PHP-IA AND PHP-IB Individuals with PHP-I, the most common of the disorders, show a deficient urinary cAMP response to administration of exogenous PTH. Patients with PHP-I are divided into type Ia and type Ib. Patients with PHP-Ia show evidence for AHO and reduced amounts of Gsα protein/activity, as determined in readily accessible tissues such as erythrocytes, lymphocytes, and fibroblasts. Patients with PHP-Ib typically lack evidence for AHO and they have normal Gsα activity. PHP-Ic, sometimes listed as a third form of PHP-I, is really a variant of PHP-Ia, although the mutant Gsα shows normal activity in certain in vitro assays.
Most patients who have PHP-Ia reveal characteristic features of AHO, which consist of short stature, round face, obesity, skeletal anomalies (brachydactyly), intellectual impairment, and/or heterotopic calcifications. Patients have low calcium and high phosphate levels, as with true hypoparathyroidism. PTH levels, however, are elevated, reflecting resistance to hormone action.
Amorphous deposits of calcium and phosphate are found in the basal ganglia in about one-half of patients. The defects in metacarpal and metatarsal bones are sometimes accompanied by short phalanges as well, possibly reflecting premature closing of the epiphyses. The typical findings are short fourth and fifth metacarpals and metatarsals. The defects are usually bilateral. Exostoses and radius curvus are frequent.
Inheritance and Genetic Defects Multiple defects at the GNAS locus have now been identified in PHP-Ia, PHP-Ib, and PPHP patients. This gene, which is located on chromosome 20q13.3, encodes the α-subunit of the stimulatory G protein (Gsα), among other products (see below). Mutations include abnormalities in splice junctions associated with deficient mRNA production, point mutations, insertions, and/or deletion that all result in a protein with defective function resulting in a 50% reduction of Gsα activity in erythrocytes or other cells.
Detailed analyses of disease transmission in affected kindreds have clarified many features of PHP-Ia, PPHP, and PHP-Ib (Fig. 424-7). The former two entities, often traced through multiple generations, have an inheritance pattern consistent with genetic imprinting. The phenomenon of gene imprinting, involving methylation of genetic loci, independent of any mutation, impairs transcription from either the maternal or the paternal allele (Chap. 82). The Gsα transcript is biallelically expressed in most tissues; expression from paternal allele is silenced through as-of-yet unknown mechanisms in some tissues including the proximal renal tubules and the thyroid; consequently, inheritance of a defective paternal allele has no implications with regard to hormonal function. Thus, females affected by either PHP-Ia or PPHP will have offspring with PHP-Ia, if these children inherit the allele carrying the GNAS mutation; in contrast, if the mutant allele is inherited from a male affected by either disorder, the offspring will exhibit PPHP. Consistent with these data in humans, gene-ablation studies in mice have shown that inheritance of the mutant Gsα allele from the female causes much reduced Gsα protein in renal cortex, hypocalcemia, and resistance to PTH. Offspring inheriting the mutant allele from the male showed no evidence of PTH resistance or hypocalcemia.
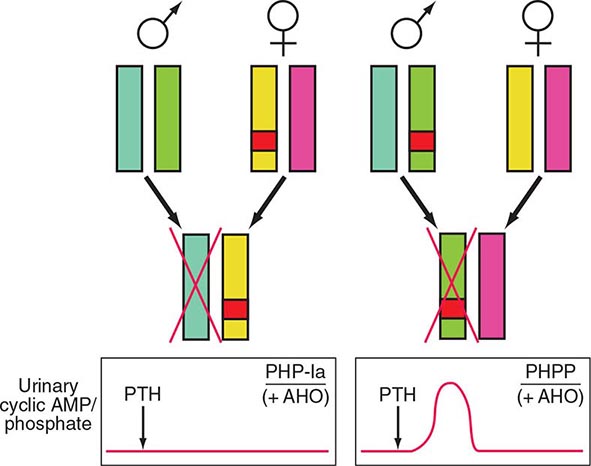
FIGURE 424-7 Paternal imprinting of renal parathyroid hormone (PTH) resistance. An impaired excretion of urinary cyclic AMP and phosphate is observed in patients with pseudohypoparathyroidism type Ia (PHP-Ia). In the renal cortex, there is selective silencing of paternal Gsα expression. The disease becomes manifest only in patients who inherit the defective gene from an obligate female carrier (left). If the genetic defect is inherited from an obligate male gene carrier, there is no biochemical abnormality; administration of PTH causes an appropriate increase in the urinary cyclic AMP and phosphate concentration (pseudo-PHP [PPHP]; right). Both patterns of inheritance lead to Albright’s hereditary osteodystrophy (AHO), perhaps because of haplotype insufficiency—i.e., both copies of Gsα must be active for normal bone development.
Imprinting is tissue selective. Paternal Gsα expression is not silenced in most tissues. It seems likely, therefore, that the AHO phenotype recognized in PPHP as well as PHP-Ia reflects Gsα haploinsufficiency during embryonic or postnatal development.
The complex mechanisms that control the GNAS gene contribute to challenges involved in unraveling the pathogenesis of these disorders, especially that of PHP-Ib. Much intensive work with families in which multiple members are affected by PHP-Ib, as well as studies of the complex regulation of the GNAS gene locus, have now shown that PHP-Ib is caused by microdeletions within or upstream of the GNAS locus, which are associated with a loss of DNA methylation at one or several loci of the maternal allele (Table 424-6). These abnormalities in methylation silence the expression of the gene. This leads in the proximal renal tubules—where Gsα appears to be expressed exclusively from the maternal allele—to PTH resistance.
PHP-Ib, lacking the AHO phenotype in most instances, shares with PHP-Ia the hypocalcemia and hyperphosphatemia caused by PTH resistance, and thus the blunted urinary cAMP response to administered PTH, a standard test to assess the presence or absence of hormone resistance (Table 424-6). Furthermore, these endocrine abnormalities become apparent only if the disease-causing mutation is inherited maternally. Bone responsiveness may be excessive rather than blunted in PHP-Ib (and in PHP-Ia) patients, based on case reports that have emphasized an osteitis fibrosa–like pattern in several PHP-Ib patients.
PHP-II refers to patients with hypocalcemia and hyperphosphatemia, who have a normal urinary cAMP but an impaired urinary phosphaturic response to PTH. In a PHP-II variant, referred to as acrodysostosis with hormonal resistance (ADOHR), patients have a defect in the regulatory subunit of PKA (PRKAR1A) that mediates the response to PTH distal to cAMP production. Acrodysostosis without hormonal resistance is caused by mutations in the cAMP-selective phosphodiesterase 4 (ADOP4). It remains unclear why the PTH-resistance in some patients, labeled as PHP-II without bony abnormalities, resolves upon treatment with vitamin D supplements.
The diagnosis of these hormone-resistant states can usually be made without difficulty when there is a positive family history for features of AHO, in association with the signs and symptoms of hypocalcemia. In both categories—PHP-Ia and PHP-Ib—serum PTH levels are elevated, particularly when patients are hypocalcemic. However, patients with PHP-Ib or PHP-II without acrodysostosis present only with hypocalcemia and high PTH levels, as evidence for hormone resistance. In PHP-Ia and PHP-Ib, the response of urinary cAMP to the administration of exogenous PTH is blunted. The diagnosis of PHP-II, in the absence of acrodysostosis, is more complex, and vitamin D deficiency must be excluded before such a diagnosis can be entertained.
|
TREATMENT |
PSEUDOHYPOPARATHYROIDISM |
Treatment of PHP is similar to that of hypoparathyroidism, except that calcium and vitamin D doses are usually higher. Patients with PHP show no PTH-resistance in the distal tubules—hence, urinary calcium clearance is typically reduced, and they are not at risk of developing nephrocalcinosis as are patients with true hypoparathyroidism, unless overtreatment occurs, for example, after the completion of pubertal development and skeletal mutation, when calcium and 1,25(OH)2D treatment should be reduced. Variability in response makes it necessary to establish the optimal regimen for each patient, based on maintaining appropriate blood calcium level and urinary calcium excretion and keeping the PTH level within or slightly above the normal range.
PTH OVERWHELMED
Occasionally, loss of calcium from the ECF is so severe that PTH cannot compensate. Such situations include acute pancreatitis and severe, acute hyperphosphatemia, often in association with renal failure, conditions in which there is rapid efflux of calcium from the ECF. Severe hypocalcemia can occur quickly; PTH rises in response to hypocalcemia but does not return blood calcium to normal.
Severe, Acute Hyperphosphatemia Severe hyperphosphatemia is associated with extensive tissue damage or cell destruction (Chap. 423). The combination of increased release of phosphate from muscle and impaired ability to excrete phosphorus because of renal failure causes moderate to severe hyperphosphatemia, the latter causing calcium loss from the blood and mild to moderate hypocalcemia. Hypocalcemia is usually reversed with tissue repair and restoration of renal function as phosphorus and creatinine values return to normal. There may even be a mild hypercalcemic period in the oliguric phase of renal function recovery. This sequence, severe hypocalcemia followed by mild hypercalcemia, reflects widespread deposition of calcium in muscle and subsequent redistribution of some of the calcium to the ECF after phosphate levels return to normal.
Other causes of hyperphosphatemia include hypothermia, massive hepatic failure, and hematologic malignancies, either because of high cell turnover of malignancy or because of cell destruction by chemotherapy.
|
TREATMENT |
SEVERE, ACUTE HYPERPHOSPHATEMIA |
Treatment is directed toward lowering of blood phosphate by the administration of phosphate-binding antacids or dialysis, often needed for the management of CKD. Although calcium replacement may be necessary if hypocalcemia is severe and symptomatic, calcium administration during the hyperphosphatemic period tends to increase extraosseous calcium deposition and aggravate tissue damage. The levels of 1,25(OH)2D may be low during the hyperphosphatemic phase and return to normal during the oliguric phase of recovery.
Osteitis Fibrosa after Parathyroidectomy Severe hypocalcemia after parathyroid surgery is rare now that osteitis fibrosa cystica is an infrequent manifestation of hyperparathyroidism. When osteitis fibrosa cystica is severe, however, bone mineral deficits can be large. After parathyroidectomy, hypocalcemia can persist for days if calcium replacement is inadequate. Treatment may require parenteral administration of calcium; addition of calcitriol and oral calcium supplementation is sometimes needed for weeks to a month or two until bone defects are filled (which, of course, is of therapeutic benefit in the skeleton), making it possible to discontinue parenteral calcium and/or reduce the amount.
DIFFERENTIAL DIAGNOSIS OF HYPOCALCEMIA
Care must be taken to ensure that true hypocalcemia is present; in addition, acute transient hypocalcemia can be a manifestation of a variety of severe, acute illnesses, as discussed above. Chronic hypocalcemia, however, can usually be ascribed to a few disorders associated with absent or ineffective PTH. Important clinical criteria include the duration of the illness, signs or symptoms of associated disorders, and the presence of features that suggest a hereditary abnormality. A nutritional history can be helpful in recognizing a low intake of vitamin D and calcium in the elderly, and a history of excessive alcohol intake may suggest magnesium deficiency.
Hypoparathyroidism and PHP are typically lifelong illnesses, usually (but not always) appearing by adolescence; hence, a recent onset of hypocalcemia in an adult is more likely due to nutritional deficiencies, renal failure, or intestinal disorders that result in deficient or ineffective vitamin D. Neck surgery, even long past, however, can be associated with a delayed onset of postoperative hypoparathyroidism. A history of seizure disorder raises the issue of anticonvulsive medication. Developmental defects may point to the diagnosis of PHP. Rickets and a variety of neuromuscular syndromes and deformities may indicate ineffective vitamin D action, either due to defects in vitamin D metabolism or to vitamin D deficiency.
A pattern of low calcium with high phosphorus in the absence of renal failure or massive tissue destruction almost invariably means hypoparathyroidism or PHP. A low calcium and low phosphorus pattern points to absent or ineffective vitamin D, thereby impairing the action of PTH on calcium metabolism (but not phosphate clearance). The relative ineffectiveness of PTH in calcium homeostasis in vitamin D deficiency, anticonvulsant therapy, gastrointestinal disorders, and hereditary defects in vitamin D metabolism leads to secondary hyperparathyroidism as a compensation. The excess PTH on renal tubule phosphate transport accounts for renal phosphate wasting and hypophosphatemia.
Exceptions to these patterns may occur. Most forms of hypomagnesemia are due to long-standing nutritional deficiency as seen in chronic alcoholics. Despite the fact that the hypocalcemia is principally due to an acute absence of PTH, phosphate levels are usually low, rather than elevated, as in hypoparathyroidism. Chronic renal failure is often associated with hypocalcemia and hyperphosphatemia, despite secondary hyperparathyroidism.
Diagnosis is usually established by application of the PTH immunoassay, tests for vitamin D metabolites, and measurements of the urinary cAMP response to exogenous PTH. In hereditary and acquired hypoparathyroidism and in severe hypomagnesemia, PTH is either undetectable or inappropriately in the normal range (Fig. 424-4). This finding in a hypocalcemic patient is supportive of hypoparathyroidism, as distinct from ineffective PTH action, in which even mild hypocalcemia is associated with elevated PTH levels. Hence a failure to detect elevated PTH levels establishes the diagnosis of hypoparathyroidism; elevated levels suggest the presence of secondary hyperparathyroidism, as found in many of the situations in which the hormone is ineffective due to associated abnormalities in vitamin D action. Assays for 25(OH)D can be helpful. Low or low-normal 25(OH)D indicates vitamin D deficiency due to lack of sunlight, inadequate vitamin D intake, or intestinal malabsorption. Recognition that mild hypocalcemia, rickets, and hypophosphatemia are due to anticonvulsant therapy is made by history.
|
TREATMENT |
HYPOCALCEMIC STATES |
The management of hypoparathyroidism, PHP, chronic renal failure, and hereditary defects in vitamin D metabolism involves the use of vitamin D or vitamin D metabolites and calcium supplementation. Vitamin D itself is the least expensive form of vitamin D replacement and is frequently used in the management of uncomplicated hypoparathyroidism and some disorders associated with ineffective vitamin D action. When vitamin D is used prophylactically, as in the elderly or in those with chronic anticonvulsant therapy, there is a wider margin of safety than with the more potent metabolites. However, most of the conditions in which vitamin D is administered chronically for hypocalcemia require amounts 50–100 times the daily replacement dose because the formation of 1,25(OH)2D is deficient. In such situations, vitamin D is no safer than the active metabolite because intoxication can occur with high-dose therapy (because of storage in fat). Calcitriol is more rapid in onset of action and also has a short biologic half-life.
Vitamin D (at least 1000 U/d [2–3 μg/d] [higher levels required in older persons]) or calcitriol (0.25–1 μg/d) is required to prevent rickets in normal individuals. In contrast, 40,000–120,000 U (1–3 mg) of vitamin D2 or D3 is typically required in hypoparathyroidism. The dose of calcitriol is unchanged in hypoparathyroidism, because the defect is in hydroxylation by the 25(OH)D-1α-hydroxylase. Calcitriol is also used in disorders of 25(OH)D-1α-hydroxylase; vitamin D receptor defects are much more difficult to treat.
Patients with hypoparathyroidism should be given 2–3 g of elemental calcium PO each day. The two agents, vitamin D or calcitriol and oral calcium, can be varied independently. Urinary calcium excretion needs to be monitored carefully. If hypocalcemia alternates with episodes of hypercalcemia in high-brittleness patients with hypoparathyroidism, administration of calcitriol and use of thiazides, as discussed above, may make management easier. Clinical trials with PTH(1–34) or PTH(1–84) are promising, but these alternative treatments have not yet been approved.
425 |
Osteoporosis |
Osteoporosis, a condition characterized by decreased bone strength, is prevalent among postmenopausal women but also occurs in men and women with underlying conditions or major risk factors associated with bone demineralization. Its chief clinical manifestations are vertebral and hip fractures, although fractures can occur at almost any skeletal site. Osteoporosis affects almost 10 million individuals in the United States, but only a small proportion are diagnosed and treated.
DEFINITION
Osteoporosis is defined as a reduction in the strength of bone that leads to an increased risk of fractures. Loss of bone tissue is associated with deterioration in skeletal microarchitecture. The World Health Organization (WHO) operationally defines osteoporosis as a bone density that falls 2.5 standard deviations (SD) below the mean for young healthy adults of the same sex—also referred to as a T-score of –2.5. Postmenopausal women who fall at the lower end of the young normal range (a T-score <–1.0) are defined as having low bone density and are also at increased risk of osteoporosis. Although risk is lower in this group, more than 50% of fractures among postmenopausal women, including hip fractures, occur in this group with low bone density, because the number of individuals in this category is so much larger than that in the osteoporosis range. As a result, there are ongoing attempts to identify individuals within the low bone density range who are at high risk of fracture and might benefit from pharmacologic intervention. Furthermore, some have advocated using fracture risk as the “diagnostic” criterion for osteoporosis.
EPIDEMIOLOGY
In the United States, as many as 9 million adults have osteoporosis (T-score <–2.5 in either spine or hip), and an additional 48 million individuals have bone mass levels that put them at increased risk of developing osteoporosis (e.g., bone mass T-score <–1.0). Osteoporosis occurs more frequently with increasing age as bone tissue is lost progressively. In women, the loss of ovarian function at menopause (typically about age 50) precipitates rapid bone loss so that most women meet the diagnostic criterion for osteoporosis by age 70–80. As the population continues to age, the number of individuals with osteoporosis and fractures will also continue to increase, despite a recognized reduction in age-specific risk. It is estimated that about 2 million fractures occur each year in the United States as a consequence of osteoporosis, and that number is expected to increase as the population continues to age.
The epidemiology of fractures follows the trend for loss of bone density, with exponential increases in both hip and vertebral fractures with age. Fractures of the distal radius have a somewhat different epidemiology, increasing in frequency before age 50 and plateauing by age 60, with only a modest age-related increase thereafter. In contrast, incidence rates for hip fractures double every 5 years after age 70 (Fig. 425-1). This distinct epidemiology may be related to the way the elderly fall as they age, with fewer falls on an outstretched hand and more falls directly on the hip. About 300,000 hip fractures occur each year in the United States, most of which require hospital admission and surgical intervention. The probability that a 50-year-old white individual will have a hip fracture during his or her lifetime is 14% for women and 5% for men; the risk for African Americans is lower (about one-half those rates), and the risk for Asians is roughly equal to that for whites. Hip fractures are associated with a high incidence of deep vein thrombosis and pulmonary embolism (20–50%) and a mortality rate between 5 and 20% during the year after surgery. there is also significant morbidity, with about 20–40% of survivors requiring long-term care, and many who are unable to function as they did before the fracture.
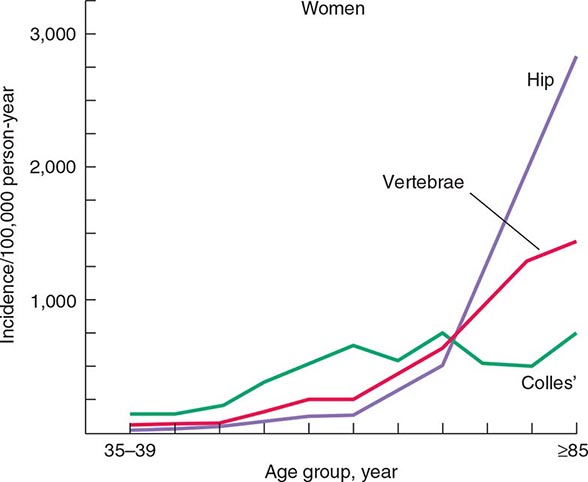
FIGURE 425-1 Epidemiology of vertebral, hip, and Colles’ fractures with age. (Adapted from C Cooper, LJ Melton III: Trends Endocrinol Metab 3:224, 1992; with permission.)
There are about 550,000 vertebral crush fractures per year in the United States. Only a fraction (estimated to be one-third) of them are recognized clinically, because many are relatively asymptomatic and are identified incidentally during radiography for other purposes (Fig. 425-2). Vertebral fractures rarely require hospitalization but are associated with long-term morbidity and a slight increase in mortality rates, primarily related to pulmonary disease. Multiple vertebral fractures lead to height loss (often of several inches), kyphosis, and secondary pain and discomfort related to altered biomechanics of the back. Thoracic fractures can be associated with restrictive lung disease, whereas lumbar fractures are associated with abdominal symptoms that include distention, early satiety, and constipation.
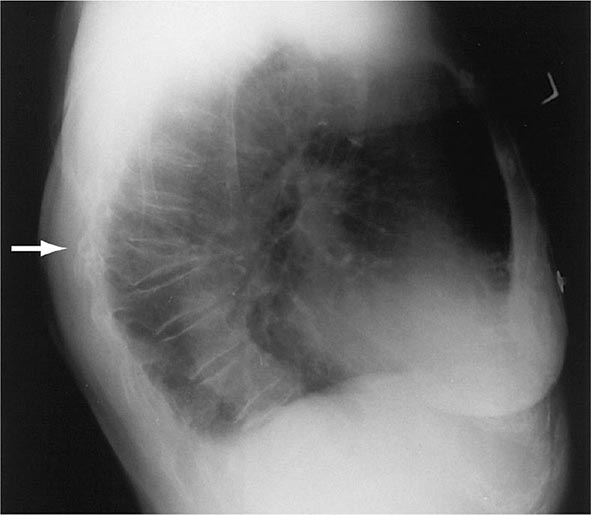
FIGURE 425-2 Lateral spine x-ray showing severe osteopenia and a severe wedge-type deformity (severe anterior compression).
Approximately 400,000 wrist fractures and 135,000 pelvic fractures occur in the United States each year. Fractures of the humerus and other bones (estimated to be about 675,000 per year) also occur with osteoporosis; this is not surprising in light of the fact that bone loss is a systemic phenomenon. Although some fractures result from major trauma, the threshold for fracture is reduced for an osteoporotic bone (Fig. 425-3). In addition to bone density, there are a number of risk factors for fracture; the common ones are summarized in Table 425-1 Age, prior fractures (especially recent fractures), a family history of osteoporosis-related fractures, low body weight, smoking, and excessive alcohol use are all independent predictors of fracture. Chronic diseases with inflammatory components that increase skeletal remodeling such as rheumatoid arthritis, increase the risk of osteoporosis, as do diseases associated with malabsorption. Chronic diseases that increase the risk of falling or frailty, including dementia, Parkinson’s disease, and multiple sclerosis, also increase fracture risk.
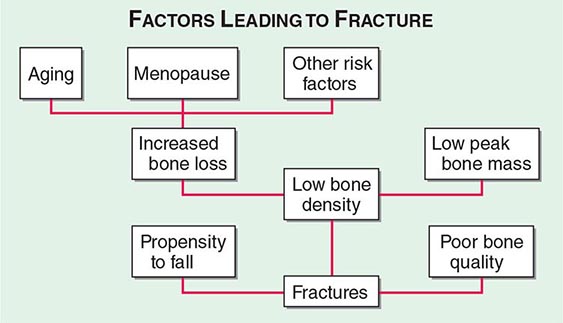
FIGURE 425-3 Factors leading to osteoporotic fractures.
|
CONDITIONS, DISEASES, AND MEDICATIONS THAT CONTRIBUTE TO OSTEOPOROSIS AND FRACTURES |
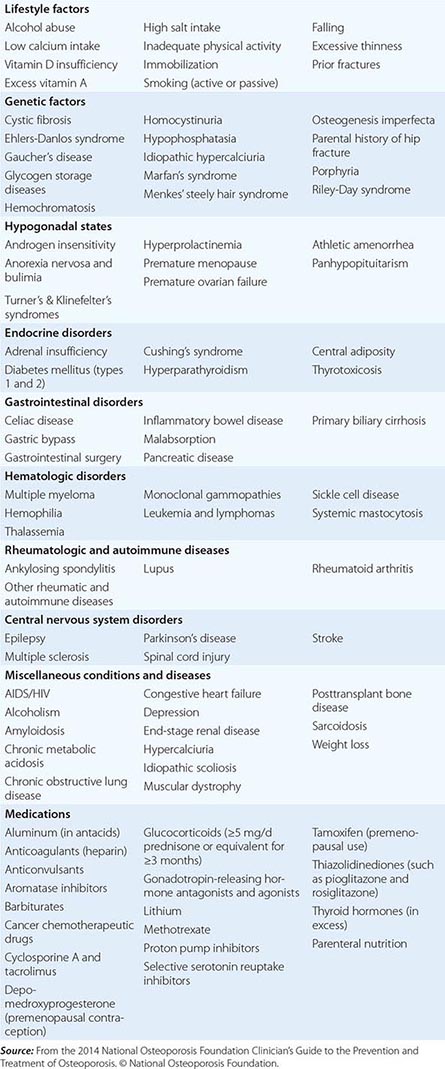
In the United States and Europe, osteoporosis-related fractures are more common among women than men, presumably due to a lower peak bone mass as well as postmenopausal bone loss in women. However, this sex difference in bone density and age-related increase in hip fractures is not as apparent in some other cultures, possibly due to genetics, physical activity level, or diet.
Fractures are themselves risk factors for future fractures (Table 425-1). Vertebral fractures increase the risk of other vertebral fractures as well as fractures of the peripheral skeleton such as the hip and wrist. Wrist fractures also increase the risk of vertebral and hip fractures. The risk for subsequent fractures is particularly high in the first several years after the first fracture, and the risk wanes considerably thereafter. Consequently, among individuals over age 50, any fracture should be considered as potentially related to osteoporosis regardless of the circumstances of the fracture. Osteoporotic bone is more likely to fracture than normal bone at any level of trauma, and a fracture in a person over 50 should trigger evaluation for osteoporosis. This often does not occur because postfracture care is not always well coordinated.
PATHOPHYSIOLOGY
BONE REMODELING
Osteoporosis results from bone loss due to age-related changes in bone remodeling as well as extrinsic and intrinsic factors that exaggerate this process. These changes may be superimposed on a low peak bone mass. Consequently, understanding the bone remodeling process is fundamental to understanding the pathophysiology of osteoporosis (Chap. 423). During growth, the skeleton increases in size by linear growth and by apposition of new bone tissue on the outer surfaces of the cortex (Fig. 425-4). The latter process is called modeling, a process that also allows the long bones to adapt in shape to the stresses placed on them. Increased sex hormone production at puberty is required for skeletal maturation, which reaches maximum mass and density in early adulthood. It is around puberty that the sexual dimorphism in skeletal size becomes obvious, although true bone density remains similar between the sexes. Nutrition and lifestyle also play an important role in growth, although genetic factors primarily determine peak skeletal mass and density. Numerous genes control skeletal growth, peak bone mass, and body size, as well as skeletal structure and density. Heritability estimates of 50–80% for bone density and size have been derived on the basis of twin studies. Although peak bone mass is often lower among individuals with a family history of osteoporosis, association studies of candidate genes (vitamin D receptor; type I collagen, the estrogen receptor [ER], and interleukin 6 [IL-6]; and insulin-like growth factor I [IGF-I]) and bone mass, bone turnover, and fracture prevalence have been inconsistent. Linkage studies suggest that a genetic locus on chromosome 11 is associated with high bone mass. Families with high bone mass and without much apparent age-related bone loss have been shown to have a point mutation in LRP5, a low-density lipoprotein receptor–related protein. The role of this gene in the general population is not clear, although a nonfunctional mutation results in osteoporosis-pseudoglioma syndrome, and LRP5 signaling appears to be important in controlling bone formation. LRP5 acts through the Wnt signaling pathway. With LRP5 and Wnt activation, beta-catenin is translocated to the nucleus, allowing stimulation of osteoblast formation, activation, and life span as well as suppression of osteoclast activity, thereby increasing bone formation. The osteocyte product, sclerostin, is a negative inhibitor of Wnt signaling.
