Chapter 13 Pancreatic Neuroendocrine Tumors
Introduction
A number of neuroendocrine tumors (NETs) exist. This chapter discusses pancreatic neuroendocrine tumors (PNETs) such as insulinomas, gastrinomas, VIPomas (vasoactive intestinal polypeptidomas), glucagonomas, and nonfunctional PNETs. PNETs are a subgroup of gastroenteropancreatic NETs.1 The term neuroendocrine is derived from the similarity to neural cells in the expression of proteins such as synaptophysin, neuron-specific enolase, and chromogranin A.2 Previously, PNETs were often referred to as “carcinoid tumor” or “islet cell tumor.” However, with the World Health Organization (WHO) classification of 2000,2 these have been replaced by the more generic term pancreatic neuroendocrine tumor (PNET). The WHO classification reserves the term carcinoid for well-differentiated NETs of purely gastroenteric origin.
PNETs are thought to arise from a common precursor neuroendocrine cell that shares features with similar such cells throughout the body constituting the “neuroendocrine system” of the body.3 The embryologic origin of this common precursor cell is controversial. Masson and coworkers4 suggested that the neuroendocrine cell was an endocrine cell derived from the gastrointestinal epithelium, suggesting an endodermal origin. However, in 1968, Pearse5 showed that all the neuroendocrine cells throughout the body shared various features including the amine precursor uptake and decarboxylation (APUD) capacity and postulated that the neural crest was the common origin for these cells called the APUD cells. These cells have pluripotential capabilities including differentiating into various types of NETs.6 More recently, an endodermal origin to the pluripotent cells has been favored.7
Epidemiology and Risk Factors
The data on the incidence of PNETs in autopsy series are limited and varied. PNETs have been reported to occur in between 2% of autopsies8 and 10% in another autopsy series.9 However, the annual incidence is reported to be only between 2.5 and 5 per 100,000 per year, suggesting that this apparent lower incidence may be related to the detection of predominantly functional tumors.10 The incidence of PNETs is increasing.11 These still represent a small percentage of pancreatic tumors, accounting for 1% to 2% of all primary pancreatic tumors.9 Recent literature indicates that the majority of PNETs are actually nonfunctional, ranging between 50% and 80% of all PNETs.12–14 Overall, PNETs have a better long-term survival and overall prognosis when compared with ductal adenocarcinoma of the pancreas. The 5-year survival for PNETs ranges from 30% in nonfunctional PNETs to 97% in a subgroup of functional PNETs, the insulinoma.15
Four syndromes have been associated with a higher incidence of PNETs. These are the multiple endocrine neoplasia type I (MEN I or Wermer’s syndrome); von Hippel–Lindau (vHL) syndrome; neurofibromatosis-1 (NF-1 or von Recklinghausen’s syndrome), and tuberous sclerosis (TS).16 The relative frequency with which patients with these disorders develop PNETs is MEN I > vHL > NF-1 > TS.
MEN I is a syndrome characterized by tumors/hyperplasia of endocrine glands. This is inherited as an autosomal dominant trait. MEN I gene mutations can be identified in 78% to 93% of MEN I patients.17 MEN I patients usually have a family history of MEN I. The syndrome is characterized primarily by tumors of the parathyroid glands, endocrine components of the gastrointestinal and pancreatic systems, and the pituitary gland (Figure 13-1). The different incidences of PNETs in patients with MEN I are nonfunctional tumors (80-100%), gastrinomas (20-61%), insulinomas (7-31%), and other functional PNETs (<5%). With increasing control of symptoms of excess hormone production, PNETs have become an important factor in determining survival in patients with MEN I. Patients with MEN I syndrome have a higher incidence of premature death and PNETs have been attributed to be one of the causes for this.18,19 Thus, identifying and treating PNETs in this subgroup of patients may have a significant impact on survival.
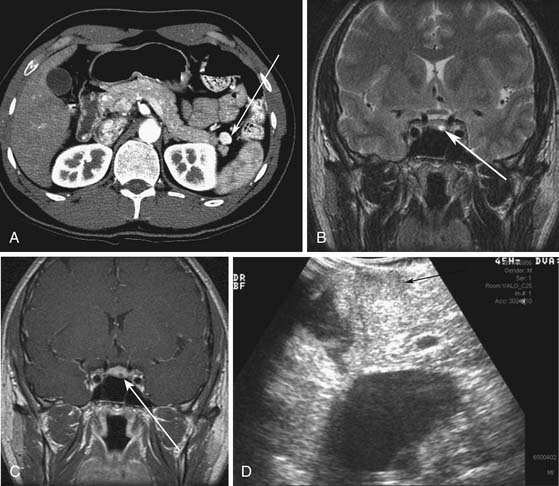
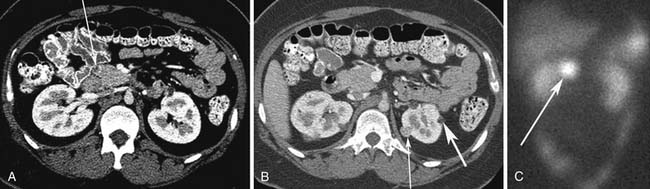
vHL is a syndrome characterized by autosomal dominant inheritance. vHL patients develop hemangioblastomas of the central nervous system (prevalence 44-72%) including retinal hemangioblastomas and cerebellar hemangioblastomas, renal cysts and renal cell carcinomas (25-60%), endolymphatic sac tumors (10%), and pheochromocytomas (10-20%).20 Pancreatic tumors or cysts develop in 35% to 70% of patients (Figure 13-2). Pancreatic cysts or serous cystadenomas develop in 17% to 56% of patients and PNETs occur in 8% to 17% of patients.21 The majority of the PNETs are nonfunctional.
NF-1 is more common than MEN I and vHL syndrome. Duodenal somatostatinomas (0-10%) and ampullary carcinoid tumors can occur in NF-1 patients. Pancreatic somatostatinomas (functional PNET) occur in patients with NF-1 at a much lower prevalence than the duodenal somatostatinoma.22 Insulinomas and nonfunctional PNETs are less common but can also be present in patients with NF-1.23
TS also has autosomal dominant inheritance. Patients with TS have been reported to have both functional and nonfunctional PNETs (<1%) especially patients with the TSC-2 gene mutation.16
Anatomy and Pathology
Anatomy
The pancreas is an elongated organ varying from 12.5 to 15 cm with a weight from 60 to 100 g. It is composed of both exocrine portions and endocrine portions. Multiple fat lobulations are seen on the surface due to invaginating adipose tissue.24 The portion of the pancreas located to the right side of the abdomen is recognized as the head and lies within the curve of the duodenum. A conical elongation of the pancreas extending posterior to the superior mesenteric vessels is the uncinate process. A narrow neck, anterior to the superior mesenteric vein (SMV), connects the head to an elongated body that tapers in the left upper quadrant to form the tail. The pancreas is located in the retroperitoneum except for a portion of the tail of the pancreas, which may be intraperitoneal along the gastrosplenic ligament.
The arterial supply for the pancreas is from the pancreaticoduodenal arteries and the splenic artery. The celiac trunk gives rise to the common hepatic artery, splenic artery, and the left gastric artery. The common hepatic artery gives off the gastroduodenal artery (GDA) and becomes the proper hepatic artery. The GDA divides into the superior pancreaticoduodenal arteries (anterior and posterior) and the right gastroepiploic artery. The anterior and posterior superior pancreaticoduodenal arteries supply the cranial aspect of the pancreas and descend between the duodenum and the pancreas, supplying both these organs. These arteries anastomose with the anterior and posterior inferior pancreaticoduodenal arteries and the pancreatic branches of the splenic artery. The anterior and posterior inferior pancreaticoduodenal arteries are branches of the inferior pancreaticoduodenal artery arising from the superior mesenteric artery (SMA). The splenic artery gives rise to numerous small branches that supply the body and tail of the pancreas. One branch, the pancreatica magna artery, runs along the body of the pancreas. The dorsal pancreatic artery is another branch usually of the splenic artery but can have a variable origin. It is usually located posterior to the portal venous confluence, passing behind the splenic vein.25 This artery divides into two terminal bifurcating branches.
The venous drainage of the pancreas parallels the arterial supply. Four major pancreaticoduodenal veins drain the pancreas in addition to multiple smaller pancreatic veins that drain the pancreas and enter the splenic vein directly.26 The inferior pancreaticoduodenal veins drain the inferior portion of the pancreas and enter the first jejunal vein, which drains into the SMV. The posterior superior pancreaticoduodenal vein drains directly into the caudal portion of the portal vein. The anterior superior pancreaticoduodenal vein runs horizontally and drains into either the gastrocolic branch of the SMV or the right gastroepiploic vein, which drains into the SMV. The SMV and the splenic veins join posterior to the neck of the pancreas to form the portal vein. The inferior mesenteric vein may enter at this confluence in one third of patients, enter the splenic vein close to this confluence in one third of patients, or enter the SMV in one third of patients.
