CHAPTER 35 Overdose of Cardiotoxic Drugs
We begin with a review of poisoning due to calcium channel antagonists and β-adrenergic receptor antagonists (β-blockers). These two primary cardiovascular drug classes account for well more than half of the life-threatening events and deaths due to cardiovascular agents reported to the American Association of Poison Control Centers each year.1 Digitalis poisoning is also discussed. Finally, agents that produce cardiotoxicity primarily through sodium channel blockade and those with prominent sympathomimetic toxicity are also reviewed.
Calcium Channel Antagonists
Pharmacology
The calcium channel blocking drugs are a heterogeneous class of drugs that block the inward movement of calcium into cells from extracellular sites through “slow channels.”2 There are three major classes of these agents: phenylalkylamines (e.g., verapamil), benzothiazepines (e.g., diltiazem), and dihydropyridines (e.g., nifedipine, amlodipine, nicardipine, nimodipine, felodipine). They are used in the treatment of coronary vasospasm, supraventricular dysrhythmias, hypertension, migraine headache, Raynaud phenomenon, subarachnoid hemorrhage, and many other disease states.3 In general, calcium channel antagonists are rapidly and completely absorbed from the gastrointestinal tract and, with the exception of nifedipine, undergo extensive first-pass hepatic metabolism yielding low systemic bioavailability. The volume of distribution is large for all but nifedipine, and protein binding is high (>90% for all but diltiazem). Elimination is almost entirely by the liver; impaired renal function does not affect clearance with the exception of a somewhat pharmacologically active metabolite of verapamil that is renally excreted.4 Terminal half-lives are generally from 3 to 10 hours, but all three classes of calcium channel antagonists are available in sustained-release preparations, which can result in greatly prolonged half-lives.
Pathophysiology
Electrophysiologic effects are most prominent for verapamil and diltiazem and are seen much less often with nifedipine and other dihydropyridines, which work primarily on the peripheral vasculature. Sinus node function may be significantly altered by verapamil and diltiazem in patients with underlying sinus node disease; in excess, these agents may prolong AV nodal conduction sufficient to produce advanced heart block. Depression of myocardial contractility by impeding phase 2 calcium influx is most pronounced in overdose or in patients who already have depressed myocardial function from underlying disease or concomitant drugs. Contraction of vascular smooth muscle, particularly arterial smooth muscle, is also mediated by calcium influx that is inhibited by calcium antagonists. In overdose, the effect of vasodilation on systemic blood pressure may be profound. However, in some cases, especially those involving the dihydropyridines, vasodilation may be ameliorated by a reflex increase in sympathetic activity, with increased heart rate and cardiac output.
Clinical Manifestations
The most serious consequences of calcium antagonist toxicity result from their effects on the cardiovascular system. Generally, these effects are an extension of the pharmacodynamic effects of the specific agent, although unique features of the different agents’ specificity profiles may be lost in overdose.5 Clinical features are summarized in Table 35-1. Bradycardia and conduction defects are among the most frequent findings in overdose of verapamil or diltiazem. Additionally, hypotension is present in most significant exposures to any calcium antagonist. These features generally develop within 1 to 2 hours of exposure, but the onset of moderate to severe cardiovascular manifestations may be delayed for more than 12 hours when a sustained release preparation has been ingested.6
Table 35–1 Clinical Features of Calcium Antagonist and β-Blocker Overdose
| Cardiovascular |
| Hypotension, shock |
| Dysrhythmias |
Patients at particular risk for toxicity from calcium antagonists include those with sinus node dysfunction, AV nodal conduction disease, severe myocardial dysfunction, obstructive valvular disease, hypertrophic cardiomyopathy, hepatic failure (leading to impaired elimination), and combined treatment of a calcium antagonist with β-blockers or digoxin.7 In addition, verapamil may dangerously accelerate conduction through accessory pathways when administered intravenously to patients with accessory or anomalous AV connections such as in Wolff-Parkinson-White syndrome.8 It should not be given to patients with atrial fibrillation and evidence of pre-excitation on electrocardiography.
Profound hypotension is the major manifestation of overdose with nifedipine and may produce reflex tachycardia, flushing, and palpitations. Conduction defects are rare unless there is an underlying conduction disease, a very large ingestion, or the presence of coingestants such as β-blockers.5,7
Management
Hypotension should be addressed based on the pathophysiology discussed earlier. Intravenous fluids and vasoconstriction with agents such as norepinephrine, epinephrine, phenylephrine, or dopamine may be successful in hypotension primarily due to peripheral vasodilation. Hypotension due to depressed myocardial contractility may be responsive to intravenous administration of calcium salts (calcium chloride 10% solution, 10 to 20 mL, or calcium gluconate 10% solution, 30 mL, followed by continuous infusion). The optimal dose of calcium is unclear from the available literature, and the danger of hypercalcemia-induced impairment of myocardial contractility and vascular tone must be kept in mind.10 However, calcium levels have been elevated to as high as 15 to 20 mg/dL in previous case reports without any adverse effects, and with an improvement in blood pressure.11
Glucagon has had some anecdotal success in cases of calcium antagonist overdose, and several animal models have shown its efficacy in this setting.12 Its use is discussed further in the section on treatment of β-blocker toxicity. Calcium antagonists are generally both highly protein bound and extensively distributed in tissue. Therefore, enhanced elimination techniques such as hemodialysis and hemoperfusion are unlikely to be of benefit, and clinical reports have failed to support a role in either therapeutic or overdose settings.13,14
Finally, a newer treatment using a hyperinsulinemia/euglycemia protocol has shown impressive results in case reports of calcium antagonist poisoning.15 Laboratory research in this area has also been promising.16 Numerous reports of the success of this treatment, along with published reviews of the management of calcium channel blocker toxicity, support its use early in the management of these poisonings. Insulin is thought to improve ionotropy and increase peripheral vascular resistance. Although the mechanisms are not completely known, it is thought to have a direct ionotropic effect on cells and to improve calcium pumps in myocardial cells.16,17,19 The most common insulin dosing regimen is 0.5 to 1 unit/kg/hr, along with 0.5 g/kg/hr of glucose using D5, D10, D25, or D50 (the latter two typically require central venous access due to their vascular irritant effects). In general, however, these patients are often already hyperglycemic and may not require the glucose component of the regimen while they remain toxic from the poisoning. Serum glucose concentrations should be checked hourly while the patient is on this therapy.
In severe refractory cases, cardiovascular bypass remains a viable option. Implementation has revealed successful results in previous reports, as patients are supported through the toxic effects of their poisoning.20,21 If patients can survive through the metabolism of the medication, they can often demonstrate a full recovery, both cardiovascularly and neurologically.
β-Adrenergic Antagonists
Clinical Manifestations
β-blocker toxicity is most commonly due either to administration to patients with underlying cardiac disease or to acute massive overdose. In the setting of acute overdose with a nonsustained release product, the onset of symptoms can be expected to occur within 6 hours of ingestion.22
Generally, poisoning due to β-blockers shares many features of clinical presentation with poisoning due to calcium channel antagonists (see Table 35-1), but the hallmark of β-blocker poisoning is hypotension, due predominantly to impaired contractility. Sinus node depression and conduction abnormalities are also common. As noted earlier, membrane-stabilizing properties seen most prominently with propranolol may lead to impaired conduction, QRS prolongation, and ventricular dysrhythmias23,24 Highly β-selective agents (atenolol, nadolol) may produce hypotension with a normal heart rate, but selectivity is frequently lost in large overdose. Overdose of agents with intrinsic sympathomimetic activity, most notably pindolol, may actually manifest with hypertension and tachycardia due to a stimulation. Sotalol is a unique agent that possesses some class III antiarrhythmic properties and therefore may produce Q–T interval prolongation, ventricular tachycardia, and torsades de pointes.25
Lethargy and coma may be present in patients with β-blocker poisoning. Seizures are rare manifestations of β-blocker poisoning, except for propranolol. This appears to correspond with CNS effects of the drug rather than to hypoperfusion of the CNS.23,24 Bronchospasm and respiratory depression may occur from overdose with β-blockers, but are infrequent. Hypoglycemia may also occur in contradistinction to calcium channel antagonists that result in hyperglycemia.26
Management
Glucagon is the mainstay of antidotal therapy for symptomatic β-blocker toxicity. Glucagon is a polypeptide hormone that appears to bypass the β-adrenergic receptor on a cardiac myocyte and increases intracellular levels of cyclic AMP by stimulating a distinct glucagon receptor on the membrane. The resultant promotion of transmembrane calcium flux and intracellular calcium release leads to restoration of chronotropy and inotropy.27 Although not universally effective, glucagon is of benefit in the majority of β-blocker overdoses. The initial dose of glucagon for a symptomatic β-blocker poisoning in the average adult is 3 to 5 mg bolused intravenously. The bolus may be repeated, and a continuous infusion of 2 to 5 mg/hr or higher may be necessary to maintain conduction and contractility. Mild nausea and vomiting, along with mild hyperglycemia, may occur with these doses, but otherwise the use of glucagon is without significant side effects.
As with calcium channel antagonist toxicity, calcium salts have been reported to be useful in β-blocker toxicity. In studies, calcium infusion can increase blood pressure in hypotensive β-blocker poisonings without any concomitant effect on heart rate.28 Thus calcium therapy may augment glucagon treatment in these cases. Recommended starting doses are 1 to 3 grams of calcium chloride 10% solution (10 to 30 mL) given intravenously. If central line access is not available, calcium gluconate should be used, as calcium chloride can be irritating to peripheral veins.
Some β-blocking agents, such as propranolol and acebutolol, can also act as membrane-stabilizing drugs, and can cause QRS prolongation in overdose. When the QRS duration is widened to greater than 120 milliseconds, treatment with sodium bicarbonate boluses may be required (see later). Some animal models and case reports have shown proven benefit with sodium bicarbonate in such circumstances.29
Phosphodiesterase inhibitors such as amrinone have not been shown to be of any additional benefit when compared with glucagon for management of β-blocker overdose, but their use might be considered if other therapy is failing.30,31 These agents may vasodilate and should be discontinued if blood pressure does not immediately respond.
Successful use of an intra-aortic balloon pump support in patients in whom other measures were unsuccessful has been reported.32 This may allow sufficient time for elimination of the toxicant and should be considered when the patient remains profoundly hypotensive despite glucagon and high-dose vasopressors.