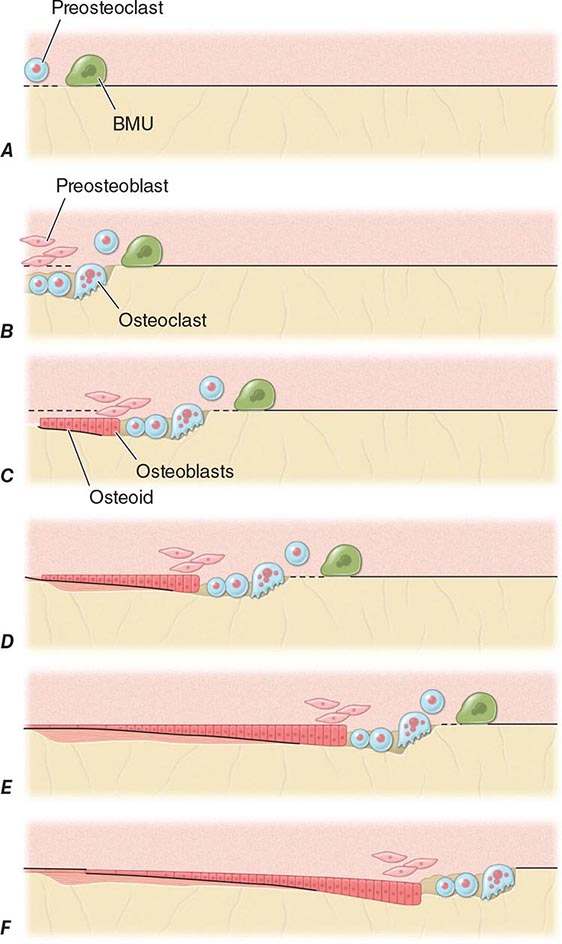
FIGURE 425-4 Mechanism of bone remodeling. The basic molecular unit (BMU) moves along the trabecular surface at a rate of about 10 μm/d. The figure depicts remodeling over ~120 days. A. Origination of BMU-lining cells contracts to expose collagen and attract preosteoclasts. B. Osteoclasts fuse into multinucleated cells that resorb a cavity. Mononuclear cells continue resorption, and preosteoblasts are stimulated to proliferate. C. Osteoblasts align at bottom of cavity and start forming osteoid (black). D. Osteoblasts continue formation and mineralization. Previous osteoid starts to mineralize (horizontal lines). E. Osteoblasts begin to flatten. F. Osteoblasts turn into lining cells; bone remodeling at initial surface (left of drawing) is now complete, but BMU is still advancing (to the right). (Adapted from SM Ott, in JP Bilezikian et al [eds]: Principles of Bone Biology, vol. 18. San Diego, Academic Press, 1996, pp 231–241.)
Genome-wide scans for low bone mass suggest multiple genes are involved, many of which are also implicated in control of body size.
In adults, bone remodeling, not modeling, is the principal metabolic skeletal process. Bone remodeling has two primary functions: (1) to repair microdamage within the skeleton to maintain skeletal strength and ensure the relative youth of the skeleton and (2) to supply calcium from the skeleton to maintain serum calcium. Remodeling may be activated by microdamage to bone as a result of excessive or accumulated stress. Acute demands for calcium involve osteoclast-mediated resorption as well as calcium transport by osteocytes. Chronic demands for calcium result in secondary hyperparathyroidism, increased bone remodeling, and overall loss of bone tissue.
Bone remodeling also is regulated by several circulating hormones, including estrogens, androgens, vitamin D, and parathyroid hormone (PTH), as well as locally produced growth factors such as IGF-I and immunoreactive growth hormone II (IGH-II), transforming growth factor β (TGF-β), parathyroid hormone–related peptide (PTHrP), interleukins (ILs), prostaglandins, and members of the tumor necrosis factor (TNF) superfamily. These factors primarily modulate the rate at which new remodeling sites are activated, a process that results initially in bone resorption by osteoclasts, followed by a period of repair during which new bone tissue is synthesized by osteoblasts. The cytokine responsible for communication between the osteoblasts, other marrow cells, and osteoclasts is RANK ligand (RANKL; receptor activator of nuclear factor-κB [NF-κB]). RANKL, a member of the TNF family, is secreted by osteoblasts and certain cells of the immune system (Chap. 423). The osteoclast receptor for this protein is referred to as RANK. Activation of RANK by RANKL is a final common path in osteoclast development, activation, and life span. A humoral decoy for RANKL, also secreted by osteoblasts, is osteoprotegerin (Fig. 425-5). Modulation of osteoclast recruitment and activity appears to be related to the interplay among these three factors. It appears that estrogens are pivotal in modulating secretion of osteoprotegerin (OPG) and perhaps also RANKL. Additional influences include nutrition (particularly calcium intake) and physical activity level.
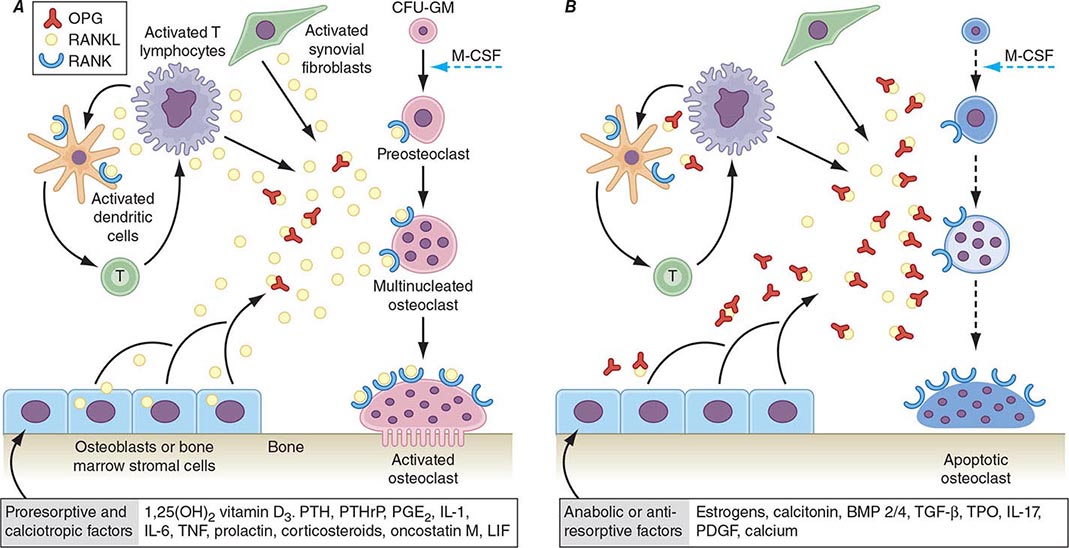
FIGURE 425-5 Hormonal control of bone resorption. A. Proresorptive and calciotropic factors. B. Anabolic and antiosteoclastic factors. RANK ligand (RANKL) expression is induced in osteoblasts, activated T cells, synovial fibroblasts, and bone marrow stromal cells. It binds to membrane-bound receptor RANK to promote osteoclast differentiation, activation, and survival. Conversely, osteoprotegerin (OPG) expression is induced by factors that block bone catabolism and promote anabolic effects. OPG binds and neutralizes RANKL, leading to a block in osteoclastogenesis and decreased survival of preexisting osteoclasts. CFU-GM, colony-forming units, granulocyte macrophage; IL, interleukin; LIF, leukemia inhibitory factor; M-CSF, macrophage colony-stimulating factor; OPG-L, osteoprotegerin-ligand; PDGF, platelet-derived growth factor; PGE2, prostaglandin E2; PTH, parathyroid hormone; RANKL, receptor activator of nuclear factor nuclear factor-κB; TGF-β, transforming growth factor β; TNF, tumor necrosis factor; TPO, thrombospondin. (From WJ Boyle et al: Nature 423: 337, 2003.)
In young adults, resorbed bone is replaced by an equal amount of new bone tissue. Thus, the mass of the skeleton remains constant after peak bone mass is achieved in adulthood. After age 30–45, however, the resorption and formation processes become imbalanced, and resorption exceeds formation. This imbalance may begin at different ages and varies at different skeletal sites; it becomes exaggerated in women after menopause. Excessive bone loss can be due to an increase in osteoclastic activity and/or a decrease in osteoblastic activity. In addition, an increase in remodeling activation frequency, and thus the number of remodeling sites, can magnify the small imbalance seen at each remodeling unit. Increased recruitment of bone remodeling sites produces a reversible reduction in bone tissue but also can result in permanent loss of tissue and disrupted skeletal architecture. In trabecular bone, if the osteoclasts penetrate trabeculae, they leave no template for new bone formation to occur, and, consequently, rapid bone loss ensues and cancellous connectivity becomes impaired. A higher number of remodeling sites increases the likelihood of this event. In cortical bone, increased activation of remodeling creates more porous bone. The effect of this increased porosity on cortical bone strength may be modest if the overall diameter of the bone is not changed. However, decreased apposition of new bone on the periosteal surface coupled with increased endocortical resorption of bone decreases the biomechanical strength of long bones. Even a slight exaggeration in normal bone loss increases the risk of osteoporosis-related fractures because of the architectural changes that occur, and osteoporosis is primarily a disease of disordered skeletal architecture. The main clinically available tool (dual-energy x-ray absorptiometry) measures mass not architecture. Emerging data from high-resolution peripheral quantitative computed tomography (CT) scans suggest that aging is associated with changes in microstructure of bone tissue, including increased cortical porosity and reduced cortical thickness.
CALCIUM NUTRITION
Peak bone mass may be impaired by inadequate calcium intake during growth among other nutritional factors (calories, protein, and other minerals), leading to increased risk of osteoporosis later in life. During the adult phase of life, insufficient calcium intake contributes to relative secondary hyperparathyroidism and an increase in the rate of bone remodeling to maintain normal serum calcium levels. PTH stimulates the hydroxylation of vitamin D in the kidney, leading to increased levels of 1,25-dihydroxyvitamin D [1,25(OH)2D] and enhanced gastrointestinal calcium absorption. PTH also reduces renal calcium loss. Although these are all appropriate compensatory homeostatic responses for adjusting calcium economy, the long-term effects are detrimental to the skeleton because the increased remodeling rates and the ongoing imbalance between resorption and formation at remodeling sites combine to accelerate loss of bone tissue.
Total daily calcium intakes <400 mg are detrimental to the skeleton, and intakes in the range of 600–800 mg, which is about the average intake among adults in the United States, are also probably suboptimal. The recommended daily required intake of 1000–1200 mg for adults accommodates population heterogeneity in controlling calcium balance (Chap. 95e). Such intakes should preferentially come from dietary sources, and supplements should be used only when dietary intakes fall short. The supplement should contain enough calcium to bring total intake to about 1200 mg/d.
VITAMIN D
(See also Chap. 423) Severe vitamin D deficiency causes rickets in children and osteomalacia in adults. However, there is accumulating evidence that vitamin D insufficiency may be more prevalent than previously thought, particularly among individuals at increased risk such as the elderly; those living in northern latitudes; and individuals with poor nutrition, malabsorption, or chronic liver or renal disease. Dark-skinned individuals are also at high risk of vitamin D deficiency. There is controversy regarding optimal levels of serum 25-hydroxy-vitamin D [25(OH)D], with some advocating levels >20 ng/mL and others advocating optimal targets >75 nmol/L (30 ng/mL). To achieve this level for most adults requires an intake of 800–1000 units/d, particularly in individuals who avoid sunlight or routinely use ultra-violet-blocking lotions. Vitamin D insufficiency leads to compensatory secondary hyperparathyroidism and is an important risk factor for osteoporosis and fractures. Some studies have shown that >50% of inpatients on a general medical service exhibit biochemical features of vitamin D deficiency, including increased levels of PTH and alkaline phosphatase and lower levels of ionized calcium. In women living in northern latitudes, vitamin D levels decline during the winter months. This is associated with seasonal bone loss, reflecting increased bone turnover. Even among healthy ambulatory individuals, mild vitamin D deficiency is increasing in prevalence, in part due to decreased exposure to sunlight coupled with increased use of potent sunscreens. Treatment with vitamin D can return levels to normal and prevent the associated increase in bone remodeling, bone loss, and fractures. Improved muscle function and gait associated with reduced falls and fracture rates also have been documented among individuals in northern latitudes who have greater vitamin D intake and higher 25(OH)D levels (see below). Vitamin D adequacy also may affect risk and/or severity of other diseases, including cancers (colorectal, prostate, and breast), autoimmune diseases, and diabetes; however, many observational studies suggesting these potential extraskeletal benefits have not been confirmed with randomized controlled trials.
ESTROGEN STATUS
Estrogen deficiency probably causes bone loss by two distinct but interrelated mechanisms: (1) activation of new bone remodeling sites and (2) exaggeration of the imbalance between bone formation and resorption. The change in activation frequency causes a transient bone loss until a new steady state between resorption and formation is achieved. The remodeling imbalance, however, results in a permanent decrement in mass. In addition, the very presence of more remodeling sites in the skeleton increases the probability that trabeculae will be penetrated, eliminating the template on which new bone can be formed and accelerating the loss of bony tissue.
The most common estrogen-deficient state is the cessation of ovarian function at the time of menopause, which occurs on average at age 51 (Chap. 413). Thus, with current life expectancy, an average woman will spend about 30 years without an ovarian supply of estrogen. The mechanism by which estrogen deficiency causes bone loss is summarized in Fig. 425-5. Marrow cells (macrophages, monocytes, osteoclast precursors, mast cells) as well as bone cells (osteoblasts, osteocytes, osteoclasts) express ERs α and β. Loss of estrogen increases production of RANKL and may reduce production of OPG, increasing osteoclast recruitment. Estrogen also may play an important role in determining the life span of bone cells by controlling the rate of apoptosis. Thus, in situations of estrogen deprivation, the life span of osteoblasts may be decreased, whereas the longevity and activity of osteoclasts are increased. The rate and duration of bone loss after menopause are heterogeneous and unpredictable. Once surfaces are lost in cancellous bone, the rate of bone loss must decline. In cortical bone, loss is slower but continues for a longer time period.
Because remodeling is initiated at the surface of bone, it follows that trabecular bone—which has a considerably larger surface area (80% of the total) than cortical bone—will be affected preferentially by estrogen deficiency. Fractures occur earliest at sites where trabecular bone contributes most to bone strength; consequently, vertebral fractures are the most common early consequence of estrogen deficiency.
PHYSICAL ACTIVITY
Inactivity, such as prolonged bed rest or paralysis, results in significant bone loss. Concordantly, athletes have higher bone mass than does the general population. These changes in skeletal mass are most marked when the stimulus begins during growth and before the age of puberty. Adults are less capable than children of increasing bone mass after restoration of physical activity. Epidemiologic data support the beneficial effects on the skeleton of chronic high levels of physical activity. Fracture risk is lower in rural communities and in countries where physical activity is maintained into old age. However, when exercise is initiated during adult life, the effects of moderate exercise on the skeleton are modest, with a bone mass increase of 1–2% in short-term studies of <2 years in duration. It is argued that more active individuals are less likely to fall and are more capable of protecting themselves upon falling, thereby reducing fracture risk.
CHRONIC DISEASE
Various genetic and acquired diseases are associated with an increase in the risk of osteoporosis (Table 425-1). Mechanisms that contribute to bone loss are unique for each disease and typically result from multiple factors, including nutrition, reduced physical activity levels, and factors that affect rates of bone remodeling. In most, but not all, circumstances the primary diagnosis is made before osteoporosis presents clinically.
MEDICATIONS
A large number of medications used in clinical practice have potentially detrimental effects on the skeleton (Table 425-1). Glucocorticoids are the most common cause of medication-induced osteoporosis. It is often not possible to determine the extent to which osteoporosis is related to glucocorticoids or to other factors, because treatment is superimposed on the effects of the primary disease, which in itself may be associated with bone loss (e.g., rheumatoid arthritis). Excessive doses of thyroid hormone can accelerate bone remodeling and result in bone loss.
Other medications have less detrimental effects on the skeleton than pharmacologic doses of glucocorticoids. Anticonvulsants are thought to increase the risk of osteoporosis, although many affected individuals have concomitant insufficiency of 1,25(OH)2D, as some anticonvulsants induce the cytochrome P450 system and vitamin D metabolism. Patients undergoing transplantation are at high risk for rapid bone loss and fracture not only from glucocorticoids but also from treatment with other immunosuppressants such as cyclosporine and tacrolimus (FK506). In addition, these patients often have underlying metabolic abnormalities, such as hepatic or renal failure, that predispose to bone loss.
Aromatase inhibitors, which potently block the aromatase enzyme that converts androgens and other adrenal precursors to estrogen, reduce circulating postmenopausal estrogen levels dramatically. These agents, which are used in various stages for breast cancer treatment, also have been shown to have a detrimental effect on bone density and risk of fracture. More recently a variety of agents have been implicated in increased bone loss and fractures. These include selective serotonin reuptake inhibitors, proton pump inhibitors, and thiazolidinediones. It is difficult in some cases to separate the risk accrued by the underlying disease from that attributable to the medication. For example, both depression and diabetes are risk factors for fracture by themselves.
CIGARETTE CONSUMPTION
The use of cigarettes over a long period has detrimental effects on bone mass. These effects may be mediated directly by toxic effects on osteoblasts or indirectly by modifying estrogen metabolism. On average, cigarette smokers reach menopause 1–2 years earlier than the general population. Cigarette smoking also produces secondary effects that can modulate skeletal status, including intercurrent respiratory and other illnesses, frailty, decreased exercise, poor nutrition, and the need for additional medications (e.g., glucocorticoids for lung disease).
MEASUREMENT OF BONE MASS
Several noninvasive techniques are available for estimating skeletal mass or density. They include dual-energy x-ray absorptiometry (DXA), single-energy x-ray absorptiometry (SXA), quantitative CT, and ultrasound (US). DXA is a highly accurate x-ray technique that has become the standard for measuring bone density. Although it can be used for measurement in any skeletal site, clinical determinations usually are made of the lumbar spine and hip. DXA also can be used to measure body composition. In the DXA technique, two x-ray energies are used to estimate the area of mineralized tissue, and the mineral content is divided by the area, which partially corrects for body size. However, this correction is only partial because DXA is a two-dimensional scanning technique and cannot estimate the depth or posteroanterior length of the bone. Thus, small slim people tend to have lower than average bone mineral density (BMD), a feature that is important in interpreting BMD measurements when performed in young adults, and something that must be taken into account at any age. Bone spurs, which are common in osteoarthritis, tend to falsely increase bone density of the spine and are a particular problem in measuring the spine in older individuals. Because DXA instrumentation is provided by several different manufacturers, the output varies in absolute terms. Consequently, it has become standard practice to relate the results to “normal” values by using T-scores (a T-score of 1 equals 1 SD), which compare individual results to those in a young population that is matched for race and sex. Z-scores (also measured in SD) compare individual results to those of an age-matched population that also is matched for race and sex. Thus, a 60-year-old woman with a Z-score of –1 (1 SD below mean for age) has a T-score of –2.5 (2.5 SD below mean for a young control group) (Fig. 425-6). A T-score below –2.5 in the lumbar spine, femoral neck, or total hip has been defined as a diagnosis of osteoporosis. As noted above, because more than 50% of fractures occur in individuals with low bone mass rather than BMD osteoporosis, attempts are ongoing to redefine the disease as a fracture risk rather than a specific BMD. Consistent with this concept, fractures of the spine and hip that occur in the absence of major trauma would be considered to be sufficient to diagnose osteoporosis, regardless of BMD. Fractures of other sites, such as pelvis, proximal humerus, and wrist, would be tantamount to an osteoporosis diagnosis in the presence of low BMD. CT can also be used to measure the spine and the hip, but is rarely used clinically, in part because of higher radiation exposure and cost, in addition to a lesser body of data confirming its ability to predict fracture risk, compared with BMD by DXA. High-resolution peripheral CT is used to measure bone in the forearm or tibia as a research tool to noninvasively provide some measure of skeletal architecture. Magnetic resonance imaging (MRI) can also be used in research settings to obtain some architectural information on the forearm and perhaps the hip.

FIGURE 425-6 Relationship between Z-scores and T-scores in a 60-year-old woman. BMD, bone mineral density; SD, standard deviation.
DXA equipment can also be used to obtain lateral images of the spine, from T4 through L4, a technique called vertebral fracture assessment (VFA). Although not as definitive as radiography, it is a useful screening tool when height loss, back pain, or postural change suggests the presence of an undiagnosed vertebral fracture. Furthermore, because vertebral fractures are so prevalent with advancing age, screening vertebral imaging is recommended in women and men with low bone mass (T-score <1) by age 70 and 80, respectively.
US is used to measure bone mass by calculating the attenuation of the signal as it passes through bone or the speed with which it traverses the bone. It is unclear whether US assesses properties of bone other than mass (e.g., quality), but this is a potential advantage of the technique. Because of its relatively low cost and mobility, US is amenable for use as a screening procedure in stores or at health fairs.
All of these techniques for measuring BMD have been approved by the U.S. Food and Drug Administration (FDA) on the basis of their capacity to predict fracture risk. The hip is the preferred site of measurement in most individuals, because it predicts the risk of hip fracture, the most important consequence of osteoporosis, better than any other bone density measurement site. When hip measurements are performed by DXA, the spine can be measured at the same time. In younger individuals such as perimenopausal or early postmenopausal women, spine measurements may be the most sensitive indicator of bone loss. A risk assessment tool (FRAX) incorporates femoral neck BMD to assess 10-year fracture risk (see below).
WHEN TO MEASURE BONE MASS
Clinical guidelines have been developed for the use of bone densitometry in clinical practice. The original National Osteoporosis Foundation guidelines recommend bone mass measurements in postmenopausal women, assuming they have one or more risk factors for osteoporosis in addition to age, sex, and estrogen deficiency. The guidelines further recommend that bone mass measurement be considered in all women by age 65, a position ratified by the U.S. Preventive Health Services Task Force. Criteria approved for Medicare reimbursement of BMD are summarized in Table 425-2.
|
INDICATIONS FOR BONE DENSITY TESTING |
Source: From the 2014 National Osteoporosis Foundation Clinician’s Guide to the Prevention and Treatment of Osteoporosis. © National Osteoporosis Foundation.
WHEN TO TREAT BASED ON BONE MASS RESULTS
Most guidelines suggest that patients be considered for treatment when BMD is >2.5 SD below the mean value for young adults (T-score ≤–2.5), in either spine, total hip, or femoral neck. Treatment also should also be considered in postmenopausal women with fracture risk factors even if BMD is not in the osteoporosis range. Risk factors (age, prior fracture, family history of hip fracture, low body weight, cigarette consumption, excessive alcohol use, steroid use, and rheumatoid arthritis) can be combined with BMD to assess the likelihood of a fracture over a 5- or 10-year period. Treatment threshold depends on cost-effectiveness analyses but probably is ~1% per year of risk in the United States.
APPROACH TO THE PATIENT:
Osteoporosis
The perimenopausal transition is a good opportunity to initiate a discussion about risk factors for osteoporosis and consideration of indications for a BMD test. A careful history and physical examination should be performed to identify risk factors for osteoporosis. A low Z-score increases the suspicion of a secondary disease. Height loss >2.5–3.8 cm (>1–1.5 in.) is an indication for VFA by DXA or radiography to rule out asymptomatic vertebral fractures, as is the presence of significant kyphosis or back pain, particularly if it began after menopause. In appropriate individuals, screening BMD and screening vertebral imaging should be recommended as above, even in the absence of any specific risk factors (Table 425-3). For patients who present with fractures, it is important to ensure that the fractures are not caused by an underlying malignancy. Usually this is clear on routine radiography, but on occasion, CT, MRI, or radionuclide scans may be necessary.
|
INDICATIONS FOR VERTEBRAL IMAGING |
Consider vertebral imaging tests in the following individuals:
• In all women age 70 and older and all men age 80 and older if bone mineral density (BMD) T-score is –1.0 or below
• In women age 65–69 and men age 75–79 if BMD T-score is –1.5 or below
• In postmenopausal women age 50–64 and men age 50–69 with specific risk factors:
![]() Low-trauma fracture
Low-trauma fracture
![]() Historical height loss of 1.5 in. or more
Historical height loss of 1.5 in. or more
(4 cm)
![]() Prospective height loss of 0.8 in. or more (2 cm)
Prospective height loss of 0.8 in. or more (2 cm)
![]() Recent or ongoing long-term glucocorticoid treatment
Recent or ongoing long-term glucocorticoid treatment
Source: From the 2014 National Osteoporosis Foundation Clinician’s Guide to the Prevention and Treatment of Osteoporosis. © National Osteoporosis Foundation.
ROUTINE LABORATORY EVALUATION
There is no established algorithm for the evaluation of women who present with osteoporosis. A general evaluation that includes complete blood count, serum and 24-h urine calcium, renal and hepatic function tests, and a 25(OH)D level is useful for identifying selected secondary causes of low bone mass, particularly for women with fractures or very low Z-scores. An elevated serum calcium level suggests hyperparathyroidism or malignancy, whereas a reduced serum calcium level may reflect malnutrition and osteomalacia. In the presence of hypercalcemia, a serum PTH level differentiates between hyperparathyroidism (PTH↑) and malignancy (PTH↓), and a high PTHrP level can help document the presence of humoral hypercalcemia of malignancy (Chap. 424). A low urine calcium (<50 mg/24 h) suggests osteomalacia, malnutrition, or malabsorption; a high urine calcium (>300 mg/24 h) is indicative of hypercalciuria and must be investigated further. Hypercalciuria occurs primarily in three situations: (1) a renal calcium leak, which is more common in males with osteoporosis; (2) absorptive hypercalciuria, which can be idiopathic or associated with increased 1,25(OH)2D in granulomatous disease; or (3) hematologic malignancies or conditions associated with excessive bone turnover such as Paget’s disease, hyperparathyroidism, and hyperthyroidism. Renal hypercalciuria is treated with thiazide diuretics, which lower urine calcium and help improve calcium economy.
