Orbital Neoplasia
Ocular Lesions
Approximately two thirds of all retinoblastoma cases are unilateral, and one third are bilateral. Several classification systems have been developed for intraocular retinoblastoma, all of which are based on expected results of therapy and predicted salvage of the globe. The rapid evolution of newer retinoblastoma treatment has resulted in the replacement of the previously widely accepted Reese-Ellsworth classification, which is based on intraocular tumor staging and is used in tumor management after external beam radiation. A new International Classification of Retinoblastoma is based mainly on the natural history of retinoblastoma (early disease [group A] to late disease [group E]) and upon the extent of tumor seeding within the vitreous and subretinal space (Box 7-1). This classification is more applicable for patients treated with chemotherapy.
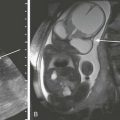
Leukocoria in a young child that is confirmed by an ophthalmoscopic examination requires further evaluation. Imaging may reliably distinguish retinoblastoma from a host of other conditions that also may present with leukocoria (e.g., persistent hyperplastic primary vitreous, Toxascaris infection, Coats disease, and medulloepithelioma). In many retinoblastoma centers, eye ultrasonography has replaced orbital CT for initial disease assessment. Three-dimensional high-frequency ultrasound is sufficient for assessment of tumor and calcifications but is less suitable for the evaluation of extraocular spread. Retinoblastoma is readily visible on ultrasound as an echogenic irregular retinal mass with focal acoustic shadows. CT is an excellent tool for depicting ocular calcifications (Fig. 7-1, A), but its use in retinoblastoma evaluation has markedly decreased because of the associated radiation risks. MRI has become a relatively quick and convenient modality for the evaluation of retinoblastoma. A variety of tailored MR sequences are beneficial in illustrating the different features of these masses. Specifically, intraocular hemorrhages and calcifications are depicted best on gradient sequences, whereas the malignant, markedly cellular nature of these tumors (composed of immature retinoblasts) is confirmed on T2-weighted and DWI sequences (Fig. 7-1, B and C). Contrast-enhanced sequences provide important information that may affect prognosis and treatment options with respect to tumor spread into the optic nerve (Fig. 7-1, D), anterior chamber, or other adjacent orbital structures. In addition, MRI serves as a surveillance examination of the brain when monitoring for possible leptomeningeal spread of tumor or for the presence of retinoblastoma in more remote locations.
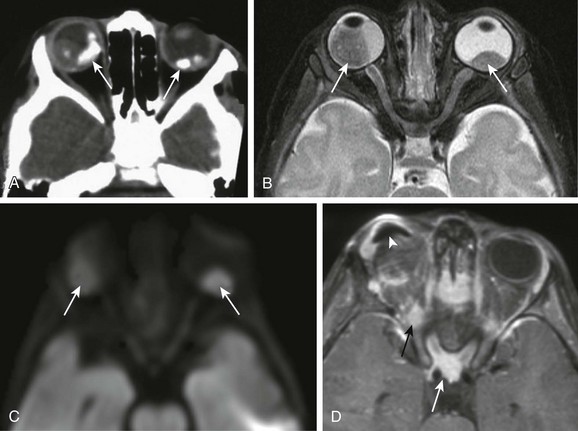
Figure 7-1 Retinoblastoma of the orbits in three different patients.
A, An orbital computed tomography scan demonstrates bilateral, markedly calcified orbital masses (arrows). Orbital magnetic resonance images (MRI), including an axial fat-saturated T2-weighted image (B) and a diffusion-weighted image (C), demonstrate bilateral, lobulated, T2-hypointense, retinal-based masses that exhibit diffusion restriction (arrows). D, In another patient with known recurrent retinoblastoma who has had right enucleation and prosthesis insertion (arrowhead), postcontrast T1-weighted fat-saturated MRI demonstrates fusiform enlargement and enhancement of the stump of the optic nerve sheath throughout its intraorbital (black arrow) and intracanalicular components, with associated posterior extension to the optic chiasm (white arrow).