Tumor Types
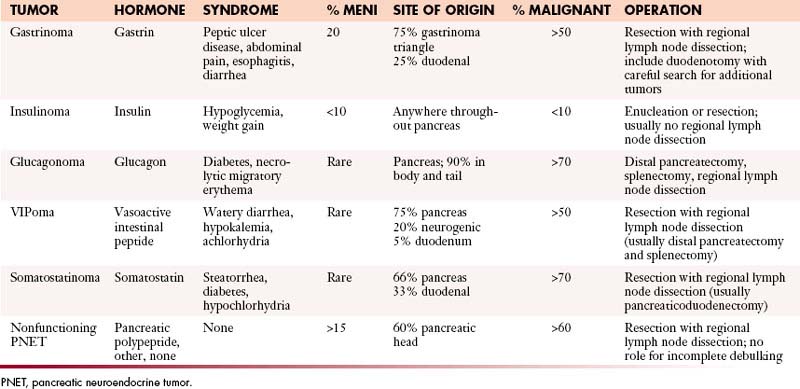
PNETs can be divided into the functional and nonfunctional. The main functional PNETs include insulinomas, gastrinomas, glucagonomas, somatostatinomas, VIPomas, growth hormone releasing factor (GRFomas). The nonfunctional PNETs are named as such owing to either lack of hormone production by the tumor or lack of clinically identifiable symptoms related to the tumor, as shown in Table 13-1.
| GRADE | MITOSES | KI-67 INDEX |
|---|---|---|
| 1 | <2 | ≤2% |
| 2 | 2-20 | 3-20% |
| 3 | >20 | >20% |
PNETs are typically well-circumscribed solitary masses within the pancreas.27 They can vary in size from less than 1 cm to large tumors greater than 15 cm. They can be white to yellow or pink-brown in color.
The WHO classification has differentiated PNETs into well-differentiated tumors with benign, benign or low malignant potential; well-differentiated carcinomas with low-grade malignant potential with local invasion or metastases; and poorly differentiated carcinomas with high-grade malignant potential (Table 13-2).28 Well-differentiated tumors or carcinomas are characterized by monomorphic tumor cells with abundant cytoplasm and low mitotic index. They tend to have a well-developed stroma with well-formed blood vessels and may exhibit a trabecular, glandular, or acinar pattern. Poorly differentiated carcinomas are characterized by solid appearance with large areas of necrosis and cells demonstrating a round small tumor cell pattern with high mitotic index and cellular atypia.29
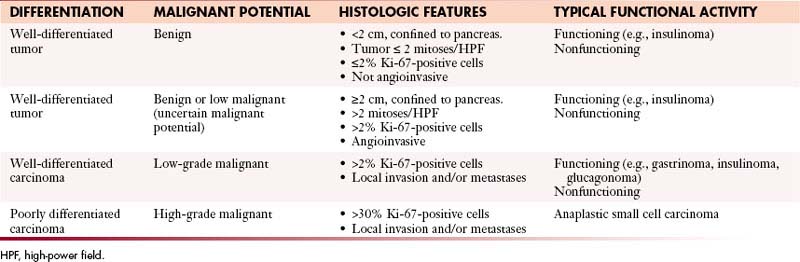
Rindi and coworkers30 and the ENETS (European Neuroendocrine Tumor Society) consensus statement address the issue of tumor grading. The proliferative activity of tumor cells can be assessed by counting mitoses or by immunostaining for Ki-67 (using the MIB1 antibody), which is a cell-cycle–dependent marker as shown in Table 13-1. In a spot where there is high mitotic density, mitoses can be counted on 10 high-power fields (2-mm2 area) at 40 times the magnification and the Ki-67 index can be determined by assessing 2000 tumor cells in an area of high nuclear labeling.
Clinical Presentation
The clinical presentation of PNETs is variable depending on whether these tumors are functional or nonfunctional as shown in Table 13-2. Functional tumors including insulinomas, gastrinomas, glucagonomas, and VIPomas are discussed here.
Patterns of Tumor Spread
The important factors to be defined in a patient with a new diagnosis of PNET are
1. Whether the tumor is functional or not.
2. If the tumor is functional, which subtype does it belong to?
These factors will help in predicting the pattern of spread.
Staging
The WHO classification has differentiated PNETs into well-differentiated tumors with benign, benign or low malignant potential that are confined to the pancreas28; well-differentiated carcinomas with low-grade malignant potential with local invasion or metastases; and poorly differentiated carcinomas with high-grade malignant potential with local invasion or metastases (see Table 13-2).
The ENETS met to define further standards in stratification of the gastroenteropancreatic NETs and proposed tumor-node-metastasis (TNM) staging for different NETs.30 These guidelines were evaluated for consensus among the 62 experts from different countries.
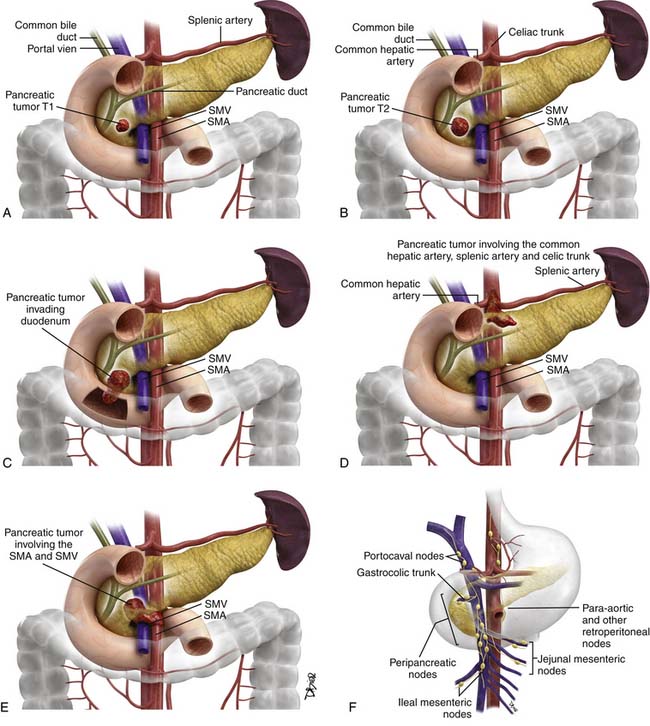
The proposed TNM staging for PNETs is shown in Table 13-3 and Figure 13-3.
| DESCRIPTION | |
|---|---|
| T Stage | |
| Tx | Primary tumor cannot be assessed |
| T0 | No evidence of primary tumor |
| T1 | Primary tumor limited to the pancreas and < 2 cm in size |
| T2 | Primary tumor limited to the pancreas and 2-4 cm in size |
| T3 | Primary tumor limited to the pancreas and > 4 cm in size OR primary tumor is invading the duodenum or bile duct |
| T4 | Primary tumor is invading adjacent organs or the walls of large vessels such as the celiac trunk or superior mesenteric artery |
| N Stage | |
| Nx | Regional lymph nodes cannot be assessed |
| N0 | Regional nodes not involved |
| N1 | Regional nodes involved |
| M Stage | |
| Mx | Distant metastases cannot be assessed |
| M0 | No distant metastases |
| M1 | Distant metastases |
| Proposed Group Stages | |
| STAGE | TNM |
| I | T1N0M0 |
| IIa | T2N0M0 |
| IIb | T3N0M0 |
| IIIa | T4N0M0 |
| IIIb | Any T N1M0 |
| IV | Any T Any N M1 |
Key Points Staging
Grouped Staging
| Stage I | T1N0M0 |
| Stage IIa | T2N0M0 |
| Stage IIb | T3N0M0 |
| Stage IIIa | T4N0M0 |
Imaging
Ultrasound
Ultrasound is a noninvasive and widely available modality that may be the first imaging tool used especially in evaluation of patients with nonfunctional PNETs who present with symptoms related to mass effect. Owing to operator variability and patient variability, a wide range of sensitivities and specificities have been reported. According to a recent consensus statement, the mean detection rate of the primary tumor by ultrasound is 39%, ranging from 17% to 79%.31
For optimal detection of PNETs, the patient should be asked to drink water before the ultrasound to provide a good window through the stomach for evaluation of the pancreas. The patient can be positioned in the supine, left lateral decubitus, or standing position. The PNET is typically a well-defined hypoechoic mass when compared with the rest of the pancreas. They tend to be vascular on Doppler imaging and may demonstrate a hyperechoic halo. Tumors at the edge of the pancreas or located close to the duodenal wall may not be well visualized.32 Intraoperative ultrasound has a higher detection rate with a mean detection rate of 92% (range 74-96%) according to the consensus statement.31
For the detection of lymph node metastases, ultrasound showed a detection rate of 18% according to one study.33 For the detection of liver metastases, one study showed that ultrasound had a sensitivity of 88% and a specificity of 95%.34 Liver metastases may be hypoechoic when small (<1 cm) and tend to be hyperechoic as they get larger. Involvement of the common bile duct, pancreatic duct, and vessel can be assessed well in patients by ultrasound. Patients with large body habitus are not good candidates for sonographic evaluation and should be referred to computed tomography (CT) or magnetic resonance imaging (MRI) owing to poor penetrability by ultrasound.
