CHAPTER 124 Other Diseases of the Colon and Rectum
LYMPHOCYTIC AND COLLAGENOUS COLITIS
BACKGROUND
The term collagenous colitis was used first in 1976 by Lindstrom to describe the findings in a middle-aged woman with chronic diarrhea in whom evaluation was normal except for colonic biopsies that showed a thickened band of subepithelial collagen and increased lymphocytes in the lamina propria.1 Histologically, the subepithelial collagen deposits resembled those in the small intestine of patients with collagenous sprue. The term microscopic colitis was used first in 1980 by Read and associates, who detailed a group of patients with chronic idiopathic diarrhea, a subset of which had a normal-appearing colon at colonoscopy but abnormal histopathology on biopsy.2 Subsequent review showed that most of these patients had collagenous colitis, but some had increased lymphocytes in the lamina propria in the absence of a thickened collagen band. The term lymphocytic colitis was proposed in 1989 by Lazenby and associates3 as a more-specific histopathologic diagnosis to distinguish this entity from patterns of microscopic colitis in which other cellular elements such as eosinophils, mast cells, or neutrophils predominate.
EPIDEMIOLOGY
Collagenous and lymphocytic colitis occur most commonly between ages 50 and 70 years. Both have a strong female predominance and frequent association with arthritis, celiac disease, and autoimmune disorders. In a large population-based study in Spain, the demographic features of collagenous and lymphocytic colitis were similar: The disorders were found in 9.5 of every 100 patients with chronic watery diarrhea and normal-appearing mucosa on colonoscopy, of whom 61% had lymphocytic colitis; the incidence rates in the general population of lymphocytic colitis and collagenous colitis were 3.1/100,000 and 1.1/100,000, respectively.4 This latter observation contrasts strikingly with published reports of more than 400 cases of collagenous colitis compared with more than 60 cases of lymphocytic colitis, a finding that suggests there may be a publication bias to explain the discrepancy. The overall mean annual incidence of both colitides was 4.2 per 100,000 inhabitants in Spain, similar to the rates from an epidemiologic study conducted in Sweden5 but lower than the 8.6 cases for 100,000 person years in the United States.6 Although the incidence of these microscopic colitides clearly is higher in older age groups, both entities have been reported in children and teenagers, in whom the clinical presentation is similar to that of adults.
PATHOLOGY
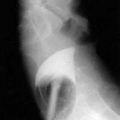
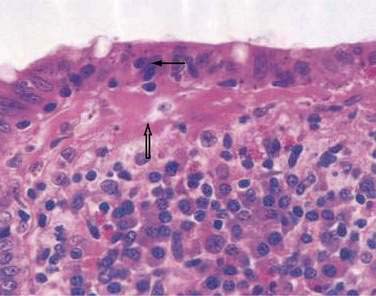
In both collagenous and lymphocytic colitis, there is a modest increase of mononuclear cells within the lamina propria and between crypt epithelial cells, consisting mainly of CD8+ T lymphocytes, plasma cells, and macrophages.3 There may be flattening of the surface epithelial cells, a mild decrease in the number of goblet cells, hyperplasia of Paneth cells, and an increased number of intraepithelial lymphocytes (Fig. 124-1). Neutrophils are not prominent, and cryptitis and crypt distortion are unusual.
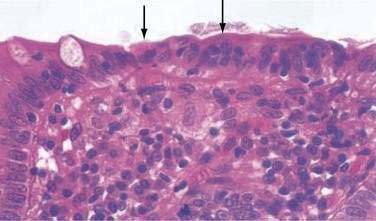
In collagenous colitis, there is a thickened subepithelial collagen layer, which may be continuous or patchy (Fig. 124-2). In normal colon, the width of this collagen band is less than 4 to 5 µm and consists predominantly of type IV collagen, whereas in collagenous colitis, it is greater than 10 µm, averages 20 to 60 µm,7 and is composed of type VI collagen and tenascin as well as lesser amounts of types I and III collagen. Tenascin is a glycoprotein that is a marker of matrix remodeling and is a product of intestinal subepithelial myofibroblasts.8 These changes are absent in lymphocytic colitis and suggest that the two main forms of microscopic colitis should be considered as separate disease entities.
Although inflammatory changes occur diffusely throughout the colon in collagenous colitis, the characteristic collagen band thickening is highly variable, occurring in the cecum and transverse colon in more than 80% of cases and in the rectum in less than 30% of cases. Although involvement of the left colon appears to be less intense than involvement of the right colon, multiple biopsies of the left colon taken above the rectosigmoid during flexible sigmoidoscopy are sufficient to make the diagnosis in approximately 90% of cases.7 The diagnosis of collagenous colitis requires both mucosal inflammation and a thickened collagen band; artifact resulting from poor orientation can give the mistaken appearance of a thickened basement membrane. It has been suggested that tenascin immunohistochemistry be used as a routine test in the diagnosis of microscopic colitis.8
ETIOLOGY AND PATHOGENESIS
The cause(s) of collagenous and lymphocytic colitis is (are) unknown. The most widely held hypothesis is that they are inflammatory disorders arising from epithelial immune responses to intraluminal dietary or bacterial contents. This hypothesis is supported by the regression of inflammation following diversion of the fecal stream and recurrence of inflammation following restoration of intestinal continuity in three patients.9 The identity of the inciting antigenic factors is uncertain, although medications,10,11 bile salts, toxins, and infectious agents12 have been postulated.
The strong association of rheumatologic diseases with microscopic colitis has raised the possibility that nonsteroidal anti-inflammatory drugs (NSAIDs) might play an etiologic role. One well-controlled study13 found that chronic NSAID use occurred more frequently in patients with collagenous colitis than in age- and gender-matched controls (61% vs. 13%; P < 0.02), a finding confirmed by a case-control study.10 One postulated mechanism by which NSAIDs might damage the colon is by increasing colonic permeability to allow luminal antigens to enter the lamina propria and promote inflammation. Other medications implicated include selective serotonin reuptake inhibitors (SSRIs), specifically sertraline, for collagenous colitis; and SSRIs, beta-blockers, statins, and bisphosphonates for lymphocytic colitis.10 Because many patients with collagenous or lymphocytic colitis have not used these medications, and because use of these drugs in older adults is common but these disorders are uncommon, other causes have been invoked, including genetic susceptibility. Genetic susceptibility is supported by the finding that 12% of patients with microscopic colitis have a family history of inflammatory bowel disease (IBD).11
Approximately 20% to 30% of patients with celiac disease have been reported to have lymphocytic colitis, raising the possibility of similar pathogenetic mechanisms.14 In one study, 40% of patients with collagenous colitis had small intestinal biopsies that were compatible with celiac disease,15 although in another study, the frequency of celiac disease was only 2% of 45 patients with collagenous colitis16 and 9% of 199 patients with lymphocytic colitis.11 Furthermore, patients with microscopic colitis do not respond to a gluten-free diet, and neither collagenous nor lymphocytic colitis is associated with human leukocyte antigens (HLAs) B8 and DR3, as is celiac disease. Finally, CD8+ T intraepithelial cells are predominant in both collagenous and lymphocytic colitis, in contrast to celiac disease, in which CD3 and CD8 predominate.
Because autoimmune disorders such as arthritis and thyroid abnormalities have been described in patients with collagenous and lymphocytic colitis,11 there have been continued efforts to associate microscopic colitis with various autoimmune HLA haplotypes and serum markers. One small study showed that HLA-A1 antigens were expressed with increased frequency in lymphocytic but not collagenous colitis,17 and another study showed similar abnormal expressions of HLA-DR antigens by mucosal epithelial cells in both conditions.18 Whether such abnormalities are the cause or the result of these disorders is unknown. Another study found similarities in HLA-DQ loci between patients with celiac disease and patients with either collagenous or lymphocytic colitis.19 Although gluten is not the inciting antigen in microscopic colitis, similar immune mechanisms may be involved in celiac disease and microscopic colitis.
The pathogenesis of the increased collagen band in collagenous colitis is unclear. Initially, it had been assumed that collagen synthesis is increased,1 but colonic biopsies from patients with the disease showed decreased levels of interstitial collagenase, suggesting that reduced matrix degradation might contribute to the accumulation of matrix proteins.20
The mechanism of diarrhea in microscopic colitis is related to the severity of inflammation and not the extent or thickness of the collagen band. Perfusion studies have demonstrated defective active and passive absorption of sodium and chloride and reduced chloride-bicarbonate exchange in the colon21; two of six subjects had coexisting abnormalities of small intestinal fluid and electrolyte absorption. Other investigations have correlated colonic fluid absorption with the severity of inflammation.22 A potential role for soluble mediators is suggested by a report that diarrhea was resolved by a histamine H1 antagonist in a patient with microscopic colitis characterized by increased numbers of mast cells.23 It has been suggested that bile acid malabsorption might contribute to diarrhea in patients with collagenous colitis and that treatment with a bile acid-binding resin such as cholestyramine might lead to a reduction in diarrhea. Bile acids are unlikely to cause the histologic changes observed in collagenous colitis, however, and a reduction in diarrhea with cholestyramine was not associated with a decrease in colitis. Successful treatment of collagenous colitis with budesonide, however, was associated with increased bile acid absorption and normalization of the 75SeHCAT (selenium-75–labeled homocholyltaurine) test for bile acid malabsorption.24
CLINICAL AND LABORATORY FEATURES
Patients with collagenous and lymphocytic colitis usually present with chronic watery diarrhea, with an average of eight stools each day ranging in volume from 300 to 1700 g per 24 hours21 and associated with occasional fecal incontinence and abdominal cramps. Symptoms decrease with fasting.5 Nausea, weight loss, and fecal urgency are variably present. Diarrhea generally is long-standing, lasting from months to years, with a fluctuating course of remissions and exacerbations. In one series of 172 patients, the median time from the onset of symptoms to diagnosis was 11 months,16 whereas in another smaller series, the median time to diagnosis was 5.4 years.25 Physical examination usually is unremarkable, and blood is not detected in the stool. Routine laboratory studies also are normal.
Examination of fresh stools showed fecal leukocytes in 55% of 116 patients with collagenous colitis.16 Mild steatorrhea, mild anemia, low serum vitamin B12 levels, and hypoalbuminemia have been reported in varying numbers of patients and are not characteristic. Autoimmune markers that have been identified in patients with collagenous colitis include antinuclear antibodies (in up to 50%), perinuclear antineutrophil cytoplasmic antibodies (pANCAs) (in 14%), rheumatoid factor, and increased C3 and C4 complement levels,22 but none of these markers is of diagnostic value.
Colonoscopic examination usually is normal. Nonspecific abnormalities including patchy edema, erythema, friability, and an abnormal vascular pattern were reported in one study,26 whereas mucosal lacerations in the ascending and transverse colons have been reported in a few patients with collagenous colitis.27
TREATMENT
There have been few controlled trials of treatment for either collagenous or lymphocytic colitis, and therapy is largely empiric. Evaluation of therapy is difficult, because both disorders usually exhibit a relapsing and remitting course over many years. No single agent works in all cases.28
About one third of patients respond to antidiarrheal agents, such as loperamide or diphenoxylate with atropine, as well as bulking agents such as psyllium or methylcellulose; clinical response is not associated with improvement of inflammation or collagen thickness. In an open-label trial of bismuth subsalicylate (eight chewable tablets per day for eight weeks) in 12 patients, diarrhea resolved and stool weight was reduced within two weeks; in nine patients colitis resolved with disappearance of the collagen band thickening.29 Over a seven- to 28-month follow-up, nine patients remained well, two were well but required retreatment, and one had persistent diarrhea. Both collagenous and lymphocytic colitis responded similarly, and there were no side effects of treatment; a subsequent controlled trial by the same investigators published only in abstract form confirmed these findings. Although the basis for its efficacy is unknown, bismuth subsalicylate possesses antidiarrheal, antibacterial, and anti-inflammatory properties; bismuth enemas have been reported to be effective in ulcerative colitis and chronic pouchitis.30
Other treatment trials for collagenous and lymphocytic colitis have studied 5-aminosalicylate (mesalamine) compounds, glucocorticoids, and bile acid resins, alone or in combination; these agents appear to improve diarrhea and inflammation in some, but certainly not all, patients.28 Although glucocorticoids given by either the oral or the rectal route provide symptomatic improvement and decrease inflammation in more than 80% of cases, relapse usually occurs quickly after the drug is stopped.5,31 Moreover, long-term use of glucocorticoids has undesirable effects, especially in older patients. Other immunosuppressants, such as azathioprine and 6-mercaptopurine, have been reported to be effective, but there are no sizable studies using these agents.29,32
Budesonide has been reported to be highly effective over a six- to eight-week period in three placebo-controlled trials in patients with collagenous colitis.33–35 Budesonide is a topically acting synthetic corticosteroid with both a high receptor-binding affinity in the mucosa and a high first-pass effect in the liver. In view of its proven efficacy, budesonide should be considered over 5-aminosalicylates or bile acid resins in patients who do not respond to antidiarrheal agents and bismuth subsalicylate.36,37
The only report of surgery for collagenous colitis involved nine patients who underwent ileostomy for disabling refractory collagenous colitis, after which all had symptomatic and histologic remission.28 In patients in whom intestinal continuity was restored, the disease recurred, and of three patients who underwent proctocolectomy with ileal pouch-anal anastomosis, problematic diarrhea occurred. Ileostomy should be considered only as a last resort, but it appears to be effective in patients with disabling and refractory symptoms.