Medulloepithelioma
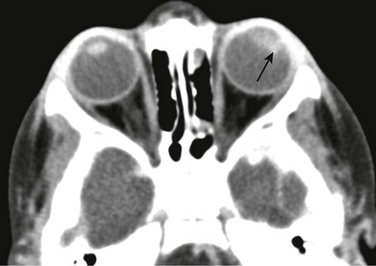
A medulloepithelioma is another pediatric malignant intraocular tumor that may present with leukocoria. This tumor is a rare embryonal type of neoplasm arising from the nonpigmented epithelial lining of the ciliary body. Patients usually are diagnosed in the first decade of life (at a mean age of 6 years); only rarely is medulloepithelioma seen in adults. Many imaging features of medulloepithelioma may closely resemble those of a retinoblastoma, such as presentation with a nodular enhancing intraocular mass, which occasionally will have calcifications (Fig. 7-2). A medulloepithelioma differs from a retinoblastoma mainly by its anterior location, but it may appear identical to a retinoblastoma when it is located in the vicinity of the optic nerve. Medulloepitheliomas have been divided into two types, teratoid and nonteratoid (diktyoma), based on their histologies. More complex teratoid medulloepitheliomas are composed of heteroplastic elements, including cartilage, which may have associated calcifications, whereas the nonteratoid diktyoma presents as a well-defined, noncalcified mass with associated diffuse contrast enhancement.
Hereditary Orbital Hamartomatosis
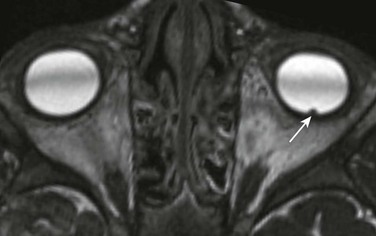
Ocular manifestations of tuberous sclerosis include astrocytic hamartomas of the retina and optic disk. These lesions have a typical appearance on ophthalmologic examination; however, they may calcify as the patient ages and may in fact resemble drusen when located on the optic disk. On thin-section high-resolution T2-weighted imaging, hamartomas of tuberous sclerosis are visible as small hypointense nodules within the posterior globe (e-Fig. 7-3).
Drusen
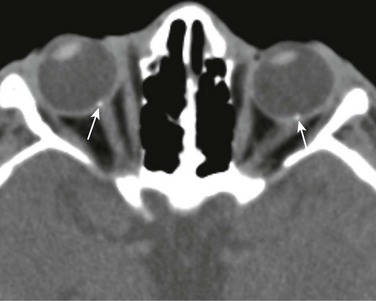
Disc drusen are nonhamartomatous subretinal lesions without astrocytic hyperplasia that are associated with the presence of intrapapillary, partially calcified hyaline bodies that form concretions of unknown nature. Drusen likely are the most common etiology for congenital bilateral elevation of the optic nerve discs. Drusen may be detected on funduscopic evaluation or may be seen as an incidental finding on imaging. In both scenarios, it is important to establish the benign nature of disk elevation so as not to confuse drusen (which cause pseudopapilledema) with true papilledema. Equivocal results of ophthalmoscopic examination may lead to orbital imaging, which will either confirm the presence of disc drusen or detect the source of the increased intracranial pressure. Drusen may be diagnosed with use of ultrasound, appearing as foci of increased echogenicity, or drusen can be seen on noncontrast CT most often as bilateral punctuate calcifications within the optic nerve heads (e-Fig. 7-4). MRI demonstrates isolated mild protrusion of the optic discs into the vitreous without perioptic cerebrospinal fluid space enlargement or other imaging features of papilledema. Clinically, drusen are usually asymptomatic and only rarely may be associated with slowly progressive visual loss.
Optic Nerve-Sheath Complex Tumors
Optic Nerve Glioma
Optic nerve glioma (ONG) is the most common primary neoplasm of the optic nerve in children. ONG may be seen in the setting of NF type 1 (Fig. 7-5, A) or may present as an isolated tumor (nonsyndromic) (Fig. 7-5, B). ONGs, which are associated with NF, most often are bilateral lesions that may involve the nerve and surrounding subarachnoid space and are remarkable for their low-grade nature and favorable prognosis. Nonsyndromic ONGs are usually unilateral and histologically are either pilocytic or fibrillary astrocytomas. The mortality rate for these tumors is approximately 5% when the tumor involves only the optic nerve, but hypothalamic involvement portends a more ominous prognosis; despite their benign histology, some series report mortality rates approaching 50%.
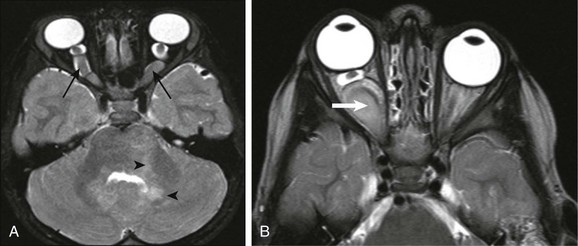
Figure 7-5 Optic glioma.