Endoscopic Ultrasound
A mean detection rate of 90% (range 77-100%) was shown in 10 studies according to the consensus statement.31 EUS also had a mean detection rate of 92% (range 88-94%) in the detection of insulinomas alone. The insulinomas can be difficult to diagnose on cross-sectional imaging owing to their size and may not be well seen on somatostatin (SST) receptor scanning because of small size or a lack of SST receptor subtypes that bind the radiolabeled octreotide with high affinity. EUS has been shown to have a role in patients with MEN I and also vHL in which the PNETs may be less than 1 cm and can be difficult to detect by other imaging modalities.20,35–37 EUS can be used along with fine-needle aspiration (FNA) to obtain tissue samples and confirm the diagnosis. However, EUS is not widely available and is operator-dependent. The entire liver and portions of the tail of the pancreas may not be well evaluated by EUS alone. The role of EUS is complementary to that of other cross-sectional imaging in evaluation of PNETs.
Computed Tomography
Multidetector row computed tomography (MDCT) is available in most radiology practices and has a key role in the evaluation of patients with PNETs.
Imaging
The consensus statement reports a mean sensitivity and detection rate of 73% (range for sensitivity 63-82%) with a higher specificity of 96% for PNETs based on CT.31 Liver metastases can be evaluated with a sensitivity of 82% and a mean specificity of 92%.
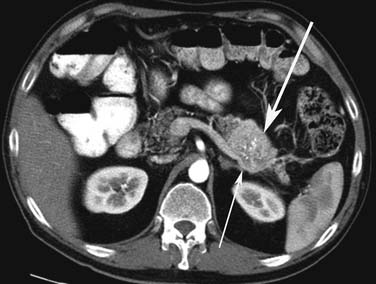
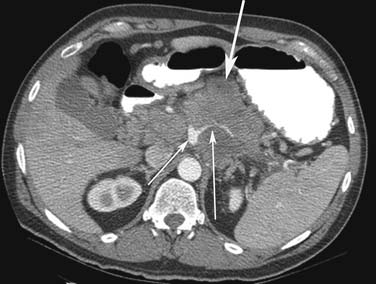
Small PNETs (usually functional tumors) tend to be uniformly hypervascular lesions enhancing in the early phase of enhancement.38 In patients with MEN I syndrome, they are multiple in number and the pancreas should be carefully assessed to localize all the tumors present. They usually tend to be hypervascular to the rest of the pancreas on the portal venous phase of enhancement. Larger PNETs tend to have heterogeneous enhancement. Some of the PNETs may show central areas of necrosis or calcifications (Figure 13-4). Tumor enhancement has been shown to be related to microvascular density according to one study.39 The relationship of these tumors to the surrounding vasculature should be carefully assessed to conform with the proposed TNM staging. Presence of celiac or SMA involvement can preclude surgery (Figure 13-5). The management of venous involvement with narrowing or with tumor thrombus also needs to be addressed preoperatively, especially as related to the technical aspect of venous resection and reconstruction (Figure 13-6). Primary tumors can be hypovascular in up to 40% of cases. Less commonly, primary tumors can be cystic (5-17%).
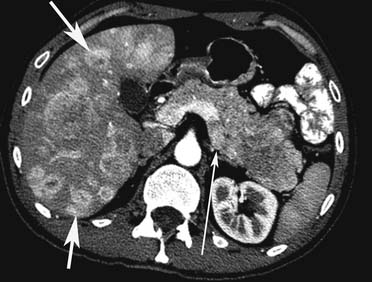
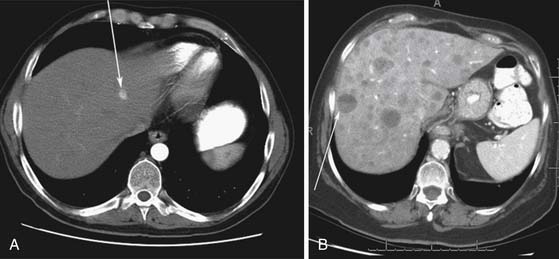
Liver metastases can have a variable appearance. In hypervascular PNETs, the liver metastases are typically hypervascular and are seen in the arterial phase of images well (Figure 13-7A). However, like the primary tumors that are hypovascular, the liver metastases are frequently hypovascular and are seen better in the portal venous phase of enhancement. These metastases may have areas of central necrosis. Some cystic primary tumors can have cystic liver metastases with a fluid-fluid level (see Figure 13-7B).
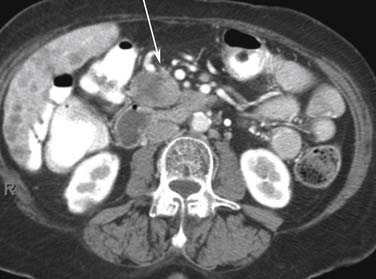
Nodal metastases may show heterogeneous enhancement (Figure 13-8). They are typically located in the peripancreatic, periportal, and retroperitoneal locations. Assessment of treatment response is by the RECIST criteria (Response Evaluation Criteria In Solid Tumors).
Magnetic Resonance Imaging
Imaging
A mean sensitivity of 93% and a specificity of 88%40 has been reported in two studies.41 However, other studies report a mean detection rate of 73% comparable with CT in the detection of the primary tumor in PNETs.42,43 MRI may also be more sensitive than CT for the detection of liver metastases.44,45
PNETs are of low T1 and high T2 signal intensity. Small PNETs may demonstrate uniform hypervascular enhancement. Larger PNETs can demonstrate heterogeneous enhancement similar to CT enhancement characteristics (Figure 13-9). Common bile duct and pancreatic ductal dilatation can be well evaluated by MRI on the T2-weighted sequences. Liver metastases are of low T1 signal intensity and can be of high T2 signal intensity similar to hemangiomas. However, the enhancement pattern of these metastases can be heterogeneous with areas of central necrosis different from the classic enhancement pattern of hemangiomas involving the peripheral puddling of contrast with gradual fill in of contrast. The enhancement of the nodes is similar to that on CT demonstrating significant enhancement.
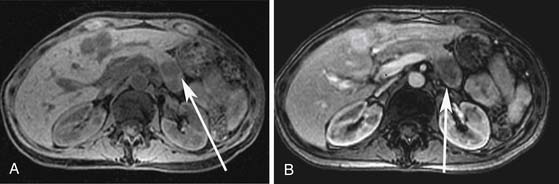
Vascular involvement should be carefully assessed as described in “Computed Tomography,” earlier.
Radionuclide Imaging
Technique
SST is a 14 amino acid peptide hormone present in many organs including the gastrointestinal tract and pancreas. There are five types of SST receptors, which are membrane proteins. These receptors are expressed in the thyroid, islet cells of the pancreas, cells of neuroendocrine origin, kidneys, and prostate. However, SST has a short biologic half-life of 2 to 4 minutes.46 A synthetic eight peptide analogue called octreotide is used to bind to tissues expressing SST receptors. Octreotide has a biologic half-life of 1.7 to 1.9 hours. Octreotide binds to tissues expressing SST receptors 2 and 5. 111In-labeled octreotide is administered at a typical dose of 5 to 6 mCi. The patient is then imaged with a large field of view gamma camera with single-photon emission computed tomography (SPECT) images. The patient is imaged at 4 and 24 hours. Physiologic uptake is seen in the pituitary gland, salivary glands, thyroid, liver, spleen, kidneys, and bladder.
Use of 123I-labeled metaiodobenzylguanidine (MIBG) is still used in the evaluation of NETs but predominantly in the evaluation of pheochromocytomas, neuroblastomas, and paragangliomas. The sensitivity of 123I-labeled MIBG for PNET detection has been reported as low as 10%.47
Imaging
The sensitivity of octreotide scans in the detection of PNETs has been reported to be between 70% and 90%.48,49 PNETs smaller than 1 cm will not be detected by this technique. The type of receptor expressed by the PNET may also affect detection. Insulinomas that do not express SST subtype 2 receptors in abundance may not be detected. The primary tumor and metastases appear as “hot spots” or areas of increased uptake of the octreotide. Liver metastases are also optimally evaluated on SPECT images (Figure 13-10).
