Analysis of the IgG subclass distribution and inflammatory infiltrates in patients with anti-Hu–associated paraneoplastic encephalomyelitis. Neurology. 1994;44:140-147.
12 Benyahia B, Liblau R, Merle-Béral H, et al. Cell-mediated autoimmunity in paraneoplastic neurologic syndromes with anti-Hu antibodies. Ann Neurol. 1999;45:162-167.
13 Albert ML, Austin LM, Darnell RB. Detection and treatment of activated T cells in the cerebrospinal fluid of patients with paraneoplastic cerebellar degeneration. Ann Neurol. 2000;47:9-17.
14 Tanaka M, Tanaka K, Shinozawa K, et al. Cytotoxic T cells react with recombinant Yo protein from a patient with paraneoplastic cerebellar degeneration and anti-Yo antibody. J Neurol Sci. 1998;161:88-90.
15 Tanaka K, Tanaka M, Inuzuka T, et al. Cytotoxic T lymphocyte–mediated cell death in paraneoplastic sensory neuronopathy with anti-Hu antibody. J Neurol Sci. 1999;163:159-162.
16 Pellkofer H, Schubart AS, Hoftberger R, et al. Modelling paraneoplastic CNS disease: T-cells specific for the onconeuronal antigen PNMA1 mediate autoimmune encephalomyelitis in the rat. Brain. 2004;127:1822-1830.
17 Dalmau J, Graus F, Rosenblum MK, et al. Anti-Hu–associated paraneoplastic encephalomyelitis/sensory neuronopathy. A clinical study of 71 patients. Medicine. 1992;71:59-72.
18 Graus F, Keime-Guibert F, Rene R, et al. Anti-Hu–associated paraneoplastic encephalomyelitis: analysis of 200 patients. Brain. 2001;124:1138-1148.
19 Sillevis SP, Grefkens J, De Leeuw B, et al. Survival and outcome in 73 anti-Hu positive patients with paraneoplastic encephalomyelitis/sensory neuronopathy. J Neurol. 2002;249:745-753.
20 Hetzel DJ, Stanhope CR, O’Neill BP, et al. Gynecologic cancer in patients with subacute cerebellar degeneration predicted by anti–Purkinje cell antibodies and limited in metastatic volume. Mayo Clin Proc. 1990;65:1558-1563.
21 Rojas I, Graus F, Keime-Guibert F, et al. Long-term clinical outcome of paraneoplastic cerebellar degeneration and anti-Yo antibodies. Neurology. 2000;55:713-715.
22 Rojas-Marcos I, Rousseau A, Keime-Guibert F, et al. Spectrum of paraneoplastic neurologic disorders in women with breast and gynecologic cancer. Medicine (Baltimore). 2003;82:216-223.
23 Lucchinetti CF, Kimmel DW, Lennon VA. Paraneoplastic and oncologic profiles of patients seropositive for type 1 antineuronal nuclear autoantibodies. Neurology. 1998;50:652-657.
24 Vernino S, O’Neill BP, Marks RS, et al. Immunomodulatory treatment trial for paraneoplastic neurological disorders. Neuro-oncol. 2004;6:55-62.
25 Keime-Guibert F, Graus F, Broet P, et al. Clinical outcome of patients with anti-Hu–associated encephalomyelitis after treatment of the tumor. Neurology. 1999;53:1719-1723.
26 Posner JB. Neurologic Complications of Cancer. Philadelphia: FA Davis, 1995.
27 Gultekin SH, Rosenfeld MR, Voltz R, et al. Paraneoplastic limbic encephalitis: neurological symptoms, immunological findings and tumour association in 50 patients. Brain. 2000;123:1481-1494.
28 Crotty E, Patz EFJr. FDG-PET imaging in patients with paraneoplastic syndromes and suspected small cell lung cancer. J Thorac Imaging. 2001;16:89-93.