A, An optic glioma in a patient with neurofibromatosis (NF) type 1. An axial fat-saturated T2-weighted image of the orbits shows bilaterally symmetrically enlarged intracanalicular portions of the optic nerves associated with marked tortuosity (arrows), which commonly is seen in patients with NF type 1. An additional clue to the diagnosis is the areas of signal abnormality within the cerebellar white matter, in keeping with spongiform changes of NF type 1 (arrowheads). B, An isolated right optic nerve glioma in a different patient. Magnetic resonance imaging shows an optic nerve glioma with similar expansion and tortuosity of the optic nerve (arrow) but with unilateral involvement of the right optic nerve and without additional imaging findings of NF type 1.
Orbital Meningioma
Orbital meningioma is not a common pediatric tumor but has been reported in patients in the first decade of life. Meningiomas of the orbits may be perioptic (e-Fig. 7-6, A-C), arising from the optic nerve sheath, or may extend through the optic canal from an intracranial origin, that is, arising from the area of anterior clinoid process of the sphenoid bone or from the tuberculum sella, with extension anteriorly into the orbit (e-Fig. 7-6, D).
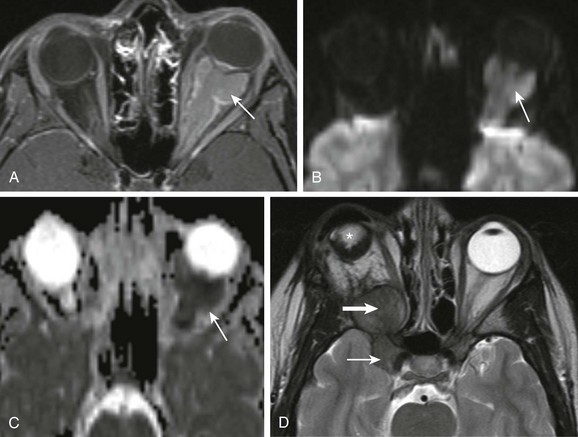
e-Figure 7-6 Optic meningioma.
A, Axial fat-saturated T1-weighted magnetic resonance imaging shows a pyramidal-shaped mass (optic meningioma) within the left orbit extending from the apex through to the posterior globe with demonstrable contrast enhancement that surrounds the optic nerve itself. B and C, A diffusion-weighted image (B) and an apparent diffusion coefficient image (C) show restricted diffusion. D, An axial T2-weighted image of another patient with a perioptic meningioma shows the mass extending posteriorly from the orbital apex on the right and into the adjacent cavernous sinus and pericarotid region (arrows). Note the orbital implant (star) from prior enucleation.
In general, CT is useful in the detection of the orbital portion of some meningiomas but not in differentiating whether the tumor originates from the optic nerve itself or from the optic nerve sheath. These tumors are usually hyperattenuating on CT, often with calcifications (but not in the early stages), seen as the so-called “rail road track sign,” which implies that the surrounding tumor of the nerve sheath is enhancing, not the optic nerve itself. Meningiomas ideally should be imaged with MRI. They most often appear hypointense on T1-weighted and T2-weighted sequences and exhibit avid gadolinium enhancement and restricted diffusion on DWI because of their high cellularity (e-Fig. 7-6, A-C).
Primitive Neuroectodermal Tumor
A PNET is a small, round, blue cell malignant tumor of neuroectodermal origin. PNETs that present outside of the central nervous system are referred to as peripheral PNET. It appears that PNET is the least aggressive subtype of tumor among other similar small cell tumors, with a favorable prognosis seen after complete tumor resection. This tumor, similar to Ewing sarcoma, expresses the MIC-2 gene (CD99) on cell membranes, which allows for their differentiation from other tumors. This rare tumor of the orbit presents as an enhancing, heterogeneous, T2-hypointense, intraconal lesion with associated restricted diffusion on DWI (Fig. 7-7, A-C). DWI characteristics are important imaging features that favor a highly cellular lesion, which includes PNET within the limited differential diagnosis. Treatment usually includes globe enucleation, high-dose chemotherapy, and stem cell transplantation.
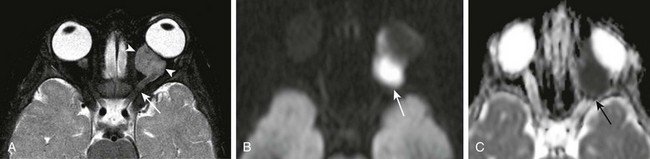
Figure 7-7 Primitive neuroectodermal tumor.
An axial fat-saturated T2-weighted image (A), an axial diffusion-weighted image (B), and an axial apparent diffusion coefficient image (C) of a patient with a left orbital primitive neuroectodermal tumor (PNET). Note the left-sided posteriorly located intraconal T2-hypointense lesion with associated marked restricted diffusion (arrows). The restricted diffusion suggests a densely cellular tumor, a finding almost invariably seen with PNETs.