Individuals who have osteoporosis-related fractures or bone density in the osteoporotic range should have a measurement of serum 25(OH)D level, because the intake of vitamin D required to achieve a target level >20–30 ng/mL is highly variable. Vitamin D levels should be optimized in all individuals being treated for osteoporosis. Hyperthyroidism should be evaluated by measuring thyroid-stimulating hormone (TSH).
When there is clinical suspicion of Cushing’s syndrome, urinary free cortisol levels or a fasting serum cortisol should be measured after overnight dexamethasone. When bowel disease, malabsorption, or malnutrition is suspected, serum albumin, cholesterol, and a complete blood count should be checked. Asymptomatic malabsorption may be heralded by anemia (macrocytic—vitamin B12 or folate deficiency; microcytic—iron deficiency) or low serum cholesterol or urinary calcium levels. If these or other features suggest malabsorption, further evaluation is required. Asymptomatic celiac disease with selective malabsorption is being found with increasing frequency; the diagnosis can be made by testing for antigliadin, antiendomysial, or transglutaminase antibodies but may require endoscopic biopsy. A trial of a gluten-free diet can be confirmatory (Chap. 349). When osteoporosis is found associated with symptoms of rash, multiple allergies, diarrhea, or flushing, mastocytosis should be excluded by using 24-h urine histamine collection or serum tryptase.
Myeloma can masquerade as generalized osteoporosis, although it more commonly presents with bone pain and characteristic “punched-out” lesions on radiography. Serum and urine electrophoresis and or serum free light chains are required to exclude this diagnosis. More commonly, a monoclonal gammopathy of unclear significance (MGUS) is found, and the patient is subsequently monitored to ensure that this is not an incipient myeloma. Approximately 1% of patients with MGUS progress to myeloma each year. A bone marrow biopsy may be required to rule out myeloma (in patients with equivocal electrophoretic results) and also can be used to exclude mastocytosis, leukemia, and other marrow infiltrative disorders such as Gaucher’s disease. MGUS syndromes, although benign, may also be associated with reduced bone mass and elevated bone turnover.
BONE BIOPSY
Tetracycline labeling of the skeleton allows determination of the rate of remodeling as well as evaluation for other metabolic bone diseases. The current use of BMD tests, in combination with hormonal evaluation and biochemical markers of bone remodeling, has largely replaced the clinical use of bone biopsy, although it remains an important tool in clinical research and assessment of mechanism of action of medication for osteoporosis.
BIOCHEMICAL MARKERS
Several biochemical tests are available that provide an index of the overall rate of bone remodeling (Table 425-4). Biochemical markers usually are characterized as those related primarily to bone formation or bone resorption. These tests measure the overall state of bone remodeling at a single point in time. Clinical use of these tests has been hampered by biologic variability (in part related to circadian rhythm) as well as analytic variability, although the latter is improving.
|
INDICATIONS FOR BIOCHEMICAL MARKERS |
Abbreviations: BMD, bone mineral density; FDA, U.S. Food and Drug Administration.
Source: Adapted from the 2014 National Osteoporosis Foundation Clinician’s Guide to the Prevention and Treatment of Osteoporosis. © National Osteoporosis Foundation.
Biochemical markers of bone resorption may help in the prediction of fracture risk, independently of bone density, particularly in older individuals. In women ≥65 years, when bone density results are greater than the usual treatment thresholds noted above, a high level of bone resorption should prompt consideration of treatment. The primary use of biochemical markers is for monitoring the response to treatment. With the introduction of antiresorptive therapeutic agents, bone remodeling declines rapidly, with the fall in resorption occurring earlier than the fall in formation. Inhibition of bone resorption is maximal within 3 months or so. Thus, measurement of bone resorption (C-telopeptide [CTX] is the preferred marker) before initiating therapy and 3–6 months after starting therapy provides an earlier estimate of patient response than does bone densitometry. A decline in resorptive markers can be ascertained after treatment with potent antiresorptive agents such as bisphosphonates, denosumab, or standard-dose estrogen; this effect is less marked after treatment with weaker agents such as raloxifene or intranasal calcitonin. A biochemical marker response to therapy is particularly useful for asymptomatic patients and may help ensure long-term adherence to treatment. Bone turnover markers are also useful in monitoring the effects of osteoanabolic agents such as 1-34hPTH, or teriparatide, which rapidly increases bone formation (P1NP is preferred, but osteocalcin is a reasonable alternative) and later bone resorption. The recent suggestion of “drug holidays” (see below) has created another use for biochemical markers, allowing evaluation of the off effect of drugs such as bisphosphonates.
|
TREATMENT |
OSTEOPOROSIS |
MANAGEMENT OF PATIENTS WITH FRACTURES
Treatment of a patient with osteoporosis frequently involves management of acute fractures as well as treatment of the underlying disease. Hip fractures almost always require surgical repair if the patient is to become ambulatory again. Depending on the location and severity of the fracture, condition of the neighboring joint, and general status of the patient, procedures may include open reduction and internal fixation with pins and plates, hemiarthroplasties, and total arthroplasties. These surgical procedures are followed by intense rehabilitation in an attempt to return patients to their prefracture functional level. Long bone fractures (e.g., wrist) often require either external or internal fixation. Other fractures (e.g., vertebral, rib, and pelvic fractures) usually are managed with supportive care, requiring no specific orthopedic treatment.
Only ~25–30% of vertebral compression fractures present with sudden-onset back pain. For acutely symptomatic fractures, treatment with analgesics is required, including nonsteroidal anti-inflammatory agents and/or acetaminophen, sometimes with the addition of a narcotic agent (codeine or oxycodone). A few small, randomized clinical trials suggest that calcitonin may reduce pain related to acute vertebral compression fracture. Percutaneous injection of artificial cement (polymethylmethacrylate) into the vertebral body (vertebroplasty or kyphoplasty) may offer significant immediate pain relief in patients with severe pain from acute or subacute vertebral fractures. Safety concerns include extravasation of cement with neurologic sequelae and increased risk of fracture in neighboring vertebrae due to mechanical rigidity of the treated bone. Exactly which patients are the optimal candidates for this procedure remains unknown. Short periods of bed rest may be helpful for pain management, but in general, early mobilization is recommended because it helps prevent further bone loss associated with immobilization. Occasionally, use of a soft elastic-style brace may facilitate earlier mobilization. Muscle spasms often occur with acute compression fractures and can be treated with muscle relaxants and heat treatments.
Severe pain usually resolves within 6–10 weeks. More chronic severe pain might suggest the possibility of multiple myeloma or underlying metastatic disease. Chronic pain following vertebral fracture is probably not bony in origin; instead, it is related to abnormal strain on muscles, ligaments, and tendons and to secondary facet-joint arthritis associated with alterations in thoracic and/or abdominal shape. Chronic pain is difficult to treat effectively and may require analgesics, sometimes including narcotic analgesics. Frequent intermittent rest in a supine or semireclining position is often required to allow the soft tissues, which are under tension, to relax. Back-strengthening exercises (paraspinal) may be beneficial. Heat treatments help relax muscles and reduce the muscular component of discomfort. Various physical modalities, such as US and transcutaneous nerve stimulation, may be beneficial in some patients. Pain also occurs in the neck region, not as a result of compression fractures (which almost never occur in the cervical spine as a result of osteoporosis) but because of chronic strain associated with trying to elevate the head in a person with a significant thoracic kyphosis.
Multiple vertebral fractures often are associated with psychological symptoms; this is not always appreciated. The changes in body configuration and back pain can lead to marked loss of self-image and a secondary depression. Altered balance, precipitated by the kyphosis and the anterior movement of the body’s center of gravity, leads to a fear of falling, a consequent tendency to remain indoors, and the onset of social isolation. These symptoms sometimes can be alleviated by family support and/or psychotherapy. Medication may be necessary when depressive features are present. Multiple thoracic vertebral fractures may be associated with restrictive lung disease symptoms and increased pulmonary infections. Multiple lumbar vertebral fractures are often associated with abdominal pain, constipation, protuberance, and early satiety. Multiple vertebral fractures are associated with greater age-specific mortality.
Multiple studies show that the majority of patients presenting in adulthood with fractures are not evaluated or treated for osteoporosis. Estimates suggest only about 20% of fracture patients receive follow-up care. Patients who sustain acute fractures are at dramatically elevated risk for more fractures, particularly within the first several years, and pharmacologic intervention can reduce that risk substantially. Recently, several studies have demonstrated the effectiveness of a relatively simple and inexpensive program that reduces the risk of subsequent fractures. In the Kaiser system, it is estimated that a 20% decline in hip fracture occurrence was seen with the introduction of what is called a fracture liaison service. This typically involves a health care professional (usually a nurse) whose job is to coordinate follow-up care and education of fracture patients. If the Kaiser experience can be repeated, there would be significant savings of health care dollars, as well as a dramatic drop in hip fracture incidence and a marked improvement in morbidity and mortality among the aging population.
MANAGEMENT OF THE UNDERLYING DISEASE
Patients presenting with typical osteoporosis-related fractures (certainly hip and spine) can be assumed to have osteoporosis and can be treated appropriately. Patients with osteoporosis by BMD are handled in a similar fashion. Other fracture patients and those with reduced bone mass can be classified according to their future risk of fracture and treated if that risk is sufficiently high. It must be emphasized, however, that risk assessment is an inexact science when applied to individual patients. Fractures are chance occurrences that can happen to anyone. Patients often do not understand the relative benefits of medications, compared to the perceived risks of the medications themselves.
Risk Factor Reduction Several tools exist for risk assessment. The most commonly available is the FRAX tool, developed by a working party for the WHO, and available as part of the report from many DXA machines. It is also available online (http://www.shef.ac.uk/FRAX/tool.jsp?locationValue=9) (Fig. 425-7). In the United States, it has been estimated that it is cost-effective to treat a patient if the 10-year major fracture risk (including hip, clinical spine, proximal humerus, and tibia) from FRAX is ≥20% and/or the 10-year risk of hip fracture is ≥3%. FRAX is an imperfect tool because it does not include any assessment of fall risk and secondary causes are excluded when BMD is entered. Moreover, it does not include any term for multiple fractures or recent versus remote fracture. Nonetheless, it is useful as an educational tool for patients.
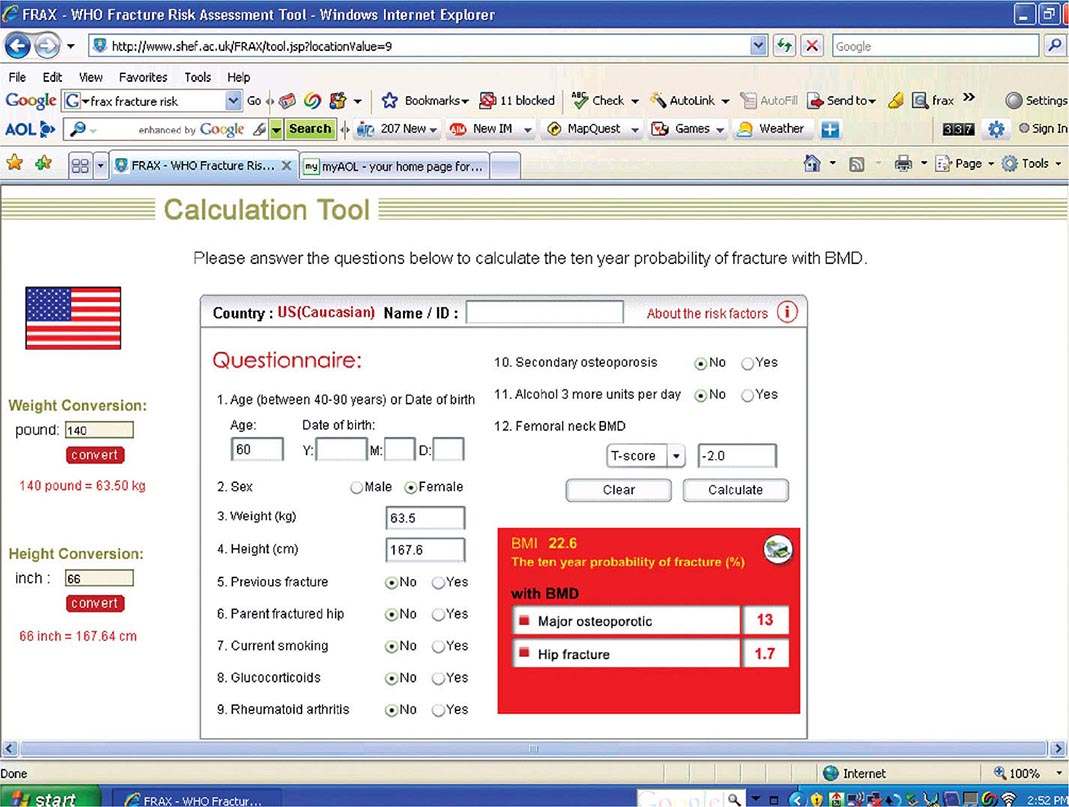
FIGURE 425-7 FRAX calculation tool. When the answers to the indicated questions are filled in, the calculator can be used to assess the 10-year probability of fracture. The calculator (available online at http://www.shef.ac.uk/FRAX/tool.jsp?locationValue=9) also can risk adjust for various ethnic groups.
After risk assessment, patients should be thoroughly educated to reduce the impact of modifiable risk factors associated with bone loss and falling. All medications that increase risk of falls, bone loss, or fractures should be reviewed to ensure that they are necessary and being used at the lowest required dose. For those on thyroid hormone replacement, TSH testing should be performed to confirm that an excessive dose is not being used, because biochemical and symptomatic thyrotoxicosis can be associated with increased bone loss. In patients who smoke, efforts should be made to facilitate smoking cessation. Reducing risk factors for falling also include alcohol abuse treatment and a review of the medical regimen for any drugs that might be associated with orthostatic hypotension and/or sedation, including hypnotics and anxiolytics. If nocturia occurs, the frequency should be reduced, if possible (e.g., by decreasing or modifying diuretic use), because arising in the middle of sleep is a common precipitant of a fall. Patients should be instructed about environmental safety with regard to eliminating exposed wires, curtain strings, slippery rugs, and mobile tables. Avoiding stocking feet on wood floors, checking carpet condition (particularly on stairs), and providing good light in paths to bathrooms and outside the home are important preventive measures. Treatment for impaired vision is recommended, particularly a problem with depth perception, which is specifically associated with increased falling risk. Elderly patients with neurologic impairment (e.g., stroke, Parkinson’s disease, Alzheimer’s disease) are particularly at risk of falling and require specialized supervision and care.
Nutritional Recommendations • CALCIUM A large body of data indicates that optimal calcium intake reduces bone loss and suppresses bone turnover. Recommended intakes from an Institute of Medicine report are shown in Table 425-5.
|
ADEQUATE CALCIUM INTAKE |
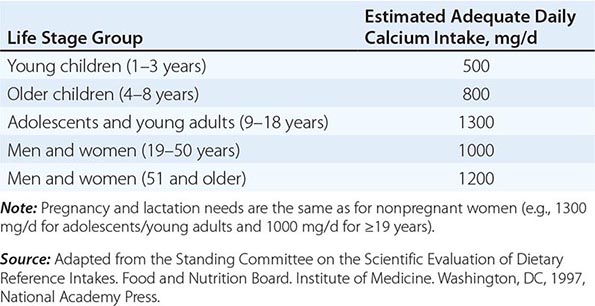
The National Health and Nutrition Examination Surveys (NHANES) have consistently documented that average calcium intakes fall considerably short of these recommendations. Food sources of calcium are dairy products (milk, yogurt, and cheese) and fortified foods such as certain cereals, waffles, snacks, juices, and crackers. Some of these fortified foods contain as much calcium per serving as milk. Green leafy vegetables and nuts, particularly almonds, are also sources of calcium, although their bioavailability may be lower than with dairy products. Calcium intake from the diet can also be assessed (Table 425-6) and calculators are available at NOF.org or NYSOPEP.org.
|
SIMPLE METHOD FOR CALCULATING DIETARY CALCIUM INTAKE |
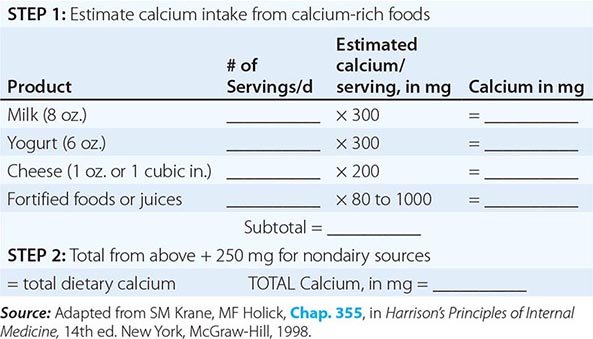
If a calcium supplement is required, it should be taken in doses sufficient to supplement dietary intake to bring total intake to the required level (1000–1200 mg/d). Doses of supplements should be ≤600 mg at a time, because the calcium absorption fraction decreases at higher doses. Calcium supplements should be calculated on the basis of the elemental calcium content of the supplement, not the weight of the calcium salt. Calcium supplements containing carbonate are best taken with food because they require acid for solubility. Calcium citrate supplements can be taken at any time. To confirm bioavailability, calcium supplements can be placed in distilled vinegar. They should dissolve within 30 min.
Several controlled clinical trials of calcium, mostly plus vitamin D, have confirmed reductions in clinical fractures, including fractures of the hip (~20–30% risk reduction). All recent studies of pharmacologic agents have been conducted in the context of calcium replacement (± vitamin D). Thus, it is standard practice to ensure an adequate calcium and vitamin D intake in patients with osteoporosis whether they are receiving additional pharmacologic therapy or not. A systematic review confirmed a greater BMD response to antiresorptive therapy when calcium intake was adequate.
Although side effects from supplemental calcium are minimal (eructation and constipation mostly with carbonate salts), individuals with a history of kidney stones should have a 24-h urine calcium determination before starting increased calcium to avoid significant hypercalciuria. Many studies confirm a small but significant increase in the risk of renal stones with calcium supplements, but not dietary calcium. A recent analysis of published data has suggested that high intakes of calcium from supplements are associated with an increase in the risk of heart disease. This is an evolving story with additional studies that confirm or refute this finding. Because high calcium supplement intakes increase the risk of renal stones and confer no extra benefit to the skeleton, the recommendation that total intakes should be between 1000 and 1200 mg/d is reasonable.
VITAMIN D Vitamin D is synthesized in skin under the influence of heat and ultraviolet light (Chap. 423). However, large segments of the population do not obtain sufficient vitamin D to maintain what is now considered an adequate supply [serum 25(OH)D consistently >75 μmol/L (30 ng/mL)]. Because vitamin D supplementation at doses that would achieve these serum levels is safe and inexpensive, the Institute of Medicine (based on obtaining a serum level of 20 ng/mL) recommends daily intakes of 200 IU for adults <50 years of age, 400 IU for those 50–70 years, and 600 IU for those >70 years. Multivitamin tablets usually contain 400 IU, and many calcium supplements also contain vitamin D. Some data suggest that higher doses (≥1000 IU) may be required in the elderly and chronically ill. The Institute of Medicine report suggests that it is safe to take up to 4000 IU/d. For those with osteoporosis or those at risk of osteoporosis, 1000–2000 IU/d can usually maintain serum 25(OH)D above 30 ng/mL.
OTHER NUTRIENTS Other nutrients such as salt, high animal protein intakes, and caffeine may have modest effects on calcium excretion or absorption. Adequate vitamin K status is required for optimal carboxylation of osteocalcin. States in which vitamin K nutrition or metabolism is impaired, such as with long-term warfarin therapy, have been associated with reduced bone mass. Research concerning cola intake is controversial but suggests a possible link to reduced bone mass through factors that are independent of caffeine. Although dark green leafy vegetables such as spinach and kale contain a fair amount of calcium, the high oxalate content reduces absorption of this calcium (but does not inhibit absorption of calcium from other food eaten simultaneously).
Magnesium is abundant in foods, and magnesium deficiency is quite rare in the absence of a serious chronic disease. Magnesium supplementation may be warranted in patients with inflammatory bowel disease, celiac disease, chemotherapy, severe diarrhea, malnutrition, or alcoholism. Dietary phytoestrogens, which are derived primarily from soy products and legumes (e.g., garbanzo beans [chickpeas] and lentils), exert some estrogenic activity but are insufficiently potent to justify their use in place of a pharmacologic agent in the treatment of osteoporosis.
Patients with hip fractures are often frail and relatively malnourished. Some data suggest an improved outcome in such patients when they are provided calorie and protein supplementation. Excessive protein intake can increase renal calcium excretion, but this can be corrected by an adequate calcium intake.
Exercise Exercise in young individuals increases the likelihood that they will attain the maximal genetically determined peak bone mass. Meta-analyses of studies performed in postmenopausal women indicate that weight-bearing exercise helps prevent bone loss but does not appear to result in substantial gain of bone mass. This beneficial effect wanes if exercise is discontinued. Most of the studies are short term, and a more substantial effect on bone mass is likely if exercise is continued over a long period. Exercise also has beneficial effects on neuromuscular function, and it improves coordination, balance, and strength, thereby reducing the risk of falling. A walking program is a practical way to start. Other activities, such as dancing, racquet sports, cross-country skiing, and use of gym equipment, are also recommended, depending on the patient’s personal preference and general condition. Even women who cannot walk benefit from swimming or water exercises, not so much for the effects on bone, which are quite minimal, but because of effects on muscle. Exercise habits should be consistent, optimally at least three times a week.
PHARMACOLOGIC THERAPIES
Before the mid-1990s, estrogen treatment, either by itself or in concert with a progestin, was the primary therapeutic agent for prevention or treatment of osteoporosis. There are now a number of new medications approved for osteoporosis and more under development. Some are agents that specifically treat osteoporosis (bisphosphonates, calcitonin, denosumab, and teriparatide [1-34hPTH]); others, such as selective estrogen response modulators (SERMs) and, most recently, an estrogen/SERM combination medication, have broader effects. The availability of these drugs allows therapy to be tailored to the needs of an individual patient.
Estrogens A large body of clinical trial data indicates that various types of estrogens (conjugated equine estrogens, estradiol, estrone, esterified estrogens, ethinyl estradiol, and mestranol) reduce bone turnover, prevent bone loss, and induce small increases in bone mass of the spine, hip, and total body. The effects of estrogen are seen in women with natural or surgical menopause and in late postmenopausal women with or without established osteoporosis. Estrogens are efficacious when administered orally or transdermally. For both oral and transdermal routes of administration, combined estrogen/progestin preparations are now available in many countries, obviating the problem of taking two tablets or using a patch and oral progestin.
Dose of Estrogen For oral estrogens, the standard recommended doses have been 0.3 mg/d for esterified estrogens, 0.625 mg/d for conjugated equine estrogens, and 5 μg/d for ethinyl estradiol. For transdermal estrogen, the commonly used dose supplies 50 μg estradiol per day, but a lower dose may be appropriate for some individuals. Dose-response data for conjugated equine estrogens indicate that lower doses (0.3 and 0.45 mg/d) are effective. Doses even lower have been associated with bone mass protection.
FRACTURE DATA Epidemiologic databases indicate that women who take estrogen replacement have a 50% reduction, on average, of osteoporotic fractures, including hip fractures. The beneficial effect of estrogen is greatest among those who start replacement early and continue the treatment; the benefit declines after discontinuation to the extent that there is no residual protective effect against fracture by 10 years after discontinuation. The first clinical trial evaluating fractures as secondary outcomes, the Heart and Estrogen-Progestin Replacement Study (HERS) trial, showed no effect of hormone therapy on hip or other clinical fractures in women with established coronary artery disease. These data made the results of the Women’s Health Initiative (WHI) exceedingly important (Chap. 413). The estrogen-progestin arm of the WHI in >16,000 postmenopausal healthy women indicated that hormone therapy reduces the risk of hip and clinical spine fracture by 34% and reduces the risk of all clinical fractures by 24%. There was similar antifracture efficacy seen with estrogen alone in women who had had a hysterectomy.
A few smaller clinical trials have evaluated spine fracture occurrence as an outcome with estrogen therapy. They have consistently shown that estrogen treatment reduces the incidence of vertebral compression fracture.
The WHI has provided a vast amount of data on the multisystemic effects of hormone therapy. Although earlier observational studies suggested that estrogen replacement might reduce heart disease, the WHI showed that combined estrogen-progestin treatment increased risk of fatal and nonfatal myocardial infarction by ~29%, confirming data from the HERS study. Other important relative risks included a 40% increase in stroke, a 100% increase in venous thromboembolic disease, and a 26% increase in risk of breast cancer. Subsequent analyses have confirmed the increased risk of stroke and, in a substudy, showed a twofold increase in dementia. Benefits other than the fracture reductions noted above included a 37% reduction in the risk of colon cancer. These relative risks have to be interpreted in light of absolute risk (Fig. 425-8). For example, out of 10,000 women treated with estrogen-progestin for 1 year, there will be 8 excess heart attacks, 8 excess breast cancers, 18 excess venous thromboembolic events, 5 fewer hip fractures, 44 fewer clinical fractures, and 6 fewer colorectal cancers. These numbers must be multiplied by years of hormone treatment. There was no effect of hormone treatment on the risk of uterine cancer or total mortality.
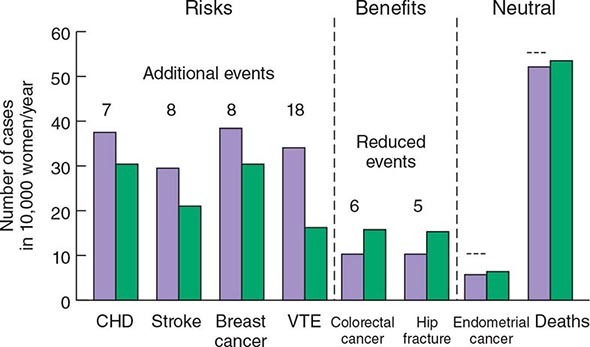
FIGURE 425-8 Effects of hormone therapy on event rates: green, placebo; purple, estrogen and progestin. CHD, coronary heart disease; VTE, venous thromboembolic events. (Adapted from Women’s Health Initiative. WHI HRT Update. Available at http://www.nhlbi.nih.gov/health/women/upd2002.htm.)
It is important to note that these WHI findings apply specifically to hormone treatment in the form of conjugated equine estrogen plus medroxyprogesterone acetate. The relative benefits and risks of unopposed estrogen in women who had hysterectomies vary somewhat. They still show benefits against fracture occurrence and increased risk of venous thrombosis and stroke, similar in magnitude to the risks for combined hormone therapy. In contrast, though, the estrogen-only arm of WHI indicated no increased risk of heart attack or breast cancer. The data suggest that at least some of the detrimental effects of combined therapy are related to the progestin component. In addition, there is the possibility, suggested by primate data, that the risk accrues mainly to women who have some years of estrogen deficiency before initiating treatment. (The average woman in the WHI was more than 10 years from the last menstrual period). Nonetheless, there is reluctance among women to use estrogen/hormone therapy, and the U.S. Preventive Services Task Force has specifically suggested that estrogen/hormone therapy not be used for disease prevention.