29 Dadparvar S, Anderson GS, Bhargava P, et al. Paraneoplastic encephalitis associated with cystic teratoma is detected by fluorodeoxyglucose positron emission tomography with negative magnetic resonance image findings. Clin Nucl Med. 2003;28:893-896.
30 Henson RA, Urich HE. Cancer and the Nervous System: The Neurological Manifestations of Systemic Malignant Disease. London: Blackwell Scientific, 1982.
31 Younes-Mhenni S, Janier MF, Cinotti L, et al. FDG-PET improves tumour detection in patients with paraneoplastic neurological syndromes. Brain. 2004;127:2331-2338.
32 Graus F, Dalmau J, Rene R, et al. Anti-Hu antibodies in patients with small-cell lung cancer: association with complete response to therapy and improved survival. J Clin Oncol. 1997;15:2866-2872.
33 Bataller L, Wade DF, Graus F, et al. Antibodies to Zic4 in paraneoplastic neurologic disorders and small-cell lung cancer. Neurology. 2004;62:778-782.
34 Graus F, Lang B, Pozo-Rosich P, et al. P/Q type calcium-channel antibodies in paraneoplastic cerebellar degeneration with lung cancer. Neurology. 2002;59:764-766.
35 Dalmau J, Graus F, Villarejo A, et al. Clinical analysis of anti-Ma2–associated encephalitis. Brain. 2004;127:1831-1844.
36 Alamowitch S, Graus F, Uchuya M, et al. Limbic encephalitis and small cell lung cancer. Clinical and immunological features. Brain. 1997;120:923-928.
37 Keime-Guibert F, Graus F, Fleury A, et al. Treatment of paraneoplastic neurological syndromes with antineuronal antibodies (Anti-Hu, anti-Yo) with a combination of immunoglobulins, cyclophosphamide, and methylprednisolone. J Neurol Neurosurg Psychiatry. 2000;68:479-482.
38 Lawn ND, Westmoreland BF, Kiely MJ, et al. Clinical, magnetic resonance imaging, and electroencephalographic findings in paraneoplastic limbic encephalitis. Mayo Clin Proc. 2003;78:1363-1368.
39 Vincent A, Buckley C, Schott JM, et al. Potassium channel antibody–associated encephalopathy: a potentially immunotherapy-responsive form of limbic encephalitis. Brain. 2004;127:701-712.
40 Mason WP, Graus F, Lang B, et al. Small-cell lung cancer, paraneoplastic cerebellar degeneration and the Lambert-Eaton myasthenic syndrome. Brain. 1997;120:1279-1300.
41 Shams’ili S, Grefkens J, De Leeuw B, et al. Paraneoplastic cerebellar degeneration associated with antineuronal antibodies: analysis of 50 patients. Brain. 2003;126:1409-1418.
42 Bernal F, Shams’ili S, Rojas I, et al. Anti-Tr antibodies as markers of paraneoplastic cerebellar degeneration and Hodgkin’s disease. Neurology. 2003;60:230-234.
43 Bataller L, Graus F, Saiz A, et al. Clinical outcome in adult onset idiopathic or paraneoplastic opsoclonus-myoclonus. Brain. 2001;124:437-443.
44 Russo C, Cohn SL, Petruzzi MJ, et al. Long-term neurologic outcome in children with opsoclonus-myoclonus associated with neuroblastoma: a report from the Pediatric Oncology Group. Med Pediatr Oncol. 1997;28:284-288.
45 Sutton IJ, Barnett MH, Watson JD, et al. Paraneoplastic brainstem encephalitis and anti-Ri antibodies. J Neurol. 2002;249:1597-1598.
46 Brown P, Marsden CD. The stiff man and stiff man plus syndromes. J Neurol. 1999;246:648-652.
47 De Camilli P, Thomas A, Cofiell R, et al. The synaptic vesicle-associated protein amphiphysin is the 128-kD autoantigen of stiff-man syndrome with breast cancer. J Exp Med. 1993;178:2219-2223.