Orbital Lesions
Orbital Hemangioma
These lobulated lesions often appear bright on T2-weighted imaging, avidly take up gadolinium, and may have multiple flow voids and increased vascularity seen on perfusion imaging (Fig. 7-8, A-C). Although usually nonsyndromic, some hemangiomas are known to occur in conjunction with other systemic abnormalities, such as PHACES (posterior fossa abnormalities, hemangiomas of the cervical facial region, arterial anomalies, cardiac defects, eye anomalies, and sternal defects) (e-Fig. 7-9).
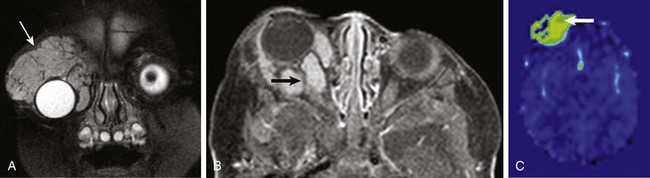
Figure 7-8 Hemangioma.
A coronal fat-saturated T2-weighted image (A), an axial postcontrast fat-saturated T1-weighted image (B), and an axial arterial spin labeling perfusion color map (C) from a patient with a right orbital region hemangioma. Note the fleshy mass surrounding the right globe in the supraorbital region (arrow in A) with associated flow voids, avid contrast enhancement (arrow in B), and increased blood flow (arrow in C).
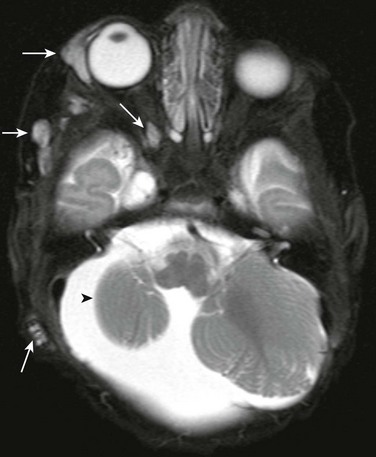
e-Figure 7-9 Multiple hemangiomata in a patient with PHACES syndrome (posterior fossa abnormalities, hemangiomas of the cervical facial region, arterial anomalies, cardiac defects, eye anomalies, and sternal defects).
An axial fat-saturated T2-weighted image of the orbit shows multiple areas of T2 prolongation within the soft tissues about the right orbit, temporal region, and mastoid region (arrows), in keeping with multiple hemangiomata. In addition, associated right cerebellar hypoplasia is present (arrowhead).
Venolymphatic Malformation
Imaging features may reflect the dual nature of these vascular malformations, with multicystic lymphatic components often containing blood-fluid levels and enhancing solid venous components, which may contain phleboliths. Uncomplicated lymphatic malformations usually do not demonstrate contrast enhancement, or at most may show some peripheral enhancement of the septations. On ultrasound, lymphangiomas exhibit no Doppler flow and demonstrate cystic spaces filled with blood and fluid. On CT their appearance may be inconclusive, not fully reflecting the internal complexity of the lesion (Fig. 7-10, A), but they can appear as a soft tissue mass with associated proptosis. MRI is capable of fully evaluating their true nature and distinguishing them from masses such as RMS lesions (Fig. 7-10, B).
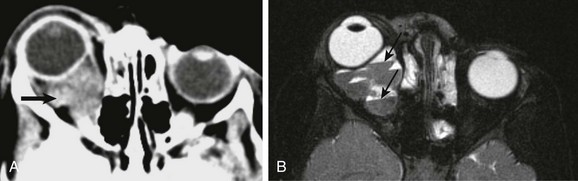
Figure 7-10 Venolymphatic malformation.
An axial contrast-enhanced computed tomography (CT) scan (A) and an axial fat-saturated T2-weighted magnetic resonance image (MRI) of the orbit (B) show a soft tissue intraconal mass posterior to the right globe (arrow in A), which is associated with anterior displacement of the globe. Although the finding on CT is nonspecific, the multiple fluid-fluid levels on MRI (arrows in B) are highly suggestive of the diagnosis.
Orbital Varix
Primary congenital venous varices (uncommonly affecting children) represent distensible venous malformations in the retrobulbar compartment of the orbit (e-Fig. 7-11), which may enlarge with dependent posture. The varix may consist of marked enlargement of a single orbital vein or may present as a tangled multivascular mass. Clinically these lesions may present with globe displacement that may augment and increase in conspicuity when the Valsalva maneuver is performed, but at times the lesions are collapsed and may be undetectable. Occasionally these lesions may lead to spontaneous orbital hemorrhage. Secondary varices may occur in the setting of intracranial dural venous thrombosis or arteriovenous shunting.