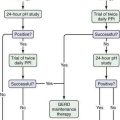
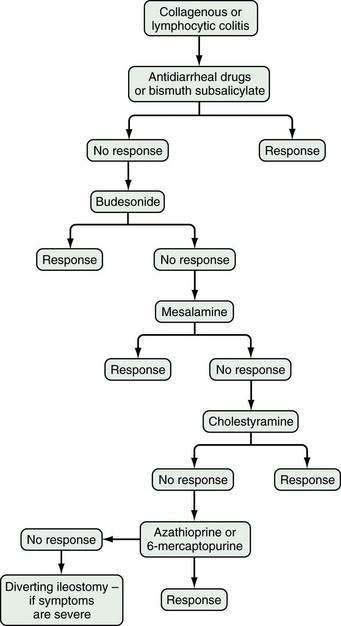
Based on available data, the treatment algorithm shown in Figure 124-3 is proposed.
DIVERSION COLITIS
BACKGROUND AND EPIDEMIOLOGY
Diversion colitis is an inflammatory process that occurs in the diverted segment of colon and rectum after surgical diversion of the fecal stream. The entity was first reported in 1981 by Glotzer and colleagues in 10 patients who had undergone ileostomy or colostomy for various indications other than IBD.38 Since then, diversion colitis has been found in patients who have undergone surgical diversion for many indications, although it has been reported to occur more commonly in patients with IBD (87%) than in those with noninflammatory conditions (28%).39,40 The prevalence of diversion colitis has been underestimated because many patients are asymptomatic, even though histologic changes are likely to occur in diverted segments of the colon within months of surgical diversion.
PATHOLOGY
A spectrum of histologic changes has been described in diversion colitis, ranging from lymphoid follicular hyperplasia and mixed mononuclear and neutrophilic infiltration to severe inflammation with crypt abscesses, mucin granulomas, and Paneth cell metaplasia41,42; however, large ulcers and transmural changes are absent and crypt architecture generally is preserved. Endoscopic findings include erythema, friability, nodularity, edema, aphthous ulcerations, exudates, and frank bleeding, as in idiopathic IBD. After extended periods following diversion, inflammatory pseudopolyps and strictures can develop.
PATHOGENESIS
Diversion colitis appears to be caused largely by luminal nutrient deficiency of the colonic epithelium. The principal nutrient substrates of the colonocytes are luminal short-chain fatty acids (SCFAs), which are metabolic products of carbohydrate and peptide fermentation by anaerobic bacteria.43,44 Roediger demonstrated that SCFAs are the major and preferred energy source for colonic epithelium and that the distal colon is more dependent on SCFAs for its metabolic needs than is the proximal colon.45 Butyrate supplies the bulk of oxidative energy to the distal colon, and acetate, glutamine, and ketones provide alternative sources of energy. Harig and associates demonstrated that the excluded segments of colon contain negligible amounts of SCFAs and that infusion of glucose results in no appreciable anaerobic fermentation.46 The number of obligate anaerobes is reduced in the excluded colon, consistent with reduced production of SCFAs.47 Further support for the SCFA nutrient deficiency hypothesis is from the report that instillation of enemas containing SCFAs resulted in disappearance of endoscopic changes within four to six weeks in four patients with diversion colitis, although resolution of histologic abnormalities was slower and incomplete.46
Although SCFA deficiency has been widely accepted as the cause of diversion colitis, other observations suggest that this might not be the entire etiologic explanation. First, studies in children indicate that SCFA enemas are not universally successful in treating diversion colitis.48 Second, in germ-free rodents with surgical diversion and in patients receiving long-term parenteral nutrition or elimination diets (circumstances in which luminal SCFA concentrations are low), mucosal atrophy occurs rather than inflammation.49 Third, inflammation does not occur in urinary colon conduits from which the fecal stream has been diverted, and urine does not contain measurable SCFAs.50 Finally, in a prospective, randomized, double-blind study of 13 patients with diversion colitis, butyrate enemas given for 14 days provided no improvement in either endoscopic or histologic parameters.51 In a subsequent study by the same group, administration of SCFAs did not affect the bacterial population in the excluded colon.52 Other luminal elements besides SCFA deficiency are likely to play a role, but the nature of such factors is unknown.
DIAGNOSIS
The diagnosis of diversion colitis is based on the clinical picture, endoscopic findings, and histology. Diagnosis is relatively straightforward in a patient without preexisting IBD, and stool specimens for C. difficile toxin,53 ova and parasites, and cultures usually are adequate to exclude other etiologies.
In patients with a preoperative diagnosis of Crohn’s disease, diversion colitis must be distinguished from recurrent IBD. Colonoscopic findings such as linear ulcers and possibly strictures are said to favor Crohn’s disease, as do transmural inflammation, marked crypt architectural abnormalities, and epithelioid granulomas.39 Lymphoid hyperplasia occurs in both disorders but tends to be more prominent in diversion colitis.54 If rectal involvement with Crohn’s disease is absent before diversion, rectal inflammation is more likely to be caused by diversion than Crohn’s disease.2,39,55
TREATMENT
The preferred treatment of diversion colitis is surgical restoration of colonic continuity, which rapidly reverses symptoms and histologic changes. If symptoms are moderate to severe and reanastomosis is not feasible, SCFA enemas in a volume of 60 mL and containing a mixture of 60 mmol/L of acetate, 30 mmol/L of propionate, and 40 mmol/L of butyrate with 22 mmol/L of sodium chloride per liter are administered through the anus or mucous fistula twice daily for four weeks and then decreased to once or twice weekly.46 Such preparations are not commercially available and must be formulated by compounding pharmacies, making it the most expensive of the nonsurgical options.56 There are anecdotal reports that 5-aminosalicylate and hydrocortisone retention enemas are effective as well.57 Because they are available commercially, these agents are considered first-line therapies for most patients. One report suggested that intraluminal irrigation with soluble and insoluble fiber solutions improved endoscopic and histologic abnormalities and might be useful to reduce inflammation before surgical restoration of bowel continuity.58
NONSPECIFIC COLONIC ULCERS
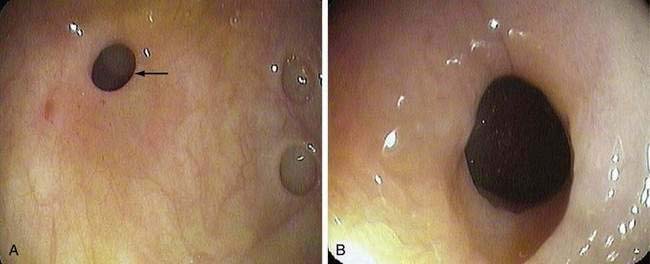
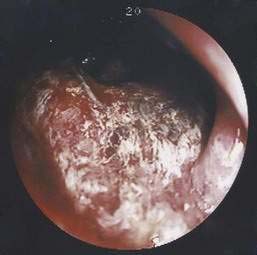
Benign nonspecific ulcers of the colon are uncommon. The most recent large review of the literature encompassed 127 patients and indicated that such nonspecific ulcers occur at any age, with a peak incidence in the 4th and 5th decades and a slight female predominance.59 Most of these ulcers occur in the proximal colon, virtually all are solitary and located on the antimesenteric side of the colon, and most are round and sharply demarcated from relatively normal surrounding mucosa (Fig. 124-4).59,60 Histologically, there is nonspecific acute and chronic inflammation.59
PATHOGENESIS
The causes of nonspecific colonic ulcers are unknown. Potential etiologies that have been suggested, albeit with little or no supporting evidence, include ischemia and cecal diverticulosis. Correlations with the use of drugs such as glucocorticoids, NSAIDs,61,62 oral contraceptives, and oxyphenbutazone have been suggested, but cause has not been established and these drugs have not been implicated in most of the ulcer cases reported. It is likely that no single causative agent explains all cases. There have been reports of associations of nonspecific colon ulcers with chronic renal failure and renal transplantation,63 Churg-Strauss syndrome,64 Wegener’s granulomatosis,65 Behçet’s disease, essential mixed cryoglobulinemia,66 and systemic lupus erythematosus. Perhaps a mechanism common to all exists, but none has yet been identified.
CLINICAL FEATURES
The most common presenting symptoms are abdominal pain and bleeding. More than half of patients with nonspecific colon ulcers present with acute or chronic abdominal pain, often in the right lower abdomen and mimicking appendicitis.59 One third have lower gastrointestinal bleeding with hematochezia, and 16% present with an abdominal mass, most often when the ulcer is located in the left or sigmoid colon. A cecal ulcer should be suspected in a patient with gastrointestinal bleeding when the clinical picture is otherwise consistent with appendicitis or in a patient with symptoms suggesting pelvic inflammatory disease, ovarian disease, or Crohn’s disease in the absence of these diseases.
DIAGNOSIS
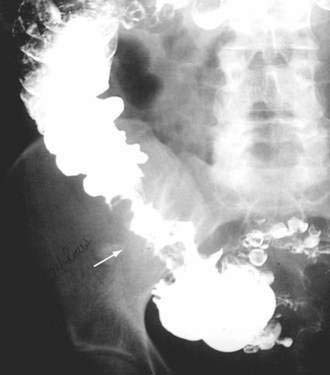
Colonoscopy currently is the diagnostic test of choice.54 Flexible sigmoidoscopy is inadequate because most colonic ulcers are beyond the reach of the instrument. Abnormalities have been described in up to 75% of air-contrast barium enemas59 and include mucosal irregularities, intraluminal filling defects or narrowing, a mass effect, or localized colonic spasm (Fig. 124-5). Roentgen findings are nonspecific, however, and are diagnostically inferior to direct inspection by colonoscopy. Computed tomographic (CT) scans are most helpful in the presence of perforation or associated abscess formation.
TREATMENT
Surgery is recommended for patients with ulcers complicated by perforation or by significant gastrointestinal bleeding and for those with persistent symptoms and failure of the ulcer to heal. In uncomplicated cases, however, an expectant approach has been advocated, with colonoscopy every six weeks to monitor healing. The most common surgical procedures are local excision of the ulcer, oversewing of the ulcer if there is significant bleeding, and occasionally, segmental colectomy.54,67
NSAID-INDUCED COLONIC LESIONS
In contrast to nonspecific colonic ulcers, lesions that are now considered to be pathognomonic for NSAID-induced colitis are known as diaphragm disease68 of the colon. Although it is well known that small bowel strictures are associated with use of NSAIDs, only a small number of cases have been reported since their first published description in 1989.69 The presence of diaphragms with ulcers distinguish these lesions from other nonspecific ulcers.
It is believed that the diaphragm-like strictures are due to scarring following an ulcerating injury. Strictures are concentric, often with a pinhole-size lumen. Lesions in the colon are similar to those found in the small intestine (see Chapter 115). The mucosa between diaphragms (if multiple) is normal. Strictures are characterized by submucosal fibrosis with normal overlying epithelium.
PATHOGENESIS
The pathogenesis of these uncommon lesions is unknown but is thought to involve direct contact of the NSAID with colonic mucosa. The vast majority of cases involve oral intake of NSAIDs, and most of the patients in one study were known to have taken extended-release formulations.68 Half had used diclofenac, and most patients had lesions in the proximal colon. The injury is presumed to involve a high local concentration of active NSAID, which increases intestinal permeability, a prerequisite for NSAID-induced enterocolopathy. This is in contrast to gastroduodenal injury, which is thought to be mediated more often by systemic effects. Postulated mechanisms of injury include prostaglandin inhibition alterations in mucosal blood flow and increased permeability to injurious luminal substances. The risk of lower gastrointestinal toxicity is reduced by the use of selective COX-2 inhibitors.70
CLINICAL FEATURES
The most common clinical presentations include occult blood in the stool, iron-deficiency anemia or frank bleeding, abdominal pain, or change in bowel habits. Other findings can include intermittent bowel obstruction, diarrhea, or colonic perforation with an acute abdomen. Many patients have an underlying rheumatologic disorder and have been using NSAIDs chronically, although lesions have been reported after using NSAIDs for only a few days.71
DIAGNOSIS
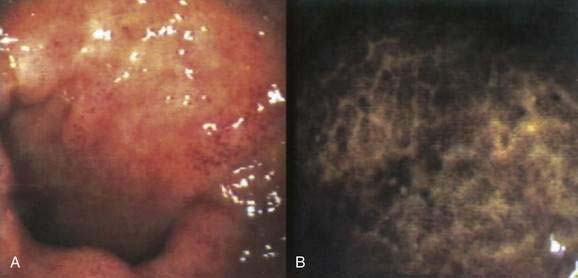
As with nonspecific colonic ulcers, colonoscopy is currently the diagnostic test of choice; flexible sigmoidoscopy is inadequate because most diaphragm disease colon ulcers are proximal to the descending colon (Fig. 124-6). Whereas the differential diagnosis of nonspecific colonic ulcers and NSAID-associated colitis and erosions is broad, the presence of characteristic diaphragmatic webs with normal intervening mucosa in a setting of NSAID use is virtually pathognomonic.