Digoxin
Clinical Manifestations
There are no dysrhythmias diagnostic of digoxin toxicity. Several rare dysrhythmias, such as ventricular bigeminy and bidirectional ventricular tachycardia, are highly suggestive of poisoning by this agent. The classic early cardiac presentation of chronic toxicity is the appearance of premature ventricular contractions in a patient with atrial fibrillation whose ventricular response rate had been previously well controlled. Most patients with chronic, unintentional toxicity will complain first of anorexia and fatigue and will often have nausea and vomiting. Neurologic symptoms can begin subtly as visual changes, described as blurred vision, decreased visual acuity, or yellow halos, and progress on to confusion, hallucinations, seizures, or coma.33,34
Management
Digoxin-specific Fab fragments (Fab) are ovine IgG antibodies to digoxin that have had the Fc portion removed by papain digestion to reduce immunogenicity. When administered intravenously into a victim with digoxin toxicity, Fab fragments reverse conduction disturbances, restore contractility, and re-establish sodium-potassium ATPase activity by removing digoxin off receptor sites.35 Hyperkalemia is also reversed after Fab administration. Signs and symptoms of toxicity should resolve in less than an hour but are often gone within 10 minutes. Patients with severe hypotension or cardiac arrest may not be able to circulate the antibody fragments and may therefore be refractory to treatment.36
The dose of Fab fragment recommended to reverse digoxin toxicity is an equimolar dose to that of the ingested cardiac glycoside. A dose of 50 to 100 mg will neutralize 1 mg of digoxin. One vial contains 40 mg, and the manufacturer recommends a starting dose of 10 vials when the amount ingested or the level is unknown. Tables are available in the Fab package insert or through regional poison control centers to relate the dose of Fab to the measured serum digoxin concentration. Allergic reactions to Fab are extremely rare, and skin testing is unnecessary.35 Fab has also been shown to be effective in the treatment of severe cardiac glycoside cardiotoxicity from plants such as oleander containing similar compounds, but larger doses of the Fab may be required.37
Sodium Channel Blocking Agents
Of all categories of cardiotoxic drugs, perhaps the most heterogeneous contains those that impair sodium conduction through membrane channels (Table 35-2). These substances are commonly described as having “quinidine-like” or “membrane stabilizing” effects on the myocardial cell. Substances exhibiting these properties include analgesics, antihistamines, psychotropics, antidepressants, antidysrhythmics, anticonvulsants, and local anesthetics. Many of these medications have unique clinical effects at therapeutic doses, but in overdose, each can produce similar cardiotoxicity. The most common group of sodium channel blocking drugs, and the one to which all others are compared, is the class I antiarrhythmic agents.
| Class Ia antiarrhythmics |
| Class Ib antiarrhythmics |
| Class Ic antiarrhythmics |
| Chloroquine |
| Quinine |
| Propoxyphene |
| Cyclic antidepressants |
| Phenothiazines |
| Antihistamines (sedating and nonsedating H, antagonists) |
| Cocaine |
| Propranolol |
| Carbamazepine |
Pathophysiology
The subclassification of class I agents is partly based on the effect of these agents on potassium channels during cell repolarization. Blockade of potassium channels, most commonly displayed by class Ia drugs, leads to prolongation of repolarization and a subsequent increase in Q–T interval duration.39 As the duration of repolarization and therefore the Q–T interval lengthens, the opportunity for early afterpolarizations during this relative refractory period increases. Episodes of polymorphic ventricular tachycardia (torsades de pointes) can occur in this situation, especially in the presence of low potassium or magnesium concentrations.40 Class Ib agents shorten repolarization and reduce the duration of the action potential, while leaving potassium channels open and the Q–T intervals unaffected.39 Class Ic drugs are the most potent sodium channel blockers38 but have little effect on the repolarization phase of the action potential.
Pharmacology and Clinical Manifestations
Class Ia Antiarrhythmics
As noted earlier, all drugs in this class inhibit fast sodium channels in a dose-dependent manner. Generally, class Ia drugs are high potency sodium channel blockers.38 Depression of slow inward calcium and outward potassium movement may account for reduced action potential plateau and prolonged repolarization. The result is prolongation of the relative refractory period, decreased pacemaker automaticity, and a generalized slowing of conduction through the heart.
Quinidine
Quinidine, the prototype of class Ia antiarrhythmics, was released in the United States in the early 1900s. Orally ingested quinidine has good bioavailability. The sulfate reaches peak plasma concentrations within 90 minutes, while the absorption of gluconate and polygalacturonate salts may be delayed 3 to 6 hours.41 Quinidine is highly protein-bound, with a large volume of distribution throughout the body (3.0 L/kg).41 Up to 40% of an ingested dose of quinidine may be eliminated by the kidneys, but the remainder is metabolized to inactive products in the liver. High “therapeutic” plasma concentrations of quinidine were found in some individuals that developed both QRS and Q–T interval prolongation, and a sudden loss of consciousness associated with its use was soon described.41 These symptoms, referred to as “quinidine syncope,” were found to be caused by ventricular tachydysrhythmias.38 The incidence of these attacks is estimated to be 2% to 4%, and they are usually associated with polymorphic ventricular tachycardia.38 This dysrhythmia is often related to a prolonged Q–T interval, but some studies have determined that quinidine-associated ventricular tachycardia often does not present as torsades de pointes and may not be associated with a prolonged Q–T interval.42 Studies have also demonstrated little relationship between quinidine concentrations and the incidence of this dysrhythmia.44,48 Hypokalemia, however, is frequently found in patients with quinidine-associated syncope.40,45
As quinidine serum concentrations increase, Q–T interval prolongation is the earliest and most predictable electrocardiographic effect, 46 followed closely by QRS widening. In overdose, QRS widening is almost always present, with bundle branch blocks, sinoatrial and AV blocks, sinus arrest, and junctional or ventricular escape rhythms noted at high concentrations.39,40
Hypotension from quinidine, like many other of the drugs discussed in this section, is multifactorial. Unlike quinidine syncope, quinidine-induced generalized myocardial depression is dose-dependent.40,47 At low doses, especially when administered intravenously, quinidine exerts little negative inotropic effect but is an antagonist of peripheral α-receptors, leading to vasodilation.40 This effect can result in orthostatic syncope in some patients. At toxic concentrations, quinidine causes circulatory collapse due to a profound negative inotropic effect.47 In addition to shock, severely poisoned patients can have recurrent dysrhythmias, central nervous system depression, and renal failure.
The optical isomer of quinidine is quinine, and this compound has the capability to produce the same signs and symptoms in overdose.48,49 Toxic doses of either of these agents can also lead to cinchonism, a condition named after the tree from which these compounds are derived.49 This syndrome results in tinnitus, blurred vision, photophobia, confusion, delirium, and abdominal pain.49 Quinine amblyopia may result from large ingestions of these compounds, and the visual loss may be complete and sudden. Although vision returns in some patients as toxicity resolves, the loss may be permanent.50 Coma and seizures can occur with toxic concentrations of these drugs, even in hemodynamically stable individuals.40 Cinchonism is not reported with poisonings of the other class Ia agents.
Quinidine also has antimuscarinic effects and it may exacerbate the ventricular response to atrial flutter via enhanced conduction of the atrioventricular (AV) node. Furthermore, its potassium channel blockade may cause increased insulin release in the pancreatic islet cells, leading to hypoglycemia.51
Procainamide
A therapeutic oral dose of procainamide reaches peak plasma concentration within an hour, but massive ingestions can greatly delay absorption and prolong toxicity.49 Like quinidine, up to 40% of a given dose of procainamide may be eliminated unchanged in the urine. Unlike quinidine, procainamide is metabolized to a compound with cardiac activity similar to that of the parent drug, N-acetylprocainamide (NAPA), which may complicate the correlation of plasma levels of the parent compound with clinical effects.49 The therapeutic volume of distribution of procainamide is 2.0 L/kg, with a plasma half-life of 3 to 4 hours. The plasma half-life in overdose may increase significantly.49
Cardiotoxicity from procainamide is mechanistically similar to that described from quinidine. Myocardial depression, polymorphic ventricular tachycardia, and other cardiac dysrhythmias are all expected at high serum concentrations of procainamide.49 However, procainamide exerts a less negative inotropic effect, and a lower incidence of ventricular dysrhythmia than quinidine.52,53 Hypotension is mostly seen with intravenous use and usually only during infusions faster than 20 mg/min.40 Procainamide overdose can result in severe hypotension and dysrhythmias identical to those described with quinidine. Inability to electrically pace the heart of a procainamide-intoxicated patient due to high pacing thresholds has been described.54 Serious toxicity from procainamide includes lethargy, confusion, and depressed mentation along with the cardiotoxicity.39–41 Other adverse events in acute overdoses include seizures and antimuscarinic effects.55 Hematologic abnormalities such as agranulocytosis, thrombocytopenia, and hemolytic anemia have been reported in long-term use of procainamide.56 Procainamide may also produce a lupus-like syndrome.
Disopyramide
Peak serum concentrations of disopyramide may be delayed up to several hours in toxic ingestions owing to its antimuscarinic effects on intestinal motility.40 The protein binding (50% to 60%) and volume of distribution (<1 L/kg) of disopyramide are less than those of quinidine or procainamide and suggest the possibility that hemodialysis could be effective in removing toxic concentrations.40,57 A therapeutic dose of disopyramide has a mean plasma half-life of 6 to 8 hours and is 40% to 60% eliminated by the kidneys.49 The main hepatic metabolite, mono-N-dealkylated disopyramide, has little cardiac activity, but produces more antimuscarinic effects than the parent compound.
Disopyramide is the newest of the Class Ia antiarrhythmic agents and demonstrates electrophysiologic effects similar to quinidine and procainamide. Although Q–T interval prolongation does not usually occur with therapeutic concentrations of disopyramide, syncopal episodes have been reported.58 Of all class Ia agents, the negative inotropic effects of disopyramide are most pronounced, and hypotension can be seen in disopyramide poisoning without concomitant electrocardiographic changes.40,59,60 This may in part be related to its ability to block myocardial calcium channels.61 Although mild antimuscarinic effects can be noted in poisonings of all class Ia agents, those following disopyramide toxicity are the most clinically significant40 and can result in sinus tachycardia, blurred vision, altered mental status, seizures, urinary retention, and ileus, at times without accompanying serious cardiotoxicity.40 Overdose experience with disopyramide is limited in the United States.