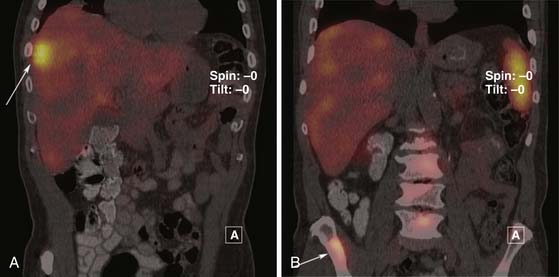
Positron-Emission Tomography
According to one study, results with 11C-labeled 5-hydroxy tryptophan indicate a greater than 90% detection of PNETs.50 Another study also demonstrated a higher detection rate of 84% with 11C-labeled 5-hydroxy tryptophan compared with a detection rate of 47% with SRS.51
Angiography
For arterial stimulation, calcium gluconate is administered via the catheter placed in either the splenicartery, the GDA, or the SMA.52 Venous sampling from the hepatic veins is performed to document a greater than twofold increase in the level of the hormone produced by the tumor. A sensitivity of greater than 90% in the detection of functional tumors such as insulinomas has been reported.53
Insulinoma
Clinical Presentation
Insulinomas are PNETs that release inappropriately high amounts of insulin, resulting in a hyperinsulinemic hypoglycemic state.37 Whipple’s triad includes the symptoms of fasting hypoglycemia, documented fasting hypoglycemia with a serum glucose level less than 45 mg/dL, and the reversal of hypoglycemic symptoms after glucose administration. Insulinomas are the most common functional PNETs and occur mainly in the pancreas. The patients typically present with fasting hypoglycemia and may present with neuroglycopenic symptoms such as blurred vision, diplopia, confusion, and abnormal behavior (Table 13-4). This can progress to loss of consciousness, coma, or even permanent brain damage. Symptoms related to release of catecholamines following the hypoglycemia such as weakness, sweating, hunger, tremors, and palpitations may also occur.
Insulinomas can be diagnosed during the supervised fasting of patients. A serum glucose level less than 45 mg/dL associated with a plasma insulin level of greater than or equal to 3 µU/mL is found.37,54 For symptomatic patients during supervised fasting, 1 mg of glucagon can be administered intravenously to reverse the hypoglycemia.
Primary Tumor and Patterns of Tumor Spread
They are typically hypervascular and solitary tumors. Ninety percent of the tumors are less than 2 cm and 30% of the tumors are less than 1 cm. They tend to be benign in a majority of cases with malignant presentation in about 10% of cases. They may be associated with MEN I in 7% to 31% of cases and can be malignant in 25% of these cases.55 The malignant insulinomas typically present with liver metastases by hematogenous spread.
Surgical Treatment
The management of insulinomas is surgical. Although nearly all sporadic insulinomas are solitary, careful exploration at the time of surgery is essential in order to identify the presence of multiple tumors; intraoperative ultrasound is often a valuable tool. Ideal management of insulinomas involves enucleation of the tumor. However, tumors that lie close to the main pancreatic duct may be more safely managed by formal pancreatic resection (pancreaticoduodenectomy for pancreatic head tumors or distal pancreatectomy for tumors in the pancreatic tail). If enucleation is performed and a pancreatic duct leak is identified intraoperatively, a formal resection can then be performed, or alternatively, a Roux limb of jejunum can be anastomosed to the pancreatic defect. Distal pancreatectomy can almost always be spleen-sparing because there is little need to remove lymph nodes in this disease that is almost always benign. Laparoscopic operations for insulinoma are often appropriate and are becoming routine.56
Gastrinoma
Clinical Presentation
Gastrinomas are functional PNETs characterized by high levels of gastrin production leading to Zollinger-Ellison syndrome (ZES). The classic triad of ZES consists of severe peptic ulcer disease, gastric acid hypersecretion, and non-beta islet cell tumors of the pancreas. Gastrinomas can occur in the duodenum (more common) or within the pancreas (especially in the head/uncinate process). The most common symptoms include abdominal pain, peptic ulcers, and are found in 90% to 95% of these patients (see Table 13-4).
The upper limit of serum gastrin level is 100 pg/mL. A serum gastrin level of 1000 pg/mL or greater and a gastric pH of 2 or less is diagnostic of a gastrinoma.57 In patients with serum gastrin levels between 100 and 1000 pg/mL and a gastric pH of 2 or less, a secretin stimulation test should be performed. A positive secretin test is performed by administering 0.4 µg/kg of secretin by a subcutaneous injection. A postinjection serum gastrin level increase of greater than 120 pg/mL has been shown to have a sensitivity of 94% and a specificity of 100%.58
Primary Tumor and Patterns of Tumor Spread
These tumors often occur in the gastrinoma triangle, the area between the confluence of the cystic and common bile duct, the junction of the second and third portions of the duodenum, and the junction of the neck and body of the pancreas. The remainder of these tumors are located in the body of the pancreas and the distal duodenum. Overall, duodenal tumors are 3 to 10 times more common than pancreatic tumors. In contrast to insulinomas, the majority of gastrinomas tend to be malignant. They can present with lymph node (nodal spread) and liver metastases (hematogenous spread) at the time of diagnosis in 75% to 80% of the cases and with bone metastases in 12% of all of the cases.59 The presence of liver metastases at the time of diagnosis is the most important determinant of survival and is found more often in patients with pancreatic than with duodenal gastrinomas. The presence of lymph node metastases does not appear to have a significant impact on survival.60
They are associated with MEN I syndrome in 20% to 60% of cases. These tumors tend to be predominantly small and difficult to visualize by imaging. They also tend to present earlier in life and can be multiple. The overall survival (OS) of MEN I–associated gastrinomas is similar to that of the sporadic form and is determined by the presence of liver metastases.61
Surgical Treatment
There is evidence to support routine surgical management of gastrinoma in sporadic cases; surgery is associated with little morbidity and can provide a postoperative cure rate of 60%, with a third of patients achieving long-term cure (10 yr).62,63 The goal of surgery is to perform a complete resection of disease and preserve the maximal amount of pancreas. Tumors in the tail of the pancreas can be managed by distal pancreatectomy. Tumors in the head of the pancreas can often be managed by enucleation. Duodenal tumors can be managed by full-thickness excision. Duodenotomy with careful palpation of the duodenum or endoscopic transillumination should be performed routinely, even for patients who do not have a diagnosed duodenal primary, because duodenal tumors in patients with gastrinoma are extremely common and can be multiple and very small. Routine peripancreatic lymph node dissection should also be performed because of the high incidence of lymph node metastasis at the time of diagnosis. There is growing evidence to support more extensive surgery (pancreaticoduodenectomy) in certain cases: the presence of a large duodenal or pancreatic head tumor that is not amenable to enucleation; the presence of multiple duodenal tumors or multiple enlarged lymph nodes; and failure of cure after routine surgical management.64 Minimally invasive options, such as endoscopic resection of duodenal tumors, or laparoscopic pancreatic procedures can also play a role in selected cases.
VIPoma
Clinical Presentation
VIPomas are due to VIP overproduction by PNETs. The classic triad in Verner-Morrison syndrome is watery diarrhea, hypokalemia, and achlorhydria. These patients experience dehydration, hyperglycemia, and flushing.65 The large volume diarrhea seen is at least 700 mL/day with 70% to 80% of patients having greater than 3 L/day. Most VIPomas arise from the pancreas in adults but can be extrapancreatic in children, arising in ganglioneuromas, ganglioneuroblastomas, and neurofibromas involving the retroperitoneum and mediastinum. Fluid and electrolyte replacement is often needed at the time of diagnosis.
An elevated VIP level (>500 pg/mL) in the presence of a secretory diarrhea is highly suggestive of a VIPoma.66
Primary Tumor and Patterns of Tumor Spread
They are of pancreatic origin in over 80% of all cases. The rest of the cases are of neural origin. The pancreatic tumors present as larger masses and tend to be malignant. They metastasize to the liver (hematogenous spread) and lymph nodes (nodal spread) in the majority of cases.67
Glucagonoma
Clinical Presentation
Glucagonomas cause hypersecretion of glucagon. Glucagonomas can cause glucose intolerance, weight loss, diarrhea, migratory necrolytic erythema, glossitis, deep vein thrombi, and stomatitis (see Table 13-4). Migratory necrolytic erythema is a skin condition characterized by erythematous macules, which become papules and heal with necrosis and pigmented scarring.68 Serum glucagon levels of 500 to 1000 pg/mL are diagnostic of glucagonomas.
Primary Tumor and Patterns of Tumor Spread
Glucagonomas are typically sporadic and are pancreatic in origin. Glucagonomas can be large and are typically malignant. The majority of cases tend to have liver metastases (hematogenous spread) at the time of diagnosis.69 They may be associated with MEN I syndrome in about 5% of cases. The patients with MEN I syndrome and glucagonomas tend to present earlier than the patients with sporadic cases.