TREATMENT
For nonstricturing NSAID lesions, discontinuing the NSAID often is curative and is essential to management; obstructive symptoms associated with strictures, however, require more-aggressive management. For strictures that are easily accessible, endoscopic dilation with a through-the-scope (TTS) balloon has been reported to be safe and effective.72 If endoscopic dilation is not possible, the area should be marked endoscopically to guide the surgeon, because there may be no serosal abnormalities or palpable areas of transition to guide resection. Surgery also is indicated for significant bleeding or perforation and when carcinoma cannot be excluded with confidence.
DIEULAFOY-TYPE COLONIC ULCERATION
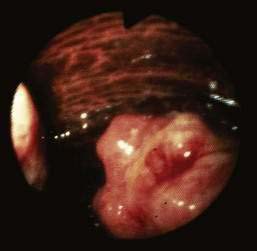
In 1897, Georges Dieulafoy described massive gastrointestinal bleeding emanating from a relatively enlarged (“persistent calibre”) submucosal artery by way of a minute mucosal ulcer at the most superficial point of the vessel.73 Although originally described in the stomach and most commonly occurring in the gastric fundus, identical lesions have been described in other gastrointestinal organs, including the colon and rectum.74–76 In the colon, Dieulafoy-type lesions appear to have a strong male predominance and have been reported in all age groups.
The clinical picture is one of acute and massive bleeding. Colonoscopy can identify the lesion in some cases, but in most cases identification is often difficult or impossible, especially when bleeding continues or thorough cleansing of the colon cannot be accomplished. Selective mesenteric angiography is the diagnostic study of choice, and surgical resection has been the principal form of therapy. Even after angiographic detection of the bleeding site, precise localization of the lesion is usually difficult, and extended resection often is required. In some cases, colonic lesions appear as pseudopolyps,74 and successful treatment with sclerotherapy, electrocautery, or endoscopic hemoclipping might obviate the need for surgery.76,77
CATHARTIC COLON
Cathartic colon is an uncommon and severe sequela of chronic irritant laxative abuse. In 1943, Heilbrun first described radiologic abnormalities of the colon and terminal ileum associated with prolonged abuse of irritant cathartics.78 Fewer than 50 cases have been reported in the literature, all in women with a duration of laxative abuse ranging from 10 to 70 years. It is important to emphasize that the term cathartic colon is based on barium enema characteristics and is not synonymous with prolonged use of laxatives or with laxative abuse. Indeed, misuse of the term cathartic colon has led to inappropriate concerns over the chronic use of laxatives which, when appropriate, is not associated with structural or functional damage to the colon. Cathartic colon is not the inevitable consequence of chronic laxative abuse, which may be associated with a variety of reversible symptoms as well as fluid and electrolyte abnormalities. In a review of 240 cases of chronic laxative abuse published in more than 70 reports, no case of cathartic colon was demonstrated.79,80
RADIOLOGIC AND PATHOLOGIC FEATURES
Heilbrun originally described the following characteristics in his original case report: loss of haustrations, pseudostrictures, dilated colon and terminal ileum, and gaping of the ileocecal valve78; similarity to the radiologic appearance of chronic ulcerative colitis was noted in this and subsequent studies. Characteristic changes are not always found throughout the colon, and there is a predilection for involvement of the ascending colon. Pathologic changes in resected specimens of cathartic colon have included mucosal atrophy, chronic inflammation with thickening of the muscularis mucosae, submucosal fatty infiltration, and mild fibrosis. Irreversible strictures and degenerative changes in intestinal neurons are absent. Neuronal changes have been found in patients with chronic laxative abuse, but these patients did not exhibit cathartic colon as defined here.81
CHANGING ROLE OF LAXATIVES IN COLON DAMAGE
The original suggestion that irritant laxatives, predominantly anthraquinones, damage the colon was based on studies in laboratory animals and in colons resected from laxative abusers.82 Although mucosal atrophy and abnormalities of the enteric nervous system were described, the identities of the laxatives were not documented, nor was there information concerning preexisting conditions that might have prompted chronic use of laxatives.
Subsequent studies have reported changes in colonic epithelial cells and the submucosa in patients with long-term laxative abuse, and both anthraquinones and bisacodyl have been implicated.81 The unclear nature and duration of laxative use and the inability to exclude preexisting conditions, however, make the significance of these observations uncertain. Studies in rodents and in chronically constipated women do not support the deleterious effect of anthraquinones on the ultrastructure of colonic nerves,83,84 nor is there evidence to suggest that sennosides, bisacodyl, or related substances cause significant morphologic damage to the colonic enteric nervous system in either experimental animals or humans. Perhaps one or more laxatives that are no longer in use, such as podophyllin, might have accounted for cases of cathartic colon, because no case of cathartic colon has been reported in persons who began to use or abuse irritant laxatives after 1960.85
PSEUDOMELANOSIS COLI
Melanosis coli is a brownish discoloration of the colon mucosa caused by the accumulation of pigment in macrophages within the lamina propria (Fig. 124-7). First described in the early 19th century, the term melanosis coli was coined by Virchow in 1857, because the pigment was thought to be melanin or a melanin-like substance. Subsequently, the pigment proved to be lipofuscin, both histochemically and ultrastructurally.86,87 The term pseudomelanosis coli, though more accurate than melanosis coli, has not been adopted widely.
The association between pseudomelanosis coli and chronic use of anthraquinone laxatives is established firmly and is supported further by the development of characteristic pigmentation in laboratory animals after administration of anthraquinones.88 Pseudomelanosis develops in more than 70% of persons who use anthraquinone laxatives (cascara sagrada, aloe, senna, rhubarb, and frangula), often within four months of use, with an average of nine months. The condition is widely regarded as benign and reversible, and the pigment generally disappears within one year of stopping laxatives.89 Pseudomelanosis coli probably can result from other factors or exposure to other laxatives, however, and so its presence is not specific for anthraquinone use.
The pigment in pseudomelanosis coli is thought to originate from either macrophages or organelles within epithelial cells after cell damage and apoptosis; such a sequence of damage has been demonstrated in guinea pigs exposed to anthraquinones.90 Histologically, the number and size of macrophages within the lamina propria are increased, and the greatest amount of pigment is found in macrophages farthest from the lumen. Abnormalities of colonic epithelial cells are noted on electron microscopy but not on light microscopy.91
Concern about a possible relationship between pseudomelanosis coli and the development of colonic neoplasia has not been substantiated in a prospective case-control study.92 Other confounding factors such as chronic constipation or dietary intake might account for the increased risk of colon cancer suggested by earlier studies.93,94 Colonic neoplasms lack pigment-containing macrophages and therefore are identified more easily in patients with pseudomelanosis coli (Fig. 124-8).95 Biopsies should be taken of any nonpigmented area of the colon in a patient with pseudomelanosis coli who undergoes colonoscopy.
CHEMICAL COLITIS
ETIOLOGY AND PATHOGENESIS
Damage to the colon has been reported after exposure to a number of rectally administered agents (Table 124-1), the better known of which are soaps and detergents used as “cleansing” enemas.96,97 Other offending substances include hydrogen peroxide,98 water-soluble contrast agents such as sodium diatrizoate (Hypaque, Gastrografin),99 vinegar, potassium permanganate, herbal medications,100 glutaraldehyde,101,102 formalin,103 and alcohol.104 Milder damage to the mucosa occurs after use of monobasic or dibasic sodium phosphate enemas,105 bisacodyl suppositories, and oral henna.106 Colonic damage presumably occurs from a detergent, hypertonic, or direct toxic effect on the mucosa. The severity of the reaction depends on the type and concentration of the substance, the duration and extent of its contact with the mucosa, and perhaps the presence of underlying colonic disease.107
Table 124-1 Chemical Agents That Cause Colitis When Given as Enemas
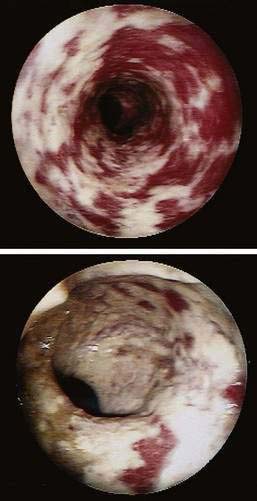
Hydrogen peroxide enemas no longer are commonly used, but at one time they were employed to relieve meconium ileus and to remove fecal impactions. There are reports of severe damage associated with use of hydrogen peroxide, including severe colitis, pneumatosis coli, perforation, sepsis, and death.98 Within minutes of contact, diffuse mucosal emphysema occurs, and after about an hour, the colon can become ischemic and eventually ulcerate (Fig. 124-9). Acute colitis also has been reported after using a colonoscope that had been disinfected with glutaraldehyde (Fig. 124-10).101,102
Colitis also has followed the use of several hyperosmolar water-soluble contrast materials that often are employed to opacify the colon in cases of partial obstruction and to treat fecal impactions in adults.99 Damage is believed to occur because of the hypertonicity of these agents, but the addition of Tween 80 to hyperosmolar agents to improve mucosal contrast might have contributed to the mucosal damage because of its detergent properties. Most reports of injury have occurred in the colon proximal to an obstruction and mainly in the right colon, suggesting that prolonged contact with these agents predisposes to mucosal injury.
PREVENTION AND TREATMENT
Proper cleaning and rinsing of endoscopes are required to minimize exposure of the patient to injurious disinfecting chemicals. Protocols require strict adherence to proper maintenance and adjustments in the rinse cycle of disinfecting machines.102 Forced-air drying and rinsing of endoscope channels and the exterior of the instrument should ensure a chemical-free procedure.
PNEUMATOSIS COLI (PNEUMATOSIS CYSTOIDES INTESTINALIS)
The term pneumatosis coli is synonymous with pneumatosis cystoides intestinalis when the latter disorder is limited to the colon. This uncommon entity is characterized by multiple gas-filled cysts located in the submucosa and subserosa of the intestine. Pneumatosis cystoides intestinalis must be distinguished from pneumatosis linearis, or gas within the wall of the bowel, which usually is associated with bowel necrosis, signifies loss of bowel viability, and mandates surgery. In acquired immunodeficiency syndrome (AIDS), pneumatosis linearis may be associated with opportunistic infections of the colon and can resolve without surgery if the infection is treated successfully.108
Numerous conditions have been associated with pneumatosis coli, including appendicitis, Crohn’s disease,109 ulcerative colitis, diverticular disease, necrotizing enterocolitis, pseudomembranous colitis,110 ileus,111 and sigmoid volvulus. Pneumatosis also has been associated with nongastrointestinal conditions, including emphysema, collagen vascular diseases,111,112 transplantation,112 AIDS,108 glucocorticoid use, chemotherapy,111 and certain medications. In approximately 20% of cases, there are no identified associated medical conditions, and pneumatosis is considered primary.113
ETIOLOGY
According to the mechanical theory, intraluminal gas enters the bowel wall under pressure through a defect or potential defect in the intestinal mucosa. The mucosal defect can result from direct trauma or increased intraluminal pressure. This hypothesis could account for reports of pneumatosis after sigmoidoscopy without biopsy; in cases of colitis, perforated duodenal ulcers, and jejunal diverticula; and after intestinal anastomoses. The plausibility of this theory is diminished by the absence of a connection between the mucosa and the cysts and the presence of elevated levels of hydrogen gas in the cysts.114
The bacterial theory suggests that the cystic gas collections are the by-products of bacteria, specifically those that produce hydrogen. This theory has been supported by clinical observations and laboratory experiments. In laboratory animals, pneumatosis coli can be induced by injecting gas-forming bacteria into the bowel wall. In addition to local invasion of the intestinal wall, bacteria can produce gas cysts by manufacturing large amounts of hydrogen gas as a result of the fermentation of carbohydrates. Levitt and Olsson theorized that the high hydrogen tension in the colonic lumen leads to rapid diffusion of this gas into an intramural gas bubble and can cause N2, O2, and CO2 to diffuse from the circulation into the bubble.115 According to their theory, the gas bubble enlarges if there is continued diffusion of hydrogen into it. Indeed, high hydrogen content in the cysts has been documented,116 and cysts regress in patients fed an elemental diet to decrease carbohydrate substrate for colonic bacteria. Two major observations argue against the bacterial theory of pneumatosis: bacteria are not cultured from cysts, and with cyst rupture and pneumoperitoneum, peritonitis is not seen.