Class Ib Antiarrhythmics
Drugs in this class suppress automaticity similarly to the class Ia agents but shorten the action potential refractory phase and increase conduction through hypoxic myocardial tissue.62 Class Ib drugs have rapid “on-off” binding kinetics for myocardial sodium channels and possess the highest affinity for sodium channels that are in the inactivated state.49 At therapeutic concentrations, these compounds moderately depress phase 0 of the myocyte action potential. The resultant effects on the electrocardiogram include a normal or shortened Q–T interval and an unchanged QRS duration.63
Lidocaine
A large first-pass effect is seen with oral dosing of lidocaine, and only 30% to 35% of an ingested dose is bioavailable.64 However, large ingestions have resulted in significant absorption, resulting in toxicity.65 Lidocaine is well absorbed topically through abraded epithelium and from the trachea and bronchi after endotracheal administration.66 The apparent volume of distribution of lidocaine is 1.3 L/kg, but it is significantly reduced in the presence of congestive heart failure.67 The liver metabolizes virtually all of a lidocaine dose, with an elimination half-life in therapeutic concentrations of about 2 hours.49 The most significantly active metabolite is monoethylglycinexylidide (MEGX), with a half-life also of 2 hours.68
Lidocaine is the prototype of class Ib antiarrhythmics, and in poisoning it causes cardiovascular and CNS toxicity. Lidocaine is primarily metabolized in the liver to MEGX. Both compounds are neurotoxic at high concentrations and can cause seizures and apnea.63 Some individuals experience mild neurologic symptoms at therapeutic plasma concentrations.63 Since lidocaine rapidly passes through the blood-brain barrier, patients with toxicity usually manifest CNS dysfunction as initial symptoms.69 Early signs of CNS toxicity from lidocaine include lightheadedness, agitation, confusion, hallucinations, and dysarthria. Some individuals initially complain of tongue or perioral numbness. Progression to convulsions or coma can be rapid. Cardiovascular toxicity occurs primarily in massive overdose. Following central nervous system dysfunctions, intrinsic cardiac pacemakers are depressed, conduction is delayed, and myocardial contractility is impaired.70,71 Large intravenous lidocaine doses greater than 1 g have resulted in asystole, complete heart block, and refractory hypotension.63,70,72
Phenytoin
Phenytoin absorption from the gastrointestinal tract can be erratic, and peak levels after an oral overdose can be delayed up to 24 hours or more.73 Phenytoin is 90% protein bound, and the volume of distribution of the free drug is 0.5 L/kg.49,73 Signs of toxicity correlate better with free phenytoin levels, but most laboratories still assay for both bound and unbound fractions. Phenytoin is metabolized to nontoxic metabolites by the liver. At high concentrations, these enzymes become saturated, and the half-life may increase to several days as elimination kinetics change from first order to zero order.74 Patients with low serum albumin concentrations may be particularly prone to chronic phenytoin toxicity, owing to the higher relative fraction of free drug.49
Phenytoin poisoning primarily causes neurotoxicity, manifested as drowsiness, ataxia, dysarthria, and nystagmus. In massive poisonings, coma and seizures can be seen, but respiratory depression is infrequent. Cardiac dysrhythmias and hypotension are rarely reported with toxic phenytoin ingestions but are often encountered during rapid intravenous infusions.75 Although phenytoin does have sodium channel blocking effects on myocardial tissue, the diluent of the intravenous solution, propylene glycol, has been implicated as the source of myocardial depression and dysrhythmias associated with intravenous infusion.76 Slowing the rate of infusion prevents the majority of these complications.
Tocainide, Mexilitine
Overdose experience with other class Ib drugs has been limited. Tocainide toxicity has resulted in gastrointestinal symptoms, seizures, and cardiac dysrhythmias similar to other class I agents.77 Therapeutic use of tocainide resulting in blood dyscrasias78 and pulmonary fibrosis79 has also been reported. Mexilitine poisoning has caused seizures, ventricular dysrhythmias, and impaired myocardial conduction.80 Other adverse effects of mexilitine are primarily neurologic and comparable to symptoms that occurred with lidocaine.
Class Ic Antiarrhythmics
Class Ic drugs (encainide, flecainide, propafenone) bind to sodium channels in the activated state and have the most potent sodium channel blocking effects of all class I antiarrhythmics.49 Therapeutic concentrations of class Ic agents produce little effect on myocyte repolarization compared to other class I antiarrhythmics.81 At higher concentrations, these agents exert a significant negative inotropic effect.82 Overdose experience with these drugs is limited, but they would be expected to produce similar cardiovascular effects as other class I compounds, with no antimuscarinic features. Seizures, hypotension, and dysrhythmias have been reported after a 3- to 4-g overdose of encainide.82
Class III Antiarrhythmics
Class III agents (amiodarone, ibutilide, dofetilide) block the rapidly activating component of the delayed rectifier potassium channels.49 These potassium channels are responsible for phase 3 repolarization in cardiac action potentials. Therefore Class III drugs delay repolarization and cause an increase in duration of the action potential and an increase in the effective refractory period. On the electrocardiogram, this results in prolonged Q–T intervals. By increasing the refractory period, these drugs are very useful in terminating re-entrant dysrhythmias.
Amiodarone
Amiodarone is an antidysrhythmic with a multitude of pharmacologic effects. It undergoes hepatic metabolism through cytochrome oxidase systems to produce another pharmacologically active but less potent metabolite, desethylamiodarone.83 The main mechanism of action of amiodarone is prolongation of cardiac myocyte repolarization through blockade of the rapidly activating delayed rectifier potassium channel. Collectively this represents a class III antidysrhythmic effect. However, amiodarone is also a weak α- and β-adrenergic receptor antagonist and may block inactivated sodium channels and L-type calcium channels.84 Expected electrocardiographic outcomes are prolonged Q–T intervals, ventricular dysrhythmias, and conduction blocks. Amiodarone is used to terminate re-entrant atrial or ventricular dysrhythmias and it has recently been added to the Advanced Cardiac Life Support (ACLS) tachydysrhythmia guidelines.85 Despite its more frequent use today, there are few reported cases of oral amiodarone overdose resulting in significant cardiac toxicity. The low toxicity is likely due to its low and unpredictable oral bioavailability (22% to 86%) and large volume of distribution (>6.0 L/kg).86 Intravenous overdose of amiodarone has not been reported although hypotension, bronchospasms, and hepatitis from therapeutic doses have been described. Most of the toxic effects reported from amiodarone in the literature are from long-term treatment and appear to be dose-related. Chronic use of amiodarone has been associated with pneumonitis, hypothyroidism, thyrotoxicosis, hepatitis, skin discoloration, and corneal damage.87 Although not well studied, cholestyramine has been suggested as treatment for both acute and chronic amiodarone toxicity by its gastrointestinal binding of unabsorbed amiodarone and blockade of enterohepatic circulation of the drug.88
Ibutilide, Dofetilide
Both ibutilide and dofetilide are newer class III antiarrhythmics used for chemical cardioversion of atrial fibrillation or flutter. Because of their effect on the rapidly activating delayed rectifier potassium channels, both drugs can delay action potentials and prolong Q–T intervals.49 Experiences with overdoses of either drug are limited but expected toxicity would be induction of ventricular dysrhythmias. Toxic effect of either drug occurs within 60 minutes of administration and therapeutic use has resulted in torsades de pointes.84,89 Therefore a reasonable observational period of 4 to 6 hours is recommended in all patients who received ibutilide or dofetilide.
Cyclic Antidepressants
Toxicity from cyclic antidepressants is perhaps the most well-described of all fast sodium channel blocking compounds. These substances are responsible for more fatalities each year than any of the other drugs in this group.90 After being originally studied for their antihistaminic properties in the 1940s, cyclic antidepressants were introduced into the pharmaceutical market in the United States in the 1960s, rapidly replacing electroshock therapy as a more “humane” treatment for severe depression.
Pharmacology
The “first generation” of cyclic antidepressant compounds, known as the tertiary amines, includes amitriptyline, imipramine, doxepine, and trimipramine. All are potent inhibitors of myocyte sodium channels. Each undergoes metabolism in the liver. Amitriptyline and imipramine are hepatically converted to the active secondary amines nortriptyline and desipramine, respectively, and these latter compounds were soon marketed as second-generation therapies for depression.49 The membrane-stabilizing effects of these second-generation agents were soon appreciated as being similar to that of their parent compounds.49
In therapeutic doses, cyclic antidepressant drugs are rapidly absorbed. Although one might expect delayed gastric emptying as a result of an antimuscarinic effect in large ingestions, clinical signs of cyclic antidepressant toxicity usually appear within 6 hours.91 Asymptomatic patients without concomitant ingestions should not develop toxicity after 6 hours. Antimuscarinic effects on the central and peripheral nervous systems usually precede cardiotoxicity, but large ingestions of agents with less muscarinic receptor activity, such as imipramine, may present initially with hypotension and dysrhythmias. Cardiotoxicity, mental status depression, and seizures resulting from these agents can proceed rapidly, necessitating continuous monitoring.
Cyclic antidepressants are significantly protein bound in circulation and widely distributed in the body (40 L/kg).49 Hepatic metabolism is the major route of elimination for these compounds, with some, such as amitriptyline and imipramine, producing active metabolites. Elimination half-life can be prolonged in overdose due to enzyme saturation.
Pathophysiology
The pathophysiology of cyclic antidepressant cardiotoxicity results from four main properties: (1) fast sodium channel or membrane-stabilizing effects, (2) muscarinic receptor blockade, (3) α-receptor blockade, and (4) norepinephrine reuptake blockade. Animal data also suggest blockade of delayed rectifier potassium channel and the γ-aminobutyric acid (GABA) receptor complex in the brain.92,93
Cyclic antidepressants block fast sodium channels in a manner similar to quinidine and other class Ia antiarrhythmics, markedly decreasing the slope of phase 0 of the action potential.49 Prolongation of both the QRS and Q–T interval durations have been demonstrated clinically in overdose, but in vitro experiments show that cyclic antidepressants block primarily fast depolarizing sodium currents and actually shorten repolarization.94,95 The Q–T prolongation resulting from cyclic antidepressant toxicity is mainly attributed to the result of progressive QRS widening with globally impaired myocardial conduction.95 However, the potential blockade of the delayed rectifier potassium channels may also contribute to the Q–T prolongation.93 The cardiotoxic effects include ventricular dysrhythmias and depressed myocardial contractility.95
Cyclic antidepressants block several receptor sites in both the central and peripheral nervous system, including H1 and H2 receptors, dopamine receptors, and muscarinic receptors.96 This effect on the autonomic nervous system produces a clinical syndrome of dry mouth, blurred vision, sinus tachycardia, altered mental status (ranging from confusion and hallucination to seizures and coma), ileus, urinary retention, and anhidrosis. These signs and symptoms frequently precede the sodium channel blocking effects and are more common with doxepin and amitriptyline than imipramine.49,96
Cyclic antidepressants are potent α-receptor antagonists. This effect is responsible for the orthostatic hypotension often experienced by patients at the initiation of therapy with these compounds.49 The resulting vasodilation can be severe in overdose and combined with the impaired cardiac output from myocardial depression can lead to refractory hypotension and cardiovascular collapse.97 α-Receptor blockade of the pupil in patients with cyclic antidepressant toxicity can prevent the anticipated antimuscarinic mydriasis, resulting in an unanticipated miotic pupillary effect.