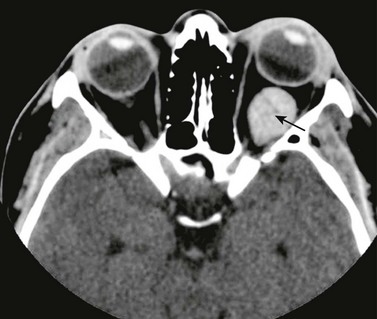
e-Figure 7-11 Venous varix.
An axial contrast-enhanced computed tomography scan of the orbit in a patient with left-sided venous varix shows tubular enhancement within the posterior half of the orbits (arrow) indicating a vascular lesion that is not specific for, but is suggestive of, a venous varix.
Nonvascular Lesions
Orbital Tumors
Orbital Teratoma: Orbital teratoma is a very rare congenital tumor that often is diagnosed prenatally with ultrasound and fetal MRI (Fig. 7-12). These tumors usually present as a large heterogeneous mass containing multiple tissue types, including fat, calcium, and bone. Teratomas of the orbit are most often benign and well differentiated; however, they may lead to significant proptosis when located at the orbital apex. The imaging appearance of a teratoma is quite distinctive, with the presence of a very large multicystic mass that includes a solid component and calcification. The solid components may exhibit enhancement after administration of a contrast agent. The enhancement pattern may help to distinguish a teratoma from a dermoid cyst, which, when present, tends to have peripheral enhancement.
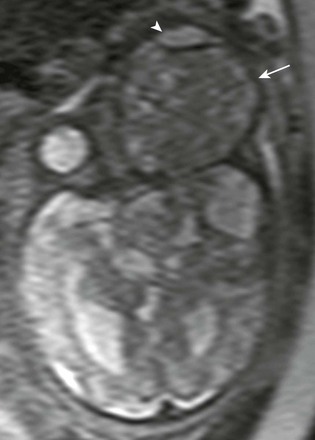
Figure 7-12 Fetal teratoma.
An axial half-Fourier acquisition single-shot turbo spin-echo magnetic resonance image of the orbital region shows a very large, slightly T2-hypointense lesion in the region of the left orbit (arrow), with associated marked anterior/ventral displacement of the globe (arrowhead) representing a teratoma.
Rhabdoid tumors of the orbital region occur in young children (with a mean age of 15 months at presentation) and can be highly vascular and mimic more benign lesions such as hemangiomas. These rare malignant tumors are highly aggressive and lethal (death occurs within 12 months of presentation). Their dense cellularity can produce restricted diffusion on DWI (e-Fig. 7-13, A and B).
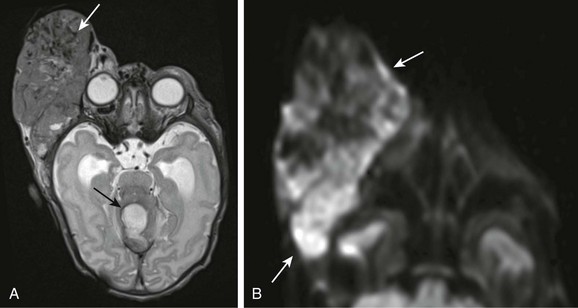
e-Figure 7-13 A congenital rhabdoid tumor.
An axial T2-weighted image (A) and an axial diffusion-weighted image (B) from a patient with a right-sided rhabdoid tumor show a lobulated, heterogeneous, extremely vascular mass along the right lateral orbit with numerous intratumoral flow voids indicating marked vascularity (white arrow in A). In addition, the lesion demonstrates restricted diffusion (arrows in B). An additional area of abnormality (evolving hematoma) is seen within the supracerebellar cistern (black arrow in A) as a result of bleeding diathesis, thought to be secondary to Kasabach-Merritt phenomenon.
Rhabdomyosarcoma: RMS is the most common malignant soft tissue tumor in childhood. The orbits and paranasal sinuses are the second most common location. The embryonal type is the most common variety of orbital RMS; the alveolar and pleomorphic varieties occur rarely. The mean age at presentation is 6 years.
Both CT and MR imaging play important roles in the preoperative evaluation, staging, and follow-up of orbital RMS tumors. On T2-weighted imaging, these lesions may appear as hypointense, isointense, and even hyperintense with respect to both extraocular muscles and orbital fat (Fig. 7-14, A and B). Because of the high degree of RMS cellularity, the lesions may be predominantly hypointense on T2-weighted imaging, hyperattenuating on CT, and exhibit restricted diffusion on DWI. On T1-weighted contrast-enhanced images, RMS will have moderate to marked enhancement, and in some cases a component of highly vascular internal tissue may mimic the appearance of a hemangioma. Meticulous attention is required in the assessment of local invasion because RMS may invade the paranasal sinuses, as well as for more remote invasion because RMS can spread to the intracranial space via the orbital fissures and then into cavernous sinuses and even the middle cranial fossa. Favorable prognostic factors include lack of distant metastases, a primary site in the orbit, disease confined to the orbit, gross total surgical resection, a patient age of younger than 10 years, an embryonal histologic type, hyperdiploid deoxyribonucleic acid content, and a tumor size of 5 cm or less. The most important prognostic factor is response to therapy, which is assessed with follow-up imaging.