MODE OF ACTION Two subtypes of ERs, α and β, have been identified in bone and other tissues. Cells of monocyte lineage express both ERα and ERβ, as do osteoblasts. Estrogen-mediated effects vary with the receptor type. Using ER knockout mouse models, elimination of ERα produces a modest reduction in bone mass, whereas mutation of ERβ has less of an effect on bone. A male patient with a homozygous mutation of ERα had markedly decreased bone density as well as abnormalities in epiphyseal closure, confirming the important role of ERα in bone biology. The mechanism of estrogen action in bone is an area of active investigation (Fig. 425-5). Although data are conflicting, estrogens may inhibit osteoclasts directly. However, the majority of estrogen (and androgen) effects on bone resorption are mediated through paracrine factors produced by osteoblasts and osteocytes. These actions include decreasing RANKL production and increasing OPG production by osteoblasts.
Progestins In women with a uterus, daily progestin or cyclical progestins at least 12 days per month are prescribed in combination with estrogens to reduce the risk of uterine cancer. Medroxyprogesterone acetate and norethindrone acetate blunt the high-density lipoprotein response to estrogen, but micronized progesterone does not. Neither medroxyprogesterone acetate nor micronized progesterone appears to have an independent effect on bone; at lower doses of estrogen, norethindrone acetate may have an additive benefit. On breast tissue, progestins may increase the risk of breast cancer.
SERMs
Two SERMs are used currently in postmenopausal women: raloxifene, which is approved for the prevention and treatment of osteoporosis as well as the prevention of breast cancer, and tamoxifen, which is approved for the prevention and treatment of breast cancer. A third SERM, bazedoxifene, has been complexed with conjugated estrogen, creating a tissue selective estrogen complex (TSEC). This agent has been approved for prevention of osteoporosis.
Tamoxifen reduces bone turnover and bone loss in postmenopausal women compared with placebo groups. These findings support the concept that tamoxifen acts as an estrogenic agent in bone. There are limited data on the effect of tamoxifen on fracture risk, but the Breast Cancer Prevention Study indicated a possible reduction in clinical vertebral, hip, and Colles’ fractures. The major benefit of tamoxifen is on breast cancer occurrence. The breast cancer prevention trial indicated that tamoxifen administration over 4–5 years reduced the incidence of new invasive and noninvasive breast cancer by ~45% in women at increased risk of breast cancer. The incidence of ER-positive breast cancers was reduced by 65%. Tamoxifen increases the risk of uterine cancer and increases risk of venous thrombosis, cataracts, and possibly stroke in postmenopausal women, limiting its use for breast cancer prevention in women at low or moderate risk.
Raloxifene (60 mg/d) has effects on bone turnover and bone mass that are very similar to those of tamoxifen, indicating that this agent is also estrogenic on the skeleton. The effect of raloxifene on bone density (+1.4–2.8% vs placebo in the spine, hip, and total body) is somewhat less than that seen with standard doses of estrogens. Raloxifene reduces the occurrence of vertebral fracture by 30–50%, depending on the population; however, there are no data confirming that raloxifene can reduce the risk of nonvertebral fractures over 8 years of observation.
Raloxifene, like tamoxifen and estrogen, has effects in other organ systems. The most beneficial effect appears to be a reduction in invasive breast cancer (mainly decreased ER-positive) occurrence of ~65% in women who take raloxifene compared to placebo. In a head-to-head study, raloxifene was as effective as tamoxifen in preventing breast cancer in high-risk women, and raloxifene is now FDA approved for this indication. In a further study, raloxifene had no effect on heart disease in women with increased risk for this outcome. In contrast to tamoxifen, raloxifene is not associated with an increase in the risk of uterine cancer or benign uterine disease. Raloxifene increases the occurrence of hot flashes but reduces serum total and low-density lipoprotein cholesterol, lipoprotein(a), and fibrinogen. Raloxifene, with positive effects on breast cancer and vertebral fractures, has become a useful agent for the treatment of the younger asymptomatic postmenopausal woman. In some women, a recurrence of menopausal hot flashes may occur. Usually this is evanescent, but occasionally, it is sufficiently impactful on daily life and sleep that the drug must be withdrawn. Raloxifene increases the risk of deep vein thrombosis and may increase the risk of death from stroke among older women. Consequently, it is not usually recommended for women over 70 years of age.
The main advantage of the bazedoxifene/conjugated estrogen compound is that the bazedoxifene protects uterine tissue from the effects of estrogen and makes it possible to avoid taking a progestin, while using an estrogen primarily for control of menopausal symptoms. The TSEC prevents bone loss somewhat more potently than raloxifene alone and appears safe for the breast.
MODE OF ACTION OF SERMS All SERMs bind to the ER, but each agent produces a unique receptor-drug conformation. As a result, specific co-activator or co-repressor proteins are bound to the receptor (Chap. 400e), resulting in differential effects on gene transcription that vary depending on other transcription factors present in the cell. Another aspect of selectivity is the affinity of each SERM for the different ERα and ERβ subtypes, which are expressed differentially in various tissues. These tissue-selective effects of SERMs offer the possibility of tailoring estrogen therapy to best meet the needs and risk factor profile of an individual patient.
Bisphosphonates Alendronate, risedronate, ibandronate, and zoledronic acid are approved for the prevention and treatment of postmenopausal osteoporosis. Alendronate, risedronate, and zoledronic acid are also approved for the treatment of steroid-induced osteoporosis, and risedronate and zoledronic acid are approved for prevention of steroid-induced osteoporosis. Alendronate, risedronate, and zoledronic acid are approved for treatment of osteoporosis in men.
Alendronate has been shown to decrease bone turnover and increase bone mass in the spine by up to 8% versus placebo and by 6% versus placebo in the hip. Multiple trials have evaluated its effect on fracture occurrence. The Fracture Intervention Trial provided evidence in >2000 women with prevalent vertebral fractures that daily alendronate treatment (5 mg/d for 2 years and 10 mg/d for 9 months afterward) reduces vertebral fracture risk by about 50%, multiple vertebral fractures by up to 90%, and hip fractures by up to 50%. Several subsequent trials have confirmed these findings (Fig. 425-9). For example, in a study of >1900 women with low bone mass treated with alendronate (10 mg/d) versus placebo, the incidence of all nonvertebral fractures was reduced by ~47% after only 1 year. In the United States, the 10-mg dose is approved for treatment of osteoporosis and 5 mg/d is used for prevention.
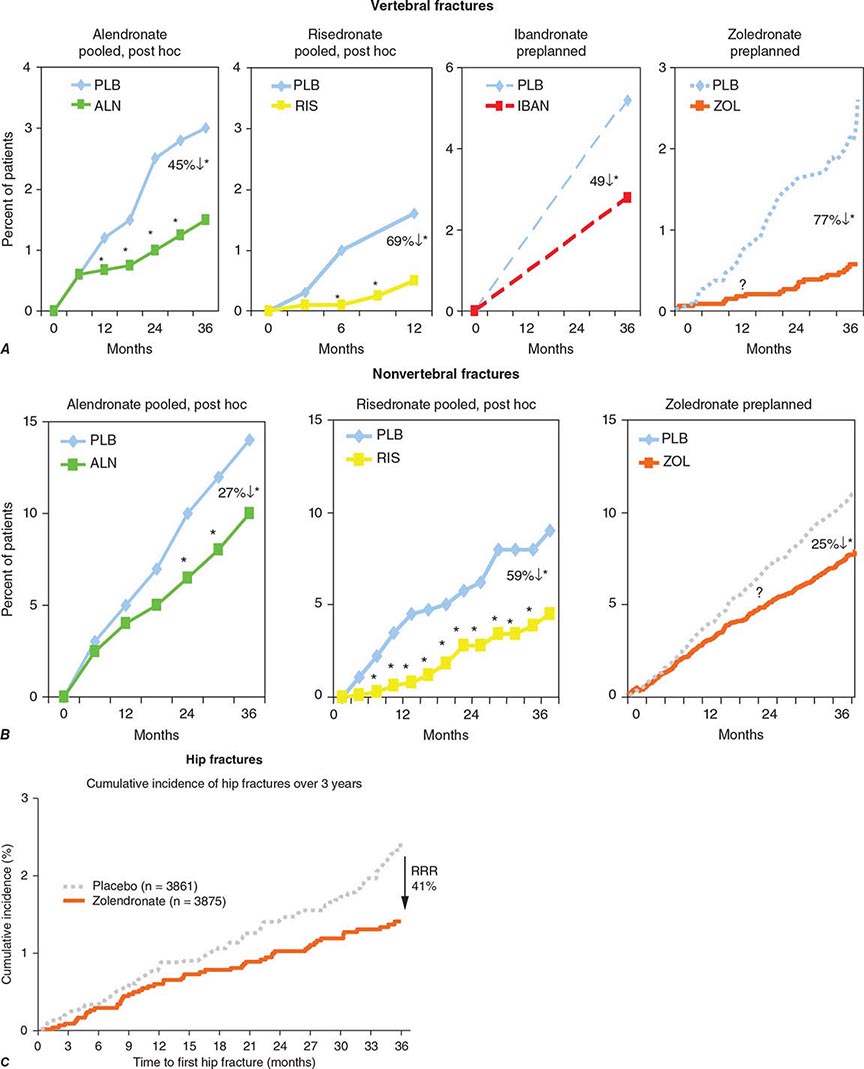
FIGURE 425-9 Effects of various bisphosphonates on clinical vertebral fractures (A), nonvertebral fractures (B), and hip fractures (C). PLB, placebo; RRR, relative risk reduction. (After DM Black et al: J Clin Endocrinol Metab 85:4118, 2000; C Roux et al: Curr Med Res Opin 4:433, 2004; CH Chesnut et al: J Bone Miner Res 19:1241, 2004; DM Black et al: N Engl J Med 356:1809, 2007; JT Harrington et al: Calcif Tissue Int 74:129, 2003.)
Trials comparing once-weekly alendronate, 70 mg, with daily 10-mg dosing have shown equivalence with regard to bone mass and bone turnover responses. Consequently, once-weekly therapy generally is preferred because of the low incidence of gastrointestinal side effects and ease of administration. Alendronate should be given with a full glass of water before breakfast, because bisphosphonates are poorly absorbed. Because of the potential for esophageal irritation, alendronate is contraindicated in patients who have stricture or inadequate emptying of the esophagus. It is recommended that patients remain upright for at least 30 min after taking the medication to avoid esophageal irritation. Cases of esophagitis, esophageal ulcer, and esophageal stricture have been described, but the incidence appears to be low. In clinical trials, overall gastrointestinal symptomatology was no different with alendronate than with placebo. Alendronate is also available in a preparation that contains vitamin D.
Risedronate also reduces bone turnover and increases bone mass. Controlled clinical trials have demonstrated 40–50% reduction in vertebral fracture risk over 3 years, accompanied by a 40% reduction in clinical nonspine fractures. The only clinical trial specifically designed to evaluate hip fracture outcome (HIP) indicated that risedronate reduced hip fracture risk in women in their seventies with confirmed osteoporosis by 40%. In contrast, risedronate was not effective at reducing hip fracture occurrence in older women (80+ years) without proven osteoporosis. Studies have shown that 35 mg of risedronate administered once weekly is therapeutically equivalent to 5 mg/d and that 150 mg once monthly is therapeutically equivalent to 35 mg once weekly. Patients should take risedronate with a full glass of plain water to facilitate delivery to the stomach and should not lie down for 30 min after taking the drug. The incidence of gastrointestinal side effects in trials with risedronate was similar to that of placebo. A new preparation, which allows risedronate to be taken with food, was recently approved.
Etidronate was the first bisphosphonate to be approved, initially for use in Paget’s disease and hypercalcemia. This agent has also been used in osteoporosis trials of smaller magnitude than those performed for alendronate and risedronate but is not approved by the FDA for treatment of osteoporosis. Etidronate probably has some efficacy against vertebral fracture when given as an intermittent cyclical regimen (2 weeks on, 2.5 months off). Its effectiveness against nonvertebral fractures has not been studied.
Ibandronate is the third amino-bisphosphonate approved in the United States. Ibandronate (2.5 mg/d) has been shown in clinical trials to reduce vertebral fracture risk by ~40% but with no overall effect on nonvertebral fractures. In a post hoc analysis of subjects with a femoral neck T-score of –3 or below, ibandronate reduced the risk of nonvertebral fractures by ~60%. In clinical trials, ibandronate doses of 150 mg/month PO or 3 mg every 3 months IV had greater effects on turnover and bone mass than did 2.5 mg/d. Patients should take oral ibandronate in the same way as other bisphosphonates, but with 1 h elapsing before other food or drink (other than plain water).
Zoledronic acid is a potent bisphosphonate with a unique administration regimen (5 mg by slow IV infusion annually). The data confirm that it is highly effective in fracture risk reduction. In a study of >7000 women followed for 3 years, zoledronic acid (three annual infusions) reduced the risk of vertebral fractures by 70%, nonvertebral fractures by 25%, and hip fractures by 40%. These results were associated with less height loss and disability. In the treated population, there was an increased risk of transient postdose symptoms (acute-phase reaction) manifested by fever, arthralgia, myalgias, and headache. The symptoms usually last less than 48 h. An increased risk of atrial fibrillation and transient but not permanent reduction in renal function was seen in comparison to placebo. Detailed evaluation of all bisphosphonates failed to confirm that these agents increased the risk of atrial fibrillation. Zoledronic acid is the only osteoporosis agent that has been studied in the elderly with a prior hip fracture. The risk of all clinical fractures was reduced significantly by about 35%, and there was a trend toward reduced risk of a second hip fracture (effect size similar to that seen above). There was also a reduction in mortality of about 30% that was not completely accounted for the reduced hip fracture risk.
Recently there has been concern about two potential side effects associated with bisphosphonate use. The first is osteonecrosis of the jaw (ONJ). ONJ usually follows a dental procedure in which bone is exposed (extractions or dental implants). It is presumed that the exposed bone becomes infected and dies. It is not uncommon among cancer victims with multiple myeloma or patients receiving high doses of bisphosphonates for skeletal metastases, but is rare among persons with osteoporosis on usual doses of bisphosphonates. The second side effect is called atypical femur fracture. These are unusual fractures that occur distal to the lesser trochanter and anywhere along the femoral shaft. They are often preceded by pain in the lateral thigh or groin that can be present for weeks or months before the fracture. The fractures occur with trivial trauma, sometimes completely spontaneously, and are primarily transverse, with a medial break when complete and minimally comminuted. A localized periosteal reaction, consistent with a stress fracture, is often seen in the lateral cortex (Fig. 425-10). The overall risk is low (suggested to be about one-one hundredth to one-tenth that of hip fracture) but appears to increase in incidence with long-term use of bisphosphonates. Although the fractures may be bisphosphonate related in many individuals, they clearly occur in patients with no prior bisphosphonate exposure. When complete, they require surgical fixation and may be difficult to heal. Anabolic medication may accelerate healing of these fractures in some patients, and surgery can sometimes be avoided. Patients initiating bisphosphonates need to be warned that if they develop thigh or groin pain they must notify their physician. Routine x-rays will sometimes pick up cortical thickening or even a stress fracture, but more commonly MRI or technetium bone scan is required. The presence of an abnormality requires at minimum a period of modified weight bearing and may need prophylactic rodding of the femur. It is important to realize that these fractures may be bilateral, and when an abnormality is found, the other femur should be investigated.
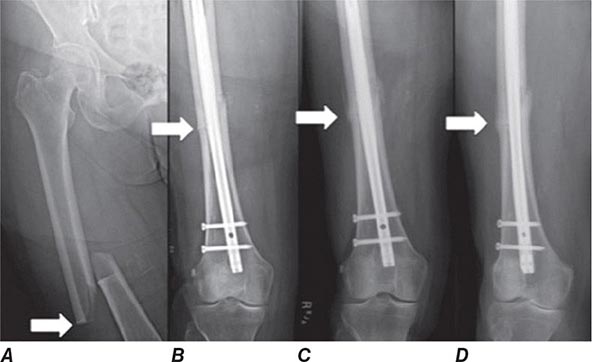
FIGURE 425-10 An atypical femur fracture (AFF) of the femoral diaphysis. A. Note the transverse fracture line in the lateral cortex that becomes oblique as it progresses medially across the femur (white arrow). B. On radiograph obtained immediately after intramedullary rod placement, a small area of periosteal thickening of the lateral cortex is visible (white arrow). C. On radiograph obtained at 6 weeks, note callus formation of the fracture site (white arrow). D. On radiograph obtained at 3 months, there is a mature callus that has failed to bridge the cortical gap (white arrow). Note the localized periosteal and/or endosteal thickening of the lateral cortex at the fracture site (white arrow). (From E Shane et al: J Bone Min Res 29:1-23, 2014. Courtesy of Fergus McKiernan.)
MODE OF ACTION Bisphosphonates are structurally related to pyrophosphates, compounds that are incorporated into bone matrix. Bisphosphonates specifically impair osteoclast function and reduce osteoclast number, in part by inducing apoptosis. Recent evidence suggests that the nitrogen-containing bisphosphonates also inhibit protein prenylation, one of the end products in the mevalonic acid pathway, by inhibiting the enzyme farnesyl pyrophosphate synthase. This effect disrupts intracellular protein trafficking and ultimately may lead to apoptosis. Some bisphosphonates have very long retention in the skeleton and may exert long-term effects. The consequences of this, if any, are unknown.
Calcitonin Calcitonin is a polypeptide hormone produced by the thyroid gland (Chap. 424). Its physiologic role is unclear because no skeletal disease has been described in association with calcitonin deficiency or excess. Calcitonin preparations are approved by the FDA for Paget’s disease, hypercalcemia, and osteoporosis in women >5 years past menopause. Concerns have been raised about an increase in the incidence of cancer associated with calcitonin use. Initially, the cancer noted was of the prostate, but an analysis of all data suggested a more general increase in cancer risk. In Europe, the European Medicines Agency (EMA) has removed the osteoporosis indication, and an FDA Advisory Committee has voted for a similar change in the United States.
Injectable calcitonin produces small increments in bone mass of the lumbar spine. However, difficulty of administration and frequent reactions, including nausea and facial flushing, make general use limited. A nasal spray containing calcitonin (200 IU/d) is available for treatment of osteoporosis in postmenopausal women. One study suggests that nasal calcitonin produces small increments in bone mass and a small reduction in new vertebral fractures in calcitonin-treated patients versus those on calcium alone. There has been no proven effectiveness against nonvertebral fractures.
Calcitonin is not indicated for prevention of osteoporosis and is not sufficiently potent to prevent bone loss in early postmenopausal women. Calcitonin might have an analgesic effect on bone pain, both in the subcutaneous and possibly the nasal form.
MODE OF ACTION Calcitonin suppresses osteoclast activity by direct action on the osteoclast calcitonin receptor. Osteoclasts exposed to calcitonin cannot maintain their active ruffled border, which normally maintains close contact with underlying bone.
Denosumab A novel agent that was given twice yearly by SC administration in a randomized controlled trial in postmenopausal women with osteoporosis has been shown to increase BMD in the spine, hip, and forearm and reduce vertebral, hip, and nonvertebral fractures over a 3-year period by 70, 40, and 20%, respectively (Fig. 425-11). Other clinical trials indicate ability to increase bone mass in postmenopausal women with low bone mass (above osteoporosis range) and in postmenopausal women with breast cancer treated with hormonal agents. Furthermore, a study of men with prostate cancer treated with gonadotropin-releasing hormone (GnRH) agonist therapy indicated the ability of denosumab to improve bone mass and reduce vertebral fracture occurrence. Denosumab was approved by the FDA in 2010 for the treatment of postmenopausal women who have a high risk for osteoporotic fractures, including those with a history of fracture or multiple risk factors for fracture, and those who have failed or are intolerant to other osteoporosis therapy. Denosumab is also approved for the treatment of osteoporosis in men at high risk, men with prostate cancer on GnRH agonist therapy, and women with breast cancer on aromatase inhibitor therapy.
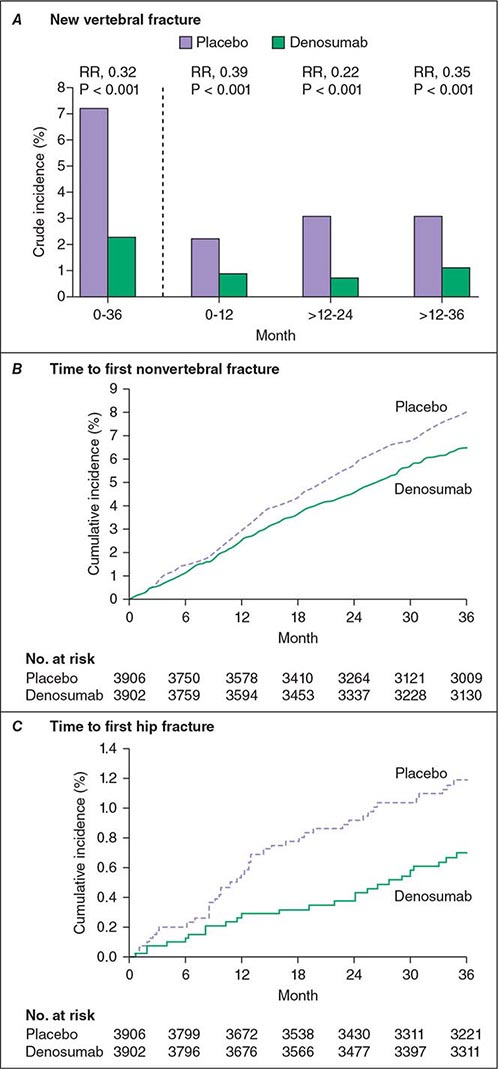
FIGURE 425-11 Effects of denosumab on new vertebral fractures (A) and times to nonvertebral and hip fracture (B and C). RR, relative risk. (After SR Cummings et al: N Engl J Med 361:756, 2009.)
MODE OF ACTION Denosumab is a fully human monoclonal antibody to RANKL, the final common effector of osteoclast formation, activity, and survival. Denosumab binds to RANKL, inhibiting its ability to initiate formation of mature osteoclasts from osteoclast precursors and to bring mature osteoclasts to the bone surface and initiate bone resorption. Denosumab also plays a role in reducing the survival of the osteoclast. Through these actions on the osteoclast, denosumab induces potent antiresorptive action, as assessed biochemically and histomorphometrically, and may contribute to the occurrence of ONJ. Atypical femur fractures have also been noted. Serious adverse reactions include hypocalcemia, skin infections (usually cellulitis of the lower extremity), and dermatologic reactions such as dermatitis, rashes, and eczema. The effects of denosumab are rapidly reversible. If denosumab is stopped, bone will be lost rapidly if another agent is not used.
Parathyroid Hormone Endogenous PTH is an 84-amino-acid peptide that is largely responsible for calcium homeostasis (Chap. 424). Although chronic elevation of PTH, as occurs in hyperparathyroidism, is associated with bone loss (particularly cortical bone), PTH when given exogenously as a daily injection exerts anabolic effects on bone. Teriparatide (1-34hPTH) is approved for the treatment of osteoporosis in both men and women at high risk for fracture. In a pivotal study (median time of treatment, 19 months’ duration), 20 μg of teriparatide daily by SC injection reduced vertebral fractures by 65% and nonvertebral fractures by 45% (Fig. 425-12). Treatment is administered as a single daily injection given for a maximum of 2 years. Teriparatide produces increases in bone mass and mediates architectural improvements in skeletal structure. These effects are lower when patients have been exposed previously to bisphosphonates, possibly in proportion to the potency of the antiresorptive effect. When teriparatide is being considered for treatment-naive patients, it is best administered as monotherapy and followed by an antiresorptive agent such as a bisphosphonate. If teriparatide treatment is not followed by an antiresorptive agent, the bone gained is rapidly lost.
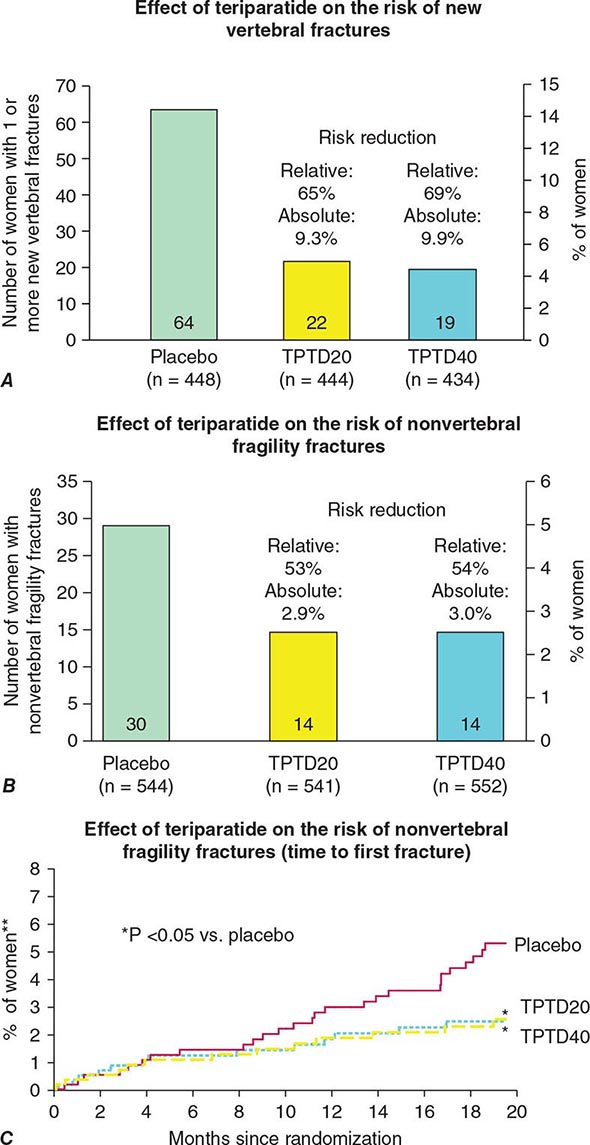
FIGURE 425-12 Effects of teriparatide (TPTD) on new vertebral fractures (A) and nonvertebral fragility fractures (B and C). (After RM Neer et al: N Engl J Med 344:1434, 2001.)
Side effects of teriparatide are generally mild and can include leg cramps, muscle pain, weakness, dizziness, headache, and nausea. Rodents given prolonged treatment with PTH in relatively high doses developed osteogenic sarcomas. Long-term surveillance studies suggest no association between 2 years of teriparatide administration and osteosarcoma risk in humans.