The hypothesized mechanism of antidepressant action of cyclic antidepressants lies in their ability to block the catecholamine reuptake pump on the presynaptic terminal of neurons.96 In overdose, this effect can deplete presynaptic catecholamine concentrations, thought to contribute to dysrhythmias and hypotension.49
Hypotension can be present in cyclic antidepressant poisoning without significant dysrhythmias. Sinus tachycardia is usually present before the development of ventricular dysrhythmias or heart block.98 Several studies have evaluated the electrocardiographic abnormalities predictive of toxicity from these agents. One review found that 33% of patients with QRS intervals greater than 100 ms developed seizures and 14% developed ventricular dysrhythmias.99 There was also a 50% incidence of ventricular dysrhythmias in patients with QRS duration exceeding 160 ms.99 Unfortunately, up to 25% of normal individuals may have a QRS duration greater than 0.10 second.49 Another study suggested that a rightward terminal vector, best seen in the R wave of lead aVR, may correlate with the degree of cyclic antidepressant toxicity.100,101 In this study, an R wave of lead aVR greater than 3 mm notably predicted toxicity. Although having electrocardiographic abnormalities can be useful in assessing potential toxicity, none of the findings is 100% sensitive. Absence of these concerning findings is more indicative that cardiac toxicity is not developing.
Antipsychotics (Phenothiazines, Butyrophenones, and Atypical Agents)
Phenothiazine derivative compounds such as Thorazine, thioridazine, and prochlorperazine may have clinical effects similar to the cyclic antidepressants. Antimuscarinic toxicity is frequently more pronounced in poisonings from these drugs, rather than their membrane-stabilizing cardiac effects. However, heart block and wide complex tachycardias and refractory hypotension are occasionally reported in overdoses of these agents.102,103 Fatalities are rare, even in massive ingestions of these drugs. The most common clinical presentation of phenothiazine overdose is neurotoxicity, manifesting as delirium, agitation, coma, seizures, and other antimuscarinic effects.
Haloperidol and droperidol are butyrophenone antipsychotic and antiemetic compounds available in the United States. Although these drugs share the potent dopamine receptor blocking properties of the phenothiazines, overdoses of these agents lack the prolonged sedation and antimuscarinic effects most often seen with phenothiazines. Torsades de pointes has been reported with the butyrophenones but usually follows large parenteral dosing.104
Antihistamines
Many H1 receptor antagonists have been found to exert similar effects on the myocardial action potential as the class Ia antidysrhythmic compounds. The most commonly used medication in this class, diphenhydramine, has been shown to effectively block fast sodium channels at high concentrations.105
In massive poisonings, antihistamines can impair fast sodium channel conduction, resulting in dysrhythmias and hypotension similar to that seen with other sodium channel blocking agents.105, 106 Dysrhythmias and cardiovascular collapse from these drugs are exacerbated by acidosis and, therefore, often occur after seizures, which frequently cause a sudden decline in the serum pH.105, 106
Propoxyphene
Propoxyphene is an opioid analgesic found in combination with acetaminophen in Darvocet, and with salicylates in Darvon. Hepatic metabolism of propoxyphene produces an active metabolite known as norpropoxyphene. Both agents block fast sodium channels, causing QRS prolongation in toxic serum concentrations.107 Overdoses of propoxyphene can result in hypotension and widening of the QRS complex, which can lead to ventricular dysrhythmias.108 It is this membrane-stabilizing effect that is also thought to be responsible for the greater incidence of seizures from propoxyphene poisonings than from other opioids. As with other sodium channel blocking agents, widening of the QRS complex with propoxyphene poisoning has been shown to respond to therapy with sodium bicarbonate (see later discussion).109 Propoxyphene-induced seizures can be refractory to conventional anticonvulsants, such as phenytoin, necessitating use of benzodiazepines and phenobarbital for control.
Carbamazepine
Carbamazepine is an anticonvulsant with structural similarity to the cyclic antidepressants. In vitro, carbamazepine cardiotoxicity resembles that of the class Ia antiarrhythmics. However, carbamazepine rarely causes significant quinidine-like effects in poisoning, even with massive ingestions.110,111 Carbamazepine toxicity often results in mental status changes and occasionally respiratory depression.110,111 In addition, toxic carbamazepine concentrations may produce blockade of muscarinic receptors, resulting in the classic antimuscarinic syndrome with effects such as sinus tachycardia, dry mouth, and mydriasis.
Chloroquine
Chloroquine is a common antimalarial agent that often results in severe toxicity when taken in overdose. Chloroquine is structurally related to quinine and quinidine, and cardiotoxicity resulting from any of these agents can be indistinguishable.111 The toxic to therapeutic ratio of chloroquine is low, and ingestions of as little as 300 mg by children have been fatal.112 Seizures commonly and rapidly accompany the cardiotoxicity of this drug and appear to be unrelated to hypoxia.111 A combination of intensive supportive care, intravenous boluses of benzodiazepines for convulsions, and vasopressor use for cardiovascular support has been shown to decrease mortality in animal models and human case reports of chloroquine poisoning.113
Management of Sodium Channel Blocking Drug Toxicity
As in the treatment of other cardiotoxic drug overdoses discussed in this chapter, the initial management of poisonings involving sodium channel blocking medications should begin with airway and circulatory support (Table 35-3). Any patient who is not breathing or in whom a patent airway is of question should receive endotracheal intubation and mechanical ventilation. Combative patients may require sedation and paralysis before an endotracheal tube can be placed.
Table 35–3 Outline of General Management of Sodium Channel Blocking Agent Toxicity
|
2. Control breathing and hyperventilate patient if head injury is possible, or if cardiotoxicity is present and intravenous access is not established.
|
Resuscitating the patient poisoned with sodium channel blocking drugs can be challenging. The hypotension resulting from massive ingestions of these agents is multifactorial and often refractory to intravenous fluid boluses. Vasopressors may be necessary in some situations, and vasopressor choice may be important. Although dopamine is employed with success in many cases of poisoning-related hypotension, it may be ineffective or even exacerbate the hypotension associated with cyclic antidepressants, phenothiazines, and other α-receptor blocking agents.114 Dopamine is the precursor of norepinephrine and requires uptake into the presynaptic terminals for activation; thus it may be ineffective with these agents, which block the catecholamine reuptake pump. At higher doses, the vasoconstrictive effects of dopamine are antagonized by the α-blocking effects of some of these drugs, leading to unopposed β-receptor stimulation of blood vessels and worsening of hypotension. For these reasons, vasopressors with more direct α-receptor stimulation, such as norepinephrine or phenylephrine, may be required for consistent blood pressure support.
The most helpful adjunct in the management of sodium channel blockade from drug poisoning has proved to be sodium bicarbonate. Sodium bicarbonate has been shown to be effective in raising the blood pressure and treating dysrhythmias associated with cyclic antidepressants, cocaine, flecainide, quinidine, chloroquine, and diphenhydramine.115–121 Sodium bicarbonate has been shown in both animal models and human cases of toxicity from these agents to increase blood pressure and improve conduction through the heart. Studies suggest that this is the result of the concomitant effects of both the bicarbonate and sodium components because they offer a synergistic benefit together when compared to each alone. Thus the beneficial effects are likely secondary to both an increase in the blood pH and an increase in the extracellular sodium concentration.
In some severe cases of sodium channel blocking drug toxicity, ventricular dysrhythmias may be refractory to the aforementioned management. Oxygenation should be maintained and resuscitative efforts continued as long as cardiac activity is present. Patients have survived neurologically intact after over an hour of advanced cardiac life support following massive cyclic antidepressant poisoning.122 Prolonged resuscitation may allow enough drug redistribution from cardiac receptors to restore conduction. The use of lidocaine has been effective in improving cardiac performance after overdose of membrane-stabilizing drugs,116 and can be administered in cases refractory to sodium bicarbonate.
Several therapeutic considerations should be addressed with regard to the antimuscarinic toxicity resulting from agents such as cyclic antidepressants, antihistamines, and phenothiazines. The antimuscarinic signs and symptoms that usually predominate are seldom life-threatening. Seizures from these agents, in the absence of severe hypotension or hypoxia, are most likely related to blockade of muscarinic receptors in the brain. They are usually self-limited and easily controlled with benzodiazepines or barbiturates. Sinus tachycardia produced by reduced vagal tone does not require specific treatment. Physostigmine, a short-acting carbamate cholinesterase inhibitor, can reverse the CNS toxicity and the sinus tachycardia associated with these agents, but may exacerbate impaired conduction throughout the heart, occasionally resulting in asystole.123 For this reason, physostigmine is best reserved for cases with altered mental status in whom no cardiac conduction delays are present.
Gut decontamination may prevent absorption after ingestions of any of the agents discussed earlier. If benefit is to be derived from gastric decontamination, it should be undertaken soon after ingestion. The longer the delay, the more the drug escapes into the small intestine where most absorption occurs. Gastric decontamination more than 1 hour after ingestion may be of little benefit unless drugs that delay gastric emptying are involved in the poisoning. Large studies have found no effect on outcome when gastric decontamination is avoided altogether.124 Syrup of ipecac is no longer recommended as a method of decontamination. Furthermore, gastric lavage is no longer routinely advocated, with the exception of life-threatening poisonings that present within 1 hour of ingestion.
Activated charcoal has been found to bind most cardiotoxic drugs and may be of benefit in limiting absorption. The timing of activated charcoal administration is also important, but charcoal administration may still be effective in binding a drug that has entered the duodenum. It is therefore rational to administer a dose of activated charcoal to most patients with a history or clinical evidence of ingesting a cardiotoxic substance. The recommended dose of activated charcoal is 1 g/kg, and patients may either drink the aqueous charcoal suspension (which has no taste) or have the dose administered through a nasogastric tube when intubated or uncooperative. Caution should be used when using activated charcoal in obtunded patients or in those who are unable to protect their airway because this poses a risk of aspiration. Multiple doses of activated charcoal have been found to be of benefit in poisonings with agents such as theophylline and phenobarbital, but this therapy is not likely to benefit humans poisoned with any of the cardiotoxic agents listed in this chapter. In addition, those drugs that slow gastrointestinal motility may predispose the patient to charcoal bezoar formation when multiple doses are administered.125
Illicit Drugs
Cocaine
Pharmacology
Cocaine is well absorbed from the mucous membranes of the nose, lung, genitourinary, and gastrointestinal tract. Administration can occur through the intravenous, respiratory, intramuscular, and rectal routes. The method and dose of administration determine the onset of action. The “high” from intravenous administration of cocaine peaks within a few minutes after injection. Inhalation of the drug will produce effects within 1 to 3 minutes, but oral ingestion may delay symptoms up to 60 to 90 minutes. Plasma concentrations after intranasal use peak within 20 to 30 minutes and gradually decline over the next 60 minutes.126
Cocaine is metabolized by nonenzymatic hydrolysis and liver esterases, including plasma cholinesterase. The two major metabolites include benzoylecgonine and ecgonine methyl ester, neither of which crosses the blood-brain barrier. Both of these compounds are water-soluble and are excreted in urine. Cocaine metabolites may be detected in urine up to 72 hours after an exposure, although heavy users may have positive urine screens for up to 3 weeks.127 When a user of cocaine also coingests ethanol, hepatic transesterification will create another pharmacologically active metabolite, cocaethylene. Cocaethylene is not on routine urine screens for cocaine metabolites.
