29 Organ Transplantation
Liver Transplantation
The first successful liver transplant was performed by Dr. Tom Starzl in a child in 1967, but the history of liver transplantation begins in 1955, with Stuart Welch in Albany, and Jack Cannon at UCLA. Welch was the first to describe auxiliary liver transplantation in the dog and Cannon was the first to attempt orthotopic liver transplantation in dogs. Unfortunately, none of the dogs survived the operation.1 Francis Moore and Tom Starzl continued the research in the dog model. From 1958 to 1959, they each were able to successfully transplant the liver in the dog but the animals all died from rejection within 4 to 20 days. These deaths highlighted the barriers that prevented the first application in humans.
In the early stages of animal experimentation with liver transplantation, the main barriers to success involved surgical technique, organ preservation, and immunosuppression. Regarding organ preservation, the liver used to be preserved with chilled electrolyte solutions like lactated ringers and normal saline. Preservation time with these solutions was only 5 to 6 hours. In 1987, the University of Wisconsin developed a solution which increased the preservation time of livers to 18 to 24 hours. The third barrier, immunosuppression, was the most significant and likely explained most of the canine deaths. Medawar discovered the role of the immune system in organ rejection in 1944. Since that time, several unsuccessful attempts to deliberately weaken the immune system and control rejection failed. It was not until an animal model demonstrated that the combination of azathioprine and prednisone were synergistic and ameliorated rejection. This combination was first used in human kidney transplants, and then expanded to liver transplantation. Later, in 1967, antilymphocyte globulin was introduced, providing lymphoid depletion, and supplemented azathioprine and prednisone, providing better immunosuppression.2
Despite the initial success of the first pediatric liver transplant, the 1-year survival rate in subsequent transplant patients remained no greater than 50%. In 1979, with the introduction of cyclosporine, the 1-year patient survival increased to 70%.3,4 FK-506 (tacrolimus) was introduced in 1989, replacing cyclosporine,5 and the 1-year survival further increased to approximately 80%.
Demographics and Epidemiology
Indications for liver transplantation in children include the presence of an underlying primary liver pathology with acute or chronic liver failure caused by cholestatic liver disease, acute hepatic failure, metabolic disorders, cirrhosis, tumors, toxins, and other derangements (i.e., Budd-Chiari syndrome) (E-Table 29-1). The most common cause for liver transplantation in children is cholestatic liver disease secondary to biliary atresia, particularly in children less than 1 year of age, where it accounts for 50% or more of transplants.6 Biliary atresia continues to be the most common overall cause for liver transplantation, and the most common cholestatic cause, but cholestatic liver disease secondary to total parenteral nutrition has become more prominent over the past 10 years and accounts for just over 4% of all pediatric liver transplants. After cholestatic liver disease, acute hepatic failure and metabolic disorders are the next most common causes for pediatric liver transplantation. In the past, the most common metabolic disorders, in decreasing frequency, were α1-antitrypsin deficiency, tyrosinemia, Wilson disease, oxalosis, and glycogen storage diseases. The three most common disorders that lead to liver transplantation have changed, with cystic fibrosis now the second most common metabolic indication for pediatric liver transplantation, according to the United Network for Organ Sharing/Organ Procurement and Transplantation Network (UNOS/OPTN.org).

E-TABLE 29-1 Primary Diagnosis of Liver Disease in Pediatric Patients, 1988 to 2011
| Primary Diagnosis | |
| Total | 12760 |
| Cholestatic (Total) | 6675 (52%) |
| Biliary atresia | 4673 |
| TPN-induced cholestasis | 659 |
| Alagille syndrome | 386 |
| Primary sclerosing cholangitis | 254 |
| Secondary biliary cirrhosis | 218 |
| Familiar cholestasis (Byler, other) | 180 |
| Biliary hypoplasia | 114 |
| Primary biliary cirrhosis | 33 |
| Neonatal cholestatic disease | 14 |
| Other cholestasis | 144 |
| Acute Hepatic Necrosis (Total) | 1732 (13.5%) |
| Neonatal hepatitis | 207 |
| Drug induced | 119 |
| Hepatitis A | 50 |
| Hepatitis B | 34 |
| Hepatitis C | 14 |
| Unknown | 938 |
| Other | 370 |
| Metabolic Disorder (Total) | 1651 (12.9%) |
| α1-antitrypsin | 465 |
| Cystic fibrosis | 198 |
| Wilson disease | 163 |
| Tyrosinemia | 128 |
| Oxalosis | 111 |
| Glycogen storage disease | 74 |
| Maple syrup urine disease | 59 |
| Hemochromatosis | 50 |
| Other | 403 |
| Cirrhosis (Total) | 1027 (8.0%) |
| Idiopathic | 391 |
| Autoimmune | 330 |
| Hepatitis C | 92 |
| Chronic active hepatitis | 25 |
| Hepatitis B | 24 |
| Drug or toxin | 13 |
| Hepatitis A | 5 |
| Combined exposure (alcohol, hepatitis) | 2 |
| Alcoholic | 2 |
| Other | 143 |
| Hepatic Tumors (Total) | 601 (4.7%) |
| Hepatoblastoma | 370 |
| Hepatocellular carcinoma | 78 |
| Hemangioendothelioma | 50 |
| Benign tumor | 41 |
| Other | 62 |
| Other (Total) | 1071 (8.4%) |
| Congenital hepatic fibrosis | 127 |
| Budd-Chiari syndrome | 55 |
| Graft vs. host, secondary to nonliver tx | 24 |
| Trauma | 10 |
| Other | 855 |
TPN, Total parenteral nutrition; tx, transplantation.
Data from the United Network for Organ Sharing (UNOS) Scientific Registry 2011. Available at UNOS.org.
The cause for acute or fulminant hepatic failure is not known in the majority of patients. Neonatal hepatitis is the primary cause for acute hepatic failure in children. Drugs and toxins are the second leading cause of acute hepatic failure and viral hepatitis is third. Acetaminophen is the most common cause of drug- or toxin-induced liver failure.7
Allocation of the available livers to the appropriate recipients has been a challenge. Initially liver transplant candidates were prioritized based on geographic location and medical condition, as defined by Child-Turcotte-Pugh (CTP) score. Patients were ranked as status 1, 2a, 2b, or 3. Status 1 patients received the highest priority and were defined by the presence of acute liver failure of less than 6 weeks or a failed liver transplant within 1 week. Status 2a, 2b, and 3 were defined by their CTP score and time on the wait list.8 Efforts by the UNOS/OPTN Liver Disease Severity Scale (LDSS) committee to identify predictors of mortality in patients with chronic liver disease resulted in the implementation of the model for end-stage liver disease (MELD) and the pediatric end-stage liver disease (PELD) severity score in 2002.9 The PELD score incorporates variables for age, growth failure, serum albumin, bilirubin, and international normalized ratio (INR) (E-Table 29-2). In 2005, the cutoff for using the PELD score was revised to include only children 12 years of age or younger; the MELD score was extended downwards to include those as young as 12 years of age.10 Serum creatinine is incorporated in the MELD score because it predicts mortality for adult patients waiting for liver transplantation. Although this value may predict mortality after liver transplantation in adults, it is not predictive in children.11

E-TABLE 29-2 Pediatric End-Stage Liver Disease (PELD) Severity Score Determination
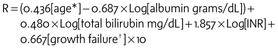
INR, International normalized ratio; R, PELD severity score.
*If younger than 1 year, use 1; if older than 1 year, use 0.
†If more than 2 standard deviations below the mean for age, use 1; if less than 2 standard deviations below the mean for age, use 0.