Surgical Treatment
More than 80% of glucagonomas ultimately display malignant behavior; over 50% of patients have regional or distant metastatic disease at the time of diagnosis.70 Glucagonomas are always located in the pancreas, and 90% are located in the body and tail. They are often very large tumors that are best managed by distal pancreatectomy and splenectomy, with regional lymph node dissection.
Nonfunctional Pancreatic Neuroendocrine Tumors
Clinical Presentation
These are PNETs not associated with any syndrome. They are usually found related to mass effect caused by the pancreatic tumor on adjacent organs or, less often, are found incidentally. They typically present with larger primary tumors and advanced disease.14 The most common symptoms include nonspecific abdominal pain (40-60%), weight loss (25-50%), or jaundice (30-40%). The diagnosis for a nonfunctional PNET is based on biopsy of the tumor.
Primary Tumor and Patterns of Tumor Spread
These tend to be large and solitary pancreatic masses and present from symptoms of local mass effect. The majority of tumors are malignant.71 They can present with lymph node (nodal spread) and liver metastases (hematogenous spread) at the time of diagnosis.
Surgical Treatment
Nonfunctional PNETs are usually diagnosed because of the mechanical effects from local growth or metastatic disease (e.g., pain, jaundice). Thus, most nonfunctioning PNETs are diagnosed when the primary tumor is large or has already metastasized; only 25% of patients are candidates for a potentially curative resection at the time of diagnosis.72 These tumors are malignant in more than 60% of cases, with metastasis most commonly to lymph nodes and liver.
Approximately 60% of nonfunctioning PNETs are located in the pancreatic head.73 Surgical resection (pancreaticoduodenectomy or distal pancreatectomy) with lymph node dissection is indicated for localized resectable tumors. However, only approximately 50% of those patients experience long-term cure.72 There is no survival benefit to incomplete resection of a primary tumor (cytoreduction or debulking), and patients experience considerable morbidity and mortality.74
The surgical management of patients who present with a localized but unresectable tumor is controversial. Because the majority of nonfunctioning PNETs are located in the pancreatic head, local growth will eventually result in bile duct and duodenal obstruction. Although endoscopic stents can be used to manage these situations in patients with localized but unresectable tumors, we prefer surgical biliary and gastric bypass. Such patients typically have a median survival of 5 years and surgical bypass provides a much more durable response than endoscopic stenting.72
The management of an intact primary tumor in the setting of metastatic disease is also controversial. A small number of patients (<5%) will be candidates for complete resection of the primary tumor and all metastatic disease and may experience a survival benefit.72 There is no evidence that surgical resection of the primary tumor without complete resection of all metastatic disease results in increased survival.73 The only indication for surgery in this subpopulation of patients is symptom palliation. In the case of tumors in the body and tail of the pancreas, symptoms are unusual, even with tumors that become very large. However, surgical resection may be considered for symptom palliation of tumors in the pancreatic head, especially in the setting of low-volume metastatic disease and a low-risk patient. In such a situation, biliary and duodenal obstruction or gastrointestinal hemorrhage from erosion into the duodenum may be managed better by pancreaticoduodenectomy than by palliative bypass alone.
Chemotherapy
Cytotoxic Chemotherapy
Advanced PNETs are generally not curable. Median survival among those with distant metastatic low- to intermediate-grade PNETs in the Surveillance, Epidemiology, and End Results (SEER) is 27 months.75 Although some patients with limited liver disease may benefit from surgical intervention, most patients will have more extensive disease needing systemic therapy.
Systemic chemotherapy for advanced PNETs has been studied in many clinical trials since the early 1970s. Despite this and approval of streptozocin by the U.S. Food and Drug Administration (FDA) for this indication, the role of chemotherapy continues to be debated. A number of problems make the interpretation of results in this area difficult. Many (older and current) studies combine PNETs with NETs of other primary sites in the same study. This tended to lower the response rates because more common small bowel NETs tend to be more resistant. Another issue is the lack of standard response criteria in older studies. Many such trials counted either a response by cross-sectional imaging, a decrease in biochemical markers, or even shrinkage by physical examination as evidence of objective response.76,77 Finally, no study has attempted to document improvements in progression-free survival (PFS) or OS against best supportive care.76,77
Single-agent Chemotherapy
The antitumor activity of streptozocin in PNETs was first reported in 1973.78 A response rate of 50% was observed among 52 patients, leading to the only FDA approval in 1976. Streptozocin’s single-agent activity was subsequently confirmed in a study comparing that agent alone with streptozocin plus fluorouracil for PET.76 In that study, a response rate of 36% was reported for streptozocin alone. Doxorubicin was studied in a small phase II trial that reported responses in 20% of patients.79 Dacarbazine has also been studied in PNETs. A small initial study in 11 patients with PNETs reported a response rate of 9%.80 However, in a subsequent phase II trial that treated 42 patients with measurable disease, a higher response rate of 33% was observed.81
Whereas fluorouracil (5-FU) has often been included in combination chemotherapy regimens for PNETs, its activity as a single agent has not been documented in prospective studies. Etoposide, carboplatin, and docetaxel have also been studied in PNETs without evidence of single-agent clinical activity.82,83
Combination Chemotherapy
With several agents showing modest clinical activity, it is only natural that combinations be explored. The most extensively studied of these have been based on streptozocin. The combinations of 5-FU plus streptozocin and 5-FU plus doxorubicin achieve response rates ranging from 27% to 69%.76,77,84–86 However, in a retrospective study of 16 patients who received streptozocin and doxorubicin at Memorial Sloan-Kettering Cancer Center, only 1 response (6%) was observed.87 Similarly, a retrospective review of 16 patients treated with streptozocin and doxorubicin at Dana Farber Cancer Center found only 1 objective response (6%). Triplet chemotherapy consisting of all three drugs has reported response rates of 40% and 55% in two small series.88 We reviewed the records of 84 consecutive patients with islet cell carcinoma treated with fluorouracil, doxorubicin, and streptozocin (FAS).89 Using the criteria proposed by the RECIST committee, a response rate of 39% was observed. For this regimen, the 2-year PFS rate was 41% and the 2-year OS rate was 74%.
More recently, temozolomide, an oral analogue of dacarbazine, has also shown promising results in PNETs. In two phase II clinical studies, response rates of 45% and 24% were observed among patients with PNET treated with temozolomide and thalidomide or with temozolomide and bevacuzimab, respectively.90 However, the numbers of NPET patients included were small. Temozolomide has not been studied as a single agent.
Biologics
Somatostatin Analogues
SST analogues bind to specific receptors on neuroendocrine cells to block the release of bioactive peptides and amines. In the United States, octreotide is the only SST analogue currently approved for the treatment of hormone-related symptoms from NETs. In Europe and other parts of the world, lanreotide is also approved. The role of a SST analogue as an antitumor agent is less well defined. Although there have been anecdotal reports of tumor shrinkage among patients with PNETs, in large series, tumor shrinkage occurs in only 2% to 3% of patients.91 In 2009, a multi-institutional German research group reported the result of the PROMID (Placebo controlled double blind prospective Randomized study on the effect of Octreotide LAR [long acting release] in the control of tumor growth in patients with metastatic neuroendocrine MID gut tumors) study, which prospectively compared octreotide LAR versus placebo among treatment-naïve patients with midgut NETs. The study showed a significant improvement in time to progression (TTP) among patients treated with octreotide LAR.92 Similar data for PNETs are not available.