Pathophysiology
The pharmacologic effects of cocaine in humans include the ability to stabilize membranes and block nerve conduction. The resulting effects on myocardial tissue cause blockade of fast sodium channels, leading to widening of the QRS complex with subsequent dysrhythmias. The sympathomimetic effects of cocaine are caused by impaired catecholamine reuptake and enhanced catecholamine release at nerve terminals.126 The increased synaptic concentrations of neurotransmitters stimulate α- and β-receptors throughout the autonomic nervous system resulting in a cascade of clinical effects. Cocaine may also enhance the release of norepinephrine and dopamine in the CNS.128 The unique ability of cocaine to inhibit nerve conduction while enhancing vasoconstriction is primarily responsible for its cardiovascular toxicity.126
Cocaethylene is also a potent sodium channel blocking agent and appears to prolong the recovery time for the channel compared with cocaine.129 In animal models, cocaine plus ethanol depressed myocardial contractility more than either agent given alone.130,131 Once formed, cocaethylene has a longer half-life than cocaine.
The mechanism of cocaine-induced myocardial ischemia is thought to be multifactorial. Cocaine increases myocardial oxygen demand while increasing heart rate and blood pressure. Usually, myocardial oxygen demand results in coronary vasodilation; however, cocaine taken by some routes can induce coronary vasospasm.132 Coronary artery thrombus formation has also been implicated as a cause of cocaine-induced myocardial ischemia. Thrombus formation leading to myocardial infarction has been associated with coronary artery vasospasm.133 The vasospasm may damage the endothelium and cause release of vasoactive substances precipitating platelet aggregation. Cocaine may enhance this effect because in vitro studies have demonstrated that cocaine alone may directly stimulate platelet aggregation and platelet thromboxane production.134 Cocaine activates platelets in whole blood by inducing the release of platelet granule contents and by promoting the binding of fibrinogen to the surface of the platelet.134
Clinical Manifestations
The clinical effects of cocaine result from diffuse hyperadrenergic stimulation both centrally and peripherally. The peripheral sympathomimetic effects include tremor, mydriasis, urinary retention, and ileus. Adrenergic stimulation of the CNS leads to agitation, hallucinations, seizures, and coma.135 Patients may experience psychosis, paranoia, and anxiety due to increased dopaminergic transmission.136 Cerebrovascular complications from cocaine-induced vasospasm and a hyperadrenergic state include cerebral infarctions, transient ischemic attacks, and subarachnoid and intracranial hemorrhages.135,136
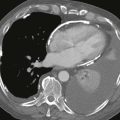
Myocardial ischemia and infarction are well-documented complications of cocaine use. Ischemia of the myocardium does not require a massive exposure to cocaine and occurs commonly in the young adult with no history of cardiac risk factors. Symptoms of chest pain may be typical, atypical, or absent. A delay of several hours in the onset of chest discomfort may occur after exposure to the drug.137–139 Electrocardiograms from patients with cocaine-associated chest pain may demonstrate a variety of abnormalities, including classic findings of myocardial injury such as ST segment elevation but may also be normal or have only nonspecific findings. A study of 42 cocaine users with chest pain and normal or nondiagnostic electrocardiograms documented 8 of these patients as having acute myocardial infarctions, defined by total creatinine kinase and myocardial isoenzyme levels.140 Thus single or even serial electrocardiograms may not be useful in ruling out cocaine-induced ischemia.
Cocaine has been associated with a variety of dysrhythmias. Sinus tachycardia is common owing to the sympathomimetic effects. Atrial fibrillation, premature ventricular contractions, ventricular tachycardia, and ventricular fibrillation have all been described.141 Dysrhythmias may occur with or without underlying ischemia. Since cocaine can cause sodium channel blockade, this can lead to widening of the QRS complex and can precipitate associated dysrhythmias. Sodium channel blockade should be treated with sodium bicarbonate, as previously discussed.
Hypertension is also common with cocaine poisoning. The elevation in blood pressure combined with tachycardia may increase shear forces on the great vessels, resulting in aortic dissection,142 and coronary artery dissection.143 The intestinal vasculature is susceptible to α-stimulating effects of catecholamines, and ischemic colitis has been described in adults144 and in neonates after in utero exposure to cocaine.145 The uteroplacental vasculature may also respond to cocaine exposure with diminished uterine blood flow after maternal cocaine use.146
Extreme hyperthermia is often documented in cocaine overdose. Temperatures are frequently reported in excess of 106° F and are thought to result from a disturbance in thermoregulation due to excess dopaminergic stimulation, combined with excessive musculoskeletal activity and agitation.147,148 Although cocaine-induced hyperthermia may occur independent of seizure activity, it can be exacerbated by concomitant convulsions.149 An acute rise in the central body temperature has also occurred after rupture of bags of cocaine ingested by body packers.150
Rhabdomyolysis also contributes to the morbidity and mortality of cocaine poisoning. All routes of cocaine exposure have been associated with a rise in serum creatinine phosphokinase level from direct myotoxicity.150–152 An association has been made between drug-induced hyperthermia and rhabdomyolysis, but observations suggest that cocaine can also induce rhabdomyolysis independently of hyperthermia.150 Cocaine-induced rhabdomyolysis is associated with myoglobinuric renal failure.152 Cocaine is not known to be directly toxic to the renal tubules; however, the effects of cocaine on renal blood flow may exacerbate the effects of myoglobinuria.
Amphetamine Derivatives
Amphetamine derivatives, such as methamphetamine, are popular agents of abuse available as many different “designer” drugs in the phenylethylamine family (Table 35-4). Substitutions of the phenylethylene ring can result in many compounds with similar effects. The only approved uses of phenylethylamines in the United States at this time are for treatment of narcolepsy, attention deficit disorder, and short-term use for weight loss.
| Chemical Name | Nickname |
|---|---|
| 3,4-Methylenedioxymethamphetamine | MDMA, Adam, Ecstasy, XTC |
| 3,4-Methylenedioxyethamphetamine | MDEA, Eve |
| 3,4-Methylenedioxyamphetamine | MDA, Love Drug |
| 4-Methyl-2,5-dimethoxyamphetamine | DOM/STP, Serenity, Tranquility |
Amphetamines have been recognized for their stimulant properties for centuries and continue to be abused by various routes including intravenous and oral administration. “Ice” is a pure preparation of methamphetamine and is marketed in a solid form, hence its nickname. This preparation is volatile and can be smoked, resulting in rapid absorption and effect. This form of methamphetamine rapidly became one of the leading drugs of abuse in Hawaii and California in the 1980s.153,154 Illicit laboratories are able to produce large quantities of methamphetamines because of easy availability of most reagents.
Pharmacology
The volume of distribution of amphetamines tends to be large, and the half-life ranges from 8 to 30 hours.155 Elimination is primarily through hepatic transformation, but renal excretion results in significant elimination of certain members of the amphetamine family, such as methamphetamine.156 Although acidification of the urine may enhance the excretion of some amphetamine derivatives, it may also exacerbate renal toxicity in the presence of rhabdomyolysis and is therefore not recommended.157
Pathophysiology
The pharmacologic mechanisms of action of amphetamines are diverse but are thought to rely on indirect effects on catecholamine receptors. These compounds act by entering presynaptic neurons and stimulating the release of endogenous catecholamines such as norepinephrine and dopamine. Amphetamines also inhibit the reuptake of catecholamines and their breakdown by the monoamine oxidase enzyme system. These effects may last for hours, whereas those of cocaine may resolve within several minutes.155
Increased catecholamine release results in stimulation of α- and β-receptors, both peripherally and centrally. Dopaminergic and serotonergic receptor stimulation may contribute to the behavioral disturbances and hyperthermic effects that are common with these poisonings.158 The release of dopamine may be responsible for the pleasurable effects reported with these drugs. Although all members of the amphetamine family may produce a generalized hyperadrenergic state, the pattern of effects with these compounds differs with modification of the parent phenylethylamine molecule, resulting in different anorectic, cardiovascular, and hallucinogenic properties.159
Clinical Manifestations
Physical findings in amphetamine poisoning are similar to those seen with other sympathomimetic drugs. The cardiovascular toxicity of amphetamines manifests most commonly as tachycardia and hypertension. Dysrhythmias are a common cause of death and can include ventricular tachycardia and ventricular fibrillation.160 Hypertensive emergencies with intracranial hemorrhages and cerebrovascular accidents may be more common with amphetamine and methamphetamine than cocaine abuse.161 Acute myocardial ischemia, infarction, aortic dissection, and dilated cardiomyopathy are also known to occur in the setting of amphetamine use.162 Diffuse vascular spasm has also been reported with amphetamine poisoning, and may result in death.163,164
CNS toxicity is the most common reason for amphetamine-poisoned patients to present to a hospital. Most victims are agitated, anxious, and can become volatile and violent. Tactile and visual hallucinations may contribute to patient agitation, and psychoses similar to paranoid schizophrenia are frequently observed in these patients. Mydriasis and diaphoresis are common. Seizures often complicate amphetamine poisoning.164
As in acute cocaine intoxication, hyperthermia is well documented in amphetamine poisoning and is associated with increased morbidity and mortality. Hyperthermia may occur independent of seizures and has been associated with rhabdomyolysis, coagulopathy, renal failure, and death.164–166
Management
Decontamination of the patient who ingested sympathomimetics or bags containing these drugs begins with the administration of activated charcoal as described earlier. Whole-bowel irrigation with an iso-osmotic, isoelectric lavage solution (e.g., Go-Lytely) may enhance the removal of bags from the gastrointestinal tract.167 Due to the large volume of distribution of these agents, hemodialysis and hemoperfusion are not effective in their removal; however, hemodialysis may be required if acute renal failure develops as a complication of rhabdomyolysis.
Rapid and effective control of hypertension from sympathomimetic poisoning is imperative. The use of β-blockers to control the hypertension associated with sympathomimetics is controversial because these compounds may potentiate both coronary and peripheral vasoconstriction due to unopposed α-agonist activity.168,169 Case reports have suggested the use of labetalol as an alternative to nonselective β-blocking agents,170 but labetalol is a more potent β than α antagonist. One study of patients given intranasal cocaine while undergoing angiography demonstrated that labetalol reduced the mean arterial pressure but had no effect on coronary artery vasoconstriction.171 Use of direct vasodilators such as nitroglycerin, nitroprusside, or phentolamine is optimal.172,173
Cocaine or amphetamine-related chest pain must be considered to represent active myocardial ischemia. Therapy should initially include the application of oxygen and the reduction of central sympathomimetic effects with the liberal use of benzodiazepines. Nitroglycerin has been demonstrated to be effective in alleviating cocaine-induced vasoconstriction in diseased and nondiseased coronary arteries.173 An antiplatelet drug such as aspirin may be administered because platelets are activated by cocaine. Heparin or thrombolytics may be considered when ischemia is refractory to more conservative management.