The allocation of deceased liver donors has changed with the new MELD/PELD policy. Before this policy, organs from donors younger than 18 years old were distributed only to those younger than 18 years old. With the new policy, the donor graft is first allocated to a status 1 child (less than 12 years of age) in the local region. If none is available, it is offered to the first status 1 adult in the region. If no status 1 adult is available, the liver is made available to children with more than 50% risk of mortality. Adults with mortality risk above 50% are next, and then all children are offered the graft over all other adult candidates. If there are no appropriate pediatric recipients in the region, the donor organ is offered to the national pool.12 The introduction of the MELD/PELD score appears to have decreased the wait time for deceased donor liver grafts. Analysis of prescore and postscore MELD/PELD data indicates that the median time to transplant, defined as the number of days for half of the new registrants to receive organs, has significantly decreased from 981 days in 2002 to 361 days in 2007.10
Survival of deceased-donor organs is age dependent. Infants less than 1 year old have the lowest 3-month and 1-year survival, at 88% and 83%, respectively, when compared with other pediatric groups. If the infant recipient of a transplanted liver survives the first year, the survival for this age group increases. In fact the 5-year survival is the greatest for infants less than 1 year old, at 84%. The 10-year survival for infants less than 1 year old is 77%, for children 1 to 5 years old, 79%, and for children 6 to 11 years old, 81%.10 However, outcomes other than survival, such as growth and cognitive function, should also be taken into consideration.13
Pathophysiology of Liver Disease
Cardiac Manifestations
Cardiac disturbances occur in children with liver disease because of altered physiology, congenital heart defects, and toxic side effects. A hyperdynamic circulation secondary to vasodilation characterizes the altered cardiac physiology from liver disease, with a compensatory increase in cardiac output (CO). Vasodilation is central to the hyperdynamic circulation that accompanies portal hypertension. It likely occurs because of the presence of vasoactive mediators. These mediators or gut-derived “humoral factors” (e.g., nitric oxide [NO], tumor necrosis factor α, endocannabinoids) enter the systemic circulation through portosystemic collaterals and bypass hepatic detoxification.14 Shunting also occurs at the level of the skin and the lungs. Mixed venous saturation increases in children with liver disease because of poor tissue oxygen extraction. Arterial-venous oxygen difference is reduced because of the combination of decreased oxygen consumption and hypoxia from arterial-venous shunting.
Cardiomyopathy associated with portal hypertension is well described in adults but is not well characterized in children with liver disease. However, children with liver disease can develop a cardiomyopathy for other reasons. Inborn errors of metabolism and other syndromes are associated with cardiomyopathies and cardiac anomalies. Some of the inborn errors include Wilson disease, oxalosis, glycogen storage disease type III, and Gaucher disease.15 Tacrolimus and cyclosporine A have also been associated with hypertrophic cardiomyopathy in animal studies and in pediatric liver transplant recipients.16,17,18 Echocardiographic assessment of cardiac function is generally well preserved in children receiving tacrolimus, but there may be evidence of subtle cardiovascular changes, which predispose a small percentage of children to develop hypertrophic cardiomyopathy.19
QT prolongation has also been described in adults with alcoholic liver disease and may be associated with sudden cardiac death.20 A decrease in K+ currents observed in rat cardiomyocytes with cirrhosis may provide a possible mechanism for the QT prolongation. Children with liver failure have also been shown to have an increase in QT interval (QTc greater than 450 msec in 18% of children with liver disease), possibly increasing the risk of ventricular arrhythmias, however, there is no evidence of an increased dispersion of repolarization (see Chapter 14). These changes appear to be transient and reversible after liver transplantation.21 Nonselective β-adrenergic blockade has also been shown to reduce the QT prolongation, but it is unclear if this reduces the risk of arrhythmias or improves survival.22 Although previous data suggested that prolonged QT did not predict decreased survival,23 more recent evidence suggests that the presence of a prolonged QT was associated with an increased PELD score and portal hypertension. Children with chronic liver disease and prolonged QT may be at increased risk of mortality while waiting for a transplant.24
Pulmonary Manifestations
The hallmarks of the pulmonary manifestations of liver disease are hypoxia and pulmonary hypertension. Hypoxia is secondary to hepatopulmonary syndrome (HPS) and ventilation/perfusion (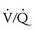 ) mismatch from atelectasis caused by tense ascites, hepatosplenomegaly, and/or pleural effusions. HPS is characterized by hypoxia from intrapulmonary arteriovenous shunting and intrapulmonary vascular dilatation.25 The diagnosis is predicated on either arterial hypoxia (Pao2 less than 70 mm Hg in room air) or an increased alveolar-arterial gradient of more than 20 mm Hg in the setting of pulmonary vascular dilatation. Intrapulmonary vascular dilatation can best be demonstrated on echocardiography or lung perfusion scan with macroaggregated albumin.26 HPS occurs in 15% to 20% of adults and in 0.5% to 20% of infants and children with cirrhosis27 as young as 6 months of age. It appears to be more prevalent in children with biliary atresia and polysplenia syndrome.28,29
) mismatch from atelectasis caused by tense ascites, hepatosplenomegaly, and/or pleural effusions. HPS is characterized by hypoxia from intrapulmonary arteriovenous shunting and intrapulmonary vascular dilatation.25 The diagnosis is predicated on either arterial hypoxia (Pao2 less than 70 mm Hg in room air) or an increased alveolar-arterial gradient of more than 20 mm Hg in the setting of pulmonary vascular dilatation. Intrapulmonary vascular dilatation can best be demonstrated on echocardiography or lung perfusion scan with macroaggregated albumin.26 HPS occurs in 15% to 20% of adults and in 0.5% to 20% of infants and children with cirrhosis27 as young as 6 months of age. It appears to be more prevalent in children with biliary atresia and polysplenia syndrome.28,29
Treatment for hypoxia is long-term supplemental oxygen; definitive treatment is liver transplantation. In a case series of seven children with HPS who were successfully transplanted, all recovered postoperatively with their hypoxia resolving within an average of 24 weeks.30
Portopulmonary hypertension (PPH) is defined by the World Health Organization as pulmonary artery hypertension (pulmonary systolic pressure of 25 mm Hg or greater) in the setting of a normal pulmonary capillary wedge pressure and portal hypertension.31 The incidence of PPH is 0.2% to 0.7% in adults with cirrhosis but increase to 3% to 9% in adults presenting for liver transplantation.32 The incidence in children is unknown, with accounts limited to case reports and one case series. Signs and symptoms on presentation are new heart murmurs, dyspnea, and syncope. Echocardiography can successfully identify pulmonary hypertension in pediatric and adult patients with PPH33; the severity of PPH predicts mortality. In a retrospective review, mild PPH did not increase mortality; however, those children who underwent liver transplantation with moderate PPH (pulmonary artery pressure [PAP] is 35 to 45 mm Hg) had a 50% mortality rate and those with severe PPH (PAP greater than 50 mm Hg) had a 100% mortality rate.34
There are no definitive guidelines for the management of children with PPH. Early identification is essential, and this may be accomplished with echocardiography. If PPH appears likely, cardiac catheterization should be performed to confirm the diagnosis, measure pulmonary artery pressures, and assess the response to NO and epoprostenol. Children who respond to medical management may be candidates for liver transplantation.35 Otherwise, severe PPH is generally a contraindication for liver transplantation because of the increased risk of mortality.
Neurologic Manifestations
Hepatic encephalopathy (HE) is a significant neurologic complication that is classified as either acute (seen in fulminant hepatic failure) or chronic (seen in chronic cirrhosis or chronic portal hypertension). The classification of the severity of acute and chronic HE is similar and is shown in Table 29-1. The pathophysiology is not entirely known, but cerebral edema appears to be a feature of both acute and chronic HE. Cerebral edema is more severe in acute HE and can result in increased intracranial pressure. Ammonia is repeatedly implicated in the pathogenesis of HE and may participate in the process by causing astrocyte swelling and low-grade cerebral edema.36,37 The two major sources of ammonia in humans are catabolism of endogenous protein and gastrointestinal absorption. Bacterial breakdown of nitrogen-containing products in the gut results in ammonia formation, which is then absorbed in the portal circulation. Factors that increase blood ammonia concentrations can exacerbate the signs and symptoms of HE. These typically include increased catabolism from infection, increased gut absorption from high-protein diets, constipation, and gastrointestinal bleeding. Other neurotoxins that have been implicated in the exacerbation of HE include endogenous production of benzodiazepines, hyponatremia, and inflammatory cytokines; these may all share the final common pathway of increasing cerebral edema.