PTH use may be limited by its mode of administration; alternative modes of delivery are being investigated. The optimal frequency of administration also remains to be established, and it is possible that PTH might be effective when used intermittently. Cost also may be a limiting factor. In some settings, the effect of PTH might be enhanced by combination with an antiresorptive agent. This might be particularly important in patients who have been treated previously with bisphosphonate medications.
MODE OF ACTION Exogenously administered PTH appears to have direct actions on osteoblast activity, with biochemical and histomorphometric evidence of de novo bone formation early in response to PTH, before activation of bone resorption. Subsequently, PTH activates bone remodeling but still appears to favor bone formation over bone resorption. PTH stimulates Wnt signaling, IGF-I, and collagen production and appears to increase osteoblast number by stimulating replication, enhancing osteoblast recruitment, and inhibiting apoptosis. Unlike all other treatments, PTH produces a true increase in bone tissue and an apparent restoration of bone microarchitecture (Fig. 425-13).
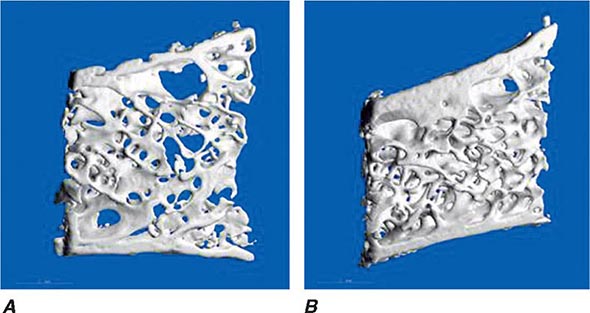
FIGURE 425-13 Effect of parathyroid hormone (PTH) treatment on bone microarchitecture. Paired biopsy specimens from a 64-year-old woman before (A) and after (B) treatment with PTH. (From DW Dempster et al: J Bone Miner Res 16:1846, 2001.)
Fluoride Fluoride has been available for many years and is a potent stimulator of osteoprogenitor cells when studied in vitro. It has been used in multiple osteoporosis studies with conflicting results, in part because of the use of varying doses and preparations. Despite increments in bone mass of up to 10%, there are no consistent effects of fluoride on vertebral or nonvertebral fracture; the latter may actually increase when high doses of fluoride are used. Fluoride remains an experimental agent despite its long history and multiple studies.
Strontium Ranelate Strontium ranelate is approved in several European countries for the treatment of osteoporosis. It increases bone mass throughout the skeleton; in clinical trials, the drug reduced the risk of vertebral fractures by 37% and that of nonvertebral fractures by 14%. It appears to be modestly antiresorptive while at the same time not causing as much of a decrease in bone formation (measured biochemically). Strontium is incorporated into hydroxyapatite, replacing calcium, a feature that might explain some of its fracture benefits. Small increased risks of venous thrombosis, sometimes severe dermatologic reactions, seizures, and abnormal cognition have been seen and require further study. An increase in risk of cardiovascular disease has also been associated with use of strontium, such that the EMA has restricted its use at present.
Other Potential Anabolic Agents Several small studies of growth hormone (GH), alone or in combination with other agents, have not shown consistent or substantial positive effects on skeletal mass. Many of these studies have been relatively short term, and the effects of GH, growth hormone–releasing hormone, and the IGFs are still under investigation. Anabolic steroids, mostly derivatives of testosterone, act primarily as antiresorptive agents to reduce bone turnover but also may stimulate osteoblastic activity. Effects on bone mass remain unclear but appear weak in general, and use is limited by masculinizing side effects. Several observational studies suggested that the statin drugs, used to treat hypercholesterolemia, may be associated with increased bone mass and reduced fractures, but conclusions from clinical trials have been largely negative. Early studies with sclerostin antibodies, which inhibit sclerostin, activate Wnt, and might be highly anabolic to bone, are under development. Odanacatib is a mixed antiresorptive, partial bone formation stimulator that is currently in the late stages of development.
NONPHARMACOLOGIC APPROACHES
In some early studies, protective pads worn around the outer thigh, which cover the trochanteric region of the hip, were able to prevent hip fractures in elderly residents in nursing homes. Randomized controlled trials of hip protectors have been unable to confirm these early findings. Therefore, the efficacy of hip protectors remains controversial at this time.
Kyphoplasty and vertebroplasty are also useful nonpharmacologic approaches for the treatment of painful vertebral fractures. However, no long-term data are available.
TREATMENT MONITORING
There are currently no well-accepted guidelines for monitoring treatment of osteoporosis. Because most osteoporosis treatments produce small or moderate bone mass increments on average, it is reasonable to consider BMD as a monitoring tool. Changes must exceed ~4% in the spine and 6% in the hip to be considered significant in any individual. The hip is the preferred site due to larger surface area and greater reproducibility. Medication-induced increments may require several years to produce changes of this magnitude (if they do at all). Consequently, it can be argued that BMD should be repeated at intervals >2 years. Only significant BMD reductions should prompt a change in medical regimen, because it is expected that many individuals will not show responses greater than the detection limits of the current measurement techniques.
Biochemical markers of bone turnover may prove useful for treatment monitoring, but little hard evidence currently supports this concept; it remains unclear which endpoint is most useful. If bone turnover markers are used, a determination should be made before therapy is started and repeated ≥4 months after therapy is initiated. In general, a change in bone turnover markers must be 30–40% lower than the baseline to be significant because of the biologic and technical variability in these tests. A positive change in biochemical markers and/or bone density can be useful to help patients adhere to treatment regimens.
GLUCOCORTICOID-INDUCED OSTEOPOROSIS
Osteoporotic fractures are a well-characterized consequence of the hypercortisolism associated with Cushing’s syndrome. However, the therapeutic use of glucocorticoids is by far the most common form of glucocorticoid-induced osteoporosis. Glucocorticoids are used widely in the treatment of a variety of disorders, including chronic lung disorders, rheumatoid arthritis and other connective tissue diseases, inflammatory bowel disease, and after transplantation. Osteoporosis and related fractures are serious side effects of chronic glucocorticoid therapy. Because the effects of glucocorticoids on the skeleton are often superimposed on the consequences of aging and menopause, it is not surprising that women and the elderly are most frequently affected. The skeletal response to steroids is remarkably heterogeneous, however, and even young, growing individuals treated with glucocorticoids can present with fractures.
The risk of fractures depends on the dose and duration of glucocorticoid therapy, although recent data suggest that there may be no completely safe dose. Bone loss is more rapid during the early months of treatment, and trabecular bone is affected more severely than cortical bone. As a result, fractures have been shown to increase within 3 months of steroid treatment. There is an increase in fracture risk in both the axial skeleton and the appendicular skeleton, including risk of hip fracture. Bone loss can occur with any route of steroid administration, including high-dose inhaled glucocorticoids and intraarticular injections. Alternate-day delivery does not appear to ameliorate the skeletal effects of glucocorticoids.
PATHOPHYSIOLOGY
Glucocorticoids increase bone loss by multiple mechanisms, including (1) inhibition of osteoblast function and an increase in osteoblast apoptosis, resulting in impaired synthesis of new bone; (2) stimulation of bone resorption, probably as a secondary effect; (3) impairment of the absorption of calcium across the intestine, probably by a vitamin D–independent effect; (4) increase of urinary calcium loss and perhaps induction of some degree of secondary hyperparathyroidism; (5) reduction of adrenal androgens and suppression of ovarian and testicular secretion of estrogens and androgens; and (6) induction of glucocorticoid myopathy, which may exacerbate effects on skeletal and calcium homeostasis as well as increase the risk of falls.
EVALUATION OF THE PATIENT
Because of the prevalence of glucocorticoid-induced bone loss, it is important to evaluate the status of the skeleton in all patients starting or already receiving long-term glucocorticoid therapy. Modifiable risk factors should be identified, including those for falls. Examination should include testing of height and muscle strength. Laboratory evaluation should include an assessment of 24-h urinary calcium. All patients on long-term (>3 months) glucocorticoids should have measurement of bone mass at both the spine and the hip using DXA. If only one skeletal site can be measured, it is best to assess the spine in individuals <60 years and the hip in those >60 years.
PREVENTION
Bone loss caused by glucocorticoids can be prevented and the risk of fractures significantly reduced. Strategies must include using the lowest dose of glucocorticoid for disease management. Topical and inhaled routes of administration are preferred, where appropriate. Risk factor reduction is important, including smoking cessation, limitation of alcohol consumption, and participation in weight-bearing exercise, when appropriate. All patients should receive an adequate calcium and vitamin D intake from the diet or from supplements.
|
TREATMENT |
GLUCOCORTICOID-INDUCED OSTEOPOROSIS |
Several bisphosphonates (alendronate, risedronate, and zoledronic acid) have been demonstrated in large clinical trials to reduce the risk of vertebral fractures in patients being treated with glucocorticoids, as well as improve bone mass in spine and hip. Teriparatide also improves bone mass and reduces fracture risk in glucocorticoid-treated osteoporosis compared to an active comparator (alendronate).
426e |
Paget’s Disease and Other Dysplasias of Bone |
PAGET’S DISEASE OF BONE
Paget’s disease is a localized bone-remodeling disorder that affects widespread, noncontiguous areas of the skeleton. The pathologic process is initiated by overactive osteoclastic bone resorption followed by a compensatory increase in osteoblastic new bone formation, resulting in a structurally disorganized mosaic of woven and lamellar bone. Pagetic bone is expanded, less compact, and more vascular; thus, it is more susceptible to deformities and fractures. Although most patients are asymptomatic, symptoms resulting directly from bony involvement (bone pain, secondary arthritis, fractures) or secondarily from the expansion of bone causing compression of surrounding neural tissue are not uncommon.
 Epidemiology There is a marked geographic variation in the frequency of Paget’s disease, with high prevalence in Western Europe (Great Britain, France, and Germany, but not Switzerland or Scandinavia) and among those who have immigrated to Australia, New Zealand, South Africa, and North and South America. The disease is rare in native populations of the Americas, Africa, Asia, and the Middle East; when it does occur, the affected subjects usually have evidence of European ancestry, supporting the migration theory. For unclear reasons, the prevalence and severity of Paget’s disease are decreasing, and the age of diagnosis is increasing.
Epidemiology There is a marked geographic variation in the frequency of Paget’s disease, with high prevalence in Western Europe (Great Britain, France, and Germany, but not Switzerland or Scandinavia) and among those who have immigrated to Australia, New Zealand, South Africa, and North and South America. The disease is rare in native populations of the Americas, Africa, Asia, and the Middle East; when it does occur, the affected subjects usually have evidence of European ancestry, supporting the migration theory. For unclear reasons, the prevalence and severity of Paget’s disease are decreasing, and the age of diagnosis is increasing.
The prevalence is greater in males and increases with age. Autopsy series reveal Paget’s disease in about 3% of those over age 40. Prevalence of positive skeletal radiographs in patients over age 55 is 2.5% for men and 1.6% for women. Elevated alkaline phosphatase (ALP) levels in asymptomatic patients have an age-adjusted incidence of 12.7 and 7 per 100,000 person-years in men and women, respectively.
Etiology The etiology of Paget’s disease of bone remains unknown, but evidence supports both genetic and viral etiologies. A positive family history is found in 15–25% of patients and, when present, raises the prevalence of the disease seven- to tenfold among first-degree relatives.
A clear genetic basis has been established for several rare familial bone disorders that clinically and radiographically resemble Paget’s disease but have more severe presentation and earlier onset. A homozygous deletion of the TNFRSF11B gene, which encodes osteoprotegrin (Fig. 426e-1), causes juvenile Paget’s disease, also known as familial idiopathic hyperphosphatasia, a disorder characterized by uncontrolled osteoclastic differentiation and resorption. Familial patterns of disease in several large kindred are consistent with an autosomal dominant pattern of inheritance with variable penetrance. Familial expansile osteolysis, expansile skeletal hyperphosphatasia, and early-onset Paget’s disease are associated with mutations in TNFRSF11A gene, which encodes RANK (receptor activator of nuclear factor-κB), a member of the tumor necrosis factor superfamily critical for osteoclast differentiation (Fig. 426e-1). Finally, mutations in the gene for valosin-containing protein cause a rare syndrome with autosomal dominant inheritance and variable penetrance known as inclusion body myopathy with Paget’s disease and frontotemporal dementia (IBMPFD). The role of genetic factors is less clear in the more common form of late-onset Paget’s disease. Although a few families with mutations in the gene encoding RANK have been reported, the most common mutations identified in familial and sporadic cases of Paget’s disease have been in the SQSTM1 gene (sequestasome-1 or p62 protein) in the C-terminal ubiquitin-binding domain. The p62 protein is involved in nuclear factor κB (NF-κB) signaling and regulates osteoclastic differentiation. The phenotypic variability in patients with SQSTM1 mutations suggests that additional factors, such as other genetic influences or viral infection, may influence clinical expression of the disease.
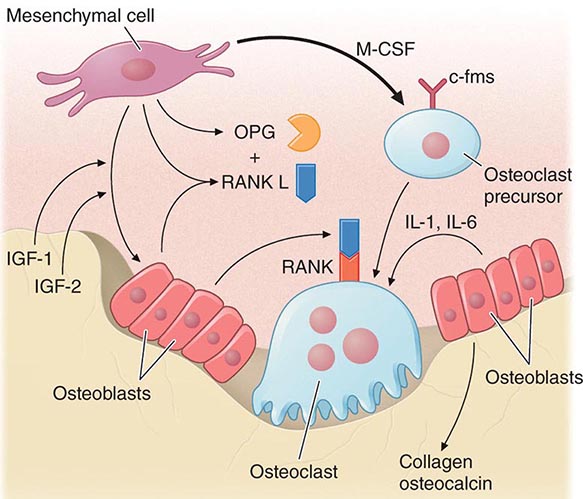
FIGURE 426e-1 Diagram illustrating factors that promote differentiation and function of osteoclasts and osteoblasts and the role of the RANK pathway. Stromal bone marrow (mesenchymal) cells and differentiated osteoblasts produce multiple growth factors and cytokines, including macrophage colony-stimulating factor (M-CSF), to modulate osteoclastogenesis. RANKL (receptor activator of nuclear factor-κB ligand) is produced by osteoblast progenitors and mature osteoblasts and can bind to a soluble decoy receptor known as OPG (osteoprotegerin) to inhibit RANKL action. Alternatively, a cell-cell interaction between osteoblast and osteoclast progenitors allows RANKL to bind to its membrane-bound receptor, RANK, thereby stimulating osteoclast differentiation and function. RANK binds intracellular proteins called TRAFs (tumor necrosis factor receptor–associated factors) that mediate receptor signaling through transcription factors such as NF-κB. M-CSF binds to its receptor, c-fms, which is the cellular homologue of the fms oncogene. See text for the potential role of these pathways in disorders of osteoclast function such as Paget’s disease and osteopetrosis. IL, interleukin; IGF, insulin-like growth factor.
Several lines of evidence suggest that a viral infection may contribute to the clinical manifestations of Paget’s disease, including (1) the presence of cytoplasmic and nuclear inclusions resembling paramyxoviruses (measles and respiratory syncytial virus) in pagetic osteoclasts and (2) viral mRNA in precursor and mature osteoclasts. The viral etiology is further supported by conversion of osteoclast precursors to pagetic-like osteoclasts by vectors containing the measles virus nucleocapsid or matrix genes. However, the viral etiology has been questioned by the inability to culture a live virus from pagetic bone and by failure to clone the full-length viral genes from material obtained from patients with Paget’s disease.
Pathophysiology The principal abnormality in Paget’s disease is the increased number and activity of osteoclasts. Pagetic osteoclasts are large, increased 10- to 100-fold in number, and have a greater number of nuclei (as many as 100 compared to 3–5 nuclei in the normal osteoclast). The overactive osteoclasts may create a sevenfold increase in resorptive surfaces and an erosion rate of 9 μg/d (normal is 1 μg/d). Several causes for the increased number and activity of pagetic osteoclasts have been identified: (1) osteoclastic precursors are hypersensitive to 1,25(OH)2D3; (2) osteoclasts are hyperresponsive to RANK ligand (RANKL), the osteoclast stimulatory factor that mediates the effects of most osteotropic factors on osteoclast formation; (3) marrow stromal cells from pagetic lesions have increased RANKL expression; (4) osteoclast precursor recruitment is increased by interleukin (IL) 6, which is increased in the blood of patients with active Paget’s disease and is overexpressed in pagetic osteoclasts; (5) expression of the protooncogene c-fos, which increases osteoclastic activity, is increased; and (6) the antiapoptotic oncogene Bcl-2 in pagetic bone is overexpressed. Numerous osteoblasts are recruited to active resorption sites and produce large amounts of new bone matrix. As a result, bone turnover is high, and bone mass is normal or increased, not reduced, unless there is concomitant deficiency of calcium and/or vitamin D.
The characteristic feature of Paget’s disease is increased bone resorption accompanied by accelerated bone formation. An initial osteolytic phase involves prominent bone resorption and marked hypervascularization. Radiographically, this manifests as an advancing lytic wedge, or “blade of grass” lesion. The second phase is a period of very active bone formation and resorption that replaces normal lamellar bone with haphazard (woven) bone. Fibrous connective tissue may replace normal bone marrow. In the final sclerotic phase, bone resorption declines progressively and leads to a hard, dense, less vascular pagetic or mosaic bone, which represents the so-called burned-out phase of Paget’s disease. All three phases may be present at the same time at different skeletal sites.
Clinical Manifestations Diagnosis is often made in asymptomatic patients because they have elevated ALP levels on routine blood chemistry testing or an abnormality on a skeletal radiograph obtained for another indication. The skeletal sites most commonly involved are the pelvis, vertebral bodies, skull, femur, and tibia. Familial cases with an early presentation often have numerous active sites of skeletal involvement.
The most common presenting symptom is pain, which may result from increased bony vascularity, expanding lytic lesions, fractures, bowing, or other deformities. Bowing of the femur or tibia causes gait abnormalities and abnormal mechanical stresses with secondary osteoarthritis of the hip or knee joints. Long bone bowing also causes extremity pain by stretching the muscles attached to the bone softened by the pagetic process. Back pain results from enlarged pagetic vertebrae, vertebral compression fractures, spinal stenosis, degenerative changes of the joints, and altered body mechanics with kyphosis and forward tilt of the upper back. Rarely, spinal cord compression may result from bone enlargement or from the vascular steal syndrome. Skull involvement may cause headaches, symmetric or asymmetric enlargement of the parietal or frontal bones (frontal bossing), and increased head size. Cranial expansion may narrow cranial foramens and cause neurologic complications including hearing loss from cochlear nerve damage from temporal bone involvement, cranial nerve palsies, and softening of the base of the skull (platybasia) with the risk of brainstem compression. Pagetic involvement of the facial bones may cause facial deformity; loss of teeth and other dental conditions; and, rarely, airway compression.
Fractures are serious complications of Paget’s disease and usually occur in long bones at areas of active or advancing lytic lesions. Common fracture sites are the femoral shaft and subtrochanteric regions. Neoplasms arising from pagetic bone are rare (<0.5%). The incidence of sarcoma appears to be decreasing, possibly because of earlier, more effective treatment with potent antiresorptive agents. The majority of tumors are osteosarcomas, which usually present with new pain in a long-standing pagetic lesion. Osteoclast-rich benign giant cell tumors may arise in areas adjacent to pagetic bone, and they respond to glucocorticoid therapy.
Cardiovascular complications may occur in patients with involvement of large (15–35%) portions of the skeleton and a high degree of disease activity (ALP four times above normal). The extensive arteriovenous shunting and marked increases in blood flow through the vascular pagetic bone lead to a high-output state and cardiac enlargement. However, high-output heart failure is relatively rare and usually develops in patients with concomitant cardiac pathology. In addition, calcific aortic stenosis and diffuse vascular calcifications have been associated with Paget’s disease.
Diagnosis The diagnosis may be suggested on clinical examination by the presence of an enlarged skull with frontal bossing, bowing of an extremity, or short stature with simian posturing. An extremity with an area of warmth and tenderness to palpation may suggest an underlying pagetic lesion. Other findings include bony deformity of the pelvis, skull, spine, and extremities; arthritic involvement of the joints adjacent to lesions; and leg-length discrepancy resulting from deformities of the long bones.
Paget’s disease is usually diagnosed from radiologic and biochemical abnormalities. Radiographic findings typical of Paget’s disease include enlargement or expansion of an entire bone or area of a long bone, cortical thickening, coarsening of trabecular markings, and typical lytic and sclerotic changes. Skull radiographs (Fig. 426e-2) reveal regions of “cotton wool,” or osteoporosis circumscripta, thickening of diploic areas, and enlargement and sclerosis of a portion or all of one or more skull bones. Vertebral cortical thickening of the superior and inferior end plates creates a “picture frame” vertebra. Diffuse radiodense enlargement of a vertebra is referred to as “ivory vertebra.” Pelvic radiographs may demonstrate disruption or fusion of the sacroiliac joints; porotic and radiodense lesions of the ilium with whorls of coarse trabeculation; thickened and sclerotic iliopectineal line (brim sign); and softening with protrusio acetabuli, with axial migration of the hips and functional flexion contracture. Radiographs of long bones reveal bowing deformity and typical pagetic changes of cortical thickening and expansion and areas of lucency and sclerosis (Fig. 426e-3). Radionuclide 99mTc bone scans are less specific but are more sensitive than standard radiographs for identifying sites of active skeletal lesions. Although computed tomography (CT) and magnetic resonance imaging (MRI) studies are not necessary in most cases, CT may be useful for the assessment of possible fracture, and MRI is necessary to assess the possibility of sarcoma, giant cell tumor, or metastatic disease in pagetic bone. Definitive diagnosis of malignancy often requires bone biopsy.
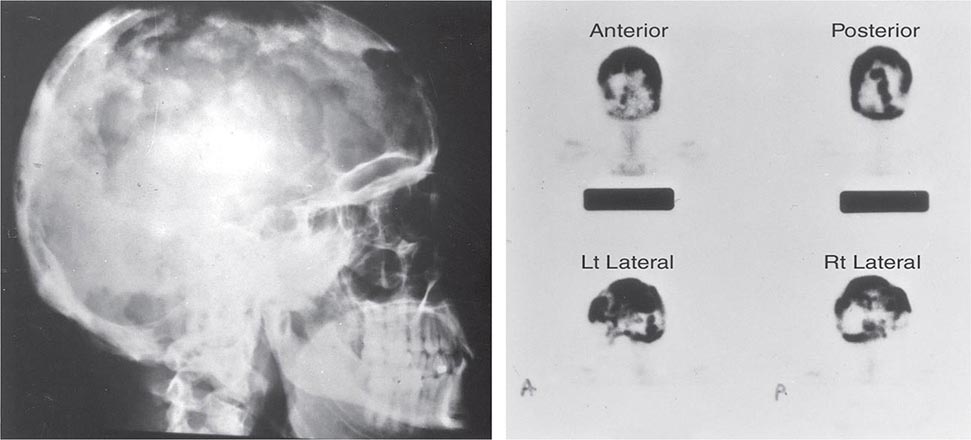
FIGURE 426e-2 A 48-year-old woman with Paget’s disease of the skull. Left. Lateral radiograph showing areas of both bone resorption and sclerosis. Right. 99mTc HDP bone scan with anterior, posterior, and lateral views of the skull showing diffuse isotope uptake by the frontal, parietal, occipital, and petrous bones.
FIGURE 426e-3 Radiograph of a 73-year-old man with Paget’s disease of the right proximal femur. Note the coarsening of the trabecular pattern with marked cortical thickening and narrowing of the joint space consistent with osteoarthritis secondary to pagetic deformity of the right femur.
Biochemical evaluation is useful in the diagnosis and management of Paget’s disease. The marked increase in bone turnover can be monitored using biochemical markers of bone formation and resorption. The parallel rise in markers of bone formation and resorption confirms the coupling of bone formation and resorption in Paget’s disease. The degree of bone marker elevation reflects the extent and severity of the disease. Patients with the highest elevation of ALP (10 × the upper limit of normal) typically have involvement of the skull and at least one other skeletal site. Lower values suggest less extensive involvement or a quiescent phase of the disease. For most patients, serum total ALP remains the test of choice both for diagnosis and assessing response to therapy. Occasionally, a symptomatic patient with evidence of progression at a single site may have a normal total ALP level but increased bone-specific ALP. For unclear reasons, serum osteocalcin, another marker of bone formation, is not always elevated and is not recommended for use in diagnosis or management of Paget’s disease. Bone resorption markers (serum or urine N-telopeptide or C-telopeptide measured in the blood or urine) are also elevated in active Paget’s disease and decrease more rapidly in response to therapy than does ALP.
Serum calcium and phosphate levels are normal in Paget’s disease. Immobilization of a patient with active Paget’s disease may rarely cause hypercalcemia and hypercalciuria and increase the risk for nephrolithiasis. However, the discovery of hypercalcemia, even in the presence of immobilization, should prompt a search for another cause of hypercalcemia. In contrast, hypocalcemia or mild secondary hyperparathyroidism may develop in Paget’s patients with very active bone formation and insufficient calcium and vitamin D intake, particularly during bisphosphonate therapy when bone resorption is rapidly suppressed and active bone formation continues. Therefore, adequate calcium and vitamin D intake should be instituted prior to administration of bisphosphonates.
|
TREATMENT |
PAGET’S DISEASE OF BONE |
The development of effective and potent pharmacologic agents (Table 426e-1) has changed the treatment philosophy from treating only symptomatic patients to treating asymptomatic patients who are at risk for complications. Pharmacologic therapy is indicated in the following circumstances: to control symptoms caused by metabolically active Paget’s disease such as bone pain, fracture, headache, pain from pagetic radiculopathy or arthropathy, or neurologic complications; to decrease local blood flow and minimize operative blood loss in patients who need surgery at an active pagetic site; to reduce hypercalciuria that may occur during immobilization; and to decrease the risk of complications when disease activity is high (elevated ALP) and when the site of involvement involves weight-bearing bones, areas adjacent to major joints, vertebral bodies, and the skull. Whether or not early therapy prevents late complications remains to be determined. A randomized study of over 1200 patients from the United Kingdom showed no difference in bone pain, fracture rates, quality of life, and hearing loss between patients who received pharmacologic therapy to control symptoms (bone pain) and those receiving bisphosphonates to normalize serum ALP. However, the most potent agent (zoledronic acid) was not used, and the duration of observation (mean of 3 years with a range of 2 to 5 years) may not be long enough to assess the impact of treatment on long-term outcomes. It seems likely that the restoration of normal bone architecture following suppression of pagetic activity will prevent further deformities and complications.
|
PHARMACOLOGIC AGENTS APPROVED FOR TREATMENT OF PAGET’S DISEASE |
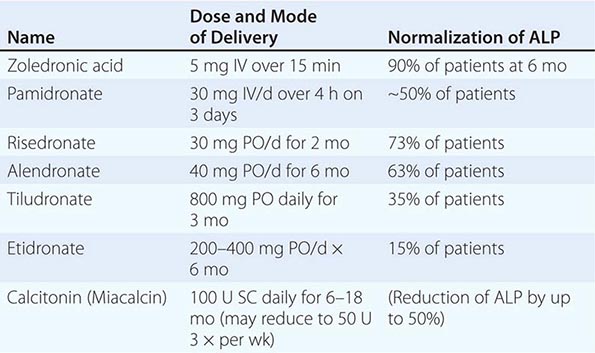
Agents approved for treatment of Paget’s disease suppress the very high rates of bone resorption and secondarily decrease the high rates of bone formation (Table 426e-1). As a result of decreasing bone turnover, pagetic structural patterns, including areas of poorly mineralized woven bone, are replaced by more normal cancellous or lamellar bone. Reduced bone turnover can be documented by a decline in serum ALP and urine or serum resorption markers (N-telopeptide, C-telopeptide).