It has been hypothesized that gas cysts can form by counterperfusion supersaturation of H2 gas in which super H2 production by colonic bacteria provides the condition for H2 tension in the colonic lumen to approach the level of N2 tension in the blood.117 One such mechanism is by exposure to certain drugs, such as chloral hydrate, that inhibit the growth of H2-consuming methanogenic bacteria in the colon and thereby increase net H2 production.117 Another possible etiologic setting is the administration of a nonabsorbable carbohydrate such as lactulose, thereby increasing colonic hydrogen production, in a setting where bacteria metabolizing H2 are deficient.118
Successful treatment with antibiotics116,118 and colonic washouts also supports a bacterial etiology for pneumatosis coli.119 In one study, stools from patients with pneumatosis coli were demonstrated to lack two major species of hydrogen-consuming bacteria.114 Because hydrogen normally is produced only in the colon and not in the small intestine, pneumatosis coli may differ from pneumatosis intestinalis with respect to pathogenic mechanisms.
CLINICAL FEATURES AND DIAGNOSIS
The incidence of pneumatosis coli is highest in the sixth decade, with equal frequency in men and women.113 In most cases, pneumatosis is an unexpected finding on abdominal plain films. The most common symptoms are diarrhea (68%), mucus discharge (68%), rectal bleeding (60%), and constipation (48%).113 Approximately 3% of patients present with a complication of pneumatosis coli, including pneumoperitoneum, volvulus, intestinal obstruction, intussusception, tension pneumoperitoneum, and intestinal perforation. Physical examination might detect an abdominal mass, and rectal examination might reveal the cystic lesions.
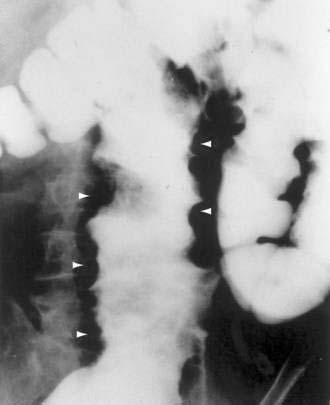
A plain abdominal film can show radiolucent clusters or streaks along the bowel wall with pneumoperitoneum, if a cyst has ruptured. A markedly redundant sigmoid colon as well as the outline of the cysts or linear streaks may be seen on barium enema (Fig. 124-11).113 Endoscopic examination with biopsy is necessary for definitive diagnosis, to exclude carcinoma and to differentiate pneumatosis from familial adenomatosis polyposis120 and from the thumbprinting of colon ischemia. The endoscopic appearance is of multiple cysts, which vary in size from a few millimeters to several centimeters (Fig. 124-12) and which, on puncture with a needle, rapidly deflate. Endoscopic ultrasonography also has been used to establish the diagnosis in pneumatosis.121

PATHOLOGY
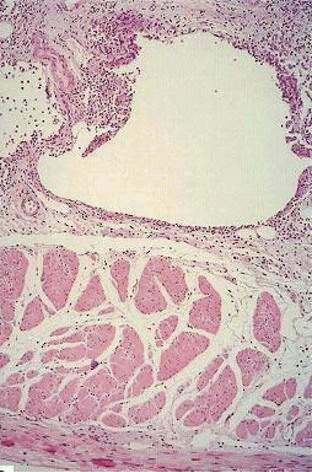
The cysts of pneumatosis cystoids resemble soap bubbles. They usually are thin-walled and unilocular and can occur separately or in clusters. They do not communicate with the intestinal lumen or with each other and have a spongy consistency that pops like a balloon when compressed. On cross section they appear shiny and honeycombed, and they range in size from a few millimeters to several centimeters. Microscopically, the cysts have an endothelial lining that tends to gather and coalesce, forming multinucleate giant cells that increase in number as the cysts collapse, undergo fibrosis (Fig. 124-13), and eventually are sloughed, leaving the cysts without a lining. Progressive fibrosis leads to a decrease in the size of the cysts and ultimately to their obliteration. The connective tissue surrounding the cysts can show a granulomatous inflammatory reaction made up of eosinophils, lymphocytes, macrophages, and plasma cells. Subserosal cysts are surrounded by fibrous connective tissue and can produce adhesion of adjacent bowel loops. The mucosa in pneumatosis may be normal, or it may be thinned and with or without ulcerations and inflammation where it is stretched over a cyst. Mucosal changes vary from mild focal abnormalities to extensive changes including granulomas, abnormal crypts with branching, shortening, cryptitis and abscesses, dilatation, and rupture.
TREATMENT
Because the natural history of pneumatosis is one of spontaneous regression in up to 50% of cases and because cysts can reappear after surgery, specific treatment is not recommended in asymptomatic patients. Symptomatic patients may be treated successfully by breathing high-flow oxygen for several days122 or by using hyperbaric oxygen, especially in resistant cases121; high oxygen levels lead to replacement of hydrogen within the cysts and a corresponding reduction in the size of the cysts. Because cysts can recur after oxygen therapy,122 a minimum of 48 hours of oxygen therapy is recommended to maximize the success rate. Metronidazole also has been used to treat pneumatosis coli, an observation that suggests that anaerobic bacteria play a role in the genesis of the disorder. Because cysts have been reported to recur after short courses of metronidazole,119 treatment should continue until complete resolution of the cysts is documented. In general, colonic resection is reserved for patients with complications such as intestinal obstruction and massive bleeding.
MALAKOPLAKIA
Malakoplakia is a rare chronic granulomatous disease first named by von Hansemann in 1903123 after being reported by Michaelis and Gutmann in 1902.124 The term malakoplakia is derived from the Greek malakos, “soft,” and plakos, “plaque” and reflects its usual appearance as a friable yellow mucosal lesion. Microscopically, coliform bacteria are located in the cytoplasm of macrophages (von Hansemann cells), and laminated intracytoplasmic inclusion bodies (Michaelis-Gutmann bodies) are considered the diagnostic features of this disorder.125
Malakoplakia can affect many organs, including lung, brain, adrenal glands, pancreas, bone, and the genitourinary tract. The most common site of gastrointestinal involvement is the colon, with the rectum, sigmoid, and right colon, involved in descending order of frequency.126
ETIOLOGY
The pathogenesis of malakoplakia is unknown. Proposed etiologies are infection, immunosuppression, systemic illness, neoplasia, and a genetic disorder. Evidence for an infectious etiology is based on the finding that some patients with malakoplakia have associated chronic infections. This was first described in the urologic form of malakoplakia in which more than 75% of patients were infected with Escherichia coli. This finding led to the belief that E. coli might be a primary cause of malakoplakia; however, other organisms also have been isolated, including Klebsiella, Proteus, Mycobacterium, Staphylococcus, and fungi,127 suggesting that one infection is not the primary cause of the disease.
Other evidence points to a defect in macrophage killing as the cause of malakoplakia. Nondigested microorganisms are found within the lysosomes of macrophages in affected persons. Macrophages from these patients show a decrease in cyclic guanosine monophosphate, resulting in impaired bactericidal activity.128 Peripheral blood monocytes also are found to have decreased bactericidal activity in malakoplakia. The defect in macrophage dysfunction may be reversed with the addition of a cholinergic agonist, both in vitro and in vivo.129
Malakoplakia has been reported in patients receiving chemotherapy and immunosuppressive therapy for organ transplantation130; reversal of macrophage abnormalities and resolution of clinical symptoms has been documented after discontinuation of glucocorticoids and azathioprine.131 Malakoplakia has been reported in various immune deficiency states such as primary hypogammaglobulinemia and AIDS,132,133 and it also has been associated with chronic systemic diseases such as systemic lupus erythematosus, ulcerative colitis, and sarcoidosis.126
There have been a substantial number of cases associated with colorectal cancer, to perhaps suggest a neoplastic etiology for at least one form of malakoplakia.134,135 A possible genetic etiology was suggested by one report of colonic malakoplakia that clustered in a family.136
CLINICAL FEATURES AND DIAGNOSIS
Patients with malakoplakia usually present with abdominal pain, diarrhea, hematochezia, and fever.126 Physical findings may include a rectal mass on digital examination, abdominal mass, and weight loss. Diagnosis is by colonoscopy (and biopsy), which generally reveals one of the following three patterns of disease:
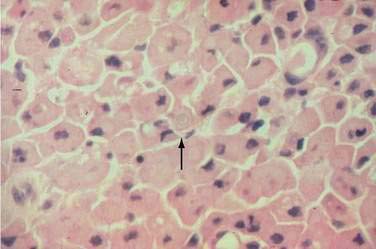
Biopsy is essential to confirm the diagnosis and to exclude an underlying colonic malignancy (Fig. 124-14). Histology reveals the characteristic macrophages with voluminous cytoplasm (von Hansemann cells) containing the classic Michaelis-Gutmann bodies (intracytoplasmic concentric laminated inclusion bodies). The histiocytes (von Hansemann cells) must be distinguished from those found in fungal disease, leprosy, Whipple’s disease, reticulum cell sarcoma, and macrophages harboring Mycobacterium avium complex.
TREATMENT
Patients with newly diagnosed malakoplakia should undergo a thorough medical evaluation to determine if they are taking immunosuppressive medications or have coexisting medical illnesses. Tests of immune function and screening for associated bladder malakoplakia and colorectal cancer are prudent. Patients receiving immunosuppressive medications might improve after these medications are discontinued. Antibiotics such as trimethoprim-sulfamethoxazole and ciprofloxacin have been successful in treating malakoplakia.137,138 Both antibiotics appear to kill the bacteria associated with malakoplakia and can penetrate the defective host macrophages. Cholinergic agents also may be useful in treating children with malakoplakia.129 Surgical resection of the involved colon is recommended for cases associated with carcinoma or severe bleeding.
COLITIS CYSTICA PROFUNDA AND SUPERFICIALIS
Colitis cystica profunda is a rare disease characterized by mucin-filled cysts located in the submucosa of the large intestine (Fig. 124-15). Colitis cystica profunda is to be distinguished from colitis cystica superficialis in which there are numerous small cysts, located superficially in the mucosa of the colon and not penetrating beyond the muscularis mucosa. Colitis cystica superficialis occurs mainly in patients with pellagra or advanced celiac disease, usually causes no symptoms, and resolves following nutritional repletion. Colitis cystica profunda was first described in 1766 by Stark, who reported two cases associated with dysentery.139 There are three patterns of disease: localized with a polypoid lesion, diffuse with multiple polypoid lesions, and diffuse with a confluent sheet of cysts.
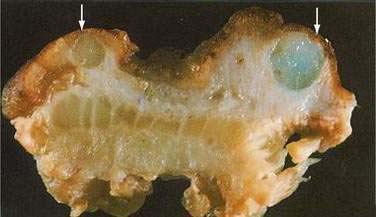
Figure 124-15. Resection specimen of colitis cystica profunda. Several submucosal cysts are filled with mucinous material (arrows).
(Courtesy of Feldman M, Boland CR, editors. Slide Atlas of Gastroenterology and Hepatology. Philadelphia: Current Medicine; 1996.)
ETIOLOGY
The etiology of colitis cystica profunda is unknown, but several theories have been proposed. A possible congenital etiology is supported by several findings. In embryologic examinations, submucosal cysts have been found in multiple gastrointestinal locations. The occurrence of colitis cystica profunda in children and its association with other congenital conditions such as Peutz-Jeghers syndrome140 also support a congenital origin for this disease; the absence of submucosal cysts in large autopsy series of infants and children reduces the plausibility of this etiology.
Colitis cystica profunda has been associated with acquired diseases that predispose to mucosal ulceration and inflammation, including ulcerative colitis,141 Crohn’s disease,142 and infectious colitis.143 Submucosal cysts also have been reported in areas exposed to local trauma, such as an intestinal anastomosis or colostomy.144 Proctitis cystica profunda developed in rats treated with irradiation145 and at small bowel stomas also created in rats.146
Colitis cystica profunda has been found in association with adenocarcinoma of the colon, suggesting a neoplastic etiology. Several cases of adenocarcinoma of the stomach associated with gastritis cystica profunda have been reported.147 In some reports, there is strong evidence of a causal link between cancer and colitis cystica profunda, because the submucosal cysts are often found adjacent to the adenocarcinoma, whereas adjacent benign mucosa is devoid of submucosal cysts.