TABLE 29-1 West Haven Staging Classification of the Severity of Acute and Chronic Hepatic Encephalopathy
| Grade | Description |
|---|---|
| 0 | Detectable only by neuropsychological testing |
| 1 | Lack of awareness, euphoria, or anxiety; shortened attention span; impaired addition and subtraction |
| 2 | Lethargy, minimal disorientation to time, personality change, inappropriate behavior |
| 3 | Somnolence but responsive to verbal stimuli, confusion, gross disorientation, bizarre behavior |
| 4 | Comatose |
Management of HE should begin with assessing the child’s ability to manage their airway. Children with grade 3 (somnolence to semi-stupor, responsive to verbal stimuli but confused) and 4 (coma, unresponsive to verbal or noxious stimuli) HE may require tracheal intubation to protect the airway and to provide adequate oxygenation and ventilation. Otherwise, management typically focuses on reducing gastrointestinal production and absorption of ammonia. Lactulose is often prescribed to create an osmotic gastrointestinal diuresis and to acidify the lumen of the gut to trap ammonia and minimize absorption. Antibiotics (e.g., neomycin and metronidazole) kill the gastrointestinal bacteria that are involved in metabolizing nitrogen products to ammonia. Other medications include sodium benzoate, which combines in the liver with ammoniagenic amino acids, like glycine, to facilitate their excretion.38 Ornithine aspartate may also provide a substrate to the liver for enhancing urea cycle and glutamine synthesis, and reduce ammonia levels. Flumazenil may reduce the symptoms of HE by inhibiting endogenous benzodiazepines and γ-aminobutyric acid, although 0.01 mg/kg in children with fulminant hepatic failure failed to correct the HE.38
Children with fulminant hepatic failure can have increased intracranial pressure (ICP), which is the major cause of mortality and may be a contraindication for liver transplantation. Intracranial hypertension occurs in 38% to 81% of adult patients with fulminant hepatic failure39 and is often monitored in those with fulminant hepatic failure (grade 3 to 4 HE). The risk of intracranial hemorrhage secondary to coagulopathy can be reduced by replacing clotting factors and platelets, and by placing an epidural rather than a subdural monitor.40 Management strategies for children with increased ICP should focus on maintaining cerebral perfusion pressure above 60 mm Hg, and ICP at less than 20 mm Hg. Management strategies often include tracheal intubation and ventilation with the head in midline position and slightly elevated to 30 degrees to facilitate venous drainage. Ventilation is adjusted to achieve a Paco2 of 30 to 35 mm Hg with minimal positive end-expiratory pressure (PEEP). Medical management to reduce ICP includes administering barbiturates or propofol to minimize stimulation and to directly reduce ICP.41 Mannitol can be administered if ICP remains increased. Hypothermia has also been described, and in a small trial with 14 patients with fulminant hepatic failure, ICP was reduced by maintaining core body temperature at 32° C to 33° C.42 Orthotopic liver transplantation is the definitive treatment for children with acute or chronic HE.
Renal Manifestations
Renal failure is common in children with acute and chronic liver disease and its cause is multifactorial. Renal failure can be classified as prerenal azotemia, acute tubular necrosis (ATN), or hepatorenal syndrome (HRS). Prerenal azotemia from hypovolemia is a common cause for renal failure and occurs secondary to diuretic therapy, gastrointestinal bleeding, splanchnic pooling, and sepsis. ATN occurs because of decreased central blood volume secondary to central splanchnic pooling, and decreased prostaglandin synthesis. HRS is characterized by renal failure in the setting of liver failure and portal hypertension. The incidence in adults with chronic liver disease is approximately 10% to 15%; in children the incidence is even less, at approximately 5%. The reduced incidence in children may reflect the lack of definitive criteria for diagnosis of HRS in children.43 HRS occurs secondary to intense renal vasoconstriction from activation of the renin-angiotensin, arginine vasopressin, and sympathetic nervous systems. This activation is a homeostatic response to the profound splanchnic vasodilation that occurs with portal hypertension.44 HRS presents similarly to prerenal azotemia (increased creatinine, decreased urine sodium [UNa less than 10 mM, FeNa less than 1%]), but is differentiated from azotemia by its lack of response to a fluid challenge. HRS has been classified based on the rate of progression of renal failure into types 1 and 2: type 1, which has a worse prognosis, is characterized by a rapid progression of renal failure with a 100% increase in creatinine in less than 2 weeks. It usually occurs in patients with acute liver failure. In type 2, renal failure progresses over weeks to months, and usually occurs in patients with chronic liver disease. Regardless of the type, prognosis is poor in children with HRS, with a mortality of 80% to 95%.45 The definitive treatment for HRS is liver transplantation, because the renal failure is reversible if the liver is replaced.46
Pretransplant renal function predicts mortality in adult patients undergoing transjugular intrahepatic shunt and liver transplantation, and serum creatinine is used in the MELD score. Preexisting renal failure is also a major determinant of survival after liver transplantation in adults. Efforts to improve renal function pretransplant may improve posttransplantation outcome.47 At this time it is not clear that serum creatinine is a predictor of mortality in children with liver disease.48 Before transplantation, type 1 HRS can be managed with vasoconstrictors (vasopressin analogs, norepinephrine), although there are limited data on their use in children. Critically ill children may require continuous renal replacement therapy (continuous venovenous hemofiltration or hemodiafiltration) as a bridge to transplantation.
Metabolic Manifestations
The metabolic derangements seen in liver disease include glucose, ammonia, electrolyte, and acid-base disturbances. Electrolyte abnormalities include hyponatremia, hypo- and hyperkalemia, hypocalcemia, and hypomagnesemia. Hypoglycemia may occur in children with fulminant hepatic failure or abrupt discontinuation of total parenteral nutrition, but hyperglycemia is more common in the perioperative period. During the dissection and anhepatic phase there are several causes of hyperglycemia. Typically glucose concentrations will increase immediately after reperfusion. Serum glucose increases if there is an exogenous source of glucose or if there is altered glucose metabolism. Exogenous sources include glucose from blood products,49 dextrose containing intravenous (IV) fluids, and damaged hepatocytes from the liver graft.50 Glucose uptake is altered by methylprednisolone steroid-induced insulin resistance. Hepatic denervation likely results in alterations in insulin and glucose clearance during the postoperative period, and may explain the frequent occurrence of impaired glucose tolerance and diabetes in liver transplant recipients.51
Acid-base disturbances commonly occur during liver transplantation. Children with renal disease may have a preexisting metabolic acidosis from bicarbonate elimination. A metabolic acidosis is typically present during the dissection and anhepatic phase, but it is usually most pronounced immediately after reperfusion. Lactic acid and citrate (from blood products) are not metabolized during the anhepatic phase and contribute to the acidosis. Cross clamping of the inferior vena cava (IVC) and aorta alters blood flow to gut and lower extremity tissue beds and may also contribute to the lactic acidosis. Once the liver graft begins to function, the metabolism of lactate and citrate may lead to a metabolic alkalosis.49
Preoperative Evaluation
The preoperative evaluation begins with a history and physical examination to identify the primary cause of liver failure and to identify liver- and non–liver-related alterations in physiology that may affect the anesthetic and surgical plan. A complete review of the systems will identify most of the perioperative concerns (Table 29-2).
TABLE 29-2 Preoperative Evaluation of Liver Transplant Candidates
HPS, Hepatopulmonary syndrome.
The pulmonary concerns include hypoxia and PPH. Hypoxia can occur from 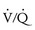 mismatch caused by pleural effusions and ascites, and HPS. Oxygen saturation on room air and with oxygen will identify those who are hypoxic and whether they respond to oxygen. Children with significant intrapulmonary shunts from HPS will not increase their oxygen saturation significantly while receiving oxygen. HPS can be diagnosed by demonstrating intrapulmonary vascular dilatation on echocardiography or lung perfusion scan with macroaggregated albumin.52
mismatch caused by pleural effusions and ascites, and HPS. Oxygen saturation on room air and with oxygen will identify those who are hypoxic and whether they respond to oxygen. Children with significant intrapulmonary shunts from HPS will not increase their oxygen saturation significantly while receiving oxygen. HPS can be diagnosed by demonstrating intrapulmonary vascular dilatation on echocardiography or lung perfusion scan with macroaggregated albumin.52
PPH can usually be identified on echocardiography (tricuspid regurgitation). Children suspected of having PPH may undergo a cardiac catheterization to define the severity of pulmonary hypertension and to assess the response to therapeutic agents (NO, epoprostenol). Children with severe pulmonary hypertension (PAP greater than 50 mm Hg) are at increased risk of perioperative mortality and liver transplantation is contraindicated.34
Renal failure is common and in adults predicts decreased survival in the period after transplantation.53 Both a blood urea nitrogen and creatinine measurement should be obtained preoperatively.