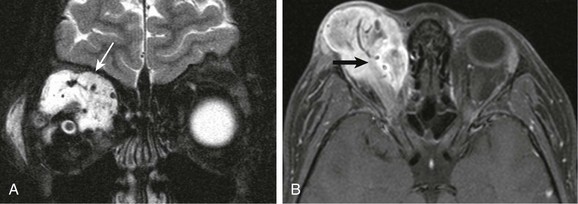
Figure 7-14 Rhabdomyosarcoma.
A coronal fat-saturated T2-weighted image (A) and an axial fat-saturated postcontrast T1-weighted image (B) in a patient with a right orbital rhabdomyosarcoma show a large, mainly extraconal lesion with flow voids seen on the T2-weighted image (white arrow) and with associated enhancement within the superior aspect of the right orbit (black arrow) indicative of the highly vascular nature of this tumor.
Lesions that Secondarily Involve the Orbit
Metastatic Lesions Involving the Orbits
Neuroblastoma
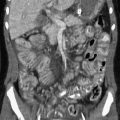
Neuroblastoma is the most common metastatic tumor of young pediatric patients (Figs. 7-15 and 7-16). In 8% of cases, neuroblastoma first presents with acute orbital symptoms related to tumoral hemorrhage, sudden proptosis, and ecchymosis (raccoon eyes). Neuroblastoma metastases to the head and neck favors the bony skull base and the cranial sutures, at times leading to sutural diastasis on conventional radiographs, with the additional extraaxial soft tissue masses only discernable on cross-sectional CT and MR imaging.
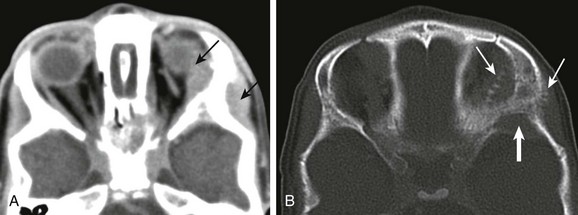
Figure 7-15 Neuroblastoma metastases.
Axial contrast-enhanced computed tomography scans of the orbits with soft tissue (A) and bone windows (B) in a patient with known metastatic neuroblastoma show enhancing soft tissue masses on either side of the lateral orbital wall (arrows in A). In addition, the bone surrounding the orbit has a moth-eaten appearance, which is a typical appearance of neuroblastoma that has metastasized to the orbits/skull base (arrows in B).
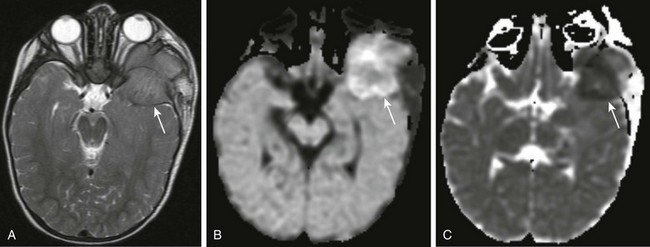
Figure 7-16 Neuroblastoma metastases.
Axial T2-weighted magnetic resonance imaging (A), axial diffusion-weighted imaging (DWI) (B), and axial apparent diffusion coefficient (ADC) imaging (C) of the orbit in a patient with known metastatic neuroblastoma. A large, nearly isointense mass is seen on the T2-weighted image at the level of the mid orbit; it is extraaxial in location and produces posterior displacement of the ipsilateral temporal lobe (arrow in A). Corresponding restricted diffusion (arrows) is identified on DWI (B) and ADC (C) images at the same level, indicating marked cellularity of the tumor.
Leukemia and Lymphoma
NHL (a tumor composed of B cells) often presents with extranodal disease. Most patients with orbital lymphoma also have systemic disease. The locations involved may include the lacrimal glands, anterior orbit, retrobulbar region, and the superior orbital compartment. CT may demonstrate a hyperattenuating mass with sharply defined margins. MRI findings include hypointense masses on T2-weighted imaging, with associated restricted diffusion on DWI (e-Fig. 7-17, A and B).