The localized form of colitis cystica profunda is associated with rectal prolapse and solitary rectal ulcer syndrome.139 Mucosal prolapse has been found in more than 50% of patients with the localized form of the disease. Trauma or ischemia caused by chronic traction on the mucosa and intramural vessels might play a role in the development of the submucosal cysts. Microscopic features of the localized form of the disease often include fibrosis of the lamina propria and hypertrophic muscle fibers, changes that are characteristic of solitary rectal ulcer syndrome (see Chapter 115).148
CLINICAL FEATURES AND DIAGNOSIS
Colitis cystica profunda affects men and women equally. The most common symptoms are rectal bleeding, mucus discharge, and diarrhea139; less common are tenesmus, abdominal pain, and rectal pain. Rarely, the patient presents with intestinal obstruction caused by the cysts.149
At endoscopy, most lesions are located on the anterior rectal wall 6 to 7 cm from the anal verge. The lesions appear as polyps with overlying mucosa that may be normal, inflamed, or ulcerated. Endoscopy might disclose an associated rectal prolapse in some cases. The endoscopic appearance of the lesions may be indistinguishable from a variety of lesions, including adenocarcinoma, adenomatous polyps, submucosal lipoma, neurofibroma, inflammatory pseudopolyps, pneumatosis coli, and endometriosis.139 Barium enema can reveal radiolucent filling defects. Transrectal ultrasound may be useful in differentiating this disease from cancer and reveals hypoechoic cysts that may be surrounded by intact submucosa, unlike invasive cancer.150 A characteristic finding consisting of noninfiltrating submucosal cysts also has been described on magnetic resonance imaging (MRI).151
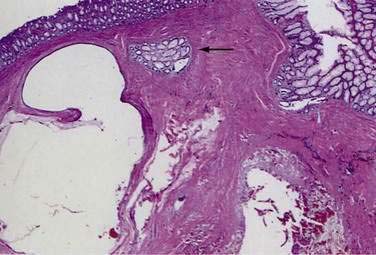
Biopsy is necessary to differentiate this lesion from a variety of inflammatory, neoplastic, and infectious conditions. On biopsy, the submucosa is seen to be thickened by the presence of the mucus-filled cysts (Fig. 124-16). The cysts usually communicate with the lumen through small openings in the mucosa. Although usually confined to the submucosa, cysts involving the muscularis propria and serosa have been reported. The surrounding connective tissue often shows chronic inflammation, and there may be extensive replacement of the lamina propria by fibroblasts.148 As with solitary rectal ulcer syndrome, misplaced glands can cause confusion with invasive adenocarcinoma (see Fig. 124-16).
TREATMENT
A high-fiber diet and bowel retraining to avoid straining have led to regression of this disease in a few cases.150 If fiber is not effective, polyethylene glycol solutions may be tried.152 Glucocorticoid enemas also have been used with some success.139 Most patients have been treated with surgery. In patients with associated rectal prolapse, repair of the prolapse alone may treat the colitis cystica profunda successfully, whereas for disease localized to the rectum and in the absence of procidentia, local excision through a transanal approach is efficacious.152 When the disease is localized to the rectum but is circumferential, total excision may be accomplished by mucosal sleeve resection and coloanal pull-through.152 More-diffuse lesions have been removed by segmental resection. Segmental resection also may be necessary for large obstructing lesions and for lesions that cause hypokalemia, hypoalbuminemia, or severe anemia from chronic blood loss. A diverting colostomy can lead to regression of this disease and may be the best option for a patient with significant comorbidities.
NEUTROPENIC ENTEROCOLITIS (TYPHLITIS)
Neutropenic enterocolitis (typhlitis) is a potentially life-threatening condition described by Wagner in 1970 in children undergoing chemotherapy for leukemia.153 Neutropenic enterocolitis subsequently has been described after organ transplantation, with AIDS, in patients with leukemia treated with cytosine arabinoside, and in patients with solid tumors treated with combination chemotherapy154–158; the frequency in persons at risk varies from 1% to 46%.159 The disease commonly affects the ileum and cecum and can result in intestinal perforation.
ETIOLOGY
The cause of neutropenic enterocolitis may be multifactorial. The initial injury is an ulceration of the bowel mucosa with no associated inflammatory response. Mucosal injury can occur from leukemic infiltration, stasis of bowel contents, or mucosal ischemia from splanchnic vasoconstriction resulting from sepsis.155,157,158 Certain drugs also can contribute to mucosal damage. Cytosine arabinoside can cause necrosis and delayed regeneration of intestinal glandular epithelium.160 Vinca alkaloids used to treat leukemia also can contribute to cecal distention by damaging the myenteric plexus of the intestine. With mucosal injury in the setting of impaired host defenses, infectious colitis subsequently occurs. The infection is often polymicrobial; causative bacteria include Escherichia coli and Staphylococcus, Streptococcus, Enterococcus, and Klebsiella species161–162; fungal organisms such as Aspergillus and Candida also have been isolated.161 In addition to transmural infection of the intestine, the cecum can become gangrenous and perforate as a result of increased distention and ischemia. The process can involve the ileum alone or both the ileum and cecum.161
CLINICAL FEATURES AND DIAGNOSIS
The most common presentation is with fever, diarrhea, nausea, vomiting, and abdominal pain in a patient who is receiving antineoplastic drugs.162 Abdominal tenderness typically is localized to the right lower quadrant of the abdomen, but it may be absent or masked by drugs such as prednisone; localized tenderness can progress rapidly to diffuse signs of peritonitis as a result of intestinal perforation. Shock can occur as a result of bacteremia or intestinal perforation. On occasion, the sigmoid colon may be affected, further complicating the diagnosis.163 Neutropenia is common but was absent in 12% of children in a review of typhlitis in childhood cancer162; blood cultures are positive in up to 50% of cases.155,161 Differential diagnosis includes appendicitis, pseudomembranous colitis, ischemic colitis, volvulus, diverticulitis, and drug-induced diarrhea.
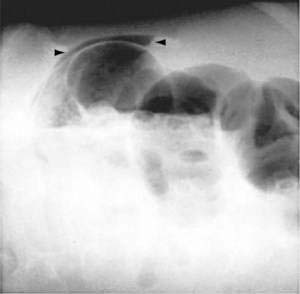
Diagnostic evaluation should include a radiologic evaluation to exclude other diseases, confirm the diagnosis, and determine the severity of illness. Abdominal films can demonstrate dilated loops of small bowel with decreased air in the right lower quadrant and free intraperitoneal air if intestinal perforation has occurred (Fig. 124-17).155 CT scans are most sensitive for establishing the diagnosis and help to exclude other conditions such as appendicitis and diverticulitis. The CT scan might reveal a thickened bowel wall, pneumatosis intestinalis, ascites, and free air.164 One study suggested that ultrasonography is superior to CT scan in predicting the outcome of typhlitis.162 Barium enema should be avoided because of the potential risk of perforation. Stool assay for C. difficile toxin should be performed routinely to exclude pseudomembranous C. difficile colitis.
TREATMENT
Management of neutropenic enterocolitis has varied and is controversial because there are no prospective or high-quality retrospective studies concerning medical or surgical therapies.165 Approaches have included supportive measures alone, aggressive initial surgical resection, and combined medical and surgical treatment; successes and failures have been documented with all these approaches. In two studies, all patients treated medically recovered, whereas in another similar series, all patients managed medically died.166 Clearly, successful management of patients with neutropenic enterocolitis needs to be individualized to optimize outcome.
In general, medical management includes broad-spectrum antibiotics, nasogastric suction, and bowel rest. Fluid resuscitation with isotonic solutions is critical for maintaining renal perfusion in the face of decreased systemic vascular resistance from sepsis and intra-abdominal fluid sequestration. Close observation and serial abdominal and radiologic examinations are necessary to monitor the response to medical treatment. Antibiotics should have activity against enteric Gram-negative organisms, Gram-positive organisms, and anaerobes. Causative microorganisms include Enterococcus species, S. aureus, E. coli, group A Streptococcus, and Klebsiella.161,162 For patients who do not respond to antibacterial agents, amphotericin should be considered, because fungemia is common.161 Blood transfusions may be necessary because the diarrhea often is bloody. Granulocyte-macrophage colony-stimulating factor to correct the neutropenia may be a useful adjunct to medical therapy.167
Early surgical intervention has been recommended in persons with a rapidly deteriorating course despite maximal medical therapy. Two series have shown a decreased mortality rate in patients with severe disease who are treated surgically compared with those treated medically.166 For patients with complications such as frank gangrene, intestinal perforation, and shock despite vasopressor support, surgical intervention is mandatory.
Controversy surrounds the choice of operation. Gangrenous or perforated bowel should be resected. When the bowel is edematous with no vascular compromise and no signs of perforation, successful management has included no resection,168 intestinal diversion with no resection,166 and resection of the involved bowel. If resection is performed, construction of an ileostomy and mucous fistula may be the safest option, because intestinal anastomoses may be prone to breakdown in patients with neutropenia.169 Because recurrences of neutropenic enterocolitis are common when chemotherapy is restarted, right hemicolectomy is recommended before chemotherapy is resumed.157
ENDOMETRIOSIS
Endometriosis, defined as the presence of endometrial tissue outside the uterine cavity and musculature, was first described by von Rokitansky in 1860. Most often, ectopic endometrial tissue lies in the vicinity of the uterus. Endometriosis occurs in up to 15% of menstruating women and up to 30% of infertile women.170 The initial description of nonpigmented endometriosis in 1986 resulted in increased recognition and a much higher prevalence of this disorder than appreciated previously.171 In contrast to endometrial involvement of the female reproductive organs, gastrointestinal involvement by endometriosis is less common, usually asymptomatic, and clinically less important.172 The intestinal organs most commonly involved are the rectosigmoid (96%), appendix (10%), and ileum (5%), with other organs involved uncommonly.173 Intestinal endometriosis can mimic a wide variety of inflammatory, infectious, and neoplastic digestive disorders.174
ETIOLOGY AND PATHOGENESIS
Several hypotheses have been advanced to explain the ectopic location of endometrial tissue.175–177 The most commonly accepted explanation is that of retrograde passage of endometrial tissue, which then implants and grows on pelvic organs and the peritoneum. From these sites, more distant implants arise via hematogenous or lymphatic dissemination; further dissemination can occur during surgical interventions. A less-accepted hypothesis, with fewer supporting data, is that of endometrial metaplasia, in which multipotential peritoneal mesothelial cells are induced by unknown factors to undergo metaplastic transformation to endometrial tissue.
CLINICAL FEATURES
Endometriosis is found almost exclusively in women of childbearing age, with clinical onset usually between the ages of 20 and 45 years.173 Women who experience symptoms or who undergo surgery for endometriosis beyond menopause presumably have chronic fibrosis or exacerbations induced by exogenous estrogen.
Although most women with endometrial implants on intestinal structures have no symptoms, those with serosal implants may complain of localized tenderness, low backache, or abdominal pain. Penetration of endometrial tissue into the bowel wall can produce constipation, diarrhea, and partial obstruction, resulting in intermittent abdominal pain. Contrary to popular thinking, symptoms are not always cyclical and might not fluctuate with hormonal levels; nor are gastrointestinal symptoms necessarily associated with gynecologic symptoms. Rarely, hematochezia occurs when endometrial implants penetrate to the mucosa or when severe colonic fibrosis results in ischemia.172 Less common presentations occur with involvement of the more proximal colon or small intestine and include acute appendicitis caused by an obstructing endometrioma, small bowel intussusception, and volvulus.174,178
DIAGNOSIS
It is rare to see endometrial implants on the colonic mucosa except when there is hematochezia. Thus, colonoscopy is often normal except for areas of extrinsic compression or strictures with intact mucosa.179 More helpful is an air-contrast barium enema, which demonstrates submucosal polypoid masses or areas of noncircumferential narrowing of the lumen (Fig. 124-18). Diagnostic yield and accuracy may be enhanced by performing these tests just before the onset of menses. CT scans (Fig. 124-19), ultrasonography, and MRI all have been reported to assist with the diagnosis or assessment of the extent of endometrial involvement. High-resolution transvaginal and transrectal ultrasonography also may be useful in detecting small endometrial implants, particularly in the retroperitoneal pelvis.180
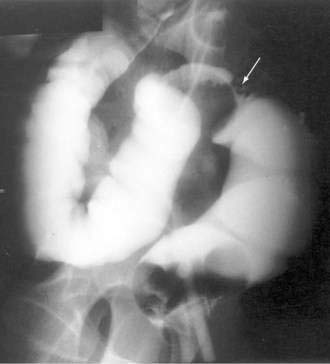
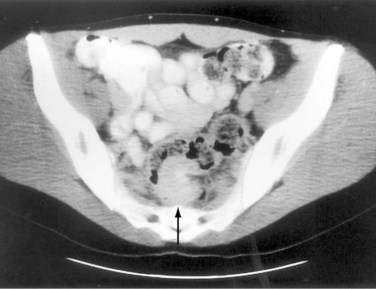
Figure 124-19. Computed tomography scan in the same patient as in Figure 124-18 showing the endometrial mass in the cul-de-sac (arrow) extending into the colon.