Treatment of sympathomimetic-induced dysrhythmias begins with the administration of benzodiazepines to sedate the patient and reduce catecholamine release.174 Wide complex tachydysrhythmias from cocaine have been effectively treated by administering intravenous sodium bicarbonate.175 Lidocaine may be considered for treatment of dysrhythmias secondary to ischemia or refractory to sodium bicarbonate but should be used with caution because it has potentiated cocaine-induced seizures and death in rats.176
Benzodiazepines are the mainstay of treatment for CNS effects of sympathomimetic poisonings. These sedative-hypnotics have been demonstrated to reduce the lethality of both cocaine and amphetamines.174,177 Butyrophenones have also been effective in reducing the dopaminergic-based delirium associated with amphetamine use.178 Butyrophenones must be administered cautiously to patients with either cocaine or amphetamine toxicity because most antipsychotic medications may lower seizure thresholds, alter temperature regulation, and cause acute dystonias.
1. AAPCC: AAPCC annual reports, 2005. Available at http://www.aapcc.org/dnn/NPDSPoisonData/AnnualReports/tabid/125/Default.aspx.Accessed November 29, 2009.
2. Katz A.M. Cardiac ion channels. N Engl J Med. 1993;328:1244-1251.
3. Braunwald E. Mechanism of action of calcium-channel blocking agents. N Engl J Med. 1982;307:1618-1627.
4. McAllister R.G., Hamann S.R., Blouin R.A. Pharmacokinetics of calcium-entry blockers. Am J Cardiol. 1985;55:30B-40B.
5. Ramoska E.A., Spiller H.A., Winter M., et al. A one-year evaluation of calcium channel blocker overdoses: toxicity and treatment. Ann Emerg Med. 1993;22:196-200.
6. Tom P.A., Morrow C.T., Kelen G.D. Delayed hypotension after overdose of sustained release verapamil. J Emerg Med. 1994;12:621-625.
7. Pearigen P.D., Benowitz N.L. Poisoning due to calcium antagonists: experience with verapamil, diltiazem and nifedipine. Drug Saf. 1991;6:408-430.
8. McGovern B., Garan H., Ruskin J.N. Precipitation of cardiac arrest by verapamil in patients with Wolff-Parkinson-White syndrome. Ann Intern Med. 1986;104:791-794.
9. Spurlock B.W., Virani N.A., Henry C.A. Verapamil overdose. West J Med. 1991;154:51-60.
10. Dolan D.L. Intravenous calcium before verapamil to prevent hypotension. Ann Emerg Med. 1991;20:588-589.
11. Hung Y.M., Olsen K.R. Acute amlodipine overdose treated by high dose intravenous calcium in a patient with severe renal insufficiency. Clin Toxicol. 2007;45:301-303.
12. Bailey B. Glucagon in beta-blocker and calcium channel blocker overdoses: a systematic review. J Toxicol Clin Toxicol. 2003;41:595-602.
13 ter Wee P.M., Hovinga T.K.K., Uges D.R.A., et al. 4-Aminopyridine and haemodialysis in the treatment of verapamil intoxication. Hum Toxicol. 1985;4:327-329.
14. Schiffl H., Ziupa J., Schollmeyer P. Clinical features and management of nifedipine overdosage in a patient with renal insufficiency. J Toxicol Clin Toxicol. 1984;22:387-395.
15. Yuan T.H., Kerns W.P., Tomaszewski C.A., et al. Insulin-glucose as adjunctive therapy for severe calcium channel antagonist poisoning. J Toxicol Clin Toxicol. 1999;37:463-474.
16. Kline J.A., Leonova E., Raymond R.M. Beneficial myocardial metabolic effects of insulin during verapamil toxicity in the anesthetized canine. Crit Care Med. 1995;23:1251-1263.
17. Lheureux P.E., Zahir S., Gris M., et al. Bench-to-bedside review: hyperinsulinaemia/euglycaemia therapy in the management of overdose of calcium-channel blockers. Crit Care. 2006;212:1-6.
18. Megarbane B., Karyo S., Baud F. The role of insulin and glucose (hyperinsulinaemia/euglycaemia) therapy in acute calcium channel antagonist and beta-blocker poisoning. Toxicol Rev. 2004;23:215-222.
19. Kline J.A., Raymond R.M., Leonova E.D., et al. Insulin improves heart function and metabolism during non-ischemic cardiogenic shock in awake canines. Cardiovasc Res. 1997;34:289-298.
20. Durward A., Guerguerian A.M., Lefebvre M., et al. Massive diltiazem overdose treated with extracorporeal membrane oxygenation. Pediatr Crit Care Med. 2003;4:372-376.
21. Holzer M., Sterz F., Schoerkhuber W., et al. Successful resuscitation of a verapamil-intoxicated patient with percutaneous cardiopulmonary bypass. Crit Care Med. 1999;27:2818-2823.
22. Love J.N. Beta blocker toxicity after overdose: when do symptoms develop in adults? J Emerg Med. 1994;12:799-802.
23. Henry J.A., Cassidy S.L. Membrane stabilizing activity: a major cause of fatal poisoning. Lancet. 1986;1:1414-1417.
24. Love J.N., Litovitz T.L., Howell J.M., et al. Characterization of fatal beta blocker ingestion: a review of the American Association of Poison Control Centers data from 1985 to 1995. J Toxicol Clin Toxicol. 1997;35:353-359.
25. Hohnloser S.H., Woosley R.L. Sotalol. N Engl J Med. 1994;331:31-38.
26. Kerns W., Kline J., Ford M.D. β-Blocker and calcium channel blocker toxicity. Emerg Med Clin North Am. 1994;12:365-390.
27. Smith R.C., Wilkinson J., Hull R.L. Glucagon for propranolol overdose. JAMA. 1985;254:2412.
28. Love J.N., Hanfling J.M., Howell J.M. Hemodynamic effects of calcium chloride in a canine model of acute propranolol intoxication. Ann Emerg Med. 1996;28:1-6.
29. Donovan K.D., Gerace R.V., Dreyer J.F. Acebutolol-induced ventricular tachycardia reversed with sodium bicarbonate. J Toxicol Clin Toxicol. 1999;37:481-484.
30. Love J.N., Leasure J.A., Mundt D.J. A comparison of combined amrinone and glucagon therapy to glucagon alone for cardiovascular depression associated with propranolol toxicity in a canine model. Am J Emerg Med. 1993;11:360-363.
31. Sato S., Tsuji M.H., Okubo N., et al. Milrinone versus glucagons: comparative hemodynamic effects in canine propranolol poisoning. J Toxicol Clin Toxicol. 1994;32:277-289.
32. Lane A.S., Woodward A.C., Goldman M.R. Massive propranolol overdose poorly responsive to pharmacologic therapy: use of the intra-aortic balloon pump. Ann Emerg Med. 1987;16:1381-1383.
33. Piltz J.R., Wertenbaker C., Lance S.E., et al. Digoxin toxicity: recognizing the varied visual presentations. J Clin Neuroophthalmol. 1993;13:275-280.
34. Cooke D. The use of central nervous system manifestations in the early detection of digitalis toxicity. Heart Lung. 1993;22:477-481.
35. Mauskopf J.A., Wenger T.L. Cost-effectiveness analysis of the use of digoxin immune Fab (ovine) for treatment of digoxin toxicity. Am J Cardiol. 1991;68:1709-1714.
36. Rose S.R., Gorman R.L., McDaniel J. Fatal digoxin poisoning: an unsuccessful resuscitation with use of digoxin-immune Fab. Am J Emerg Med. 1987;5:509-511.
37. Clark R.F., Selden B.S., Curry S.C. Digoxin-specific Fab fragments in the treatment of oleander toxicity in a canine model. Ann Emerg Med. 1991;20:1073-1077.
38. Vaughan-Williams E.M. Classifying antiarrhythmic actions: by facts or speculation. J Clin Pharmacol. 1992;32:964-977.
39. Roden D.M. Antiarrhythmic drugs. In: Hardman J.G., Limbird L.E., editors. The Pharmacologic Basis of Therapeutics. 10th ed. New York: McGraw-Hill; 2001:933-970.
40. Kim S.Y., Benowitz N.L. Poisoning due to Class Ia antiarrhythmic drugs. Drug Saf. 1990;5:393-420.
41. Grace A.A., Camm A.J. Quinidine. N Engl J Med. 1998;338:35-43.
42. Nguyen P.T., Scheinman M.M., Seger J. Polymorphous ventricular tachycardia: characterization, therapy, and the QT interval. Circulation. 1986;74:340-349.
43. Thompson K.A., Murray J.J., Blair I.A., et al. Plasma concentrations of quinidine, its major metabolites and dihydroquinidine in patients with torsades de pointes. Clin Pharmacol Ther. 1988;43:636-642.
44. Bauman J.L., Bauernfeind R.A., Hoff J.V., et al. Torsades de pointes due to quinidine: observations in 31 patients. Am Heart J. 1984;107:425-430.
45. Roden D.M., Woosley R.L., Primm R.K. Incidence and clinical features of the quinidine-associated long QT syndrome: implications for patient care. Am Heart J. 1986;111:1088-1093.
46. Mathis A.S., Gandhi A.J. Serum quinidine concentration and effect on QT dispersion and interval. Ann Pharmacother. 2002;36:1156-1161.
47. Woie L., Oyri A. Quinidine intoxication treated with hemodialysis. Acta Med Scand. 1974;195:237-239.
48. Dyson E.H., Proudfoot A.T., Prescott L.F., et al. Death and blindness due to overdose of quinine. BMJ. 1985;291:31-33.
49. Lewin N.A., Nelson L.S. Goldfrank’s Toxicologic Emergencies, 8th ed. New York: McGraw-Hill; 2006.
50. Murray S.B., Jay J.L. Loss of sight after self poisoning with quinine. BMJ (Clin Res Ed). 1983;287:1700.
51. Phillps R.E., Looareesuwan S., White N.J. Hypoglycemia and antimalarial drugs: quinidine and release of insulin. BMJ. 1986;292:1319-1321.
52. Hemmermeister K.E., Boerth R.C., Warbasse J.R. The comparative inotropic effects of six clinically used antiarrhythmic agents. Am Heart J. 1972;84:643-652.
53. Au P.K., Bhandari A.K., Bream R., et al. Proarrhythmic effects of antiarrhythmic drugs during programmed ventricular stimulation in patients without ventricular tachycardia. J Am Coll Cardiol. 1987;9:389-397.
54. Gay R.J., Brown D.F. Pacemaker failure due to procainamide toxicity. Am J Cardiol. 1974;34:728-732.
55. White S.R., Dy G., Wilson J.M. The case of the slandered Halloween cupcake: survival after massive pediatric procainamide overdose. Pediatr Emerg Care. 2002;18:185-188.