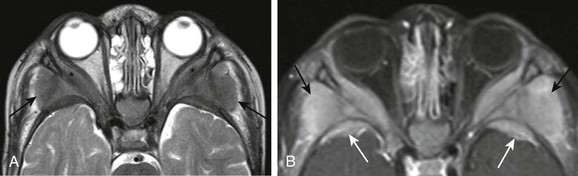
e-Figure 7-17 Acute myelogenous leukemia (AML).
An axial T2-weighted image (A) and an axial postcontrast T1-weighted image (B) with fat saturation in a patient with known AML show bilateral symmetric involvement of the lateral orbital walls by soft tissue masses from leukemic infiltration. Extraconal intraorbital soft tissue involvement is present (asterisks in A), along with osseous and extraosseous soft tissue masses along the posterior lateral margins of the lateral orbital walls (black arrows in A). In addition, note the bilateral osseous and periosteal masses along the anterior margins of the middle cranial fossa (white arrows in B), with associated posterior displacement of the temporal lobes.
Dermoid and Epidermoid Cysts
Dermoid and epidermoid cysts may occur anywhere within the orbit, but most reside in the superolateral aspect of the orbit, at the frontozygomatic suture, or in a superonasal position at the frontolacrimal suture. Osseous remodeling may occur but without associated periosteal reaction. These lesions are well-demarcated, cystic-appearing extraconal masses with differing degrees of fatty content. Uncomplicated superficial dermoid cysts are well depicted on orbital CT without the need for IV contrast because these lesions should not enhance (Fig. 7-18, A). MRI is superior for more detailed depiction of possible intracranial connection and the presence of a potential sinus tract. These lesions are hyperintense on T1-weighted imaging because of their fatty content. Mild rim enhancement occasionally may be seen and is considered normal, whereas the appearance of a more irregular type of enhancement is suggestive of prior rupture with a secondary inflammatory reaction. Epidermoid cysts are hyperintense on fluid attenuated inversion recovery sequences and exhibit restricted diffusion on DWI (Fig. 7-18, B and C).
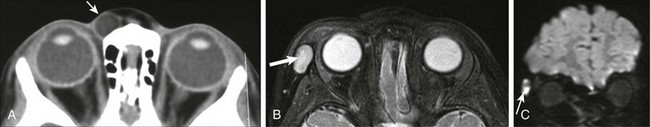
Figure 7-18 Epidermoid.
A, An axial noncontrast computed tomography scan shows an ovoid area of low-attenuation (fat) within the anterior soft tissues medial to the right globe (arrow). In another patient, an axial fat-saturated T2-weighted image (B) and coronal diffusion-weighted image (C) show a similar lesion along the right lateral orbital soft tissues, which is bright on the T2-weighted fat-saturated axial image (arrow in B) but exhibits restricted diffusion on the coronal diffusion-weighted image (arrow in C), as typically is seen with epidermoid cysts.
Langerhans Cell Histiocytosis
LCH is a granulomatous disease resulting from proliferation and infiltration of abnormal histiocytes within various tissues. Orbital lesions occur mainly in young children ages 1 to 4 years. Orbital disease may accompany widespread disease, but occasionally it may be the first manifestation of LCH. The lesions arise from the orbital bone or bone marrow and spread directly to the orbit. Orbital disease may present clinically with proptosis and rapidly enlarging periorbital masses. CT demonstrates expansile soft tissue masses associated with smoothly marginated (“punched out”) areas of osseous destruction in the posterolateral orbits, with a predilection for the frontosphenoid sutures (e-Fig. 7-19, A). MR imaging demonstrates periorbital heterogeneous lesions that may demonstrate blood-fluid levels and restricted diffusion (e-Fig. 7-19, B). The masses exhibit diffuse enhancement on CT and MRI. Evaluation for intracranial extension is imperative on postcontrast sequences (e-Fig 7-19, C). Discovery of a solitary orbital LCH lesion necessitates additional examination for other possible sites of involvement. Multiple soft tissue masses may resemble neuroblastoma metastases by certain imaging features; that is, both lesions may demonstrate restricted diffusion, diffuse enhancement, and intraosseous and extraosseous components. The type of osseous involvement may help to distinguish the “clear-cut” margins of LCH from the permeative pattern and aggressive periosteal reaction of a neuroblastoma. Orbital LCH also can simulate the imaging appearance of an RMS tumor with bone invasion, although bone destruction with LCH typically is more pronounced.