48 Chalk CH, Windebank AJ, Kimmel DW, et al. The distinctive clinical features of paraneoplastic sensory neuronopathy. Can J Neurol Sci. 1992;19:346-351.
49 Camdessanche JP, Antoine JC, Honnorat J, et al. Paraneoplastic peripheral neuropathy associated with anti-Hu antibodies. A clinical and electrophysiological study of 20 patients. Brain. 2002;125:166-175.
50 Oh SJ, Gurtekin Y, Dropcho EJ, et al. Anti-Hu antibody neuropathy: a clinical, electrophysiological, and pathological study. Clin Neurophysiol. 2005;116:28-34.
51 Antoine JC, Honnorat J, Camdessanche JP, et al. Paraneoplastic anti-CV2 antibodies react with peripheral nerve and are associated with a mixed axonal and demyelinating peripheral neuropathy. Ann Neurol. 2001;49:214-221.
52 Oh SJ, Dropcho EJ, Claussen GC. Anti-Hu–associated paraneoplastic sensory neuropathy responding to early aggressive immunotherapy: report of two cases and review of literature. Muscle Nerve. 1997;20:1576-1582.
53 Antoine JC, Mosnier JF, Absi L, et al. Carcinoma associated paraneoplastic peripheral neuropathies in patients with and without anti-onconeural antibodies. J Neurol Neurosurg Psychiatry. 1999;67:7-14.
54 Sodhi N, Camilleri M, Camoriano JK, et al. Autonomic function and motility in intestinal pseudoobstruction caused by paraneoplastic syndrome. Dig Dis Sci. 1989;34:1937-1942.
55 Chinn JS, Schuffler MD. Paraneoplastic visceral neuropathy as a cause of severe gastrointestinal motor dysfunction. Gastroenterology. 1988;95:1279-1286.
56 Gosney MA, Gosney JR, Lye M. Orthostatic hypotension and vasodilatory peptides in bronchial carcinoma. J Clin Pathol. 1995;48:1102-1105.
57 Ashraf W, Farrow LJ. Severe orthostatic hypotension in carcinoma of the pancreas. Br J Clin Pract. 1992;46:278-279.
58 Zacharias AS, Brashear HR, Munoz EL, et al. Paraneoplastic encephalomyeloneuritis obscured by coma. Neurology. 1996;46:1177-1179.
59 Angelini P, Holoye PY. Neurocardiogenic syncope and Prinzmetal’s angina associated with bronchogenic carcinoma. Chest. 1997;111:819-822.
60 Veilleux M, Bernier JP, Lamarche JB. Paraneoplastic encephalomyelitis and subacute dysautonomia due to an occult atypical carcinoid tumour of the lung. Can J Neurol Sci. 1990;17:324-328.
61 Dropcho EJ. Neurotoxicity of cancer chemotherapy. Semin Neurol. 2004;24:419-426.
62 Ropper AH, Gorson KC. Neuropathies associated with paraproteinemia. N Engl J Med. 1998;338:1601-1607.
63 Antoine JC, Camdessanche JP, Ferraud K, et al. Antiganglioside antibodies in paraneoplastic peripheral neuropathies. J Neurol Neurosurg Psychiatry. 2004;75:1765-1767.
64 Kloos L, Sillevis SP, Ang CW, et al. Paraneoplastic ophthalmoplegia and subacute motor axonal neuropathy associated with anti-GQ1b antibodies in a patient with malignant melanoma. J Neurol Neurosurg Psychiatry. 2003;74:507-509.
65 Vernino S, Low PA, Fealey RD, et al. Autoantibodies to ganglionic acetylcholine receptors in autoimmune autonomic neuropathies. N Engl J Med. 2000;343:847-855.
66 Oh SJ. Paraneoplastic vasculitis of the peripheral nervous system. Neurol Clin. 1997;15:849-863.
67 Wirtz PW, Smallegange TM, Wintzen AR, et al. Differences in clinical features between the Lambert-Eaton myasthenic syndrome with and without cancer: an analysis of 227 published cases. Clin Neurol Neurosurg. 2002;104:359-363.