The first clinically useful agent, etidronate, is now rarely used because the doses required to suppress bone resorption may impair mineralization, necessitating that the drug be given for a maximum of 6 months followed by a 6-month drug-free period. The second-generation oral bisphosphonates—tiludronate, alendronate, and risedronate—are more potent than etidronate in controlling bone turnover and, thus, induce a longer remission at a lower dose. The lower doses reduce the risks of impaired mineralization and osteomalacia. Oral bisphosphonates should be taken first thing in the morning on an empty stomach, followed by maintenance of upright posture with no food, drink, or other medications for 30–60 min. The efficacy of different agents, based on their ability to normalize or decrease ALP levels, is summarized in Table 426e-1, although the response rates are not comparable because they are obtained from different studies.
Intravenous bisphosphonates approved for Paget’s disease include pamidronate and zoledronic acid. Although the recommended dose for pamidronate is 30 mg dissolved in 500 mL of normal saline or dextrose IV over 4 h on 3 consecutive days, a more commonly used simpler regimen is a single infusion of 60–90 mg in patients with mild elevation of serum ALP and multiple 90-mg infusions in those with higher levels of ALP. In many patients, particularly those who have severe disease or need rapid normalization of bone turnover (neurologic symptoms, severe bone pain due to a lytic lesion, risk of an impending fracture, or pretreatment prior to elective surgery in an area of active disease), treatment with zoledronic acid is the first choice. It normalizes ALP in about 90% of patients by 6 months, and the therapeutic effect persists for at least 6 more months in most patients. About 10–20% of patients experience a flulike syndrome after the first infusion, which can be partly ameliorated by pretreatment with acetaminophen or nonsteroidal anti-inflammatory drugs (NSAIDs). In patients with high bone turnover, vitamin D and calcium should be provided to prevent hypocalcemia and secondary hyperparathyroidism. Remission following treatment with IV bisphosphonates, particularly zoledronic acid, may persist for well over 1 year. Bisphosphonates should not be used in patients with renal insufficiency (glomerular filtration rate <35 mL/min).
The subcutaneous injectable form of salmon calcitonin is approved for the treatment of Paget’s disease. The common side effects of calcitonin therapy are nausea and facial flushing. Secondary resistance after prolonged use may be due to either the formation of anticalcitonin antibodies or downregulation of osteoclastic cell–surface calcitonin receptors. The lower potency and injectable mode of delivery make this agent a less attractive treatment option that should be reserved for patients who either do not tolerate bisphosphonates or have a contraindication to their use. In early reports, denosumab, an antibody to RANKL, has shown promise but has not been approved for this indication.
SCLEROSING BONE DISORDERS
OSTEOPETROSIS
Osteopetrosis refers to a group of disorders caused by severe impairment of osteoclast-mediated bone resorption. Other terms that are often used include marble bone disease, which captures the solid x-ray appearance of the involved skeleton, and Albers-Schonberg disease, which refers to the milder, adult form of osteopetrosis also known as autosomal dominant osteopetrosis type II. The major types of osteopetrosis include malignant (severe, infantile, autosomal recessive) osteopetrosis and benign (adult, autosomal dominant) osteopetrosis types I and II. A rare autosomal recessive intermediate form has a more benign prognosis. Autosomal recessive carbonic anhydrase (CA) II deficiency produces osteopetrosis of intermediate severity associated with renal tubular acidosis and cerebral calcification.
Etiology and Genetics Naturally occurring and gene-knockout animal models with phenotypes similar to those of the human disorders have been used to explore the genetic basis of osteopetrosis. The primary defect in osteopetrosis is the loss of osteoclastic bone resorption and preservation of normal osteoblastic bone formation. Osteoprotegerin (OPG) is a soluble decoy receptor that binds osteoblast-derived RANK ligand, which mediates osteoclast differentiation and activation (Fig. 426e-1). Transgenic mice that overexpress OPG develop osteopetrosis, presumably by blocking RANK ligand. Mice deficient in RANK lack osteoclasts and develop severe osteopetrosis.
Recessive mutations of CA II prevent osteoclasts from generating an acid environment in the clear zone between its ruffled border and the adjacent mineral surface. Absence of CA II, therefore, impairs osteoclastic bone resorption. Other forms of human disease have less clear genetic defects. About one-half of the patients with malignant infantile osteopetrosis have a mutation in the TCIRG1 gene encoding the osteoclast-specific subunit of the vacuolar proton pump, which mediates the acidification of the interface between bone mineral and the osteoclast ruffled border. Mutations in the CICN7 chloride channel gene cause autosomal dominant osteopetrosis type II.
Clinical Presentation The incidence of autosomal recessive severe (malignant) osteopetrosis ranges from 1 in 200,000 to 1 in 500,000 live births. As bone and cartilage fail to undergo modeling, paralysis of one or more cranial nerves may occur due to narrowing of the cranial foramens. Failure of skeletal modeling also results in inadequate marrow space, leading to extramedullary hematopoiesis with hypersplenism and pancytopenia. Hypocalcemia due to lack of osteoclastic bone resorption may occur in infants and young children. The untreated infantile disease is fatal, often before age 5.
Adult (benign) osteopetrosis is an autosomal dominant disease that is usually diagnosed by the discovery of typical skeletal changes in young adults who undergo radiologic evaluation of a fracture. The prevalence is 1 in 100,000 to 1 in 500,000 adults. The course is not always benign, because fractures may be accompanied by loss of vision, deafness, psychomotor delay, mandibular osteomyelitis, and other complications usually associated with the juvenile form. In some kindred, nonpenetrance results in skip generations, while in other families, severely affected children are born into families with benign disease. The milder form of the disease does not usually require treatment.
Radiography Typically, there are generalized symmetric increases in bone mass with thickening of both cortical and trabecular bone. Diaphyses and metaphyses are broadened, and alternating sclerotic and lucent bands may be seen in the iliac crests, at the ends of long bones, and in vertebral bodies. The cranium is usually thickened, particularly at the base of the skull, and the paranasal and mastoid sinuses are underpneumatized.
Laboratory Findings The only significant laboratory findings are elevated serum levels of osteoclast-derived tartrate-resistant acid phosphatase (TRAP) and the brain isoenzyme of creatine kinase. Serum calcium may be low in severe disease, and parathyroid hormone and 1,25-dihydroxyvitamin D levels may be elevated in response to hypocalcemia.
PYKNODYSOSTOSIS
This is an autosomal recessive form of osteosclerosis that is believed to have affected the French impressionist painter Henri de Toulouse-Lautrec. The molecular basis involves mutations in the gene that encodes cathepsin K, a lysosomal metalloproteinase highly expressed in osteoclasts and important for bone-matrix degradation. Osteoclasts are present but do not function normally. Pyknodysostosis is a form of short-limb dwarfism that presents with frequent fractures but usually a normal life span. Clinical features include short stature; kyphoscoliosis and deformities of the chest; high arched palate; proptosis; blue sclerae; dysmorphic features including small face and chin, frontooccipital prominence, pointed beaked nose, large cranium, and obtuse mandibular angle; and small, square hands with hypoplastic nails. Radiographs demonstrate a generalized increase in bone density, but in contrast to osteopetrosis, the long bones are normally shaped. Separated cranial sutures, including the persistent patency of the anterior fontanel, are characteristic of the disorder. There may also be hypoplasia of the sinuses, mandible, distal clavicles, and terminal phalanges. Persistence of deciduous teeth and sclerosis of the calvarium and base of the skull are also common. Histologic evaluation shows normal cortical bone architecture with decreased osteoblastic and osteoclastic activities. Serum chemistries are normal, and unlike osteopetrosis, there is no anemia. There is no known treatment for this condition, and there are no reports of attempted bone marrow transplant.
PROGRESSIVE DIAPHYSEAL DYSPLASIA
Also known as Camurati-Engelmann disease, progressive diaphyseal dysplasia is an autosomal dominant disorder that is characterized radiographically by diaphyseal hyperostosis and a symmetric thickening and increased diameter of the endosteal and periosteal surfaces of the diaphyses of the long bones, particularly the femur and tibia, and, less often, the fibula, radius, and ulna. The genetic defect responsible for the disease has been localized to the area of chromosome 19q13.2 encoding tumor growth factor (TGF) β1. The mutation promotes activation of TGF-β1. The clinical severity is variable. The most common presenting symptoms are pain and tenderness of the involved areas, fatigue, muscle wasting, and gait disturbance. The weakness may be mistaken for muscular dystrophy. Characteristic body habitus includes thin limbs with little muscle mass yet prominent and palpable bones and, when the skull is involved, large head with prominent forehead and proptosis. Patients may also display signs of cranial nerve palsies, hydrocephalus, central hypogonadism, and Raynaud’s phenomenon. Radiographically, patchy progressive endosteal and periosteal new bone formation is observed along the diaphyses of the long bones. Bone scintigraphy shows increased radiotracer uptake in involved areas.
Treatment with low-dose glucocorticoids relieves bone pain and may reverse the abnormal bone formation. Intermittent bisphosphonate therapy has produced clinical improvement in a limited number of patients.
HYPEROSTOSIS CORTICALIS GENERALISATA
This is also known as van Buchem’s disease; it is an autosomal recessive disorder characterized by endosteal hyperostosis in which osteosclerosis involves the skull, mandible, clavicles, and ribs. The major manifestations are due to narrowed cranial foramens with neural compressions that may result in optic atrophy, facial paralysis, and deafness. Adults may have an enlarged mandible. Serum ALP levels may be elevated, which reflect the uncoupled bone remodeling with high osteoblastic formation rates and low osteoclastic resorption. As a result, there is increased accumulation of normal bone. Endosteal hyperostosis with syndactyly, known as sclerosteosis, is a more severe form. The genetic defects for both sclerosteosis and van Buchem’s disease have been assigned to the same region of the chromosome 17q12-q21. It is possible that both conditions may have deactivating mutations in the BEER (bone-expressed equilibrium regulator) gene.
MELORHEOSTOSIS
Melorheostosis (Greek, “flowing hyperostosis”) may occur sporadically or follow a pattern consistent with an autosomal recessive disorder. The major manifestation is progressive linear hyperostosis in one or more bones of one limb, usually a lower extremity. The name comes from the radiographic appearance of the involved bone, which resembles melted wax that has dripped down a candle. Symptoms appear during childhood as pain or stiffness in the area of sclerotic bone. There may be associated ectopic soft tissue masses, composed of cartilage or osseous tissue, and skin changes overlying the involved bone, consisting of scleroderma-like areas and hypertrichosis. The disease does not progress in adults, but pain and stiffness may persist. Laboratory tests are unremarkable. No specific etiology is known. There is no specific treatment. Surgical interventions to correct contractures are often unsuccessful.
OSTEOPOIKILOSIS
The literal translation of osteopoikilosis is “spotted bones”; it is a benign autosomal dominant condition in which numerous small, variably shaped (usually round or oval) foci of bony sclerosis are seen in the epiphyses and adjacent metaphyses. The lesions may involve any bone except the skull, ribs, and vertebrae. They may be misidentified as metastatic lesions. The main differentiating points are that bony lesions of osteopoikilosis are stable over time and do not accumulate radionucleotide on bone scanning. In some kindred, osteopoikilosis is associated with connective tissue nevi known as dermatofibrosis lenticularis disseminata, also known as Buschke-Ollendorff syndrome. Histologic inspection reveals thickened but otherwise normal trabeculae and islands of normal cortical bone. No treatment is indicated.
HEPATITIS C–ASSOCIATED OSTEOSCLEROSIS
Hepatitis C–associated osteosclerosis (HCAO) is a rare acquired diffuse osteosclerosis in adults with prior hepatitis C infection. After a latent period of several years, patients develop diffuse appendicular bone pain and a generalized increase in bone mass with elevated serum ALP. Bone biopsy and histomorphometry reveal increased rates of bone formation, decreased bone resorption with a marked decrease in osteoclasts, and dense lamellar bone. One patient had increased serum OPG levels, and bone biopsy showed large numbers of osteoblasts positive for OPG and reduced osteoclast number. Empirical therapy includes pain control, and there may be beneficial response to bisphosphonate. Long-term antiviral therapy may reverse the bone disease.
DISORDERS ASSOCIATED WITH DEFECTIVE MINERALIZATION
HYPOPHOSPHATASIA
This is a rare inherited disorder that presents as rickets in infants and children or osteomalacia in adults with paradoxically low serum levels of ALP. The frequency of the severe neonatal and infantile forms is about 1 in 100,000 live births in Canada, where the disease is most common because of its high prevalence among Mennonites and Hutterites. It is rare in African Americans. The severity of the disease is remarkably variable, ranging from intrauterine death associated with profound skeletal hypomineralization at one extreme to premature tooth loss as the only manifestation in some adults. Severe cases are inherited in an autosomal recessive manner, but the genetic patterns are less clear for the milder forms. The disease is caused by a deficiency of tissue nonspecific (bone/liver/kidney) ALP (TNSALP), which, although ubiquitous, results only in bone abnormalities. Protein levels and functions of the other ALP isozymes (germ cell, intestinal, placental) are normal. Defective ALP permits accumulation of its major naturally occurring substrates including phosphoethanolamine (PEA), inorganic pyrophosphate (PPi), and pyridoxal 5′-phosphate (PLP). The accumulation of PPi interferes with mineralization through its action as a potent inhibitor of hydroxyapatite crystal growth.
Perinatal hypophosphatasia becomes manifest during pregnancy and is often complicated by polyhydramnios and intrauterine death. The infantile form becomes clinically apparent before the age of 6 months with failure to thrive, rachitic deformities, functional craniosynostosis despite widely open fontanels (which are actually hypomineralized areas of the calvarium), raised intracranial pressure, and flail chest with predisposition to pneumonia. Hypercalcemia and hypercalciuria are common. This form has a mortality rate of about 50%. Prognosis seems to improve for the children who survive infancy. Childhood hypophosphatasia has variable clinical presentation. Premature loss of deciduous teeth (before age 5) is the hallmark of the disease. Rickets causes delayed walking with waddling gait, short stature, and dolichocephalic skull with frontal bossing. The disease often improves during puberty but may recur in adult life. Adult hypophosphatasia presents during middle age with painful, poorly healing metatarsal stress fractures or thigh pain due to femoral pseudofractures.
Laboratory investigation reveals low ALP levels and normal or elevated levels of serum calcium and phosphorus despite clinical and radiologic evidence of rickets or osteomalacia. Serum parathyroid hormone, 25-hydroxyvitamin D, and 1,25-dihydroxyvitamin D levels are normal. The elevation of PLP is specific for the disease and may even be present in asymptomatic parents of severely affected children. Because vitamin B6 increases PLP levels, vitamin B6 supplements should be discontinued 1 week before testing. Clinical testing is available to detect loss-of-function mutation(s) within the ALPL gene that encodes TNSALP.
There is no established medical therapy. In contrast to other forms of rickets and osteomalacia, calcium and vitamin D supplementation should be avoided because they may aggravate hypercalcemia and hypercalciuria. A low-calcium diet, glucocorticoids, and calcitonin have been used in a small number of patients with variable responses. Because fracture healing is poor, placement of intramedullary rods is best for acute fracture repair and for prophylactic prevention of fractures.
AXIAL OSTEOMALACIA
This is a rare disorder characterized by defective skeletal mineralization despite normal serum calcium and phosphate levels. Clinically, the disorder presents in middle-aged or elderly men with chronic axial skeletal discomfort. Cervical spine pain may also be present. Radiographic findings are mainly osteosclerosis due to coarsened trabecular patterns typical of osteomalacia. Spine, pelvis, and ribs are most commonly affected. Histologic changes show defective mineralization and flat, inactive osteoblasts. The primary defect appears to be an acquired defect in osteoblast function. The course is benign, and there is no established treatment. Calcium and vitamin D therapies are not effective.
FIBROGENESIS IMPERFECTA OSSIUM
This is a rare condition of unknown etiology. It presents in both sexes; in middle age or later; and with progressive, intractable skeletal pain and fractures; worsening immobilization; and a debilitating course. Radiographic evaluation reveals generalized osteomalacia, osteopenia, and occasional pseudofractures. Histologic features include a tangled pattern of collagen fibrils with abundant osteoblasts and osteoclasts. There is no effective treatment. Spontaneous remission has been reported in a small number of patients. Calcium and vitamin D have not been beneficial.
FIBROUS DYSPLASIA AND MCCUNE-ALBRIGHT SYNDROME
Fibrous dysplasia is a sporadic disorder characterized by the presence of one (monostotic) or more (polyostotic) expanding fibrous skeletal lesions composed of bone-forming mesenchyme. The association of the polyostotic form with café au lait spots and hyperfunction of an endocrine system such as pseudoprecocious puberty of ovarian origin is known as McCune-Albright syndrome (MAS). A spectrum of the phenotypes is caused by activating mutations in the GNAS1 gene, which encodes the α subunit of the stimulatory G protein (Gsα). As the postzygotic mutations occur at different stages of early development, the extent and type of tissue affected are variable and explain the mosaic pattern of skin and bone changes. GTP binding activates the Gsα regulatory protein and mutations in regions of Gsα that selectively inhibit GTPase activity, which results in constitutive stimulation of the cyclic AMP–protein kinase A signal transduction pathway. Such mutations of the Gsα protein–coupled receptor may cause autonomous function in bone (parathyroid hormone receptor); skin (melanocyte-stimulating hormone receptor); and various endocrine glands including ovary (follicle-stimulating hormone receptor), thyroid (thyroid-stimulating hormone receptor), adrenal (adrenocorticotropic hormone receptor), and pituitary (growth hormone–releasing hormone receptor). The skeletal lesions are composed largely of mesenchymal cells that do not differentiate into osteoblasts, resulting in the formation of imperfect bone. In some areas of bone, fibroblast-like cells develop features of osteoblasts in that they produce extracellular matrix that organizes into woven bone. Calcification may occur in some areas. In other areas, cells have features of chondrocytes and produce cartilage-like extracellular matrix.
Clinical Presentation Fibrous dysplasia occurs with equal frequency in both sexes, whereas MAS with precocious puberty is more common (10:1) in girls. The monostotic form is the most common and is usually diagnosed in patients between 20 and 30 years of age without associated skin lesions. The polyostotic form typically manifests in children <10 years old and may progress with age. Early-onset disease is generally more severe. Lesions may become quiescent in puberty and progress during pregnancy or with estrogen therapy. In polyostotic fibrous dysplasia, the lesions most commonly involve the maxilla and other craniofacial bones, ribs, and metaphyseal or diaphyseal portions of the proximal femur or tibia. Expanding bone lesions may cause pain, deformity, fractures, and nerve entrapment. Sarcomatous degeneration involving the facial bones or femur is infrequent (<1%). The risk of malignant transformation is increased by radiation, which has proven to be ineffective treatment. In rare patients with widespread lesions, renal phosphate wasting and hypophosphatemia may cause rickets or osteomalacia. Hypophosphatemia may be due to production of a phosphaturic factor by the abnormal fibrous tissue.
MAS patients may have café au lait spots, which are flat, hyperpigmented skin lesions that have rough borders (“coast of Maine”) in contrast to the café au lait lesions of neurofibromatosis that have smooth borders (“coast of California”). The most common endocrinopathy is isosexual pseudoprecocious puberty in girls. Other less common endocrine disorders include thyrotoxicosis, Cushing’s syndrome, acromegaly, hyperparathyroidism, hyperprolactinemia, and pseudoprecocious puberty in boys.
Radiographic Findings In long bones, the fibrous dysplastic lesions are typically well-defined, radiolucent areas with thin cortices and a ground-glass appearance. Lesions may be lobulated with trabeculated areas of radiolucency (Fig. 426e-4). Involvement of facial bones usually presents as radiodense lesions, which may create a leonine appearance (leontiasis osea). Expansile cranial lesions may narrow foramens and cause optic lesions, reduce hearing, and create other manifestations of cranial nerve compression.
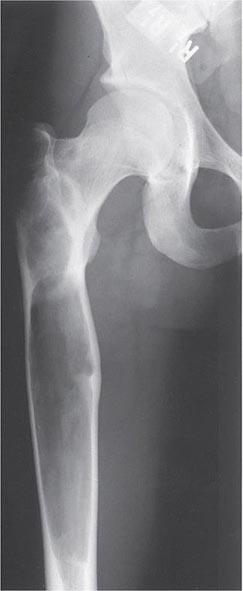
FIGURE 426e-4 Radiograph of a 16-year-old male with fibrous dysplasia of the right proximal femur. Note the multiple cystic lesions, including the large lucent lesion in the proximal midshaft with scalloping of the interior surface. The femoral neck contains two lucent cystic lesions.
Laboratory Results Serum ALP is occasionally elevated but calcium, parathyroid hormone, 25-hydroxyvitamin D, and 1,25-dihydroxy-vitamin D levels are normal. Patients with extensive polyostotic lesions may have hypophosphatemia, hyperphosphaturia, and osteomalacia. The hypophosphatemia and phosphaturia are directly related to the levels of fibroblast growth factor 23 (FGF23). Biochemical markers of bone turnover may be elevated.
OTHER DYSPLASIAS OF BONE AND CARTILAGE
PACHYDERMOPERIOSTOSIS
Pachydermoperiostosis, or hypertrophic osteoarthropathy (primary or idiopathic), is an autosomal dominant disorder characterized by periosteal new bone formation that involves the distal extremities. The lesions present as clubbing of the digits and hyperhidrosis and thickening of the skin, primarily of the face and forehead. The changes usually appear during adolescence, progress over the next decade, and then become quiescent. During the active phase, progressive enlargement of the hands and feet produces a paw-like appearance, which may be mistaken for acromegaly. Arthralgias, pseudogout, and limited mobility may also occur. The disorder must be differentiated from secondary hypertrophic osteopathy that develops during the course of serious pulmonary disorders. The two conditions can be differentiated by standard radiography of the digits in which secondary pachydermoperiostosis has exuberant periosteal new bone formation and a smooth and undulating surface. In contrast, primary hypertrophic osteopathy has an irregular periosteal surface.
There are no diagnostic blood or urine tests. Synovial fluid does not have an inflammatory profile. There is no specific therapy, although a limited experience with colchicine suggests some benefit in controlling the arthralgias.
OSTEOCHONDRODYSPLASIAS
These include several hundred heritable disorders of connective tissue. These primary abnormalities of cartilage manifest as disturbances in cartilage and bone growth. Selected growth-plate chondrodysplasias are described here. For discussion of chondrodysplasias, see Chap. 427.
Achondrodysplasia This is a relatively common form of short-limb dwarfism that occurs in 1 in 15,000 to 1 in 40,000 live births. The disease is caused by a mutation of the fibroblast growth factor receptor 3 (FGFR3) gene that results in a gain-of-function state. Most cases are sporadic mutations. However, when the disorder appears in families, the inheritance pattern is consistent with an autosomal dominant disorder. The primary defect is abnormal chondrocyte proliferation at the growth plate that causes development of short, but proportionately thick, long bones. Other regions of the long bones may be relatively unaffected. The disorder is manifest by the presence of short limbs (particularly the proximal portions), normal trunk, large head, saddle nose, and an exaggerated lumbar lordosis. Severe spinal deformity may lead to cord compression. The homozygous disorder is more serious than the sporadic form and may cause neonatal death. Pseudoachondroplasia clinically resembles achondrodysplasia but has no skull abnormalities.
Enchondromatosis This is also called dyschondroplasia or Ollier’s disease; it is also a disorder of the growth plate in which the primary cartilage is not resorbed. Cartilage ossification proceeds normally, but it is not resorbed normally, leading to cartilage accumulation. The changes are most marked at the ends of long bones, where the highest growth rates occur. Chondrosarcoma develops infrequently. The association of enchondromatosis and cavernous hemangiomas of the skin and soft tissues is known as Maffucci’s syndrome. Both Ollier’s disease and Maffucci’s syndrome are associated with various malignancies, including granulosa cell tumor of the ovary and cerebral glioma.
Multiple Exostoses This is also called diaphyseal aclasis or osteochondromatosis; it is a genetic disorder that follows an autosomal dominant pattern of inheritance. In this condition, areas of growth plates become displaced, presumably by growing through a defect in the perichondrium. The lesion begins with vascular invasion of the growth-plate cartilage, resulting in a characteristic radiographic finding of a mass that is in direct communication with the marrow cavity of the parent bone. The underlying cortex is resorbed. The disease is caused by inactivating mutations of the EXT1 and EXT2 genes, whose products normally regulate processing of chondrocyte cytoskeletal proteins. The products of the EXT gene likely function as tumor suppressors, with the loss-of-function mutation resulting in abnormal proliferation of growth-plate cartilage. Solitary or multiple lesions are located in the metaphyses of long bones. Although usually asymptomatic, the lesions may interfere with joint or tendon function or compress peripheral nerves. The lesions stop growing when growth ceases but may recur during pregnancy. There is a small risk for malignant transformation into chondrosarcoma.
EXTRASKELETAL (ECTOPIC) CALCIFICATION AND OSSIFICATION
Deposition of calcium phosphate crystals (calcification) or formation of true bone (ossification) in nonosseous soft tissue may occur by one of three mechanisms: (1) metastatic calcification due to a supranormal calcium × phosphate concentration product in extracellular fluid; (2) dystrophic calcification due to mineral deposition into metabolically impaired or dead tissue despite normal serum levels of calcium and phosphate; and (3) ectopic ossification, or true bone formation. Disorders that may cause extraskeletal calcification or ossification are listed in Table 426e-2.
|
DISEASES AND CONDITIONS ASSOCIATED WITH ECTOPIC CALCIFICATION AND OSSIFICATION |
METASTATIC CALCIFICATION
Soft tissue calcification may complicate diseases associated with significant hypercalcemia, hyperphosphatemia, or both. In addition, vitamin D and phosphate treatments or calcium administration in the presence of mild hyperphosphatemia, such as during hemodialysis, may induce ectopic calcification. Calcium phosphate precipitation may complicate any disorder when the serum calcium × phosphate concentration product is >75. The initial calcium phosphate deposition is in the form of small, poorly organized crystals, which subsequently organize into hydroxyapatite crystals. Calcifications that occur in hypercalcemic states with normal or low phosphate have a predilection for kidney, lungs, and gastric mucosa. Hyperphosphatemia with normal or low serum calcium may promote soft tissue calcification with predilection for the kidney and arteries. The disturbances of calcium and phosphate in renal failure and hemodialysis are common causes of soft tissue (metastatic) calcification.