(Courtesy of Mark Peterson, MD, Pittsburgh, Pa.)
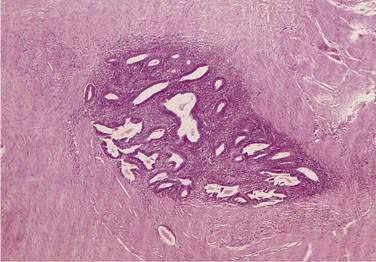
Definitive diagnosis of endometriosis often is made by laparoscopy or laparotomy with biopsy and is especially useful in patients with intestinal implants without pelvic involvement. The classic peritoneal implant appears as a bluish-black “powder-burn” lesion with variable degrees of pigmentation and surrounding fibrosis, the dark coloration resulting from hemosiderin deposition. Most peritoneal implants, however, appear as subtle, nonpigmented lesions. The appreciation that endometrial tissue may be nonpigmented has increased the yield of these procedures considerably.171
The differential diagnosis of intestinal endometriosis includes inflammatory disorders such as Crohn’s disease and ulcerative colitis with stricture, diverticulitis, infectious diseases such as ileocolonic tuberculosis and schistosomiasis, benign and malignant neoplastic disorders, and colon ischemia.174 It is important to emphasize that no radiologic or imaging finding is pathognomonic of endometriosis; mucosal abnormalities that permit positive biopsies are rare; and tissue for a definitive diagnosis usually is obtained only at laparotomy (Fig. 124-20).
TREATMENT
In general, when a diagnosis of serosal intestinal endometriosis is made, hormonal therapy is often the first therapeutic option, similar to the standard approach to pelvic endometriosis.181,182 Low-dose estrogen-progestin compounds cause a pseudopregnancy state that results in decidualization of endometrial tissue and often relieves dysmenorrhea. Their use in more-severe disease is questionable, however, and they generally are not recommended for symptomatic intestinal disease.
The most effective agents currently available are the synthetic androgen danazol and the gonadotropin-releasing hormone agonists.183,184 Both act to decrease ovarian steroid synthesis by inhibiting pituitary release of follicle-stimulating hormone and luteinizing hormone. Although both are effective in decreasing pelvic pain associated with endometriosis and appear to decrease the size of endometrial implants, there are no studies of these agents in endometriosis-associated intestinal disease, and there is some concern that treatment can result in increased fibrosis and inadequate resolution of symptoms.185 Ablation of endometrial implants on surfaces that can be visualized laparoscopically can be accomplished using carbon dioxide laser.186
For endometriosis that causes partial obstruction of the colon or small intestine, segmental resection of the involved area provides the best results and also serves to exclude an underlying carcinoma.187–189 Resection can be performed by laparoscopy or by open surgery, according to available expertise.190 If the patient is postmenopausal or if future pregnancies are not wanted, hysterectomy and bilateral salpingo-oophorectomy can be done at the time of resective surgery to minimize the risk of symptomatic disease in the future. Similar surgery also can be performed in premenopausal women who, despite medical therapy, have intractable symptoms.
Abdou N, NaPombejara C, Sagawa A, et al. Malacoplakia: Evidence for monocyte lysosomal abnormality correctable by cholinergic agonist in vitro and in vivo. N Engl J Med. 1977;297:1413-19. (Ref 129.)
Beck DE. Surgical treatment for colitis cystica profunda and solitary rectal syndrome. Curr Treat Options Gastroenterol. 2002;5:231-37. (Ref 152.)
Cappell MS. Colonic toxicity of administered drugs and chemicals. Am J Gastroenterol. 2004;99:1175-90. (Ref 107.)
Chande N, McDonald JWD, MacDonald JK. Interventions for treating lymphocytic colitis. Cochrane Database Sys Rev 2008; (2):CD006096. (Ref 36.)
Chande N, McDonald JWD, MacDonald JK. Interventions for treating collagenous colitis. Cochrane Database Sys Rev 2008; (2):CD003575. (Ref 37.)
Davila ML. Neutropenic enterocolitis: Current issues in diagnosis and management. Curr Infect Dis Rep. 2007;9:116-20. (Ref 165.)
deOliviera-Neto JP, de Aguilar-Nascomento JE. Intraluminal irrigation with fibers improves mucosal inflammation and atrophy in diversion colitis. Nutrition. 2004;20:197-99. (Ref 58.)
Eggenberger JC, Farid A. Diversion colitis. Curr Treat Options Gastroenterol. 2001;4:255-9. (Ref 56.)
Fernandez-Banares F, Esteve M, Espinos JC, et al. Drug consumption and the risk of microscopic colitis. Am J Gastroenterol. 2007;102:324-30. (Ref 10.)
Florin THJ. Alkyl halides, super hydrogen production, and the pathogenesis of pneumatosis cystoides coli. Gut. 1997;41:778-84. (Ref 117.)
Gadenstatter M, Wetscher G, Crookes PF, et al. Dieulafoy’s disease of the large and small bowel. J Clin Gastroenterol. 2002;27:169-72. (Ref 72.)
Gopal DV, Katon RM. Endoscopic balloon dilation of multiple NSAID-induced colonic strictures: case report and review of literature on NSAID-related colopathy. Gastrointest Endosc. 1999;50:120-23. (Ref 72.)
McCarville MB, Adelman CS, Chenghong L, et al. Typhlitis in childhood cancer. Cancer. 2005;104:380-87. (Ref 162.)
Muller-Lissner SA, Kamm MA, Scarpignato C, Wald A. Myths and misconceptions about chronic constipation. Am J Gastroenterol. 2005;100:232-42. (Ref 85.)
Olive DL, Pritts EA. Treatment of endometriosis. N Engl J Med. 2001;345:266-75. (Ref 181.)
1. Lindstrom CG. “Collagenous colitis” with watery diarrhea: A new entity. Pathol Eur. 1976;11:87.
2. Read NW, Krejs GJ, Read MG, et al. Chronic diarrhea of unknown origin. Gastroenterology. 1980;68:264.
3. Lazenby AJ, Yardley JH, Giardiello FM, et al. Lymphocytic (“microscopic”) colitis: A comparative histopathologic study with particular reference to collagenous colitis. Hum Pathol. 1989;20:18.
4. Fernandez-Banares F, Salas A, Forne M, et al. Incidence of collagenous and lymphocytic colitis: A 5-year population-based study. Am J Gastroenterol. 1999;94:418.
5. Olesen M, Eriksson S, Bohr J, et al. Microscopic colitis: A common diarrheal disease: An epidemiologic study in Orebro, Sweden, 1993-1998. Gut. 2004;53:346.
6. Pardi DS, Loftus EVJr, Smyrk TC, et al. The epidemiology of microscopic colitis: A population based study in Olmsted County, Minnesota. Gut. 2007;56:504.
7. Tanaka M, Mazzoleni G, Riddell RH. Distribution of collagenous colitis: Utility of flexible sigmoidoscopy. Gut. 1992;33:65.
8. Salas A, Fernandez-Banares F, Casalots J, et al. Subepithelial myofibroblasts and tenascin expression in microscopic colitis. Histopathology. 2003;43:48.
9. Jarnerot G, Tysk C, Bohr J, Ericksson S. Collagenous colitis and fecal stream diversion. Gastroenterology. 1995;109:449.
10. Fernandez-Banares F, Esteve M, Espinos JC, et al. Drug consumption and the risk of microscopic colitis. Am J Gastroenterol. 2007;102:324.
11. Olesen M, Eriksson S, Bohr J, et al. Lymphocytic colitis: A retrospective clinical study of 199 Swedish patients. Gut. 2004;53:536.
12. Anderson T, Anderson JR, Tvede M, Franzmann MB. Collagenous colitis: Are bacterial cytotoxins responsible? Am J Gastroenterol. 1993;88:375.
13. Riddell RH, Tanaka M, Mazzoleni G. Non-steroidal anti-inflammatory drugs as a possible cause of collagenous colitis: A case control study. Gut. 1992;33:683.
14. Wolber R, Owen D, Freeman H. Colonic lymphocytosis in patients with celiac sprue. Hum Pathol. 1990;21:1092.
15. Armes J, Gee DC, MaCrae FA, et al. Collagenous colitis: Jejunal and colorectal pathology. J Clin Pathol. 1992;45:784.
16. Zins BJ, Tremaine WJ, Carpenter HA. Collagenous colitis: Mucosal biopsies and association with fecal leukocytes. Mayo Clin Proc. 1995;70:430.
17. Giardiello FM, Lazenby AJ, Yardley JH, et al. Increased HLA A1 and diminished HLA A3 in lymphocytic colitis compared to controls and patients with collagenous colitis. Dig Dis Sci. 1992;37:496.
18. Sylwestrowicz T, Kelly JK, Hwang WS, et al. Collagenous colitis and microscopic colitis: The watery diarrhea–colitis syndrome. Am J Gastroenterol. 1989;84:763.
19. Fine KD, Do K, Schulte K, et al. High prevalence of celiac sprue-like HLA-DQ genes and enteropathy in patients with the microscopic colitis syndrome. Am J Gastroenterol. 2000;95:1974.
20. Aigner T, Neureiter D, Muller S, et al. Extracellular matrix composition and gene expression in collagenous colitis. Gastroenterology. 1997;113:136.
21. Bo-Linn GW, Vendrell DD, Lee E, Fordtran JS. An evaluation of the significance of microscopic colitis in patients with chronic diarrhea. J Clin Invest. 1985;75:1559.
22. Burgel N, Bojarski C, Mankertz J, et al. Mechanisms of diarrhea in collagenous colitis. Gastroenterology. 2002;123:433.
23. Baum CA, Bhatia P, Miner PB. Increased colonic mucosal mast cells associated with severe watery diarrhea and microscopic colitis. Dig Dis Sci. 1998;34:1462.
24. Bajor A, Kilander A, Galman C, et al. Budesonide treatment is associated with increased bile acid absorption in collagenous colitis. Aliment Pharmacol Ther. 2006;24:1643.
25. Goff JS, Barnett JL, Pelke T, Appelman HD. Collagenous colitis: Histopathology and clinical course. Am J Gastroenterol. 1997;92:57.
26. Richieri JP, Bonneau HP, Cano N, et al. Collagenous colitis: An unusual endoscopic appearance. Gastrointest Endosc. 1993;39:192.
27. Wickbom A, Lindqvist M, Bohr J, et al. Colonic mucosal tears in collagenous colitis. Scand J Gastroenterol. 2006;41:726.
28. Schiller LR. Diagnosis and management of microscopic colitis syndrome. J Clin Gastroenterol. 2004;38(Suppl 1):527.
29. Fine KD, Lee EL. Efficacy of open-label bismuth subsalicylate for the treatment of microscopic colitis. Gastroenterology. 2002;114:2.
30. Ryder SD, Walker RJ, Jones H, Rhodes JM. Rectal bismuth subsalicylate as therapy for ulcerative colitis. Aliment Pharmacol Ther. 1990;4:333.
31. Munck LK, Kjeldsen J, Philipsen E, et al. Incomplete remission with short-term prednisolone treatment in collagenous colitis: A randomized study. Scand J Gastroenterol. 2003;38:606.
32. Pardi DS, Loftus EVJr, Tremaine WJ, Sandborn WJ. Treatment of refractory microscopic colitis with azathioprine and 6-mercaptopurine. Gastroenterology. 2001;120:1483.
33. Baert F, Schmit A, D’Haens G, et al. Budesonide in collagenous colitis: A prospective double-blind, placebo-controlled trial with histological follow-up. Gastroenterology. 2002;122:20.
34. Miehlke S, Heymer P, Bethke B, et al. Budesonide treatment for collagenous colitis: A randomized double-blind, placebo-controlled multicenter study. Gastroenterology. 2002;123:978.
35. Bonderup OK, Hansen JB, Birket-Smith L, et al. Budesonide treatment of collagenous colitis: A randomized, double-blind, placebo-controlled trial with morphometric analysis. Gut. 2003;52:248.
36. Chande N, McDonald JWD, MacDonald JK. Interventions for treating lymphocytic colitis. Cochrane Database Sys Rev 2008; (2):CD006096.
37. Chande N, McDonald JWD, MacDonald JK. Interventions for treating collagenous colitis. Cochrane Database Sys Rev 2008; (2):CD003575.
38. Glotzer DJ, Glick ME, Goldman H. Proctitis and colitis following diversion of the fecal stream. Gastroenterology. 1981;24:211.