56. Shields A.F., Berenson J.A. Procainamide-associated pancytopenia. Am J Hematol. 1998;27:299-304.
57. Horn J.R., Hughes M.L. Disopyramide dialysability [Letter]. Lancet. 1978;2:214.
58. Nicholson W.J., Martin C.E., Gracey J.G., et al. Disopyramide-induced ventricular fibrillation. Am J Cardiol. 1979;43:1053-1055.
59. Hoffmeister H.M., Hepp A., Seipel L. Negative inotropic effect of class 1 antiarrhythmic drugs: comparison of flecainide with disopyramide and quinidine. Eur Heart J. 1987;8:1126-1132.
60. Hayler A.M., Medd R.K., Holt D.W., et al. Experimental disopyramide poisoning: treatment by cardiovascular support and with charcoal hemoperfusion. J Pharm Exp Ther. 1979;211:491-495.
61. Hiroka M., Kuga K., et al. New observations on the mechanism of antiarrhythmic actions of disopyramide on cardiac membranes. Am J Cardiol. 1989;64:15J-19J.
62. Kupersmith J., Antman E.M., Hoffman B.F. In vivo electrophysiological effects of lidocaine in canine acute myocardial infarction. Circ Res. 1976;36:84-91.
63. Denaro C.P., Benowitz N.L. Poisoning due to class 1b antiarrhythmic drugs. Med Toxicol Adverse Drug Exp. 1989;4:412-428.
64. Colburn W.A. First pass clearance of lidocaine in healthy volunteer and epileptic patients: influence of effective liver volume. J Pharm Sci. 1981;70:969-971.
65. Fruncillo R.J., Gibbons W., Bowman S.M. CNS toxicity after ingestion of topical lidocaine. N Engl J Med. 1982;306:426-427.
66. Lie R.L., Vermeer B.J., Edelbroek P.M. Severe lidocaine toxicity by cutaneous absorption. J Am Acad Dermatol. 1990;23:1026-1028.
67. Cusson J., Rattel S., Matthew C., et al. Age dependent lidocaine disposition in patients with acute myocardial infarction. Clin Pharmacol Ther. 1985;37:381-386.
68. Blumer J., Strong J.M., Atkinson A.J. The convulsant potency of lidocaine and its N-dealkylated metabolites. J Pharmacol Exp Ther. 1973;186:31-36.
69. Edgren B., Tilelli J., et al. Intravenous lidocaine overdosage in a child. J Toxicol Clin Toxicol. 1986;24:51-58.
70. Badui E., Garcia-Rubi D., Estanol B. Inadvertent massive lidocaine overdose causing temporary complete heart block in myocardial infarction. Am Heart J. 1981;102:801-803.
71. Brown D.L., Skiendzielewski J.J. Lidocaine toxicity. Ann Emerg Med. 1980;9:627-629.
72. Finkelstein F., Kreeft J. Massive lidocaine poisoning. N Engl J Med. 1979;301:50.
73. Chaikin P., Adir J. Unusal absorption profile of phenytoin in a massive overdose case. J Clin Pharmacol. 1987;27:70-73.
74. Hardman J.G., Ruddon R.W., et al. Goodman and Gilman’s Pharmacological Basis of Therapeutics, 10th ed. New York: McGraw-Hill; 2001.
75. York R.T., Coleridge S.T. Cardiopulmonary arrest following intravenous phenytoin loading. Am J Emerg Med. 1988;6:255-259.
76. Wyte C.D., Berk W.A. Severe oral phenytoin overdose does not cause cardiovascular morbidity. Ann Emerg Med. 1991;20:508-512.
77. Barnfield C., Kemmenoe A.V. A sudden death due to tocainide overdose. Hum Toxicol. 1986;5:337-340.
78. Soff G.A., Kadin M.E. Tocainide induced reversible agranulocytosis and anemia. Arch Intern Med. 1987;147:598-599.
79. Feinberg L., Bernton H.F., et al. Pulmonary fibrosis associated with tocainide: report of a case with literature review. Am Rev Respir Dis. 1990;141:505-508.
80. Nora M.O., Chandrasekaran K., Hammill S.C., et al. Prolongation of ventricular depolarization: ECG manifestation of mexiletine toxicity. Chest. 1989;95:925-928.
81. Kreeger R.W., Hammill S.C. New antiarrhythmic drugs: tocainide, mexiletine, flecainide, encainide, and amiodarone. Mayo Clin Proc. 1987;62:1033-1050.
82. Pentel P.R., Goldsmith S.R., Salerno D.M., et al. Effects of hypertonic sodium bicarbonate on encainide overdose. Am J Cardiol. 1986;57:878-880.
83. Kodama I., Toyama J., et al. Amiodarone: ionic and cellular mechanisms of action of the most promising class III agent. Am J Cardiol. 1999;84:20R-28R.
84. Kowey P.R., Bharucha D., et al. Pharmacologic and pharmacokinetic profile of class III antiarrhythmic drugs. Am J Cardiol. 1997;80:16G-23G.
85. American Heart Association. 2005 American Heart Association guidelines for cardiopulmonary resuscitation and emergency cardiovascular care. Circulation. 2005;112(Suppl I):IV67-IV77.
86. Kowey P.R., Bharucha D., et al. Pharmacologic and pharmacokinetic profile of class III antiarrhythmic drugs. Am J Cardiol. 1997;80:16G-23G.
87. Loke Y.K., Aronson J.K. A comparison of three different sources of data in assessing the frequencies of adverse reactions to amiodarone. Br J Clin Pharmacol. 2004;57:616-621.
88. Gooddard C.J., Whorwell P.J. Amiodarone overdose and its management. Br J Clin Pharmacol. 1989;43:184-186.
89. Gowda R.M., Sacchi T.J., et al. Ibutilide induced long QT syndrome and torsades de pointes. Am J Ther. 2002;9:527-529.
90. Litovitz T.L., Clark L.R., Soloway R.A. 1993 Annual report of the American Association of Poison Control Centers toxic exposures surveillance system. Am J Emerg Med. 1994;12:546-584.
91. Callaham M., Kassel D. Epidemiology of fatal tricyclic antidepressant ingestion: implications for management. Ann Emerg Med. 1985;14:1-9.
92. Nakashita M., Sakai N., et al. Effects of tricyclic and tetracyclic antidepressants on the three subtypes of GABA transporter. Neurosci Res. 1997;29:87-91.
93. Sala M., De Ferrari G.M., et al. Antidepressants: their effects on cardiac channels, QT prolongation and torsades de pointes. Curr Opin Investig Drugs. 2006;7:256-263.
94. Miur W.W., Strauch S.M., Schaal S.F. Effects of tricyclic antidepressant drugs on the electrophysiologic properties of dog Purkinje fibers. J Cardiovasc Pharmacol. 1982;4:82-90.
95. Ansel G.M., Coyne K., Arnold S., et al. Mechanisms of ventricular dysrhythmia during amitriptyline toxicity. J Cardiovasc Pharmacol. 1993;22:798-803.
96. Baldessarini R.J., et al. Drugs and the treatment of psychiatric disorders. In Depression and anxiety disorders. Hardman J.G., Limbird L.E., editors: The Pharmacologic Basis of Therapeutics, 10th ed., New York: McGraw-Hill, 2001. pp 447-484
97. Frommer D.A., Kulig K.W., Marx J.A., et al. Tricyclic antidepressant overdose. JAMA. 1987;257:521-526.
98. Foulke G.E., Albertson T.E., Walby W.F. Tricyclic antidepressant overdose: emergency department findings as predictors of clinical course. Am J Emerg Med. 1986;4:496-500.
99. Boehnert M.T., Lovejoy F.H. Value of QRS duration versus the serum drug level in predicting seizures and ventricular dysrhythmias after an acute overdose of tricyclic antidepressants. N Engl J Med. 1985;313:474-479.
100. Wolfe T.R., Caravati E.M., Rollins D.E. Terminal 4-ms frontal plane QRS axis as a marker for tricyclic antidepressant overdose. Ann Emerg Med. 1989;18:348-351.
101. Liebelt E.L., Woolf A.D. ECG lead aVR versus QRS interval in predicting seizures and dysrhythmias in acute tricyclic antidepressant toxicity. Ann Emerg Med. 1995;26:195-201.
102. Burda C.D. Electrocardiographic abnormalities induced by thioridazine (Mellaril). Am Heart J. 1968;76:153-156.
103. Ray W.A., Meredith S., Thapa P.B., et al. Antipsychotics and the risk of sudden cardiac death. Arch Gen Psychiatry. 2001;12:1161-1167.
104. Wilt J.L., Minnema A.M., Johnson R.F., et al. Torsades de pointes associated with the use of intravenous haloperidol. Ann Intern Med. 1993;119:391-394.
105. Neto F.R. Effects of cyproheptadine on electrophysiological properties of isolated cardiac muscle of dogs and rabbits. Br J Pharmacol. 1983;80:335-341.
106. Clark R.F., Vance M.V. Massive diphenhydramine poisoning resulting in a wide complex tachycardia: successful treatment with sodium bicarbonate. Ann Emerg Med. 1992;21:318-321.
107. Freedberg R.S., Friedman G.R., Palu R.N., et al. Cardiogenic shock due to antihistamine overdose. JAMA. 1987;257:660-661.
108. Amsterdam E.A., Rendig S.V., Henderson G.L., et al. Depression of myocardial contractility function by propoxyphene and norpropoxyphene. J Cardiovasc Pharmacol. 1981;3:129-138.
109. Stork C.M., Redd J.T., Fine K., et al. Propoxyphene-induced wide QRS complex dysrhythmia responsive to sodium bicarbonate–a case report. J Toxicol Clin Toxicol. 1995;33(2):179-183.
110. Apfelbaum J.D., Caravati E.M., Kerns W.P., et al. Cardiovascular effects of carbamazepine toxicity. Ann Emerg Med. 1995;25:631-635.
111. Seymour J.F. Carbamazepine overdose: features of 33 cases. Drug Saf. 1993;8:81-88.
112. Kelley J.C., Wasserman G.S., Bernard W.D., et al. Chloroquine poisoning in a child. Ann Emerg Med. 1990;19:47-50.
113. Riou B., Barriot P., Rimailho A., et al. Treatment of severe chloroquine poisoning. N Engl J Med. 1988;318:1-6.
114. Smilkstein M.J. Reviewing cyclic antidepressant cardiotoxicity: wheat and chaff. J Emerg Med. 1990;8:645-648.
115. Lovecchio F., Berlin R., Brubacher J.R., et al. Hypertonic sodium bicarbonate in an acute flecainide overdose. Am J Emerg Med. 1998;16:534-537.
116. Winecoff A.P., Hariman R.J., Grawe J.J., et al. Reversal of electrophysiological effects of cocaine by lidocaine: I. Comparison with sodium bicarbonate and quinidine. Pharmacotherapy. 1994;14:698-703.
117. Kerns W.2nd, Garvey L., Owens J. Cocaine-induced wide complex dysrhythmia. J Emerg Med. 1997;15:321-329.
118. Hoffman J.R., Votey S.R., Bayer M., et al. Effect of hypertonic sodium bicarbonate in the treatment of moderate-to-severe cyclic antidepressant overdose. Am J Emerg Med. 1993;11:336-341.