Interferon
Interferon also has been extensively studied in NETs. However, most of the studies have included a variety of NETs and only a small number of PNETs. In a larger series using interferon-alpha at doses of 5 to 6 million units given three to five times per week, radiologic responses have been reported in 12% of patients with PNETs.93
Radiation Therapy
Wherever possible, surgical resection is the preferred and only potentially curative therapy for PNETs. Given their relatively indolent natural history and better prognosis than exocrine carcinomas of the pancreas, surgical resection is advocated as a reasonable strategy even for patients with metastatic disease. Nevertheless, as a matter of routine clinical practice, a majority of patients do not undergo surgical resection as documented by an analysis of the National Cancer Data Base, in which only 61% of patients with newly diagnosed PNETs underwent resections.94 The role of external beam radiation therapy in the management of PNETs is inadequately defined. These tumors are relatively radiosensitive, as demonstrated by case reports describing response to external beam radiation therapy and small clinical trials describing response to radionuclide therapy.95–98
Potential scenarios in which external beam radiation is an attractive therapeutic modality are palliative therapy of symptomatic primary or metastatic disease, primary tumor-directed therapy of unresectable disease (with or without metastatic disease), and consolidation therapy after systemic chemotherapy. Palliation of pain, gastric outlet/duodenal obstruction, and bleeding at the primary site or similar pain, compressive symptoms, and bleeding at a site of metastasis are achieved using fewer fractions of large doses administered with or without concurrent chemotherapy. In a recent report, among patients with progressive disease, radiation therapy produced high rates of symptomatic palliation for both intra-abdominal and distant sites of disease, yielding an approximate 90% clinical response rate.99 In this study, the actuarial 3-year local freedom from progression rate was 49% for unresectable PNETs; higher doses (biologically equivalent dose > 32 Gy) resulted in higher rates of freedom from local progression. Despite this being a heavily pretreated population of patients, the radiographic response rate of 39% (13% complete response, 26% partial response, 56% stable disease, and 4% progressive disease) is comparable with responses produced by traditional chemotherapy regimens and argues for consideration of radiation therapy in these patients.77,81 This is particularly true in an era in which promising newer biologic agents and SST-tagged radionuclides are likely to result in better control of oligometastatic disease and primary tumor control becomes more of a clinical priority.100 Lastly, because 50% of patients who undergo curative intent surgical resections develop a recurrence within 5 years, adjuvant radiation therapy is worth exploring prospectively, at least in the subset of patients with a positive surgical resection margin that portends a poor prognosis.72,101 In summary, radiation therapy is an appealing treatment option in the multidisciplinary management of PNETs.102
Surveillance
Owing to the indolent nature of the disease, patients need not be followed at short intervals but need to be followed for a longer period of time. We recommend that patients be reassessed once between 3 to 6 months after complete resection. Subsequently, patients should be assessed every 6 to 12 months for at least 7 years. Follow-up should consist of interval history, physical, chromogranin A, and 5-HIAA at minimum. Multiphasic CT or MRI is recommended by many experts. 111In-diethylenetriamine-penta-acetic acid (DTPA) octreotide scintigraphy can be performed as clinically indicated.
Key Points Detection of recurrence
New Therapies
A number of novel therapeutic strategies are under development. Peptide receptor radiotherapy takes advantage of the presence of SST receptors on low-grade neuroendocrine carcinoma. Compounds using indium-111, yttrium-90, or luticium-177 attached to SST analogues have been designed and studied in clinical trials. Only limited activity with the indium- and yttrium-based compounds have been observed in earlier studies. Luticium-177 octreotate has been more extensively studied. Promising activity has been reported in a large recent phase II study.103 Among patients with PNETs, the reported response rate was 42%. However, the study failed to follow intent-to-treat principles, 39% of treated patients were not included in the efficacy analyses. Had these been included, the response rate would likely be significantly lower. The reported median TTP was, unfortunately, not informative because only patients “who did not have progressive disease” as best response were included.
The clinical introduction of molecularly targeted therapeutics has brought renewed clinical research activity in this area. Low-grade neuroendocrine carcinomas are vascular tumors; therapeutic strategies targeting vascular endothelial growth factor (VEGF) signaling have been under development. Phase II studies of VEGF receptor tyrosine kinase inhibitors that have stratified patients by pancreatic and nonpancreatic primary site have generally reported higher objective response rates (ORRs) among patients with pancreatic primary sites. A recent phase III study compared sunitinib with placebo in PNETs.104 Results of an unplanned analysis after 81 PFS events showed significantly improved median PFS duration by investigator review.105
Mammalian target of rapamycin (mTOR) is an intracellular protein that has a central role in cellular function. It acts as a nutrient sensor and mediates signaling downstream of receptor tyrosine kinases. mTOR controls cell growth, protein synthesis, autophagy, and angiogenesis. The association between aberrant mTOR pathway signaling and the pathogenesis of PNETs is suggested by the development of PNETs in patients with inherited genetic mutations in TSC2, NF-1, and vHL genes. The combination of octreotide LAR and everolimus was studied in 60 patients with NETs. The intent-to-treat response rate was 20%.106 Median PFS duration was 60 weeks. In a follow-up, multinational phase II study of everolimus in advanced PNETs with progression after chemotherapy, 160 patients were treated in two strata, with everolimus (N = 115) or everolimus plus octreotide (N = 45) based on whether patients were on octreotide at study entry.107 Durable disease stabilizations were observed among patients with progression at study entry. The median PFS for patients receiving everolimus or everolimus plus octreotide were 9.7 and 16.7 months, respectively. A large pivotal phase III study (RADIANT-3) has now completed enrollment of over 400 patients.108
1. Ehehalt F., Saeger H.D., Schmidt C.M., Grutzmann R. Neuroendocrine tumors of the pancreas. Oncologist. 2009;14:456-467.
2. Kloppel G., Perren A., Heitz P.U. The gastroenteropancreatic neuroendocrine cell system and its tumors: the WHO classification. Ann N Y Acad Sci. 2004;1014:13-27.
3. Regitnig P., Spuller E., Denk H. Insulinoma of the pancreas with insular-ductular differentiation in its liver metastasis—indication of a common stem-cell origin of the exocrine and endocrine components. Virchows Arch. 2001;438:624-628.
4. Thompson M., Fleming K.A., Evans D.J., et al. Gastric endocrine cells share a clonal origin with other gut cell lineages. Development. 1990;110:477-481.
5. Pearse A.G. The cytochemistry and ultrastructure of polypeptide hormone-producing cells of the APUD series and the embryologic, physiologic and pathologic implications of the concept. J Histochem Cytochem. 1969;17:303-313.
6. Heitz P.U., Kasper M., Polak J.M., Kloppel G. Pancreatic endocrine tumors. Hum Pathol. 1982;13:263-271.
7. Andrew A., Kramer B., Rawdon B.B. The origin of gut and pancreatic neuroendocrine (APUD) cells—the last word? J Pathol. 1998;186:117-118.
8. Kimura W., Kuroda A., Morioka Y. Clinical pathology of endocrine tumors of the pancreas. Analysis of autopsy cases. Dig Dis Sci. 1991;36:933-942.
9. Oberg K., Eriksson B. Endocrine tumours of the pancreas. Best Pract Res Clin Gastroenterol. 2005;19:753-781.
10. Modlin I.M., Lye K.D., Kidd M.A. 5-decade analysis of 13,715 carcinoid tumors. Cancer. 2003;97:934-959.
11. Halfdanarson T.R., Rabe K.G., Rubin J., Petersen G.M. Pancreatic neuroendocrine tumors (PNETs): incidence, prognosis and recent trend toward improved survival. Ann Oncol. 2008;19:1727-1733.
12. Zerbi A., Falconi M., Rindi G., et al. Clinicopathological features of pancreatic endocrine tumors: a prospective multicenter study in Italy of 297 sporadic cases. Am J Gastroenterol. 2010;105:1421-1429.
13. Panzuto F., Nasoni S., Falconi M., et al. Prognostic factors and survival in endocrine tumor patients: comparison between gastrointestinal and pancreatic localization. Endocr Relat Cancer. 2005;12:1083-1092.
14. Falconi M., Plockinger U., Kwekkeboom D.J., et al. Well-differentiated pancreatic nonfunctioning tumors/carcinoma. Neuroendocrinology. 2006;84:196-211.
15. Bilimoria K.Y., Bentrem D.J., Merkow R.P., et al. Application of the pancreatic adenocarcinoma staging system to pancreatic neuroendocrine tumors. J Am Coll Surg. 2007;205:558-563.
16. Jensen R.T., Berna M.J., Bingham D.B., Norton J.A. Inherited pancreatic endocrine tumor syndromes: advances in molecular pathogenesis, diagnosis, management, and controversies. Cancer. 2008;113:1807-1843.
17. Lemos M.C., Thakker R.V. Multiple endocrine neoplasia type 1 (MEN1): analysis of 1336 mutations reported in the first decade following identification of the gene. Hum Mutat. 2008;29:22-32.
18. Wilkinson S., Teh B.T., Davey K.R., et al. Cause of death in multiple endocrine neoplasia type 1. Arch Surg. 1993;128:683-690.
19. Dean P.G., van Heerden J.A., Farley D.R., et al. Are patients with multiple endocrine neoplasia type I prone to premature death? World J Surg. 2000;24:1437-1441.
20. Lonser R.R., Glenn G.M., Walther M., et al. von Hippel–Lindau disease. Lancet. 2003;361:2059-2067.
21. Libutti S.K., Choyke P.L., Bartlett D.L., et al. Pancreatic neuroendocrine tumors associated with von Hippel–Lindau disease: diagnostic and management recommendations. Surgery. 1998;124:1153-1159.
22. Mao C., Shah A., Hanson D.J., Howard J.M. Von Recklinghausen’s disease associated with duodenal somatostatinoma: contrast of duodenal versus pancreatic somatostatinomas. J Surg Oncol. 1995;59:67-73.
23. Fujisawa T., Osuga T., Maeda M., et al. Malignant endocrine tumor of the pancreas associated with von Recklinghausen’s disease. J Gastroenterol. 2002;37:59-67.