Intraoperative Care
Anesthesia is typically maintained with an inhalational agent, an opioid, and a neuromuscular blocking drug. Isoflurane, sevoflurane or desflurane may be used because they are readily available, undergo minimal hepatic metabolism, and appear to have minimal adverse effects on the liver, although all three have been associated with isolated case reports of hepatotoxicity.54,55,56 Propofol is also an option to maintain anesthesia during liver transplantation. It is relatively short-acting and even though the primary metabolic pathway is hepatic, there appears to be extrahepatic metabolism in the lung, kidney, and intestine.57,58 However, when used for a prolonged period, the pharmacokinetics of propofol switch from those of a short-acting to those of a long-acting anesthetic, and the risk of propofol infusion syndrome becomes a substantive concern (see Chapter 6). Neuromuscular blockade can be maintained with a variety of agents. Rocuronium, vecuronium, pancuronium, atracurium, and cisatracurium have all been described. Pancuronium may have the added advantages of increased heart rate, long duration, and reduced cost. The disadvantage of pancuronium, rocuronium, and vecuronium is their partial hepatic metabolism, but this can be overcome with appropriate monitoring and dose adjustments. Dose requirements of continuous infusions of rocuronium, vecuronium, and pancuronium are reduced during the anhepatic phase of liver transplantation, but return to initial values after reperfusion.59 There is no change in the dose requirements of atracurium during the anhepatic phase.60 Atracurium or cisatracurium may be ideal in patients with combined hepatic and renal insufficiency, because they are cleared by nonspecific esterases in tissues, and erythrocytes and do not rely on hepatic or renal function.
The liver metabolizes all opioids, with the exception of remifentanil, which is metabolized by plasma and tissue esterases. The metabolic pathway for most opioids is oxidation, although morphine undergoes glucuronidation.61 There is evidence that the elimination half-life and clearance of alfentanil and fentanyl are not dramatically altered in patients with cholestatic and cirrhotic liver disease.62,63 Fentanyl, sufentanil, alfentanil, and morphine have all been used during liver transplantation. Fentanyl is commonly selected and is usually administered as a bolus during induction (2 to 10 µg/kg) and maintained as an infusion throughout the anesthetic and the immediate postoperative period (2 to 5 µg/kg/hr).
Vascular access is important for resuscitation and monitoring. At least two peripheral upper extremity IV catheters should be placed, along with a central venous line. The central venous line is used to monitor trends in central venous pressure and measure superior vena cava oxygen saturation (a surrogate marker for venous oxygen saturation). Larger children tolerate rapid infusion catheters placed in the upper extremities. Blood loss can be significant, with estimates between 0.5 to 25 blood volumes (mean is approximately four blood volumes).49 Fluid warmers and rapid infusion devices (e.g., Level 1 Rapid Infuser, SIMS Level One INC, Rockland, Mass.) need to be available to facilitate resuscitation if significant hemorrhage occurs (see Chapters 10 and 51). The Level 1 Rapid Infuser has been associated with massive air emboli, and all air must be removed from the infusion bags before using the device, or the device should be used with air-detection add-on modifications.64 The Rapid Infusion System (RIS, Haemonetics, Braintree, Mass.) is no longer manufactured and has been replaced by the Belmont FMS (Belmont Instrument Corp., Billerica, Mass.). This device is not routinely used in small children (less than 30 kg) but may be indicated in larger children and adolescents (see Chapter 51). Choice of resuscitation fluids should be limited to 0.9% normal saline and Plasmalyte. Lactated ringers is not recommended because the lactate will accumulate unmetabolized during the anhepatic stage. Many children with liver disease are hypoalbuminemic, and the use of 5% albumin is appropriate.
Standard monitoring should include electrocardiogram, pulse oximetry (×2), noninvasive blood pressure, invasive arterial blood pressure, central venous pressure, and temperature. Other high-technology monitoring that is commonly employed in adult liver transplantation includes transesophageal echocardiography (TEE), continuous cardiac output (CCO) catheter, bispectral index (BIS), venovenous bypass (VVBP), and more than one arterial catheter. In children, there are limitations to the use of these monitors because of patient size. A United States–based utilization survey found that TEE, CCO, BIS, and VVBP are used in 0%, 7.7%, 15.4%, and 7.7% of pediatric transplant centers, respectively.65
Hematologic and electrolyte changes are common and measurements of arterial blood gases, sodium, potassium, calcium, magnesium, glucose, hemoglobin, platelets, and coagulation parameters (PT, PTT, fibrinogen, and D-dimers) need to be performed frequently throughout the procedure. Most centers use either portable devices or an operating room (OR) laboratory to obtain these data. Assessment of coagulation variables can be obtained with thromboelastography. However, only 28% of pediatric transplant centers used TEG routinely65 (E-Fig. 29-1). Positioning is critical to prevent soft tissue and peripheral nerve injuries. All extremities should be padded and all cables and wires need to be wrapped to protect the skin. The head should be rotated and repositioned periodically to prevent pressure sores and alopecia. To minimize the risk of peripheral neuropathy, the upper extremities should not be abducted more than 90 degrees and the wrists should not be hyperextended for the arterial catheter.

Surgical Technique
Hepatectomy (Stage 1)
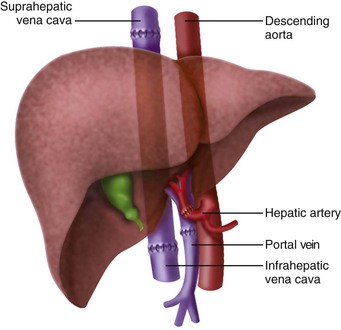
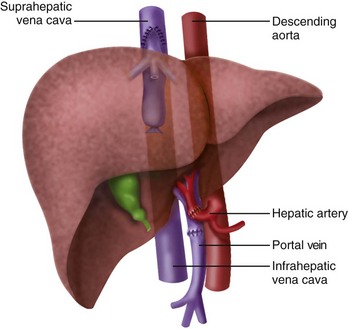
The initial description of orthotopic liver transplantation is referred to as the “classic” technique. In the “classic” approach the liver is dissected to its vascular supply and the supra- and infrahepatic IVC are clamped, along with the portal vein and hepatic artery. The liver is removed enbloc (Fig. 29-1). The disadvantage of this approach is that the IVC is cross clamped, thus reducing preload. The piggyback technique was described 1989 and is the preferred approach for pediatric transplants, because there is more flexibility with the organ size and it only requires partial clamping of the IVC.66 The liver is dissected away from the IVC, the short hepatic veins, portal vein, and left, right, middle hepatic vein. The infrahepatic vena cava of the donor is oversewn and the suprahepatic vena cava is anastomosed to the native hepatic veins (Figs. 29-2 and 29-3). This only requires partial clamping of the IVC. A portacaval shunt can be established for children who do not tolerate clamping of the portal vein (see Fig. 29-3). Typically these are recipients who have not developed collateral flow secondary to portal hypertension (e.g., maple syrup urine disease).
There are several physiologic considerations that take place during the hepatic dissection that affect the anesthetic management. Hypotension is common and can occur for a variety of reasons. These include changes to the cardiovascular, hematologic, and metabolic systems. The most common cause of hypotension includes hypovolemia secondary to hemorrhage and third space volume losses. Resuscitation with citrated blood products can result in hypocalcemia and surgical manipulation can cause mechanical compression of the IVC or right ventricle. Bleeding occurs from fragile collaterals, adhesions from prior surgery (Kasai procedure), and coagulopathy.67 Blood conservation during this portion of the surgery by maintaining low central venous pressure (decrease of 30% from baseline) has been described in adults. The reported benefit of a low central venous pressure is less bleeding, with subsequent decrease in allogeneic blood requirements and decreased morbidity. Some studies have suggested an overall reduction in 1-year mortality.68,69 The technique is controversial and the potential risks include end-organ injury, such as renal or graft failure.70 This technique has not been described in the pediatric liver transplant population.