39. Korelitz BI, Cheskin LJ, Sohn N, Sommers SC. The fate of the rectal segment after diversion of the fecal stream in Crohn’s disease: Its implications for surgical management. J Clin Gastroenterol. 1985;7:37.
40. Edwards CM, George B, Warren B. Diversion colitis: New light through old windows. Histopathology. 1999;34:1.
41. Yeong ML, Bethwaite PB, Prasad J, Isbister WH. Lymphoid follicular hyperplasia: A distinctive feature of diversion colitis. Histopathology. 1991;19:55.
42. Haque S, Eisen RN, West AB. The morphologic features of diversion colitis: Studies of a pediatric population with no other disease of the intestinal mucosa. Hum Pathol. 1993;24:211.
43. Cook SI, Sellin JH. Review article: Short-chain fatty acids in health and disease. Aliment Pharmacol Ther. 2002;12:507.
44. Mortensen PB, Clausen MR. Short-chain fatty acids in the human colon: Relation to gastrointestinal health and disease. Scand J Gastroenterol. 1996;216:132.
45. Roediger WEW. Utilization of nutrients by isolated epithelial cells of the rat colon. Gastroenterology. 1982;83:424.
46. Harig JM, Soergel KH, Komorowski RA, Wood CM. Treatment of diversion colitis with short-chain fatty acid irrigation. N Engl J Med. 1989;320:23.
47. Neut C, Colombel JF, Guillemot F, et al. Impaired bacterial flora in human excluded colon. Gut. 1989;30:1094.
48. Ordein JJ, DiLorenzo CD, Flores A, Hyman PE. Diversion colitis in children with severe gastrointestinal motility disorders. Am J Gastroenterol. 1992;87:88.
49. Morin CL, Ling V, Bourassa D. Small intestinal and colonic changes induced by a chemically defined diet. Dig Dis Sci. 1980;25:123.
50. Tomasino RM, Morello V, Latteri MA, et al. Histological and histochemical changes in the colon mucosa after ureterosigmoidostomy or colon conduit. Eur Urol. 1988;15:248.
51. Guillemot F, Colombel JF, Neut C, et al. Treatment of diversion colitis by short-chain fatty acids: Prospective and double-blind study. Dis Colon Rectum. 1991;34:861.
52. Neut C, Guillemot F, Gowerrousseau C, et al. Treatment of diversion colitis by short-chain fatty acids: Prospective and double-blind study. Gastroenterol Clin Biol. 1995;19:871.
53. Tsironi E, Irving PM, Feakins RM, Rampton DS. “Diversion” colitis caused by Clostridium difficile infection: Report of a case. Dis Colon Rectum. 2006;49:1074.
54. Geraghty JM, Talbot IC. Diversion colitis: Histological features in the colon and rectum after defunctioning colostomy. Gut. 1991;32:1020.
55. Haas PA, Fox TA, Szilag EJ. Endoscopic examination of the colon and rectum distal to a colostomy. Am J Gastroenterol. 1990;85:850.
56. Eggenberger JC, Farid A. Diversion colitis. Curr Treat Options Gastroenterol. 2001;4:255.
57. Sartor RB, Murphy ME, Rydzak E. Miscellaneous inflammatory and structural disorders of the colon. In: Yamada T, editor. Textbook of gastroenterology. 4th ed. Philadelphia: Lippincott Williams & Wilkins; 2003:1791-816.
58. deOliviera-Neto JP, de Aguilar-Nascomento JE. Intraluminal irrigation with fibers improves mucosal inflammation and atrophy in diversion colitis. Nutrition. 2004;20:197.
59. Ona FV, Allende HD, Vivenio R, et al. Diagnosis and management of nonspecific colon ulcer. Arch Surg. 1982;117:888.
60. Khawaja FI, Vakil N. Colonoscopy as an aid in the diagnosis of nonspecific ulcers of the colon. Gastrointest Endosc. 1987;33:43.
61. Kaufman HL, Fischer AH, Carroll M, Becker JM. Colonic ulceration associated with nonsteroidal anti-inflammatory drugs: Review of three cases. Dis Colon Rectum. 1999;39:705.
62. Buchman AL, Schwartz MR. Colonic ulceration associated with the systemic use of nonsteroidal anti-inflammatory medication. J Clin Gastroenterol. 1996;22:224.
63. Mills B, Zuckerman G, Sicard G. Discrete colon ulcers as a cause of lower gastrointestinal bleeding and perforation in endstage renal disease. Surgery. 1981;89:548.
64. Shimamoto C, Hirata I, Ohshiba S, et al. Churg-Strauss syndrome (allergic granulomatous angiitis) with peculiar multiple colonic ulcers. Am J Gastroenterol. 1990;85:316.
65. Wilson RT, Dean PJ, Upshaw JD, Wruble LD. Endoscopic appearance of Wegener’s granulomatosis involving the colon. Gastrointest Endosc. 1987;33:388.
66. Baxter R, Nino-Murcia M, Bloom RJ, Kosek J. Gastrointestinal manifestations of essential mixed cryoglobulinemia. Gastrointest Radiol. 1988;13:160.
67. Ong J, Lim JF, Lim HK, Eu KW. Benign solitary cecal ulcer syndrome: Series of eight cases and literature review. Dis Colon Rectum. 2008. [Epub ahead of print]
68. Smith JA, Pineau BC. Endoscopic therapy of NSAID-induced colonic diaphragm disease: Two cases and a review of published reports. Gastrointest Endoscopy. 2000;52:120.
69. Sheers R, Williams WR. NSAIDs and gut damage [letter]. Lancet. 1989;2:1154.
70. Laine L, Connors LG, Reicin A, et al. Serious lower gastrointestinal clinical events with nonselective NSAID or coxib use. Gastroenterology. 2003;124:288.
71. Schiffmann L, Kahran S, Berger G, Buhr HJ. Colon perforation in an adolescent after short-term diclofenac intake. ANZ J Surg. 2005;75:726.
72. Gopal DV, Katon RM. Endoscopic balloon dilation of multiple NSAID-induced colonic strictures: Case report and review of literature on NSAID-related colopathy. Gastrointest Endosc. 1999;50:120.
73. Dieulafoy G. Leçons clinique de l’Hotel-Dieu de Paris. Paris: Masson. 1897;2:1.
74. Gadenstatter M, Wetscher G, Crookes PF, et al. Dieulafoy’s disease of the large and small bowel. J Clin Gastroenterol. 2002;27:169.
75. Nozae T, Kitamura M, Matsumata T, Sugimachi K. Dieulafoy-like lesions of colon and rectum in patients with chronic renal failure on long-term hemodialysis. Hepatogastroenterology. 1999;46:3121.
76. Abdulian JD, Santoro MJ, Chen YK, Collen MJ. Dieulafoy-like lesion of the rectum presenting with exsanguinating hemorrhage: Successful endoscopic therapy. Am J Gastroenterol. 1993;88:1939.
77. Katsinelos P, Pilpilidis I, Paroutoglou G, et al. Dieulafoy-like lesion of the colon presenting with massive lower gastrointestinal bleeding. Surg Endosc. 2004;18:346.
78. Heilbrun N. Roentgen evidence suggesting enterocolitis associated with prolonged cathartic abuse. Radiology. 1943;41:486.
79. Leng-Peschlow E. Senna and its rational use. Pharmacology. 1992;44(Suppl 1):1.
80. Muller-Lissner S. What has happened to the cathartic colon? Gut. 1996;39:486.
81. Rieman JF, Zimmerman W. Ultrastructural studies of colonic nerve plexuses in chronic laxative abuse. Gastroenterology. 1978;74:1085.
82. Smith B. Effect of irritant purgatives on the myenteric plexus in man and the mouse. Gut. 1969;139:43.
83. Riecken EO, Zertz M, Ende C, et al. The effect of an anthraquinone laxative on colonic nerve tissue: A controlled trial in constipated women. Z Gastroenterol. 1990;28:660.
84. Wald A. Is chronic use of stimulant laxatives harmful to the colon? J Clin Gastroenterol. 2003;36:380.
85. Muller-Lissner SA, Kamm MA, Scarpignato C, Wald A. Myths and misconceptions about chronic constipation. Am J Gastroenterol. 2005;100:232.
86. Ghadially FN, Walley VM. Pigments of the gastrointestinal tract: A comparison of light microscopic and electron microscopic findings. Ultrastruct Pathol. 1995;19:213.
87. Benavides SR, Morgante PE, Monserrat AJ, et al. The pigment of melanosis coli: A lectin histochemical study. Gastrointest Endosc. 1997;46:131.
88. Walker NI, Bennett RE, Axelson RA. Melanosis coli: A consequence of anthraquinone-induced apoptosis of colonic epithelial cells. Am J Pathol. 1988;131:465.
89. Speare GS. Melanosis coli: Experimental observations of its production and elimination in twenty-three cases. Am J Surg. 1951;82:631.
90. Mengs U, Rudolph RL. Light and electron-microscopic changes in the colon of the guinea pig after treatment with anthranoid and non-anthranoid laxatives. Pharmacology. 1993;47(Suppl 1):172.
91. Balazs M. Melanosis coli: Ultrastructural study in 45 patients. Dis Colon Rectum. 1986;29:839.
92. Nusko G, Schneider B, Wittekind C, Hahn EG. Anthranoid laxative use is not a risk factor for colorectal neoplasia: Results of a prospective case control study. Gut. 2000;46:651.
93. Jacobs EJ, White E. Constipation, laxative use, and colon cancer among middle-aged adults. Epidemiology. 2002;9:385.
94. Roberts MC, Millikan RC, Galanko JA, et al. Constipation, laxative use, and colon cancer in a North Carolina population. Am J Gastroenterol. 2003;98:857.
95. Morganstern L, Shemen L, Allen W, et al. Melanosis coli: Changes in appearance when associated with colonic neoplasia. Arch Surg. 1983;118:62.
96. Hardin RD, Tedesco FJ. Colitis after Hibiclens enema. J Clin Gastroenterol. 1986;8:572.
97. Pike BF, Phillippi PJ, Lawson EHJr. Soap colitis. N Engl J Med. 1971;285:217.
98. Bollen P, Goossens A, Hauser B, Vandenplas Y. Colonic ulcerations caused by an enema containing hydrogen peroxide. J Pediatr Gastroenterol Nutr. 2002;26:232.
99. Creteur V, Douglas D, Galante M, et al. Inflammatory colonic changes produced by contrast material. Radiology. 1983;147:77.
100. Segal I, Tim LO, Hamilton DG, et al. Ritual-enema-induced colitis. Dis Colon Rectum. 1979;22:195.
101. Stein BL, Lamoreux E, Miller M, et al. Glutaraldehyde-induced colitis. Can J Surg. 2001;44:113.
102. Fukunaga K, Khatibi A. Glutaraldehyde colitis: A complication of screening flexible sigmoidoscopy in the primary care center. Ann Intern Med. 2000;133:315.
103. Munoz-Navas M, Garcia-Villareal L. Caustic colitis due to formalin enema. Gastrointest Endosc. 1992;38:521.
104. Michopoulos S, Bouzakis H, Sotiropoulou M, et al. Colitis due to accidental alcohol enema: Clinicopathological presentation and outcome. Dig Dis Sci. 2000;45:1188.
105. Chan A, Depew W, Vanner S. Use of oral sodium phosphate colonic lavage solution by Canadian colonoscopists: Pitfalls and complications. Can J Gastroenterol. 1997;11:334.
106. Uvgur-Bayramicli O, Dabak R, Sargin M. Acute chemical colitis resulting from oral intake of henna. J Clin Gastroenterol. 2005;39:920.
107. Cappell MS. Colonic toxicity of administered drugs and chemicals. Am J Gastroenterol. 2004;99:1175.
108. Gelman SF, Brandt LJ. Pneumatosis intestinalis and AIDS: A case report and review of the literature. Am J Gastroenterol. 2002;93:646.
109. John A, Dickey K, Fenwick J, et al. Pneumatosis intestinalis in Crohn’s disease. Dig Dis Sci. 1992;37:813.
110. Tak P, Van Duinen C, Bun P, et al. Pneumatosis cystoides intestinalis with intestinal pseudo-obstruction: Resolution after metronidazole. Dig Dis Sci. 1992;37:949.
111. Knetchle S, Davidoff A, Rice R. Pneumatosis intestinalis: Clinical management and surgical outcome. Ann Surg. 1990;212:160.
112. Sequeira W. Pneumatosis cystoides intestinalis in systemic sclerosis and other diseases. Semin Arthritis Rheum. 1990;19:269.
113. Gagliardi G, Thompson I, Hershman M, et al. Pneumatosis coli: A proposed pathogenesis based on study of 25 cases and review of the literature. Int J Colorectal Dis. 1996;11:11.
114. Christl S, Gibson G, Murgatroyd P, et al. Impaired hydrogen metabolism in pneumatosis cystoides intestinalis. Gastroenterology. 1993;104:392.