24. Gray H. Anatomy of the Human Body. Churchill-Livingstone, Elsevier; 2008. 1576
25. Witte B., Frober R., Linss W. Unusual blood supply to the pancreas by a dorsal pancreatic artery. Surg Radiol Anat. 2001;23:197-200.
26. Crabo L.G., Conley D.M., Graney D.O., Freeny P.C. Venous anatomy of the pancreatic head: normal CT appearance in cadavers and patients. AJR Am J Roentgenol. 1993;160:1039-1045.
27. Frankel W.L. Update on pancreatic endocrine tumors. Arch Pathol Lab Med. 2006;130:963-966.
28. Ong S.L., Garcea G., Pollard C.A., et al. A fuller understanding of pancreatic neuroendocrine tumours combined with aggressive management improves outcome. Pancreatology. 2009;9:583-600.
29. Kloppel G., Couvelard A., Perren A., et al. ENETS Consensus Guidelines for the Standards of Care in Neuroendocrine Tumors: towards a standardized approach to the diagnosis of gastroenteropancreatic neuroendocrine tumors and their prognostic stratification. Neuroendocrinology. 2009;90:162-166.
30. Rindi G., Kloppel G., Alhman H., et al. TNM staging of foregut (neuro)endocrine tumors: a consensus proposal including a grading system. Virchows Arch. 2006;449:395-401.
31. Sundin A., Vullierme M.P., Kaltsas G., Plockinger U. ENETS Consensus Guidelines for the Standards of Care in Neuroendocrine Tumors: radiological examinations. Neuroendocrinology. 2009;90:167-183.
32. Rockall A.G., Reznek R.H. Imaging of neuroendocrine tumours (CT/MR/US). Best Pract Res Clin Endocrinol Metab. 2007;21:43-68.
33. De Angelis C., Carucci P., Repici A., Rizzetto M. Endosonography in decision making and management of gastrointestinal endocrine tumors. Eur J Ultrasound. 1999;10:139-150.
34. Chiti A., Fanti S., Savelli G., et al. Comparison of somatostatin receptor imaging, computed tomography and ultrasound in the clinical management of neuroendocrine gastro-entero-pancreatic tumours. Eur J Nucl Med. 1998;25:1396-1403.
35. Langer P., Kann P.H., Fendrich V., et al. Prospective evaluation of imaging procedures for the detection of pancreaticoduodenal endocrine tumors in patients with multiple endocrine neoplasia type 1. World J Surg. 2004;28:1317-1322.
36. Wamsteker E.J., Gauger P.G., Thompson N.W., Scheiman J.M. EUS detection of pancreatic endocrine tumors in asymptomatic patients with type 1 multiple endocrine neoplasia. Gastrointest Endosc. 2003;58:531-535.
37. Metz D.C., Jensen R.T. Gastrointestinal neuroendocrine tumors: pancreatic endocrine tumors. Gastroenterology. 2008;135:1469-1492.
38. Buetow P.C., Parrino T.V., Buck J.L., et al. Islet cell tumors of the pancreas: pathologic-imaging correlation among size, necrosis and cysts, calcification, malignant behavior, and functional status. AJR Am J Roentgenol. 1995;165:1175-1179.
39. Rodallec M., Vilgrain V., Couvelard A., et al. Endocrine pancreatic tumours and helical CT: contrast enhancement is correlated with microvascular density, histoprognostic factors and survival. Pancreatology. 2006;6:77-85.
40. Thoeni R.F., Mueller-Lisse U.G., Chan R., et al. Detection of small, functional islet cell tumors in the pancreas: selection of MR imaging sequences for optimal sensitivity. Radiology. 2000;214:483-490.
41. Semelka R.C., Custodio C.M., Cem Balci N., Woosley J.T. Neuroendocrine tumors of the pancreas: spectrum of appearances on MRI. J Magn Reson Imaging. 2000;11:141-148.
42. Owen N.J., Sohaib S.A., Peppercorn P.D., et al. MRI of pancreatic neuroendocrine tumours. Br J Radiol. 2001;74:968-973.
43. Ichikawa T., Peterson M.S., Federle M.P., et al. Islet cell tumor of the pancreas: biphasic CT versus MR imaging in tumor detection. Radiology. 2000;216:163-171.
44. Dromain C., de Baere T., Baudin E., et al. MR imaging of hepatic metastases caused by neuroendocrine tumors: comparing four techniques. AJR Am J Roentgenol. 2003;180:121-128.
45. Debray M.P., Geoffroy O., Laissy J.P., et al. Imaging appearances of metastases from neuroendocrine tumours of the pancreas. Br J Radiol. 2001;74:1065-1070.
46. Tamm E.P., Kim E.E., Ng C.S. Imaging of neuroendocrine tumors. Hematol Oncol Clin North Am. 2007;21:409-432. vii
47. Kaltsas G., Korbonits M., Heintz E., et al. Comparison of somatostatin analog and meta-iodobenzylguanidine radionuclides in the diagnosis and localization of advanced neuroendocrine tumors. J Clin Endocrinol Metab. 2001;86:895-902.
48. Kubo H., Chijiiwa Y., Akahoshi K., et al. Pre-operative staging of ampullary tumours by endoscopic ultrasound. Br J Radiol. 1999;72:443-447.
49. Kwekkeboom D.J., Krenning E.P. Somatostatin receptor imaging. Semin Nucl Med. 2002;32:84-91.
50. Eriksson B., Bergstrom M., Sundin A., et al. The role of PET in localization of neuroendocrine and adrenocortical tumors. Ann N Y Acad Sci. 2002;970:159-169.
51. Orlefors H., Sundin A., Garske U., et al. Whole-body (11)C-5-hydroxytryptophan positron emission tomography as a universal imaging technique for neuroendocrine tumors: comparison with somatostatin receptor scintigraphy and computed tomography. J Clin Endocrinol Metab. 2005;90:3392-3400.
52. Aoki T., Sakon M., Ohzato H., et al. Evaluation of preoperative and intraoperative arterial stimulation and venous sampling for diagnosis and surgical resection of insulinoma. Surgery. 1999;126:968-973.
53. Brandle M., Pfammatter T., Spinas G.A., et al. Assessment of selective arterial calcium stimulation and hepatic venous sampling to localize insulin-secreting tumours. Clin Endocrinol (Oxf). 2001;55:357-362.
54. Grant C.S. Insulinoma. Best Pract Res Clin Gastroenterol. 2005;19:783-798.
55. Service F.J., McMahon M.M., O’Brien P.C., Ballard D.J. Functioning insulinoma—incidence, recurrence, and long-term survival of patients: a 60-year study. Mayo Clin Proc. 1991;66:711-719.
56. Fernandez-Cruz L., Saenz A., Astudillo E., et al. Outcome of laparoscopic pancreatic surgery: endocrine and nonendocrine tumors. World J Surg. 2002;26:1057-1065.
57. Berna M.J., Hoffmann K.M., Serrano J., et al. Serum gastrin in Zollinger-Ellison syndrome: I. Prospective study of fasting serum gastrin in 309 patients from the National Institutes of Health and comparison with 2229 cases from the literature. Medicine (Baltimore). 2006;85:295-330.
58. Frucht H., Howard J.M., Slaff J.I., et al. Secretin and calcium provocative tests in the Zollinger-Ellison syndrome. A prospective study. Ann Intern Med. 1989;111:713-722.
59. Tomassetti P., Migliori M., Lalli S., et al. Epidemiology, clinical features and diagnosis of gastroenteropancreatic endocrine tumours. Ann Oncol. 2001;12(Suppl 2):S95-S99.
60. Weber H.C., Venzon D.J., Lin J.T., et al. Determinants of metastatic rate and survival in patients with Zollinger-Ellison syndrome: a prospective long-term study. Gastroenterology. 1995;108:1637-1649.
61. Skogseid B., Eriksson B., Lundqvist G., et al. Multiple endocrine neoplasia type 1: a 10-year prospective screening study in four kindreds. J Clin Endocrinol Metab. 1991;73:281-287.
62. Fraker D.L., Norton J.A., Alexander H.R., et al. Surgery in Zollinger-Ellison syndrome alters the natural history of gastrinoma. Ann Surg. 1994;220:320-328. discussion 328–330
63. Norton J.A., Fraker D.L., Alexander H.R., et al. Surgery to cure the Zollinger-Ellison syndrome. N Engl J Med. 1999;341:635-644.
64. Norton J.A., Jensen R.T. Resolved and unresolved controversies in the surgical management of patients with Zollinger-Ellison syndrome. Ann Surg. 2004;240:757-773.
65. Nikou G.C., Toubanakis C., Nikolaou P., et al. VIPomas: an update in diagnosis and management in a series of 11 patients. Hepatogastroenterology. 2005;52:1259-1265.