Anhepatic Stage (Stage 2)
Cardiovascular changes that occur during this stage can result in hypotension. This occurs from the IVC cross-clamp, which causes a decrease in preload. Hemodynamically, there is a decrease in cardiac output, central venous pressure, and pulmonary artery pressure, with an increase in systemic vascular resistance.67 Preload should be gently augmented to maintain mean arterial pressure (MAP) with the lowest filling pressures possible. Hypervolemia may cause hepatic congestion during reperfusion. Inotropic agents like dopamine or epinephrine may need to be used to maintain MAP. A portacaval shunt, if performed, may moderate some of these hemodynamic changes by preserving preload from the portal vein (if venovenous bypass is going to be used, it is initiated during this period). Unmetabolized citrate causes hypocalcemia and hypomagnesemia because it chelates both. Acidosis occurs from the unmetabolized citrate, lactate, and other acids. Bicarbonate may be administered if there is a significant metabolic acidosis (base deficit less than 5 mEq/L). Once the liver is on the surgical field warm ischemia time begins. The time to reperfusion should be brief, and steps should be in place for reperfusion. Before reperfusion, potassium should be in the low-normal range and calcium and bicarbonate should be in the high-normal range. Hemoglobin should be maintained between 9 and 10 g/dL. If the potassium is greater than 5 mEq/L, steps need to be taken to reduce it. This may be achieved by increasing the serum pH with hyperventilation, and the administration of sodium bicarbonate (1 to 3 mEq/kg). Glucose and insulin can be administered to acutely reduce potassium (see Chapter 8). Potassium-wasting diuretics like furosemide (0.5 to 1 mg/kg) can also be used. Administering fresh blood or washed red blood cells will minimize the increase in potassium during transfusion therapy. β2-Adrenoceptor agonists may decrease potassium and can be used. Calcium and epinephrine should be immediately available for reperfusion.
Reperfusion (Stage 3)
The liver graft can be reperfused once the hepatic and portal vein anastomosis are complete. Before reestablishing hepatic blood flow, the graft is flushed to remove the preservation solution to minimize the reperfusion syndrome. Many changes can occur acutely during the reperfusion period secondary to cardiovascular, hematologic, and metabolic derangements. Postreperfusion syndrome is characterized by a decrease in MAP of greater than 30%.71 Several factors participate in this event, including myocardial dysfunction, arrhythmias, and bleeding. The myocardial dysfunction is thought to occur from release of NO and tumor necrosis factor α.72 Cardiovascular collapse can occur and the child may require epinephrine to correct the hemodynamic effects of reperfusion.71 Hyperkalemia is a common event immediately after reperfusion and may cause ventricular arrhythmias. It should be treated with calcium chloride (10 to 30 mg/kg) initially to stabilize the cardiac membrane and then insulin and dextrose, hyperventilation, furosemide, β2-adrenoceptor agonists, and sodium bicarbonate to decrease the serum potassium concentration (see previous section). The high potassium content of the preservation solution is the cause of the hyperkalemia. University of Wisconsin (UW) solution, a very commonly used preservation solution contains large amounts of potassium (120 mmol/L). Histidine-tryptophan-ketoglutarate (HTK) solution was introduced in 1980 as a cardioplegic solution, and contains substantially less potassium (10 mmol/L) than the UW solution.73 A recent study comparing the two found equal 1-month and 1-year graft survival with HTK and UW solutions. The viscosity is reduced with HTK and may introduce itself more easily into the vascular spaces in the donor liver.74 Although hyperkalemia is the hallmark electrolyte disturbance in the immediate reperfusion period, hypokalemia is more common in children throughout the intraoperative period and may require correction.75
Fibrinolysis can occur after reperfusion, and in one study it occurred in 60% of children and 80% of adults.76 This results from increased tissue plasminogen activator activity and decreased synthesis of fibrinolysis inhibitors. Heparin effect is produced by endogenous heparinoids from the graft, residual heparin from the preservation, and release of tissue plasminogen activator from endothelial cells of the revascularized graft. Aprotinin blunts this response by inhibiting kallikrein and plasmin, which led to the wide spread use of aprotinin. The beneficial effects of aprotinin to reduce blood loss in orthotopic liver transplant in adults and children had been considered inconclusive until the European Multicenter Study on the Use of Aprotinin in Liver Transplantation (EMSALT) study in 2000.77 However, the benefits of aprotinin in pediatric patients are not as clear. A retrospective study in 18 pediatric transplant patients receiving a high or regular dose of aprotinin demonstrated a reduction of red cell and fresh frozen plasma transfusion by half, but it was not found to be statistically significant. There is concern that there may be an association between aprotinin and intraoperative thrombotic events (hepatic artery and portal vein thrombosis) in pediatric recipients of liver transplants. Children may be at increased risk for developing intraoperative thrombi or emboli. Children are hypercoagulable after liver transplantation because of a decrease in protein C and antithrombin III,78 and they may be more likely to develop hepatic artery thrombosis.79,80,81 Aprotinin has been removed from the market because of the increased risk of death in adult cardiac patients. Tranexamic acid, an alternative antifibrinolytic is being used in adults for liver transplants; a nonrandomized prospective study did not find a difference in transfusion requirements or thromboembolic events after changing from aprotinin to tranexamic acid.82 Currently there are no pediatric data to support or refute the use of tranexamic acid or ε-aminocaproic acid.
Biliary and Hepatic Artery Reconstruction (Stage 4)
During biliary reconstruction, metabolic and hematologic alterations are addressed. As the liver graft begins to function, the citrate administered during the previous three phases is metabolized, and metabolic alkalosis may develop. The hemodynamic goals include maintaining a normal central venous pressure. If the central venous pressure is high (greater than 8 mm Hg), there is a concern that the liver graft can become congested and may not function normally. The risk of hepatic artery thrombosis ranges from 0% to 25% and is greater in infants and children.80,81 This risk increases if the PT and PPT are corrected. Also, the viscosity from a greater hematocrit may increase the risk of hepatic artery or portal vein thrombosis. The hematocrit does not need to be corrected to normal values. In fact, maintaining the hematocrit at 8 to 9 g/dL is safe and reasonable. Anticoagulation strategies with heparin, dextran, aspirin, and alprostadil may reduce the risk of hepatic artery thrombosis.
Split Liver Techniques and Living Donor Liver Transplants
Advances in surgical technique, tissue preservation, and immunosuppression have resulted in improved survival in children undergoing liver transplantation. The result is more children are awaiting liver transplantation, but the number of available organs has not increased significantly. Children are at a particular disadvantage because of size limitations. Two techniques have attempted to address these issues. The reduced liver technique does not increase the number of available grafts, and efforts were made to perform split liver techniques to make two grafts from one adult donor. The initial results were poor, with an increase in complications and mortality.83,84 The technique has evolved, and today the graft is split while still in vivo (in the heart-still-beating donor), as compared to ex vivo (splitting performed after the graft is removed from the donor). This decreases cold ischemia time and facilitates hemostasis of the liver edge. The result is improved child and graft survival,85 which has increased from 60% to 70% in the 1990’s to 80% to 90% in 2003. In one series, 218 split-liver grafts were transplanted between 1995 and 2002. Overall child survival at 1 year was 81.7%, and overall graft survival was 75.8%. Surgical complications that caused a return to the OR were bleeding (9.2%), bowel perforation (8.3%), and biliary problems (7.5%); hepatic artery complications occurred in 6.7%.85
Living-donor liver transplantation was first described in 1989.86 This approach has reduced mortality among children awaiting liver transplantation. The benefit of a living donor (especially if related), is improved posttransplant results because of better graft quality, smaller ischemic times, and better immune compatibility. In fact, these improved results have been observed in several institutions that perform this procedure.87 In a recent study, the 1- and 5-year patient survivals were 94% and 92% respectively.88 The left hepatic segment is removed for pediatric recipients, whereas the right hepatic lobe is removed for adult recipients. The regenerative capacity of the liver allows the donor to regenerate the liver without hepatic insufficiency. Despite the success of this technique for the recipients, there is considerable risk to the donor. Complications include exposure to blood products, peripheral nerve injuries, biliary leakage, abdominal wall defects, pleural effusions, pneumonia, pulmonary emboli, and death.88,89,90 Other than death, these complications were without long-term sequelae, except for a peripheral nerve injury.88
Outcomes
The Studies of Pediatric Liver Transplantation (SPLIT) registry was initiated in 1995 and consists of 38 centers in the United States and Canada. These centers contributed 85% of the pediatric liver transplants in 2002. Transplants performed more recently had greater survival rates. In the past, age less than 1 year was considered an increased risk factor for mortality, but over the past 20 years there is little mortality difference between children less than 2 years of age and those greater than 2 years of age at transplant.91
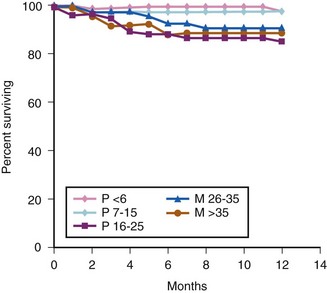
Survival also depends on the preoperative MELD/PELD score. Patients stratified to status 1 had a significantly smaller 1-year survival rate when compared to other transplant recipients (76% vs. 87%). Adults with MELD scores greater than 35 demonstrated decreased 1-year patient and graft survival. There is a suggestion that pediatric patients with greater PELD scores have decreased 1-year patient and graft survival, but that association was not statistically significant. The overall 1-year survival remained excellent, at more than 85%92 (E-Fig. 29-2).