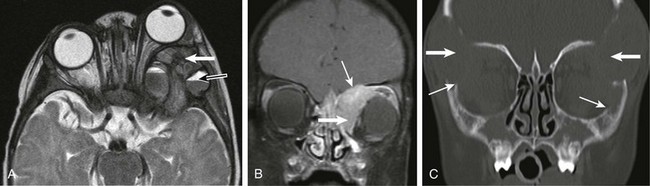
e-Figure 7-19 Langerhans cell histiocytosis (LCH).
An axial T2-weighted image (A), a coronal postcontrast fat-saturated T1-weighted image (B), and coronal bone windows from computed tomography (CT) (C) show the orbit in three different patients with known LCH. Note the marked proptosis on the axial T2-weighted image, associated with a posterior bony lesion along the greater wing of the sphenoid (white arrow in A) and with associated fluid-fluid levels (black arrow in A). Although these findings are more common in aneurysmal bone cysts and telangiectatic osteosarcomas, they also are seen in lesions of LCH. Lesions of LCH avidly enhance (thick arrow in B) with a propensity for invasion of the skull base and thickening and enhancement of the adjacent dura (thin arrow in B). Bone lesions on CT often are large and bilateral with an ominous appearance (thick arrows in C). However, the bony margins often have a sharp appearance that appears to be an attempt at maintaining the integrity of the bony margins (thin arrows in C).
Orbital Pseudotumor (Idiopathic Orbital Inflammation)
An orbital pseudotumor, which is a condition of unknown etiology, represents a noninfectious infiltration of the orbital structures by lymphocytes and plasma cells, usually without associated osseous involvement. The disease may present as unilateral, isolated involvement of the extraocular muscles but also may manifest with a more diffuse pattern that involves multiple orbital sites. It also can present with neuritis or an extrabulbar mass. CT and MRI are equally effective in depicting the extent of disease (Fig. 7-20).
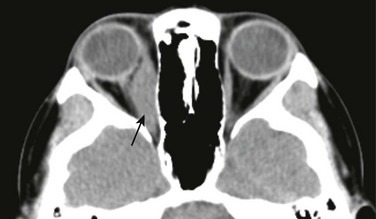
Figure 7-20 Inflammatory orbital pseudotumor.
A contrast-enhanced computed tomography scan of the orbit in a patient with a known right-sided inflammatory orbital pseudotumor. Note the fusiform enhancement and enlargement of the right medial rectus muscle (arrow) with extension to the myotendinous junction.
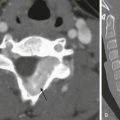
Osteoma
Osteomas within the orbits demonstrate typical imaging features of a broad-based expansile osseous mass, sometimes with an inner ground-glass lucent area, surrounded by dense compact bone. Osteomas usually do not cause bony destruction, even when the lesions are large in size. CT is the best imaging modality to delineate the lesion’s characteristics and extension (e-Fig. 7-21). MRI findings may be confusing, because the mature dense bone of the osteoma (and its absence of protons), composing most of the lesion’s volume, may not be distinguishable from the air in the paranasal sinuses.
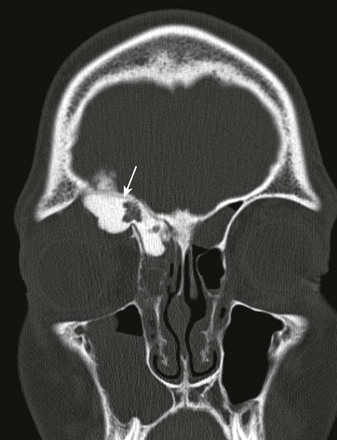
e-Figure 7-21 Osteoma.
A coronal noncontrast computed tomography scan of the orbit and sinus region in a patient with a known right frontal sinus osteoma shows a markedly dense lesion emanating from the right frontal sinus, similar in attenuation to that of enamel and dense bone (arrow), in keeping with a right frontal osteoma, with extension into the right orbit via a breach of the right orbital roof.
Bilaniuk, LT. Vascular lesions of the orbit in children. Neuroimaging Clin North Am. 2005;15:107–120.
Brennan, RC, Wilson, MW, Kaste, S, et al. US and MRI of pediatric ocular masses with histopathological correlation. Pediatr Radiol. 2012;42:738–749.
Chung, EM, Smirniotopoulos, JC, Specht, CS, et al. Pediatric orbit tumors and tumor like lesions: nonosseous lesions of the extraocular orbit. RadioGraphics. 2007;27:1777–1799.
Razek, A, Elkhamary, S, Mousa, A. Differentiation between benign and malignant orbital tumors at 3T diffusion MR imaging. Neuroradiology. 2011;53:517–522.
Rootman, J. Distribution and differential diagnosis of orbital disease. In Diseases of the orbit: a multidisciplinary approach, 2nd ed, Philadelphia: Lippincott Williams & Wilkins; 2003:52–84.