66. Ghaferi A.A., Chojnacki K.A., Long W.D., et al. Pancreatic VIPomas: subject review and one institutional experience. J Gastrointest Surg. 2008;12:382-393.
67. Soga J., Yakuwa Y. VIPoma/diarrheogenic syndrome: a statistical evaluation of 241 reported cases. J Exp Clin Cancer Res. 1998;17:389-400.
68. van Beek A.P., de Haas E.R., van Vloten W.A., et al. The glucagonoma syndrome and necrolytic migratory erythema: a clinical review. Eur J Endocrinol. 2004;151:531-537.
69. Wermers R.A., Fatourechi V., Wynne A.G., et al. The glucagonoma syndrome. Clinical and pathologic features in 21 patients. Medicine (Baltimore). 1996;75:53-63.
70. Soga J., Yakuwa Y. Glucagonomas/diabetic-dermatogenic syndrome (DDS): a statistical evaluation of 407 reported cases. J Hepatobiliary Pancreat Surg. 1998;5:312-319.
71. Bartsch D.K., Schilling T., Ramaswamy A., et al. Management of nonfunctioning islet cell carcinomas. World J Surg. 2000;24:1418-1424.
72. Solorzano C.C., Lee J.E., Pisters P.W., et al. Nonfunctioning islet cell carcinoma of the pancreas: survival results in a contemporary series of 163 patients. Surgery. 2001;130:1078-1085.
73. Evans D.B., Skibber J.M., Lee J.E., et al. Nonfunctioning islet cell carcinoma of the pancreas. Surgery. 1993;114:1175-1181. discussion 1181–1182
74. Bloomston M., Muscarella P., Shah M.H., et al. Cytoreduction results in high perioperative mortality and decreased survival in patients undergoing pancreatectomy for neuroendocrine tumors of the pancreas. J Gastrointest Surg. 2006;10:1361-1370.
75. Yao J.C., Hassan M., Phan A., et al. One hundred years after “carcinoid”: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008;26:3063-3072.
76. Moertel C.G., Hanley J.A., Johnson L.A. Streptozocin alone compared with streptozocin plus fluorouracil in the treatment of advanced islet-cell carcinoma. N Engl J Med. 1980;303:1189-1194.
77. Moertel C.G., Lefkopoulo M., Lipsitz S., et al. Streptozocin-doxorubicin, streptozocin-fluorouracil or chlorozotocin in the treatment of advanced islet-cell carcinoma. N Engl J Med. 1992;326:519-523.
78. Broder L.E., Carter S.K. Pancreatic islet cell carcinoma. II. Results of therapy with streptozotocin in 52 patients. Ann Intern Med. 1973;79:108-118.
79. Moertel C.G., Lavin P.T., Hahn R.G. Phase II trial of doxorubicin therapy for advanced islet cell carcinoma. Cancer Treat Rep. 1982;66:1567-1569.
80. Altimari A.F., Badrinath K., Reisel H.J., Prinz R.A. DTIC therapy in patients with malignant intra-abdominal neuroendocrine tumors. Surgery. 1987;102:1009-1017.
81. Ramanathan R.K., Cnaan A., Hahn R.G., et al. Phase II trial of dacarbazine (DTIC) in advanced pancreatic islet cell carcinoma. Study of the Eastern Cooperative Oncology Group-E6282. Ann Oncol. 2001;12:1139-1143.
82. Saltz L., Lauwers G., Wiseberg J., Kelsen D. A phase II trial of carboplatin in patients with advanced APUD tumors. Cancer. 1993;72:619-622.
83. Kulke M.H., Kim H., Stuart K., et al. A phase II study of docetaxel in patients with metastatic carcinoid tumors. Cancer Invest. 2004;22:353-359.
84. Chernicoff D., Bukowski R.M., Groppe C.W.Jr., Hewlett J.S. Combination chemotherapy for islet cell carcinoma and metastatic carcinoid tumors with 5-fluorouracil and streptozotocin. Cancer Treat Rep. 1979;63:795-796.
85. Eriksson B., Skogseid B., Lundqvist G., et al. Medical treatment and long-term survival in a prospective study of 84 patients with endocrine pancreatic tumors. Cancer. 1990;65:1883-1890.
86. Eriksson B., Oberg K. An update of the medical treatment of malignant endocrine pancreatic tumors. Acta Oncol. 1993;32:203-208.
87. Cheng P.N., Saltz L.B. Failure to confirm major objective antitumor activity for streptozocin and doxorubicin in the treatment of patients with advanced islet cell carcinoma. Cancer. 1999;86:944-948.
88. von Schrenck T., Howard J.M., Doppman J.L., et al. Prospective study of chemotherapy in patients with metastatic gastrinoma. Gastroenterology. 1988;94:1326-1334.
89. Kouvaraki M.A., Ajani J.A., Hoff P., et al. Fluorouracil, doxorubicin, and streptozocin in the treatment of patients with locally advanced and metastatic pancreatic endocrine carcinomas. J Clin Oncol. 2004;22:4762-4771.
90. Kulke M.H., Stuart K., Enzinger P.C., et al. Phase II study of temozolomide and thalidomide in patients with metastatic neuroendocrine tumors. J Clin Oncol. 2006;24:401-406.
91. Schnirer I.I., Yao J.C., Ajani J.A. Carcinoid—a comprehensive review. Acta Oncol. 2003;42:672-692.
92. Rinke A., Muller H.H., Schade-Brittinger C., et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. J Clin Oncol. 2009;27:4656-4663.
93. Oberg K., Eriksson B., Janson E.T. Interferons alone or in combination with chemotherapy or other biologicals in the treatment of neuroendocrine gut and pancreatic tumors. Digestion. 1994;55(Suppl 3):64-69.
94. Bilimoria K.Y., Tomlinson J.S., Merkow R.P., et al. Clinicopathologic features and treatment trends of pancreatic neuroendocrine tumors: analysis of 9,821 patients. J Gastrointest Surg. 2007;11:1460-1467. discussion 1467–1469
95. Moore F.T., Nadler H., Radefeld D.A., Zollinger R.M. Prolonged remission of diarrhea due to nonbeta islet cell tumor of the pancreas by radiotherapy. Am J Surg. 1968;115:854-855.
96. Torrisi J.R., Treat J., Zeman R., Dritschilo A. Radiotherapy in the management of pancreatic islet cell tumors. Cancer. 1987;60:1226-1231.
97. Gery B., Roussel A., Valla A. Usefulness of radiotherapy in the treatment of advanced gastrinomas. Radiother Oncol. 1993;27:259-260.
98. van Essen M., Krenning E.P., Kam B.L., et al. Peptide-receptor radionuclide therapy for endocrine tumors. Nat Rev Endocrinol. 2009;5:382-393.
99. Contessa J.N., Griffith K.A., Wolff E., et al. Radiotherapy for pancreatic neuroendocrine tumors. Int J Radiat Oncol Biol Phys. 2009;75:1196-1200.
100. Modlin I.M., Oberg K., Chung D.C., et al. Gastroenteropancreatic neuroendocrine tumours. Lancet Oncol. 2008;9:61-72.
101. Phan G.Q., Yeo C.J., Hruban R.H., et al. Surgical experience with pancreatic and peripancreatic neuroendocrine tumors: review of 125 patients. J Gastrointest Surg. 1998;2:473-482.
102. Modlin I.M., Moss S.F., Chung D.C., et al. Priorities for improving the management of gastroenteropancreatic neuroendocrine tumors. J Natl Cancer Inst. 2008;100:1282-1289.
103. Kwekkeboom D.J., de Herder W.W., Kam B.L., et al. Treatment with the radiolabeled somatostatin analog [177 Lu-DOTA 0, Tyr3]octreotate: toxicity, efficacy, and survival. J Clin Oncol. 2008;26:2124-2130.
104. Kulke M.H., Lenz H.J., Meropol N.J., et al. Activity of sunitinib in patients with advanced neuroendocrine tumors. J Clin Oncol. 2008;26:3403-3410.
105. Raymond E., Raoul J., Niccoli P., et al. Phase III, randomized, double-blind trial of sunitinib versus placebo in patients with progressibe well-differentiated pancreatic islet cell tumours. Ann Oncol. 2009;20(Suppl 7):11. vii
106. Yao J.C., Phan A.T., Chang D.Z., et al. Efficacy of RAD001 (everolimus) and octreotide LAR in advanced low- to intermediate-grade neuroendocrine tumors: results of a phase II study. J Clin Oncol. 2008;26:4311-4318.
107. Yao J., Van Cutsem E., Lombard-Bohas C. Chromogranin A and neuron-specific enolase as biomarkers for everolimus efficacy in patients with advanced pancreatic neuroendocrine tumors after failure of chemotherapy. Ann Oncol. 2009;20(Suppl 7):10. vii
108. Yao J.C., Shah M.H., Ito T., et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011;364:514-523.