Immediate Postoperative Care
Hemodynamically, patients after liver transplantation will continue to lose intravascular volume because of bleeding and third-space losses. These losses need to be replaced to maintain a normal central venous pressure and adequate urine output (0.5 to 1 mL/kg/hr). Replacement with a lactate free isotonic solution (0.9% normal saline or Plasmalyte) and albumin is appropriate. Particular attention should be focused on children with underlying ventricular dysfunction or PPH because they tolerate fluid overload poorly. In adults, evidence suggests that fluid overload was responsible for ICU readmission,93 whereas hypovolemia could lead to renal insufficiency and increase the risk of hepatic artery thrombosis. Preexisting pulmonary hypertension will not dissipate immediately, and children that were previously on prostaglandins will need to continue these infusions in the OR and in the postoperative period. Systemic hypertension is common after liver transplantation and has been described in as many as one-third of children94; this is typically attributed to cyclosporine therapy.
Most children require tracheal intubation, mechanical ventilation, and sedation in the immediate postoperative period. This is particularly true for smaller pediatric patients who may have received a relatively large graft and in children with underlying lung disease (HPS). Ascites, pulmonary edema, and pleural effusions may prolong the period of mechanical ventilation.95 Efforts to minimize atelectasis include positive-pressure ventilation with PEEP. Diuretics may be required on the second or third postoperative day to treat edema and effusions. There is speculation that prolonged mechanical ventilation may have a negative effect on the hemodynamics of transplant patients and may contribute to overall morbidity and mortality.96 High levels of PEEP may play a role in this morbidity. Some have advocated for early extubation to decrease the incidence of pulmonary complications and to facilitate discharge from the ICU.97
Neurologic complications after liver transplantation are common. In adults, 10% to 40% develop these complications.98 Comparable pediatric data are lacking. In adults, neurologic complications manifest as encephalopathy, seizures, or coma. The causes of encephalopathy and coma include drugs (immunosuppressive agents like tacrolimus and OKT3), infection (meningitis and brain abscess), strokes (bleeding), and hyponatremia with central pontine myelinolysis (CPM). The most common cause of seizures is an adverse drug reaction associated with immunosuppressant agents.99 Hyponatremia, which can contribute to neurologic complications, should be corrected slowly to prevent CPM. Correcting the hyponatremia no faster than 0.5 mEq/L/hr is considered safe. If the correction proceeds faster than recommended, then evidence from animal models indicates that dexamethasone administered within 6 hours of the correction may minimize the risk of CPM.100
Surgical complications after transplantation include vascular complications, acute rejection, and infections; frequent monitoring for their occurrence is important to ensure prompt treatment. Vascular complications include hepatic artery and portal vein thrombosis, bleeding, and bowel perforation.94 Hepatic artery thrombosis is identified with frequent hepatic Doppler flow imaging. Children may be anticoagulated with aspirin, heparin, dextran, and alprostadil to reduce the risk of thrombosis.101 Infections are common and contribute to significant morbidity. The primary source for infections appears to be central lines, percutaneous catheter drainage, and mechanical ventilation. Acute rejection should be suspected in children with fever and elevated liver enzymes. The diagnosis is made by histologic examination.94
Rejection is an immune response, and efforts to understand and control this immune response lie at the heart of transplant medicine. Initial efforts to control the response involved suppressing the recipient’s immune system. There has been a move from immunosuppression to immunotolerance. Immunotolerance describes the concept of immune cells from both the recipient and the donor coexisting without attacking each other.102 The goal of managing immunosuppression medication is to get to this state of tolerance. In this state of immunotolerance, minimal immunosuppression can be used. The benefits of decreasing immunosuppression include the reduced risk of infection, hypercholesterolemia, malignancy, hypertension, and diabetes mellitus. Protocols to induce tolerance include exposing patients to lymphoid-depleting agents (antilymphoid antibodies), like antithymocyte globulin, before liver engraftment, to reduce the antidonor response to a more controllable or “deletable” range and allow maintenance therapy (e.g., tacrolimus) to begin with one agent. Other agents are added only when there is evidence of rejection.103 The immunosuppressant agents currently used include calcineurin inhibitors (CNIs) like tacrolimus and cyclosporine, and they provide the mainstay of therapy. Other options include azathioprine or mycophenolate mofetil, for children who cannot tolerate the CNIs because of toxicity. Adverse effects of the CNIs include hypertension, tremor, and renal failure. Other agents being used include monoclonal antibodies against interleukin-2 (IL-2).104
Long-Term Issues
The cardiovascular effects of immunosuppressant agents includes hypertension from cyclosporine and cardiomyopathy (rare) from tacrolimus.105,106 Renal insufficiency can occur secondary to cyclosporine, diuretics, or hypertension. Baseline blood urea nitrogen and creatinine should be obtained preoperatively in children with a history of renal insufficiency, and medications that are renally cleared (morphine, rocuronium, vecuronium) should be adjusted or avoided. Hyperkalemia may accompany renal failure and should be evaluated before induction of anesthesia.
The endocrine effects of chronic steroid exposure include diabetes, growth retardation, and adrenal insufficiency. Children receiving insulin for diabetes need intraoperative blood glucose monitoring and dextrose-containing IV fluids if they are hypoglycemic or are at risk of developing hypoglycemia. Children receiving chronic steroids need stress-dose steroids during the perioperative period (see Chapter 25).
Most recipients of a liver transplant have been exposed to multiple procedures and may benefit from an anxiolytic preoperatively.107 Midazolam is safe, provided there is no evidence of residual HE. Anesthesia can be induced with an inhalational technique, provided the liver graft is functioning normally and nothing-by-mouth (NPO) guidelines have been followed. Children who are hospitalized, septic, bleeding, encephalopathic, or rejecting the graft should have an IV induction and their airways should be secured with an endotracheal tube. Isoflurane, sevoflurane, and desflurane are suitable for maintenance of anesthesia.
Kidney Transplantation
Demographics and Epidemiology
Renal transplantation improves development, quality of life, and survival108,109 in children with chronic renal failure. The OPTN maintains the national transplant registry in the United States; the OPTN data as of 2011 shows 826 children under the age of 18 on the active waiting list; this represents fewer than 1% of the 89,169 patients in the US waiting for a kidney.110 Characterization of such a small patient population is challenging. The North American Pediatric Renal Trials and Collaborative Studies (NAPRTCS) group has obtained the voluntary participation of all centers in the United States and Canada performing renal transplants on more than four children per year. Since the advent of the NAPRTCS in 1987, it has registered 11,603 pediatric renal transplants in 10,632 recipients.111 This represents a capture rate of approximately 68% of the 16,000 kidney transplants that have been performed in the United States in children younger than 18 years of age110 (E-Table 29-3). The inclusion into the NAPRTCS of pediatric patients requiring dialysis began in 1992, and the inclusion of children in chronic renal failure (creatinine clearance of 75 mL/min/1.73 m2 or less) began in 1994. Currently, there are 16,874 children in the NAPRTCS registry based on the above indications (E-Table 29-4).