115. Levitt M, Olsson S. Pneumatosis cystoides intestinalis and high breath H2 excretion: Insights into the role of H2 in this condition. Gastroenterology. 1995;108:1560.
116. Read N, Al-Janabi N, Cann P. Is raised breath hydrogen related to the pathogenesis of pneumatosis coli? Gut. 1984;25:839.
117. Florin THJ. Alkyl halides, super hydrogen production, and the pathogenesis of pneumatosis cystoides coli. Gut. 1997;41:778.
118. Goodman RA, Riley TR. Lactulose-induced pneumatosis intestinalis and pneumoperitoneum. Dig Dis Sci. 2001;46:2549.
119. Ellis B. Symptomatic treatment of primary pneumatosis coli with metronidazole. BMJ. 1980;280:763.
120. Spigelman A, Williams C, Ansell J, et al. Pneumatosis coli: A source of diagnostic confusion. Br J Surg. 1990;77:155.
121. Shimada M, Ina K, Takahashi H, et al. Pneumatosis cystoides intestinalis treated with hyperbaric oxygen therapy: Usefulness of an endoscopic ultrasonic catheter probe for diagnosis. Intern Med. 2001;40:896.
122. Miralbes M, Hinojosa J, Aconso J, Berenguer J. Oxygen therapy in pneumatosis coli: What is the minimum oxygen requirement? Dis Colon Rectum. 1983;26:458.
123. von Hansemann D. Ulser Malakoplakia der Harnblase. Virch Arch Pathol Anat. 1903;173:302.
124. Michaelis L, Gutmann C. Ueber Einschlusse in Blasentumoren. Z Clin Med. 1902;47:208.
125. Lewin K, Harell G, Lee A, Crowley L. Malacoplakia—an electron-microscopic study: Demonstration of bacilliform organisms in malacoplakic macrophages. Gastroenterology. 1974;66:28.
126. Cipolletta L, Bianco M, Fumo F, et al. Malacoplakia of the colon. Gastrointest Endosc. 1995;41:225.
127. Stevens S, McClure J. The histochemical features of the Michaelis-Gutmann body and a consideration of the pathophysiological mechanisms of its formation. J Pathol. 1982;137:119.
128. Thorning D, Vaco R. Malacoplakia: Defect in digestion of phagocytosed material due to impaired vacuolar acidification? Arch Pathol. 1975;99:456.
129. Abdou N, NaPombejara C, Sagawa A, et al. Malacoplakia: Evidence for monocyte lysosomal abnormality correctable by cholinergic agonist in vitro and in vivo. N Engl J Med. 1977;297:1413.
130. Kim PT, Davis JE, Erb SR, Yoshida EM, Steinbrecher VP. Colonic malakoplakia in a liver transplant recipient. Can J Gastroenterol. 2007;21:753.
131. Biggar W, Crawford L, Cardella C, et al. Malacoplakia and immunosuppressive therapy: Reversal of clinical and leukocyte abnormalities after withdrawal of prednisone and azathioprine. Am J Pathol. 1985;119:5.
132. Mir-Madjlessi S, Tavassolie H, Kamalian N. Malakoplakia of the colon and recurrent colonic strictures in a patient with primary hypogammaglobulinemia: An association not previously described. Dis Colon Rectum. 1982;25:723.
133. Kianifar H, Sharifi N, Talebi S, Kiani MA. Malakoplakia of colon in a child with celiac disease and chronic granulomatous disease. Ind J Gastroenterol. 2006;25:163.
134. Pillay K, Chetty R. Malakoplakia in association with colorectal carcinoma: A series of four cases. Pathology. 2002;34:332.
135. Asiyanbola B, Camuto P, Mansourian V. Malakoplakia occurring in association with colon carcinoma. J Gastrointest Surg. 2006;10:657.
136. El-Mouzan M, Satti M, Al-Quorain A, El-Ageb A. Colonic malacoplakia—occurrence in a family: Report of cases. Dis Colon Rectum. 1988;31:390.
137. Van Furth R, Van’t Wout J, Wertheimer P, Zwartendijk J. Ciprofloxacin for treatment of malacoplakia. Lancet. 1992;339:148.
138. Maderazo E, Berlin B, Marhardt C. Treatment of malacoplakia with trimethoprim-sulfamethoxazole. Urology. 1979;13:70.
139. Guest CB, Reznick RK. Colitis cystica profunda: Review of the literature. Dis Colon Rectum. 1989;32:983.
140. Anderson N, Rivera E, Flores D. Peutz-Jeghers syndrome with cervical adenocarcinoma and enteritis cystica profunda. West J Med. 1984;141:242.
141. Castleman B, McNeely BU. Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. N Engl J Med. 1972;286:147.
142. Madan A, Minocha A. First reported case of colitis cystica profunda in association with Crohn’s disease. [letter]. Am J Gastroenterol. 2002;97:2472.
143. Wayte D, Helwig E. Colitis cystica profunda. Am J Clin Pathol. 1967;48:159.
144. Rosen Y, Vallant J, Yermakov V. Submucosal cysts at a colostomy site: Relationship to colitis cystica profunda and reopening of a case. Dis Colon Rectum. 1976;19:453.
145. Hubmann F. Proctitis cystica profunda and radiation fibrosis in the rectum of the female Wistar rat after x-irradiation: A histopathological study. J Pathol. 1982;138:193.
146. Brynjolfsson G, Haley H. Experimental enteritis cystica in rats. Am J Clin Pathol. 1967;47:69.
147. Franzin G, Novelli P. Gastritis cystica profunda. Histopathology. 1981;5:535.
148. Levin D. “Solitary” rectal ulcer syndrome: Are “solitary” rectal ulcer syndrome and “localized” colitis cystica profunda analogous syndromes caused by rectal prolapse? Gastroenterology. 1987;92:243.
149. Bentley E, Chandrasoma P, Cohen H, et al. Colitis cystica profunda presenting with complete intestinal obstruction and recurrence. Gastroenterology. 1985;89:1157.
150. Petritsch W, Hinterleitner T, Aichbichler B, et al. Endosonography in colitis cystica profunda and solitary rectal ulcer syndrome. Gastrointest Endosc. 1995;41:382.
151. Inau N, Arslau AS, Akansel G, et al. Colitis cystica profunda: MRI appearance. Abdom Imaging. 2007;32:239.
152. Beck DE. Surgical treatment for colitis cystica profunda and solitary rectal syndrome. Curr Treat Options Gastroenterol. 2002;5:231.
153. Wagner M, Rosenberg H, Ferbach D, Singleton E. Typhlitis: A complication of leukemia in childhood. AJR Am J Roentgenol. 1972;109:341.
154. Petruzzelli G, Johnson J, de Vries E. Neutropenic enterocolitis: A new complication of the head and neck cancer chemotherapy. Arch Otolaryngol Head Neck Surg. 1990;116:209.
155. Otaibi AA, Barker C, Anderson R, Sigalet DL. Neutropenic enterocolitis (typhlitis) after pediatric bone marrow transplant. J Pediatr Surg. 2002;37:770.
156. Till M, Lee N, Soper W, Murphy R. Typhlitis in patients with HIV-1 infection. Ann Intern Med. 1992;116:998.
157. Keidan R, Fanning J, Gatenby R, Weese JL. Recurrent typhlitis: A disease resulting from aggressive chemotherapy. Dis Colon Rectum. 1989;32:206.
158. Paulino A, Kenney R, Forman E, Medeiros L. Typhlitis in a patient with acute lymphoblastic leukemia prior to the administration of chemotherapy. Am J Pediatr Hematol Oncol. 1994;16:348.
159. Moir D, Bale P. Necropsy findings on childhood leukemia emphasizing neutropenic enterocolitis and cerebral calcification. Pathology. 1976;8:247.
160. Slavin R, Dias M, Saral R. Cytosine arabinoside–induced gastrointestinal toxic alterations in sequential chemotherapeutic protocols. Cancer. 1978;42:1747.
161. Katz J, Wagner M, Gresik M, et al. Typhlitis: An 18-year experience and postmortem review. Cancer. 1990;65:1041.
162. McCarville MB, Adelman CS, Chenghong L, et al. Typhlitis in childhood cancer. Cancer. 2005;104:380.
163. Abbasoglu O, Cakmakci M. Neutropenic enterocolitis in patients without leukemia. Surgery. 1993;113:113.
164. Vas W, Seeling R, Manhanta B, et al. Neutropenic colitis: Evaluation with computed tomography. CT J Comput Tomogr. 1988;12:211.
165. Davila ML. Neutropenic enterocolitis: Current issues in diagnosis and management. Curr Infect Dis Rep. 2007;9:116.
166. Moir C, Scudamore C, Benny W. Typhlitis: Selective surgical management. Am J Surg. 1986;151:563.
167. Kouroussis C, Samonis G, Androulakis N, et al. Successful conservative treatment of neutropenic enterocolitis complicating taxane-based chemotherapy. Am J Clin Oncol. 2000;23:309.
168. Mower W, Hawkins J, Nelson E. Neutropenic enterocolitis in adults with acute leukemia. Arch Surg. 1986;121:571.
169. Villar H, Warneke J, Peck M, et al. Role of surgical treatment in the management of complications of the gastrointestinal tract in patients with leukemia. Surg Gynecol Obstet. 1987;165:217.
170. Olive DL, Schwartz LB. Endometriosis: Medical progress. N Engl J Med. 1993;328:1759.
171. Jansen RP, Russel P. Nonpigmented endometriosis: Clinical, laparoscopic, and pathological definition. Am J Obstet Gynecol. 1986;155:1154.
172. Zwas FR, Lyon DT. Endometriosis: An important condition in clinical gastroenterology. Dig Dis Sci. 1991;36:353.
173. Weed JC, Ray JE. Endometriosis of the bowel. Obstet Gynecol. 1987;69:727.
174. Shah M, Tager D, Feller E. Intestinal endometriosis masquerading as common digestive disorders. Arch Intern Med. 1995;155:977.
175. Rock JA, Markham SM. Pathogenesis of endometriosis. Lancet. 1992;240:1264.
176. Bontis JN, Vavilis DT. Etiopathology of endometriosis. Ann N Y Acad Sci. 1997;816:305.
177. Oral E, Arici A. Pathogenesis of endometriosis. Obstet Gynecol Clin North Am. 1997;24:219.
178. Ferguson CM. Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 28-1996. A 45-year-old woman with abdominal pain and a polypoid mass in the colon. N Engl J Med. 1996;335:807.
179. Bozdech JM. Endoscopic diagnosis of colonic endometriosis. Gastrointest Endosc. 1992;38:568.
180. Brosens I, Puttemans P, Campo R, et al. Noninvasive methods of diagnosis of endometriosis. Curr Opinion Obstet Gynecol. 2003;15:519.
181. Olive DL, Pritts EA. Treatment of endometriosis. N Engl J Med. 2001;345:266.
182. Davis L, Kennedy SS, Moore J, Prentice A. Modern combined oral contraceptives for pain associated with endometriosis. Cochrane Database Sys Rev 2007; (3):CD00101.
183. Berquist IA. Hormonal regulation of endometriosis and the rationales and effects of gonadotropin-releasing hormone agonist treatment: a review. Hum Reprod. 1995;10:446.
184. Hornstein MD, Yuzpe AA, Burry KA, et al. Prospective randomized double-blind trial of three versus six months of nafarelin therapy for endometriosis-associated pelvic pain. Fertil Steril. 1995;63:955.
185. Hall LLH, Malone JM, Ginsburg KA. Flare-up of endometriosis induced by gonadotropin-releasing hormone agonist leading to bowel obstruction. Fertil Steril. 1995;64:1204.
186. Sutton CJ, Ewen SP, Whitelaw N, Haines P. Prospective, randomized, double-blind, controlled trial of laser laparoscopy in the treatment of pelvic pain associated with minimal, mild, and moderate endometriosis. Fert Steril. 1994;62:696.
187. Cameron IC, Rogers S, Collins MC, Reed MW. Intestinal endometriosis: Presentation, investigation, and surgical management. Int J Colorectal Dis. 1995;10:83.
188. Tran KT, Kuijpers HC, Willemsen WN, Bulten H. Surgical treatment of symptomatic rectosigmoid endometriosis. Eur J Surg. 1996;162:139.
189. Nozhat C, Nezhat F, Pennington E. Laparoscopic treatment of infiltrative rectosigmoid colon and rectovaginal septum endometriosis by the technique of videolaparoscopy and the CO2 laser. Br J Obstet Gynaecol. 1992;99:664.
190. Redwine DB, Koning M, Sharpe DR. Laparoscopically assisted transvaginal segmental resection of the rectosigmoid colon for endometriosis. Fertil Steril. 1996;65:198.