TUMORAL CALCINOSIS
This is a rare genetic disorder characterized by masses of metastatic calcifications in soft tissues around major joints, most often shoulders, hips, and ankles. Tumoral calcinosis differs from other disorders in that the periarticular masses contain hydroxyapatite crystals or amorphous calcium phosphate complexes, while in fibrodysplasia ossificans progressiva (below), true bone is formed in soft tissues. About one-third of tumoral calcinosis cases are familial, with both autosomal recessive and autosomal dominant modes of inheritance reported. The disease is also associated with a variably expressed abnormality of dentition marked by short bulbous roots, pulp calcification, and radicular dentin deposited in swirls. The primary defect responsible for the metastatic calcification appears to be hyperphosphatemia resulting from the increased capacity of the renal tubule to reabsorb filtered phosphate. Spontaneous soft tissue calcification is related to the elevated serum phosphate, which, along with normal serum calcium, exceeds the concentration product of 75.
All of the North American patients reported have been African American. The disease usually presents in childhood and continues throughout the patient’s life. The calcific masses are typically painless and grow at variable rates, sometimes becoming large and bulky. The masses are often located near major joints but remain extracapsular. Joint range of motion is not usually restricted unless the tumors are very large. Complications include compression of neural structures and ulceration of the overlying skin with drainage of chalky fluid and risk of secondary infection. Small deposits not detected by standard radiographs may be detected by 99mTc bone scanning. The most common laboratory findings are hyperphosphatemia and elevated serum 1,25-dihydroxyvitamin D levels. Serum calcium, parathyroid hormone, and ALP levels are usually normal. Renal function is also usually normal. Urine calcium and phosphate excretions are low, and calcium and phosphate balances are positive.
An acquired form of the disease may occur with other causes of hyperphosphatemia, such as secondary hyperparathyroidism associated with hemodialysis, hypoparathyroidism, pseudohypoparathyroidism, and massive cell lysis following chemotherapy for leukemia. Tissue trauma from joint movement may contribute to the periarticular calcifications. Metastatic calcifications are also seen in conditions associated with hypercalcemia, such as in sarcoidosis, vitamin D intoxication, milk-alkali syndrome, and primary hyperparathyroidism. In these conditions, however, mineral deposits are more likely to occur in proton-transporting organs such as kidney, lungs, and gastric mucosa in which an alkaline milieu is generated by the proton pumps.
DYSTROPHIC CALCIFICATION
Posttraumatic calcification may occur with normal serum calcium and phosphate levels and normal ion-solubility product. The deposited mineral is either in the form of amorphous calcium phosphate or hydroxyapatite crystals. Soft tissue calcification complicating connective tissue disorders such as scleroderma, dermatomyositis, and systemic lupus erythematosus may involve localized areas of the skin or deeper subcutaneous tissue and is referred to as calcinosis circumscripta. Mineral deposition at sites of deeper tissue injury including periarticular sites is called calcinosis universalis.
ECTOPIC OSSIFICATION
True extraskeletal bone formation that begins in areas of fasciitis following surgery, trauma, burns, or neurologic injury is referred to as myositis ossificans. The bone formed is organized as lamellar or trabecular, with normal osteoblasts and osteoclasts conducting active remodeling. Well-developed haversian systems and marrow elements may be present. A second cause of ectopic bone formation occurs in an inherited disorder, fibrodysplasia ossificans progressiva.
FIBRODYSPLASIA OSSIFICANS PROGRESSIVA
This is also called myositis ossificans progressiva; it is a rare autosomal dominant disorder characterized by congenital deformities of the hands and feet and episodic soft tissue swellings that ossify. Ectopic bone formation occurs in fascia, tendons, ligaments, and connective tissue within voluntary muscles. Tender, rubbery induration, sometimes precipitated by trauma, develops in the soft tissue and gradually calcifies. Eventually, heterotopic bone forms at these sites of soft tissue trauma. Morbidity results from heterotopic bone interfering with normal movement and function of muscle and other soft tissues. Mortality is usually related to restrictive lung disease caused by an inability of the chest to expand. Laboratory tests are unremarkable.
There is no effective medical therapy. Bisphosphonates, glucocorticoids, and a low-calcium diet have largely been ineffective in halting progression of the ossification. Surgical removal of ectopic bone is not recommended, because the trauma of surgery may precipitate formation of new areas of heterotopic bone. Dental complications including frozen jaw may occur following injection of local anesthetics. Thus, CT imaging of the mandible should be undertaken to detect early sites of soft tissue ossification before they are appreciated by standard radiography.
SECTION 5 |
DISORDERS OF INTERMEDIARY METABOLISM |
427 |
Heritable Disorders of Connective Tissue |
CLASSIFICATION OF CONNECTIVE TISSUE DISORDERS
Some of the most common conditions that are transmitted genetically in families are disorders that produce clinically obvious changes in the skeleton, skin, or other relatively acellular tissues that have been loosely defined as connective tissues. Because of their heritability, the disorders were recognized as potentially traceable to mutated genes soon after the principles of genetics were introduced into medicine. In the last several decades, many of these disorders have been linked to mutations in several hundred different genes. However, classifying the disorders on the basis of either their clinical presentations or the mutations causing them presents a challenge for both the clinician and the geneticist.
A major development in the field was made by McKusick, who suggested that a group of disorders that included brittle bones in children (osteogenesis imperfecta), hyperextensible skin (Ehlers-Danlos syndrome), and characteristic distortions of skeleton (Marfan’s syndrome) be considered as “heritable disorders of connective tissue” and that mutations causing the disorders would be found in the genes coding for proteins of the tissues.
The information on the disorders has continued to develop on two levels. The initial clinical classifications suggested by McKusick, and others, had to be refined as additional patients were examined. For example, some patients had skin changes similar to those commonly seen in Ehlers-Danlos syndrome, but this feature was overshadowed by other features such as extreme hypotonia or sudden rupture of large blood vessels. To account for the full spectrum of presentations in patients and families, many of the disorders have been reclassified several times, and each has been divided into a series of subtypes. For example, a recent effort to classify all the heritable disorders that alter the skeleton defined 456 distinctive conditions that were divided into 40 major groups.
The identification of mutations causing the diseases has developed on a parallel track. The first genes cloned for connective tissues were the two genes coding for type I collagen, the most abundant protein in bones, skin, tendons, and several other tissues. Some of the first assays in patients with osteogenesis imperfecta (OI) revealed mutations in type I collagen genes. Biochemical data developed using cultures of skin fibroblasts from affected patients demonstrated that the mutations dramatically altered the synthesis or structure of collagen fibers. The results stimulated efforts to identify additional mutations in genes coding for structural proteins. Genes for collagens provided an attractive paradigm to search for mutations, since a series of different types of collagens were found in different connective tissues and the collagen genes were readily isolated by their unique signature sequences. Also, the collagen genes are particularly vulnerable to a large number different mutations because of unusual structural requirements of the protein. The search for mutations in collagen genes proved fruitful in that mutations were found in most patients with OI, in many patients with hyperextensible skin, in some patients with dwarfism, and in patients with other disorders, including Alport’s syndrome, that were not initially classified as disorders of connective tissue. Also, mutations in collagen genes were found in subset of patients with a diagnosis of osteoarthritis and a subset of patients with the diagnosis of osteoporosis. However, the search for mutations quickly expanded to hundreds of other genes that included those for other structural proteins, for the posttranslational processing of structural proteins, and for growth factors and their receptors, and other genes whose functions are still not fully understood.
In many instances, the mutations helped to define the clinical subtype of the disorder. In others, however, they did not. Some patients with the same clinical presentations were found to have mutations in different genes. Also, some patients with different manifestations were found to have mutations in the same genes. In addition, it was difficult to establish whether a change in the structure of a gene caused the phenotypic changes in patients and was not simply a neutral polymorphism. Therefore, there has been a continuing debate as to whether the disorders should be classified by their clinical presentations or by the genetic abnormalities. As an illustration of the problem, mutations in 226 genes have been found to be associated with the 456 defined disorders of the skeleton, but the latest nosology remains a “hybrid” between a list of clinically defined disorders, waiting for molecular clarification, and an annotated database documenting the phenotypic spectrum produced by mutations in a given gene. A simpler system of classification proved feasible for one rare heritable disorder of skin, epidermolysis bullosa. The disorder was first defined by the presence of friction-induced blister. It was then divided into subtypes that were defined by the ultrastructural layers of the skin that cleaved and blistered. Most patients in each subtype were subsequently shown to have mutations in genes expressed in the corresponding layer of skin. Even with these patients, however, the strength of the genotype-phenotype correlation varies, and mutations have not yet been found in every patient.
In the end, consensus reports by experts in the field and sources such as the Online Mendelian Inheritance in Man database provide valuable resources for physicians searching for diagnoses of patients with unusual clinical features. However, patients with the most common forms of the disorders have mutations in a limited number of genes. This chapter will focus primarily on these more common disorders.
COMPOSITION OF CONNECTIVE TISSUES
Connective tissues such as skin, bone, cartilage, ligaments, and tendons are the critical structural frameworks of the body important for development and function. They consist of a complex interacting extracellular matrix network of collagens, proteoglycans, and a large number of noncollagenous glycoproteins and proteins. Although these precise combinations of up to ~500 potential extracellular matrix building blocks provide tissue-specific function, there are many overarching similarities in composition, such as the role of composite collagen fibrils in providing strength and form, elastin fibrils and proteoglycans and other interacting proteins, and glycoproteins that fine-tune function (Table 427-1). The most abundant components are three similar fibrillar collagens (types I, II, and III). They have a similar tensile strength as steel wires. The three fibrillar collagens are distributed in a tissue-specific manner: type I collagen accounts for most of the protein of dermis, ligaments, tendons, and demineralized bone; type I and type III are the most abundant proteins of large blood vessels; and type II is the most abundant protein of cartilage.
|
CONSTITUENTS OF CONNECTIVE TISSUES |
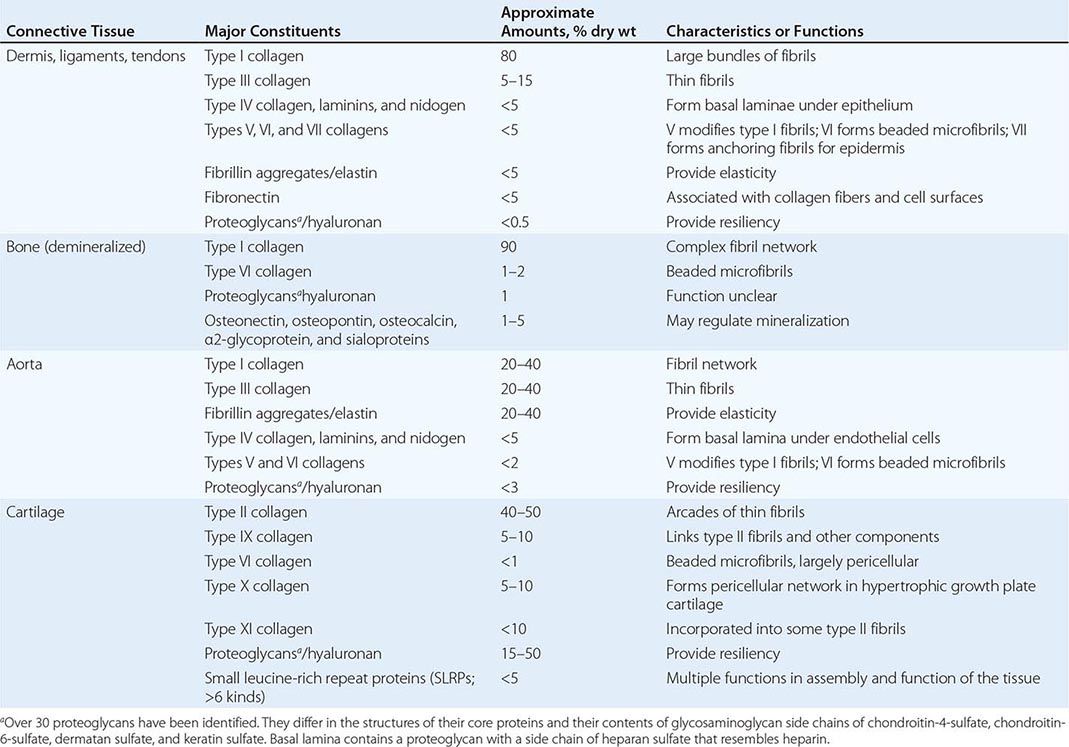
BIOSYNTHESIS AND TURNOVER OF CONNECTIVE TISSUES
Connective tissues are among the most stable components in living organisms, but they are not inert. During embryonic development, connective tissue membranes appear as early as the four-cell blastocyst to provide strength and a structural scaffold for the developing embryo. With the development of blood vessels and skeleton, there is a rapid increase in the synthesis, degradation, and resynthesis of connective tissues. The turnover continues at a slower, but still rapid pace throughout postnatal development and then spikes during the growth spurt of puberty. During adulthood, the metabolic turnover of most connective tissues is slow, but it continues at a moderate pace in bone. With age, malnutrition, physical inactivity, and low gravitational stress, the rate of degradation of most connective tissues, especially in bone and skin, begins to exceed the rate of synthesis and the tissues shrink. In starvation, a large fraction of the collagen in skin and other connective tissues is degraded and provides amino acids for gluconeogenesis (Chap. 97). In both osteoarthritis and rheumatoid arthritis, there is extensive degradation of articular cartilage collagen. Glucocorticoids weaken most tissues by decreasing collagen synthesis. In many pathologic states, however, collagen is deposited in excess. With most injuries to tissues, inflammatory and immune responses stimulate the deposition of collagen fibrils in the form of fibrotic scars. The deposition of the fibrils is largely irreversible and prevents regeneration of normal tissues in hepatic cirrhosis, pulmonary fibrosis, atherosclerosis, and nephrosclerosis.
Structure and Biosynthesis of Fibrillar Collagens The tensile strength of collagen fibers derives primarily from the self-assembly of protein monomers into large fibril structures in a process that resembles crystallization. The self-assembly requires monomers of highly uniform and relatively rigid structure. It also requires a complex series of posttranslational processing steps that maintain the solubility of the monomers until they are transported to the appropriate extracellular sites for fibril assembly. Because of the stringent requirements for correct self-assembly, it is not surprising that mutations in genes for fibrillar collagens cause many of the diseases of connective tissues.
The monomers of the three fibrillar collagens are formed from three polypeptide chains, called α chains, that are wrapped around each other into a rope-like triple-helical conformation. The triple helix is a unique structure among proteins, and it provides rigidity to the molecule. It also orients the side chains of amino acids in an “inside out” manner relative to most other proteins so that the charged and hydrophobic residues on the surface can direct self-assembly of the monomers into fibrils. The triple-helical conformation of the monomer is generated because each of the α chains has a repetitive amino acid sequence in which glycine (Gly) appears as every third amino acid. Each α chain contains about 1000 amino acids. Therefore, the sequence of each α chain can be designated as (-Gly-X-Y-)n, where × and Y represent amino acids other than glycine and n is >338. The presence of glycine, the smallest amino acid, in every third position in the sequence is critical because this residue must fit in a sterically restricted space in the middle of the helix where the three chains come together. The requirement for a glycine residue at every third position explains the severe effects of mutations that convert any of the glycine residues to an amino acid with a bulkier side chain (see below). Many of the X- and Y-position amino acids are proline and hydroxyproline, which, because of their ring structures, provide additional rigidity to the triple helix. Other X- and Y-positions are occupied by charged or hydrophobic amino acids that precisely direct lateral and longitudinal assembly of the monomers into highly ordered fibrils. Mutations that substitute amino acids in some X- and Y-positions can, in rare instances, also produce genetic diseases.
The fibers formed by the three fibrillar collagens differ in thickness and length, but they have a similar fine structure. As viewed by electron microscopy, they all have a characteristic pattern of cross-striations that are about one-quarter the length of the monomers and reflect the precise packing into fibrils. The three fibrillar collagens, however, differ in sequences found in the X- and Y-positions of the α chains and therefore in some of their physical properties. Type I collagen is composed of two identical α1(I) chains and a third α2(I) chain that differs slightly in its amino acid sequence. Type II collagen is composed of three identical α(II) chains. Type III collagen is composed of three identical α1(III) chains.
To deliver a monomer of the correct structure to the appropriate site of fibril assembly, the biosynthesis of fibrillar collagens involves a large number of unique processing steps (Fig. 427-1). The monomer, first synthesized as a soluble precursor called procollagen, contains an additional globular domain at each end. As the proα chains of procollagen are synthesized on ribosomes, the free N-terminal ends move into the cisternae of the rough endoplasmic reticulum. Signal peptides at the N-termini are cleaved, and additional posttranslational reactions begin. Proline residues in the Y-position of the repeating -Gly-X-Y- sequences are converted to hydroxyproline by the enzyme prolyl hydroxylase. The hydroxylation of prolyl residues is essential for the three α chains of the monomer to fold into a triple helix at body temperature. The enzyme requires ascorbic acid as one of its essential cofactors, an observation that explains why wounds fail to heal in scurvy (Chap. 96e). In scurvy, some of the underhydroxylated and unfolded protein accumulates in the cisternae of the rough endoplasmic reticulum and is degraded. Lysine residues in the Y-position are also hydroxylated to hydroxylysine by a separate lysyl hydroxylase. Many of the hydroxylysine residues are glycosylated with galactose or with galactose and glucose. A large mannose-rich oligosaccharide is assembled on the C-terminal propeptide of each chain. The proα chains are assembled by interactions among these C-terminal propeptides that control the selection of the appropriate partner chains to form hetero- or homotrimers and provide the correct chain registration required for subsequent formation of the collagen triple helix. After the C-terminal propeptides assemble the three proα chains, a nucleus of triple helix is formed near the C terminus, and the helical conformation is propagated toward the N terminus in a zipper-like manner that resembles crystallization. The folding into the triple helix is spontaneous in solution, but as discussed below, identification of rare mutations causing OI demonstrated that the folding in cellulo is assisted by ancillary proteins. The fully folded protein is then secreted. After secretion, procollagen is processed to collagen by cleavage of the N-propeptides and C-propeptides by two specific proteinases. The release of the propeptides decreases the solubility of the protein about 1000-fold. The entropic energy that is released drives the self-assembly of the collagen into fibrils. Self-assembled collagen fibers have considerable tensile strength, but their strength is increased further by cross-linking reactions that form covalent bonds between α chains in one molecule and α chains in adjacent molecules.
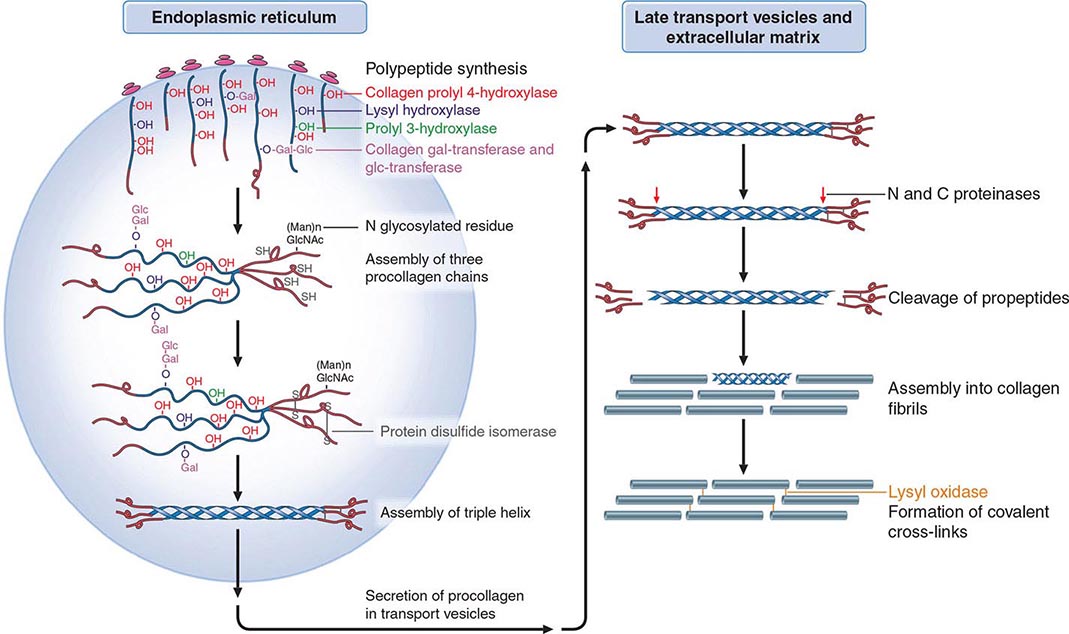
FIGURE 427-1 Schematic summary of biosynthesis of fibrillar collagens. (Modified and reproduced with permission from J Myllyharju, KI Kivirikko: Trends in Genetics 20:33, 2004.)
Although the assembly of collagen monomers into fibers is a spontaneous reaction, the process in tissues is modulated by the presence of less abundant collagens (type V with type I, and type XI with type II) and by other components such as a series of small leucine-rich proteins (SLRPs). Some of the less abundant components alter the rate of fibril assembly, whereas others change the morphology of the fibers or their interactions with cells and other molecules.
Collagen fibers are resistant to most proteases, but during degradation of connective tissues, they are cleaved by specific matrix metalloproteinases (collagenases) that cause partial unfolding of the triple helices into gelatin-like structures that are further degraded by less specific proteinases.
OTHER COLLAGENS AND RELATED MOLECULES
The unique properties of the triple helix are used to define a family of at least 28 collagens that contain repetitive -Gly-X-Y- sequences and form triple helices of varying length and complexity. The proteins are heterogeneous both in structure and function, and many are the sites of mutations causing genetic diseases. For example, the type IV collagen found in basement membranes is composed of three α chains synthesized from any of six different genes. Mutations in any of the six genes can cause Alport’s syndrome.
Fibrillin Aggregates and Elastin In addition to tensile strength, many tissues such as the lung, large blood vessels, and ligaments require elasticity. The elasticity was originally ascribed to an amorphous rubber-like protein named elastin. Subsequent analyses, largely sparked by discoveries of mutations causing the Marfan’s syndrome (MFS), demonstrated that the elasticity resided in thin fibrils composed primarily of large glycoproteins named fibrillins. The fibrillins contain large numbers of epidermal growth factor–like domains interspersed with characteristic cysteine-rich domains that are also found in latent transforming growth factor β (TGF-β) binding proteins. The fibrillins assemble into long beadlike strands that also contain numerous other components including small and variable amounts of elastin, bone morphogenic proteins (BMPs), and microfibril-associated glycoproteins (MAGPs). The principles whereby the fibrils provide elasticity to tissue and their biosynthetic assembly are still under investigation. As well as contributing to extracellular matrix structure, the fibrillins play a major role in TGF-β signaling.
Proteoglycans The resiliency to compression of connective tissues such as cartilage or the aorta is largely explained by the presence of proteoglycans. Proteoglycans are composed of a core protein to which are attached a large series of negatively charged polymers of disaccharides (largely chondroitin sulfates). At least 30 proteoglycans have been identified. They vary in their binding to collagens and other components of matrix, but specific functions have not been assigned to most. The major proteoglycan of cartilage, called aggrecan, has a core protein of 2000 amino acids that is decorated with about 100 side chains of chondroitin sulfate and keratin sulfate. The core protein, in turn, binds to long chains of the polymeric disaccharide hyaluronan to form proteoglycan aggregates, one of the largest soluble macromolecular structures in nature. Because of its highly negative charge and extended structure, the proteoglycan aggregate binds large amounts of water and small ions to distend the three-dimensional arcade of collagen fibers found in the same tissues. It thereby makes the cartilage resilient to pressure.
SPECIFIC DISORDERS
OSTEOGENESIS IMPERFECTA (OI)
The central feature of OI is a severe decrease in bone mass that makes bones brittle. The disorder is frequently associated with blue sclerae, dental abnormalities (dentinogenesis imperfecta), progressive hearing loss, and a positive family history. Most patients have mutations in one of the two genes coding for type I collagen.
Classification OI was originally classified into two subtypes of congenita and tarda depending on the age of onset of the symptoms. Sillence suggested a series of subtypes based on clinical and radiologic findings and mode of inheritance. As with the other disorders discussed here, the description of rare recessive forms of OI and discovery of mutations in new genes have opened a debate as to whether the disorders should be classified by the clinical phenotypes or by the genes at fault. For the near term, the classification based on the clinical presentations seems the most useful (Table 427-2).
|
CLASSIFICATION OF OSTEOGENESIS IMPERFECTA (OI) |
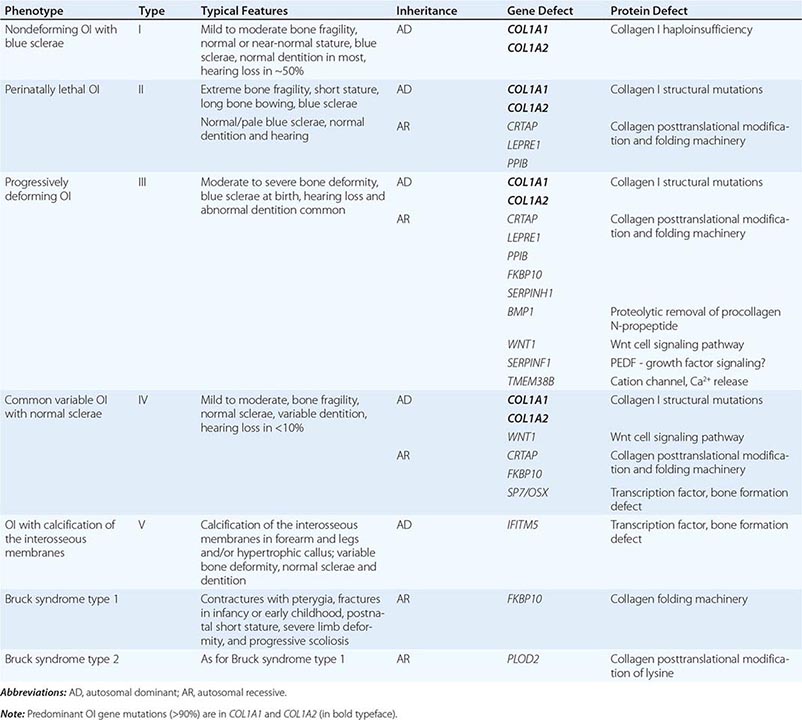
Type I is the mildest subtype and can produce either mild or no apparent deformities of the skeleton. Most patients have distinctly blue sclerae. Type II produces bone so brittle that it is lethal in utero or shortly after birth; it can be subclassified into types IIA, IIB, and IIC, depending on radiologic findings. Of the nonlethal forms, type III is progressively deforming with moderate to severe bone deformity, and type IV (common variable OI with normal sclerae) has mild to moderate bone fragility.