E-TABLE 29-3 Index Transplants: Recipient and Transplant Characteristics
| N | % | |
|---|---|---|
| TOTAL | 10,632 | 100.0 |
| Sex | ||
| Male | 6298 | 59.2 |
| Female | 4334 | 40.8 |
| Race | ||
| Caucasian | 6296 | 59.2 |
| African American | 1820 | 17.1 |
| Hispanic | 1806 | 17.0 |
| Other | 710 | 6.7 |
| Primary Diagnosis | ||
| Aplasia/hypoplasia/dysplasia kidney | 1681 | 15.8 |
| Obstructive uropathy | 1630 | 15.3 |
| Focal segmental glomerulosclerosis | 1246 | 11.7 |
| Reflux nephropathy | 549 | 5.2 |
| Chronic glomerulonephritis | 340 | 3.2 |
| Polycystic disease | 323 | 3.0 |
| Medullary cystic disease | 287 | 2.7 |
| Congenital nephritic syndrome | 277 | 2.6 |
| Hemolytic uremic syndrome | 273 | 2.6 |
| Prune belly | 268 | 2.5 |
| Familial nephritis | 241 | 2.3 |
| Cystinosis | 221 | 2.1 |
| Membranoproliferative glomerulonephritis, Type I | 186 | 1.7 |
| Pyelonephritis or interstitial nephritis | 184 | 1.7 |
| Idiopathic crescentic glomerulonephritis | 181 | 1.7 |
| SLE nephritis | 159 | 1.5 |
| Renal infarct | 140 | 1.3 |
| Berger (IgA) nephritis | 135 | 1.3 |
| Henoch-Schönlein nephritis | 113 | 1.1 |
| Membranoproliferative glomerulonephritis, Type II | 85 | 0.8 |
| Wegener granulomatosis | 66 | 0.6 |
| Wilms tumor | 56 | 0.5 |
| Drash syndrome | 55 | 0.5 |
| Oxalosis | 55 | 0.5 |
| Membranous nephropathy | 47 | 0.4 |
| Other systemic immunologic disease | 34 | 0.3 |
| Sickle cell nephropathy | 16 | 0.2 |
| Diabetic glomerulonephritis | 11 | 0.1 |
| Other | 1110 | 10.4 |
| Unknown | 663 | 6.2 |
SLE, Systemic lupus erythematosus.
Data from Exhibit 1.2 of The North American Pediatric Renal Trials and Collaborative Studies 2010 Annual Transplant Report. Available at https://web.emmes.com/study/ped/annlrept/annlrept.html (accessed June 17, 2011).
E-TABLE 29-4 Summary of North American Pediatric Renal Trials and Collaborative Studies (NAPRTCS) Patient Registrations from the NAPRTCS 2008 Annual Report
| N | % | |
|---|---|---|
| All Patients | 1684 | 100.0 |
| CKD only | 4586 | 27.2 |
| Dialysis only | 1828 | 10.8 |
| Transplant only | 4810 | 28.5 |
| CKD and dialysis | 606 | 3.6 |
| CKD and transplant | 987 | 5.8 |
| Dialysis and transplant | 3199 | 19.0 |
| Total index transplant patients | 9854 | 58.4 |
| Total index dialysis patients | 6491 | 38.5 |
| Total CKD patients | 7037 | 41.7 |
CKD, Chronic kidney disease.
Data from the North American Pediatric Renal Trials and Collaborative Studies 2008 Annual Transplant Report. Available at https://web.emmes.com/study/ped/annlrept/annlrept.html (accessed June 26, 2011).
Pathophysiology: Implications of the Patient in Renal Failure
The kidneys perform a number of vital functions; both their dysfunction and the current methods of mechanical renal-replacement therapy have systemic implications. Because renal dysfunction creates profound disturbances in normal homeostasis, morbidity and mortality is increased in children with renal failure, especially the very young (Fig. 29-4).
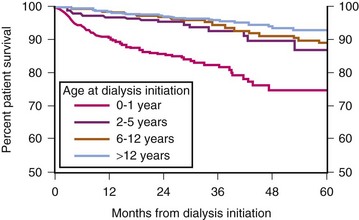
FIGURE 29-4 Patient survival while on dialysis.
(Exhibit 1.9 from The North American Pediatric Renal Trials and Collaborative Studies 2011 Annual Dialysis Report. Available at https://web.emmes.com/study/ped/annlrept/annlrept.html [accessed June 17, 2011].)
Disturbances in growth and development occur with renal failure, and depend on both the age of onset and the severity of the disease.112 Infants, in particular, are vulnerable to growth retardation caused by renal failure. Appropriate management of nutrition, with113 or without114 the provision of renal replacement therapy, is necessary to ameliorate this effect. Renal transplantation may provide for some “catch-up” growth, although this is limited to younger children; the child’s final height is a function of the height at the time of transplant, graft function, and the immunosuppression regimen.111,114,115 Growth hormone therapy can be used for children, both pretransplant and posttransplant, to manage height deficits. However, when therapy is initiated before severe growth retardation has occurred, growth hormone therapy is more effective.114,116,117,118 A possible complication of growth hormone replacement in transplant recipients has been scoliosis.119
In addition to the effects on growth, renal failure also effects intellectual and behavioral development. Children requiring renal-replacement therapy from infancy are particularly vulnerable. Nutritional invasive enteral support and renal replacement therapy, along with early transplantation, can lead to near-normal cognitive function.120 However, these children may have more problems with hyperactivity and conduct disorder. In addition, if these children are suffering from other comorbid conditions, they are very likely to have diminished IQ.121 In older children and adults with end-stage renal disease since childhood, age at diagnosis and increased duration of dialysis are risks for decreased mental capacity. Renal transplantation status did not appear to be protective from the risks of decreased mental capacity.122,123
Chronic renal failure is associated with cardiovascular changes. Between 20% and 33%116,124 of deaths in children on dialysis are cardiac in nature. Approximately 25% to 33%125,108 of deaths in children and young adults with end-stage renal disease whose disease began in childhood are from cardiac causes. The incidence of coronary artery calcifications and carotid intimal-medial thickening, two surrogate measures of coronary artery disease, are increased in dialysis-dependent children126 and in young adults with childhood-onset chronic renal failure.127 Hyperhomocysteinemia, associated with cardiovascular disease in adults, is common in children with chronic renal failure,128 as is dyslipidemia.129 Hypertension is frequent, and is a risk factor for deterioration of renal function.130,131 Left ventricular hypertrophy (LVH) is prevalent, both in children with chronic renal failure and in children on dialysis.132–134 LVH may be associated with diastolic dysfunction.135 Both LVH134 and diastolic dysfunction135 were noted to be more severe in children requiring dialysis.