119. Curry S.C., Conner D.A., Clark R.F., et al. The effect of hypertonic sodium bicarbonate on QRS duration in rats poisoned with chloroquine. J Toxicol Clin Toxicol. 1996;34:73-76.
120. Goldman M.J., Mowry J.B., Kirk M.A. Sodium bicarbonate to correct widened QRS in a case of flecainide overdose. J Emerg Med. 1997;15:183-186.
121. Bodenhamer J.E., Smilkstein M.J. Delayed cardiotoxicity following quinine overdose: a case report. J Emerg Med. 1993;11:898-905.
122. Orr D.A., Bramble M.G. Tricyclic antidepressant poisoning and prolonged external cardiac massage during asystole. BMJ. 1981;283:1107-1108.
123. Suchard J.R. Assessing physostigmine’s contraindications in cyclic antidepressant ingestions. J Emerg Med. 2003;25:185-191.
124. Merigian K.S., Woodard M., Hedges J.R., et al. Prospective evaluation of gastric emptying in the self-poisoned patient. Am J Emerg Med. 1990;8:479-483.
125. Ray M.J., Padin R., Condie J.D., et al. Charcoal bezoar small bowel obstruction secondary to amitriptyline overdose therapy. Dig Dis Sci. 1988;33:106-107.
126. Flomenbaum N.E., Howland M.N., Lewin N.A., et al. Cocaine. In Goldfrank’s Toxicologic Emergencies. 8th ed. New York: McGraw-Hill;2006:pp 1133-1146
127. Weiss R.D., Gawin F.H. Protracted elimination of cocaine metabolites in long-term, high-dose cocaine abusers. Am J Med. 1988;85:879-880.
128. Koob G.F., Bloom F.E. Cellular and molecular mechanisms of drug dependence. Science. 1988;242:715-723.
129. Xu Y.Q., Crumb W.J.Jr., Clarkson C.W. Cocaethylene, a metabolite of cocaine and ethanol, is a potent blocker of cardiac sodium channels. J Pharmacol Exp Ther. 1994;271:319-325.
130. Henning R.J., Wilson L.D., Glauser J.M. Cocaine plus ethanol is more cardiotoxic than cocaine or ethanol alone. Crit Care Med. 1994;22:1896-1906.
131. Wilson L.D., Jeromin J., Garvey L., et al. Cocaine, ethanol, and cocaethylene cardiotoxicity in an animal model of cocaine and ethanol abuse. Acad Emerg Med. 2001;8:211-212.
132. Lange R.A., Cigarroa R.G., Yancy C.W., et al. Cocaine-induced coronary-artery vasoconstriction. N Engl J Med. 1989;321:1557-1562.
133. Vincent G.M., Anderson J.L., Marshall H.W. Coronary spasm producing coronary thrombosis and myocardial infarction. N Engl J Med. 1983;309:220-239.
134. Kugelmass A.D., Oda A., Monahan K., et al. Activation of human platelets by cocaine. Circulation. 1993;88:876-883.
135. Mody C.K., Miller B.L., McIntyre H.B., et al. Neurologic complications of cocaine abuse. Neurology. 1988;38:1189-1193.
136. Levine S.R., Burst J.C.M., Futrell N., et al. Cerebrovascular complications of the use of the “crack” form of alkaloidal cocaine. N Engl J Med. 1990;323:699-704.
137. Amin M., Gabelman G., Karpel J., et al. Acute myocardial infarction and chest pain syndromes after cocaine use. Am J Cardiol. 1990;66:1434-1437.
138. Zimmerman J.L., Dellinger R.P., Majid P.A. Cocaine-associated chest pain. Ann Emerg Med. 1991;20:611-615.
139. Gitter M.J., Goldsmith S.R., Dunbar D.N., et al. Cocaine and chest pain: clinical features and outcome of patients hospitalized to rule out myocardial infarction. Ann Intern Med. 1991;115:277-282.
140. Tokarski G.F., Paganussi P., Urbanski R., et al. An evaluation of cocaine induced chest pain. Ann Emerg Med. 1990;19:1088-1092.
141. Isner J.M., Estes N.A.M., Thompson P.D., et al. Acute cardiac events temporally related to cocaine abuse. N Engl J Med. 1986;315:1438-1443.
142. Hsue P.Y., Salinas C.L., Bolger A.F., et al. Acute aortic dissection related to crack cocaine. Circulation. 2002;105:1592-1595.
143. Bizzarri F., Mondillo S., Guerrine F., et al. Spontaneous acute coronary dissection after cocaine abuse in a young woman. Can J Cardiol. 2003;19:297-299.
144. Linder J.D., Monkemuller K.E., Raijman I., et al. Cocaine-associated ischemic colitis. South Med J. 2000;93:909-913.
145. Telsey A.M., Merrit A., Dixon S.D. Cocaine exposure in a term neonate: necrotizing enterocolitis as a complication. Clin Pediatr. 1988;27:547-550.
146. Woods J.R., Plessinger M.A., Clark K.E. Effect of cocaine on uterine blood flow and fetal oxygenation. JAMA. 1987;257:957-961.
147. Merigian K.S., Roberts J.R. Cocaine intoxication: hyperpyrexia, rhabdomyolysis, and acute renal failure. Clin Toxicol. 1987;25:135-148.
148. Bauwens J.E., Boggs J.M., Hartwell P.S. Fatal hyperthermia associated with cocaine use. West J Med. 1989;150:210-212.
149. Bettinger J. Cocaine intoxication: massive oral overdose. Ann Emerg Med. 1980;9:429-430.
150. Roth D., Alarcon F.J., Fernandez J.A., et al. Acute rhabdomyolysis associated with cocaine intoxication. N Engl J Med. 1988;319:673-677.
151. Horowitz B.Z., Panecek E.A., Jouriles N.J. Severe rhabdomyolysis with renal failure after intranasal cocaine use. J Emerg Med. 1997;15:833-837.
152. Curry S.C., Chang D., Connor D. Drug- and toxin-induced rhabdomyolysis. Ann Emerg Med. 1989;18:1068-1084.
153. Miller M.A. Trends and patterns of methamphetamine smoking in Hawaii. NIDA Res Monogr. 1991;115:72-83.
154. Helschober B., Miller M.A. Methamphetamine abuse in California. NIDA Res Monogr. 1991;115:60-71.
155. Chiang W.K., Goldfrank L.R. Amphetamines. In In Goldfrank’s Toxicologic Emergencies, 8th ed, New York: McGraw-Hill; 2006:1118-1132.
156. Baselt R.C., Cravey R.H. Disposition of Toxic Drugs and Chemicals in Man, 7th ed. Foster City: Calif, Biomedical Publications; 2004.
157 Kendrick W.C., Hull A.R., Knochel J.P. Rhabdomyolysis and shock after intravenous amphetamine administration. Ann Intern Med. 1977;86:381-387.
158 Gold L.H.G., Geyer M.A., Koob G.F. Neurochemical mechanisms involved in behavioral effects of amphetamines and related designer drugs. NIDA Res Monogr. 1989;94:101-126.
159. Glennon R.A. Stimulus properties of hallucinogenic phenalkylamines and related designer drugs: formulation of structure-activity relationship. NIDA Res Monogr. 1989;94:43-67.
160. Lucas P.B., Gardner D.L., Wolkowitz O.M., et al. Methylphenidate-induced cardiac dysrhythmias. N Engl J Med. 1986;315:1485.
161. Imanse J., Vanneste J. Intraventricular hemorrhage following amphetamine abuse. Neurology. 1990;40:1318-1319.
162. Hong R., Matsuyama E., Khalid N. Cardiomyopathy associated with the smoking of crystal methamphetamine. JAMA. 1991;265:1152-1154.
163. Bowen J.S., Davis G.B., Kearney T.E., et al. Diffuse vascular spasm associated with 4-bromo-2, 5-dimethoxyamphetamine ingestion. JAMA. 1983;249:1477-1479.
164. Derlet R.W., Rice P., Horowitz B.Z., et al. Amphetamine toxicity: experience with 127 cases. J Emerg Med. 1989;7:157-161.
165. Ginsberg M.D., Hertzman M., Schmidt-Nowara W. Amphetamine intoxication with coagulopathy, hyperthermia, and reversible renal failure: a syndrome representing heatstroke. Ann Intern Med. 1970;73:81-85.
166. Simpson D.L., Rumack B.H. Methylenedioxyamphetamine: clinical description of overdose, death, and review of pharmacology. Arch Intern Med. 1981;141:1507-1509.
167. Hoffman R.S., Smilkstein M.G., Goldfrank L.R. Whole bowel irrigation and the cocaine body-packer: a new approach to the common problem. Am J Emerg Med. 1990;8:523-527.
168. Lange R.A., Cigarroa R.G., Flores E.D., et al. Potentiation of cocaine-induced coronary vasoconstriction by beta-adrenergic blockade. Ann Intern Med. 1990;112:897-903.
169. Ramoska E., Saccetti A.D. Propranolol-induced hypertension in treatment of cocaine intoxication. Ann Emerg Med. 1985;14:1112-1113.
170. Gay G.R., Loper K.A. The use of labetalol in the management of cocaine crisis. Ann Emerg Med. 1988;17:282-283.
171. Boehrer J.D., Moliterno D.J., Willard J.E., et al. Influence of labetalol on cocaine-induced coronary vasoconstriction in humans. Am J Med. 1993;94:608-610.
172. Hollander J.E., Carter W.A., Hoffman R.S. Use of phentolamine for cocaine-induced myocardial ischemia. N Engl J Med. 1992;327:361.
173. Brogan W.C., Lange R.A., Kim A.S., et al. Alleviation of cocaine-induced coronary vasoconstriction by nitroglycerin. J Am Coll Cardiol. 1991;18:581-586.
174. Derlet R.W., Albertson T.E. Diazepam in the prevention of seizures and death in cocaine-intoxicated rats. Ann Emerg Med. 1989;18:542-546.
175. Beckman K.J., Parker R.B., Hariman R.J., et al. Hemodynamic and electrophysiological actions of cocaine: effects of sodium bicarbonate as an antidote in dogs. Circulation. 1991;83:1799-1807.
176. Derlet R.W., Albertson T.E., Tharratt R.S. Lidocaine potentiation of cocaine toxicity. Ann Emerg Med. 1991;20:135-138.
177. Catravas J.D., Waters I.W., Davis W.M., et al. Haloperidol for acute amphetamine poisoning: a study in dogs. JAMA. 1975;231:1340-1341.
178. Catravas J.D., Waters I.W., Hickenbottom J.P., et al. The effects of haloperidol, chlorpromazine, and propranolol on acute amphetamine poisoning in the conscious dog. J Pharmacol Exp Ther. 1977;202:230-243.