The classifications of patients by types of OI do not consistently predict the clinical course of the disease. Some patients appear normal at birth and become progressively worse; others have multiple fractures in infancy and childhood, improve after puberty, and fracture more frequently later in life. Women are particularly prone to fracture during pregnancy and after menopause. A few women from families with mild variants of OI do not develop fractures until after menopause, and their disease may be difficult to distinguish from postmenopausal osteoporosis.
Incidence Type I OI has a frequency of about 1 in 15,000–20,000 births. Type II OI has a reported incidence of about 1 in 60,000. Only a limited number of patients with the severe forms of OI have been reported, and the combined incidence of the severe forms that are recognizable at birth (types II, III, and IV) may be higher than 1 in 60,000.
Skeletal Effects In type I OI, the fragility of bones may be severe enough to limit physical activity or be so mild that individuals are unaware of any disability. Radiographs of the skull in patients with mild disease may show a mottled appearance because of small islands of irregular ossification. In type II OI, ossification of many bones is frequently incomplete. Continuously beaded or broken ribs and crumpled long bones (accordina femora) may be present. For reasons that are not apparent, the long bones may be either thick or thin. In types III and IV, multiple fractures from minor physical stress can produce severe deformities. Kyphoscoliosis can impair respiration, cause cor pulmonale, and predispose to pulmonary infections. The appearance of “popcorn-like” deposits of mineral in x-rays of the ends of long bones is an ominous sign. Progressive neurologic symptoms may result from basilar compression and communicating hydrocephalus. Type V OI is recognized by the presence of dislocated radial heads and hyperplastic callus formation.
In all forms of OI, bone mineral density is decreased. However, the degree of osteopenia may be difficult to evaluate because recurrent fractures limit exercise and thereby diminish bone mass. Surprisingly, fractures appear to heal normally.
Ocular Features The sclerae can be normal, gray, slightly bluish, or bright blue. The color is probably caused by a thinness of the collagen layers of the sclerae that allows the choroid layers to be seen. Blue sclerae, however, are an inherited trait in some families who do not have increased bone fragility.
Dentinogenesis The teeth may be normal, moderately discolored, or grossly abnormal. The enamel generally appears normal, but the teeth may have a characteristic amber, yellowish brown, or translucent bluish gray color because of a deficiency of dentin that is rich in type I collagen. The deciduous teeth are usually smaller than normal, whereas permanent teeth are frequently bell-shaped and restricted at the base. In some patients, the teeth readily fracture and need to be extracted. Similar tooth defects, however, can be inherited without any evidence of OI.
Hearing Loss Hearing loss usually begins during the second decade of life and occurs in more than 50% of individuals over age 30. The loss can be conductive, sensorineural, or mixed, and it varies in severity. The middle ear usually exhibits maldevelopment, deficient ossification, persistence of cartilage in areas that are normally ossified, and abnormal calcium deposits.
Other Features Changes in other connective tissues can include thin skin that scars extensively, joint laxity with permanent dislocations indistinguishable from those of Ehlers-Danlos syndrome (EDS), and occasionally, cardiovascular manifestations such as aortic regurgitation, floppy mitral valves, mitral incompetence, and fragility of large blood vessels. For unknown reasons, some patients develop bouts of a hypermetabolic state with elevated serum thyroxine levels, hyperthermia, and excessive sweating.
![]() Molecular Defects Of the ~1360 unique gene mutations now described in OI, more than 90% are heterozygous mutations in either COL1A1 or COL1A2, the genes coding for the proα1 or proα2 chain of type I procollagen (Table 427-2).
Molecular Defects Of the ~1360 unique gene mutations now described in OI, more than 90% are heterozygous mutations in either COL1A1 or COL1A2, the genes coding for the proα1 or proα2 chain of type I procollagen (Table 427-2).
Most patients with type I OI and blue sclerae have mutations that reduce the synthesis of proα1 chains to about one-half. Mutations that reduce the synthesis of proα2 chains produce slightly more severe phenotypes and skin defects similar to EDS.
In contrast to the null mutations found in type I OI, most of the severe variants (types II, III, and IV) are caused by mutations that produce structurally abnormal proα chains that have compromised assembly or abnormal folding of the triple helix. As with collagen mutations in other connective tissue diseases, these structural mutations generally fall into two functional categories. First, the relatively rare mutations in the C-propeptide domain can prevent or seriously impair initial assembly of the procollagen trimers. These misfolded chains are retained in the endoplasmic reticulum (ER) and targeted for degradation by the ER-associated proteasomal pathway. Because these mutations induce an ER stress response, the unfolded protein response (UPR) may have many downstream effects on cells. ER stress is a new concept in the pathophysiology of connective tissue disease and has been best characterized for chondrodysplasias (see below).
The most common type I collagen mutations, however, are single base substitutions that introduce an amino acid with a bulky side chain for one of the glycine residues that appear as every third amino acid in the triple helix. In effect, any of the 338 glycine residues in the helical domain of either the proα1 or proα2 chain of type I procollagen is a potential site for a disease-producing mutation. These mutations compromise the structural integrity of the triple helix, causing disruption to helix folding, retention of the mutant trimers in the ER, and increased posttranslational hydroxylation and glycosylation of lysines. Collagen-containing helix mutations can form insoluble aggregates in the ER that are degraded by the autophagosome-endosome system, rather than the proteasomes.
A similar sequence of events occurs with less common mutations that produce partial gene deletions, partial gene duplications, and splicing mutations. In addition to their intracellular effects, the structurally abnormal mutant-containing collagen that is secreted by the cell can also have important extracellular affects. For example, the presence of one abnormal proα chain in a procollagen molecule can interfere with cleavage of the N-propeptide from the protein. The persistence of the N-propeptide on a fraction of the molecules interferes with the self-assembly of normal collagen so that thin and irregular collagen fibrils are formed. Furthermore, if structurally abnormal collagens are incorporated into fibrils, they may have a destabilizing effect and be selectively degraded, or they may alter the interactions of collagen with other connective tissue components, disturbing architecture and stability.
Several generalizations can be made about mutations in type I collagen genes. One is that unrelated patients rarely have the same mutation in the same gene. Glycine substitutions in the N-terminal region of the triple helix tend to produce milder phenotypes, apparently because they have less effect on the zipper-like propagation of the triple-helical conformation from the C terminus. Rare substitutions of charged amino acids (Asp, Arg) or a branched amino acid (Val) in X- or Y-positions produce lethal phenotypes, apparently because they are located at sites for lateral assembly of the monomers or binding of other components of the matrix.
The search for mutations causing the less common and autosomal recessive forms of OI identified mutations in genes for a series of proteins that are essential for the timely folding of the procollagen monomer: cartilage-associated protein (CRTAP), prolyl-3-hydroxylase (LEPRE1/P3H1), cyclophilin B (PPIB), collagen chaperone-like protein HSP47 (SERPINH1), and the procollagen chaperone protein FKBP65 (FKBP10). Recently, mutations have been characterized in additional downstream components of the collagen fibrillogenesis pathway: BMP1, the gene coding for a metalloproteinase that cleaves the C-propeptide of type I procollagen, and PLOD2 (LH2, lysyl oxidase 2), which is involved in establishing collagen cross-links. In addition to these mutations that affect the collagen assembly pathway, mutations have been characterized in genes involved in the regulation of bone formation and mineralization such as SP7 (osterix), IFITM5, WNT1, and TMEM38B (Table 427-2).
Inheritance and Mosaicism in Germline Cells and in Somatic Cells Type I OI is inherited as an autosomal dominant trait. However, some patients with type I OI appear to represent sporadic new mutations or a diagnosis that was missed in earlier generations. Most lethal OI is the result of sporadic mutations that occur in the germ line in one of the parents. Because of the possibility for germline mosaicism for newly generated mutations, there is about a 7% probability that a second child could inherit a severe variant of OI.
Diagnosis OI is usually diagnosed on the basis of clinical criteria. The presence of fractures together with blue sclerae, dentinogenesis imperfecta, or family history of the disease is usually sufficient to make the diagnosis. Other causes of pathologic fractures must be excluded, including battered child syndrome, nutritional deficiencies, malignancies, and other inherited disorders such as chondrodysplasias and hypophosphatasia that can have overlapping presentations. The absence of superficial bruises can be helpful in distinguishing OI from battered child syndrome. X-rays usually reveal a decrease in bone density that can be verified by photon or x-ray absorptiometry. Bone microscopy can be helpful in the diagnosis. The diagnosis, as in other genetic disorders, is now routinely determined using targeted candidate gene sequencing and exome sequencing, but whole-genome sequencing may be used in the future.
|
TREATMENT |
OSTEOGENESIS IMPERFECTA |
Many patients with OI lead productive and successful lives despite severe deformities. Those with mild forms of the disease may need little treatment when fractures decrease after puberty, but women require special attention during pregnancy and after menopause, when fractures again increase. More severely affected children require a comprehensive program of physical therapy and surgical management of fractures and skeletal deformities.
Many fractures are only slightly displaced and have little soft tissue swelling and, therefore, can be treated with minimal support or traction for a week or two followed by a light cast. If fractures are relatively painless, physical therapy can be initiated early. A judicious amount of exercise prevents loss of bone mass secondary to physical inactivity. Some physicians advocate insertion of steel rods into long bones to correct limb deformities; the risk/benefits and cost/benefits of such procedures are difficult to evaluate. Aggressive conventional intervention is usually warranted for pneumonia and cor pulmonale. For severe hearing loss, stapedectomy or replacement of the stapes with a prosthesis may be successful. Moderately to severely affected patients should be evaluated periodically to anticipate possible neurologic problems. About half of children have a substantial increase in growth when given growth hormone. Treatment with bisphosphonates to decrease bone loss has been introduced for moderate to severe forms of OI. Improvements in bone mineral density are consistently seen in patients. Some clinical trials observed improvements in bone pain and fracture incidence; however, there are still unresolved questions about the best delivery protocols and the risks associated with long-term use in OI patients. For these reasons, the current consensus is that bisphosphonate therapy should be restricted to moderate to severe OI, where the possible benefits outweigh risks. Also, a clinical trial was performed in which patients were treated by intravenous infusion of cells from bone marrow referred to as mesenchymal stem cells, or multipotential stromal cells (MSCs; see Chap. 90e). Promising results were obtained, but the trial required a prior bone marrow transplant with marrow from a normal donor who subsequently was used as a source of normal MSCs. As a result, the procedure has not been widely adopted. However, the results raise the possibility that it may be possible in the future to develop effective stem cell therapies for OI.
Counseling and emotional support are important for patients and their parents; lay organizations in some countries provide help in these areas. Prenatal ultrasonography will detect severely affected fetuses at about 16 weeks of pregnancy. Diagnosis is routinely performed on DNA from blood.
EHLERS-DANLOS SYNDROME
EDS is characterized by hyperextensible skin and hypermobile joints, but the category includes rare patients with other distinctive features. Mutations in different types of collagen are found in many patients, but other genes are at fault in rare forms. Contrary to initial expectations, no patients have been found with mutations in the gene for elastin in EDS.
Classification Several types of EDS have been defined, based on the extent to which the skin, joints, and other tissues are involved, mode of inheritance, and molecular and biochemical analysis (Table 427-3). Classical EDS includes a severe form of the disease (type I) and a milder form (type II), both characterized by joint hypermobility and skin that is velvety in texture, hyperextensible, and easily scarred. In hypermobile EDS (type III), joint hypermobility is more prominent than skin changes. In vascular-type EDS (type IV), the skin changes are more prominent than joint changes, and the patients are predisposed to sudden death from rupture of large blood vessels or other hollow organs. EDS type V is similar to EDS type II but is inherited as an X-linked trait. The ocular-scoliotic type of EDS (type VI) is characterized by scoliosis, ocular fragility, and a cone-shaped deformity of the cornea (keratoconus). The arthrochalasic type of EDS (type VIIA and VIIB) is characterized by marked joint hypermobility that is difficult to distinguish from EDS III except by the specific molecular defects in the processing of type I procollagen to collagen. The periodontotic-type EDS (type VIII) is distinguished by prominent periodontal changes. EDS types IX, X, and XI were defined on the basis of preliminary biochemical and clinical data. EDS due to tenascin × deficiency has not been assigned a type; it is an autosomal recessive form of the syndrome similar to EDS II. The cardiac valvular form of EDS has similar features to EDS II, but also involves severe changes to the aorta. The progeroid form of EDS displays features of both EDS and progeria. Because of overlapping signs and symptoms, many patients and families with some of the features of EDS cannot be assigned to any of the defined types.
|
DIFFERENT FORMS OF EHLERS-DANLOS SYNDROME |
Incidence The overall incidence of EDS is about 1 in 5000 births, with a higher rate for blacks. Classical and hypermobile types of EDS are the most common. Patients with milder forms frequently do not seek medical attention.
Skin Skin changes vary from thin and velvety to skin that is either dramatically hyperextensible (“rubber person” syndrome) or easily torn or scarred. Patients with classical EDS develop characteristic “cigarette-paper” scars. In vascular-type EDS, extensive scars and hyperpigmentation develop over bony prominences, and the skin may be so thin that subcutaneous blood vessels are visible. In the periodontotic type of EDS, the skin is more fragile than hyperextensible, and it heals with atrophic, pigmented scars. Easy bruisability occurs in several types of EDS.
Ligament and Joint Changes Laxity and hypermobility of joints vary from mild to unreducible dislocations of hips and other large joints. In mild forms, patients learn to avoid dislocations by limiting physical activity. In more severe forms, surgical repair may be required. Some patients have progressive difficulty with age.
Other Features Mitral valve prolapse and hernias occur, particularly with type I. Pes planus and mild to moderate scoliosis are common. Extreme joint laxity and repeated dislocations may lead to degenerative arthritis. In the ocular-scoliotic type of EDS, the eye may rupture with minimal trauma, and kyphoscoliosis can cause respiratory impairment. Also, sclerae may be blue.
![]() Molecular Defects Subsets of patients with different types of EDS have mutations in the structural genes for collagens (Table 427-3). These include mutations in the COL1A1 gene in a few patients with moderately severe classical EDS (type I); mutations in COL1A2 in rare patients with an aortic valvular form of EDS; mutations in two of the three genes (COL5A1 and COL5A2) for type V collagen, a minor collagen found in association with type I collagen, in about half the patients with classical EDS (types I and II); and mutations in the COL3A1 gene for type III collagen, which is abundant in the aorta in patients with the frequently lethal vascular EDS (type IV).
Molecular Defects Subsets of patients with different types of EDS have mutations in the structural genes for collagens (Table 427-3). These include mutations in the COL1A1 gene in a few patients with moderately severe classical EDS (type I); mutations in COL1A2 in rare patients with an aortic valvular form of EDS; mutations in two of the three genes (COL5A1 and COL5A2) for type V collagen, a minor collagen found in association with type I collagen, in about half the patients with classical EDS (types I and II); and mutations in the COL3A1 gene for type III collagen, which is abundant in the aorta in patients with the frequently lethal vascular EDS (type IV).
Some of the type I collagen-related mutations alter processing of the protein or genes for the processing enzymes. Arthrochalasic EDS (type VII) is caused by mutations in the amino acid sequence that make type I procollagen resistant to cleavage by procollagen N-proteinase or by mutations that decrease the activity of the enzyme. The persistence of the N propeptide causes the formation of collagen fibrils that are thin and irregular. Some of the patients have fragile bones and therefore a phenotype that overlaps with OI. The ocular-scoliotic type of EDS (type VI) is caused by homozygous or compound heterozygous mutations in the PLOD1 gene, which encodes procollagen-lysine 5-dioxygenase (lysyl hydroxylase 1), an enzyme required for formation of stable cross-links in collagen fibers.
Some patients with the hypermobile EDS (type III) and a few with mild EDS (type II) have mutations in the TNXB gene, which encodes tenascin X, another minor component of connective tissue that appears to regulate the assembly of collagen fibers. Mutations in proteoglycans have been found in a few patients. The progeroid form of EDS results from autosomal recessive mutations in B4GALT7, the gene for β-1,4-galactosyltransferase 7, a key enzyme in the addition of glycosaminoglycan chains to proteoglycans.
Diagnosis The diagnosis is based on clinical criteria and increasingly on DNA sequencing. Correlations between genotype and phenotype can be challenging, but gene or biochemical tests are particularly useful for the diagnosis of vascular type IV EDS with its dire prognosis.
As with other heritable diseases of connective tissue, there is a large degree of variability among members of the same family carrying the same mutation. Some patients have increased fractures and are difficult to distinguish from OI. A few families with heritable aortic aneurysms have mutations in the gene for type III collagen without any evidence of EDS or OI.
|
TREATMENT |
EHLERS-DANLOS SYNDROME |
Surgical repair and tightening of joint ligaments require careful evaluation of individual patients, as the ligaments frequently do not hold sutures. Patients with easy bruisability should be evaluated for bleeding disorders. Patients with type IV EDS and members of their families should be evaluated at regular intervals for early detection of aneurysms, but surgical repair may be difficult because of friable tissues. Also, women with type IV EDS should be counseled about the increased risk of uterine rupture, bleeding, and other complications of pregnancy.
CHONDRODYSPLASIAS
(See also Chap. 426e) Chondrodysplasias (CDs), also referred to as skeletal dysplasias, are heritable skeletal disorders that are characterized by dwarfism and abnormal body proportions. The category also includes some individuals with normal stature and body proportions who have features such as ocular changes or cleft palate, which are common in more severe CDs. Many patients develop degenerative joint changes, and mild CD in adults may be difficult to differentiate from primary generalized osteoarthritis. An undefined number of patients have mutations in either the most abundant collagen in cartilage (type II) or the less abundant collagens (types × or XI). Other patients have mutations in genes that code for other components of cartilage or for proteins required for the embryonic development of cartilage, including a common mutation in a gene for a fibroblast growth factor receptor.
Classification Over 200 distinct types and subtypes have been defined based on criteria such as “bringing death” (thanatophoric), causing “twisted” bones (diastrophic), affecting metaphyses (metaphyseal), affecting epiphyses (epiphyseal), affecting spine (spondylo-), and producing histologic changes such as an apparent increase in the fibrous material in the epiphyses (fibrochondrogenesis). Also, a number of eponyms are based on the first or most comprehensive case reports. Severe forms of the diseases produce dwarfism with gross distortions of most cartilaginous structures and of other structures including the eye. Mild forms are more difficult to classify. Among the features are cataracts, degeneration of the vitreous, retinal detachment, high forehead hypoplastic facies, cleft palate, short extremities, and gross distortions of the epiphyses, metaphyses, and joint surfaces. Patients with Stickler’s syndrome (hereditary arthro-ophthalmopathy) have been classified into three types based on a combination of the ocular phenotype and mutated genes.
INCIDENCE
The overall incidence of all forms of CD ranges from 1 per 2500 to 1 per 4000 births. Data on the frequency of individual CDs are incomplete, but the incidence of Stickler’s syndrome is 1 in 10,000. Therefore, the disease is probably among the more common heritable disorders of connective tissue.
![]() Molecular Defects Mutations in the COL2A1 gene for the type II collagen of cartilage are found in a fraction of patients with both mild and severe CDs. For example, a mutation in the gene substituting a cysteine residue for an arginine was found in three unrelated families with spondyloepiphyseal dysplasia (SED) and precocious generalized osteoarthritis (OA). Mutations in the gene, often glycine substitution mutations with the collagen II triple helix, were also found in some lethal CDs characterized by gross deformities of bones and cartilage, such as those found in spondyloepiphyseal dysplasia congenita, spondyloepimetaphyseal dysplasia congenita, hypochondrogenesis/achondrogenesis type II, and Kniest’s syndrome. The highest incidence of COL2A1 mutations, however, occurs in patients with the distinctive features of Stickler’s syndrome, which is characterized by skeletal changes, orofacial abnormalities, and auditory abnormalities. Most of the mutations in COL2A1 are premature stop codons that produce haploinsufficiency. In addition, some patients with Stickler’s syndrome or a closely related syndrome have mutations in two genes specific for type XI collagen, which is an unusual heterotrimer formed from α chains encoded by the gene for type II collagen (COL2A1) and two distinctive genes for type XI collagen (COL11A1 and COL11A2). Mutations in the COL11A1 gene are also found in patients with Marshall’s syndrome, which is similar to classic Stickler’s syndrome, but with more severe hearing loss and dysmorphic features, such as a flat or retracted midface with a flat nasal bridge, short nose, anteverted nostrils, long philtrum, and large-appearing eyes.
Molecular Defects Mutations in the COL2A1 gene for the type II collagen of cartilage are found in a fraction of patients with both mild and severe CDs. For example, a mutation in the gene substituting a cysteine residue for an arginine was found in three unrelated families with spondyloepiphyseal dysplasia (SED) and precocious generalized osteoarthritis (OA). Mutations in the gene, often glycine substitution mutations with the collagen II triple helix, were also found in some lethal CDs characterized by gross deformities of bones and cartilage, such as those found in spondyloepiphyseal dysplasia congenita, spondyloepimetaphyseal dysplasia congenita, hypochondrogenesis/achondrogenesis type II, and Kniest’s syndrome. The highest incidence of COL2A1 mutations, however, occurs in patients with the distinctive features of Stickler’s syndrome, which is characterized by skeletal changes, orofacial abnormalities, and auditory abnormalities. Most of the mutations in COL2A1 are premature stop codons that produce haploinsufficiency. In addition, some patients with Stickler’s syndrome or a closely related syndrome have mutations in two genes specific for type XI collagen, which is an unusual heterotrimer formed from α chains encoded by the gene for type II collagen (COL2A1) and two distinctive genes for type XI collagen (COL11A1 and COL11A2). Mutations in the COL11A1 gene are also found in patients with Marshall’s syndrome, which is similar to classic Stickler’s syndrome, but with more severe hearing loss and dysmorphic features, such as a flat or retracted midface with a flat nasal bridge, short nose, anteverted nostrils, long philtrum, and large-appearing eyes.
CDs are also caused by mutations in the less abundant collagens found in cartilage. For example, patients with Schmid metaphyseal CD have mutations in the gene for type × collagen, a short, network-forming collagen found in the hypertrophic zone of endochondral cartilage. The syndrome is characterized by short stature, coxa vara, flaring metaphyses, and waddling gait. As with other collagen genes, the most common mutations are of two types: nonsense mutations that lead to haploinsufficiency and structural mutations that compromise collagen assembly. In type × collagen, all the structural mutations detected occur in the C-terminal NC1 domain that coordinates the formation of the trimers. This NC1 domain is functionally equivalent to the C-propeptide of the fibrillar collagens. These mutations disturb the structure of the NC1 domain, leading to misfolding and initiation of cellular ER stress via the unfolded protein response (UPR). While the UPR evolved to allow cells to adjust their ER folding capacity to differing protein folding loads, it is deployed by cells when mutant misfolded proteins accumulate in the ER. Activation of the UPR attenuates protein translation and activates mutant protein degradation pathways such as ER-associated degradation. If these strategies do not sufficiently reduce the stress response, cell death may occur. In Schmid metaphyseal CD, mutant misfolded type × collagen induces the UPR, resulting in downstream consequences that contribute to the pathophysiology. This general mechanism may also contribute to pathology in other CDs (and in other connective tissues disorders) where gene mutations lead to protein structural abnormalities.
Some patients have mutations in genes for proteins that interact with collagens. Patients with pseudoachondroplasia or autosomal dominant multiple epiphyseal dysplasia have mutations in the gene for the cartilage oligomeric matrix protein (COMP), a protein that interacts with both collagens and proteoglycans in cartilage. However, some families with multiple epiphyseal dysplasia have a defect in one of the three genes for type IX collagen (COL9A1, COL9A2, and COL9A3) or in matrilin-3, another extracellular protein found in cartilage. With misfolding mutations in COMP and matrilin-3, the activation of the UPR has been described, providing further evidence that the UPR is a component of pathology of these conditions.
Some CDs are caused by mutations in genes that affect early development of cartilage and related structures. The most common form of short-limbed dwarfism, achondroplasia, is caused by mutations in the gene for a receptor for a fibroblastic growth factor (FGFR3). The mutations in the FGFR3 gene causing achondroplasias are unusual in several respects. The same single-base mutation in the gene that converts glycine to arginine at position 380 in the FGFR3 gene is present in over 90% of patients. Most patients harbor sporadic new mutations, and therefore, this nucleotide change must be one of the most common recurring mutations in the human genome. The mutation causes unregulated signal transduction through the receptor and inappropriate development of cartilage. Mutations that alter other domains of FGFR3 have been found in patients with the more severe disorders of hypochondroplasia and thanatophoric dysplasia and in a few families with a variant of craniosynostosis. However, most patients with craniosynostosis appear to have mutations in the related FGFR2 gene. The similarities between the phenotypes produced by mutations in genes for FGF receptors and mutations in structural proteins of cartilage are probably explained by the observation that the activity of FGFs is regulated in part by binding of FGFs to proteins sequestered in the extracellular matrix. Therefore, the situation parallels the interactions between transforming growth factors (TGFs) and fibrillin in MFS (see below).
Other mutations involve the proteoglycans of cartilage, aggrecan (AGC1), and perlecan (HSPG2) and the proteoglycan posttranslational sulphation pathway (DTDST, PAPSS2, and CHST3). Mutations in more than 45 other genes have been defined in chondrodysplasias.
Diagnosis The diagnosis of CDs is made on the basis of the physical appearance, slit-lamp eye examinations, x-ray findings, histologic changes, and clinical course. Evaluation of patients by specialists in the field is usually required for a diagnosis. Targeted gene and exome sequencing or more global sequencing strategies are used for molecular diagnosis. Given the wide spectrum of CD phenotypes, these gene tests are becoming critical diagnostic tools. For Stickler’s syndrome, more precise diagnostic criteria have made it possible to identify type I variants with mutations in the COL2A1 gene with a high degree of accuracy. It has been suggested that the type II variant with mutations in the COL11A1 gene can be identified on the basis of a “beaded” vitreous phenotype, and the type III variant with mutations in the COL11A2 gene can be identified on the basis of the characteristic systemic features without the ocular involvement. Prenatal diagnosis based on analysis of DNA obtained from chorionic villus or amniotic fluid is possible.
|
TREATMENT |
CHONDRODYSPLASIAS |
The treatment is symptomatic and is directed to secondary features such as degenerative arthritis. Many patients require joint replacement surgery and corrective surgery for cleft palate. The eyes should be monitored carefully for the development of cataracts and the need for laser therapy to prevent retinal detachment. In general, patients should be advised to avoid obesity and contact sports. Counseling for the psychological problems of short stature is critical, and support groups have formed in many countries.