In transplanted children, cardiac deaths constitute 15% of the causes of death.104 Renal transplantation does decrease the risk of cardiac death, compared to that of children in chronic renal failure or dialysis-dependent children.108,125 However, transplanted children have a persistence of pretransplant risk factors. LVH is still found in 50% to 75% of transplant recipients.136,137 Risk factors for LVH in the posttransplant patient include a history of dialysis, anemia, and hypertension.136 LVH can occur in renal transplant patients with normal 24-hour ambulatory blood pressure measurements.137 In addition to LVH, 10 of 73 posttransplant patients in one study136 demonstrated left ventricular dilation with systolic dysfunction. The incidence of coronary artery calcification in young adults who were transplanted as children is greater than in control patients,127,138 and may be associated with carotid intimal-medial thickening.127 Hyperhomocysteinemia is present in 63% to 77%138,139 of transplanted children, and its presence is associated with poor graft function.139 Folate supplementation may normalize homocysteine levels.140 Even with good graft function following transplantation, dyslipidemia persists and is generally related to immunosuppression medications.129 The incidence of diabetes requiring insulin treatment is 2.6%. Tacrolimus use and African American race are the two major risk factors for insulin requiring diabetes.141
Anemia is common both before and after transplantation. Anemia is caused by a decrease in erythropoietin production and iron deficiency. In posttransplant patients, CNIs, especially tacrolimus, are additional risk factors for anemia.142 In pediatric patients requiring dialysis for the first time, anemia was associated with a greater risk of death and prolonged hospitalization.109 Treatment of anemia has been shown to retard the progression of renal disease in adults.143 In children with chronic renal failure, a hematocrit less than 33% is a risk factor for progression to end-stage renal disease.112 Anemia may be associated with LVH in children132–136 and in adult patients with chronic renal disease; erythropoietin has been shown to improve LVH.144
Surgical Technique
The surgical techniques used in the pediatric recipient differ from those used in the adult and depend on the child’s size and underlying preexisting abnormalities, as well as the approach to the transplant (intraperitoneal or extraperitoneal). The extraperitoneal approach may be more technically difficult in smaller children. Removal of native tissue, either concurrently or (ideally) previously, occurred in approximately 22% of children registered in the NAPRTCS database.111 Preservation of urine-producing native kidneys may be desirable to prevent fluid overload during dialysis, especially for small children.145
Native nephrectomy may be required for polycystic kidney disease, uncontrollable hypertension, urinary tract infection, or nephrotic syndrome.145–148 Removal of polycystic kidneys may be necessary for space reasons, especially in small children. Children with severe vesicoureteral reflux and intractable urinary tract infections may also require native nephrectomy. Nephrectomy for severe nephrosis may be necessary to resolve associated hypoalbuminemia, malnutrition, and hypercoagulability. Ideally, native nephrectomy is performed before transplant.147–149 Native nephrectomy performed at the time of transplant increases operative time and cadaveric graft ischemic time,140 and has been shown to be a risk factor for ATN in the grafted organ.111
Children Weighing More Than 20 kg
In larger children (more than 20 kg), the surgical approach is similar to that in the adult transplant patient; the kidney is placed in the iliac fossa, and the vascular anastomoses are to the common iliac vein and artery.146,147 In larger children, the external iliac vessels may be used.147 An extraperitoneal approach has the advantage of increased ease of future graft biopsy and ability to resume peritoneal dialysis in case of delayed graft failure.145
Children Weighing Less Than 20 kg
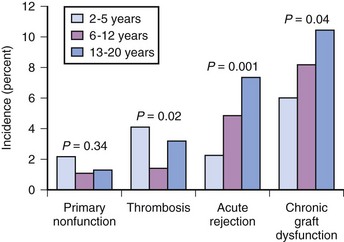
If renal transplantation in smaller children were restricted to size-compatible organs, then the surgical approach would be similar to that of the larger patient. However, a 1992 NAPRTCS report noted an increased loss of cadaveric grafts from pediatric donors. These pediatric donor organs had a greater rate of graft thrombosis and primary nonfunction, as well as a greater risk for acute rejection.150,151 Consequently, the use of pediatric donors has declined and living donors increased (Fig. 29-5). More importantly, the improvement in graft survival (Figs. 29-6 and 29-7) and patient survival (Fig. 29-8) with a living donor, although present across age-groups, is markedly greater in these young children.111
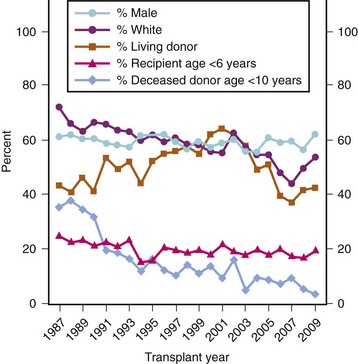
FIGURE 29-5 Patient registrations, transplants, and selected characteristics.
(Exhibit 1.1 from The North American Pediatric Renal Trials and Collaborative Studies 2010 Annual Transplant Report. Available at https://web.emmes.com/study/ped/annlrept/annlrept.html [accessed June 17, 2011].)
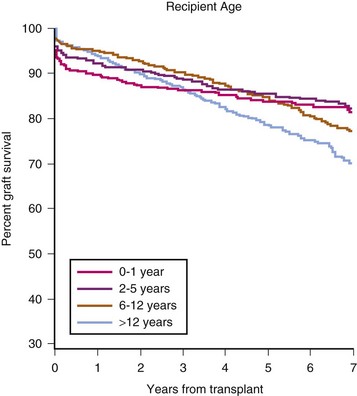
FIGURE 29-6 Living-donor graft survival by recipient age.
(Exhibit 5.4 from The North American Pediatric Renal Trials and Collaborative Studies 2010 Annual Transplant Report. Available at https://web.emmes.com/study/ped/annlrept/annlrept.html [accessed June 17, 2011].)
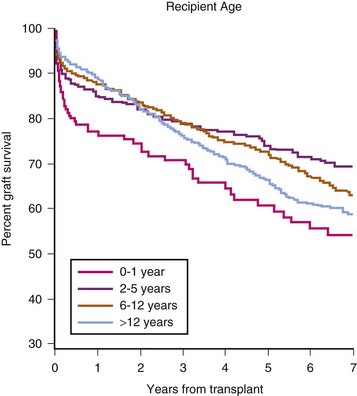
FIGURE 29-7 Deceased-donor graft survival by recipient age.
(Exhibit 5.5 from The North American Pediatric Renal Trials and Collaborative Studies 2010 Annual Transplant Report. Available at https://web.emmes.com/study/ped/annlrept/annlrept.html [accessed June 17, 2011].)
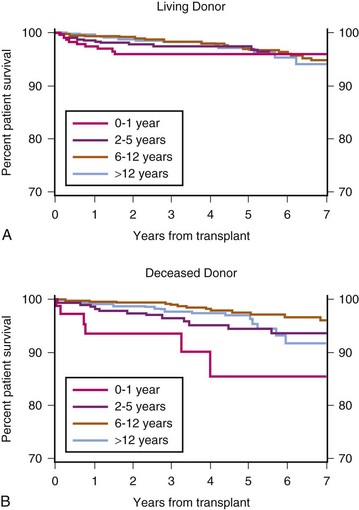
(Exhibit 7.6 from The North American Pediatric Renal Trials and Collaborative Studies 2010 Annual Transplant Report. Available at https://web.emmes.com/study/ped/annlrept/annlrept.html [accessed June 17, 2011].)
For these reasons, the surgical technique for renal transplantation in the infant and small child has been modified to accommodate the adult-sized kidney. The use of a size-discrepant organ precludes the traditional approach. As Starzl et al152 describe: “the adult organ almost completely fills a child’s right paravertebral gutter, extending from the undersurface of the liver to the pelvis.” Typically, a midline incision from pubis to xiphoid is used and the cecum and right colon are mobilized.146,149,152,153 An alternate approach used by some centers, is a right lower quadrant incision, dissecting extraperitoneally; 147,154 this may have the advantage of quicker return of bowel function. Depending on the size of the recipient’s vessels, the donor organ anastomoses may be made to the common iliac artery and vein, or directly to the aorta and vena cava.146,147,149,153
In addition to difficulties in surgical technique, adult organs in pediatric recipients present physiologic challenges. The adult-sized organ sequesters a disproportionate amount of the infant’s blood volume and cardiac output.145,146,155 In one study156 that involved nine infant recipients of adult-sized kidneys, pretransplant, early posttransplant, and late posttransplant blood flows were determined in both the infant’s aorta and the adult graft. Pretransplant graft flow was approximately 618 mL/min, whereas 8 to 12 days posttransplant, graft flow had decreased to 385 mL/min, despite aortic blood flow increasing from 331 mL/min to 761 mL/min. A further decrease was observed at 4 to 6 months, with aortic blood flow at 665 mL/min and graft flow at 296 mL/min. At 4 months the graft size had decreased by 26%. Even with excellent conditions, optimal hydration, and a doubling of aortic blood-flow, these adult grafts are still underperfused, both during transplant and in the late posttransplant period. In addition, the transplanted kidney is vulnerable to further reductions of flow resulting from gastrointestinal illness, poor oral intake, or hypotension during nontransplantation surgery.151 This may explain why early graft loss in these smaller children is either primary nonfunction or thrombosis (E-Fig. 29-3).157