68 O’Neill JH, Murray NM, Newsom-Davis J. The Lambert-Eaton myasthenic syndrome. A review of 50 cases. Brain. 1988;111:577-596.
69 Wirtz PW, Sotodeh M, Nijnuis M, et al. Difference in distribution of muscle weakness between myasthenia gravis and the Lambert-Eaton myasthenic syndrome. J Neurol Neurosurg Psychiatry. 2002;73:766-768.
70 Sanders DB, Massey JM, Sanders LL, et al. A randomized trial of 3,4-diaminopyridine in Lambert-Eaton myasthenic syndrome. Neurology. 2000;54:603-607.
71 Newsom-Davis J. Therapy in myasthenia gravis and Lambert-Eaton myasthenic syndrome. Semin Neurol. 2003;23:191-198.
72 Dalakas MC. Polymyositis, dermatomyositis and inclusion-body myositis. N Engl J Med. 1991;325:1487-1498.
73 Kissel JT, Mendell JR, Rammohan KW. Microvascular deposition of complement membrane attack complex in dermatomyositis. N Engl J Med. 1986;314:329-334.
74 Engel AG, Hohlfeld R, Banker BQ. The polymyositis and dermatomyositis syndromes. In: Engel AG, Franzini-Armstrong C, editors. Myology. New York: McGraw-Hill; 1994:1335-1383.
75 Sigurgeirsson B, Lindelof B, Edhag O, et al. Risk of cancer in patients with dermatomyositis or polymyositis. A population-based study. N Engl J Med. 1992;326:363-367.
76 Amato AA, Barohn RJ. Idiopathic inflammatory myopathies. Neurol Clin. 1997;15:615-648.
77 Dalakas MC, Illa I, Dambrosia JM, et al. A controlled trial of high-dose intravenous immune globulin infusions as treatment for dermatomyositis. N Engl J Med. 1993;329:1993-2000.
78 Levin MI, Mozaffar T, Al-Lozi MT, et al. Paraneoplastic necrotizing myopathy: clinical and pathological features. Neurology. 1998;50:764-767.
79 Antoine JC, Camdessanche JP, Absi L, et al. Devic disease and thymoma with anti–central nervous system and antithymus antibodies. Neurology. 2004;62:978-980.
80 Petzold GC, Marcucci M, Butler MH, et al. Rhabdomyolysis and paraneoplastic stiff-man syndrome with amphiphysin autoimmunity. Ann Neurol. 2004;55:286-290.
81 Bataller L, Dalmau J. Neuro-ophthalmology and paraneoplastic syndromes. Curr Opin Neurol. 2004;17:3-8.
82 Keltner JL, Thirkill CE. Cancer-associated retinopathy vs recoverin-associated retinopathy. Am J Ophthalmol. 1998;126:296-302.
83 Shiraga S, Adamus G. Mechanism of CAR syndrome: antirecoverin antibodies are the inducers of retinal cell apoptotic death via the caspase 9– and caspase 3–dependent pathway. J Neuroimmunol. 2002;132:72-82.
84 Ohguro H, Maruyama I, Nakazawa M, et al. Antirecoverin antibody in the aqueous humor of a patient with cancer-associated retinopathy. Am J Ophthalmol. 2002;134:605-607.
85 Milam AH, Saari JC, Jacobson SG, et al. Autoantibodies against retinal bipolar cells in cutaneous melanoma–associated retinopathy. Invest Ophthalmol Vis Sci. 1993;34:91-100.
86 Keltner JL, Thirkill CE, Yip PT. Clinical and immunologic characteristics of melanoma-associated retinopathy syndrome: eleven new cases and a review of 51 previously published cases. J Neuroophthalmol. 2001;21:173-187.
87 Cross SA, Salomao DR, Parisi JE, et al. Paraneoplastic autoimmune optic neuritis with retinitis defined by CRMP-5-IgG. Ann Neurol. 2003;54:38-50.






